Embed Size (px)
Citation preview

AOPIEDADES SELECTIVAS DE LA ZEOLITA Pt/MORDENITA EN LA REACCION
DE HIDROCRACKING
Tesis que presenta el
A I . Q . JORGE BERNAL NAVARRO, Para l a obtención del grado de
J~AESTRO EN QUIMICA
Noviembre,l985/

o G
i
A IRMA IRENE, mi esposa -
A m i s hi jos, Celia Isabel Irma Irene Jorge Carlos Olga Paulina

LABOR VINCI OMNIA

S U M A R I O
CAPITULO I Introducción Catálisis por zeolitas Propiedades físicas y catallticas de la zeolita
Acidez y propiedades catallticas Aplicación industrial
tipo mordenita
Cracking Hidrocracking
CAPITULO I1 Caracterización de catalizadores Preparación y activación de catalizadores Determinación de la dispersión Parte experimental
Gravimetrla Volumetría
Resultados y discusián
CAPITULO I11 Determinación de la actividad en la hidrogenación
Parte experimental Resultados y discusión
de benceno
CAPITULO IV Determinación de la actividad y l a selectivad en la
reacción de hidrocracking de n-decano, decalina y tetralina
Parte experimental Resultados y discusión Conclusiones
BIBLIOGRAFIA
1 2
6
13 2 0
2 0
21
24
24
31 32
32
4 1 47
4 8 4 9 53
57 6 0 63 8 0
8 1

CAPITULO I
INTRODUCCION
Zeolitas. Tamices Moleculares
El término Zeolita es un nombre mineraldgico que des- cribe una familia de aluminosilicatos cristalinos.
Estructuralmente las zeolitas están basadas sobre una malla tridimensional infinitamente extendida de tetraedros de (A104) y (Si04) enlazados por los átomos -de ox€geno com- partidos por dichos tetraedros.
Químicamente la fórmula estructural de una celda uni- taria cristalográfica puede ser expresada en términos de los Óxidos.
Mx/n (A102)x (Si0 1 mH20 2 Y
Donde M es un catiÓn de Valencia n, m es el número de moléculas de agua y l a suma de X y Y es el número total de tetraedros en la celda unitaria.
El esqueleto contiene canales y huecos interconecta- dos que son ocupados por cationes y moléculas de agua.
Generalmente los cationes son movibles y pueden reem- plazarse en diversos grados por intercambio coniotros ca- t i o r r e S . ( l r 2 r 3 r 4 r 5 r 6 ? l l r 1 3 r 2 2 r 2 8 r 2 9 )
El agua intracristalina puede extraerse algunas veces -

-
2
reversiblemente.
Para muchas zeolitas, la deshidratación completa pue- de ocasionar una perturbación irreversible de la estructu- ra del esqueleto y la localización de cationes metálicos, lo que se manifiesta en un colapso estructural-parcial. Es por esta razón que muchas zeolitas después de la deshi- dratacibn, contienen sistemas de canales muy pequeños.
Las estructuras de mayor interés en la catálisis de conversión de hidrocarburos, son las de las zeolitas de poro grande (ancho) tipos X, Y, L,R y mordenita.
d
h s estructuras de poro más pequeño tipos A y erioni- C ta son de interés, particularmente en el caso de Catálisis S e ~ e C ~ ~ V a ~ ~ l r 2 r 1 3 r 2 1 r 2 2 r 2 3 r 2 7 ~ I CATALISIS POR ZEOLITAS
Los progresos en los últimos años en Catálisis Hetero- génea se han asociado con el uso de las zeolitas como cata- lizadores o soportes.
Las propiedades catallticas de las zeolitas son espe- cíficas en muchos aspectos y están relacionados con las S ~ g u ~ e n ~ e s ~ ~ 5 ' 9 ' 1 0 i 2 5 f 2 7 f 3 0 ~
Características estructurales:
a). Las zeolitas tienen una red cristalina completamen- te definida.

do de la 1 b, 3
Su estructura se conserva a 700-10OO0C (dependien- composición y la naturaleza del cati6.n.
c lares constantes de 5 a 10 A. P Los poros de las zeolitas son de dimensiones mole-
n
d) Los grupos (Si04) y (A104) pueden ser reemplaza- dos por grupos estructurales que contienen otros elementos Be, Mg, B, Ga, Ge, Ti, etc.
e) En la forma catiónica, las zeolitas contienen una cantidad de cationes metálicos capaces de intercambio i6- nico.
¿Qué implicaciones tienen estas características estruc- turales de las zeolitas en catálisis?
10. La existencia de una estructura cristalina con poros de dimensiones moleculares constantes permite prepa- rar catalizadores altamente selectivos.
20. Es factible que componentes activos catalftica- mente, puedan ser introducidos en las zeolitas por inter- cambio iÓnico, los catalizadores obtenidos de este modo presentan nuevas propiedades específicas.
CARACTERISTICAS REGULARES PRINCIPALES DE LA ACCION CATALITI- CA DE LAS ZEOLITAS
1. Las zeolitas catalizan reacciones del-tipo E6n car- boni-o Red-Ox, oxidación, hidrogenacibn, etc.
2. En reacciones tipo Ion-Carbonio, las formas catió- -
- ...*

4
nicas monovalentes de las zeolitas son inactivas mientras que las formas polivalentes y también las modificaciones en las cuales los cationes monovalentes han sido extraldos, muestran un incremento en su actividad.
3 . Con el incremento de la relación Si02/A1203 en zeolitas con estructuras similares, su actividad catallti- ca en reacciones del tipo ácido base generalmente se incre- menta.
4 . Con el incremento irregular en el grado de inter- + cambio de los iones Na por cationes polivalentes, existe
un grado de intercambio límite ( 0 1 ' ) a partir del cual se presenta actividad y un rápido incremento en la misma es observado con un reemplazamiento adicional de iones Na . +
La magnitud de a' depende de la relación Si02/A1203, la naturaleza del catidn y la naturaleza de la reacción.
5 . El agua y otros donadores de protones presentan un esfecto promotor, su presencia en el catalizador en peque- ñas cantidades aumenta la velocidad de reacción que influye sobre la actividad catalltica de las forma catibnicas poli- valentes de zeolitas en reacciones tipo Ión-Carbonio; el efecto promotor depende de la composición de la zeolita, la naturaleza del catión y las condiciones de promoción.
6. Un efecto similar lo presentan varios compuestos ácidos que modifican la actividad catalftica de las zeoli- tas sbdicas.
7 . La quimisorción y la catálisis sobre zeolitas ocu-
n con la particzpación de sólo pequeñas fracc2ones arri-
_ _ . . . . . ,
. --

5
ba del 5-10% de los centros activos potenciales calculado en base a datos cristalográficos.
8 . Existen condiciones óptimas para los tratamientos térmicos preliminares (activación) de las zeolitas y son diferentes para las diversas modificaciones de las mismas y pueden depender de la naturaleza de la reacción en la que serán utilizados.
9. La selectividad de los catalizadores zeolíticos es determinada en gran parte por las propiedades de tamiz mo- lecular de las zeolitas.
10. Las reacciones catalizadas por zeolitas usualmen- te pueden ocurrir sobre aluminosilicatos amorfos pero su actividad puede diferir por órdenes de magnitud 3 - 4 .
COWARACION DE LAS PROPIEDADES DE LAS ZEOLZTAS Y LOS ALUMI-
NOSILICATOS AMORFOS
Los datos experimentales obtenidos muestran que las propiedades catalíticas de alunino-silicatos cristalinos y amorfos tiene mucho en común. Las zeolitas catalizan prác- ticamente todas las reacciones que son caracterfsticas de los alumino-silicates; las energfas de activacibn de los procesos sobre estos catalizadores de contacto no difleren grandemente, aunque las actividades de los catalizadores zeolíticos son algunas veces de 100 a 10,000 veces mayores; la modificaci6n por intercambio iónico conduce a cambios similares en l a s propiedades catallticas. Esto sugiere que la diferencia principal entre las zeolitas y los alumi-
-

6
no-silicatos radica en la diferencia del número de centros ácidos presentes en ambos tipos de materiales, sin embargo se han establecido características específicas para las zeoiitas. (4, 2 1 , 1 6 )
PROPIEDADES FISICAS Y CATALITICAS DE LA ZEOLITA TIPO MORDE- NITA
Una amplia variedad de estudios han mostrado que la mordenita es un material catalrtico versátil. Algunas de las propiedades que probablemente determinan la adsorción y difusidn en la actividad catalftica de esta zeolita son revisadas brevemente a continuación. Entre las reacciones que pueden efectuarse sobre mordenita se encuentran hidro- cracking selectivo de parafinas, hidrogenación, dismutacibn, isomerización, etc. ( 6 r 8 r 1 4 r 1 5 r 1 6 1 3 8 r 4 1 )
La importancia de estas reacciones radica en su apli-
Como ejemplo en el hidrocracking de desti- cación inmediata y potencial en la tecnologia de procesos petroqulmicos. lados de petróleo de alto punto de ebullición es posible producir diesel, combustibles, aceites lubricantes, etc.
ESTRUCTURA Y PROPIEDADES GENERALES
La mordenita tiene por celda unitaria la fórmula si- ( l r 2 I 5 r 7 r E r 5 1 r 1 6 ) guiente:
Na8 ( A l o s ) (Sios) 4 o 1 24H20

7
La estructura de la mordenita consiste de cadenas de tetrahedros de ( A 1 0 4 ) y (Si04) enlazados por l o s átomos de oxígeno compartidos por dichos tetraedros.
Cada tetraedro pertenece a uno o más anillos de cinco miembros en el esqueleto del aluminosilicato, ver Figs. 1 Y 2-
La alta estabilidad térmica de l a mordenita ha sido atribufda a l a presencia de un gran número de estos anillos de cinco miembros similares energéticamente.
La estructura porosa de la mordenita deshidratada tie- ne un sistema de poro tubular con canales de sorciQn ellp- ticos, paralelos.
Los diámetros mayor y menor de los poros son 7 y 5.8 O
A respectivamente. Cada celda unitaria contiene 2 canales de 7 . 5 2 A de longitud, los cuales están interconectados por pequeños canales laterales. Las aberturas de estas cavida- des tienen dimensiones entre 3.87 y 4.72 A. Se ha encontra- do que estas cavidades no están completamente abiertas des- de un canal a otro y previenen la difusibn cruzada, ver Fi- guras 1 y 2 y las Tablas 1 y 2 de propiedades estructurales.
O
O
La forma sintética conocida como mordenita de poro grande presenta una adsorción característica consistente con la estructura conocida (Tabla 2 ) .
La forma sintética tiene una relación molar SiO2/Al2O3 entre 4.5 y 5.
El tamaño de poro es suficientemente grande y permite -

8
la introduccibn de moléculas similares a las dimensiones de la molécula del benceno.
El diámetro efectivo de la mordenita -Na es conside- rablemente más pequeño que el de la mordenita -H.
La mordenita natural tiene un diámetro de poro efec- tivo más pequeño que la preparada sintéticamente. Esto puede ser el resultado de la presencia de impurezas que bloquean parcialmente el poro y ocasionan defectos en el material natural y también debido a baja relación de sS- lice-alúmina (Si/Al -5).
El esqueleto de la mordenita tiene una alta estabili- dad térmica y frente a los ácidos es mucho más resisten- te que la zeolita Y . Es posible extraer gran parte del aluminio estructural, cerca del 80% en peso de los tetrae- dro ( A 1 0 4 ) en mordenita -Na puede ser hidrolizado con bci- dos fuertes sin pérdida significativa en cristalinidad o contracción de los espacios interplanares (a-) .
$ La mordenita descationizada contiene sitios ácidos de Lewis y de Bronsted.
Todos los sitios Scidos en mordenita normal y desalu- minada pueden ser detectados completamente del espectro I .R. de zeolita -piridina adsorbida. A continuación anali- zaremos con más detalle las características ácidas de estos materiales.
-

9
TABLA I. Datos e s t r u c t u r a l e s d e l a z e o l i t a t i p o
mo rden i t a
Grupo e s t r u c t u r a l :
O t r a d e s i gnac i 6n :
6
P t i l o l i t a , a r d u i n i t a , f l o k i t a , d e e c k i t a
Compo s i c i 6 n Q u h i c a Fórmula t i p o ó x i d o : Na20*Al2O3*10SiO2*6H20
Contenido p o r c e l d a
u n i t a r i a :
Datos c r i s t a l o g r á f i -
cos
S i m e t r í a :
Grupo e s p a c i a l :
Constantes por c e l d a
u n i t a r i a :
Ortorrómbida Densidad: 2.13 g/cc
Cmcm
a = 18.13
b = 20.49
c = 7.52
Volumen p o r c e l d a u n i t a r i a
2794 A3
P rop i edades e s t r u c t u r a l e s : Cadenas ccwplejas de anrllos de 5
enlazados a t r a d s de aniilos de 4 Las cadenas consisten de anillos de 5
tetraedros de Si04 y un 410 tetra-
de AL04. (Figura ic) El sistema principal es paralelo a C; canales
restringido a 2.8 A paralelos ab. ( F i gura lb) O
Sistema de canales:
-~ . . . . . . . .

10
TABU 2 . Datos estructurales de l a zeolita tipo mrdenita
de poro grande
Grup estructural : 6
Otra designación: Zeolón
cbmposición química
Fórmila tipo óxido: Na20*A1203*9-10 Si02-6H20
Cbntenido por celda
unitaria -2430
Pmpiedades estructurales: Idénticas a l a mrdenita con canales grandes abiertos
Sistenia de canales: bidhsionales paralelos a b y c

I U
U Jnii 1
I
I
O
'L- b z 2 0 . 5 A- I
11
i
I I I
I b I
F IGURA 1

F I G U R A 2
12

13
ACIDEZ Y PROPIEDADES CATALITICAS
Los primeros trabajos sobre las propiedades catalfti- cas de las zeolitas fueron ampliados por investigaciones que detectaron la presencia de centros ácidos y grupos hidroxilo en estos catalizadores. Se encontró que la im- portancia de los cationes en l a fuerza de acidez es funda- mental. En 1963 Hirschler sugiri6 que la acción polarizan- te del campo del catión tiende a liberar un protdn (carác- ter ácido) de un grupo hidroxilo unido a un átomo adyacen- te de silicio o aluminio o a un protón de una molécula de agua adsorbida sobre el catión mismo. La magnitud del cam- po fuerte del catión determinará la acidez resultante.
Posteriormente Richardson consideró que el incremento del potencial iónico (e/r) del cati6n ocasiona un cambio de la distribucidn electrónica hacia la vecindad del catión vfa conducción en el modelo de bandas. El debilitamiento del enlace O-H sobre la superficie interna de l a zeolita incrementa su carácter ácido. La posibilidad de encontrar diversas fuerzas de enlace del sistema citado se manifies- ta en una amplia distribuci6n de fuerza ácida.
Es de notar que sólo un pequeño número de hidroxilos son suficientemente ácidos para ser activos en catálisis. Esta fracciÓn,depende como se mencionó anteriormente, de los potenciales iónicos del catión. Un segun& pardmetro que-puede incrementar l a fuerza ácida es la deshidroxila- cibn. Otra forma de modificar la fuerza ácida será por el movimiento de los cationes en la estructura,generando cam-
-

bios en la fuerza de campo electrostático de la superficie y consecuentemente en la fuerza de los sitios.protónicos.
Los diversos modelos presentados se clasifican como: estdticos o dinámicos. ca ha sido capaz de explicar un gran número de resultados de acidez se prefiere considerar que los sitios ácidos rea- les (sitios prot6nicos) están moviéndose contfnuamente en la estructura.
Aún cuando la aproximación estdti-
El tiempo de vida de un protón sobre un sitio fluctúa entre lo-* y segundos. Por lo que la población media de protones en cada sitio resulta de un equilibrio entre los factores de ocupación y los diversos átomos de oxíge- no. ácidas serán influenciadas por la vecindad química de los átomos de oxígeno, de la superficie interna y por la movi- lidad intrínseca del protón.
Estas consideraciones implican que las propiedades
Los sitios ácidos también pueden generarse de las si- guientes formas:
i) La descomposici6n térmica de zeolitas intercam- biadas con iones amonio producirá l a forma protónica.
ii) La ionización del agua sobre cationes polivalen- tes origina acrdez de Bronsted en estos sistemas.
iii) La reducción con hidrdgeno de cationes de meta- les de transicrón produce H - f zeolita.
iv) Los sit5os ácidos de Bronsted son también gene- rados por cationes divalentes soportados en zeolitas que
--__I . ~. ~ - .. - ~ ~, .*. . ...

15
reaccionan con compuestos haluro a temperatura ambiente y con C02 en el intervalo de temperatura de 150-400°C.
En la actualidad los trabajos publicados señalan la dificultad de encontrar una correlación directa y general entre acidez y actividad catalltica.
A continuación comentaremos algunos tópicos relacio- nados con las dificultades que se presentan al determinar la fuerza ácida.
1. Movilidad del prot6n
Las dificultades se originan de las propiedades del protón mismo y depende varios factores: pretratamiento de l a s zeolitas, especies adsorbidas, intervalo de temperatu- ra, etc.
El movimiento del protón considera dos fenómenos.
a) Los protones en cavidades inaccesibles pueden ser atraf dos.
b) La frecuencia del salto depende del reactivo titu- lante.
2. Nhero de sftgos activos
Nientras que las medidas de acidez suministran valores cercanos a lo2', 1O2l sitios por gramo, de acuerdo con el número de sitios ácidos potenciales, los estudios de la ac- t i v i d a d dan resultados cercanos a estos valores ~ 6 1 0 cuan- do se efectúa el envenenamiento con bases. -

3 . Adsorción competitiva sobre el cati6n y centros ácidos
Los resultados catallticos y de adsorción sugieren que hay una adsorción competitiva de un reactivo R sobre el catidn y los sitios ácidos de una zeolita.
Estos tres pardmetros dinámicos considerados se adi- cionan a los parsmetros estáticos utilizados para definir los centros ácidos (naturaleza, fuerza concentración, ti- po de OH).
Otros aspectos de la correlaci6n entre actividad y acidez consideran:
il Accesibilidad de sitios y cavidades
Los efectos estéricos debidos al tamaño del poro y las depósitos de alhina amorfa formados en los canales pueden modificar la actividad catalftica.
ii) Otros sitios catalfticos
La acidez de Lewis es frecuentemente considerada en procesos catalfticos. Puede incrementar la fuerza ácida protBnica vfa un efecto inductivo o actuar como un sitio activo.
iii) Propiedades catalfticas debidas a deficiencias de Aluminio.
Las propiedades catalfticas serdn mejor explicadas cuando l a localizacidn de los átomos de Aluminio sea pre-

17
cisa.
iv) Actividad catalftica de zeolitas con iones com- plejos de metales de transición.
Estos complejos presentan otras alternativas de reac- ciones que son más similares a la catálisis por conplejos metálicos en medios homogéneos que a la catálisis hetero- génea convencional.
Propiedades catailticas de las Zeolitas
Al analizar los diferentes trabajos efectuados sobre las propiedades catalfticas de las zeolitas es posible efectuar una clasificación de las mismas dentro de tres
2 4 r 3 3 )
grupos que se mencionan a continuación. ( 5 4 r 5 6 r 5 8 r 6 2 r 6 3 r 6 4 r
a) Catalizadores ácidos
b) Catalizadores monofuncionales debidos a la presen- cia de metales soportados en estos materiales.
c) Catalizadores bifuncionales
a) Catalizadores ácidos
- Es generalmente aceptado que un catalizador ácido es un catalizador capaz de producir iones carbonio sobre su superf icie. -

18
Las reacciones de iones carbonio presentan veloci- dades de reaccidn determinadas por la naturaleza del i6n carbonio formado y por el tipo y fuerza del sitio ácido donde se realiza la catálisis.
Desde hace tiempo se han presentado resultados en donde se correlacionan las propiedades ácidas con.las pro- piedades catalfticas de las zeolitas. Las más recientes han señalado que existe una relación entre el número de protones o fuerza ácida y las reacciones de iones carbo- nio.
b) Catálisis por catalizadores monofunclonales zeol€ticos con metales soportados
Bajo ciertas condiciones las zeolitas que contienen metales pueden actuar como catalizadores monofuncionales, y la zeolita puede considerarse como soporte inerte. Sin embargo, ya que el estado del componente metálico depende de la composición y estructura de,la zeolita cristalina, las propiedades de estos catalizadores dependen significa- tivamente del soporte usado.
c) Catalizadores bifuncionales
LOS catalizadores bifuncionales presentan ambas acti- vidades ácida y de hidrogenación-deshidrogenacih.
La actividad ácida se debe a la zeolita propiamente dicha y produce el ¿6n carbonio y la actividad de hidro-

genación-deshidrogenación al componente metálico, el cual es necesario para catalizar alguna de las reacciones re- queridas (e.g. hidrogenación de olefinas o hidrogenóli- cis).
INVESTIGACION CATALITICA BASICA EN LAS ZEOLITAS
Muchos progresos se han hecho por entender la activi- dad catalftica de zeolitas en diversos tipos de reacciones.
El número de reacciones catalizadas por estos mate- riales se ha ampliado y se han desarrollado nuevos catali- zadores multicomponentes y polifuncionales con propiedades especfficas.
Los estudios catallticos sobre zeolitas pueden clasi- ficarse de acuerdo a su orientación y desarrollo:
1. Establecimiento de la naturaleza de los sitios activos para varias reacciones. InvestigaciGn de simili- tudes y diferencias entre las zeolitas cristalinas y los aluminosilicatos amorfos,
2. Correlación entre estructura y composición de la zeolita y su actividad, estabilidad y selectividad, para reacciones modelo selectas.
3. Investigación de l a aplicabilidad de catalizado- res-zeollticos para reacciones nuevas y convencionales. Estudio de problemas tebrico prácticos de refinacion de petróleo y qufmica del petrbleo, de Cinética y Mecanismos de Reacciones modelo .

20
4 . Investigación de métodos de control de las propie- dades y de la composición de las zeolitas; de'la modifica- ción de estas propiedades, con el fin de obtener la más alta especificidad en su aplicación como catalizador para diversos procesos en el desarrollo de nuevos sistemas ca- talíticos polifuncionales multicomponentes.
5 . Estudio de las propiedades de tamiz molecular y su posible aplicaci6n al desarrollo de catalizadores alta- mente selectivos.
6. Estudio de las condiciones de pretratamiento y su efecto sobre la actividad, selectividad, estabilidad y habilidad para regenerar el catalizador zeolftico. ( l r 9 r l l I
1 2 r 3 0 r 3 2 r 3 5 )
APLICACION INDUSTRIAL
Las zeolitas son usadas en muchos procesos catalíti- cos industriales importantes principalmente cracking e
CRACKING
h i d r O C r a c k i n g . ( 2 6 I 3 5 r 3 4 ' 1 3 ' 3 3 f 4 ~ , 3 ~ )
El cracking catalftico utilizando zeolitas fue usado a partir de 1 9 6 2 y en los 10 años siguientes creca del 95% de la capacidad instalada que equivale aproxirnadamen- te a 5 millones de barriles por día emplean zeolitas en U . S . A .
A nivel mundial, a fines de 1975 alrededor de 50 mi- llones de barriles de crudo por dfa eran procesados en alrededor de 750 refinerías, de esta cantidad aproximada-

mente 7.5 millones de barriles por dfa puede ser catall- ticamente crackeado a capacidad de refinaciÓn.tota1.
Estas cantidades procesadas requieren la producción de aproximadamente 150,000 toneladas por año de zeolita para cracking catalltico cuyo valor es aproximado a 250 millones de dólares.
Las predicciones para 1990 estiman que alrededor de 155 millones de barriles de crudo por día serán procesa- dos,de ah€ la importancia de las zeolitas.
En general las zeolitas empleadas en el cracking ca- talítico son versiones sinteficas de la faujasita. Antes de 1964 la cantidad de faujasita sintética producida co- mercialmente era despreciable,sin embargo en la década de los 7 0 ' s la producción de faujasita sintética utilizada en cracking catalltico fluctuó alrededor de 286 millones de libras por año.
Para reacciones de hidrocracking el uso de zeolitas como catalizadores en U.S.A. en 1978 fue de 2 mi.llones de libras por año para procesar 900,000 barriles por dla con un costo del catalizador de aproximadamente 20 millo- nes de dólares.
HIDROCRACKING
L
- El hidrocracking es un proceso por medio del cual se realiza el cracking y la hidrogenación de hidrocarburos simultáneamente o secuencialrnente utilizando un cataliza-
-

22
dor bifuncional con un componente altamente ácido que fa- vorezca la reaccidn de cracking y un componente metálico activo en la hidrogenación, en el presente trabajo se uti- lizó zeolita tipo mordenita como el componente ácido y Pt como del componente metálico. ( 2 0 r 3 1 r 3 2 r 3 3 r 3 4 r 4 0 r 4 l r 4 2 r 3 8 )
El hidrocracking se emplea para degradar hidrocarbu- ros entre los que se encuentran aceites clclicos, produc- tos aromáticos superiores, naftas, hidrocarburos lineales y ramificados de cadena larga por lo que el objetivo pri- mordial del hidrocracking es convertir hidrocarburos de alto punto de ebullición a productos de bajo punto de ebu- llición por medio del rompimiento de los enlaces C-C y la hidrogenación de las olefinas producidas.. Los aromáticos policlclicos pueden hidrogenarse parcialmente antes de que se efectúe el cracking del núcleo aromático. Los átomos de nitrógeno y azufre, presentes como sulfuros simples y heterociclos complejos son convertidos en sulfuro de hi- drógeno y amonlaco.
Un papel adicional y probablemente el más importante de la hidrogenación es el de hidrogenar el coke rápidamen- te y prevenir su conversión a coke residual sobre el cata- lizador, estas caracterlsticas permiten incrementar la vi- da media del catalizador. Todas estas reacciones aumentan el consumo neto de hidrógeno.
Es interesante señalar que l a primera planta de hidro- cracking se contruyd en Luena Alemania en 1927, con el fin de hidrogenar comercialmente lignito y que elcatalizador empleado era sulfuro de tungsteno. ( 1 2 I 4 0 )
En 1937 la Esso desarrolló un catalizador de sulfuro -

2 3
de tungsteno sobre montmorilonita tratada con HF para producir gasolina de avión.
En el perlodo 1935-1940 se confirma la naturaleza bi- funcional del catalizador.
En 1939 Imperical Chemical Industries. Ltd. en Ingla- terra desarrolló un catalizador de hierro sobre HF:
Montmorilonita
En 1940-1944 la Esso utiliza un proceso en dos pasos usando en el primer paso sulfuro de tungsteno sobre HF-Montmorilonita, y en el segundo paso Níquel sobre HF- 14ontmorilonita para producir combustible de aviones.
Fue en la década de los 60's cuando se descubrió la potencialidad de l a aplicacibn de las zeolitas como cata- lizadores industriales de hidrocracking. El primer proce- so usado comercialmente fue desarrollado por la Union Oil Co. Of. California (1960) y la primera planta de hidrocrac- king que empleó catalizadors de tipo Zeolita fue, instalada por Los Angeles Refinery of Union Oil en 1964. En el año de 1973, se procesaban alrededor de 350,000 barriles por día en plantas que utilizaban estos catalizadores. La im- portancia industrial de las zeolitas-metálicas ha origina- do que se hayan efectuado estudios de la reacción de hi- drocracking de hidrocarburos individuales y de mezclas, encontrándose que esta reacción depende de la composición del catalizador, del tipo de metal y de su concentración, de la acidez de la zeolita y de los parametros de reac-
( 3 2 I 3 3 I 3 4 I 3 5 I 1 3 I 3 1 )

.-
24
CAPITULO I1
CARACTERIZACION DE CATALIZADORES
PREPARACION Y ACTIVACION DE CATALIZADORES
En general, la preparación de los catalizadores me- tal-zeolitas comprenden dos etapas:
a) El depósito del metal o compuesto metálico en o sobre los cristales de zeolita.
b) Los tratamientos que originan la dispersión fi- nal del metal.
Dado que los metales soportados en zeolitas se em- plean como catalizadores en diferentes reacciones, su pre- paración y activación dependerán del proceso en el que se- rán empleados.
Entre los métodos más usados en el depósito del me- tal en zeolitas se encuentran:
a) La impregnación bl El intercambio iónico cl La adsorción de complejos metálicos
En algunos casos dependiendo de la aplicacidn de los catalizadores, es necesario obtener selectivamente el me- tal sobre la superficie externa de la zeolita. Esto pue- de lograrse impregnando la zeolita con partlculas del me- tal o con algún compuesto de dicho metal.

2 5
El método de intercambio iónico consiste en la reac- ci6n de sustitución entre los cationes originales de las zeolitas con cationes simples o complejos en solución acuosa en las cuales el i6n metálico presente en la solu- ción sustituye al catiÓn localizado en el interior de las zeolitas.
La adsorci6n de complejos metálicos sin carga como en el caso de los carbonilos metálicos sobre zeolitas des- hidratadas constituye otro método útil, de introducir el metal en especial para aquellos elementos que no pueden ser intercambiados iónicamente con facilidad. ( 3 3 ' 4 3 r 4 6 r
4 8 1 5 2 1 4 9 r 3 6 1 3 7 )
El método utilizado para la preparación de los cata- lizadores en este trabajo fue el de intercambio iónico y se realizó de la manera que se describe a continuación:
Primero se preparó una solución acuosa de Pt(NH ) C1 3 4 2 de la concentración requerida con el fin de obtener un porcentaje determinado del metal intercambiado dentro de la zeolita mordenita.
La solución de Pt(NH3I4Cl2 es diluída hasta un volu- men de 100 ml de agua destilada y se colocan 2 gr de zeo- lita mordenita, enseguida se mantiene en agitación la mezcla durante 12 hrs. y posteriormente se filtra y se la- va con agua amoniacal para eliminar los iones ci- y pos- teriormente se ceca en la estufa durante 3 horas a una tem- peratura de llO°C y se guardan en el desecador las mues- tras.
Los catalizadores zeolitas metálicas obtenidos son

26
sometidos a tratamientos apropiados con el fin de lograr la migración y dispersión requeridos del metal.
Los cationes de metales nobles en zeolitas deshidra- tadas son fácilmente reducidos con hidrógeno, pero e s ne- cesario un estricto control de los tratamientos de reduc- ción y activación apropiados para obtener la más alta dis- persión del metal.
Rabo, et.al. ( 8 7 ) reportaron que puede obtenerse Pt
Kubo, et. al. ‘ 8 9 ) fueron los primeros en dispersado atómicamente por reducción de una zeolita 0.5% Pt/Ca Y. investigar en detalle la influencia de las condiciones de tratamiento sobre la dispersidn de Pt y encontraron que se forman partículas de 1 5 a 55 A de Pt al tratar térmicamen- te la zeolita Pt (0.24% en peso)-NaY en aire a 3OOOC se- guido de reducción a 5OOOC durante 3 horas.
O
La temperatura de calcinación dn aire previa a la reducción don hidrógeno se encontró que era el factor cla- ve que afecta la dispersión del Pt.
Se obtiene una mayor distribución del Pt dentro de una zeolita cuando se elimina totalmente el agua de l a
zeolita antes de efectuar la reducción. Una deshidratación parcial ocasiona una menor actividad. (49 r 5 0 r 5 2 )
Debido al fuerte enlace de los cationes con el agua que se encuentra fuertemente adsorbida en la zeolita antes de la reduccidn pueden formarse ciertos compuestos inesta- bles durante la reducción de los cationes metálicos y su presencia puede afectar la reducción de los cationes y por consiguiente la dispersión del metal formado. Por lo tan-
-
-

__L__
27
to la activación en flujo de O2 a 35OOC antes de la reduc- cián es una etapa esencial para obtener alta dispersión del Pt en la zeolita.
También debe considerarse un factor adicional:
En un estudio espectroscópico de IR se demostró que los cationes Pt(NH3)4 20OoC y que a partir de dicha temperatura se descomponen para dar platino enlazado idnicamente a la red liberándo- se moléculas de NH3 y H20.
2+ conservan su estructura hasta
Estas moléculas de agua o amonlaco retenidas en las cavidades internas de las zeolitas pueden favorecer una migración y aglomeración del metal.
Dalla, Betta y Boudart, ( 9 O ) señalaron la necesidad de especificar las condiciones de tratamiento para obtener catalizadores altamente dispersos.
Gallezot ( ' ' ) ha caracterizado la formación de partl- O
culas de Pt de 6 a 13 A en una zeolita 14% Pt-Na-NH4Y pre- tratada en O2 y reducida en H 2 a 300OC.
Es notable la formación después del pretratamiento y reducción a 3OOOC de átomos de Pt aislados en las cavida- des sodalita que no quimisorben hidrógeno.
Similarmente Naccache,et.al. ( 9 1 ) encontraron Pt 100% disperso en la Zeolita 3.8% Pt-NaY después de activar en O2 y reducir en atmósfera de H2 en 35OOC y cristalitos de 15 a 20 A después de activar en O2 a 500°C y reducir en
O
H2 a 400OC.

28
Wilson y Hall al tratar la zeolita en aire a 55OOC y reducirla con H a diversas temperaturas, eqcontraron que a 3OOOC el tamaño máximo de las partlculas de Pt fue de 3 a 5 A .
2
O
Un corto tratamiento con hidrógeno a 5OOOC fue sufi- ciente para favorecer el crecimiento de las partlculas de Pt las cuales presentaron un tamaño de 20-90 A encontrándo- se el Pt en la superficie externa.
O
( 4 8 , 4 9 , 5 2 )
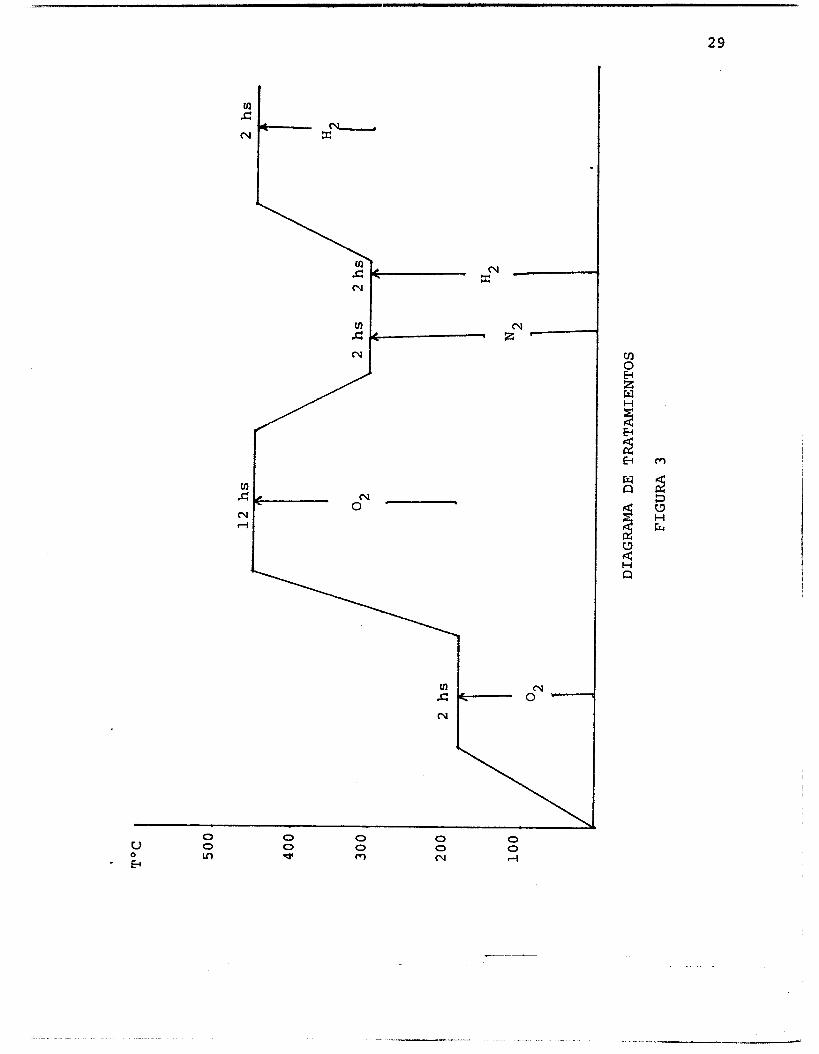
Las condiciones de tratamiento de los catalizadores utilizados en este trabajo fueron las siguientes: (Fig. 3)
2' y pos-
Esta etapa tiene como
a) Calcinación del catalizador en un flujo de O
Se realizó a la temperatura de 18OOC durante 2 hs. teriormente a 45OOC durante 1 2 hs. finalidad la descomposición del complejo de [Pt(NH3)4] 2+
b) Se somete el catalizador en un flujo de N2 a 3OOOC por 2 hs. nor del 2% en peso.
para extraer el agua residual que es me-
c) Reducción del catalizador en flujo de H2 primero durante 2 hs. a 3OOOC y posteriormente otras 2 hs. a 45OOC para obtener el Pt metálico.
El flujo de los gases en todos los casos fue de 1 mi/ seg a través de un reactor en forma de U de lecho fijo poroso en donde se deposita el catalizador (Fig. 4).
-
- El sistema se coloca dentro de un horno eléctrico programable con el fin de aumentar lentamente la temperatu- ra a una velocidad de calentamiento de 50°C/hora.
-
I - -
O O O O w m
O O O O N rl
m
k; 3 W H E

_I-
. ..
C CATALIZADOR 0 OR:F!C!O PARA
e T E R M P A R ' H HORNO
V PLACA DE VIDRiO PCROSO
f TERMOPaR
FIGURA 4
SISTEMA DE REDUCCION
. .
30

_ _ -
31
El catalizador as2 tratado estará listo para utilizar- se. En la activación de los catalizadores metal-zeolita usados para hidrocracking e hidrogenacidn deben ser deshi- dratados y reducidos a temperaturas bajas (300OC) o medias (45OOC). Los tratamientos a temperatura alta disminuyen la actividad.
DETERMINACION DE LA DISPERSION
La dispersión es una medida del área superficial es- pecffica del metal y se define:(4B'49r52'43'50,s7~s3)
"Como el número de átomos metálicos en la superfi- cie del catalizador dividido entre el número total de átomos metálicos presentes en el soporte".
En el caso de los catalizadores del tipo metal-zeolita la dispersión del metal depende del contenido del catión, del tratamiento previo a la reducción y de la temperatura de reducción.
Generalmente se utilizan técnicas de adsorción gaseo- sa para determinar el área superficial metálica.
Dentro de estas técnicas de adsorción, la quimisorcidn es de gran ayuda dado que debido a su naturaleza (implica interacciones adsorbato-adsorbente, similares a las asocia- das con la formación de un enlace qufmico) lo cual permite en un momento dado, presentar una naturaleza selectiva y
en consecuencia permite determinar el área superficial de las diferentes fases presentes en el catalizador (metal,
093050

32
soportes, etc.) . El uso de la quimisorcidn para medir áreas superfi-
ciales requiere un conocimiento de la estequiometrfa de quimisorción; en una monocapa cubierta es equivalente el promedio del número de átomos de la superficie del metal asociados con cada molécula de adsorbato.
Existe un nfhero de factores que en general influye en la naturaleza del comportamiento de la quimisorción del gas sobre metales. ( 5 7 , 5 4 , 5 5 , 5 8 )
Entre los más importantes se encuentran:
a) Presencia de impurezas en la superficie adsorbida. b) Heterogeneidad de la superficie cristalográfica. c) Reconstrucción de la superficie durante la adsor-
ción. d) ~ncorporación o solución del adsorbato. e) Efecto intrlnseco del tamaño de partícula. f) La presencia de un soporte.
Parte experimental
a) Gravimetrfa
En la caracterización de los catalizadores con el fin de determinar la dispersi6n se efectúan en primer lugar los estudios de gravimetrfa de las muestras.
El equipo utilizado en las determinaciones gravimé-
I -, .~ . . . .

33
tricas es un equipo convencional constitufdo por una elec- trobalanza CAHN-RH-UHV acoplada a un sistema de vacío cuyo rango de exactitud alcanza hasta lo-* Torr, la variación de peso es registrada eléctricamente. La determinación de isotermas es contínua y automática.
Descripción de la balanza ( 5 4 , 5 5 , 5 7 r 5 9 )
Esta balanza está basada en el principio de equilibrio conocido como "Balanza Nula" Figuras 5 y 6 en el cual la muestra se coloca en el lado A del fiel, y se equilibra del lado B con un material que no adsorba el gas de tratamiento como e1 vidrio. Esto da el fiel de la balanza una posición de equilibrio y cuando se efectúa el cambio de peso en la muestra se produce una deflexión momentánea, lo cual hace variar la posición de la bandera alternando el flujo de luz de la lámpara hacia la fotocelda, lo cual cambia la co- rriente que se estaba produciendo en ella y que es transmi- tida a un amplificador de dos pasos de aqul l a señal recibi- da se aumenta y se envía al solenoide que se encuentra en el campo magnético en el cual está soportado el fiel de la ba- lanza. Este sistema solenoide magneto actúa produciendo una fuerza (momento) proporcional a la corriente recibida según se conoce por la ley de Ampere, éste restaura al fiel a su posición original. De esta manera se tiene que cada cambio de peso en la muestra es proporcional a l cambio de fuerza electromagnética, lo que origina que el fiel se encuentre siempre en equilibrio dinámico con la suma de mo- mentos igual a cero. La velocidad a que se lleva a cabo este fenómeno es tan rápida que no se aprecia movimiento alguno.

34
La balanza se encuentra dentro de un recipiente de vidrio PYREX diseñada para trabajar en altos vaclos del orden de lo-' torr o en atmósferas corrosivas o de alta humedad. De los extremos del fiel cuelgan dos hilos de vidrio, en ellos se colocan las canastillas de vidrio don- de se ponderan la muestra y el contrapeso (Figura 5 ) .
La balanza se encuentra conectada a un monitor que sirve para ubicar los intervalos de cambio de peso y los controles de calibración de la balanza a la sensibilidad que se desea. Del monitor sale una extensión eléctrica que se conecta al registrador marca Omnigraphic tipo 3000 de dos canales que dispone de diferentes escalas en milivolts/pulgada para la apertura en el eje "Y". En el máximo intervalo de sensibilidad de la balanza y el gra- ficador disponemos de una escala de O a 1000 microgramos en toda la apertura del papel (Figura 6).
El sistema de calentamiento está formado por un hor- no, el cual tiene una resistencia en toda su extensibn de nicromel y está conectado a un programador de temperatura CAHN tipo 3600. La medida de la temperatura se hace con un termopar cromel-alumel que está conectado a un termbme- tro digital marca FLUKE.
Sistema de vaclo
El sistema de vacio está construído de vidrio PYREX de Acuerdo al diagrama de la Figura 7 y consta de:
1.- Una bomba mecánica de paletas marca WELCH tipo

35
-A VACIO - 4
I I - . 3
HüESTRA i
d
I
i
CCSTRAPESO
I
I
FIGURA 5
ESQUEMA DE LA BALANZA Y ADITAMENTO DE VIDRIO
1

36
.. O CI D a s O m r
U o O
b m r 3,
m r m
L
. . I

37 -
3
FIGURA 7
DIAGRAMA DEL SISTEPA DE VACIO ACOPLADO A LA BALANZA

3 8
Dow Seal 1400, con un desplazamiento de 25 lts./segundo y vaclo máximo de torr.
2. Una bomba de difusión de aceite marca BENDIX tipo VF-10 enfriada por agua, con vaclo máximo de torr.
3 . Trampas de nitrógeno llquido.
4 . Un medidor de vaclo tipo Mcleod, con intervalo de 0.125 x torr.
5. Válvulas para alto vacío selladas con grasa apie- zon tipo H.
La muestra de catalizador reducida es reactivada a 4OOOC durante una hora y media con un flujo de hidrógeno de 1 ml/seg y luego evacuado a y media también a 400OC.
torr durante una hora
Ya limpia la superficie de metal, se enfrla la mues- tra hasta 25OC y se procede a realizar la introducción de gases para determinar la dispersibn de acuerdo a la siguien- te secuencia.
1. Se introduce oxígeno bajo una presión de 100 torr adsorcibn total de oxlgeno (O;).
2. Se hace vacío hasta alcanzar el equilibrio, adsor- cidn irreversible de oxígeno (O2)
3 . Se introduce hidrógeno bajo una presión de 200
torr adsorción total de hidrógeno (H2')

39
4 . Se hace vacío hasta el equilibrio adsorcion irre- versible de hidregen0 (H2).
El tiempo óptimo para el equilibrio en cada una de estas etapas es de 15-20 mintuos. En la figura 8 se pre- senta una secuencia de las adsorciones ejecutadas.
Cdlculos
Ya conocida la masa de gas quimisorbido se procede a calcular la dispersibn de acuerdo a la forma siguiente:
Atomos superficiales totales = Me x N
M
Donde M es la masa molecular de H2 en gr
N es el ndmero de Avogadro
Me es la masa de gas quimisorbido en rig por gramo de catalizador
Atomos superficiales totales x 100 ATOMOS TOTALES % D =
Para conocer el tamaño del cristal se utiliza la f6r- mula siguiente:
en donde -

40 -
ADSORCIO: DE
O X X G U O
. .
.. .. *-
I
TIEMPO
FIGURA 8
DIAGRAMA DE LAS ADSORCIONES ESTANDAR.

41
-10 = 1016 x 10 KPt
Los resultados obtenidos se muestran en la Tabla 3.
b) Volumetrla
La segunda técnica de determinación de la dispersian utilizada fue la volumétrica.(54f 5 7 ’ 6 1 )
El diseño preciso del sistema usado en la determina- ción volumétrica del gas quimisorbido depende del tipo de sólido que se estudie y del método elegido’para limpiar o preparar la superficie de la muestra.
En nuestro trabajo se emple6 el sistema que se indi- ca en la Figura 9 y que consta de las siguientes partes: un instrumento para medir la presión gaseosa, una celda en donde se coloca la muestra, una sección para el almacena- miento de gases y un sistema para producir vaclo.
Todo el sistema está construfdo de vidrio PYREX.
El medidor de presión es un transductor marca Validyne
El procedimiento seguido es el siguiente:
La muestra del catalizador es pretratada a 4 O O O C du- rante 1 hora y media en f l u j o de H2 para asegurar l a eli- minaci6n de los óxidos superficiales del metal. Posterior- mente se trata bajo vacfo torr) durante 1 hora y me- dia a 4 0 O o C con el fin de eliminar el hidrógeno superfi-

4 2
cial. Y a continuación se realizan las medidas de quimi- sorcibn a 25OC usando H2 o CO a presiones de IO0 y 200 torr respectivamente.
Antes de relacionar la cantidad de CO o H2 gaseosa quimisorbida con el área superficial; debe conocerse la estequiometrla del enlace entre el adsorbato y el adsor- bente como se mencionó, en el caso de las determinaciones gravimétricas.
Cdlculos
El número de moles adsorbidas por la muestra se cal- cula haciendo un balance de masa entre el número total de moles de gas introducidas en el sistema y el número de mo- les gaseosas residuales en el sistema cuando se ha alcan- zado el equilibrio. Esta operación se realiza para dife- rentes presiones con el objeto de obtener una isoterma de adsorcibn.
Los volúmenes del gas adsorbido o reaccionado fueron calculados del cambio en la presión del gas adsorbido de- terminado con un medidor digital de presiones Validyne. El volumen de espacio muerto fue determinado con hidróge- no y la celda de adsorción puede ser evacuada en temperatu- ra alta.
El equilibrio de cada quimisorción de H2 o CO se al- canza en 10 6 15 minutos.

FIGURA 9 SISTEMA VOLUMETRIC0

__.
4 4
Las ecuaciones utilizadas para el cdlculo están basa- das en la ecuación de los gases ideales.
Para la primera inyección
Pfl (VC + vm, - 'iVc - NR1 - RT N i - - 0 RT '
Para la segunda inyección
- Na 2 - N2T - NR2
(VC + vm> - NR2 - 'f2 RT N2T = 2Ni - PflVc,
Para n inyecciones
- Nan - NnT - NRM
(VC + vm, - 'fn-1 'c ; NRn - 'fn RT NnT = "Ni -
en donde:
N = número de moles adsorbidas a j
= número de moles inyectadas Ni
= número de moles inyectadas totales jT
N

4 5
= número de moles residuales NRn
Pi = presión inicial
P = presión residual f j
Vc = volumen calibrado
Vm = volumen muerto
Para el sistema utilizado se conoce previamente:
-4 = 9.3 x 10 Lt
= 11.13 x ió3 Lt m
En las isotermas se grafican número de moles adsor- bidas contra presión y permiten conocer la cantidad de gas necesario para formar la monocapa adsorbida, con este dato es posible conocer el porciento de dispersión.
Moles adsorbidas No. Atomo-gr. de Pt %D = -
Los resultados de %D y tamaño de partfcula se encuen- tran en la Tabla 3.

46
O In
rn -r
.
rn -r rl -r
m
o'
5
z 4 l=l a
fi W m W
O co W
m CO
O N
O % e
O %3 e
k n ow
u a aND
rl rd 4J
2 ow
k O a m N -4 rl m 4J rd U
rl m
In rl
.
m In \o
O U -4 k ci \a, E -4
k F O a O 4J w) E rl a,
k O a rd a rd c -4 E k a, 4J a, a c \o -4 rn k a, a rn -4 a
II
2
W n
O u -4 k 4 m
rl O 3
O a O ci la, E rl a,
k O a rd a rd c
5
s a, ci a, r(3
c Q -4 rn k a, a rn -4 a
II
k n
O U -4 k 4J w) E -4 3 rd k tn O a O c, \a, E rl a,
k O a O a a c
3 a, 4 a, 5
id rl 1 u b( ci k rd a a, a O
I C : rd
ci
II
2
u e
O U -4 k 4J la, E 5 rl O 3 O a O 4J \a, E rl a,
k O a O a m c -4
a, c, P) a rd rl =I u bl ci k rd a a, a O IF:
m c,
II
E
E
k e
. "

47
RESULTADOS Y DISCUSION
La dispersión de los catalizadores 1 y 5% a Pt-H-mor- denita es reportada en la Tabla. De aquf se observa que para el catalizador 5% Pt-H-mordenita el tamaño de cristal determinado por gravimetrfa y volumétria de H2 es de 41.47 A y 47.50 A respectivamente, con una dispersión de 24.5% y 21.05% respectivamente. Como se ve tanto el tama- ño del cristal como el %D son muy semejantes. En cambio para el catalizador 1% Pt-H-mordenita no existe esta simi- litud siendo los valores muy divergentes. En el caso de gravimetrfa el tamaño del cristal es de 20.83 A y la dis- persión es de 48.77% y por volumetrla @ = 8.56 A y %D = 115. En este caso existe más confianza con los estu- dios volumétricos por la cantidad pequeña de metal sopor- tado.
O O
O
O
V
En las volumetrfas con CO para el catalizador 5% O
Pt-H-mordenita maño del cristal es un poco mayor que en el caso de qui- misorcidn de H2 y el %D disminuye en la misma proporción. Para el catalizador 1% Pt-H-mordenita cuando quimisorbe CO, aV = 15.31 y %D = 65.3, comparando con la quimisorcidn de H2, vemos que el tamaño del cristal es de casi el doble y el % disminuye aproximadamente la mitad.
QV = 60.97 A y %D = 16.4 como se ve el ta-

_ . ~
4 8
CAPITULO I11
DETERMINACION DE LA ACTIVIDAD CATALITICA EN LA HIDROGENACION DE BENCENO
Una reacción comGnmente seleccionada como prueba ca- talftica es la hidrogenación de benceno. ( l o ' s 5 6 r 6 3 t 3 3 t 3 2 )
Esta reacción es de una gran relevancia técnica y un modelo adecuado para estudiar la actividad catalÍtica del metal debido a las caracterfsticas siguientes:
a) Unicamente se detecta un sólo producto de reac- ción, el ciclohexano lo cual facilita el análisis croma- tográfico.
b) Su actividad catalltica es independiente o insen- sible a la estructura superficial, tamaño de cristal, arre- g l o geométrico de los átomos superficiales y capacidad de los átomos para adsorber la molécula y al modo de prepara- ción del catalizador.
c) La hidrogenación de benceno, es una reacción lenta que permite evitar las posibles limitaciones debidas a los fenómenos de transporte de masa.
d) Las zeolitas con metales soportados actúan como - catalizadores monofuncionales, siendo considerada la zeo- lit5 como soporte inerte del metal activo. el estado del componente metálico depende de la composición y estructura del cristal de zeolita.
Sin embargo,

4 9
En este trabajo se estudia la hidrogenacidn de bence- no con el propósito de calcular la actividad catalítica bajo condiciones determinadas de los catalizadores de Pt soportados en mordenita.
Parte experimental
Para la determinación de la actividad catalítica en la hidrogenacidn de benceno, se utiliza un sistema catall- tico cuyo diagrama se presenta en la Figura 10, el cual está compuesto principalmente por un saturador que contie- ne el benceno, un reactor de lecho fijo, un horno eléctri- co programable, un cromatógrafo de gases con el objeto de efectuar el análisis de los productos de reacción cua- litativa y cuantitativamente y un integrador.
Las caracterfsticas del cromatógrafo son:
cromatdgrafo de gases Perkin-Elmer modelo 4B con detector de ionización de flama. La columna es de acero inoxidable de un octavo de pulgada de diámetro y 2 metros de longitud empacada con carbowax 20M y chromosob W. La columna se mantiene a una temperatura de 7OoC y las presio- nes de los gases son: para nitrógeno 3OPsi; hidrógeno 3OPsi y aire 32Psi.
El integrador utilizado es marca Shimadsu modelo - GRIA.
-
a) .Procedimiento
- En el reactor s e coloca una masa dada del catalizador

50
' .
C. U
- 4 k U U
2 w - -
U c - l >

51
reducido y se reactiva elevando la temperatura hasta 45OOC bajo una corriente de hidrógeno durante una hQra y media, y enseguida se enfrla a temperatura ambiente; a la cual se inicia el paso del reactivo por el sistema.
El benceno en el caturador está a la temperatura de 14.OoC que corresponde a una presión de vapor del benceno de 56 torr. Con las condiciones estabilizadas se realiza la reacción de hidrogenacidn con el paso del benceno sobre el catalizador a diferentes temperaturas a intervalos re- gulares hasta 100°C realizada en flujo contlnuo.
Cálculos
Para el cálculo de la velocidad de reacción, tomando como base los moles de benceno transformados por segundo y por gramo del catalizador, se utilizó la siguiente ex- presión:
%C 100 x- x - x- x - 273 1000
T m P D
22400 760 v =
En donde: D = flujo de la corriente reaccionante en 3 cm /seg.
P = presión parcial del hidrocarburo en el saturador en mmHg
T = temperatura ambiente (298OK)-
m = masa del catalizador en gr
%C = porcentaje de conversión
0 9 3 0 5 0

52
También se hicieron cdlculos de la actividad por si- tio T.N., que es un pardmetro que expresa la relación de moléculas de benceno transformadas en la unidad de tiempo y por sitio activo de metal.
Para calcularlo se utilizó la expresión:
N V T.N. = No. de sitios activos
en donde: V = velocidad de reacción (moles/seg)
N = No. de Avogadro
Utilizando la ecuación de Arrhenius, en la que se re- laciona la constante de velocidad, K, con l a temperatura de reacción, puede calcularse la energra de activación,
Ea
k = Ae -Ea/RT
En donde: A = factor de frecuencia
= Energía de activación de la reacción Ea
Si se representa gráficamente Ink en función del in- verso de la temperatura absoluta, la pendiente de la recta nos da el valor de la energla de activación dividido entre R.

53
RESULTADOS Y DISCUSION
Las propiedades catallticas de los catalizadores 1% y 5% Pt/mordenita en la hidrogenación de Benceno se repor-
tan en las Tablas 4 y 5 I _ En la tabla se muestran los porcentajes de conversión, las velocidades y las actividades por sitio activo. Se observa claramente que para el catalizador 5% Pt/mordenita la actividad es mucho más grande alrededor de 200 veces mayor que para el catalizador 1% Pt/mordenita.
La diferencia en el valor de la actividad se puede atribuir a que en el catalizador 1% Pt/mordenita el Pt se encuentra en el interior de las cavidades de la mordenita y por consiguiente la reacción de hidrogenación de benceno únicamente se haya efectuado en el Pt fácilmente accesible o en el localizado en la superficie externa de la mordeni- ta.
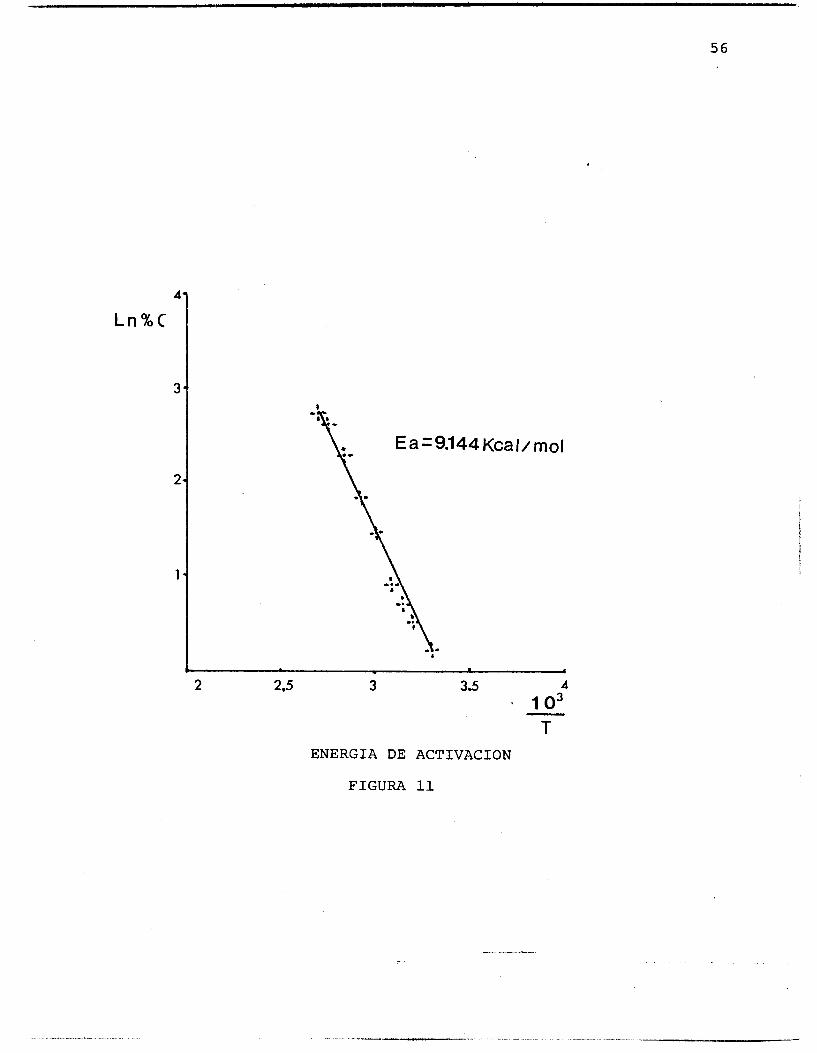
El valor de la energfa de activación fue determinada a partir de un gráfico de Ln C contra - 1 (Figura ll), h i -
T camente para el catalizador 5% Pt/mordenita resultando de 9.144 Kcal/mol°K valor que concuerda con los que apare- cen en la literatura. Debido a que la hidrogenación de benceno es una reacción altamente exotérmica, se trabajó a bajos grados de conversión, menores del 10% en el rango de 25-100°C para evitar los efectos de transferencia de calor. -

-
-r u)
a3 In
-
N W
W a2
-
a3 I-
-3 N N
- fi l- rl
I- rl
.
ln In I- N cn
O
m O rl
X
z E
W O rl
x 3
u dp
rl W
W m rl
cn m cn a3 rl
rl W
O rl
cn rl m I- In
O
In ln
I- N
rl cn O In
rl
m m m N
In In cn N
rl
rl d
Lo m
N W cn a3
rl
P W W rl
m
W fi I- W
*
P W I- m I-
-
N d ln I-
cn
O a3
I- rl In N
O rl
-
cn I- rl W
m rl
O m
cn I- m rl
N rl
-
a3 O rl rl
W d
O O rl -
a3 O W I-
O
N d m N
rl
Lo cn rl P
rl
cv m O N
*
O W
a3 W rl In
N
O In
I- a3 O N
W
O P
m O O
N
In
W N
U
E-r O O
m

U
E O
-
N 03
W
cn m N O
-3
4 * m In
O
-
N m
O m rl rl
rl 03 m P
W
rl O cn
O
-
In m
rl O
rl rl
03 m cn * W
N N W m
O
-
O * -
- N O
N rl
m m cn O
b
m rl w o\
O
O m
-
rl W
cv rl
cn m W -3
P
* O cn cn O
O cn
-
- m -3
rl rl
In W ' m b
*
-
00 W cn m O .
O O rl
-
56
4
Ln%C
3
2
1
Kca I/ mol
1
2 2.5 3 35 4 . 1 0 3 -
T ENERGIA DE ACTIVACION
FIGURA 11

57
CAPITULO IV
DETERMINACION DE LA ACTIVIDAD Y SELECTIVIDAD
EN EL HIDROCRACKING DE n-DECANO, DECALINA Y TETRALINA
Como se mencionó previamente el uso de las zeolitas como catalizadores en reacciones de hidrocracking de hi- drocarburos pesados para la producción de gasolinas ha motivado una serie de estudios sobre estas reacciones. Son de particular interes los efectuados sobre el hidro- cracking de n-decano, hexadecano, decalina. A continua- ción comentaremos algunos de los resultados más interesan- tes. Se ha encontrado que la reactividad'de las parafinas se incrementa con el aumento de la longitud de la cadena de los hidrocarburos.
(6 7 - 7 1 ) Weitkamp y Shulz estudiaron la reacción de hidro-
cracking de n-decano sobre un catalizador 0.5% Pd o Pt de- positados en zeolita CaY en el intervalo de temperatura de 300-500°C.
Su trabajo está orientado al estudio del mecanismo de esta reacción y al análisis e identificación de los inter- mediarios de la misma.
Estos autores proponen que:
El mecanismo de hidrocracking de parafinas se reali- - za de la siguiente manera: -

58
H+ + + 2 -H
+ ‘nH2n+2 4- ‘nH2n 4- ‘nH2n+i
Pt Zeolita
+ ‘nH2n+i + cracking
+ ‘mH2rn + ‘n-rnH2n-2m+l zeolita
Pt’f ‘ J - H2 Zeoiita .E J- -H+
‘mH2m+2 ‘n-m H2n-2ni
4 J -
n-m H2n-2m+2 C
El paso inicial en la reacción es la formación de una olefina a través de la deshidrogenacidn de la parafi- na. Esta olefina es entonces adsorbida sobre el sitio ácido del catalizador y convertida en un iÓn carbonio.
Este iÓn se reacomoda a una forma más estable la cual a su vez es craqueado para producir un i6n más pequeño y una olefina.
Las olefinas son hidrogenadas a parafinas sobre el componente de hidrogenación. La similitud entre el nG- mero de carbonos en la distribución de productos del crac- king catalftico y el hidrocracking de n-dodecano confirman estas suposiciones.
Burbidge, Keen y Eyles ,( “1 analizar las actividades de metales soportados en diferentes zeolitas encontraron que el Pd o Pt depositados en mordenita favorecen más el
_ Y _ . ~ - . - .... - .. . _.._-

59
hidrocracking de parafinas y ciclo parafinas que los ca- talizadores respectivos de tipo faujasita Y. . Encontrán- dose que los productos son esencialmente propano, isobu- tan0 e isopentano independientemente del tipo de reacti- vo y que las n-parafinas reaccionan más rápido que las correspondientes cicloparafinas./ Sin embargo Beecher, Voorhies y Eberly encontraron que en las mezclas binarias de hidrocarburos cíclicos y alcanos de cadena larga (Cl0), los clclicos reaccionan preferentemente debido a que es- tos hidrocarburos impiden completamente el acceso del n-decano a los sitios activos.
(7 2 )
Se ha encontrado que cuando reaccionan mezclas de n-parafinas superiores y otros hidrocarburos sobre Pt o Pd-H-mordenita las n-parafinas son hidrocraqueadas prefe- rentemente.
(6 6 1 Los trabajos de Rudham y Stockwell han proporcionado un mayor conocimiento del mecanismo de hidrocracking, los autores han encontrado que la selectividad entre cracking e isomerización es determinada por el tiempo de vida me- dia de los iones carbonio; la presencia de un mayor número de sitios ácidos ocasiona tiempos de vida media más cortos de los iones carbonio lo que se traduce en un rendimiento más alto de productos craqueados. Se ha encontrado también que la actividad catalrtica en la conversion de n-decano es función de la variacidn de la electronegatividad de la zeolita. Como lo comprueban los estudios con zeolitas Y a las cuales se han adicionado fluoruros. También Voor- hies y Kimberlik lstudiaron zeolitas de tipo faujasita impregnadas con 5% de Pd y tratados con HF encontrando ac-
tividad en la reacción de hidrocracking de n-decano a par- tir de 200OC.
7 4

60
( 7 5 ) Kruglikov, et,al., a l investigar las propiedades de
zeolitas desaluminizadas encontraron que al soportar Pt en estos matereales se incrementa considerablemente su actividad catalftica en la hidroizomerización y en el hi- drocracking, la relación entre estos dos reacciones es función de la proporción de Si02/A1203. A l introducir Pd se reduce la cantidad de isomerizaci6n incrementándose el hidrocracking.
Estudios simTlares fueron realizados por Takase,et. al. quienen prepararon catalizadores muy activos al pre- cipitar Pt o Pd en zeolitas de tipo Na-Y intercambiadas con Ca , Mg
(3 L l
2+ 2+ o NH4+ y activadas por tratamiento con HCC1F2, C12CF2 o CC14.
( 7 7 , 7 9 1 8 0 )
Steijns, et.al. estudiaron el hidrocracking del n-de- cano sobre una zeolita estabilizada que contenia 5% en peso de Pt encontránd-se que la reacci6n es un ejemplo típico de catálisis binfuncional. La distribución de productos es caracterlstica de un mecanismo de iones car- bonio con cracking primario puro.
Con el fin de comparar el efecto de tamiz molecular de las zeolitas, en este trabajo se efectuó un estudio comparativo de la actividad y selectividad de los catali- zadores Pt/H-mordenita con diferentes contenidos de pla- tino en las reacciones de hidrocracking de n-decano, de- calina y tetralina.
Parte experimental -
Para la determinación de la actividad en la reacci6n -

61
e
de hidrocracking de n-decano, decalina y tetralina, se utiliza el sistenq descrito previamente al estudiar la activfdad catallt2ca en la reacción de hidrogenacidn de benceno.
El análisis de los productos se realizó por croma- tografla de gases utilizando un cromatógrafo Perkin Elmer modelo 3920B con detector de ionización de flama, el tipo de columna es capilar empacada con escualeno y con una longitud de 50 metros, la temperatura de operación de la columna es de 100°C.
a) Procedimiento
En el reactor se coloca una masa dada de catalizador reducido y se eleva la temperatura hasta 450°C, la mues- tra se mantiene bajo una corriente de hidr6geno durante una hora y media, posteriormente se enfría a 25OOC y se inicia el paso de los reactivos sobre el catalizador. La reacción se efecttia a diferentes temperaturas hasta 450OC. El estudio de la actividad se efectuó en flujo contlnuo.
b). Cálculos
Para calcular la velocidad de reacción tomando como base los moles de n-decano, decalina o tetralina transfor- mados por segundo y por gramo de catalizador respectiva- mente, se utiliza la expresión conocida:

62
273 1000 %C = 22400 760 T m x * 100
F x D
El cdlculo de la actividad por sitio T.N. se efectuó empleando la misma expreskón que en la hidrogenación de benceno, considerando al número de átomos de platino como el número de sitios activos.
rn
N V T.N. = No. de s2tios activos
Para calcular la selectividad se utilizó l a trans- formacidn a moles de los productos lo que'se realiz6 basa- do en el hecho de que la selectividad de un determinado producto es la cantidad del mismo obtenida en relación al total de reactante transformado por medio de la siguiente ecuación :
h fi Ni j
Considerando que aij = -
Por lo tanto Ntj = k'a f ij i
Entonces :

63
En donde:
pi: j
ij a
fi
n
i
j
m
Ni j
Resultados y
Porcentaje en moles de cada producto de la mezcla
Area del pico correspondiente del producto 'i j
peso molecular del reactivo
el número de carbonos por molécula de reac- tivo
el número de carbonos por molécula de produc- to
orden de elucidn de productos
número total de constituyentes de la mezcla
nhero de moles de productos de la mezcla
discusi6n
Los resultados de las reacciones de.hidrocracking de n-decano, decalina y tetralina efectuados sobre los cata- lizadores al 1% y 5% Pt/mordenita se presentan en las Figuras 12, 13 y 14. De los resultados de la Figura 12 pa- ra la reacción de hidrocracking de n-decano se observa pa- ra el catalizador al l% en Pt/mordenita un incremento gra- dual en la velocidad especffica conforme al incremento de temperatura encontrándose que a partir de 350°C se alcanza el máximo de velocidad, el cual se mantiene constante has- ta 450°C.
En el caso del catalizador al 5% se observa un corn-

-
64
portamiento similar en el incrementQ de la velocidad de reacci6n y nuevamente un valor m$ximo de velocidad a partir de 3OOOC el cual se mantiene constante hasta 450OC. Las velocidades especlficas son superiores para el catali- zador al 1%. Esto se aprecia claramente en la Figura 12.
Para la reaccidn de hidrocracking de decalina, los resultados representados en la Figura 13 indican un incre- mento gradual de velocidad hasta 3OOOC y a partir de esta temperatura la velocidad se mantiene constante, este com- portamiento lo presentan ambos catalizadores (1% en Pt y 5% Pt/mordenita).
En el caso de la reacción de hidrocracking de tetra- lina los resultados se presentan en la Figura 14 y se no- ta que para el catalizador al 1% de Pt/nordenita la velo- cidad especlfica se mantiene prácticamente constante en el intervalo de temperatura de 250-350OC; a partir de 35OOC se presenta un incremento gradual en la velocidad en funcidn del incremento de temperatura. Para el cata- lizador al 5 % se presenta un incremento en la velocidad alcanzándose el máximo en la velocidad a 375OC.
Respecto a la selectividad en las reacciones de hi- drocracking con:
a) n-decano, los resultados de las Tablas 6 y 7 mues- tran para los dos catalizadores, una alta selectividad a pentano en el rango de temperatura de 250-450°C siendo la conversián prácticamente completa para el catalizador 5 %
Pt/mordenita como se observa en la Figura 15 y en el caso del catalizador 1% Pt/mordenita tenemos aparte del penta- no cantidades mlninas de 2-metil pentano y n-hexano prin-

65
cTpqlmente, te,comienza en 4Q% a 25OOC y alcanza el 100%.en 450°C. Estos resultados están de acuerdo con lo reportado en la literatura.
La conyers-i6n se increment6 progresivamen-
En la Figura 16 se representa una grafica de la dis- tribución de productos obtenidos con la reacci6n a 350OC.
b) Decalina, en las Tablas 8 y 9 observamos que persiste l a alta selectividad a pentano en el catalizador 5 % Pt/mordenita siendo la conversión casi del 100%. Sin embargo para el catalizador 1% Pt/mordenita a l a selecti- vidad a n-pentano se añade la alta selectividad a 2-metil pentano a bajas temperaturas, la cual disminuye conforme aumenta la temperatura, además se tiene la presencia en porcentajes bajos de n-hexano y cantidades mfnimas de is6- meros de l a decalina. A la temperatura de 275°C se detec- ta también 2,3-dimetil pentano, heptano y nonano.
La presencia de estos productos se explica dado que a esta temperatura se favorece el equilibrio hacia la reac- cidn de isomerizaci6n.
El porcentaje de conversión es de 98% como se ve en la Figura 17.
c) Tetralina, las Tablas 10 y 11 nos muestran que para los dos catalizadores la selectividad a n-pentano es completa, siendo en los dos casos el único producto,alcan- zando un valor un poco mayor al 9 6 % de conversidn de acuer- do a la Figura 18.
- -

-~
6 6
TN x 100
50
C
A c t i v i d a d en l a r e a c c i ó n d e H id r o c r a ck ing con n-decano
con l os c a t a l i z a d o r e s
\ = ;-5 % P t /No rden it a
0 1% Pt/Mordenita
3 .o
250 350 TOC
450
FIGURA 12

A c t i v i d a d en l a r e a c c i ó n d e H id r o c r a ck ing con d e c a l i n a
con
TN x l o 3 50
25
l o s c a t a l i z a d o r e s
t -;-5% Pt/mordenita
01% Pt/mordenita
I
450 TOC 250 350
FIGURA 1 3

68
A c t i v i d a d en l a r e a c c i d n d e h i d r o c r a c k i n g con t e t r a l i n a
con los c a t a l i z a d o r e s
-:-5% Pt/mordenita
01% Pt/mordenita
O 1 250 350 450
TOC
FIGURA 14

-~
69
Conversibn de n-decano con los catalizadores
-!-5 % Pt/mordenita
01% Pt/mordenita
I
o 250 350 4 5 0 TOC
FIGURA 15
%C 101
50
. . . .... * -

I
7 0
Distribución de productos can e l catalizador
5 % Pt/mordenita
% en Iuloles
5 10 No. de carbonos
FIGURA 16

Conversión de decalina con los catalizadores
-';- 5% Pt/mordenita
01% Pt/mordenita
50
O
f
250 353 450 TOC
FIGURA 17
4

72
Canversión de tetralina con los catalizadores
%c 100
50
O
250
-!-5% Pt/mordenita
01% Pt/mordenita
i
3 5 0 TOC
450
FIGURA 18

-
73
O O d
u O In -r
u O In N -r
u O O .=r
u In P m
u O In m
u In N m
u O O m
u In b N
u O In (v
m k 7 c, rd k U ) a l 0 p i + , o m a
O k pi
$ 3
I
I
a0 -r m QI
d P
m m
W m m m
O O -I
O O rl
O O d
-r O m m
O r: rd c, c al PC
I
N In
O
m N
O
d rl
O
I
I
I
I
O c rd c, C a, a rl -d c,
8 N

'i
74
U O m -3
cn m rl . (v
N
I- cn
o? rl
rl . I
I
I
I
I
I
I
m N
O .
I
r-4 -4 o 4Jc ala E4J 4 c aai
pi m N
b
1
I
I
I
I
rl rl
rl
cn v
(v
I
I
I
O c a 4J a a, X
I
-
I
U O O -3
-3 I-
W cn
u m I- m
cn Ln
I- o?
u O m m
I- m
I- o?
.
U In N m
-3 m
m cn
U O O m
I- co I- cn
O
rl
-3 a3
d
m .s rl
I
m rl
rl
W N
rl I
rl m O
rl O
rl I
-
I
m In O . -3
\c)
rl
m rl
N I I
I
W In
o?
O c a u ai
I .rl
a
O rl
O
-3 Ln
O
I- w O
I
O s4
a X ai X

75
u O O *
U m P m
u O m m
N 00
Q) m .
m cn m
m * 03 m
U in N m
O m m m .
I I I I I I
cn rl
rl
m -3
rl
rl m rl
I I
O r-i
O I I 1 I I
P m P (u t N m
I
-
I
I I
m d
m
- I
i! lit
I
m N
m
i
l! 8
I
m P
O .
m rl
O
X D
I
P 03
m m I I
% rl B % I
.rl
m
Id k 1 4J I d v l k O 0 4 J a u
7 E O
k 0.4
E a . N
. .. . -. ~ ........ - . -

76
I I
I I
I I
I I
I I
I I
- r P P N
d r l .
I I
1g 8 8
dP d
I
O 9
O
I
rD
O
W P
rl . P c-l m
Tr P
d
N N
9 4
.
.2 Y a,
d
8 d *rl
. m
O Qi
N w
In N
rl m
I
W rl
m Lo
03 m P m
cv m m
03 N
Tr m
1
d -d z I N
u In N Tr
\o O
N I
rl w W m N m rl
P
Y I m
\D VI I . m
u) P
I . m
rl d I . O
I I
I I
% % i i k d
i B 8 I m
~~
u O O w
u Lo cv m
u m P cv
u O In cv
9 N
Tr m
O 03
4 m
m m 10 c\ - m 9
N Tr
O m In m
I
I
9 m W m
I
I
I
I
I
I
co
O ?
I
. N
I
I
I
I
(u
O ?
N P
O
QJ m 3
-r
m N
.2 ! d
Y N
-4

77

1
78
id c .rl rl id k +J 0 +J
al a 0. c .rl x u (d k u O k
IC
rl al a a Id a -d 3 *rl 4J u al 4 al m
2
. rl rl
4 Gl m -4 E i
id 4J 'rl c al a k O R \ +J pi
8 rl
k O a id N .ri rl Id ci rd u rl al
c O u O a -4 c al 4J .Q O rn al r-l O E c 0
al .n (d 4J c al o k O PC
-

4
79
En l a r e a c c i ó n d e h i d r o c r a c k i n g para ambos c a t a l i z a d o -
res se encuentra una menor a c t i v i d a d en el caso d e los ca-
t a l i z a d o r e s a l I% de P t l o c u a l se a t r i b u y e que dado que en este c a t a l i z a d o r se encuentra una mayor c a n t i d a d d e me ta l
d e n t r o d e l i n t e r i o r d e l a z e o l i t a . A l e f e c t u a r l a compa-
r a c i ó n de a c t i v i d a d e s e n t r e las tres r e a c c i o n e s se encuen-
t r a una mayor a c t i v i d a d en l a r e a c c i ó n de n-decano y a me- d i d a que se comparan los r e s u l t a d o s con l a s a c t i v i d a d e s
d e d e c a l i n a y t e t r a l i n a se encuentra una n o t a b l e disminu-
c i ó n a l pasar a e s t a s d i f e r e n t e s r e a c c i o n e s comprobándose
l a impo r t anc i a d e l a p r op i edad de tamiz mo l e cu l a r d e l a
z e o l i t a morden i ta que p e r m i t e e l paso d e l a s mo lécu las li- n e a l e s d e l n-decano. En e l caso de l a s mo l é cu l a s d e d e c a l i -
na y t e t r a l i n a e l a c c e s o y l a m o v i l i d a d d e e s t a s mo lécu las e s t a r á r e s t r i n g i d a .
Con r e f e r e n c i a a los r e s u l t a d o s d e s e l e c t i v i d a d encon-
tramos que en e l caso d e l a r e a c c i ó n d e h i d r o c r a c k i n g d e
n-decano l a r e a c c i ó n e s t á o r i e n t a d a a l a producc ión d e pen-
tan0 p r i n c i p a l m e n t e .
E l rompimiento de l a mo lécu la o c u r r e en e l C5-C6 pre-
f e r e n c i a l m e n t e . En e l c a s o d e l c a t a l i z a d o r a l 1% se presen-
t a una pequeña c a n t i d a d de hexano,s in embargo no se d e t e c -
t a r o n h i d r o ca rbu ro s l i g e r o s , se propone que los compuestos
d e C4 formados p r e s en t a r on un rompimiento s u c e s i v o has t a l a
f o rmac ión d e carbbn, l o c u a l se comprueba dado que estos c a t a l i z a d o r e s presentan una gran d e s a c t i v a c i e n .
D e l a n á l i s i s d e l a s e l e c t i v i d a d de l a r e a c c i ó n d e de-
c a l i n a para ambos c a t a l i z a d o r e s p r e f e r en t emen t e se forma
pentano e i s open tano en e l i n t e r v a l o d e temperatura de 275
a 459OC. No se p r e s e n t a c r a c k i n g secundar io . A temperatu-
r a s b a j a s (menores a 275OC) l a r e a c c i b n e s t á o r i e n t a d a a

80
l a formaci6n d e isómeros de la d e c a l i n a .
En l a r e a c c i ó n d e t e t r a l i n a , l a r e a c c i ó n produce úni-
camente pentano s i n l a f o rmac ión de compuestos c lc l i cos .
CONCLUSIONES
1. S e prepararon los c a t a l i z a d o r e s a l 1 y 5% de P t
en z e o l i t a morden i ta , l o s c u a l e s fue ron a c t i v o s en l a s
r e a c c i o n e s de h i d r o g enac i ón d e benceno e h i d r o c r a c k i n g d e
n-decano, d e c a l i n a y t e t r a l i n a .
2. Se comprobó e l efecto de tamiz mo l e cu l a r d e l a
z e o l i t a morden i ta a n t e l os tres h i d r o ca rbu ro s mencionados.
3. La s e l e c t i v i d a d de los productos en l a s tres
r e a c c i o n e s e s t á o r i e n t a d a p r e f e r en t emen t e a pentano e i so- pentano.

8 1
1. B o l t o n , A.P., en "Exper imenta l Methods i n C a t a l y t i c
Research" (R.B. Anderson y P.T. Dawson ea.) Vol. 11,
p. 1, Academic ?ress 1976.
2. Breck , D.W. en "Zeolite Mo l e cu l a r S i e v e s " W i l e y Inters- cience N.Y. 1974.
3. R i c k e r t L., Adv. Ca t a l . , - 21, 281 ( 1 9 7 8 ) .
4. Minachev Kh. M., K in e t . Ca t a l . , 11, 342 ( 1 9 7 0 ) . -
5 . a r e ck , D.W., Adv. Chem. Ser . 1, 1 ( 1 9 7 1 ) .
6 . Burb idge , B.W., Keen, I.M. y E y l e s , M.K. Adv. Chem. Ser . , 71, 400 (1971).
7. Meier, W.M. y Olson,D.H., Adv. Chem. Ser . , 1 4 , 155 (1971).
8. Snerman, J.D. y Benne t t , J.M. Adv. Chem. Ser. 4, 52 ( 1 9 7 3 ) .
9. Minachev, Kh. M., e I s akov , Ya . I . , Adv Chem. Ser . 40 ,
451 (1973).
10. Eberly, P.E. e n "Zeolite Chemistry arid C a t a l y s i s (J.A. Rabo
ed.). Monograph 1 7 1 , 7, 392 A. Ch. S. Washington, D.F. 1976. - 12. P i n e s , H., "The Chemis t ry o f C a t a l y t i c Hydrocarbon Conver-
s i o n s Academic P r e s s 1981.

82
13.
14.
15 .
16.
17.
18.
19.
2 0 .
2 1 .
2 2 .
23.
2 4 .
Csycse ry , S.N. e n "Zeolite Chemistry and C a t a l y s i s
(-j.A. Rabo ed. ) ,Nonograph 171, 1 2 , 580, A. Ch. S. Washington D.C. 1976 .
Thakur, D.K. y Wller, S.W. Adv. Chem. Ser . , 5 4 , 596
(1973).
Smith, J.V. en "Zeolite Chemistry and C a t a l y s i s (J .A .
Rabo) Monograph 171, 1, 1, A. Ch. S. Washington, D.C.
1976.
Kuhn G.H., Inter. Conf. o n P lo l ecu lar S i e v e s 4 t h Mole-
c u l a r S i e v e s Ii A. Ch. s., Symp. Ser . 9, 96 (1977).
B enes i , H.A. y Winqu is t , B.H. C., Adv. Cata. 2 7 , 97 - (19)
Barthomeuf D . , Inter. Conf. on M o l e c u l a r S i e v e s 4&1Mo-
l e c u l a r S i e v e s I1 A. Ch. S., Symp. S e r . 38, 4 5 3 (1977).
Barthomeuf D. en " C a t a l y s i s by Zeolites" ( I rne l i k e t . a l . ,
e d . ) p. 5 5 E l s e v i e r , N.Y. 1980.
Whyte T.E. y D a l l a B e t t a , R.A. C a t a l . Rev. S c i . Eng. , 2 4 ( 4 ) 567 (1982).
Weisz P.B., Ann. Rev. Phys. Chem., 2 1 , 175 (1970). -
Breck, D . W . , J. Chem. Educ. 4 1 , 678 (1964). - 7
- Weisz P.B., Chem. Tech. 498 (1973).
Penchev V et. al., Adv. Chem. Ser . 4 1 , 461 (1973).
-

83
25. Minachev Kh. & I . , e t . a l . , Adv. Chem. Ser . 76, 441 (1971).
26. Arey Jr. W.F., Adv. Cnem. S e r . 77, 451 (1971).
, 27. Turkev ich , J., C a t a l . Rev. 1, 1 (1957). -
28. Venuto, P.B., Chem. Tech., 1, 215 (1971). -
29. B a r r e r , R.M., Chem. Ind. 1203 (1968).
30. Turkev i ch , J. y Onno, Y., Adv. Ca ta l . , 2, 135 (1969).
31. mho, 3.k. e n "Zeolite Chemistry and C a t a l y s i s (J.A.
Rabo ea.) Monograph 171,5, 332, A. Chi S. Washington
D.C. 1976.
32. Poustma M.L. en "Zeolite Chemistry and C a t a l y s i s (J.A.
Rabo ed.) Monograph 171, 8 , 437 A. Ch. S. Washington
D.C. 1976.
33. Minachev, Kh. M. e I s a k o v Ya.I . , e n "Zeolite Chemistry
and C a t a l y s i s (J.A. Rabo ea.) Monograph 171, 1 0 , 552,
A. Ch. S. Washington D.C. 1976.
34. B o l t o n , A.P. e n "Zeolite Chemis t ry and C a t a l y s i s " (J. A. Rabo) P-lonograph 171,13, 714 A. Ch. S. Washington
D.C. 1976.
35. Magee, J.S. y B l a z e k J.J. e n "Zeolite Chemis t ry and
C a t a l y s i s " (J.A. Rabo) Monograph 171,11, 615 A. Ch.
S. Washington D.C. 1976.
- -
. 36. Junsford J.A., Inter. Cof . on M o l e c u l a r S i e v e s , 4 t h

84
M o l e c u l a r S i e v e s I1 A. Ch. S. Symp. Ser. , 39, 473 (1977).
37. Magee J . S . , Inter. Conf . on M o l e c u l a r S i e v e s , 4 tn , Mo-
l e c u l a r S i e v e s I1 A. Ch.. S. Symp. S e r 54, 650 (1977).
38. Karge , X.G., I n t e r . Conf. on M o l e c u l a r S i e v e s , 4 th ,
M o l e c u l a r S i e v e s I1 A. Ch. S. Symp. S e r 48, (1977).
39. Guisne t , M. , et. al. , en " C a t a l y s i s by Zeolites'' ( h e -
l i k et. a l , , ed.) p. 77 E l s e v i e r N.Y. 1980.
40. M a r c i l l y C., y Franck J.P. e n " C a t a l y s i s by Zeol ites
( Ime l i k et , a l . , ed. ) p. 93 E l s e v i e r N.Y. 1980 .
41. Karge , M.G., y Ladebeck, J. en " C a t a l y s i s by Zeolites" ( Imel ik et. a l . , ea.) p. 151 E l s e v i e r N.Y. 1980.
4 2 . E c h e v s k i i , G.V. e I o n e K.G. e n " C a t a l y s i s by Zeolites"
( Ime l i k e t . a l . ed.) p. 273 E l s e v i e r N.Y. 1980.
43. Moss, R.L., Exper imenta l Methods i n C a t a l y t i c Research
(Anderson y Dawson ed.) Vol . 11, p. 43, Academic P r e s s
1976.
44. Turkev i ch J. y Onno, Y., Adv. Chem. Ser . 63, 315 (1971).
45. Beaumont, R., Barthorneuf D. y Trambouze, Y., Adv. Chem.
S e r . 64, 327 (1971).
46. Sherry , H. S., Adv. Chem. Ser . 28, 350 (1973).
47. Cremers, A., I n t . Conf. on M o l e c u l a r S i e v e s 4 th , 14olecu-
l a r S i e v e s I1 A. Ch. S. Symp. Ser . 1 6 , 179 (1977).

> , --
85
48. G a l l e z o t P., C a t a l . Rev. S c i . Eng. 20 (11, 121 (1979). -
49. G a l l e z o t P., en " C a t a l y s i s by Zeolites" ( I m e l i k e t . a l . ,
ed . ) p. 227 E l s e v i e r , N.Y. 1980.
50. R i b e i r o F., e t . a l . , e n " C a t a l y s i s by Zeolites" ( I rne l ik
et. a l . , ed. ) p. 319 E l s e v i e r N.Y. 1980.
51. B a r r e r R.M. y K l i n o w s k i , J., J. C a t a l . 39, 690 1975. _.
52. G a l l e z o t , P. AlarcÓn, A. e t . a l . , J. C a t a l . 39, 334 (1975). - 53. Innes W.B. e n "Experimenta. Methods i n C a t a l y t i c Research
(Anderson y Dawson ed. ) . Vol. 1, p. 44, Academic P r e s s 1976.
54. Thomas, J.M. y Thomas, W. J. , " I n t r o d u c t i o n t o tne P r i n c i -
ples o f Heterogeneous C a t a l y s i s " Academic P r e s s 1967.
55. Cadenhead D.A. y Wagner N.J., en "Exper imenta l Methods
i n C a t a l y t i c Research (Anderson y Dawson ea.) Vol. 11, p.
223 Academic P r e s s 1976.
56. Thompson, S. J. y Webb, G. "Heterogeneous C a t a l y s i s " , O l i -
ver and Boyd, Edinburgh and Jordan 1968.
57. Anderson J.R., "S t ruc ture of M e t a l l i c C a t a l y s t s " , Acade-
m i c P r e s s 1975.
58. Rober tson , A. J .B., " C a t a l y s i s i n Chemistry" Methuen
Educa t i ona l L td . 1972.
59. Cordoba, G., Tesis de L i c e n c i a t u r a , UAP 1983.

86
60. Portillo, R., Tesis de Maestrfa, UAMI, 1984.
61. Plerchant, I., Tesis de Licenciatura, UAP,1983.
62. Boudart, M., Adv. Catal. 20, 153 (1969). -
63. Blanco J. y Linarte, R. Catalisis, funaamentos y aplica- ciones industriales, ed. Trillas, Pléxíco, 1976.
64. Germain J.E. "Conversión Catalítica de Hidrocarburos, ed. Trillas, México, 1980.
65.,Jacobs, P.A. Catal. Rev. Sci. Eng. 24 (3) 415 (1982).
66. Rudham R., y Stockwell, A. "Catalysis by Zeolites", Vo l .
5 , p. 113 Elsevier Anst. 1980.
67. Weitkamp J. y Schulz, H.F. Ind. Eng. Chem. Prod. Res. Develop. - 11, (l), 46-53 (Eng.) 1972.
68. Weitkamp J. y Schulz, H.F. Amer. Chem. Soc. Div. Petrol. Chem. Prepr. 16 (l), A102-Al15 (1971).
69. Weitkamp J. y Hedeen, K. Chem. Ing. Tech. 47 (121, 537 - (1975) .
70. Weitkamp J. y Schulz, H.F. Compend. 74/75 Volv. Hamsttag. Dtsch. Ges. Mineralodwiss Kohlenchem., 24th 1974 (Pub. 1975) 1, 355-66. -
71. Weitkamp J. y Schulz, H.F. Erdoel Kohle, Erdgas, Petro- chemp. Brennet.-Chem. I 28 (l), 37, 1975.

4
87
72. Beecker , R. V o o r h i e s A. y E b e r l y , P. Ind. Eng. Chem.
Prod. Res. Deve lop. 7, 203 (1968). - 73. Pniak B. y Radomyski B., React . K ine t . C a t a l . Lett .
V o l . 23, 3-4, 387-91 (1973). I_
74. V o o r h i e s A, y K i m b e r l i n Ch. N., Ger. O f f en . 2, 010,
496 (C l . BOlj) O 1 oct. 1970.
75. K r u g l i k o v V. Ya., e t . a l . P revrasnch . Ugleoodorodov
Kis lo tno-Osnovn Geerogennykn Katal . , T e z i s y Dokl .
Vser . Konf.77-79 (1873.
76. Takase S. et. a l . , Ger. O f f en . 2, 156, 324 (Cl. BOlj C l o g ) 18 may 1972.
77. S t e i j n s M. et . a l . , E r d o e l Koh le , Erdgas , Petrochem.
31, (12) 581, 1978.
78. Keim W. e t . a l . , Forcnungsber. Landes Nordrhe in-West fa l en
2838, 36 (1979).
79. S t e i j n s M. e t . a l . , Ind. Eng. Chem. Prod. Res. Dev. 20 - (4), 654-60 (1981).
80. S t e i j n s M., et. al. , Ind. Eng, Chem. Prod. Res. Dev,
20 (4), 660-8 (1981). -
81. P e r o t G., e t . a l . , P roc . I n t . Conf . Zeolites, 5 t n , 640-8 (1980).
82. V o l ' f s o n I .S. y Okruzhnov A. , Ne f tpe reab . Ne f t ekh im
.(MOSCOW) 3, 32-4 (1971). -

88
83. Montes, A., P e r o t , G. y Guisne t , M. React . K i n e t .
C a t a l . Lett . - 1 3 (l), 77-81 (1983).
84. Romero, &I. et. a l . , Appl . C a t a l . 1 (5), 273-5 (1981). -
85. Montes, A., These Docteur de S p e c i a l t e , U n i v e r s i t é
de P o i t i e r s 22 jun. 1979.
86. S h e l l I n t e r n a t i o n a l Research Maatscnappy N. V., Neth.
APP l
87. Rabo, J.A., et. a l . , P r o c . I n t . Conf . C a t a l ; 3 rd
(1964) 2, 1264, (196s) .
88. Wi l son , G.R. y H a l l , W.K., J. Ca ta l . 17, 190 (1970). -
89. Kubo, T. et. a l . , B u l l . Chem. Soc. Jap 45, 607 (1972). -
90. D a l l a B e t t a , R.A. y Boudart M., C a t a l y s i s , Vol. 1, p.
1329 N o r t h H o l l a n d 1973.
91. Naccache C., et. a l . , M o l e c u l a r S i e v e s - I 1 J K a t z e r ed)
p. 538 AC'. S. Washington D.C. 1977.





























