Embed Size (px)
Citation preview

HETEROCICLOS
Un compuesto heterocíclico es una sustancia que contiene un anillo formado por más de un tipo de átomo. Existen compuestos monocíclicos, bicíclicos y mayores, todos ellos con gran interés para los químicos y bioquímicos.
Pirrol Furano Tiofeno Imidazol Oxazol Tiazol
Pirazol 3-Pirrolina Pirrolidina Piridina Pirimidina Purina
Muchos compuestos naturales presentan en su estructura anillos tipo heterocíclicos, como los que mostramos a continuación:
Cocaína, estimulante, anestésico local; se encuentra en las hojas de la coca.
Dietilamina del ácido lisérgico (LSD)psicomimético
Nicotina, se encuentra en las hojas secas del tabaco entre el 2% y el 8%.
Penicilina G,antibiótico
Como puede verse los compuestos heterocíclicos aparecen en varias moléculas de interés biológico: son heterociclos los carbohidratos, como también la clorofila y hemina, que dan el color verde a las hojas y rojo a la sangre, dándole vida a las plantas y animales. Los sitios reactivos de muchas enzimas y coenzimas son heterociclos. La herencia tiene su asiento, por último, en la secuencia de unión específica de media docena de anillos heterocíclicos a largas cadenas de ácidos nucleicos. NOMENCLATURA DE LOS HETEROCICLOS: Muchos de los heterociclos tienen nombres comunes. Además, hay varias formas alternativas de nombrar a los heterociclos que requieren memorización, no son de aplicación universal y, a veces se prestan a confusión. Para compuestos monocíclicos sencillos se utiliza prefijos para indicar la presencia y la identidad del heteroátomo: aza- para nitrógeno, oxa- para oxígeno, tio- para azufre, fosfa- para fósforo y así sucesivamente.

Oxaciclobutano Azaciclopentano Oxaciclohexano Tiociclohexano 3-metiloxaciclohexano
A continuación resumiremos las formas de preparación de los heterociclos, en forma de tabla con un respectivo ejemplo, al final de la tabla realizaremos un breve comentario de las reacciones más significativas, dado que muchas de las utilizadas ya son conocidas por el estudiante en capítulos anteriores.
PREPARACIÓN DE HETEROCICLOSREACCIÓN EJEMPLO
1.- SÍNTESIS DE AZACICLOPROPANOS
2.- SÍNTESIS DE OXACICLOPROPANOS (REACCIÓN DE EPOXIDACIÓN)
3.- SÍNTESIS DE TIOCICLOPROPANOS
4.- SÍNTESIS DE HETEROCICLOBUTANOS VÍA UNA SN2 INTRAMOLECULAR.
5.- SÍNTESIS DE HETEROCICLOPENTANOS VÍA UNA SN2 INTRAMOLECULAR

6.- SÍNTESIS DE HETEROCICLOPENTANOS POR HIDROGENACIÓN CATALITICA

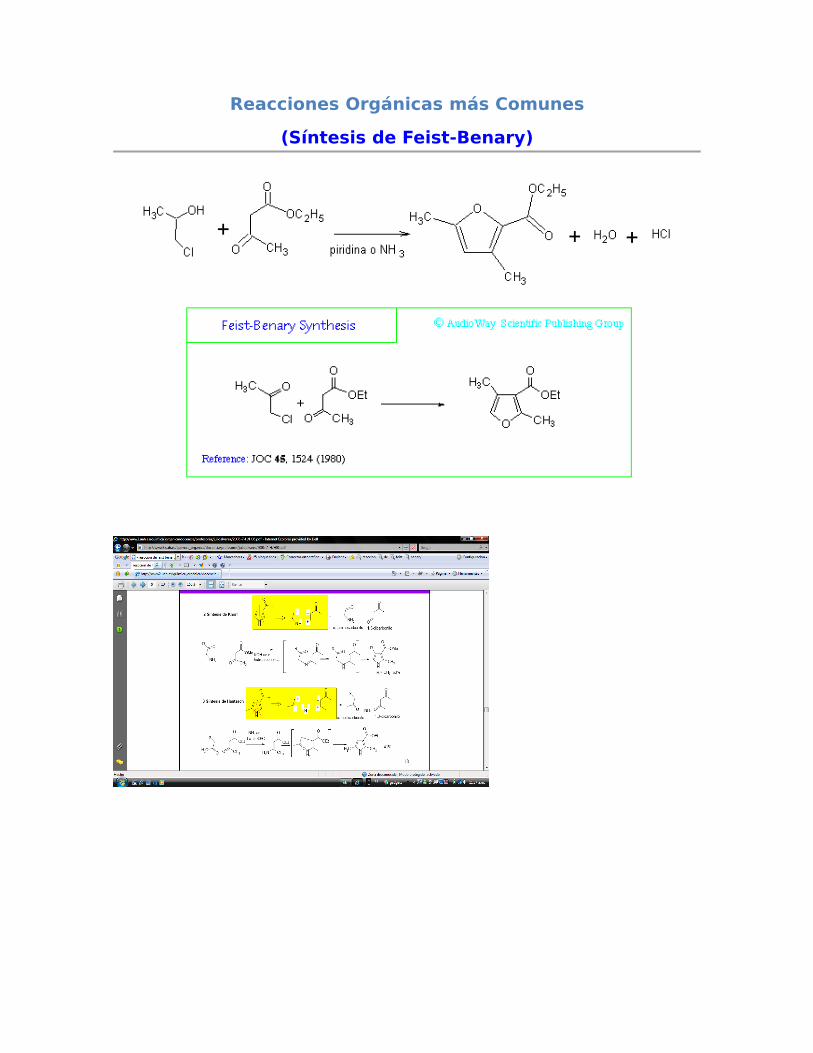
7.- SÍNTESIS DE PAAL-KNORR DE 1-HETERO- 2,4-CICLOPENTADIENOS
8.- SÍNTESIS DE HANTZSCH DE PIRIDINAS
9.- SÍNTESIS DE FISCHER DE INDOLES
10.- SÍNTESIS DE FRIEDLÄNDER DE QUINOLINAS
11.- SÍNTESIS DE BISCHLER-NAPIERALSKI DE ISOQUINOLINAS
RUTAS PARA LA PREPARACIÓN DE HETEROCICLOPROPANOS: Los azaciclopropanos pueden prepararse por adición directa de nitrenos, análogos nitrogenados de los carbenos, a alquenos. Por ejemplo la irradiación o calefacción del

azidocarboxilato de etilo se genera un nitreno que es atrapado por un alqueno para dar el azaciclopropano correspondiente con rendimientos moderados.
Un nitreno
Para el caso de la preparación de oxaciclopropanos, el método más generalizado es la epoxidación de alquenos, ya comentada con anterioridad en este libro (capítulo 13 y 14). Los tiociclopropanos se sintetizan a partir de los oxaciclopropanos correspondientes, que son fácilmente obtenibles. Un reactivo que permite la conversión directa de uno a otro es el tiocianato potásico. La transformación es estereoespecífica y transcurre con inversión en los dos carbonos que reaccionan. El mecanismo de esta reacción no será comentado en este capítulo. Para la obtención de anillos de cuatro y cinco eslabones, el método más simple entre otros es mediante una sustitución nucleofílica intramolecular (síntesis de Williamson) o por hidrogenación de los correspondientes sistemas insaturados. SÍNTESIS DE PAAL[1]-KNORR[2] DE 1-HETERO-2,4-CICLOPENTADIENOS: Un método general para éste tipo de anillos es la síntesis de Paal-Knorr. La molécula deseada se obtiene a partir de un compuesto γ-carbonílico enolizable por tratamiento con una amina (para pirroles), o P2O5 (para furanos) o P2S5 (para tiofenos). Formalmente el proceso puede considerarse como una deshidratación de un doble enol intermedio (o su equivalente con nitrógeno o azufre) para dar el heterociclo.Ejemplo:
SÍNTESIS DE HANTZSCH[3] DE PIRIDINAS: Las piridinas pueden obtenerse por condensación de productos de partida acíclicos tales como compuestos carbonílicos con amoniaco. El método más general es la síntesis de piridinas de Hantzsch. En esta reacción se combinan en varias etapas dos moléculas de un

compuesto β-dicarbonílico, un aldehído y amoniaco para dar una dihidropiridina sustituida, que se oxida fácilmente con ácido nítrico para dar el sistema aromático. Mecanismo de la síntesis de Hantzsch de piridinas: (1) Condensación de Knoevenagel del aldehído con el 3-cetoéster
Enolización del carbono α, del acetoacetato de etilo (abstracción de H), y condensación con el aldehído, con posterior deshidratación para la formación del sistema α,β-insaturado.
(2) Formación de la enamina del 3-cetoéster con amoniaco
Ataque nucleofílico del amoniaco, al carbonilo del grupo ceto, con formación de un β-hidroxi-éster, quien se deshidrata para dar la formación del sistema α,β-insaturado.
(3) Adición de Michael. Consiste en una adición de Michael vía enamina, para dar el producto de adición 1,4 sobre el producto de condensación de Knoevenagel, con posterior desplazamiento de un protón para dar como producto final una cetoenamina.
(4) Condensación intramolecular de la cetoenamina y tautomerización. Es una ciclación
intramolecular inducida por el par de electrones de la enamina, con posterior tautomerización hacia el sistema 1.4-dihidro que es mucho más estable.
(5) Oxidación y descarboxilación

El compuesto obtenido en la etapa anterior se oxida fácilmente con ácido nítrico para dar el sistema aromático. El sistema resultante es un éster 3,5-piridindicarboxílico. La hidrólisis seguida de pirólisis de la sal de calcio del ácido provoca la descarboxilación.
SÍNTESIS DE FISCHER[4] DE INDOLES: De entre los anillos de benceno condensado con otros anillos, el indol es, probablemente, el más importante ya que forma parte de muchos productos naturales. El indol está relacionado con el pirrol de la misma forma que el naftaleno está relacionado con el benceno. La forma más general de obtener indoles es la síntesis de indoles de Fischer. En esta síntesis, la arilhidrazona de un aldehído o cetona se trata con ácido polifosfórico (PPA) o un ácido mineral, o un ácido de Lewis; ello causa la extrusión de amoniaco con cierre simultáneo del anillo para dar el heterociclo deseado. Ejemplo:
El mecanismo de la síntesis de indoles de Fischer se cree que empieza con la tautomerización catalizada por ácido de la arilhidrazona, desde la forma imina a la forma enamina, tal como se ilustra a continuación.
SÍNTESIS DE BISCHLER[5]-NAPIERALSKI[6] DE ISOQUINOLINAS: Un método importante para sintetizar derivados de isoquinolina es la síntesis de Bischler-Napieralski, la cual consiste en la ciclación de derivados acilados de β-feniletilamina por tratamiento con ácidos (a menudo P2O5), lo que genera dihidroisoquinolinas que pueden aromatizarse. La síntesis en si es una ciclación intramolecular de tipo Friedel-Crafts. Ejemplo:
Mecanismo de la síntesis de Bischler-Napieralski:
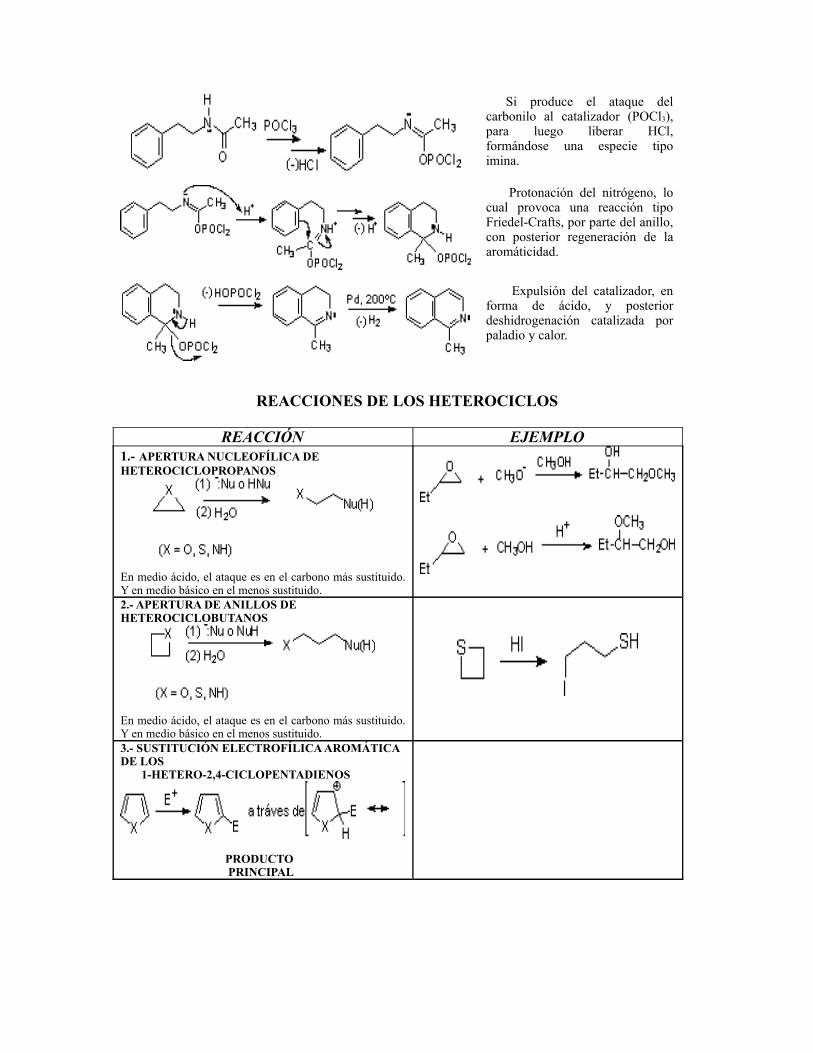
Si produce el ataque del carbonilo al catalizador (POCl3), para luego liberar HCl, formándose una especie tipo imina.
Protonación del nitrógeno, lo cual provoca una reacción tipo Friedel-Crafts, por parte del anillo, con posterior regeneración de la aromáticidad.
Expulsión del catalizador, en forma de ácido, y posterior deshidrogenación catalizada por paladio y calor.
REACCIONES DE LOS HETEROCICLOS
REACCIÓN EJEMPLO
1.- APERTURA NUCLEOFÍLICA DE HETEROCICLOPROPANOS
En medio ácido, el ataque es en el carbono más sustituido. Y en medio básico en el menos sustituido.2.- APERTURA DE ANILLOS DE HETEROCICLOBUTANOS
En medio ácido, el ataque es en el carbono más sustituido. Y en medio básico en el menos sustituido.
3.- SUSTITUCIÓN ELECTROFÍLICA AROMÁTICA DE LOS 1-HETERO-2,4-CICLOPENTADIENOS
PRODUCTO PRINCIPAL

LAS REACCIONES MÁS FRECUENTES SON:BROMACIÓNCLORACIÓNNITRACIÓNACILACIÓN DE FRIEDEL-CRAFTS
4.- SUSTITUCIÓN ELECTROFÍLICA AROMÁTICA EN EL ANILLO DE PIRIDINA
PRODUCTO PRINCIPAL LAS REACCIONES MÁS FRECUENTES SON:NITRACIÓNSULFONACIÓNBROMACIÓNALQUILACIÓN Y ACILACIÓN NO OCURREN CON LA PIRIDINA.
5.- SUSTITUCIÓN NUCLEOFÍLICA AROMÁTICA EN EL ANILLO DE PIRIDINA
(A) REACCIÓN DE CHICHIBABIN
(B) CON REACTIVOS ORGANOMETÁLICOS (R-LI)
(C) SUSTITUCIÓN NUCLEOFÍLICA DE HALOPIRIDINAS

6.- REACCIONES DE LA QUINOLINA E ISOQUINOLINA (A) SUSTITUCIÓN ELECTROFÍLICA AROMÁTICA
(B) SUSTITUCIÓN NUCLEOFÍLICA AROMÁTICA
REACCIÓN DE NITRACIÓN
REACCIÓN DE CHICHIBABIN
APERTURA DE ANILLOS TIPO HETEROCÍCLICOS DE TRES O MÁS ESLABONES: Los heterociclopropanos son relativamente reactivos ya que la tensión anular se libera en las aperturas nucleófilas del anillo. Recuérdese que en condiciones básicas la reacción tiene lugar sobre el centro menos sustituido, y en condiciones ácidas la reacción tiene lugar en el centro más sustituido (ver apertura de epóxidos, capítulo 13).

La reactividad de los heterocicloalcanos con anillos de cuatro o cinco eslabones está de acuerdo con lo esperado en base a la tensión del anillo: sólo los anillos tensionados son reactivos, y sus reacciones dan lugar a la apertura del anillo. En cualquier caso la reacción puede considerarse una sustitución nucleofílica. SUSTITUCIÓN ELECTROFÍLICA AROMÁTICA DE LOS 1-HETERO-2,4-CICLOPENTADIENOS. El pirrol, furano y tiofeno forman productos de sustitución electrofílica porque son aromáticos. Cada uno tiene seis electrones π en un sistema conjugado cíclico. La química de estos compuestos es similar a la de los anillos aromáticos bencenoides activados. Al igual que el benceno, los heterociclos aromáticos de cinco miembros experimentan reacciones de sustitución electrofílica en lugar de reacciones de adición. Si se eligen las condiciones de reacción apropiadas, pueden efectuarse halogenaciones, nitraciones, sulfonaciones y acilaciones de friedel-Cratfs
CONSECUENCIAS DEL ATAQUE ELECTRÓFILO EN C2 Y C3 DE HETEROCICLOPENTADIENOS AROMÁTICOS:
En donde X = O, N, S
La sustitución electrofilica de estos anillos aromáticos suele ocurrir preferentemente en el carbono-2, la posición siguiente al heteroátomo, debido a que es la más rica en electrones (más nucleófila) del anillo. En otras palabras, nótese que el ataque electrofílico en C2 forma un intermedio catiónico que es más estable (con más formas resonantes) que el ataque en C3, que forma un catión con sólo dos formas resonantes. Normalmente se encuentra que orden de reactividad es furano > pirrol > tiofeno >> benceno. Ejemplos:

2-NITROPIRROL (83%) 2-CLOROFURANO (65%) 2-ETANOIL-5-METILTIOFENO (70%)
SUSTITUCIÓN ELECTROFÍLICA AROMÁTICA EN LA PIRIDINA: La piridina experimenta la sustitución electrófilica muy lentamente. Reacciona forma muy similar a un anillo bencénico desactivado. Además, la sustitución esperada para la sustitución electrófila sobre el anillo de la piridina. La halogenación y la sulfonación pueden efectuarse en condiciones drásticas, pero la nitración tiene muy bajo rendimiento, y las reacciones de Friedel-Crafts son fallidas; por lo que regular se forma el producto en la posición 3. La baja reactividad de la piridina hacia las sustituciones electrofílicas aromáticas se debe a una combinación de factores. El más importante es el hecho de que la densidad electrónica del anillo se encuentra disminuida, por el efecto inductivo atrayente de electrones del átomo de nitrógeno, electronegativo. Un segundo factor que reduce la reactividad del anillo de piridina hacia el ataque electrofílico es el hecho de que la formación de complejos ácido-base entre el átomo de nitrógeno del anillo (básico) y el electófilo atacante coloca una carga positiva en el anillo y por tanto lo desactiva aún más.
Sustitución electrofílica en posición-2(La tercera estructura es muy inestable, contribuye poco a la estabilidad) Sustitución electrofílica en posición-3 Sustitución electrofílica en posición-4(La segunda estructura es muy inestable, idems a la posición 2)
La desactivación con respecto al benceno puede atribuirse parcialmente al efecto inductivo atrayente de electrones del átomo de nitrógeno que desestabiliza el catión intermedio. De las tres estructuras resonantes que pueden dibujarse para cada posición del ataque electrófilo, tanto la sustitución en la posición 2 como en la posición 4 implican una forma con contribución muy escasa. (las cuales en donde la carga positiva se encuentra sobre el átomo de nitrógeno, sin que éste tenga su octeto electrónico completo). La posición
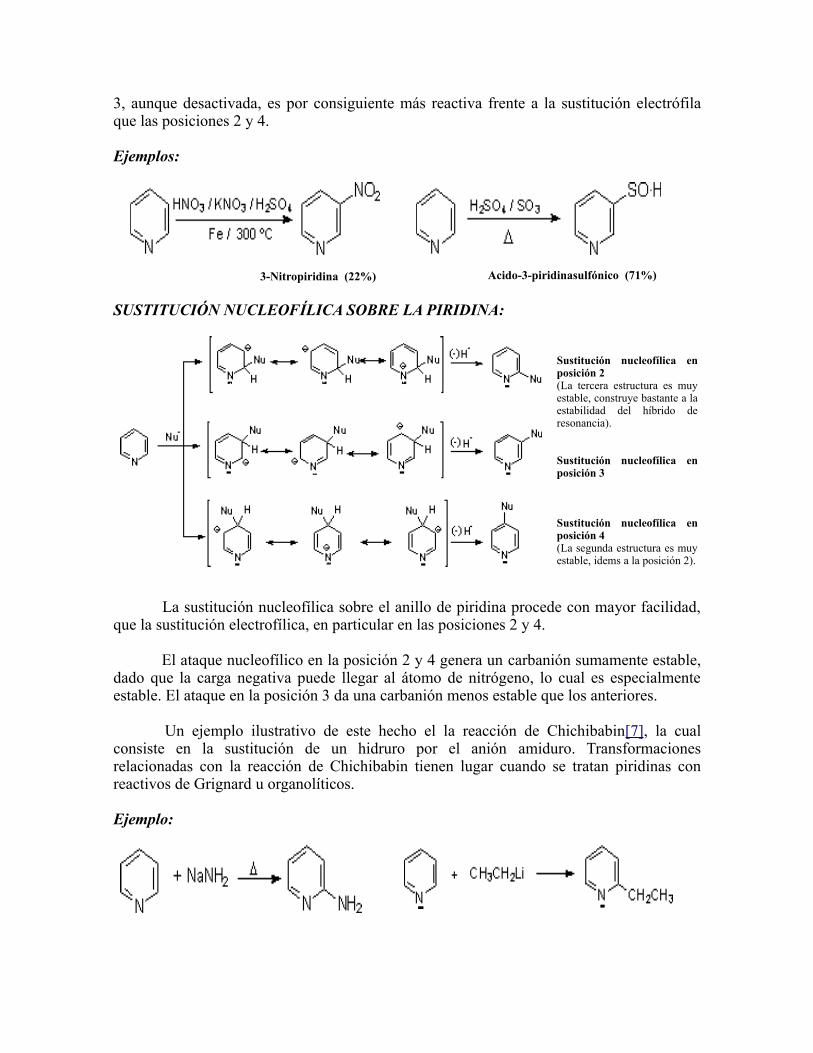
3, aunque desactivada, es por consiguiente más reactiva frente a la sustitución electrófila que las posiciones 2 y 4. Ejemplos:
3-Nitropiridina (22%) Acido-3-piridinasulfónico (71%)
SUSTITUCIÓN NUCLEOFÍLICA SOBRE LA PIRIDINA:
Sustitución nucleofílica en posición 2(La tercera estructura es muy estable, construye bastante a la estabilidad del híbrido de resonancia). Sustitución nucleofílica en posición 3 Sustitución nucleofílica en posición 4(La segunda estructura es muy estable, idems a la posición 2).
La sustitución nucleofílica sobre el anillo de piridina procede con mayor facilidad, que la sustitución electrofílica, en particular en las posiciones 2 y 4. El ataque nucleofílico en la posición 2 y 4 genera un carbanión sumamente estable, dado que la carga negativa puede llegar al átomo de nitrógeno, lo cual es especialmente estable. El ataque en la posición 3 da una carbanión menos estable que los anteriores. Un ejemplo ilustrativo de este hecho el la reacción de Chichibabin[7], la cual consiste en la sustitución de un hidruro por el anión amiduro. Transformaciones relacionadas con la reacción de Chichibabin tienen lugar cuando se tratan piridinas con reactivos de Grignard u organolíticos. Ejemplo:

Los átomos de hidrógenos en α de una cadena lateral alquílica en las posiciones 2 o 4 de la piridina tienen una acidez similar a los de una cetona. La reacción de un grupo carbonilo con el anión alquilo formado mediante una base constituye un método para la extensión de la cadena lateral de la piridina. Ejemplo:
Como otras aminas, también la piridina tiene propiedades nucleófilas: reacciona con halogenuros de alquilo, formando sales de amonio cuaternarias: Ejemplo:
Como cualquiera otra amina terciaria, puede convertirse a la piridina en su N-óxido, mediante ácido peroxibenzoico o peróxido de hidrógeno. En contraposición a la piridina misma, su óxido sufre la nitración principalmente en la posición 4. Lo cuál se puede explicar por las estructuras resonantes del N-óxido. Ejemplos:
El N-óxido puede utilizarse a menudo como una forma “activada” de la piridina. Por tratamiento del N-óxido sustituido con PCl3 se elimina el oxígeno. Ejemplos:
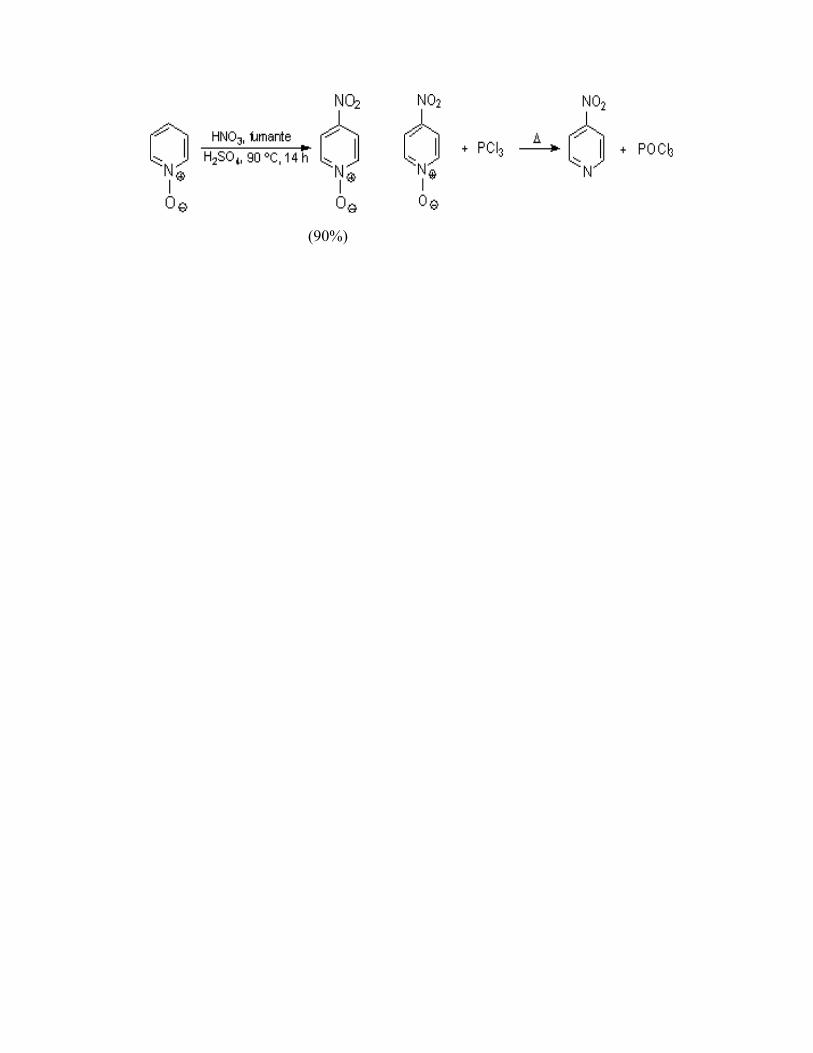
(90%)
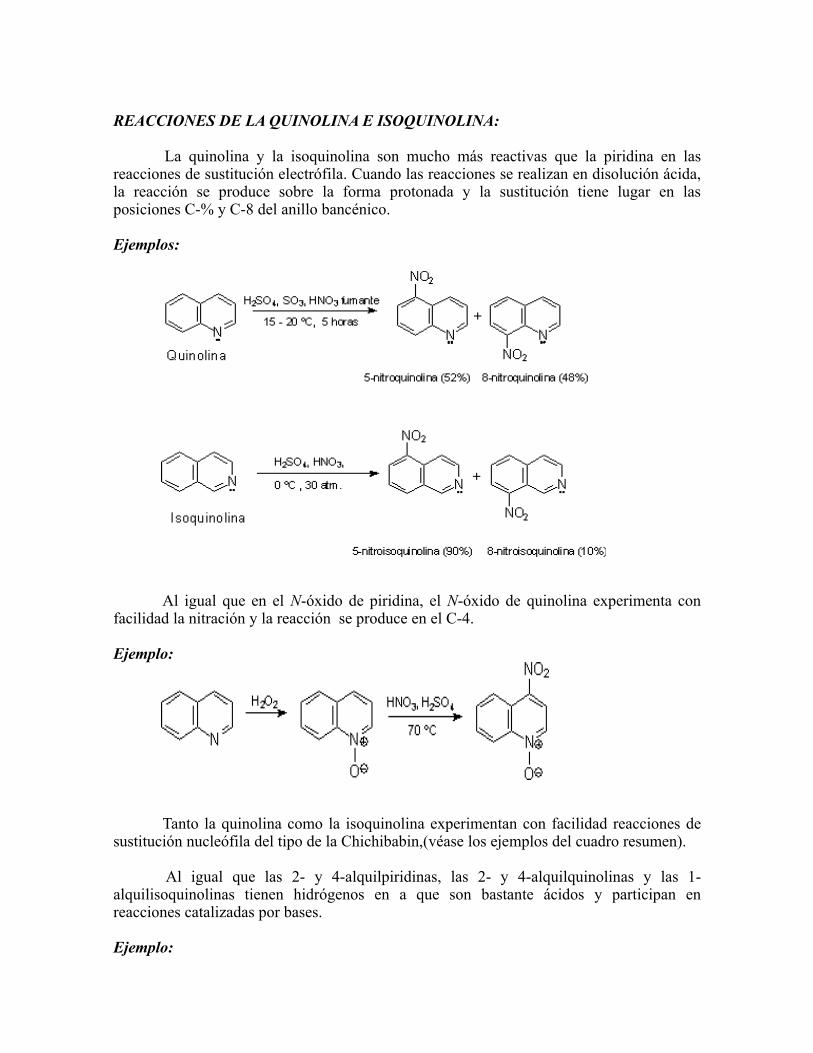
REACCIONES DE LA QUINOLINA E ISOQUINOLINA: La quinolina y la isoquinolina son mucho más reactivas que la piridina en las reacciones de sustitución electrófila. Cuando las reacciones se realizan en disolución ácida, la reacción se produce sobre la forma protonada y la sustitución tiene lugar en las posiciones C-% y C-8 del anillo bancénico. Ejemplos:
Al igual que en el N-óxido de piridina, el N-óxido de quinolina experimenta con facilidad la nitración y la reacción se produce en el C-4. Ejemplo:
Tanto la quinolina como la isoquinolina experimentan con facilidad reacciones de sustitución nucleófila del tipo de la Chichibabin,(véase los ejemplos del cuadro resumen). Al igual que las 2- y 4-alquilpiridinas, las 2- y 4-alquilquinolinas y las 1-alquilisoquinolinas tienen hidrógenos en a que son bastante ácidos y participan en reacciones catalizadas por bases. Ejemplo:

[1] Karl Paal, (1860-1935), Profesor de la Universidad de Erlangen, Alemania.[2] Ludwig Knorr, (1859-1921), Profesor de la Universidad de Jena, Alemania.[3] Arthur Hantzsch, (1857-1935), Profesor de la Universidad de Leipzig, Alemania.[4] Emil Fischer, es el mismo que propuso la proyección del carbono tetraédrico sobre un plano, (véase capítulo 6).[5] A. Bischler, Profesor de la Universidad de Zürich, Alemania.[6] B. Napieralski, Profesor de la Universidad de Zürich, Alemania.[7] Alexei E. Chichibabin, (1871-1945), Profesor de la Universidad de Moscú.
Reacciones Orgánicas más Comunes
(Síntesis de Feist-Benary)








![UNIDAD 2 - [DePa] Departamento de Programas …depa.fquim.unam.mx/amyd/archivero/2-FURANOS-PIRROLES... · Mecanismo competitivo en Feist-Benary 47 • En algunos casos como tenemos](https://img.pdfslide.es/doc/110x75/5b05c6457f8b9a5c308bfc94/unidad-2-depa-departamento-de-programas-depafquimunammxamydarchivero2-furanos-pirrolesmecanismo.jpg)







![MÉTODOS DE SÍNTESIS - [DePa] Departamento de …depa.fquim.unam.mx/amyd/archivero/SINTESISANILLOSDE5ATOMOS1... · feist f., ber., 1902, 35, 1539, 1547. benary e., ber., 1911, 44,](https://img.pdfslide.es/doc/110x75/5b05c6457f8b9a5c308bfc9d/mtodos-de-sntesis-depa-departamento-de-depafquimunammxamydarchiverosintesisanillosde5atomos1feist.jpg)








