Embed Size (px)
Citation preview

ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES
Coordinación científica:DR. ENRIQUE GRANDE PULIDO
Jefe de Servicio de Oncología médica de MD Anderson Cancer Center. Madrid

3ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES1. Epidemiología del cáncer diferenciado de tiroides en España: avanzando hacia el registro nacional2
1. Introducción
El cáncer de tiroides (CT) es el tumor más común de los tumores malignos originados en órganos endocrinos (> 92%)1, representando alrededor del 1,5% de los nuevos cánceres diagnosticados en Europa2. El CT comprende un grupo de tumo-res que varían clínicamente, epidemiológicamen-te y en cuanto a pronóstico: los originados en las células foliculares de tiroides denominados car-cinomas diferenciados de tiroides (CDT), que in-cluyen carcinoma papilar (CP), carcinoma folicu-lar (CF) y carcinoma oncocítico o de células de Hürthle (que suponen >90%), los originados en las células parafoliculares (carcinoma medular) y los carcinomas anaplásicos o indiferenciados cuyo origen también es las células foliculares.
A pesar de esta diferenciación histológica y clí-nica, desde el punto de vista epidemiológico en la mayoría de los estudios se incluye a todos los tumores, debido a la baja frecuencia de presen-tación del CT en general y de los subtipos en particular, aunque hay algunos estudios epide-miológicos cuyos datos se diferencian por tipo y características de tumor. Se excluyen de esta denominación los linfomas y los sarcomas que pueden asentar en la glándula tiroidea.
Considerando que la epidemiología estudia la frecuencia de aparición de la enfermedad y de sus determinantes en la población, se trata de conocer la frecuencia de aparición del CT (inci-
dencia), dónde es mayor esta frecuencia (distri-bución geográfica), la evolución de la misma en función del tiempo, la mortalidad que ocasiona y su evolución, y la supervivencia, además de plan-tear las posibles causas.
2. Epidemiologia del cáncer de tiroides en el mundo
El CT es un tumor relativamente infrecuente, su-pone el 3% del total de cánceres en mujeres, el 1% en varones y 1,4% en niños1,3, y actualmente según los datos de la OMS4 es el 18º tumor más frecuente en ambos sexos, con un claro predo-minio en mujeres (ratio 2,5:1)2,5. En algunos países como Japón este ratio es mucho mayor (13:1). La incidencia anual es variable en los distintos paí-ses, siendo más alta en USA (10,5/100.000)6, en Europa (3,1/100.000 en varones y 9,3/100.000 en mujeres, con las tasas más elevadas en Islan-dia, Lituania, República Checa, Bielorrusia, Aus-tria y Francia)2, en Australia y en Polinesia.
El CT afecta a un amplio espectro de población y puede aparecer a cualquier edad, desde la infan-cia a mayores de 80 años, aunque es poco fre-cuente en menores de 15 años, en los que repre-senta del 1,5-3% del total de cánceres. En USA, la incidencia anual en niños es de 0,59/100.0007. En general, el pico de incidencia se observa en edades medias de la vida, en mujeres alrededor de los 40 años y en varones 1-2 décadas más
1. Epidemiología del cáncer diferenciado de tiroides en España: avanzando hacia el registro nacional
JUAN CARLOS GALOFRÉ FERRATEREspecialista en Endocrinología y Nutrición. Clínica Universidad de Navarra. Pamplona
ELENA NAVARRO GONZÁLEZEspecialista en Endocrinología y Nutrición. Hospital Universitario Virgen del Rocío. Sevilla
ANA REYES ROMERO LLUCHEspecialista en Endocrinología y Nutrición. Hospital Universitario Virgen del Rocío. Sevilla
FRANCISCO JAVIER SANTAMARÍA SANDIEspecialista en Endocrinología y Nutrición. Hospital Universitario Cruces. Bilbao
Epidemiología del Cáncer Diferenciado de Tiroides en España: avanzando hacia el registro nacional.
JUAN CARLOS GALOFRÉ FERRATER, ELENA NAVARRO GONZÁLEZ, ANA REYES ROMERO LLUCH, FRANCISCO JAVIER SANTAMARÍA SANDI.
Biología Molecular del carcinoma diferenciado de tiroides: avanzando en la medicina de precisión en nuestros pacientes.
JAUME CAPDEVILA, JORGE HERNANDO-CUBERO.
El concepto de la refractariedad al radioyodo en los pacientes con carcinoma diferenciado de tiroides: estandarización del manejo de los pacientes.
JUAN ANTONIO VALLEJO CASAS, MARÍA VICTORIA GUIOTE MORENO, ANTONIO MARÍA SANTOS BUENO.
Conceptos prácticos del tratamiento sistémico del carcinoma diferenciado de tiroides refractario al radioyodo: ¿todos los pacientes son iguales?
BEATRIZ CASTELO FERNÁNDEZ, CRISTINA ÁLVAREZ ESCOLÁ, LORENA OSTIOS GARCÍA, ELSA BERNAL HERTFELDER, ANA PERTEJO FERNÁNDEZ, ENRIQUE ESPINOSA ARRANZ.
Lenvatinib en cáncer diferenciado de tiroides.
PAULA JIMÉNEZ FONSECA, FELIPE ÁLVAREZ-MANCECHIDO, MANUEL SÁNCHEZ-CÁNOVAS, ALBERTO CARMONA-BAYONAS, JOSÉ MARÍA VIEITEZ.
pag. 3
pag. 15
pag. 23
pag. 37
pag. 45
ÍNDICE DE CAPÍTULOS
1
3
2
4
5
Producción, diseño y edición:Medical Media, scpC/. Salut, 20. 08960 Sant Just Desvern (Barcelona)[email protected]
Toda forma de reproducción, distribución, comunicación pública o transformación de estaobra sólo puede ser realizada con la autorización de sus titulares y autores, salvo la excepciónprevista por la ley.
med
ical
med
ia

5ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES1. Epidemiología del cáncer diferenciado de tiroides en España: avanzando hacia el registro nacional4
tarde que en las mujeres y en ambos se mantiene estable hasta los 80 años3 .
Una de las características de este tumor es el incremento progresivo y significativo de la in-cidencia, que se ha observado en las últimas décadas: en USA desde 1973 al 2002 se ha in-crementado 3,8 veces8, con un porcentaje de cambio anual (PCA) del 6,6% de 1997 a 20099. Este incremento también se está observando en ambos sexos y en todos los países en Europa, Asia, Oceanía y Sudamérica10,11,12,13,14, excepto en África, donde probablemente existe un infra-diagnóstico o infra-registro.
La incidencia varía en función de varios factores, como son el género (siendo el incremento en las mujeres mucho más intenso) y la raza, ha-biéndose observado un incremento mucho más rápido en blancos (PCA 6,7%), más lento en ne-gros (4,9%), muy lento en asiáticos (2,1%) y sin cambios en hispánicos15. Este aumento de la inci-dencia se está demostrando que ocurre a todas las edades incluyendo los niños y adolescentes16.
No hay muchos estudios que describan la ten-dencia de la incidencia de los distintos subti-pos de CT, pero todos ellos demuestran que este aumento de la incidencia es fundamen-talmente a expensas del CP17,18,19 con PCA del 6,6% y específicamente de la variante clásica, aunque también está aumentando la variante folicular del CP15. Muy pocos estudios propor-cionan información acerca del CF con datos no concordantes, si bien algunos muestran un incremento en la incidencia20,15, éste es mucho menos intenso y más lento que CP18,21 (PCA 1,57%). Sin embargo, otros trabajos indican que hay una estabilización en la incidencia de este tipo de tumor22. La incidencia de cáncer me-dular se mantiene con un discreto incremen-to (PCA 1,87%)18,21 y el cáncer anaplásico tiene tendencia al descenso.
Varios estudios han puesto de manifiesto que este incremento anual en el diagnóstico de CP, se debe a tumores de pequeño tamaño, ya que más del 87% son tumores menores 2 cm de diámetro9,23. No obstante, también hay incre-mento en la incidencia de tumores de mayor tamaño, de tumores con invasión extratiroidea y con presencia de metástasis cervicales6.
En general, la mitad del incremento se debe a tumores menores de 1 cm, un 30% a tumores entre 1-2 cm y un 20% a tumores mayores de 2 cm. En las mujeres blancas el incremento en tumores mayores de 5 cm casi iguala al de los tumores pequeños. Este incremento en la inci-dencia de los tumores de todos los tamaños ha sido un hallazgo constante en varios estudios24, calculándose un incremento anual5,25 del 19% en tumores menores de 1 cm, del 12% en tumores entre 1-2 cm, del 5-10% para tumores entre 2-4 cm, y del 12% para tumores mayores de 6 cm (5,5% en >5 cm y 4,9% en >6 cm). Un estudio reciente muestra un aumento de incidencia del 3,6% anual a expensas del CP y más significativo en tumores mayores de 2 cm y entre 2-4 cm18.
A pesar de este incremento de incidencia, la mortalidad por CT se mantiene bastante esta-bilizada, al contrario que otros tipos de tumor, cuya mortalidad esta progresivamente en des-censo. En USA, la mortalidad específica por CT es de aproximadamente 0,5/100.000, e incluso con discreta tendencia al alza, ya que algunos datos sugieren un incremento de 0,8% anual en varones4.
Una publicación reciente que calcula letalidad del CT refiere un incremento de mortalidad del 1,1% anual en los CT y un 2,9% en los CT en esta-dios avanzados18.
En Europa en 1998 las tasas ajustadas de mor-talidad fueron de 0,5 y 0,7/100.000 en varones y mujeres respectivamente4 y estas tasas son similares en Oceanía, Sudamérica y Asia (entre 0,1 y 0,7/100.00)4 y en todos estos países parece mantenerse esa tendencia a la estabilización. Es decir, no hay paralelismo entre la estabilización de la mortalidad y el incremento en la incidencia, lo cual se explicaría por la baja mortalidad que per se tiene el CT. Parece que para que un incre-mento en la incidencia generase un incremento en la mortalidad deberían de transcurrir al menos 10-20 años, es decir el efecto de ese aumento de incidencia podría verse en los próximos años.
En la Unión Europea26 la supervivencia del CT a los 5 años es del 85% y se ha incrementado de 1983 a 1994 del 79% al 85%. En general, las mujeres tienen mejor supervivencia (81,4%) que los varones (71,85%), pero esta diferencia es muy
pequeña en el grupo de edad entre 15-44 años y es sobre todo mayor en los mayores de 75 años. La mayor supervivencia se observa en el grupo de 15-44 años (97% a los 5 años). Esta supervi-vencia es diferente según el país, pero sin dife-rencias significativas entre ellos.
Una publicación reciente evalúa los datos euro-peos de los años 2000-2008 y confirma la me-joría en la supervivencia que ha pasado del 85 al 88% y del 71,8 al 77% en mujeres y varones respectivamente27. Esta supervivencia está inver-samente relacionada con la edad al diagnóstico, el tamaño tumoral, el grado de invasión local, y el tipo histológico de tumor28 (mayor mortalidad en cáncer medular, en cáncer anaplásico y en cáncer poco diferenciado), pero fundamental-mente es la presencia de metástasis a distancia lo que modifica radicalmente el pronóstico de este tipo de tumor.
3. Epidemiologia del cáncer de tiroides en España
En España, conocemos datos procedentes de los registros de cáncer de población (RCP) y datos clínicos procedentes de series publicadas de dis-tintos ámbitos provinciales u hospitalarios.
Los registros poblacionales recogen todos los nuevos casos de cáncer en una población de-finida y su valor es fundamental para la identi-ficación de las tendencias de la incidencia de cáncer en diferentes localizaciones geográficas, pero nos proporcionan únicamente estadísticas sobre las características demográficas de los pacientes y en España, a diferencia de otros re-gistros como el programa del Instituto de Salud americano Surveillance, Epidemiology and End Results (SEER).
En la mayoría de los registros no se recogen da-tos específicos clínicos, información sobre los ti-pos histológicos de tumor, estadios, tratamien-tos, ni tampoco supervivencia, pues los registros de mortalidad funcionan de forma separada. Por otra parte, los registros de cáncer en España se han desarrollado de forma variable en las distin-tas comunidades autónomas o provincias, por lo que los datos que nos proporcionan son de poblaciones muy concretas, que en absoluto son
representativas de la población española, ni si-quiera de su propia comunidad, ya que no han sido elegidas como representativas de la pobla-ción española (como es el registro de la SEER en USA) sino que han ido desarrollándose en función de la atención que ha dedicado a este tema la política sanitaria de cada comunidad au-tónoma.
4. Registros poblacionales
Los dos registros de cáncer de población más antiguos en España son el Registro de Cáncer de Zaragoza, creado en el año 1960, y el de Navarra, creado en 1970.
En 1976 se puso en marcha el Plan Nacional de Registros de Cáncer de Población, añadiendo cuatro más de nueva creación: Asturias, Tenerife, Sevilla y Valladolid, si bien no todos ellos mantu-vieron su continuidad a lo largo de estos años.
En la actualidad, existen 14 RCP de Población (Albacete, Asturias, Ciudad Real, Cuenca, Gero-na, Granada, Canarias, La Rioja, Mallorca, Murcia, Navarra, País Vasco, Tarragona y Zaragoza) cu-yos datos están incluidos en la publicación de referencia Cancer Incidence in Five Continents editada por la International Agency for Research on Cancer (IARC)29. Además, existe un RCP en Castellón y un registro monográfico de Cáncer Infantil en la Comunidad Valenciana. En su con-junto, abarcan una población aproximada de 10 millones, lo que representa el 27% de la pobla-ción española, pero en una muestra que no es representativa de la población española30.
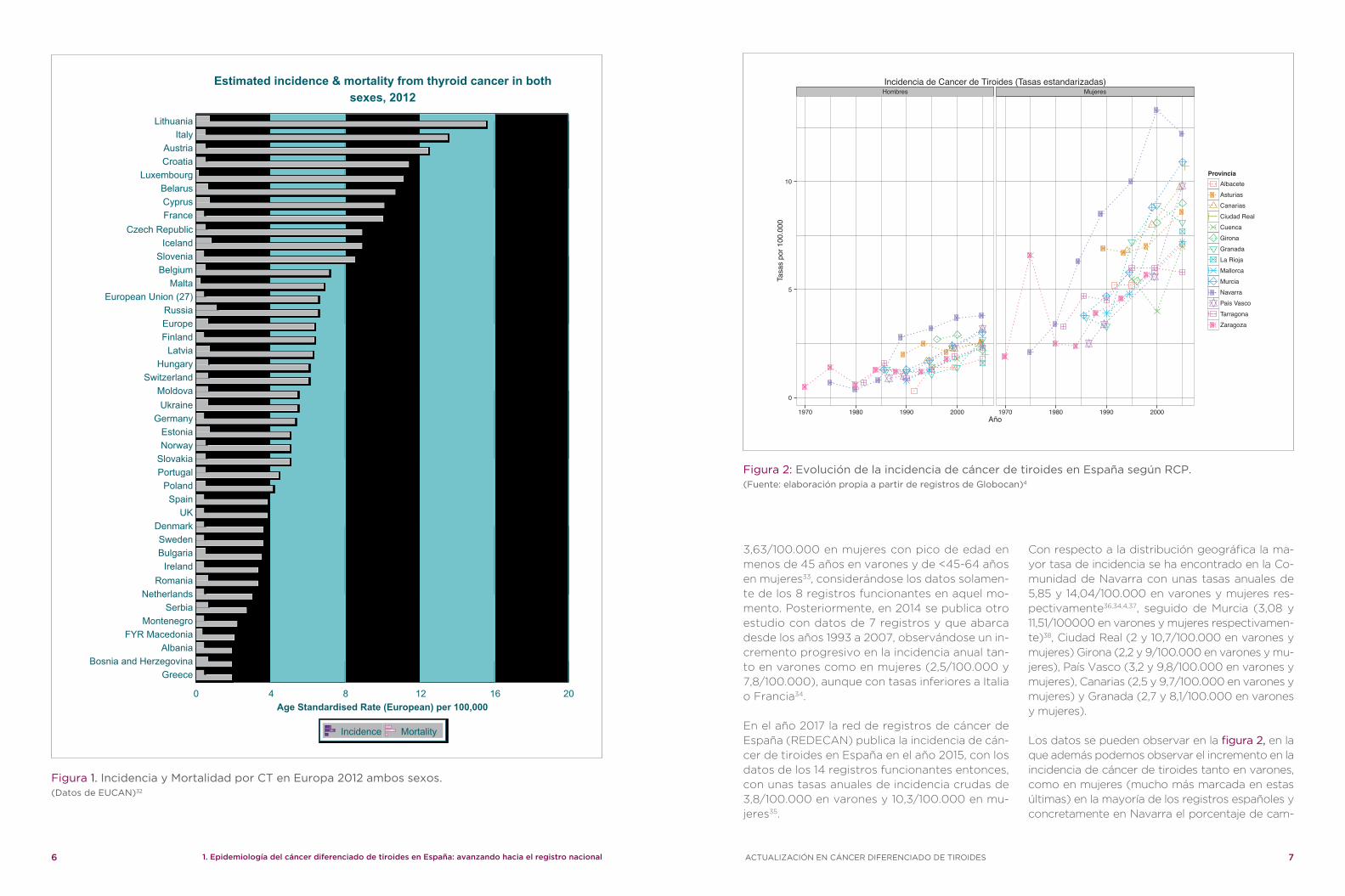
Al evaluar los datos de los registros poblacio-nales españoles, observamos una incidencia de CT en España bastante inferior a la media euro-pea, con una tasa global de incidencia anual de 3,9/100.000, ocupando el lugar 29 con respecto a los países europeos31,32. (figura 1)
La tasa como en otros países es mayor en muje-res que en hombres, va incrementándose con la edad hasta alcanzar el máximo en torno a los 45-64 años en hombres y 50-64 años en mujeres14.
En el periodo 1993-1996 se estima una incidencia anual de CT en España de 1,73 en varones y de
7ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES1. Epidemiología del cáncer diferenciado de tiroides en España: avanzando hacia el registro nacional6
3,63/100.000 en mujeres con pico de edad en menos de 45 años en varones y de <45-64 años en mujeres33, considerándose los datos solamen-te de los 8 registros funcionantes en aquel mo-mento. Posteriormente, en 2014 se publica otro estudio con datos de 7 registros y que abarca desde los años 1993 a 2007, observándose un in-cremento progresivo en la incidencia anual tan-to en varones como en mujeres (2,5/100.000 y 7,8/100.000), aunque con tasas inferiores a Italia o Francia34.
En el año 2017 la red de registros de cáncer de España (REDECAN) publica la incidencia de cán-cer de tiroides en España en el año 2015, con los datos de los 14 registros funcionantes entonces, con unas tasas anuales de incidencia crudas de 3,8/100.000 en varones y 10,3/100.000 en mu-jeres35.
Con respecto a la distribución geográfica la ma-yor tasa de incidencia se ha encontrado en la Co-munidad de Navarra con unas tasas anuales de 5,85 y 14,04/100.000 en varones y mujeres res-pectivamente36,34,4,37, seguido de Murcia (3,08 y 11,51/100000 en varones y mujeres respectivamen-te)38, Ciudad Real (2 y 10,7/100.000 en varones y mujeres) Girona (2,2 y 9/100.000 en varones y mu-jeres), País Vasco (3,2 y 9,8/100.000 en varones y mujeres), Canarias (2,5 y 9,7/100.000 en varones y mujeres) y Granada (2,7 y 8,1/100.000 en varones y mujeres).
Los datos se pueden observar en la figura 2, en la que además podemos observar el incremento en la incidencia de cáncer de tiroides tanto en varones, como en mujeres (mucho más marcada en estas últimas) en la mayoría de los registros españoles y concretamente en Navarra el porcentaje de cam-
LithuaniaItaly
AustriaCroatia
LuxembourgBelarusCyprusFrance
Czech RepublicIceland
SloveniaBelgium
MaltaEuropean Union (27)
RussiaEuropeFinland
LatviaHungary
SwitzerlandMoldovaUkraine
GermanyEstoniaNorway
SlovakiaPortugal
PolandSpain
UKDenmarkSwedenBulgaria
IrelandRomania
NetherlandsSerbia
MontenegroFYR Macedonia
AlbaniaBosnia and Herzegovina
Greece
Estimated incidence & mortality from thyroid cancer in bothsexes, 2012
0 4 8 12 16 20Age Standardised Rate (European) per 100,000
Incidence Mortality
Figura 1. Incidencia y Mortalidad por CT en Europa 2012 ambos sexos.(Datos de EUCAN)32
Figura 2: Evolución de la incidencia de cáncer de tiroides en España según RCP.(Fuente: elaboración propia a partir de registros de Globocan)4
Hombres Mujeres
0
5
10
1970 1980 1990 2000 1970 1980 1990 2000Año
Tasa
s po
r 100
.000
ProvinciaAlbaceteAsturiasCanariasCiudad RealCuencaGironaGranadaLa RiojaMallorcaMurciaNavarraPais VascoTarragonaZaragoza
Incidencia de Cancer de Tiroides (Tasas estandarizadas)

9ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES1. Epidemiología del cáncer diferenciado de tiroides en España: avanzando hacia el registro nacional8
bio anual (PCA) ha sido de 4,7 en varones y de 2,5 en mujeres39 y en Murcia el PCA ha sido de 3,3 en varones y 4,7 en mujeres42. Es decir, en nuestro país con los escasos datos publicados, se comprue-ba también la misma tendencia que en el resto del mundo, un incremento progresivo de la incidencia de CT.
Otra forma de abordar la estimación de la inci-dencia de CT en España podría ser evaluando los datos de las intervenciones quirúrgicas, ya que en el Conjunto Mínimo de datos al alta (CMDB) pode-mos conocer el número de casos intervenidos en los diferentes hospitales por patología maligna de tiroides. Este abordaje tiene varias limitaciones y sesgos, pero puede ser una aceptable aproxima-ción al problema.
En Andalucía se han evaluado los datos de CMBD desde el año 2004 al 2011 de todos los pacientes intervenidos de patología tiroidea (datos no publi-cados, comunicados al congreso de la SEEN 2013) y se comprueba la existencia de unas evidentes di-ferencias provinciales con respecto a la cirugía por neoplasia maligna de tiroides (193 CIE 9) que es de un 5,2 y 4,9/100.000 en Málaga y Sevilla respecti-vamente, frente a un 4/100.000 en Granada e infe-rior al 3/100.000 en Cádiz, Córdoba, Huelva y Jaén. También se ha observado un incremento progre-sivo durante esos años claramente más manifiesto en Sevilla y Málaga39. Esta aproximación a través de la cirugía, sin poder considerarla como incidencia, se ha realizado en otros centros, comprobándose igualmente un aumento de la frecuencia de CT que pasó de 15,2% a 24,3% de 2001 a 200340.
Nuestros registros poblacionales no recogen (o por lo menos no proporcionan información sobre ello) otras características de los tumores como son el tipo histológico, el tamaño u otras caracte-rísticas clínicas, únicamente se han publicado datos del RCP de Murcia (de 1984 a 2008) en el que se comprueba el incremento de incidencia por tipo histológico, mostrando el importante incremento a expensas del carcinoma papilar con PCA del 6,%, aunque también se incrementa el carcinoma foli-cular (PCA 1,9% anual) el carcinoma medular (PCA 3,9% anual), pero no el carcinoma anaplásico. Con respecto al tamaño se cuadriplica la tasa de micro-carcinomas en las mujeres de 2004 a 2008, de for-ma que la tasa microcarcinoma/carcinoma papilar paso de 13% a 24%42.
5.1. Conclusiones Se comprueba en España tanto por los datos de los escasos registros poblacionales, como por los escasos datos de los registros hospitalarios publicados, que existe un incremento progresivo de la incidencia de CT, que está a expensas del tipo CP, aunque también hay un menor incre-mento de CF. Es notable señalar que, si bien hay un incremento de los microcarcinomas, también lo hay de los tumores de mayor tamaño.
La supervivencia del CT en España es similar a la descrita en otros países europeos, 87% de muje-res están vivas a los 5 años y 76% en el caso de los varones, relacionándose la supervivencia con la edad del diagnóstico (a menor edad mayor supervivencia) y con el tipo de tumor25.
Con respecto a la mortalidad por CT en España está mejor cuantificada que la incidencia y dis-ponible a partir de las estadísticas de mortalidad publicadas por el Instituto Nacional de Estadís-tica, basadas en los certificados de defunción, aunque el CT es una causa de muerte infracerti-ficada en España44.
Las tasas de mortalidad son menores que la me-dia europea. Desde 1975 a 2001 la mortalidad en varones sufrió un pequeño incremento medio de un 1,21% anual, con disminución en las mujeres en promedio de un 0,39% al año45,34.
Según los datos del atlas de mortalidad en Es-paña de los años 1989-1998, el patrón geográfi-co muestra un exceso de mortalidad en el área noroccidental del país y en las Islas Canarias, es-tando los municipios con riesgo superior a 1,50 concentrados en la provincia de Lugo, zona orien-tal de A Coruña, zona occidental de Ourense y Asturias, y en Canarias el exceso se concentra en La Palma, Fuerteventura, Gomera y algunos mu-nicipios de Tenerife y Gran Canaria45.
Datos más recientes de mortalidad proporcio-nados por el Centro nacional de epidemiología del Instituto de Salud Carlos III46 muestran una tasa de mortalidad media en España similar en varones y mujeres (0,4/100.000), y con diferen-cias geográficas provinciales, demostrándose en varones una mayor incidencia que la media española y mayor de 0,55/100.000 en Ponte-vedra, Granada, Extremadura, Huesca, Lugo,
Para conocer las características clínicas y la evo-lución de las mismas tenemos que recurrir a los registros propios de las Unidades hospitalarias, estos registros permiten mejorar los sistemas de estadiaje, determinar el impacto de las terapias y evaluar morbilidad y letalidad, pero su inconvenien-te es que suelen estar limitados a centros de mayor tamaño y son registros voluntarios, por lo que no se incluyen todos los pacientes diagnosticados41.
5. Registros hospitalarios
En España se ha publicado datos de algunos de los registros hospitalarios. En el año 2009 los da-tos de Vigo42, de los años 1978 a 2001, en los que se constata un incremento en la incidencia de CT, básicamente a expensas del CP, tanto de los mi-crocarcinomas (tasas de 0,14 a 3,94) como de los tumores mayores de 1 cm (tasas de 0,87 a 4,52), comprobándose estabilidad en la incidencia en los otros tipos histológicos de CT.
En el año 2015 se comunicaron datos del Sur de Es-paña (provincias de Sevilla y Huelva)43, de los años 1970-2012 que ponen de manifiesto el incremen-to progresivo del CT desde 1970, pero sobre todo desde 1996, a expensas fundamentalmente del CP, pero con incremento también de CF y con incre-mento tanto de tumores mayores de 1 cm, como de tumores de mayor tamaño (T1b-T4). No obstan-te, en los periodos iniciales los tumores tendían a ser mayores de 4 cm y con extensión extratiroidea y N1, M1, en comparación con los años finales del periodo, en los que los tumores son menores de 4 cm, sin extensión extratiroidea y sin metástasis ganglionares.
En el año 2017 se acaban de publicar los datos del registro hospitalario de cáncer de Navarra39 desde 1986 a 2010, en el que se pone de manifiesto el in-cremento del CT fundamentalmente a expensas del CP, más frecuente la variante clásica de CP (59%), seguida de la variante folicular de CP (28%) y de los subtipos agresivos (12%), manteniéndose estable la proporción de estas variables a lo largo del tiempo. Se mostró un incremento progresivo de la propor-ción de microcarcinomas que pasaron del 8,8% al 30% en el último periodo, de forma que se observó disminución global del tamaño medio de los tumo-res de 30,9 a 24 mm, no observándose diferencias en cuanto a la estadificación al diagnóstico.
Asturias, Cantabria, Toledo, Ávila, Albacete, La Rioja y Almería. En mujeres las tasas más altas y mayores de 0,55/100.000 son en Ciudad Real, Toledo, Segovia, Palencia, Cantabria, León, Huel-va, Murcia, Vizcaya, Navarra, Guipúzcoa y Extre-madura. (figura 3)
6. Evaluación de la Epidemiología del CT en España
Hay varios déficits importantes para poder reali-zar una correcta evaluación de la epidemiología del CT en nuestro país.
En primer lugar, los registros poblacionales de cáncer son pocos y poco representativos de la población, es decir, no recogen todos los casos en todas las localizaciones geográficas: no re-cogen tipo histológico, estadio al diagnóstico, tratamientos iniciales, seguimiento del caso y letalidad del tumor, datos que si recogen otros registros como SEER.
En segundo lugar, los registros poblacionales, aún bien planteados, tienen varias limitaciones, ya que no recogen test diagnósticos, solo recogen datos del primer tratamiento, las características histológicas no están bien determinadas, no se evalúan marcadores moleculares y solo evalúan supervivencia y no la situación de la enfermedad (persistencia, recidivas, etc). Estos datos podrían estar complementados por los registros hospitalarios, que deberían ser multicéntricos y prospectivos (al menos provinciales o regionales). El escenario ideal y ambicioso sería lograr un registro nacional similar al que se está intentando desarrollar en USA41. No obstante, este tipo de registros que dependen de la voluntariedad, siempre ha de estar complementado por los registros poblacionales.
7. Cambios en la Incidencia
Hay varias hipótesis que intentan explicar el in-cremento de la incidencia del CT. Varios autores consideran que el mayor número de cánceres se debe a un “sobrediagnóstico” de los mismos, sugiriéndose que el aumento de la incidencia es debido a una mejoría en el diagnóstico de cánce-

11ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES1. Epidemiología del cáncer diferenciado de tiroides en España: avanzando hacia el registro nacional10
res de muy pequeño tamaño, gracias a las técni-cas actuales de ecografía (que se utiliza de for-ma mucho más habitual en la práctica clínica), de punción aspirado, de uso de la tomografía por emisión de positrones (PET)47,11, al aumento del número y extensión de la cirugía tiroidea y al incremento en el estudio de las piezas pato-lógicas.
Los criterios del estudio anatomopatológico han sufrido un cambio: en las guías de 1980 se precisaba estudio de 5 áreas mientras que en las guías de 2012 se definen al menos 14 áreas de estudio. Todos estos factores ocasionarían un sobrediagnóstico de este tipo de tumores4,48, habiéndose relacionado además este incremen-to con las características socioeconómicas de la población49 (a mayor nivel económico y/o edu-cativo mejor acceso a los sistemas de salud y mayor incidencia de CT), con los sistemas de salud50,51 (en los sistemas sanitarios públicos la
incidencia de CT es menor), con la densidad de endocrinólogos y el incremento del uso de la ecografía cervical13.
Otros autores opinan que, aunque no hay duda del papel que la mejora de la tecnología y del acceso a los sistemas diagnósticos puedan ju-gar, el incremento en la incidencia del CT es un incremento verdadero, que puede estar justifica-do por otros factores (carcinógenos que afecten específicamente a las células tiroideas)5,15,19, pues de otra forma resulta difícil explicar el crecimien-to de tumores de mayor tamaño y en estadios avanzados5,11, el mayor incremento en mujeres que en hombres, el claro patrón según cohor-te de nacimiento52, el aumento de la incidencia que también se está demostrando en los países en desarrollo53, los diferentes patrones de com-portamiento de los distintos tipos histológicos y el incremento que se está demostrando en la mortalidad18. Por otra parte, otros tumores para
los que se realiza cribado periódico como mama, próstata, cérvix o colon están disminuyendo su porcentaje anual de cambio.
Todos estos hechos reflejan la existencia de dis-tintos factores de riesgo resumidos en la (tabla 1) fenómenos autoinmunes, el uso de la radioterapia por patologías benignas en los niños en las dé-cadas de 1920 a 1950, la mayor exposición a ex-ploraciones radiológicas de la población, la lluvia radioactiva, mutación genética, alteraciones en la ingesta de yodo y los potenciales cancerígenos ambientales12.
Las radiaciones ionizantes suponen el único fac-tor de riesgo sobre el que hay suficiente eviden-cia, mientras que sobre el resto de los factores que pueden estar implicados (contaminantes ambientales) la evidencia existente es aún insu-ficiente y poco sólida. No debe excluirse la posi-ble existencia de carcinógenos ambientales, que
puedan ser responsables de este incremento en la incidencia de CT, en particular la exposición a sustancias químicas durante la vida intrauteri-na y en edades precoces, que puedan ocasionar cambios epigenéticos, los cuales determinarían una mayor propensión a la mutagénesis en las células tiroideas desarrollándose una glándula más sensible a la acción de carcinógenos a lo largo de la vida54. Por ello, no ha de asumirse únicamente como cierto el planteamiento más simplista del sobrediagnóstico y han de conti-nuar las investigaciones en este sentido, con idea de poder establecer medidas preventivas para evitar este incremento progresivo.
Figura 3: Mortalidad por cáncer de tiroides en hombres y mujeres año 2015.(Fuente: Centro nacional de epidemiologia. Instituto de Salud Carlos III)46
Tabla 1: Factores potenciales de riesgo para el desarrollo de Cáncer de tiroides.
FACTORES EXÓGENOS
FACTORES ENDÓGENOS
Rayos X
I 131
Yodo
Actividad volcánica
Nitratos
Contaminantes ambientales
Ocupación laboral
TSH
Tiroiditis autoinmune
Obesidad e insulinresistencia
Determinantes genéticos
Estudios diagnósticos (TAC, Rx dental)
Radioterapia terapéutica
Procedimientos Medicina nuclear
Accidentes nucleares
Dieta, profilaxis con yodo
Carcinógenos aun no descubiertos
Agua y dieta
Carcinógenos aun no descubiertos
Factor Fuente

13ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES1. Epidemiología del cáncer diferenciado de tiroides en España: avanzando hacia el registro nacional12
BIBLIOGRAFÍA
1. American Cancer Socie-ty. Cancer fast and figu-res. http://www.cancer.org (consultado 16 junio 2017).
2. Ferlay J, Steliarova-Fou-cher E, Lortet-Tiulent J, Rosso S, Coebergh JWW, Comber H et al. Cancer in-cidence and mortality pat-terns in Europe: Estimates for 40 countries in 2012. Eur J Cancer 2013; 49:1374-1403.
3. Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, et al. Cancer statistics. Can-cer J Clin 2006; 56:106-130.
4. International Agency for research of Cancer (IARC): GLOBOCAN 2012 http://gco. iarc.fr/today/home (Consultado 16 junio 2017).
5. Jemal A, Siegel R, Ward E. Cancer statistics 2010.CA: A Cancer J Clin 2010; 60:277-300.
6. Morris L, Myssiorek D. Improved detection does not fully explain the rising incidence of well differen-tiated thyroid cancer: a po-pulation based analysis. Am J Surg 2010; 200:454-461.
7. Dermody S, Walls A, Har-ley EH Jr. Pediatric thyroid cancer: An update from the SEER database 2007-2012. Int J Pediatr Otorhinolaryn-gol. 2016; 89:121-126.
8. Davies L, Welch HG. Increasing incidence of thyroid cancer in the United States, 1973-2002. JAMA 2006; 295:2164-2167.
9. Cancer of the thyroid SEER Stat Fact Sheets: http//www.seer.cancer.gov/state facts/html/thyroid.html (consultado 16 junio 2017).
10. Kilfoy B, Zheng T, Hol-ford T, Han X, Ward MH, Sjodin A, et al. Internatio-nal patterns and trends in thyroid cancer incidence 1973-2002. Cancer Causes Control 2009; 20:525-531.
11. Amphlett B, Lawson Z, Abdulrahman GO Jr, White C, Bailey R, Premawardha-na LD, et al. Recent trends in the incidence, geographi-cal distribution and survival from thyroid cancer in Wa-les 1985-2010. Thyroid 2013; 23:1470-1478.
12. Veiga L, Neta G, Asche-brook-Kilfoy B, Ron E, De-vesa S. Thyroid cancer inci-dence patterns in Sao Paulo, Brazil, and the US SEER pro-gram 1997-2008. Thyroid 2013; 23: 748-757.
13. Ismail SI, Soubani M, Ni-mri JM, Al-Zeer AH. Cancer incidence in Jordan from 1996 to 2009 - A compre-hensive study. Asian Pac J Cancer Prev. 2013; 14:3527-3534.
14. Lope Carvajal V, Pollan Santamaria M, Pérez-Gómez B, Aragonés Sanz B, Suárez Rodríguez JM, et al. Inciden-cia y mortalidad por cáncer de tiroides en España. Bo-letín epidemiológico ISCIII. Ministerio de Sanidad. 2004; 12:161-172.
15. Mao Y, Xing M. Recent incidences and differential trends of thyroid cancer in the USA. Endocr Related Cancer. 2016; 23:313-322.
16. Bonachi L, Lindsay A, Laurinavicius F, Braga K, Ro-driguez-Galindo C. Increase in the incidence of differen-tiated thyroid carcinoma in children, adolescents and young adults: A Population-Based study. J Pediatr 2014; 164:1481-1485.
17. Wiltshire JJ, Drake TM, Uttley L, Balasubramanian SP. Systematic review of trends in the incidence rates of thyroid cancer. Thyroid 2016; 26:1541-1552.
18. Hyeyeun L, Devesa S, Sosa JA, Check D, Kitahara K. Trends in thyroid inciden-ce and mortality in the Uni-ted States, 1974-2013. JAMA 2017; 317:1338-1348.
19. Kitahara CM, Sosa JA. The changing incidence of
thyroid cancer. Nat Rev En-docrinol. 2016; 12:646-653.
20. Kilfoy B, Grogan R, Ward M, Kaplan E, Devesa S. Fo-llicular thyroid cancer inci-dence patterns in the United States, 1980-2009. Thyroid 2013; 23:1015-1021.
21. SEER Cancer Statistics Review 1975-2012. National cancer Institute. https://seer.cancer.gov/archive/csr/1975_2012 (consultado 16 junio 2017).
22. Davies L, Welch HG. Cu-rrent Thyroid cancer trends in the United States. JAMA Otolaryngol Head Neck Surg. 2014;140:317-322.
23. Colonna M, Guizard AV, Schvartz C, Velten M, Raver-dy N, Molinie F, et al. A time trend analysis of papillary and folliculary cancers as a function of tumor size: A study of data from six cancer registries in France (1983-2000). Eur J Cancer. 2007; 43:891-900.
24. Chen AY, Jemal A, Ward EM. Increasing incidence of differentiated thyroid can-cer in the United States, 1988-2005. Cancer. 2009; 115:3801-3807.
25. Cramer J, Pingfu BS, Harth K, Margevicius S, Wil-helm S. Analysis of the rising incidence of thyroid cancer using the Surveillance, Epi-demiology and End Results national cancer data registry. Surgery. 2010; 148:1147-1153.
26. Sant M, Aareleid T, Berri-no F, Bielska Lasota M, Carli PM, Faivre J, et al. EUROCA-RE-3: survival of cancer pa-tients diagnosed 1990-94-re-sults and commentary. Ann Oncol 2003; 14 Suppl 5:v61-118.
27. Dal Maso L, Tavilla A, Pacini F, Serraino D, van Dijk BAC, Chirlaque MD, et al. Survival of 86,690 pa-tients with thyroid cancer: A population-based study in 29 European countries from EUROCARE-5. Eur J Cancer 2017; 77:140-152.
28. Berrino F, Capocaccia R, Coleman MP, Esteve J, Gatta G, Hakulinen T, et al. Survival of cancer patients in Europe. The Eurocare-3 Study. Ann Oncology. 2003;14 Suppl 5.
29. Bray F, Ferlay J, Laver-sanne M, Brewster DH, Gom-be Mbalawa C, Kohler B, et al. Cancer Incidence in Five Continents: Inclusion criteria, highlights from Volume X and the global status of can-cer registration Int J Cancer. 2015; 137:2060-2071.
30. Red Española de regis-tros de cáncer redecan.org (consultado 16 junio 2017).
31. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. GLOBOCAN 2008 Can-cer Incidence and mortality worldwide: IARC Cancer Base. International Agency for research on cancer; 2010 http://globocan.iarc.fr (con-sultado 16 junio 2017).
32. International agency for research on cancer WHO http://eco.iarc.fr/EUCAN/ (consultado 16 junio 2017).
33. Moreno V, González JR, Soler M, Bosch FX, Koogevi-nas M, Borras JM. Estimación de la incidencia de cáncer en España: periodo 1993-1996. Gac Sanit 2001; 15:380-388.
34. La Vecchia C, Malvez-zi M, Bosetti C, Garavello W, Betuccio P, Levi F, et al. Thyroid cancer mortality and incidence: a global overview. Int J Cancer; 2015; 136:2187-2195.
35. Galceran J, Ameijide A, Carulla M, Mateos A, Quiros JR, Rojas D, et al. Cancer in-cidence in Spain, 2015. Clin Transl Oncol. 2017; 19:799-825.
36. Lopez-Abente G, Po-llan M, Aragones N, Perez-Gómez B, Hernández V, et al. Plan Integral del cáncer: situación del cáncer en Es-paña. Madrid. Ministerio de Sanidad y Consumo 2003.
37. Rojo J, Bermejo B, Me-néndez E, Ardanaz E, Gue-
8. Conclusiones
• El cáncer de tiroides es un tumor de baja, pero creciente incidencia en todas partes del mundo, en todas las edades, en ambos sexos y a expensas en parte de carcinoma papilar de pequeño tamaño.
• En este incremento juega un importante papel la mejora en las téc-nicas diagnósticas, pero probablemente hay también un incremento verdadero de etiología desconocida, que explicaría el aumento de tumores de mayor tamaño y específicamente del carcinoma papilar.
• La mortalidad por CT está estabilizada, con discreto incremento en los últimos años.
• En España, los datos procedentes de los registros poblacionales son poco representativos de la población nacional y proporcionan es-casos datos (básicamente datos demográficos y escasos datos del tumor y de la supervivencia).
• Con los datos que disponemos en España hay una menor incidencia de CT que la media europea, con una mortalidad también inferior, pero también se observa el incremento en la incidencia en las úl-timas décadas a expensas de CP. Se observan además diferencias geográficas en las tasas de incidencia y mortalidad entre las diferen-tes provincias.
• Han de mejorarse los registros de CT, tanto los registros poblaciona-les, como los registros hospitalarios, que han de ser multicéntricos y prospectivos.

15ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES1. Epidemiología del cáncer diferenciado de tiroides en España: avanzando hacia el registro nacional14
vara M, Anda E. Aumento de la incidencia del cáncer de tiroides en Navarra. Evolu-ción y características clíni-cas, 1986-2010. Endocrinol Diabetes Nutr. 2017; 64:303-309.
38. Chirlaque MD, Molen-haur F, Salmeron D, Navarro C. Patrón evolutivo de la in-cidencia de cáncer de tiroi-des en la región de Murcia de 1984 a 2008. Gac Sanit 2014; 28:397-400.
39. Jiménez I, Romero A, Gros N, Martínez AJ, Aliaga A, González B, et al. Análi-sis de la tasa de indicación quirúrgica por patología ti-roidea en Andalucía entre los años 2004 y 2011. En-docrinol Nutr 2013 (Espec Congr).
40. Zafon C, Baena JA, Cas-tellvi J, Obiols G, González O, Fort JM, et al. Evolution of differentiated thyroid cancer: A decade of thyroi-dectomies in a single insti-tution. Eur Thyroid J. 2014; 3:197-201.
41. Mehra S, Tuttle M, Mi-las M, Orloff L, Bergman D, Vernet V, et al. Databa-se and registry research in thyroid cancer: Striving for a New and improved national thyroid database. Thyroid 2015; 25:157-168.
42. Rego-Iraeta A, Perez-Mendez L, Mantinan B, Gar-cia-Mayor R. Time trends for thyroid cancer in nor-thwestern Spain: true rise in the incidence of micro and larger forms of papillary thyroid carcinoma. Thyroid. 2009; 19:333-340.
43. Martinez Ortega AJ, Na-varro E, Guerrero R, Romero A, Martos JM, Cuenca I. Di-fferentiated thyroid cancer: tendency changes from 1970 to 2012 in the southwest of Spain. Endocrine Abstracts 2015 37EP874 DOI:10.1530/Endoabs.37. EP874.
44. Atlas municipal de la mortalidad por cáncer en España 1989-1998 Instituto de salud Carlos III. http://
www.isci i i .es/ISCII I/es/contenidos/fd-servicios-cientifico-tecnicos/fd-vigi-lancias-alertas/fd-epidemio-logia-ambiental-y-cancer/mortalidad-cancer-en-es-pana.shtml (Consultado 16 junio 2017).
45. Lope V, Pollán M, Pérez-Gómez B, Aragonés N, Ra-mis R, Gómez Barroso D, et al. Municipal mortality due to thyroid cancer in Spain. BMC Public Health 2006; 6:302-310.
46. Instituto de Salud Car-los III http://www.isciii.es/ISCIII/es/contenidos/fd-ser-vicios-cientifico-tecnicos/fd-vigilancias-alertas/fd-epidemiologia-ambiental-y-cancer/mortalidad-cancer-en-espana.shtml.
47. Udelsman R, Zhang Y. The epidemic of thyroid cancer in the United States: The role of endocrinologist and ultrasound. Thyroid 2014; 24:472-479.
48. Vaccarella S, Franceschi S, Bray F, Wild C, Plummer M, Del Maso L. Worldwide thyroid-cancer epidemic? The increasing impact of overdiagnosis. N Engl J Med. 2016; 375:614-617.
49. Li N, Du XL, Reitzel LR, Xu Li. Sturgis EM. Impact of enhanced detection on the increase in thyroid can-cer incidence in the United States: Review of incidence trends by socioeconomic status within the surveillan-ce, epidemiology and end results registry 1980-2008. Thyroid 2013; 23:103-110.
50. Lee TJ, Kim S, Cho HJ y Lee JH. The incidence of thyroid cancer is affected by the characteristics of a healthcare system. J Korean Med Sci. 2012; 27:1491-1498.
51. Morris LG, Sikora A, Tos-tesson T, Davies L. Thyroid 2013 ;23:885-891.
52. Zhu C, Zheng T, Kilfoy BA, Han X, Ma S, Ba Y, et al. A birth cohort analysis of the incidence of papillary
thyroid cancer in the United States, 1973-2004. Thyroid 2009; 19:1061-1066.
53. Wartofsky L. Increasing world incidence of thyroid cancer: increasing detection or higher radiation exposu-re? Hormones. 2010; 9:103-108.
54. Navarro González E, Guerrero Vázquez R. Epi-demiología del cáncer de tiroides. En JM Gómez Sáez (editor). Cáncer de tiroides. Ed Elsevier 2014; pp 1-10.
1. Introducción
El cáncer de tiroides es la neoplasia endocrina más frecuente, suponiendo el 1% de todos los nuevos casos de cáncer a nivel global. La mayor parte de estos tumores son tumores diferencia-dos, siendo un 80% del total carcinoma papilar (CPT) y un 15% carcinoma folicular (CFT). Estas variantes diferenciadas tienen un buen pronós-tico, con una supervivencia a 5 años del 95%1.
En los últimos años se han realizado importan-tes avances en la comprensión de la biología molecular de los carcinomas diferenciados de tiroides (CDT) que ha permitido establecer clasi-ficaciones moleculares que permiten discriminar a los pacientes mejor que las clasificaciones clí-nico-patológicas tradicionales. Tradicionalmente, la patogénesis de los CDT se ha relacionado con alteraciones genéticas y epigenéticas de la vía RAS-RAF-MAPK, además de disregulaciones en los mecanismos de neoangiogénesis y linfangio-génesis.
La publicación de los datos del The Cancer Ge-nome Atlas (TCGA) con datos de muestras de CPT, ha permitido añadir nuevas vías y altera-ciones moleculares que han reducido la fracción de estas neoplasias sin alteraciones genómicas conocidas del 25 al 3.5%. Los datos aportados por el TCGA han permitido confirmar el papel de la vía de las MAPK en la patogénesis del CDT y la baja carga mutacional del cáncer de tiroides2.
En el TCGA se estudiaron 496 muestras de pa-cientes con CPT, de las cuales el 69.4% eran his-tologías clásicas y el 21.2% variantes foliculares2. Esto ha permitido profundizar en las alteraciones genómicas del CPT, pero todavía no hemos al-canzado este grado de conocimiento en el CFT ni en variantes más agresivas del CDT como el carcinoma pobremente diferenciado.
2. Patogénesis molecular del CDT
La vía de las MAPK tiene un papel fundamental en la patogénesis del CPT. Esta vía regula el cre-cimiento, diferenciación y supervivencia celular en respuesta a factores de crecimiento, hormo-nas y citoquinas. El 70% de los CPT presentan mutaciones activadoras en BRAF ó RAS, así como fusiones de RET y NRTK1, sugiriendo que la activación de las MAPK es fundamental para la iniciación de esta neoplasia3. (figura 1)
Dentro del CPT podemos distinguir dos gran-des grupos moleculares en base a la mutación driver: BRAF V600E y RAS. Estas dos variantes presentan diferencias fenotípicas y que condi-cionan diferentes pronósticos y sensibilidad a los tratamientos. En el TCGA se desarrolló un perfil de expresión génica con 71 genes denominado BRAF-RAS-score que permite diferenciar entre el subtipo BRAF-like y RAS-like con diferencias en cuanto al status de la mutación driver y el grado de diferenciación tumoral2.
2. Biología molecular del cáncer diferenciado de tiroides: avanzando en la medicina de precisión de nuestros pacientes
JAUME CAPDEVILAJORGE HERNANDO-CUBEROUnidad de Tumores Gastrointestinales y Endocrinos, Servicio de Oncología Médica.Hospital Universitario Vall d´Hebron (Barcelona)

17ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES16 2. Biología molecular del cáncer diferenciado de tiroides: avanzando en la medicina de precisión de nuestros pacientes
Aunque menos estudiado, el CFT presenta un perfil de alteraciones genéticas diferentes al CPT. Las más frecuentes incluyen las mutaciones de RAS, la translocación de PAX8-PPARγ y las alte-raciones de la vía PI3K/AKT4.
El carcinoma de tiroides pobremente diferen-ciado es una variante poco frecuente que deriva de las células foliculares que pierden su diferen-ciación de manera progresiva. Las alteraciones genéticas más frecuentemente descritas inclu-yen las mutaciones de TP53 (15-30%)5, catenina (25%)6, RAS (18-27%)7 y BRAF (15%)8.
BRAF, que se encuentra con mayor frecuencia en casos asociados a radiación11,12.
La presencia de BRAF mutado en CDT tiene una fuerte asociación con ciertos subtipos his-tológicos. Aparece en el 60% de CPT de his-tología clásica y hasta en el 80% del subtipo de células altas13. Por el contrario, solo se ha descrito en el 10% de las variantes foliculares del CPT14,15.
Los tumores con mutación en BRAF se asocian con una mayor agresividad, incluyendo exten-sión extratiroidea, afectación ganglionar y me-tástasis a distancia13,16. También se considera un factor pronóstico independiente en pacientes con enfermedad localizada en estadios I-II, una menor sensibilidad al radioyodo y unas mayo-res tasas de recidiva17.
3.1.1. Sinergia BRAF V600E y TERTRecientemente, se han descrito las mutacio-nes C228T y C250T del promotor del gen TERT, la telomerasa transcriptasa inversa, en CDT. Estas mutaciones aparecen en el 12% de los casos, tanto en CPT como en CFT, y condiciona una mayor agresividad tumoral18.
Se ha descrito la coexistencia de mutacio-nes en BRAF y TERT en un 7% de los CPT. La presencia de ambas alteraciones con-diciona una mayor tasa de recurrencias y una peor supervivencia, significativamente más alta que la suma de los pacientes con mutaciones de BRAF o TERT de forma in-dependiente, lo que sugiere un mecanismo sinérgico de ambos genes e identifica un subgrupo de pacientes de muy mal pronós-tico19.
3.2. RASLos tres genes de la familia RAS (HRAS, KRAS, NRAS) codifican proteínas relacionadas con la propagación de señales entre los receptores de membrana y las dianas intracelulares. Las mutaciones puntuales en estos genes produ-cen una activación permanente de RAS con estimulación crónica de la vía de las MAPK. En los CDT, las mutaciones más frecuentes son la mutación NRAS en el codón 61 y la mutación de HRAS en el codón 6120,21.
3. Alteraciones genéticas en CDT
3.1. BRAFLa mutación de BRAF es la alteración genética más frecuente en el CPT, apareciendo en el 45% de los casos. La gran mayoría (>95%) de las mu-taciones consisten en la sustitución de una valina por ácido glutámico en la posición 600 (V600E). Esta mutación induce una activación permanente de la quinasa BRAF con estimulación crónica de la vía MAPK9. Otras alteraciones descritas inclu-yen la mutación K601E, relacionada con el subti-po folicular del CPT10, y la translocación AKAP9-
Estas mutaciones pueden aparecer en todos los subtipos de CDT. En el CFT suponen el 40-50% de los casos22. En el CPT se encuentra en el 10% de los casos, siendo la práctica totalidad de la variante folicular del CPT, relacionándose con una menor agresividad locorregional y una menor extensión ganglionar23,24.
3.3. RET/PTCLa translocación RET/PTC es la segunda alteración genética más frecuente en el CPT, presente en el 20% de los casos25. Esta translocación es el resulta-do de la fusión entre la porción 3´ del gen RET y la porción 5´ de varios genes, siendo los más habitua-les PTC1 (60-70% de las translocaciones) y PTC3 (20-30%). Otras translocaciones menos frecuentes como PTC2 suponen <5% de los casos26. La por-ción de RET siempre contiene el dominio tirosina quinasa intacto, por lo que la proteína de fusión es capaz de activar la vía RAS-RAF-MAPK27.
Estas translocaciones aparecen con mayor fre-cuencia en pacientes con historia de radiación (50%)28. y se relaciona con pacientes jóvenes, his-tología clásica de CPT y con la afectación ganglio-nar29. Es poco frecuente en CFT30.
3.4 PAX8-PPARγLa translocación PAX8-PPARγ se produce entre los cromosomas 2 y 3, t(2:3)(q13;p25). Incluye la fusión entre el gen PAX8 que codifica un factor de transcripción específico del tiroides y el gen PPARγ31.El resultado es una sobreexpresión de la proteína PPARγ pero todavía no se conocen con exactitud los mecanismos celulares que implica esta alteración.
Aparece en el 30-40% de los CFT y se relaciona con una presentación más temprana, con menor tamaño y con infiltración vascular32. Es poco fre-cuente en el CPT (<5%) y siempre se relaciona con la variante follicular33.
3.5 PI3K/AKTLa vía PTEN/PI3K/AKT participa en importantes procesos relacionados con la actividad celular in-cluyendo la división, supervivencia, adhesión, moti-lidad y angiogénesis. En CDT se ha observado una inactivación de PTEN y una activación de AKT, similar a otras neoplasias sólidas. Se han descrito mutaciones en el gen PIK3CA en un 6-13% de los CFT34.
Figura 1. La vía de las MAPK en el cáncer de tiroides. Adaptado de: Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer 2013;13(3):184-99

19ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES18 2. Biología molecular del cáncer diferenciado de tiroides: avanzando en la medicina de precisión de nuestros pacientes
3.6 Inestabilidad de microsatélitesLa inestabilidad de microsatélites (MSI) es una manifestación de la hipermutabilidad genética resultante de los defectos en los mecanismos de reparación del ADN. Según esta hipermutabili-dad, los tumores pueden clasificarse en: alta fre-cuencia de MSI (MSI-H), baja frecuencia de MSI (MSI-L) y estabilidad de microsatélites (MSS)35. Se ha encontrado MSI como parte de la secuen-cia de malignización de adenoma a carcinoma36.
La experiencia en la determinación de MSI en CDT avanzado es limitada en comparación con otras neoplasias como el carcinoma colorrec-tal35. En las series más recientes, la presencia de MSI aparece aproximadamente en el 60% de los CDT, tanto CPT como CFT. Todavía no se ha lo-grado establecer una relación entre la presencia de MSI-H y variables clínicas, patológicas o pro-nósticas37, probablemente debido al bajo núme-ro de casos analizados.
4. Mecanismos moleculares relacionados con la angiogénesis y linfangiogénesis
Los mecanismos de formación de nuevos vasos sanguíneos (angiogénesis) y linfáticos (linfangio-génesis) a partir de la vasculatura previa es un mecanismo fundamental en la proliferación y di-seminación del cáncer. En los últimos años se ha profundizado en los mecanismos moleculares de la angiogénesis tumoral en el CDT, que presenta algunas particularidades con respecto al resto de neoplasias, y que han permitido desarrollar nue-vas terapias dirigidas38.
4.1. AngiogénesisEl tejido tiroideo normal tiene el mayor flujo de sangre por unidad de peso de todo el organismo, gracias a una rica vasculatura finamente organiza-da. Durante los procesos de transformación que conducen al CDT, el tejido tiroideo pierde parcial-mente esta vasculatura, como se ha demostrado gracias a marcadores endoteliales como CD3439.
En este proceso de malignización se produce un disbalance entre los factores activadores e inhibi-dores de la angiogénesis40, que influyen la familia VEGF (vascular endotelial growth factor), EGF (epidermal growth factor) y PDGF (platelet deri-ved growth factor) entre otros41.
5.2. InmunoterapiaEn los últimos años se han llevado a cabo estu-dios con inmunoterapia en tumores resistentes a los tratamientos convencionales. Las familias de fármacos más desarrolladas son los anti-PD-1/PD-L1 y anti-CTLA4. Se ha demostrado que los tumores que presentan una alta carga mutacio-nal son capaces de estimular la acción del siste-ma inmune, especialmente aquellos con altera-ciones en los sistemas de reparación del DNA64. En CDT se han reportado unas tasas de positivi-dad para PD-L1 en linfocitos infiltrantes de tumor (TIL) del 28% en CPT y del 9% en CFT65.
Con respecto al CDT, los datos obtenidos en el TCGA indican que es un tumor con baja car-ga mutacional2, lo cual podría condicionar una menor sensibilidad a la inmunoterapia, aunque como hemos visto previamente podemos encon-trar MSI hasta en el 60% de los casos37.
Durante el crecimiento del CDT se produce un aumento en los niveles de expresión de VEGF-A que activa el proceso angiogénico42. Esta vas-culatura tumoral es muy abundante aunque no alcanza el grado de organización que sí encon-tramos en el tiroides normal43. Los niveles de expresión de VEGF-A condicionan un peor pro-nóstico, asociándose los niveles elevados con la diseminación linfática y el riesgo de recurren-cia44.
Se ha descrito un incremento progresivo en los niveles de FGF-2 en las lesiones benignas sobre el tejido normal y en las lesiones malignas sobre las benignas45. Se ha propuesto que la expresión de FGF-2 podría tener un valor como potencial factor pronóstico46.
4.2. LinfangiogénesisLa presencia de vasos linfáticos intratumorales, determinados mediante LYVE-1, se relacionan con las metástasis ganglionares en el momento del diagnóstico38. Además, los niveles de VEGF-C se correlacionan con la presencia de metásta-sis ganglionares al diagnóstico47.
Todos estos datos en conjunto nos sugieren que la angiogénesis y la linfangiogénesis son dos procesos complejos e interrelacionados que juegan un factor importante en el desarrollo del CDT.
5. Implicaciones terapéuticas de la biología molecular del CDT
5.1. Terapias dirigidasEl incremento en el conocimiento de la biología molecular del CDT en los últimos años ha per-mitido desarrollar múltiples terapias dirigidas en este escenario. Las familias de fármacos que han demostrado eficacia incluyen los Inhibidores de Tirosina Quinasa (ITK) con actividad antiangio-génica, los Inhibidores de BRAF y los Inhibido-res de mTOR48. Actualmente los fármacos apro-bados para el tratamiento del CDT refractario a tratamiento con yodo radiactivo en progresión son: Sorafenib y Lenvatinib49.
En la tabla 1 se describen las terapias aprobadas actualmente y otros tratamientos dirigidos en desarrollo en CDT. (tabla 1)
En el ensayo KEYNOTE-028 se exploró la efi-cacia del inhibidor de PD-1 Pembrolizumab en diferentes neoplasias sólidas con una expresión de PD-1 ≥1%. Con respecto al CDT, se trataron dentro de este estudio 22 pacientes con una tasa de respuestas del 9.1%, incluyendo 2 respuestas parciales.66
Aunque estos datos de respuesta a la inmunote-rapia no son tan llamativos como en otras neopla-sias sólidas, se ha demostrado actividad en CDT. En esta línea se está investigando la actividad de Pembrolizumab en monoterapia en CDT dentro del estudio KEYNOTE-158 (NCT02628067) y en combinación con lenvatinib (NCT02973997).
En la tabla 2 se describen otros ensayos en mar-cha con inmunoterapia en CDT. (tabla 2)
Tabla 1. Terapias dirigidas aprobadas ó en desarrollo en CDT.
Sorafenib50
Lenvatinib52
Sunitinib54
Motesanib55
Axitinib56
Gefitinib57
Pazopanib58
Nintedanib59
Dabrafenib60
Vemurafenib61
Selumetinib62
Everolimus63
Sí
Sí
No
No
No
No
No
No
No
No
No
No
VEGFR, PDGFR, c-Kit, RET
VEGFR, PDGFRβ, RET, c-Kit y FGFR
VEGFR, PDGFR, c-Kit, RET
VEGFR, PDGFR, c-Kit
VEGFR, PDGFR, c-Kit
EGFR
VEGFR, PDGFR y c-Kit
VEGFR, PDGFR, FGFR
BRAF
BRAF
MEK
mTORC1
III DECISION51
III SELECT53
II
II
II
II
II
II
II
II
II
II
Fármaco Aprobación Dianas Fase
Tabla 2. Inumonterapia en desarrollo en CDT.
Pembrolizumab
Pembrolizumab + Lenvatinib
Nivolumab + Ipilimumab
Atezolizumab
CDT
CDT
CDT
Pobremente diferenciado
PD-1
PD-1
PD-1/CTLA-4
PD-L1
II
II
II
II
NCT02628067
NCT02973997
NCT02834013
NCT03181100
Fármaco Histología Diana Fase Código

21ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES20 2. Biología molecular del cáncer diferenciado de tiroides: avanzando en la medicina de precisión de nuestros pacientes
BIBLIOGRAFÍA
1. Hay ID, Thompson GB, Grant CS, et al. Papillary thyroid carcinoma managed at the Mayo Clinic during six decades (1940-1999): tem-poral trends in initial thera-py and lonf-term outcome in 2444 consecutively trea-ted patients. World J Surg 2002;26:879-885.
2. The Cancer Genome Atlas Research Network. Integrated Genomic Cha-racterization of Papillary Thyroid Carcinoma. Cell 2014;159:676-690.
3. Nikiforova MN, Nikiforov YE. Molecular genetics of thyrod cáncer: implications for diagnosis, treatment and prognosis. Expert Rev Mol Diagn 2008;8(1):83-95.
4. Swierniak M, Pfeifer A, Stokowy T, et al. Somatic mutation profiling of folicular thyroid cáncer by next gene-ration sequencing. Mol Cel Endocrinol 2016;433:130-137.
5. Fagin FA, Matsuo K, Kar-makar A, et al. High preva-lence of mutations of the p53 gene in poorly differentiated and undifferentiated carci-nomas of the thyroid gland. J Clin Invest 1993;91:179-184.
6. Garcia-Rostan G, Camp RL, Herrero A, et al. cate-nin dysregulation in thyroid neoplasms: down-regulation, aberrant nuclear expression, and CTNNB1 exon 3 muta-tions are markers for aggres-sive tumor phenotypes and por prognosis. Am J Pathol Int 2001;51:680-685.
7. Asakawa H, Kobayashi T. Multistep carcinogenesis in anaplastic thyroid carcino-ma: a case report. Pathology 2002;34:94-97.
8. Nikiforova MN, Kimura ET, Gandhi M, et al. BRAF muta-tions in thyroid tumors are restricted to papillary car-cinomas and anaplastic or poorly differentiated carci-nomas arising from papillary carcinomas. J Clin Endocrinol Metab 2003;88:5399-5404.
9. Wan PT, Garnett MJ, Roe SM, et al. Mechanism of acti-vation of the RAF-ERK sig-naling pathway by oncoge-nic mutations of B-RAF. Cell 2004;116:855-867.
10. Trovisco V, Soares P, Preto A, et al. Type and pre-valence of BRAF mutations are closely associated with papillary thyroid carcinoma histotype and patients age but not with tumour ag-gressiveness. Virchovs Arch 2005;446:589-595.
11. Trovisco V, Vieira de Cas-tro I, Soares P, et al. BRAF mutations are associated with some histological types of papillary thyroid carcino-ma. J Pathol 2004;202:247-251.
12. Ciampi R, Knauf JA, Kerler R, et al. Oncogenic AKAP9-BRAF fusion is a novel me-chanism of MAPK pathway activation in thyroid cáncer. J Clin Invest 2005;115:94-101.
13. Nikiforova MN, Kimura ET, Gandhi M, et al. BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carci-nomas arising from papillary carcinomas. J Clin Endocri-nol Metab 2003;88:5399-5404.
14. Adeniran AJ, Zhu Z, Gan-dhi M, et al. Correlation bet-ween genetic alteartions and microscopi features, clinical manifestations and prognos-tic characteristics of thyroid papillary carcinomas. Am J Surg Pathol 2006; 30:216-222.
15. Xing M. BRAF mutation in thyroid cancer. Endocr Relat Cancer 2005;12:245-262.
16. Xing M, Westra WH, Tu-fano RP, et al. BRAF muta-tion predicts a poorer clini-cal prognosis for papillary thyroid cancer. J Clin Endo-crinol Metab 2005;90:6373-6379.
17. Kim TY, Kim WB, Rhee YS, et al. The BRAF mutation is useful for prediction of cli-
nical recurrence in low-risk patients with conventional papillary thyroid carcino-ma. Clin Endocrinol (Oxf) 2006;65:364-368.
18. Liu X, Bishop J, Shan Y, et al. Highly prevalent TERT promoter mutations in aggressive thyroide cán-cer. Endocr Relat Cancer 2013;20:603-6010.
19. Mingzhao Xing, Rengyun Liu, Xiaoli Liu, et al. BRAF V600E and TERT promo-ter mutations cooperatively identify the most aggressive papillary thyroid cáncer with highest recurrence. J Clin Oncol 2014;32(25):2718-26.
20. Suarez HG, du Villard JA, Severino M, et al. Presence of mutations in all three ras ge-nes in human thyroid tumors. Oncogene 1990;5:565-570.
21. Basolo F, Pisaturo F, Po-llina LE, et al. N-ras muta-tion in poorly differentiated thyroid carcinomas: corre-lation with bone metastases and inverse correlation to thyroglobulin expression. Thyroid 2000;10:19-23.
22. Garcia-Rostan G, Zhao H, Camp RL, et al. Ras mu-tations are associated with aggressive tumor pheno-types and por prognosis in thyroid cáncer. J Clin Oncol 2003;21:3226-3235.
23. Vasko VV, Gaudart J, Allasia C, et al. Thyroid fo-licular adenomas may dis-plasy features of follicular carcinoma and follicular variant of papillary car-cinoma. Eur J Endocrinol 2004;151:779-786.
24. Zhu Z, Gandhi M, Niki-forova MN, et al. Molecular profile and clinical-patholo-gic features of the folicular variant of papillary thryoid carcinoma. An unusually high prevalence of ras mu-tations. Am J Clin Pathol 2003;120:71-77.
25. Bongarzone I, Monzini N, Borrello MG, et al. Molecular characterization of a thyroid tumor-specific transofrming
sequence formed by the fu-sión of ret tyrosine kinase and the regulatory subunit RIα of cyclic AMP-dpendent protein kinase A. Mol Cell Biol 1993;13:385-366.
26. Tallini G, Santoro M, He-lie M, et al. RET/PTC onco-gene activation defines a subset of papillary thyroid carcinomas lacking evidence of progression to poorly di-fferentiated or undifferentia-ted tumor phenotypes. Clin Cancer Res 1998;4:287-294.
27. Saenko V, Rogounovitch T, Shimizu-Yoshida Y, et al. Novel tumorigenic rearran-gement, rfp/ret, in a papi-llary thyroid carcinoma from externally irradiated patient. Mutat Res 2003;527:81-90.
28. Rabes HM, Demidchik EP, Sidorow JD, et al. Pat-tern of radiation-induce RET and NTRK1 rearrangements in 191 post-chernobyl pa-pillary thyroid carcinomas: biological, phenotypic and clinical implications. Clin Cancer Res 2000;6:1093-1103.
29. Adeniran AJ, Zhu Z, Gandhi M, et al. Correlation between genetic alteartions and microscopi features, clinical manifestations and prognostic characteristics of thyroid papillary carci-nomas. Am J Surg Pathol 2006; 30:216-222.
30. Tallini G, Santoro M, He-lie M, et al. RET/PTC onco-gene activation defines a subset of papillary thyroid carcinomas lacking evidence of progression to poorly di-fferentiated or undifferentia-ted tumor phenotypes. Clin Cancer Res 1998;4:287-294.
31. Kroll TG, Sarraf P, Pec-ciarini L, et al. PAX8-PPARα 1 fusion oncogene in human thryoid carcinoma. Science 2000;289:1357-1360.
32. French CA, Alexander EK, Cibas ES, et al. Genetic and biological subgroups of low-stage folicular thyroid cáncer. Am J Pathol 2003;162:1053-1060.
6. Conclusiones
• El cáncer diferenciado de tiroides (CDT) es la neoplasia endocrina más frecuen-te, suponiendo el 80% de los tumores malignos de esta glándula.
• La patogénesis del CDT se relaciona fundamentalmente con la vía RAS-RAF-MAPK. El 70% de los CPT presentan mutaciones en BRAF ó RAS.
• El CPT se clasifica en dos grandes grupos moleculares según la mutación like, los BRAF-like y los RAS-like, con diferencias en cuanto a pronóstico.
• Los perfiles genómicos del CFT y del pobremente diferenciado presentan ca-racterísticas propias, siendo las alteraciones más frecuentes las mutaciones de RAS y de TP53 respectivamente.
• La mutación V600E de BRAF es la alteración genética más frecuente en el CPT y condiciona una mayor agresividad y menor sensibilidad al tratamiento.
• La presencia de mutaciones conjuntas en BRAF y TERT tienen un efecto sinér-gico e identifican un subgrupo de pacientes de muy mal pronóstico.
• La translocación de RET/PTC es la segunda alteración genética más frecuente en CPT y se relaciona con pacientes jóvenes y exposición a la radiación.
• Las alteraciones genéticas más frecuentes en el CFT son la mutación de la fa-milia de genes RAS y la translocación PAX8-PPARγ.
• Las mutaciones de la familia de genes RAS son frecuentes en el CFT, relacionán-dose con una menor agresividad locorregional.
• La inestabilidad de microsatélites (MSI) aparece en el 60% de los CDT, aunque todavía no se ha correlacionado con factores pronósticos.
• Los procesos de angiogénesis y linfangiogénesis tienen un papel fundamental en la patogénesis del CDT. Algunos de los factores pro/antiangiogénicos impli-cados incluyen VEGF, EGF y PDGF entre otros.
• Las terapias dirigidas aprobadas actualmente en CDT son sorafenib y lenvatinib.
• Las nuevas terapias dirigidas en cáncer de tiroides en desarrollo incluyen los antiangiogénicos, los inhibidores de BRAF y los inhibidores de mTOR.
• La baja carga mutacional del CDT condiciona una menor sensibilidad al tra-tamiento con inmunoterapia, aunque se ha demostrado actividad con terapia anti-PD-1.

23ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES22 2. Biología molecular del cáncer diferenciado de tiroides: avanzando en la medicina de precisión de nuestros pacientes
33. Dwight T, Thoppe SER, Foukakis T, et al. Genetic and biological subroups of low-stage folicular thyroid cáncer. Am J Endorcinol Me-tabol 2003;88:4440-4445.
34. Garcia-Rostan G, Cota AM, Pereira-Castro I, et al. Mutation of the PIK3CA gebe ub anaplastic thryoid cáncer. Cancer Res 2005;65:10199-10207.
35. Boland R, Goel A. Micro-satellite Instability in Colo-rectal Cancer. Gastroentero-logy 2010;138(6):2073-2087.
36. Soares P, dos Santos NR, Seruca R, et al. Benigna n malignant thyroid lesions show instability at micro-satellite loci. Eur J Cancer 1997;33:293.
37. Mitmaker E, Alvarado C, Bégin LR, et al. Microsatellite Instability in Benign and Ma-lignant Neoplasms. J Surg Res 2008;150:40-48.
38. Garcia de la Torre N, Buley I, Wass JAH, et al. Angiogenesis and lymphan-giogenesis in thyroid proli-ferative lesions: relationship to type and tumour beha-viour. Endocr-Rel Cancer 2006;13:931-944.
39. Garcia de la Torre N, Buley I, Wass JAH, et al. Angiogenesis and lymphan-giogenesis in thyroid proli-ferative lesions: relationship to type and tumour beha-viour. Endocr-Rel Cancer 2006;13:931-944
40. Carmeliet P, Jain RK. Angiogenesis in cáncer and other diseases. Nature 2000;407:249-257.
41. Ye L, Santarpia L, Gagel RF. Tehe evolving field of tyrosine kinase inhibitors in the treatment of endocrine tu-mors. Endocr Rev 2010;31:578-599.
42. Xie K, Wei D, Shi Q, et al. Constitutive and inducible ex-pression and regulation of vas-cular endothelial growth fac-tor. Cytokine & Growth Factor Reviews 2004;15:297-324.
43. Garcia de la Torre N, Buley I, Wass JAH, et al. Angiogenesis and lymphan-giogenesis in thyroid proli-ferative lesions: relationship to type and tumour beha-viour. Endocr-Rel Cancer 2006;13:931-944.
44. Huang SM, Lee JC, Wu TJ, et al. Clinical relevance of vascular endothelial growth factor for thyroid neoplasms. World Journal of Surgery 2001;25:302-306.
45. Eggo MC, Hopkins JM, Franklyn JA, et al. Expres-sion of fibroblast growth fac-tors in thyroid cáncer. Jour-nal of Clin Endocrinol and Metab 1995;80:1006-1011.
46. Boealert K, McCabe CJ, Tannahill LA, et al. Pituitary tumor transforming gene and fibroblast growth fac-tor-2 expression: potential prognostic indicators in di-fferentiated thyroid cáncer. J Clin Endocrin and Metabol 2003;88:2341-2347.
47. Yu XM, Lo CY, Chan WF, et al. Increased expression of vascular endothelial growth factor C in papillary thyroid carcinoma correlates with cervical lymph node me-tastases. Clin Cancer Res 2005:11:8063-8069.
48. Fallahi P, Mazzi V, Vita R, et al. New Therapies for Dedifferentiated Papillary Thyrod Cancer. Int J Mol Sci 2015;16:6153-6182.
49. Valerio L, Pieruzzi C, Giani L, et al. Targeted The-rapy in Thyroid Cancer: State of the Art. Clin Oncol 2017;29:316-324.
50. Fallahi P, Ferrari SM, San-tini F, et al. Sorafenib and thyroid cáncer. BioDrugs 2013;27:615-628.
51. Brose MS, Nutting CM, Jarzab B, et al. So-rafenib in radioactive io-dine-refractory, locally advanced or metastatic differentiated thyroid cán-cer: a randomised, double-blind, phase 3 trial. Lancet 2014;384(9940):319-28.
52. Glen H, Mason S, Patel H, et al. E7080, a multi-tar-geted tyrosine kinase inhi-bitor suppresses tumor cell migraton and invasión. BMC Cancer 2011;11:309.
53. Schlumberger M, Tehara M, Wirth LJ, et al. Lenvatinib versus placebo in radioiodi-ne-refractory thyroid cancer. N Eng J Med 2015;372:621-630.
54. Rini BI. Sunitinib. Ex-pert Opin Pharmacother 2007;8:2359-2369.
55. Polverino A, Coxon A, Starnes C, et al. AMG 706, an oral, multikinase inhibitor that selectively targets vas-cular endotelial growth fac-tor, platelet-derived growth factor, and Kit receptors, potently inhibits angiogéne-sis and induces regression in tumor xenografts.
56. Patson B, Cohen RB, Ol-szanski AJ. Pharmacokinetic evaluation of axitinib. Expert Opin Drug Metab Toxicol 2012;8:259-270.
57. Schiff BA, McMurphy AF, Jasser SA, et al. Epider-mal growth factor receptor (EGFR) is overexpressed in anaplastic thyroid cán-cer, and the EGFR inhibi-tor gefitinib inhibits the growth of anaplastic thyroid cáncer. Clin Cancer Res 2004;10:8594-8602.
58. Stenberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or me-tastatic renal cell carcino-ma: Results of a randomzied phase III trial. J Clin Oncol 2010;28:1061-1068.].
59. Cenik BK, Ostapoff KT, Gerber DE, et al. BIBF1120 (Nintedanib), a triple an-giokinase inhibitor, induces hipoxia but not EMT and blocks progression of pre-clinicals models of lung and pancreatic cáncer. Mol Can-cer Ther 2013;12(6):992-1001.
60. Falchook GS, Long GV, Kurzrock R, et al. Dabrafenib in patients with melanoma, utnreated brain metastases,
and other solid tumours: A phase 1 dose-escalation trial. Lancet 2012;379:1893-1901.
61. Sharma A, Shah SR, Illum H, et al. Targeted inhibi-tion of mutated BRAF for treatment of advanced me-lanoma and its potential in other malignancies. Drugs 2012;72:2207-2222.
62. Adjei AA, Cohen RB, Franklin W, et al. Phase I pharmacokinetic and phar-macodynamic study of the oral, small-molecule mito-gen-activated protein kina-se 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol 2008;26:2139-2146.
63. De Souza EC, Padrón AS, Braga WM, et al. MTOR down-regulates iodide up-take in thyrocytes. J Endo-crinol 2010;206:113-120.
64. Le DT, Uram JN, Wang H, et al. PD-1 blockade in tu-mors with mismatch-repair deficiency. N Eng J Med 2015;372:2509-2520
65. Ahn S, Kim TH, Kim SW, et al. Comprehensive scree-ning for PD-L1 expression in thyroid cancer. Endocr Relat Cancer 2017;24:97-106.
66. Mehnert JM, Varga A, Marcia Brose M, et al. Pem-brolizumab for advanced papillary or folicular thyroid cáncer: preliminary results from the phase 1b KEYNO-TE-028 study. J Clin Oncol 2016;34 (suppl; abstr 6091).
Introducción
El cáncer diferenciado de tiroides deriva de las células del epitelio folicular y representa más del 90% de todos los tumores tiroideos. Las histologías más frecuentes son los carcinomas papilares y foliculares, mientras que otras va-riantes como el carcinoma de células de Hürthle o los carcinomas pobremente diferencias son menos comunes1.
Globalmente, el pronóstico es excelente, alcan-zando tasas de supervivencia a 10 años del 80-95%, especialmente es caso de neoplasia papi-lar (que alcanzan cifras del 93-95%). La edad, el tamaño tumoral, la extensión de la enfermedad y la histología son factores importantes en el riesgo de recurrencia y fallecimiento. En ausen-cia de invasión local, afectación ganglionar en cadena lateral o metástasis a distancia, los pa-cientes, tras la resección quirúrgica, tiene una expectativa de vida similar a la población nor-mal2.
A pesar de la efectividad del tratamiento están-dar para el carcinoma diferenciado de tiroides (CDT), entre el 10 y 20% de los pacientes tienen una recurrencia a los 5-15 años del diagnósti-co inicial3. Así, a diferencia de otras entidades neoplásicas en las que el riesgo de recurrencia y mortalidad específica están íntimamente rela-cionados, en los casos de CDT no es infrecuente ver pacientes con alto riesgo de recurrencia,
pero con muy baja mortalidad. Esta caracterís-tica condiciona los sistemas de clasificación y estadificación de los pacientes, haciendo que los más habitualmente empleados en oncolo-gía (TNM) no se adapten a la aplicación en la práctica. La utilización de clasificaciones de riesgo basadas en guias4,5 y la evaluación de la respuesta al tratamiento (evaluación dinámica de riesgo)6 modifica el riesgo inicial de los pa-cientes en función de respuestas completas, in-completas (estructurales o bioquímicas) o pro-gresión. De este modo, pacientes inicialmente clasificados como de bajo riesgo o intermedio, pero que permanecen con enfermedad estruc-tural a los 12-14 meses modifican su riesgo de recurrencia7.
Se estima que alrededor de un 20% de los pa-cientes con enfermedad locorregional al mo-mento del diagnóstico desarrollarán metástasis en algún momento de la evolución. Las localiza-ciones más frecuentes son el pulmón y hueso. Si dichas lesiones mantienen la capacidad de cap-tación de radioyodo, la supervivencia de dichos pacientes puede alcanzar casi el 90% a 10 años8. Alrededor de un tercio de los pacientes con en-fermedad persistente o metastásica pierden la capacidad de captar radioyodo, aumentando de manera significativa la mortalidad. La mayoría de pacientes que fallecen por CDT pertenecen al grupo de radiorefractarios1, 4.
3. El concepto de la refractariedad al radioyodo en los pacientes con carcinoma diferenciado de tiroides: estandarización del manejo de los pacientes
JUAN ANTONIO VALLEJO CASASMARÍA VICTORIA GUIOTE MORENO ANTONIO MARÍA SANTOS BUENO UGC Medicina Nuclear. Hospital Universitario Reina Sofía. Instituto de Investigación Biomédica Maimónides de Córdoba (IMIBIC). Universidad de Córdoba

25ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES24 3. El concepto de la refractariedad al radioyodo en los pacientes con carcinoma diferenciado de tiroides
2. Manejo del carcinoma diferenciado de tiroides
Tras la cirugía, en base a las clasificaciones de riesgo, la ablación postquirúrgica de los rema-nentes tiroideos con radioyodo en el CDT tiene como objetivo la eliminación de cualquier resto de tejido tiroideo, facilitando la detección pre-coz de recurrencias (mediante la determinación de tiroglobulina y/o rastreo corporal con131I), así como estadificar correctamente al paciente tras el rastreo posterapia. Además, la ablación puede representar una terapia adyuvante, al eliminar focos microscópicos de tejido neoplásico en el remanente4,5,9.
Mientras que el concepto de ablación se relacio-na con el seguimiento de pacientes en función de su riesgo específico, el concepto de terapia adyuvante debe de considerarse como una he-rramienta para la reducción de recurrencia y de las tasas de mortalidad específica4,5,9.
La estratificación de riesgo es la piedra angular para las decisiones en el manejo de los pacientes con CDT. Aunque en sentido estricto las decisio-nes se toman en base al riesgo de recurrencia y de mortalidad específica, valorando el riesgo-beneficio de la terapia, la realidad es que otros muchos factores condicionan la elección, por lo que se recomienda que la evaluación de todos los parámetros se realice de manera individuali-zada y en el contexto clínico concreto4.
Habrá que considerar:
• La situación postquirúrgica (presencia o au-sencia de enfermedad persistente).
• Los niveles postquirúrgicos de tiroglobulina (nos indicarán la posibilidad de enfermedad persistente, grandes remanentes y serán predictores pronósticos). No está definido el umbral para la opción de tratar o no tratar.
• El valor del rastreo preterapéutico es limi-tado. Se realizará cuando no se pueda es-tablecer la extensión de la enfermedad tras la cirugía y de este dato pueda depender la elección del tratamiento.
3. Yodo radioactivo
El 131I es un isótopo radioactivo, que se produce en reactores nucleares mediante la irradiación de neutrones de dióxido de telurio y en el proceso de fisión del uranio.
Tiene un semiperiodo físico de 8,02 días y se desintegra mediante emisión b- (emisión de elec-trones) de una energía media de 191 keV (máxima de 606 keV) y emisión g de diferentes energías (entre 364 y 637 keV). La emisión g se utiliza para la obtención de imagen y es la que condiciona la necesidad de medidas de radioprotección.
Una vez administrado, generalmente por vía oral (en forma de cápsula o solución oral) se absorbe en el tracto gastrointestinal, pasando a la circula-ción sistémica. Rápidamente pasa al fluido extra-celular, de donde es aclarado hacia la célula tiroi-dea con una tasa aproximada de 17 ml/min. El 131I seguirá las mismas vías fisiológicas y metabólicas que el yodo no radioactivo, siendo incorporado a la célula tiroidea mediante el simportador Na/I. Este simportador es un elemento clave en el pro-ceso debiendo expresarse en la superficie de la membrana de la célula folicular tiroidea para la incorporación del yodo10.
El órgano diana será la célula tiroidea y la do-sis absorbida va a depender de la capacidad de captación de yodo (en relación a la situación de estimulación del receptor de TSH, de la expresión del simportador y del estado de los depósitos de yodo), de la masa de tejido residual tras la inter-vención y de la situación global del paciente, en cuanto a su capacidad para eliminar (vía urinaria fundamentalmente). Otros órganos que hay que considerar, porque también van a concentrar el yodo, son las glándulas salivales y el estómago. Como la vía fundamental de excreción es la uri-naria, hay que tener presente la dosimetría que puede recibir la vejiga.
La incorporación del 131I a las células del epitelio tiroideo va a producir en las mismas una lesión inducida por la radiación y finalmente la muerte celular, como suma de los procesos de genera-ción de radicales libres y las lesiones del DNA. Es importante considerar que la el tejido tiroideo normal expresa mayor avidez por el radioyodo que las células neoplásicas11.
La eliminación de los restos de tejido tiroideo normal aumentará la sensibilidad de las determi-naciones posteriores de tiroglobulina y aumenta-rá el valor de esta determinación como marcador sensible para la recurrencia. Además, el rastreo corporal que se obtiene tras la terapia permite una mejor estadificación4,12,13.
4. Aspectos moleculares en la captación de 131I
El yodo entra en la célula tiroidea a través del simportador NIS14, una proteína que se encuentra en la membrana basal y que transporta el yodo hacia la membrana apical. La función de NIS está regulada de manera positiva por la TSH (vía ac-tivación de AMPc) y negativamente por IGF1//PI3K/AKT y TGFb/Smads15.
La mutación V600E de BRAF, los reordenamien-tos RET/PTC y las mutaciones en RAS se han relacionado con la génesis de los carcinomas papilares. BRAFV600E es el evento genético más frecuente en carcinoma papilar de tiroides. Su presencia se asocia a recurrencia, ya que condu-ce a las células hacia la indiferenciación, favore-ciendo la extensión extratiroidea y la persistencia de la enfermedad.
La expresión de las vías descritas es mutuamente excluyente y las tres activan un programa común de expresión génica dependiente de la vía MEK-ERK, esencial para la proliferación y la capacidad invasiva de estos tumores. De manera similar, las mutaciones en genes de la vía PI3K, mutaciones puntuales en AKT, mutaciones y amplificaciones en PI·KCA y mutaciones inactivadoras en el gen supresor PTEN, están implicadas en la génesis de los carcinomas foliculares. Otras mutaciones en el gen de la bcatenina (CTNNB1) y de P53 se han identificado en los carcinomas más indife-renciados16.
Se ha estudiado el mecanismo subyacente a la pérdida de capacidad de captar radioyodo en algunos tumores tiroideos diferenciados y que dan lugar a metástasis. Se ha demostrado que el oncogén BRAF-V600E se asocia con metástasis refractarias al radioyodo porque reprime la ex-presión de NIS tanto a nivel transcripcional como postranscripcional, siendo este efecto MEK-ERK independiente. BRAF V600E es un factor pro-
nóstico en papilar que correlaciona con el alto riegos de recurrencias y tumores menos diferen-ciados, debido a la pérdida de captación de 131I mediada por NIS17.
Así, BRAF induce la secreción de TGFb, citoquina que a través de un bucle autocrino reprime la función de NIS y promueve la transición epitelio mesénquima y la capacidad invasiva de las célu-las. Además, se ha observado que existe una alta actividad de la vía TGFb/smads en las muestras humanas con cáncer de tiroides que se asocian con invasión extratiroidea, metástasis gangliona-res, presencia del oncogén BRAF y la pérdida de expresión de NIS18.
Aunque las células del CDT conservan la mayor parte de las propiedades bioquímicas que carac-terizan a la célula folicular tiroidea, se han de-mostrado una serie de anormalidades que con-dicionan el comportamiento.
• Las neoplasias malignas muestran áreas de hipofunción en la gammagrafía tiroidea, po-niendo de manifiesto que la pérdida de cap-tación de yodo es un marcador de carcino-génesis tiroidea19.
• Disminución de la expresión de microRNA que codifica NIS comparado con el tejido normal, que puede ser el responsable de la pérdida de capacidad de concentrar yodo20.
• Algunos estudios inmunohistoquímicos han mostrado sobreexpresión de NIS en tejido neoplásico tiroideo. De hecho la localización de NIS en estos es predominantemente in-tracelular, planteando la hipótesis del error en la localización en la membrana celular es el responsable de la pérdida de capacidad de captación de yodo21.
La hormona estimulante del tiroides (TSH) au-menta la acumulación de yodo, mediante regu-lación positiva de la expresión de NIS y mRNA, a través de la activación de AMPc. En células con respuesta normal a TSH, NIS está activo e inser-tado en la membrana basolateral del tirocito; cuando se produce una deprivación del estímulo de TSH disminuye la vida media de NIS, quizás porque la proteína se traslada desde la membra-na celular a compartimentos intracelulares, como

27ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES26 3. El concepto de la refractariedad al radioyodo en los pacientes con carcinoma diferenciado de tiroides
ocurre en algunos cáncer tiroideos21,22. En au-sencia de TSH, la cantidad de NIS disminuye por estímulo de AMPK (AMP-activated protein kinasa) que favorece la degradación en los li-sosomas de NIS. La expresión de AMPK está aumentada en el carcinoma papilar de tiroi-des23.
Además de esas alteraciones a nivel de NIS, en el carcinoma diferenciado de tiroides se han identificado anomalías en la expresión del transportador apical de yodo, disminución en la expresión de pendrinas y peroxidasas, que junto a la menor captación de yodo, conducen a una baja síntesis hormonal, a una rápida li-beración del yodo intracelular y como conse-cuencia a una disminución del tiempo efectivo de estancia intracelular (T
1/2 efectivo), lo que
condiciona menor efecto terapéutico tras la administración de radioyodo24.
En la figura 1 se resumen las principales altera-ciones que condicionan la menor captación de yodo en las células de neoplasias diferenciadas tiroideas.
5. Radiorefractariedad
El CDT tiene un pronóstico favorable, aún en las situaciones de enfermedad metastásica, siendo el 131I la principal modalidad terapéutica en aque-llos pacientes que mantienen la captación. En general, el radioyodo será más efectivo en pa-cientes jóvenes y con metástasis pequeñas8.
En la tabla 2 se recogen las principales caracte-rísticas de los tumores que no responderán a 131I.
Cuando las lesiones pierden la capacidad de cap-tar radioyodo, o no muestran respuesta al trata-
miento, disminuye de manera significativa la su-pervivencia de los pacientes, alcanzando niveles similares al linfoma de Hogdkin, el osteosarcoma o el cáncer testicular25.
Alrededor de un tercio de los pacientes con car-cinoma de tiroides con enfermedad estructural locoregional y/o enfermedad metastásica se transforman en radiorefractarios. En el caso de la enfermedad metastásica, dos tercios muestran captación de radioyodo, pero solo la mitad de estos podrían curarse con tratamiento con131I4. A pesar de esta situación, durante muchos años, el radioyodo ha sido la única opción terapéutica para estos pacientes, utilizándose de manera re-petida, aun con una más que dudosa efectividad en los casos de no respuesta26. La supervivencia a 5 años de estos pacientes que no muestran avi-dez por el radioyodo es del 66%27, disminuyendo hasta el 10% de supervivencia a los 10 años26.
La mayoría de muertes de pacientes con cáncer de tiroides corresponden a casos de radiorefrac-tariedad1, documentando en varios estudios que la supervivencia de los pacientes con neoplasia diferenciada de tiroides refractaria al radioyodo y enfermedad metastásica está entre los 2,5 y 3 años28. Sin embargo algunos de estos pacientes con baja o nula captación de 131I permanecen es-tables a los largo de años, con una buena calidad de vida26.
Varios términos se han empleado para referirse a estos pacientes con cáncer diferenciado de ti-roides con enfermedad locorregional o metásta-sis en los que la administración de radioyodo no proporciona beneficio. Los términos “radiorefrac-tario”, “resistente” o “no respondedor” implican que la célula continúa captando 131I sin que se obtenga beneficio clínico. El término “sin avidez” describe a tumores que no presentan captación de radioyodo en el estudio posterapia25.
Considerando la trascendencia de catalogar a un tumor como radiorefractario, y sobre todo, las implicaciones terapéuticas que tiene para el paciente, es imprescindible comprobar que la exploración tras la administración de radioyodo se haya realizado en las condiciones adecua-das, esto es, con una estimulación correcta de los niveles de TSH (TSH sérica por encima de 30 mUI/mL tras estimulación con tirotropina recom-
binante o deprivación hormonal) y descartando una saturación de los niveles de yodo (sobrecar-ga de yodo previa a la administración de 131I)4.
Es de vital importancia entender que una vez que un paciente se identifica como radiorefractario no está indicada la administración de nuevas do-sis de 131I aunque el tumor muestre captación por el radiofármaco.
6. Definición de radiorefractariedad
Varias han sido las definiciones y criterios que se han aplicado para clasificar las neoplasias de ti-roides como radiorefractarias. En los ensayos clí-nicos con agentes inhibidores multikinasa se han empleado diferentes situaciones, en las que se definían datos relacionados con la presentación clínica, los estudios de imagen o el incremento en los valores de tiroglobulina29-31.
En las guías ATA se recogen las categorías acep-tadas a partir de la reunión del panel de expertos que postularon las categorías que hoy acepta-mos como tipos de radiorefractariedad4,32.
6.1 Pacientes que no muestran captación de radioyodo en el momento del diagnóstico ini-cial de enfermedad locorregional o metástasisEl paciente con lesiones conocidas que no pre-senta captación de radioyodo fuera del lecho tiroideo tras el rastreo posterapia. No hay evi-dencia de que ese paciente se beneficie de un tratamiento adicional con 131I.
En este grupo se incluyen también pacientes con enfermedad estructural que no muestran cap-tación de radioyodo en el rastreo diagnóstico, aunque mostrasen captación en el estudio pos-terapia, ya que se considera que dicha captación no sería suficiente para inducir beneficio de la administración de131I33. (figura 2)
6.2 Pacientes en los que el tumor ha perdido la capacidad de captar radioyodo tras evidencia de captación previaEn este supuesto es de vital importancia la exclu-sión de contaminación por yodo estable (excluir contraste yodado reciente, dieta rica en yodo o medicaciones con alto contenido en yodo, amio-darona, por ejemplo).
Figura 1. Alteraciones a nivel de NIS y a nivel del polo apical y sus efectos en la captación de yodo en la célula tumoral tiroidea.

29ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES28 3. El concepto de la refractariedad al radioyodo en los pacientes con carcinoma diferenciado de tiroides
Ocurre en pacientes con metástasis grandes y múltiples, en las que la administración de 131I ha erradicado las células capaces de captarlo (cé-lulas más diferenciadas) y persisten los clones menos diferenciados que no captan yodo. La enfermedad progresará a partir de estos grupos celulares.
6.3 Pacientes que concentran radioyodo en una lesiones pero no en otrasEsta situación es frecuente en pacientes con me-tástasis de gran tamaño. Un estudio utilizó 124I (un trazador para PET/CT) y comparó los resul-tados con los hallazgos obtenidos en el PET/CT con 18F-FDG y en el rastreo corporal con 131I34. En estos pacientes la progresión ocurre habitual-mente a partir de las metástasis sin captación de radioyodo, especialmente cuando muestran avidez por 18F-FDG, de modo que el tratamiento con 131I no produce beneficios en términos de su-pervivencia. (figura 3)
Algunos pacientes muestran lesiones que predominantemente captan yodo y solo al-guno no lo concentran. En este caso, a pesar de que el paciente se encuadraría en la defi-nición de radiorefractario, podría beneficiarse de la combinación de tratamiento con 131I y terapia local sobre la lesión sin captación4.
6.4 Pacientes con progresión de la enfermedad metastásica a pesar de mostrar captación de radioyodoLa progresión puede objetivarse por la apari-ción de nuevas lesiones, crecimiento progre-sivo de las lesiones existentes o por la eleva-ción continua de los niveles de tiroglobulina tras la terapia (figura 4). En estos casos está demostrado que si la progresión ocurre tras un tratamiento correcto con radioyodo, tra-tamientos posteriores no aportan ningún be-neficio7.
Figura 2. Paciente que no muestra captación en el rastreo corporal con 131I. Lesiones hipermetabólicas en el estudio con 18F-FDG.
Figura 3. Paciente con lesión única en mediastino en el rastreo con 131I. Múltiples lesiones hipermetabólicas en el estudio con 18F-FDG.
Figura 4. Paciente con captaciones en rastreo con 131I inicial (A), con respuesta de la actividad en lecho tiroideo y pulmón, pero con aparición de metástasis cerebral (B y C).
A B C
6.5 Paciente con captación de radioyodo en todas las lesiones, pero sin curación a pesar de varios tratamientos, que permanece sin progresiónEs una situación más controvertida en cuanto a la actitud a tomar y a la clasificación del pacien-te como radiorefractario, especialmente si la
actividad acumulada recibida es mayor de 600 mCi. La decisión de continuar con tratamiento con 131I se basará en la respuesta a los trata-mientos previos, la intensidad de la captación de radioyodo, en la captación de 18F-FDG en los focos tumorales (una baja captación favorecería teóricamente la respuesta a radioyodo) y en la

31ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES30 3. El concepto de la refractariedad al radioyodo en los pacientes con carcinoma diferenciado de tiroides
buena tolerancia con ausencia de efectos cola-terales4,32. (figura 5)
La probabilidad de curación es baja con más tratamientos y los efectos colaterales pueden aumentarse, incluyendo el riesgo de leucemia o segunda neoplasia35.
6.6 Paciente con enfermedad avanzada sin tiroidectomía
En los casos de tumores irresecables no esta-ría indicado el tratamiento con radioyodo, por lo que deben de manejarse de entrada como radiorefractarios, dado que el tratamiento con 131I es inefectivo cuando la glándula tiroidea está presente. Esta situación podría revertirse si se re-duce la carga tumoral o es posible la intervención quirúrgica.
Además de las situaciones previamente descri-tas, otras están en la discusión sobre su conside-ración o no como radiorefractarias.
• La dosis acumulada de radioyodo superior a los 600 mCi podría ser una causa de de-finición de radiorefractariedad, pero consi-deramos que es una decisión que debe ser especialmente individualizada en cada caso y sobre todo que precisa de una evaluación del comportamiento de las lesiones, de la res-puesta anterior y de la intensidad de la cap-tación por la lesión en cuestión. Actualmen-te, la mayoría de guías reflejan la necesidad de evitar dosis acumuladas mayores de 600 mCi, de manera que cuando hay ausencia de respuesta o progresión debe de considerarse otra alternativa de tratamiento36, pero no se incluye como definición de radiorefractarie-dad en las recientes guías ATA y hay auto-res que consideran como arbitrario ese ni-vel, utilizado en varios ensayos clínicos para incluir pacientes como radiorefractarios, ya que hay ejemplos de dosis mayores sin toxi-cidad. Únicamente en casos de toxicidad cla-ramente secundaria a toxicidad se debería de suspender el tratamiento con 131I e incluir al paciente en otras alternativas37.
• Captación significativa de las lesiones con 18F-FDG en el estudio PET/CT. La captación de 18F-FDG es un factor predictivo negativo
para la captación de radioyodo y también es un factor pronóstico negativo en los pacien-tes con cáncer diferenciado de tiroides. No obstante, en aquellas lesiones que captan 131I y también tienen avidez por la 18F-FDG, a pe-sar de que es un predictor de mala respuesta, no debe de obviarse el tratamiento con ra-dioyodo únicamente en base a la captación en el PET/CT.
• Pacientes con histologías agresivas como neoplasias pobremente diferenciadas, carci-noma insular o neoplasia de células de Hür-thle que se asocian con baja probabilidad de respuesta a 131I38.
7. Factores predictores de radiorefractariedad
Algunas herramientas clínicas pueden ser útiles para evaluar la opción de que una neoplasia di-ferenciada de tiroides se transforme en radiore-fractaria durante el seguimiento.
7.1 18F-FDG PET/CTLas aplicaciones de la tomografía de emisión de positrones al estudio del cáncer de tiroides han crecido de manera significativa en los últimos años, tanto como finalidad diagnóstica como pronóstica4.
Los eventos moleculares que favorecen la cap-tación de 18F-fluorodesoxiglucosa en la célula tumoral son el incremento del transportador de glucosa (GLUT
1), sobre expresión de la enzima
hexoquinasa e incremento de la expresión del factor 1a de hipoxia inducible (HIF-1a). Pero el mecanismo molecular implicado en el cambio metabólico que incrementa la captación de glu-cosa no es bien conocido.
Así, los tumores más diferenciados mostrarán una intensa captación de radioyodo y escasa actividad metabólica con 18F-FDG. Por el contra-rio, cuando los tumores se indiferencian, crece la actividad en el estudio PET/CT y disminuye la captación de 131I, constituyendo el “fenómeno flip-flop”39. Este fenómeno describe la condición en la que tumor no tiene capacidad para cap-tar yodo o su captación es baja y muestra una elevada actividad metabólica que se traduce en captación de glucosa.
Figura 5. Paciente con captación en rastreo con 131I en pala iliaca izquierda, que ha recibido 450 mCi de 131I (actividad acumulada), mostrando actividad estable con cifras de tiroglobulina detec-tables, pero estabilizadas. Decisión de no tratar y mantener vigilancia.
La principal indicación de realizar un estudio PET/CT en pacientes con CDT es el caso de ele-vación de tiroglobulina con rastreo con 131I nega-tivo4,36.
Las guías ATA incluyen la evaluación inicial en el caso de tumores avanzados, histologías agresi-vas o carcinoma de células de Hurtle, así como en los pacientes en que se observan unas cifras inusualmente altas de tiroglobulina postquirúr-gica4.
La utilización de esta técnica como pronóstico es más reciente. Se ha demostrado que los pacien-tes con lesiones que son positivas con 18F-FDG tienen menos probabilidad de responder a 131I40. Este mismo grupo demostró que los pacientes con enfermedad metastásica con estudio positi-vo en FDG PET/CT tienen peor pronóstico que aquellos con estudio negativo, con independen-cia de su captación de 131I. Estos tumores tienden a ser más agresivos, mas invasivos y con mayor
prevalencia de mutaciones a nivel BRAF, HRAS y NRAS comparados con aquellos que son 18F-FDG negativos41,42.
7.2 HistologíaEl carcinoma papilar de tiroides es la histología más frecuente y de manera general tiene muy buen pronóstico, con una tasa de supervivencia que ronda el 95%. Algunas variantes raras de este tumor pueden ser más agresivas y trans-formarse en radiorefractarias en la evolución. De ellas la variante de células altas (tall cell) es la más común. Esta variante tiene un perfil gené-tico más agresivo (mutaciones BRAF y TERT) asociado a radiorefractariedad y a peor pronós-tico43.
Otras variantes agresivas del carcinoma papi-lar incluyen la variedad de células columnares, variedad esclerosante difusa, solida, con joro-bas (hobnail) y aquellos que corresponden a la

33ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES32 3. El concepto de la refractariedad al radioyodo en los pacientes con carcinoma diferenciado de tiroides
variedad folicular ampliamente invasiva. El peor pronóstico se debe al perfil genético, a la pre-sentación inicial y a la falta de respuesta a ra-dioyodo44.
7.3 TiroglobulinaLa determinación sérica de tiroglobulina es un elemento básico en el seguimiento de pacientes con CDT. La tiroglobulina es secretada por el tejido normal y las células neoplásicas tiroideas. Si se produce la eliminación de las células nor-males, la única fuente de producción de tiroglo-bulina serán las células tumorales. Por lo tanto, la medida de los niveles séricos de tiroglobulina proporciona una importante información sobre la presencia o ausencia de enfermedad residual, recurrencia o metástasis en pacientes con CDT45.
Un dato a considerar para evaluar los valores de tiroglobulina es la concentración de TSH que tie-ne el paciente, ya que la producción de tiroglo-bulina y su secreción son dependientes de TSH. Así, el verdadero valor de la determinación se obtiene tras estimulación con rhTSH o tras depri-vación hormonal46. Un valor indetectable tras la estimulación indica la ausencia de tejido normal y ausencia de enfermedad.
La utilización del tiempo de duplicación de la tiroglobulina, más que el incremento en sí, ha sido propuesto como herramienta para identifi-car aquellos pacientes con mayor probabilidad de desarrollar metástasis y con mayor riesgo de fallecimiento, a pesar del tratamiento con radio-yodo47.
En pacientes definidos como radiorefractarios la determinación de tiroglobulina puede ser una he-rramienta inicial para monitorizar la situación de la enfermedad, pero debe de considerarse que los tumores más indiferenciados pueden llegar a no producir tiroglobulina. Igualmente es impor-tante señalar que los valores de tiroglobulina no deben de constituir el elemento de decisión para considerar el paciente en progresión o iniciar una terapia sistémica25.
8. Terapias rediferenciadoras
En teoría, la reinducción de expresión de NIS o su relocalización en la membrana celular podría incrementar o restaurar la captación de 131I.
Clásicamente, se han utilizado los retinoides, bajo la hipótesis de sobreregulación de la expre-sión de NIS, principalmente por activación de RAR-a. A pesar de que hay varios estudios so-bre su uso, la captación no muestra correlación con el efecto terapéutico y actualmente apenas tienen validez práctica48.
La rosiglitazona, un fármaco utilizado como an-tidiabético oral, actualmente retirado del merca-do en España, se utilizó por su capacidad de ser agonista PPARg restaurando la captación me-diada por NIS en algunos pacientes49. En nues-tra experiencia se incrementaba la captación de radioyodo por las lesiones, pero no se obtenía beneficio terapéutico.
Más reciente es la utilización de inhibidores tiro-sinkinasa para incrementar o restaurar la capta-ción de 131I. Selumetinib, un inhibidor MEK, ha de-mostrado hasta un 63% de respuestas parciales, en pacientes estrictamente seleccionados, con respuestas ligadas a las mutaciones NRAS50. Un estudio ha mostrado una mejora en la captación de radioyodo de hasta el 60% en pacientes con mutaciones BRAF cuando eran tratados con da-brafenib51.
Otro mecanismo de acción es el propuesto para la romidepsina, un inhibidor de la deacetilación de las histonas, que en cultivos había demostra-do incrementar la expresión de tiroglobulina y de RNAm para NIS. En un estudio reciente se ha observado que aumenta la captación de radioyo-do, pero no se observan respuestas significativas, con eventos adversos importantes52.
BIBLIOGRAFÍA
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics 2014. CA Cancer J Clin 2014;64:9-29.
2. Verburg FA, Mader U, Tanase K, Thies ED, Diessl S, Buck AK, Luster M, Rei-ners C. Life expentacy is reduced in differentiated thyroid cáncer patients > 45 years old with extensive local tumor invasion, lateral lymph node, or distant me-tastases at diagnosis and normal y all others DTC patients. J Clin Endocrinol Metab 2013;98:172-189.
3. Jonklaas J, Sarlis NJ, Li-tofsky D et al. Outcome of
patients with differentiated thyroid carcinoma following initial therapy. Thyroid 2006;16:1229.1242.
4. Haugen BR, Alexander EK, Bible KC, Doherty GM, Mandel SJ, Nikiforov YE, et al. 2015 American Thyroid Association Management Guidelines for Adults Patiens wirt Thyroid Nodules and Di-fferentiated Thyroid Cancer: The American Thyroid Asso-ciation Guidelines Task For-ce on Thyroid Nodules and Differentiated Thyroid Can-cer. Thyroid 2016;26:1-133
5. Perros P, Colley S, Boelaert K, Evans C, Evans RM, Gerrard GE, et al. British Thyroid Association Guideli-
nes for the management of thyroid cancer. Clin Endocri-nol. 2014; 81 (suppl 1):1-136
6. Tuttle RM, Tala H, Shah J, Leboeuf R, Ghossein R, Gonen M, Brokhin M, Omry G, Fagin JA, Shaha A. Esti-mating risk of recurrence in differentiated thyroid cancer after total thyroidectomy and radioactive iodine rem-nant ablation: using respon-se to therapy variables to modify the initial risk esti-mates predicted by the new American Thyroid Associa-tion staging system. Thyoid 2010;20(12)1341-9
7. Vaisman F, Tala H, Grewal R and Tuttle RM. In diffe-rentiated thyroid cancer, an
incomplete structural res-ponse to therapy is associa-ted with significantly worse clinical outcomes than only an incomplete thyroglobulin response. Thyroid 2011;21 1317–1322.
8. Durante C, Haddy N, Bau-din E, Leboulleux S, Hartl D, Travagli JP, Caillou B, Ricard M, Lumbroso JD, De Vathai-re F et al. Long-term outco-me of 444 patients with distant metastases from pa-pillary and follicular thyroid carcinoma: benefits and li-mits of radioiodine therapy. J Clin Endocrinol and Metab 2006;91:2892–2899.
9. Pacini F, Brianzoni E, Du-rante C, Elisei R, Ferdeghini
9. Conclusiones
• El tratamiento con radioyodo constituye el principal elemento en la terapia de la neoplasia diferenciadas de tiroides tras la cirugía y la evaluación de riesgos.
• La expresión del simportador de Na/I es básica para la incorporación del radio-yodo a la célula tiroidea.
• La determinación de tiroglobulina es básica para la monitorización de los pa-cientes, aunque hay que entender que cuando el tumor se desdiferencia puede no producirla.
• El uso del PET/CT con 18F-FDG es una herramienta básica en la evaluación de las neoplasias diferenciadas de tiroides con cambios en la avidez por el radioyodo, en aquellas de mal pronóstico inicial y cuando existe afectación metastásica. Es un factor predictivo de mal pronóstico, tanto para la respuesta a radioyodo como para la supervivencia.
• La definición de radiorefractariedad es crítica, entendiéndose que tras la misma, no debemos administrar más dosis de radioyodo.
• La mayoría de los pacientes que fallecen por cáncer diferenciado de tiroides pertenecen al grupo de radiorefractarios.
• Las estrategias de rediferenciación, en especial el uso de selumetinib, plantean una opción atractiva para el futuro de los pacientes con tumores que no captan yodo.

35ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES34 3. El concepto de la refractariedad al radioyodo en los pacientes con carcinoma diferenciado de tiroides
M, Fugazzola L, et al. Reco-mendations for post-surgical thyroid ablation in differen-tiated thyroid cáncer: a 2015 position statement of the Italian Society of Endocri-nology. J Endocrinol Invest. 2016;39:3341-347.
10. Robbins RJ, Schlumber-ger MJ. The envolving role of 131-1- for treatment of diffe-rentiated thyroid carcinoma. J Nucl Med 2005; 46:28S-37S.
11. Cohen EG, Tuttle M, Kraus DH. Postoperative mana-gement of differentiated thyroid cáncer. Otolaryngol Clin N AM 2003; 36:129-157.
12. Pacini F, Schlumberger M, Dralle H, Elisei R, Smit JW, Wiersinga et al. Euro-pean consensus for the ma-nagement of patients with differentiated thyroid carci-noma of the folicular epithe-lium. Eur J Endocrinol 2006; 154(6):787-803.
13. Rutherford GC, Franc B, O’Connor A. Nuclear me-dicine in the assessment of differentiated thyroid cáncer. Clinical Radiology 2008;63:453-463.
14. Dai, G, Levy O, Carrasco N. Cloning and characteri-zation of the thyroid iodide transporter. Nature 1996; 379:458-60.
15. Riesco-Eizaguirre G, San-tisteban P. A perspective view of sodium iodide sym-porter research and its clini-cal implications. Eur J Endo-crinol 2006; 155:495-512.
16. Riesco-Eizaguirre G, San-tisteban P. New insights in thyroid folicular cell biology and its impact in thyroid cán-cer therapy. Endocrine Rela-ted Cancer 2007;14:957-977.
17. Riesco-Eizaguire G, Gar-cía-Cabezas MA, Nistal M, Gutierrez-Martínez P, San-tisteban P. The oncogene BRAFV600E is associated with high risk of recurren-ces and less differentiated papillay thyroid carcinoma due to impairment of NIS
targeting to membrane. Endocrine-RElated Cancer 2006;12:257-269.
18. Riesco-Eizaguirre G, Rodriguez I, De la Vieja A, Costamagna E, Carrasco N, Nistal M, Santisteban P. The BRAFV600E oncogene in-duces TGFbeta secretion leading to sodium ioidide symportes (NIS) repression and increased malignancy in thyroid cáncer. Cancer Re-search 2009;69:8317-8325.
19. Vaisman F, Carvalho DP, Vaisman M. A new appraisal of iodine refractory thiroid cáncer. Endocrine-Related Cancer 2015;22:R301-R310.
20. Ringel MD, AndersonJ, Souza SL, Burch HB, Tam-basxia M, Shriver CD and Tuttle RM. Expression of the sodium ioidide symporter and thyroglobulin genes are reduced in papillary thiroid cáncer. Modern Pathology 2001;14:289-296.
21. Dohan O, Baloch Z, Ban-revi Z, Livolski V and Carras-co N. Rapid comunication: Predominant intracelular overexpression of the Na+/I- symporter (NIS) in a large sampling of thyroid cáncer cases. J Clin Endocrinol Me-tab 2001;86:2697-2700.
22. Dohan O, De la Vieja A, Paroder V, Riedel C, Artami M, Reed M, Ginter CS and Carrasco N, The sodium/iodide symporte (NIS): cha-racterization, regulation, and medical significance. Endo-crine Reviews 2003;24:48-77.
23. Vidal AP, Andrade BM, Vaisma F, Cazarin J, Pinto LF, Breitenbach MM, Corbo R, Caroli-Bottino A, Soares F, Vaisman M. AMP-activa-ted protein kinsa signling is upregulate in papillary thyroid cáncer. Eur J Endo-crinol 2013;169:521-528.
24. Schlumberger M, La-croix L, Russo D, Filetti S and Bidart JM. Defect in io-dide metabolism in thyroid cáncer and implications for the follow-up and treatment
of patients. Nat Clin Prati-ce Endocrinol Metab 2007; 3(3):260-69.
25. Riesco-Eizaguirre G, Ga-lofré JC, Grande E, Zafón Llopis C, Ramon y Cajal T, Navarro Gonzalez E, et al. Spanish consensus for the management of patients with advanced radioactive iodine refractory differen-tiated thyroid cáncer. Endo-crinol Nutr 2016; 63(4):e17-24.
26. Durante C, Constante G, Filetti S. Differentiated thyroid carcinoma: defining new paradigms for posto-perative management. Endocrine-Related Cancer 2013;20:R141-R154.
27. Nixon I, Whictcher M, palmer F, Tuttle R, Shaha A, Shah J et al. The impacto of distant metatases at pre-sentation on prognosis in patients with differentiated carcinoma of the thyroid gland. Thyroid 2012;22:884-889.
28. Robbins R, Wan Q, Grewal R, Reibke R, Go-nenM, Strauss H, et al. Real-time prognosis for metasta-tic thyroid carcinoma base don 2-[18f]fluoro-2-deoxy-d-glucose-positron emis-sion tomography scanning. J Clin Endocrinol Metab 2006;91:498-505.
29. Sack W, Braunstein GD. Evolving approaches in managing radioactive io-dine-refrory differentiated thyroid cáncer. Endocrine Practice 2014;20:263-275.
30. Brose MS, Nutting CM, Sherman SI et al. Rationales and design of DECISION: a doublé-blind, randomized, placebo-controlled phase III trial evaluating the efficacy and safety of sorafenib in patients with locally advan-ced or metastatic radioac-tive iodine (RAI)-refratory, differentiated thyroid cán-cer. BMC Cancer 2011;11.
31. Schlumberger M, Tha-ra M, Wirth LJ, Robinson B, MS Brose, Elisei R, et al.
Lenvatinib versus placebo in radioiodine-refractory thyroid cáncer. N Engl J Med 2015;372:621-630.
32. Schlumberger M, Bro-se M, Elisei R, Leboulleux S, Luster M, Pitoia F, Pacini F. Definition and management of radioactive iodine-refrac-tory differentiated thyroid cáncer. Lancet Diabetes En-docrinol 2014;2:356-358.
33. Sabra MM, Grewwal RK, Tala H, Larson SM, Tuttle RM. Clinical outcome following empiric radioidine therpay in patients with strucutrally identifiable metastatic foli-cular-cell-derived thyroid carcinoma with negative diagnostic but positive post-therapy 131I whole-body scans. Thyroid 2012:22;877-883.
34. Sgouros G, Kolbert KS, Sheikh A, Pentlow KS, Mun EF, Narth A, Robbins RJ, Lar-son SM. Patient-specific do-simetry for 131I thyroid cán-cer therapy using 124I PET and 3-dimensional-internal dosimetry (3D-ID) software. J Nucl Med 2004;45:1366-1372.
35. Rubino C, de VF, Dotto-rini ME, Hall P, Schvartz C, Couette JE, Dondon MG, Ab-bas MT, Langlois C, Schlum-berger M. Second primary malignancies in thyroid can-cer patients. Br J Cancer 2003;89:1638-1644.
36. Tuttle RM, Haddad RI, Ball DW, Byrd D, Dickson P, Duh Q, Ehya H, Haymart M, Hoh C, Hunt JO, et al. Thyroid carcinoma, versión 2.2014. Journal of the Natio-nal Comprehensive Cancer Network 2014;12:1671-1680.
37. Pryma DA, Mandel SJ. Radioiodine therapy for thyroid cáncer in the era of rsik stratificacion and alter-native targeted therapies. J Nucl Med 2014;55:1485-1491 .
38. Capdevila J, Galofré JC, Grande E, Zafón Llopis C, Ramon y Cajal Asensio T, Navarro González E, Jimé-nez-Fonseca P, Santamaría
Sandi J, Gomez Saez JM and Riesco-Eizaguirre G. Consen-sus on the management of advanced radioactive iodi-ne refractory differentiated thyroi cáncer: spanish task force for thyroid cáncer on belhaf of Spanish Society of endocrinology (SEEN) and Spanish rare cáncer working group (GETHI). Clin Transl Oncol 2016. DOI: 10.1007/s12094-016-1554-5.
39. Larson SM, Robbins R. Positron emission tomogra-phy in thyroid cáncer mana-gement. Semin Roentgenol 2002;37(2):169-174.
40. Wang W, Larson SM, Tuttle RM, Kalaigian H, Kol-bert K, Sonenberg M, Rob-bins RJ. Resistance of 18F.fluorodeoxyglucose-avid-metastatic thyroid cáncer lesions to treatment with high-dose radioactive iodine. Thyroid 2001;11:1169-1175.
41. Esteva D, Muros MA, Lla-mas-Elvira JM, Jimenez Alon-so J, Villar JM, Lopez de la Torre M, Muros T. Clinical and pathological factors related to 18F-FDG_PET positivity in the diagnosis of recurrence an/or metatasis in patients with differentiated thyroid cáncer. Annal of Surgical On-cology 2009;16:2006-2013.
42. Ricarte-Filho JC, Ryder M, Chitale DA, Rivera M, He-guy A, Landanyi M, Janaki-raman M, Solit D, Knauf JA, Tuttle RM. Mutational profi-le of advanced primary and metastastic radioactive iodi-ne-refractory thyroid cáncer reveasl distinct pathogenetic roles for NRAF, PIK3CA and AKYT1. Cancer Research 2009; 69:4885-4893.
43. Liu X, Bishop J, Shan Y, Pai S, Liu D, Murugan AK, Sun H, El-Naggar A, Xing M. Highly prevalence TERR pro-moter mutations in agressive thyroid cancers. Endocrine-Related Cancer 2013;20:603-610.
44. Lastra RR, Livolsi VA, Baloch ZW. Aggressive va-riants of folicular cell-deri-ved thyroid carcinomas: a
cytopathologist’s perspec-tive. Cancer Cytopathology 2014;122:484-503.
45. Intenzo CM, Jabboir S, Dam HQ, Capuzzi Dm. Chan-ging concepts in the mana-gement of differentiated thyroid cáncer. Semin Nucl Med 2005;35(4):257-265.
46. Haugin B, Pacini F, Rei-vers C, et al. A compari-son of recombinant hum-na thyrotropin and thyroid hormone withdrawal for the detection of thyroid remnant or cáncer. J Clin Endocrinol Metab 1999;84:3877-3885.
47. Miyauchi A, Kudo T, Miya A, Kobaysahi K, Ito Y, Takamura Y, Higashiyama T, Fikushima M, Kihara M, Inoue H. Prognostic impact of serum thyroglobulin dou-bling-time under thyrotrop-pin suppression in patients with papillary thyroid car-cinoma who underwent to-tal thyroidectomy. Thyroid 2011;21:707-716.
48. Frölich E, Wahl. The cu-rrent role of targeted thera-pies to induce radioiodine uptake in the thyroid cáncer. Cancer Treatment Reviews 2014;40:665-674.
49. Kekebew E, Peng M, Reiff E, et al. A phase II trial of rosiglitazone in patients with thyroglobulin-ppositive and radioiodine-negative di-fferetiated thyroid cáncer. Surgery 2006;140:960-966
50. Ho AL, Grewal RK, Le-boeuf R, Sherman EJ, Pfis-ter DG, Deandreis D, et al. Selumetinib-enhanced ra-dioiodine uptake in advan-ced thyroid cáncer. N Engl J Med 2013;368:623-632.
51. Rothemberg S, McFadden D, Palmer E, Daniels G, Wir-th L. Re-differentiation of radioiodine-refractory BRAF V600E-mutant thyroid car-cinoma with dabrafenib: a pilot study. J Clin Oncol 2013;31(suppl: abstr 6025).
52. Sherman EJ, Su YB, Lyall A, Schöder H, Fury MG, Ghossein RA, Haque S,
Lisa D, Shaha AR, Tuttle RM, Pfister DG. Avalutaion of ro-midepsin for clinical activity and radioactive iodine reup-take in radioactive iodine-re-fractory thyroid carcinoma. Thyroid 2013;23(5):593-599.

37ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES1. Epidemiología del cáncer diferenciado de tiroides en España: avanzando hacia el registro nacional36
Introducción
El pilar del tratamiento para los pacientes con car-cinoma diferenciado de tiroides (CDT), ya sea de histología papilar o folicular, sigue siendo la cirugía, la supresión de tirotropina (TSH) y el tratamiento con radioyodo 131I, especialmente en aquellos pa-cientes con mayor riesgo de recaída o ante la pre-sencia de metástasis.
El pronóstico de los pacientes con tumores reseca-bles es, en general, excelente. Aun así, el espectro de pacientes con CDT es tan variado, desde unos con muy buen pronóstico a otros con muy mal pronóstico, que la decisión de adecuar correcta-mente el mejor tratamiento para cada paciente sin sobretratar a quién no lo necesite ni dejar de tratar a quién lo precise, debe abordarse en el seno de un manejo multidisciplinar de esta patología.
Una vez que la enfermedad alcanza criterios de refractariedad al yodo, los tratamientos activos disponibles son muy limitados, si bien en las últi-mas décadas la llegada de dos fármacos, lenvatinib y sorafenib, englobados dentro del concepto de inhibidores tirosín quinasa (ITK) ha modificado fa-vorablemente el pronóstico de nuestros pacientes hasta el punto que la mediana de supervivencia global (SG), situada en 2.3 años en pacientes con criterios de refractariedad a 131I en progresión y sin tratamiento, se ve ampliamente superada, especial-mente en los pacientes respondedores a ITK1.
En esta revisión trataremos, de un modo práctico, qué aspectos hay que tener en cuenta a la hora de plantear las opciones terapéuticas disponibles en los pacientes con CDT loco-regionalmente avan-zados irresecables o metastásicos, que cumplen criterios de refractariedad al yodo.
1. ¿Cuándo y con qué debemos empezar un trata-miento en CDT refractario a radioyodo?
Uno de los puntos más importantes es decidir el momento y la terapia más adecuados de manera individualizada. Debemos realizar una cuidadosa evaluación de los beneficios y los riesgos, enten-diendo que son tratamientos paliativos, ya que la remisión completa de la enfermedad es inusual(1).
Las opciones terapéuticas se pueden clasificar en tres grupos2: (tabla 1)
1. 1 Vigilancia activa. 1.2 Terapias locales.1.3 Terapias sistémicas.
1.1 Vigilancia activaLa llegada de lenvatinib y sorafenib, fármacos acti-vos en pacientes con CDT refractario a 131I, ha mo-dificado una conducta clásica de seguimiento del paciente basado en la pura observación clínica, sin una necesidad de incluir pruebas de imagen en di-cho seguimiento y, por tanto, en el que la decisión
4. Conceptos prácticos del tratamiento sistémico del carcinoma diferenciado de tiroides refractario al radioyodo: ¿todos los pacientes son iguales?
BEATRIZ CASTELO FERNÁNDEZServicio de Oncología Médica. Hospital Universitario La Paz. Madrid
CRISTINA ÁLVAREZ ESCOLÁServicio de Endocrinología. Hospital Universitario La Paz. Madrid
LORENA OSTIOS GARCÍAServicio de Oncología Médica. Hospital Universitario La Paz. Madrid
ELSA BERNAL HERTFELDERServicio de Oncología Médica.Hospital Universitario de Móstoles. Madrid
ANA PERTEJO FERNÁNDEZServicio de Oncología Médica. Hospital Universitario La Paz. Madrid
ENRIQUE ESPINOSA ARRANZServicio de Oncología Médica. Hospital Universitario La Paz. Madrid

39ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES38 4. Conceptos prácticos del tratamiento sistémico del CDT refractario al radioyodo: ¿todos los pacientes son iguales?
de iniciar tratamiento quedaba supeditada a la ex-clusiva aparición de síntomas.
Sabemos que el paciente con CDT refractario a 131I que progresa desde el punto de vista radio-lógico tiene muy mal pronóstico y que sin trata-miento específico su mediana de SG es inferior a los tres años. Esto ha hecho que introduzca-mos el concepto más acertado de vigilancia activa cuyo objetivo fundamental es tratar de identificar el momento en el que la enfermedad alcanza ese punto de inflexión, ese giro evoluti-vo en el que, por la disponibilidad de fármacos con impacto significativo en supervivencia libre de progresión (SLP), debe iniciarse tratamiento con un ITK3.
La vigilancia activa permite adaptar el trata-miento a cada tipo de paciente único. Es clave en ella la revisión de los pacientes cada 3-4 me-ses dentro de un equipo multidisciplinar, para detectar precozmente la progresión de la enfer-medad, incluyendo en el seguimiento tanto las determinaciones de tiroglobulina, como pruebas de imagen como la ecografía cervical (habitua-les en el seguimiento de los pacientes no clasi-ficados como radiorrefractarios), la tomografía computerizada (TC) o la TC con tomografía con
emisión de positrones con fluorodesoxiglucosa (18-FDG PET-TC). Otras pruebas más específi-cas como la gammagrafía ósea o la resonancia magnética (RM) pueden quedar a expensas de la presencia o no de síntomas clínicos de sos-pecha.
Los criterios RECIST (Response Evaluation Crite-ria in Solid Tumors) son los que utilizamos para evaluar la enfermedad, considerando que existe progresión cuando se observa un crecimiento en el diámetro mayor de las lesiones dianas superior al 20%. Si durante la vigilancia activa detecta-mos progresión radiológica de la enfermedad, es decir, ese punto de inflexión de la enfermedad, ¿tratamos a todos los pacientes?.
No existe un consenso claro de cuándo empe-zar tratamiento. Se aconseja iniciarlo cuando hay progresión radiológica, independientemente de la presencia de síntomas o de la mayor o menor carga tumoral asociada a esa progresión radio-lógica, por el mal pronóstico de estos pacientes, cuya SG no superará los tres años sin tratamien-to.
Si bien, en ocasiones, este “punto de inflexión” en la evolución de la enfermedad puede no ser el
Tabla 1. Opciones terapéuticas en pacientes con CDT refractario al radioyodo2
Criterios clínicos y radiológicos
Recomendaciones terapéuticas
Enfermedad estable o mínimamente progresiva
Ausencia de síntomas
Bajo riesgo de compresión de vía aérea, digestiva o SNC
Enfermedad no resecable
No adecuada para terapia local
Vigilancia o seguimiento cada 3-12m, Tg, eco, PET-TC u otra modalidad de imagen
Evaluación de la severidad clínica
Supresión de TSH
Enfermedad de alto riesgo o compresión local de la vía aérea, digestiva o SNC.
Metástasis única
Dolor local
Resección quirúrgica
RT externa
Ablación con radiofrecuencia
Quimioembolización hepática
Biofosfonatos (metástasis óseas)
Supresión de TSH
Enfermedad rápidamente progresiva
Enfermedad sintomática o con riesgo vital
Paciente no candidato a terapias locales
Terapia con inhibidores multiquinasa (Lenvatinib, Sorafenib)
Reinducción de la captación de I-131
Quimioterapia
Supresión de TSH
Vigilancia activa Terapias locales Terapias sistémicas único criterio a tener en cuenta. Factores como el ritmo de progresión de la enfermedad, la cer-canía a la progresión sintomática son también herramientas fundamentales para decidir cuál es el punto para empezar a tratar.
La situación clínica del paciente, así como sus comorbilidades pueden influir, también, en la decisión de instaurar tratamiento. Como tantas situaciones en oncología, la clave es la correcta selección del paciente y el detectar el momento en el que se va a beneficiar de iniciar un trata-miento4.
Podríamos identificar las siguientes situaciones como adecuadas para mantener una vigilancia activa o al menos con menor evidencia para utili-zar terapias más agresivas5:
1.1.1. Enfermedad metastásica ganglionar tras la administración de radioyodo, asintomática y de pequeño tamaño (< 1 cm)En estos casos no está claro que la cirugía local u otras técnicas locales mejoren la evo-lución de la enfermedad, especialmente en términos de recurrencia o supervivencia.
1.1.2. Enfermedad micronodular pulmonar (< 1 cm) en pacientes en los que la enfermedad no progresa o es lentamente progresiva. Podemos mantener la supresión de TSH y realizar un seguimiento.
1.1.3. Metástasis óseas asintomáticas, esta-bles y que no comprometen estructuras vi-tales.Así pues, en aquel paciente que, cumpliendo criterios de refractariedad a 131I, presente una enfermedad lentamente progresiva o estable, de poco volumen tumoral y pocas probabili-dades de presentar síntomas, podría mante-ner seguimiento mediante vigilancia activa. En esta situación, el tratamiento sólo con levoti-roxina a dosis supresoras de TSH y un segui-miento multidisciplinar sería de elección (2,5).
1.2 Terapias locales La indicación de una terapia local en pacientes con enfermedad radiorrefractariedad a 131I estará condicionada por la localización, el número de las metástasis, la velocidad de crecimiento tumoral y las opciones técnicas.
1.2.1 Recaída locorregionalHay que tener presente que incluso en si-tuaciones de sensibilidad a 131I, el tratamien-to quirúrgico debe ser la primera opción en la recaída locorregional.
No está claro el impacto de la cirugía lo-corregional en términos de supervivencia, pero también hay que considerarla como herramienta paliativa en el control sinto-mático en los casos de compromiso de es-tructuras vitales (obstrucción de vía aérea o digestiva).
La embolización o la ablación por radiofre-cuencia de adenopatías cervicales es una opción siempre que se disponga de los me-dios en el centro hospitalario6.
1.2.2 Recaída a distanciaEn determinados casos se puede plantear la metastasectomía, especialmente en lesio-nes cerebrales y óseas únicas o pulmonares limitadas.En casos de enfermedad ósea no quirúrgica con dolor, riesgo de fractura o complicaciones neurológicas, se puede va-lorar la utilización de radioterapia externa, microondas o crioterapia si bien éstas dos últimas son menos empleadas al no estar disponibles en todos los hospitales7.
Los bifosfonatos, como el ácido zoledróni-co, o denosumab se pueden emplear para el alivio del dolor óseo y mejorar la calidad de vida. Como efectos a tener en cuenta destacan la hipocalcemia y la osteonecrosis mandibular8.
Denosumab, un inhibidor del ligando RANK, ha demostrado reducir los eventos relacio-nados con el esqueleto (fracturas patológi-cas, compresión medular, necesidad de ci-rugía o radioterapia antiálgica) en tumores sólidos por lo que puede ser una alternativa a los bifosfonatos9.
1.3 Terapias sistémicasConsiderando que la gran mayoría de pacien-tes con CDT radiorrefractario metastásico falle-cerán por progresión de la enfermedad, parece una opción plausible el iniciar el tratamiento con inhibidores multiquinasas en el momento

41ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES40 4. Conceptos prácticos del tratamiento sistémico del CDT refractario al radioyodo: ¿todos los pacientes son iguales?
en el que se evidencia el punto de inflexión ha-cia la progresión. (figura 1)
Ante una enfermedad de crecimiento rápidamen-te progresivo, sintomática o con riesgo de compli-cación el tratamiento de elección es el sistémico, siendo los ITK, lenvatinib y sorafenib, los únicos que han demostrado impacto en SLP10. Ambos fármacos con efecto antiangiogénico han cam-biado, por tanto, el manejo del CDT refractario a 131I y están disponibles en nuestro país.
Los pacientes deben ser informados de estas opciones terapéuticas disponibles, sabiendo el impacto que proporcionan cada uno de ellos, deben conocer sus efectos secundarios y, en la medida de lo posible, participar en la decisión del tratamiento10.
El impacto en SLP proporcionado por cada uno de ellos, frente a placebo, alcanza una mediana
de 18,3 meses para lenvatinib y de 10,8 meses para sorafenib, en los ensayos pivotales fase III. De forma global, debido al diseño de estos estu-dios, en los que la gran mayoría de los pacientes que iniciaron tratamiento con placebo recibieron el ITK a la progresión, no se ha podido demostrar un impacto en SG con estas terapias. Detallare-mos más adelante que en el subgrupo de > 65 años, preespecificado en el diseño del estudio SELECT, lenvatinib sí impacta significativamente, frente a placebo, en SG11. La tasa de respuestas objetivas conseguida con lenvatinib es del 64.8% y del 12,2% con sorafenib12,13. En los pacientes que responden a lenvatinib, la mediana de tiempo a la progresión es de 33 m14. Este dato es de crucial importancia porque antes de la llegada de los ITK mencionábamos que la mediana de SG de los pacientes con CDT refractario a 131I era inferior a tres años, lo que viene a reforzar que claramen-te lenvatinib impacta en la supervivencia de los pacientes.
Figura 1. Distintos patrones de progresión en pacientes con CDTLos pacientes de los grupos 1, 2 y 3 corresponden a la mayoría de las situaciones de radiorrefrac-tariedad y que terminaran falleciendo por progresión de la enfermedad. Una minoría de pacientes (grupo 4) pueden tener un curso indolente de la enfermedad.
Debemos conocer el manejo de sus efectos ad-versos para incrementar la probabilidad de que el paciente reciba un tratamiento óptimo durante el mayor tiempo posible15.
Sorafenib es un inhibidor multiquinasa de BRAF, VEGFR1 y VEGFR2 actuando por ello en la prolife-ración de las células tumorales y la angiogénesis. En el ensayo Fase III que le dio la indicación en CDT, el análisis por subgrupos mostró beneficio en todos ellos. La presencia de mutaciones en BRAF o RAS no fue predictora de respuesta a tratamiento. La tasa de respuesta fue del 12% con un 54% de control de la enfermedad a los 6 meses. Ya hemos mencio-nado como la SLP, objetivo primario del estudio de registro, es de 10,8m. No ha demostrado diferen-cias estadísticamente significativas en la SG frente a placebo (HR fue de 0,884; IC del 95%: 0,633, 1,236, valor unilateral de p 0,236). La mediana de SG no se alcanzó en el brazo sorafenib y fue de 36,5 me-ses en el brazo placebo. Ciento cincuenta y siete (75%) pacientes aleatorizados a placebo y 61 (30%) pacientes aleatorizados a sorafenib lo recibieron en régimen abierto13.
La toxicidad del tratamiento obligó a reducir dosis en el 78% de los pacientes y a suspenderlo en un 19%16. Los efectos secundarios más frecuentes son la astenia, la pérdida de peso, el rash, el síndrome mano-pie, la alopecia, la diarrea, anorexia, náuseas y dolor abdominal4. Como otros inhibidores de BRAF, está descrito el desarrollo de carcinomas epider-moides cutáneos en aproximadamente 5% de los pacientes tratados, al igual que queratoacantomas (5%) y otras lesiones actínicas premalignas17.
Lenvatinib es un inhibidor de la tirosinquinasa frente a VEGFR1-3, FGFR1-4, RET. KIT y PDGFR. Es el fár-maco antiangiogénico más potente, disponible hoy en día, de ahí que su efecto secundario más común sea la hipertensión (21% grado 1-2, 41,8% grado > 3). Su acción sobre RET, KIT y PDGFR justifica su control en el crecimiento tumoral. Otros efectos se-cundarios documentados son la astenia, la diarrea, la pérdida de apetito, la pérdida de peso y la pro-teinuria4. Su impacto en la SLP es evidente, con una mediana de 18,3m frente a 3,6m del grupo con pla-cebo [HR 99% CI: 0,21, (0,14-0,31)], lo que indica que claramente los pacientes del grupo control estaban en progresión y, de una forma indirecta, podríamos decir que eran de peor pronóstico que los del ensa-yo de sorafenib.
En el estudio de registro, al 83% de los pacientes tratados con placebo, y progresión de la enferme-dad confirmada, se les brindó la oportunidad de recibir tratamiento con lenvatinib sin enmascara-miento. No hubo ninguna diferencia estadística-mente significativa en la SG entre los grupos de tratamiento en el momento del análisis principal de la eficacia (HR=0,73; IC del 95 %: 0,50-1,07, p=0,1032). Ni el grupo de lenvatinib ni el grupo transferido de placebo alcanzó la mediana de SG12.
Mención especial merece la evidencia del trata-miento con estos agentes dianas en la población anciana.
En el estudio DECISION, la mediana de edad de los pacientes fue de 63 años, tanto en el brazo con so-rafenib como en el de placebo, con un 61,4% de pa-cientes mayores de 60 años. No hubo diferencias en la supervivencia libre de progresión entre los pacientes mayores o menores de 60 años. No hay una evaluación de la toxicidad, ajuste de dosis o abandonos de tratamiento en función de la edad13.
En el estudio SELECT, la mediana de edad el grupo de tratamiento fue de 64 años y de 61 años en el grupo placebo. Hablando de beneficio en SG, en el subgrupo de pacientes mayores de 65 años, que era un subgrupo preespecificado antes del trata-miento, lenvatinib demuestra que hay beneficio en SG a pesar de que casi todos los pacientes del gru-po con placebo recibieron tratamiento específico a la progresión de placebo. Esta diferencia no ha podido ser asociada al ECOG, tratamientos o co-morbilidades previas de los pacientes. Lenvatinib consigue compensar el efecto pronóstico negativo asociado a la edad avanzada, demostrado por el hecho de que ambos grupos de edad, los menores y los mayores de 65 años, alcanzaron medianas de SG similares (22 meses)11.
Cuando se analizan los dos subgrupos de edad, se observa que no hay diferencia entre ellos en cuanto a duración de tratamiento o intensidad de dosis. La dosis media recibida por los pacientes ancianos y los jóvenes es la misma. Sí hay una ligera diferencia en el tiempo a la primera reducción de dosis que es de 3,1 meses en el brazo de pacientes jóvenes y de 1,4 meses en el brazo de pacientes mayores de 65 años, probablemente explicado debido a las comorbilidades previas de este último subgrupo. Ahora bien, no hubo diferencias en los aconteci-

43ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES42 4. Conceptos prácticos del tratamiento sistémico del CDT refractario al radioyodo: ¿todos los pacientes son iguales?
mientos adversos observados en ambos subgru-pos. Respecto al objetivo primario, supervivencia libre de enfermedad, no se observaron diferencias significativas entre los dos subgrupos de edad15. Pero sí en la tasa de respuesta global donde hay una clara diferencia a favor de los paciente jóvenes (71,6% vs 54,7%)16.
Ni las guías NCCN ni las españolas especifican un tratamiento diferente en función de la edad del pa-ciente. Se aconseja un control más estrecho para evitar complicaciones.
Otros fármacos con actividad TKI y efecto antian-giogénico como sunitinib, axitinib, cabozantinib, panzopanib y vandetanib también se han estudia-do en el contexto del tratamiento del CDT refrac-tario a 131I, pero sólo en estudios fase 2. Ninguno de ellos está actualmente aprobado en esta indi-cación19.
1.4 ¿Cuándo inciar el tratamiento con ITK?La actitud de vigilancia activa tendrá como objeti-vo anticipar el punto de inflexión de la enfermedad para determinar el mejor momento para iniciar la terapia sistémica sabiendo que retrasar el inicio de la terapia puede comprometer la supervivencia del paciente13. El criterio clave es no descuidar al pa-ciente en progresión radiológica. Se puede mante-ner la vigilancia activa con el paciente estable, con el paciente mayor que tiene poco volumen tumoral pero no en otras circunstancias13.
A día de hoy disponemos de dos fármacos apro-bados en primera línea, basados ambos en ensayos Fase III frente a placebo. Lenvatinib y sorafenib no se han comparado entre sí por lo que no podemos afirmar que uno es mejor que otro. Probablemente la población del estudio SELECT es una población más agresiva como muestra la SLP del grupo pla-cebo, de tan solo 3,6 m, y la población del estu-dio DECISION, menos agresiva. Los datos en SLP muestran a priori un mayor impacto para lenvatinib frente a placebo al igual que en términos de tasa de respuesta radiológica. Lenvatinib muestra, por tanto, una mayor potencia terapéutica en estos términos y siempre que se pueda, deberíamos em-pezar con el fármaco más potente y eficaz como es práctica habitual en el resto de tratamientos on-cológicos.
3. ¿Sigue teniendo un papel la quimioterapia?
El uso de la quimioterapia convencional está ob-soleto en el manejo de pacientes refractarios a ra-dioyodo y no debería ser tratado ningún paciente con quimioterapia antes que con un ITK como len-vatinib o sorafenib4. En el año 1974, doxorrubicina fue aprobada para su uso en pacientes con CDT refractario, sola o en combinación con taxanos o cisplatino. No en todos los estudios se ha definido la respuesta de la misma manera si bien en todos ellos las respuestas parciales se sitúan en torno al 30-40% (20). Los tratamientos combinados no han demostrado mejores resultados y sí más toxicidad que la monoterapia. Tampoco han demostrado im-pacto en SP ni SG19.
4. Retos futuros
El principal reto para el futuro sería la identificación de un marcador, biológico o genético, que permi-tiera seleccionar precozmente a los pacientes iden-tificando a aquellos que tendrán mayor riesgo de desarrollar radiorrefractariedad o tener una evolu-ción desfavorable. De ese modo, la aplicación de medidas terapéuticas más agresivas inicialmente podría mejorar la supervivencia global de este gru-po de pacientes.
La definición de radiorrefractariedad se plantea como otro de los importantes hitos en la historia natural de la enfermedad, ya que condiciona la ac-titud terapéutica. Quizá deben clarificarse los con-ceptos de pacientes que no van a responder al 131I, evitando tratamientos innecesarios y que además producen un retraso en la aplicación de la terapia correcta. La complejidad de situaciones que plan-tea este grupo de pacientes obliga, sin duda, a su manejo en el seno de comités multidisciplinares, especialmente en centros con experiencia.
La investigación futura nos deparará posiblemente herramientas para la rediferenciación y reinducción de la captación de radioyodo, optimizará el empleo de los inhibidores multiquinasa, su secuenciación o combinaciones de varias estrategias, todo ello diri-gido a cambiar la historia natural de la enfermedad y mejorar la supervivencia.
BIBLIOGRAFÍA
1. Wassermann J, Bernier MO, Spano JP, Lepotre-Lus-sey Ch, Buffet C, Simon JM, et al. Outcomes and Prog-nostic Factors in Radioiodi-ne Refractory Differentiated Thyroid Carcinomas. The on-cologist 2016;21:50-8.
2. Vaisman F, Tala H, Grewal R and Tuttle RM. In diffe-rentiated thyroid cancer, an incomplete structural res-ponse to therapy is associa-ted with significantly worse clinical outcomes than only an incomplete thyroglobu-lin response. Thyroid 2011;21 1317–1322.
3. Capdevilla J, Galofré JC, Grande E, Zafón Llopis C, Ramon y Cajal Asensio T, Navarro González E, Jimé-nez-Fonseca P, Santamaría Sandi J, Gomez Saez JM and Riesco-Eizaguirre G. Con-sensus on the management of advanced radioactive io-dine refractory differentiated thyroid cancer: Spanish task force for thyroid cancer on behalf of the Spanish Socie-ty of Endocrinology (SEEN) and Spanish rare cancer working group (GETHI). Clin Transl Oncol 2016. DOI: 10.1007/s12094- 016-1554-5.
4. Ryma DA, Mandel SJ. Ra-dioiodine therapy for thyroid cancer in the era of risk stra-tificacion and alternative tar-geted therapies. J Nucl Med 2014;55:1485-1491.
5. Dadu R, Cabanillas ME. Optimizing therapy for ra-dioactive iodine-refractory differentiated thyroid can-cer: Current state of the art and futur directions. Minerva Endocrinol 2012;37(4):335-356.
6. Riesco-Eizaguirre G, Ga-lofré JC, Grande E, Zafón Llopis C, Ramon y Cajal T, Navarro Gonzalez E, et al. Spanish consensus for the management of patients with advanced radioactive iodine refractory differentia-ted thyroid cancer. Endocri-nol Nutr 2016;63(4):e17-24.
7. Wang L, Ge M, Xu D, Chen L, Qian C, Shi K, Liu J, Chen Y. Ultrasonography-guided percutaneous radiofrequen-cy ablation for cervical lym-ph node metastasis from thyroid carcinoma. J Can-cer Res Ther. 2014 Nov;10 Suppl:C144-9.
8. Cazzato RL, Bonichon F, Buy X, Godbert Y, de Figue-reido BH, Pointillart V, Pa-lussière J. Over ten years of single-institution experience in percutaneous image-gui-ded treatment of bone me-tastases from differentiated thyroid cancer. Eur J Surg Oncol. 2015 Sep;41(9):1247-55.
9. Vitale G, Fonderico F, Mar-tignetti A, et al. Pamidronate improves the quality of life and induces clinical remis-sion of bone metastases in patients with thyroid cancer. Br J Cancer 2001; 84:1586. Muresan MM, Olivier P, Leclè-re J, et al. Bone metastases from differentiated thyroid carcinoma. Endocr Relat Cancer 2008; 15:37. Wexler JA, Sharretts J. Thyroid and bone. Endocrinol Metab Clin North Am 2007; 36:673.
10. National Comprehensi-ve Cancer Network (NCCN) guidelines. Available at: http://www.nccn.org/pro-fessionals/physician_gls/f_guidelines.asp (Accessed on September 14, 2011 no abs-tract available).
11. Brose MS, Worden FP, Newbold KL, Guo M, Hurria A. Effect of Age on the Effi-cacy and Safety of Lenvati-nib in Radioiodine-Refrac-tory Differentiated Thyroid Cancer in the Phase III SE-LECT Trial. J Clin Oncol. 2017 10;35(23):2692-9.
12. Sack W, Braunstein GD. Evolving approaches in ma-naging radioactive iodine-refractory differentiated thyroid cancer. Endocrine Practice 2014;20:263-275.
13. Schlumberger M, Tha-ra M, Wirth LJ, Robinson B, MS Brose, Elisei R, et al.
Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med 2015;372:621-630.
14. Gianoukakis AG, Mathias EG, Dutcus C, Kalantari P, Yoonet S et al. Response to lenvatinib treatment in patients with radioiodine-refractory differentiated thyroid cancer (RR-DTC): Updated results from SE-LECT. Poster presented at 2016 ASCO Annual Meeting, 3–7 June; Chicago, USA. Pos-ter 411.
15. Haugen BR, Alexander EK, Bible KC, Doherty GM, Mandel SJ, Nikiforov YE, et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Can-cer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016;26:1-1333.
16. Brose MS, Nutting CM, Jarzab B, Elisei R, Siena S, Bastholt L et al. Sorafenib in radioactive iodine-re-fractory, locally advanced or metastatic differentiated thyroid cancer: a randomi-sed, double-blind, phase 3 trial. Lancet 2014;384:319-22.
17. Dubauskas Z, Kunishige J, Prieto VG, et al. Cutaneous squamous cell carcinoma and inflammation of actinic keratoses associated with sorafenib. Clin Genitourin Cancer 2009; 7:20.
18. Brose MS, Nutting C, Shong YK, et al. Associa-tion between tumor BRAF and RAS mutation status and clinical outcomes in pa-tients with radioactive iodine (RAI)-refractory differentia-ted thyroid cancer (DTC) randomized to sorafenib or placebo: sub-analysis of the phase III DECISION trial. ECCO/ESMO/ESTRO Annual Meeting; 2013; Amsterdam.
19. Spitzweg C, Bible KC, Hofbauer LC, Morris JC. Ad-
vanced radioiodine-refrac-tory differentiated thyroid cancer: the sodium iodide symporter and other emer-ging therapeutics targets. Lancet Diabetes Endocrinol 2014;2:830-842.
20. Carter SK, Blum RH New chemotherapeutic agents--bleomycin and adriamycin. Cancer J Clin. 1974;24(6):322.
21. Haugen BR. Management of the patient with progres-sive radioiodine non-respon-sive disease. Semin Surg On-col. 1999;16(1):34.
22. Shimaoka K, Schoenfeld DA, DeWys WD, Creech RH, DeConti R. A randomized trial of doxorubicin versus doxorubicin plus cisplatin in patients with advanced thyroid carcinoma. Cancer. 1985;56(9):2155.

45ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES1. Epidemiología del cáncer diferenciado de tiroides en España: avanzando hacia el registro nacional44
1. Introducción
El abordaje del carcinoma diferenciado de ti-roides refractario al radioyodo debe ser multi-disciplinar y requiere tener en cuenta diversas características del tumor, el paciente y el centro donde se administrará el tratamiento.
En capítulos previos de esta monografía se han desarrollado los criterios de refractariedad al radioyodo y se han enumerado los aspectos fundamentales a tener en cuenta para decidir el momento de inicio de tratamiento sistémico y la mejor alternativa terapéutica para cada paciente y tumor.
En la actualidad, sorafenib y lenvatinib son los dos fármacos que han confirmado su actividad en ensayos clínicos fase III y, por tanto, son dos agentes aprobados para su administración en el carcinoma diferenciado de tiroides refractario al radioyodo, irresecable y en progresión por la Agencia Española del Medicamento y Productos Sanitarios (AEMPS).
2. Aspectos fundamentales a tener en cuenta antes de iniciar lenvatinib
Los carcinomas diferenciados de tiroides tienen un alto riesgo de recurrencia tras cirugías con intención curativa y de progresión y pérdida de
avidez por el radioyodo1. Sin embargo, su pro-nóstico es más favorable que el de otros cán-ceres de tiroides y el de cánceres de otras lo-calizaciones incluso en estadio avanzado. Dicho pronóstico empeora cuando aparece la refracta-riedad al radioyodo con una supervivencia global media en torno a 3 años, aunque existe hasta un 10-20% de pacientes con progresiones lentas y supervivencias prolongadas2.
Los ensayos clínicos fase III con los agentes in-hibidores multiquinasa y antiangiogénicos so-rafenib (estudio DECISION, ClinicalTrials.gov: NCT00984282)3 y lenvatinib (estudio SELECT, ClinicalTrials.gov: NCT01321554)4 han confirma-do un claro beneficio en su objetivo primario, prolongar la supervivencia libre de progresión comparado con placebo. Además, estos fárma-cos logran una reducción clara y duradera del tamaño tumoral. En contraposición, su impac-to en calidad de vida y en supervivencia no se conoce suficientemente y son fármacos asocia-dos con efectos secundarios que requieren una profilaxis, una concienciación del paciente y un manejo precoz y experto.
Por ello, antes de iniciar tratamiento sistémico con lenvatinib y sorafenib, el paciente debe re-cibir información sobre las ventajas e inconve-nientes del fármaco de un especialista experto y entrenado en esta patología. Además, es reco-mendable que el caso se comente en un comité
5. Lenvatinib en cáncer diferenciado de tiroides
PAULA JIMÉNEZ FONSECAServicio de Oncología MédicaHospital Universitario Central de Asturias. Oviedo
FELIPE ÁLVAREZ-MANCECHIDOServicio de Farmacia HospitalariaHospital Universitario Central de Asturias. Oviedo
MANUEL SÁNCHEZ-CÁNOVASServicio de Oncología MédicaHospital General Universitario Morales Meseguer. Murcia
ALBERTO CARMONA-BAYONASServicio de Oncología MédicaHospital General Universitario Morales Meseguer. Murcia
JOSÉ MARÍA VIEITEZServicio de Oncología MédicaHospital Universitario Central de Asturias. Oviedo

47ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES46 5. Lenvatinib en cáncer diferenciado de tiroides
multidisciplinar en el que se evalúen todas las al-ternativas disponibles incluyendo la participación del paciente en un ensayo clínico. (figura 1)
Así, la toma de decisiones compartidas entre el paciente y su médico se hará individualizada, te-niendo en cuenta las expectativas del primero y la opinión y experiencia del segundo.
En cuanto a las características del paciente que deben influir en la decisión sobre el inicio de len-vatinib, el ajuste de dosis y la periodicidad en el seguimiento estarían las habituales en otros tipos de cánceres:
• Edad del paciente5 y expectativa de vida, no sólo la edad cronológica sino también bioló-gica, condicionada por el estado funcional.
• Estado general, la capacidad del paciente para realizar sus actividades diarias, para deambular y ser independiente pueden con-dicionar la administración de fármacos aso-ciados con una toxicidad potencialmente más grave en pacientes frágiles.
• Comorbilidades, algunas patologías previas del paciente pueden contraindicar la admi-nistración de lenvatinib y, en el caso de pato-logías agudas, demorar el inicio del fármaco, por ejemplo, en el caso de fístulas, abscesos o cicatrices postquirúrgicas.
• Deseo y opinión del paciente. Habrá pa-cientes que prefieran mantener una buena calidad de vida e independencia de un cen-tro hospitalario; otros que solo admitirán tratamientos que les permitan llevar a cabo una vida activa y plena mientras que otros tendrán como objetivo controlar el tumor y estarán dispuestos a sobrellevar los efectos adversos del fármaco.
Las características del tumor que se asocian con agresividad, alto riesgo de progresión y, por tan-to, que apoyarían el inicio precoz de lenvatinib son1,2:
• Metástasis de gran tamaño o alta carga tu-moral.
• Progresión rápida.
• Paciente con síntomas derivados del creci-miento tumoral.
• Riesgo de desarrollo de complicaciones clí-nicamente significativas.
También se podría valorar tratamiento sistémi-co con lenvatinib en tumores irresecables con extensión metastásica limitada inoperables al diagnóstico pero candidatos a cirugía si, tras el tratamiento, redujeran su carga tumoral al punto de cumplir criterios quirúrgicos.
La mayoría de los ensayos clínicos en cáncer de tiroides diferenciado han requerido la confir-mación de progresión tumoral por criterios ra-diológicos Response Evaluation Criteria In Solid Tumors (RECIST) en los 12-14 meses previos a la inclusión y éste es un criterio extendido para iniciar la terapia sistémica en pacientes asinto-máticos y sin localizaciones tumorales de riesgo6.
Las peculiaridades del centro donde es atendi-do el paciente también influirán en la toma de decisiones. Si bien, es interesante incidir en que, independientemente de la carga asistencial, la disponibilidad de recursos y técnicas y la expe-riencia del especialista que tomará la decisión sobre el tratamiento sistémico a administrar, se deben tener en cuenta los siguientes aspectos:
• Resecabilidad y abordaje locorregional de las metástasis, valorando la derivación a cen-tros expertos si fuese necesario.
• Terapias sistémicas disponibles. Lenvatinib y sorafenib están aprobadas para su adminis-tración en España a pacientes con carcino-ma diferenciado de tiroides yodorefractario estadio IV irresecable en progresión3,4. Por tanto, fuera de la alternativa de un ensayo clínico, deberían ser los primeros fármacos a considerar.
• Perfil molecular. Los estudios fase III con len-vatinib4 o sorafenib3 no limitan su empleo a tumores con unos rasgos moleculares concre-tos. Sin embargo, en pacientes refractarios, el mejor conocimiento de las características ge-néticas y moleculares del tumor podría ayudar a seleccionar tratamientos como es el caso de la inmunoterapia y su diana, PD-1.
• Derivación a centro experto y de referencia. Aunque no es una práctica común en nuestro país, la elección del mejor tratamiento para cada paciente, tumor y momento suele re-querir de un abordaje multidisciplinar en el seno de un comité de tumores7.
3. Ensayo clínico fase III SELECT
A continuación, se recogen las características del en-sayo clínico SELECT publicado en 20154:
• Estudio fase III: randomizado, doble ciego, multi-céntrico, comparado con placebo en asignación
Figura 1. Algoritmo de toma de decisiones en pacientes con un carcinoma de tiroides diferenciado, metastásico y refractario al radioyodo.

49ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES48 5. Lenvatinib en cáncer diferenciado de tiroides
2:1, en el grupo placebo estaba permitido el cruce a lenvatinib a la progresión.
• Periodo de inclusión: agosto 2011 a octubre 2012.
• Objetivos: principal, supervivencia libre de progresión y secundarios, tasa de respues-ta, supervivencia global, seguridad (efectos adversos). No se aplicaron tests de calidad de vida (si se aplicaron en el estudio de so-rafenib3).
• Criterios de inclusión: refractariedad al yodo; progresión radiológica en los 13 meses pre-vios con determinación centralizada y enfer-medad medible. Se permitía haber recibido un tratamiento previo con un inhibidor mul-tiquinasa o con quimioterapia (principal dife-rencia con el estudio DECISION3). Los pacien-tes debían ser ≥ 18 años.
• Población y subgrupos: 391 pacientes. Sub-grupos: edad (≤65 vs >65 años), sexo, región geográfica (Europa, Norte América, otras), tipo histológico (papilar, pobremente dife-renciado, folicular, y células de Hürthle), ni-veles de tirotropina (TSH) (≤0.5, >0.5-2.0 y >2.0-5.5 mIU/L), líneas previas (ninguna vs una).
4. Dianas y posología de lenvatinib
Lenvatinib es un inhibidor de los receptores ti-rosina-cinasa que inhibe selectivamente la acti-vidad cinasa de los receptores del factor de cre-cimiento del endotelio vascular 1 a 3 (VEGFR-1 [FLT1], -2[KDR], -3 [FLT4]), el receptor del fac-tor de crecimiento derivado de las plaquetas (PDGFR-α), RET, c-KIT y los receptores del fac-tor de crecimiento fibroblástico 1 a 4 (FGFR1-4). (figura 2)
Lenvatinib se administra a dosis de 24 mg/día de forma continuada mientras no exista una progresión radiológica franca o una toxicidad inaceptable1,2. En pacientes con insuficiencia hepática o renal grave se debe iniciar a dosis de 14 mg/día sin necesidad de ajustar dosis en insuficiencia hepática o renal leve o moderada. No existe suficiente evidencia de su efecto en
jóvenes hasta 18 años y la evidencia disponible es limitada en >75 años.
Si aparece toxicidad es fundamental tratar de forma precoz y enérgica los efectos adversos (ver punto 6 de este capítulo). Cuando la toxici-dad es grave o persiste a pesar del tratamiento puede ser necesario detener el fármaco y rei-niciarlo cuando la toxicidad descienda a grado 1-2. Al reiniciar el tratamiento suele ser necesario reducir la dosis a 20 mg/día. Ante nuevas toxi-cidades graves la dosis se puede reducir a 14 y 10 mg/día4.
Se necesita más información para determinar cuándo iniciar lenvatinib y para elegir la secuen-cia y combinación más efectiva de fármacos sin causar una toxicidad aditiva con otros agentes multiquinasa y antiangiogénicos.
No está claro cuando interrumpir el tratamiento ya que existe la posibilidad de que el beneficio clínico se prolongue más allá de la progresión de la enfermedad, especialmente en aquellos casos de progresión lenta, sin afectación de estructuras vitales, con buen estado general del pacientes y buena tolerancia al fármaco3,4,8.
5. Actividad de lenvatinib
La supervivencia libre de progresión de lenva-tinib versus placebo, objetivo principal del es-tudio fase III, fue de 18.3 vs. 3.6 meses [hazard ratio (HR) 0.21; intervalo de confianza (IC) 99%, 0,14–0,31; p< 0.001] y el beneficio se mantuvo en todos los subgrupos predeterminados4 (ver apartado 3 de este capítulo). Cabe destacar la prolongada supervivencia libre de progresión de los pacientes que alcanzaron una respuesta por criterios RECIST que fue de 33 meses (último corte de base de datos: Agosto 2015).
En el análisis de subgrupos hubo una tenden-cia a un incremento de la supervivencia libre de progresión en los pacientes que recibieron len-vatinib en primera línea de tratamiento sistémico que, comparada con placebo, fue de 18,7 vs 3,6 meses, HR 0,21, p<0,0001. La supervivencia libre de progresión en 2ª línea, después de un agente antiangiogénico, comparada con placebo fue de 15,1 vs 3,6 meses, HR 0,22, p<0,0001. Un 25% de
los pacientes habían recibido un agente antidia-na previo que en el 77% fue sorafenib, en el 9% sunitinib y en el 5% pazopanib9.
El beneficio en supervivencia libre de progresión asociado con lenvatinib fue independiente de la localización de las metástasis, con la excepción de pacientes con metástasis cerebrales en los que cayó a 8,8 meses en pacientes tratados con lenvatinib frente a 3,7 meses en los que recibie-ron placebo10.
El control del crecimiento tumoral fue4:
• Respuestas objetivas radiológicas por crite-rios radiológicos RECIST de lenvatinib frente a placebo: 64,8% vs 1,5% (odds ratio 28.87; IC 95%, 12,46- 66,86; p <0.001). 4 pacientes (1,5%) alcanzaron una respuesta completa. La mediana de tiempo que se tardó en alcanzar la respuesta fue de 2 meses (IC 95%, 1,9-3,5).
• La tasa de respuestas fue > 50% indepen-dientemente de la localización de las me-tástasis: cerebro, hueso, hígado, pulmones y ganglios10.
• 29,8% de los pacientes lograron una estabili-zación tumoral mantenida más de 23 sema-nas.
• Los pacientes que consiguieron una estabi-lización de la enfermedad, sin respuesta ob-jetiva, la duración de la respuesta fue de 8 meses (último corte de base de datos: Agos-to 2015).
En cuanto a la supervivencia global:
• El análisis inicial de los resultados en 2014, mostró una tendencia a incrementar la super-vivencia global sin alcanzar la significación estadística probablemente debido al diseño
Figura 2. Dianas moleculares de los principales fármacos con actividad en cáncer de tiroides.

51ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES50 5. Lenvatinib en cáncer diferenciado de tiroides
del estudio en el que se permitía recibir len-vatinib a la progresión en el grupo placebo y también en relación con que los datos eran inmaduros para esta variable en el momento del análisis4. En análisis posteriores se docu-mentaron los siguientes resultados:
• Aplicando el modelo estadístico que tiene en cuenta el cruzamiento a la progresión (rank-preserving structural failure time model) se demostró un incremento en la tasa de super-vivencia global con lenvatinib (HR 0,53; IC 95%: 0,34-0,82, p =0,0051)11.
• El análisis de supervivencia global en los distintos subgrupos de pacientes y tumo-res, confirmó un aumento estadísticamente significativo de la supervivencia en pacientes mayores de 65 años respecto a los jóvenes5 y en subtipo histológico folicular respecto al papilar12.
6. Toxicidad y manejo de los efectos adversos
En el estudio SELECT, lenvatinib se asoció con aparición de toxicidad grado ≥3 en un 75% de los casos (efectos adversos acumulativos a lo largo de más de 2 años de tratamiento) y cuya tem-poralidad fue variable. Por lo tanto, es necesario que antes de iniciar el tratamiento, los pacientes sean informados de los riesgos y de cómo mane-jar las posibles toxicidades. Así mismo, debe rea-lizarse un seguimiento estrecho y una vigilancia de la toxicidad más frecuente o potencialmente grave. Para ello, los pacientes deben acudir pe-riódicamente a consulta donde se les interrogue sobre la toxicidad y se realice una exploración fí-sica con toma de tensión, determinación de pro-teinuria y de peso corporal. En dicha consulta se debe realizar una analítica que incluya pruebas hepáticas, renales, iones, hemograma y niveles de TSH. (figura 3)
El efecto secundario más común, efecto de cla-se asociado a su mecanismo de acción, es la hipertensión, presente en un 67,8% y grado ≥3 en el 41,8% en el estudio SELECT, incluyendo un 56% de pacientes con hipertensión basal alea-torizados a lenvatinib. Por esto, la presencia de hipertensión antes de iniciar el fármaco no con-traindica su administración pero es importante
asegurarse de un buen control previo a la toma de lenvatinib. La tensión se debe monitorizar se-manalmente al menos durante las 8 primeras se-manas. El tratamiento requerirá dieta hiposódica, elevada ingesta de agua, ejercicio físico e inicio precoz de un antihipertensivo preferiblemente de un calcio antagonista, inhibidor del sistema renina/angiotensina o ambos en caso de hiper-tensión grado 2-3. Las medidas higiénico-dietéti-cas para control de la tensión se deben instaurar desde el inicio del tratamiento con lenvatinib es-pecialmente en pacientes con antecedentes de hipertensión, patología cardiovascular o edad avanzada. Así mismo, existe evidencia sobre el efecto predictor de respuesta de la hipertensión y su aparición como consecuencia del daño del endotelio vascular mediado por la inhibición de VEGFR-213,14.
Otros efectos secundarios documentados y su frecuencia, documentada en el estudio fase III, fueron4:
• Diarrea: 59,4%, grado ≥3, 8%. Tratamiento médico con dieta astringente: arroz cocido, pan blanco, patata cocida, pasta, puré de ca-labacín, calabaza o zanahoria, jamón cocido, queso fresco, pollo hervido, pescado blanco hervido, yogur con zumo de limón, plátano, manzana rayada y pelada. Asegurar el apor-te de líquidos con limonada alcalina, caldos e infusiones de té o tomillo. Como fármacos antidiarreicos se suelen emplear la loperami-da, el racecadotrilo y, en casos graves o re-fractarios, se pueden valorar opiáceos.
• Astenia: 59%, grado ≥3, 9,2%. Se debe infor-mar al paciente de este efecto para evitar que lo confunda con una mala evolución del cáncer. Ciertos alimentos como los ricos en vitamina B (levadura de cerveza, nuez, pi-ñón), los germinados, los cereales integrales, el sésamo, el germen de trigo, el plátano, el polen y el yogur, pueden reducir la debilidad y la pérdida de peso, otro efecto adverso co-mún del lenvatinib.
• Pérdida de apetito: 50,2%, grado ≥3, 5,4%. El ejercicio físico moderado, la ingesta de zumos ácidos (limón, piña) o de pequeñas cantidades de caldo antes de las comidas, el laurel y otras hierbas aromáticas empleadas
como condimento pueden aumentar el ape-tito. El acetato de megestrol es un fármaco indicado en el tratamiento de la anorexia aso-ciada al cáncer o a sus tratamientos.
• Pérdida de peso: 46,4%, grado ≥3, 9,6%. Los alimentos enumerados en la astenia pueden ayudar a combatir tanto la debilidad como la pérdida ponderal. Es importante instruir al paciente en realizar unas 6 comidas ligeras al día y en aumentar el aporte de nutrientes en los momentos de mayor apetito. Si el pa-ciente pierde más del 5-10% de su peso se pueden instaurar suplementos nutricionales y se pueden asociar con acetato de megestrol para combatir la anorexia.
• Náuseas: 41%, grado ≥3, 2,3%. Los especia-listas deben prescribir profilácticamente fármacos antieméticos a los pacientes an-tes del inicio de lenvatinib por el riesgo de desarrollar este efecto adverso y para su abordaje precoz. En momentos de náuseas los alimentos que mejor se toleran son los de fácil digestión, textura blanda, sabor suave y preparación sencilla como los batidos de yogur y frutas, el queso fresco, las natillas, las cuajadas, el helado cremoso, la tortilla francesa, la patata cocida o en puré, el pollo. Antes de las comidas, se puede calmar el es-tómago y evitar regustos desagradables en la boca chupando polos o rodajas de fruta. El agua favorece los vómitos y las náuseas, se
Figura 3. Medidas encaminadas a la prevención y tratamiento precoz de los efectos adversos.

53ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES52 5. Lenvatinib en cáncer diferenciado de tiroides
puede sustituir por infusiones de manzanilla, menta, anís o jengibre. Los fármacos antie-méticos que se pueden dar o combinar según la intensidad de los vómitos son fundamen-talmente los procinéticos (domperidona o metroclopramida) y los setrones.
• Mucositis: 35,6%, grado ≥3, 4,2%. El daño de la mucosa oral se puede prevenir con enjua-gues de agua con sal o bicarbonato sódico al 9% o con ambos alternándolos o con tomi-llo. Existen diversos preparados de farmacia para el tratamiento de las aftas y las heridas en la boca. Se debe explorar en la consulta la cavidad oral para detectar infecciones. Si la mucositis es muy intensa se pueden pautar analgésicos para que el paciente logre comer sin sentir dolor.
• Síndrome palmoplantar: 31,8%, grado ≥3, 3,4%. Es importante instruir al paciente so-bre el cuidado de la piel especialmente la de manos y pies:
- La hidratación con crema que contenga urea 1-2 veces al día.
- Evitar la exposición al sol sin protección.
- Evitar el contacto de manos y pies con tem-peraturas extremas y con agua muy calien-te.
- Empleo de calzado cómodo, de piel y calce-tines de algodón .
- Evitar ejercicios vigorosos que puedan ma-cerar la piel por sudor o humedad.
- Si aparecen ampollas, inflamación o signos de infección debe comentárselo a su mé-dico.
En caso de inflamación muy severa de palmas y/o plantas puede ser necesario el uso de corti-coides sistémicos.
Proteinuria: 31%, grado ≥3, 10%. Se debe vigilar la aparición de proteinuria mediante su medi-ción con tira o análisis de orina especialmente en pacientes que hayan desarrollado hipertensión arterial con lenvatinib o en aquellos que tengan
antecedentes de hipertensión arterial, diabetes o nefropatía.
Cefalea: 27,6%, grado ≥3, 2,7%. Se debe informar al paciente del riesgo de cefalea asociado a la toma de lenvatinib. El abordaje farmacológico puede ser con el fármaco que ya tomase el pa-ciente si tiene antecedentes de cefalea o elegirlo en función de la intensidad del dolor y de las co-morbilidades del paciente.
Otras toxicidades graves (grado ≥3) como los eventos tromboembólicos arteriales y venosos, la insuficiencia renal aguda, el alargamiento del intervalo QT en el electrocardiograma, la fístula gastrointestinal y el fallo hepático tuvieron una frecuencia del 2,7%, 3,8%, 1,9%, 1,5%, 0,8% y 0,4%, respectivamente.
Los efectos adversos condicionaron la interrup-ción del tratamiento en un 82,4% fundamental-mente por hipertensión y astenia; disminuciones de dosis en un 67,8% y suspensiones del fármaco en un 14,2%. La mediana de dosis administrada fueron 17,2 mg/día que corresponde al 72% de la dosis inicial, 24 mg/día6. La mediana de tiempo de tratamiento fue de 13,8 meses a pesar de los 18,3 meses de mediana de supervivencia libre de progresión, es decir, muy probablemente la toxi-cidad evitó el tratamiento hasta progresión4.
7. Situaciones especiales
7.1. Personas mayores de 65 años5 En el estudio fase III SELECT, el grupo de mayo-res de 65 años tenía una mediana de edad de 71 años.
El subanálisis comparando el beneficio según la edad demostró un incremento de la superviven-cia libre de progresión con lenvatinib vs placebo en ambos grupos de edad, jóvenes y mayores, siendo de 20,2 vs 3,2 meses (HR 0,19; IC 95%, 0,13-0,27; p <0.001) y 16,7 vs 3.7 meses (HR 0,27; IC 95%, 0,17-0,43; p <0.001), respectivamente.
Los pacientes con una edad ≤ 65 años mostra-ron mayor tasa de respuesta (72% vs 55%; p =0.0038) y mejor tolerancia, con mayor tiempo hasta la primera reducción de dosis (3,7 vs 1,5 meses), menor toxicidad grave, grado ≥ 3 (67% vs 89%; p <0.001) comparado con los >65 años.
A pesar del cruzamiento en el grupo placebo después de la progresión, se encontró un bene-ficio en supervivencia en los pacientes de más edad (HR 0,53; IC 95%, 0,31-0,91; p =0.020).
Por todo lo anterior, lenvatinib debe ser conside-rado en el tratamiento de los pacientes de cual-quier edad teniendo mayor precaución y reali-zando un seguimiento más estrecho en mayores de 65 años.
7.2. Pacientes jóvenes con deseos genésicos Las mujeres en edad fértil deben evitar quedarse embarazadas por el riesgo teratógeno del fár-maco y deben utilizar métodos anticonceptivos altamente efectivos, preferiblemente de barrera, durante el tratamiento con lenvatinib y como mí-nimo hasta un mes tras finalizar el tratamiento.
Existe evidencia de que al detener un agente in-hibidor multiquinasa, la progresión de la enfer-medad puede ser rápida. Esto es de particular importancia en los pacientes, mujeres o hombres, que tienen la intención de tener hijos. Estos pa-cientes deben dejar de tomar el fármaco duran-te al menos 12-14 y 3-6 meses, respectivamente. Todavía no hay pruebas suficientes para predecir hasta qué punto una interrupción durante este tiempo de lenvatinib afecta a la progresión de la enfermedad. En pacientes en edad fértil y con deseo de tener hijos, debe tenerse en cuenta la preservación de los gametos antes de iniciar el tratamiento sistémico puesto que lenvatinib po-dría ser tóxico sobre ovarios y testículos8.
8. Alternativas a lenvatinib
No existen estudios que hayan comparado lenva-tinib con el otro fármaco aprobado en carcinoma de tiroides bien diferenciado refractario a radio-yodo, el sorafenib.
Por tanto, no existen criterios ni recomendacio-nes en las guías de práctica clínica o consensos que permitan seleccionar cual es el mejor tra-tamiento para cada paciente ni la secuencia de tratamientos.
Teniendo en cuenta las poblaciones de los en-sayos clínicos SELECT-lenvatinib4 y DECISION-sorafenib3, los factores que pueden ayudar a la
selección del fármaco, lenvatinib o sorafenib, se-rían:
• Subgrupos moleculares: el beneficio de len-vatinib no estuvo condicionado por la varian-te molecular siendo igualmente eficaz en to-dos los subgrupos, mientras que el beneficio de sorafenib en supervivencia parecería estar limitado a los pacientes con tumores BRAF mutado (HR 0,32, p =0.03).
• Estado del tumor: en ambos estudios el tu-mor debía de estar en progresión en los 13 (lenvatinib) y 14 (sorafenib) meses previos. La determinación se hizo a nivel local en el caso de sorafenib y centralizada en el caso de lenvatinib.
• Tratamientos previos: en el estudio DECI-SION se excluyeron pacientes que habían recibido otro tratamiento sistémico, estos pacientes se incluyeron en el estudio SELECT siendo hasta el 25% los que habían recibido previamente un agente inhibidor multiquina-sa.
• Búsqueda o necesidad de alcanzar una res-puesta tumoral. Si este fuera el objetivo, lenvatinib ha mostrado mayor tasa de res-puestas que sorafenib, 64,8% vs 12,2% y de respuestas completas, 1,5% vs 0 %. Además, lenvatinib obtuvo una reducción de la masa tumoral muy importante y de forma muy rá-pida (mediana de 2 meses).
• Perfil de efectos adversos del fármaco y co-morbilidades del paciente. La hipertensión grado ≥3 fue más frecuente con lenvatinib que con sorafenib, 41% vs 9% mientras que el rash cutáneo grado ≥3 fue más común con sorafenib que con lenvatinib, 5% vs <1%. Con lenvatinib se documentaron un 8% de casos de alargamiento del intervalo QT, 1% grado ≥3, y ninguno con sorafenib8.
En el estudio DECISION, sorafenib se asoció con un leve descenso, pero estadísticamente signi-ficativo de la calidad de vida al inicio del trata-miento debido a los efectos adversos y se man-tuvo estable durante el período de tratamiento3. En el estudio SELECT no se analizó la calidad de vida4.

55ACTUALIZACIÓN EN CÁNCER DIFERENCIADO DE TIROIDES54 5. Lenvatinib en cáncer diferenciado de tiroides
Todavía no se conoce como pueden influir en la elección de uno u otro fármaco y en la secuencia, las dianas que solamente bloquea uno de ellos como FGFR1-3 en el caso de lenvatinib y RAF y FLT3 en el caso de sorafenib. No obstante, una de las dianas moleculares de lenvatinib es el FGFR-4 que se ha descrito como esencial en los procesos de resistencia a los tratamientos anti-angiogénicos con TKIs.
Tras la progresión a lenvatinib y sorafenib, la guía americana ATA recoge la posibilidad de admi-nistrar otros inhibidores tirosin kinasa con bue-nos resultados en ensayos fase II como sunitinib, axitinib, pazopanib, vandetanib, cabozantinib, everolimus1, si bien todos ellos sólo disponen de resultados de estudios fase 2.
La quimioterapia, doxorrubicina, ha demostrado una actividad modesta pudiendo ser una alterna-tiva en cánceres más agresivos, indiferenciados, de rápido crecimiento, sintomáticos, en progre-sión a lenvatinib y sorafenib o cuando estos es-tán contraindicados15.
En la actualidad, otras alternativas como la inmu-noterapia, los inhibidores de BRAF como dabra-fenib o vemurafenib, de MEK como selumetinib16 y los reinductores de la expresión de NIS todavía requieren una confirmación de su eficacia en en-sayos clínicos fase III17.
9. Conclusiones
• Lenvatinib es, hasta la actualidad, el fármaco que ha demostrado mejores re-sultados en tasa de respuestas y supervivencia libre de progresión en cáncer diferenciado de tiroides yodorrefractario en progresión.
• Lenvatinib es, junto con sorafenib, el único agente inhibidor multiquinasa apro-bado en España para el tratamiento del cáncer diferenciado de tiroides estadio IV irresecable.
• Lenvatinib es ligeramente más eficaz cuando se administra en primera línea de tratamiento sistémico a pesar de que también es activo en segunda línea y en todos los subgrupos analizados, incluidos los pacientes ancianos y los tumores pobremente diferenciados.
• Lenvatinib se asocia con toxicidades que requieren ofrecer una información específica para el paciente, un manejo experto y una vigilancia estrecha funda-mentalmente de la hipertensión, proteinuria, eventos tromboembólicos, toxici-dad digestiva, cutánea, renal y hepática.
BIBLIOGRAFÍA
1. Haugen BR. 2015 Ame-rican Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: What is new and what has changed? Cancer. 2017;123(3):372-81.
2. Capdevila J, Galofré JC, Grande E, Zafón Llopis C, Ramón Y Cajal Asensio T, et al. Consensus on the man-agement of advanced ra-dioactive iodine-refractory differentiated thyroid can-cer on behalf of the Spanish Society of Endocrinology Thyroid Cancer Working Group (GTSEEN) and Span-ish Rare Cancer Working Group (GETHI). Clin Transl Oncol. 2017;19(3):279-87.
3. Brose MS, Nutting CM, Jarzab B, Elisei R, Siena S, Bastholt L, et al. Sorafenib in radioactive iodine-re-fractory, locally advanced or metastatic differentiat-ed thyroid cancer: a ran-domised, double-blind, phase 3 trial. Lancet. 2014;384(9940):319-28.
4. Schlumberger M, Taha-ra M, Wirth LJ, Robinson B, Brose MS, Elisei R, et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med. 2015; 372(7): 621-30.
5. Brose MS, Worden FP, Newbold KL, Guo M, Hur-ria A. Effect of Age on the Efficacy and Safety of Lenvatinib in Radioio-dine-Refractory Differen-tiated Thyroid Cancer in the Phase III SELECT Tri-al. J Clin Oncol. 2017 Jun 14:JCO2016716472. [Epub ahead of print].
6. Narayanan S, Colevas AD. Current Standards in Treatment of Radioiodine Refractory Thyroid Cancer. Curr Treat Options Oncol. 2016;17(6):30.
7. Díez JJ, Galofré JC, Olea-ga A, Grande E, Mitjavila M, Moreno P, et al. Consensus statement for accreditation
of multidisciplinary thyroid cancer units. Endocrinol Nutr. 2016;63(3):e1-15.
8. Viola D, Valerio L, Moli-naro E, Agate L, Bottici V, Biagini A, et al. Treatment of advanced thyroid can-cer with targeted thera-pies: ten years of experi-ence. Endocr Relat Cancer. 2016;23(4):R185-205.
9. Yeung KT, Cohen EE. Lenvatinib in Advanced, Radioactive Iodine-Refrac-tory, Differentiated Thyroid Carcinoma. Clin Cancer Res. 2015;21(24):5420-6.
10. Habra MA, Song JH, Ri-etschel P. Outcomes by site of metastasis for patients with radioiodine-refractory differentiated thyroid can-cer treated with lenvatinib versus placebo: results from a phase 3, randomized trial. 15th International Thyroid Congress, 18–23 October 2015, Orlando, Florida, USA. Thyroid 25 (supplement 1) A-23 abstract 55.
11. Guo M, Sherman L, Wirth L, Schlumberger M, Dutcus C, Robinson B, et al. Overall survival gain with lenvatinib vs. placebo in radioactive iodine refractory differen-tiated thyroid cancer (RR-DTC): An updated analysis. European Cancer Congress, 25–29 September 2015, Vienna, Austria. Abstract 2805.
12. Elisei R, Schlumberger M, Tahara M, Robinson B, Brose M, Dutcus C, et al. Subgroup analysis according to differ-entiated thyroid cancer his-tology in phase 3 (SELECT) trial of lenvatinib. Oncology Research and Treatment. 2015; 38 (Supplement 5) 1–270 (abstract 91).
13. Choi J, Abouzaid X, Li X, Rietschel P. Characteristics of patients on lenvatinib with treatment-emergent hypertension in the select trial. 15th International Thy-roid Congress, 18–23 Octo-ber 2015, Orlando, Florida, USA. Thyroid 25 (Supple-ment 1) A-250, abstract 629.
14. Sueta D, Suyama K, Sue-ta A, Tabata N, Yamashita T, Tomiguchi M, et al. Lenva-tinib, an oral multi-kinases inhibitor, -associated hy-pertension: Potential role of vascular endothelial dys-function. Atherosclerosis. 2017;260:116-20.
15. Ho AL, Grewal RK, Leb-oeuf R, Sherman EJ, Pfister DG, Deandreis D, et al. Sel-umetinib-enhanced radioio-dine uptake in advanced thy-roid cancer. N Engl J Med. 2013;368(7):623-32.
16. Bible KC, Ryder M. Evolving molecularly tar-geted therapies for ad-vanced-stage thyroid can-cers. Nat Rev Clin Oncol. 2016 Mar 1. [Epub ahead of print]
17. Albero A, Lopéz JE, Tor-res A, de la Cruz L, Martín T. Effectiveness of chemother-apy in advanced differenti-ated thyroid cancer: a sys-tematic review. Endocr Relat Cancer. 2016; 23(2):R71-84.

1. Epidemiología del cáncer diferenciado de tiroides en España: avanzando hacia el registro nacional56 LE
N17
21.
Fech
a d
e p
rep
ara
ció
n: N
ovie
mb
re 2
017





























