Embed Size (px)
Citation preview

C A R L O S N A V A R R O
M A R I O N A H E R R E R A
M I R - 3 C S R A F A L A F E N A
T U T O R E S : M ª D O L O R E S A I C A R T
M A N U E L B A T A L L A
Distrofia muscular de Duchenne Diagnóstico y manejo

¿Qué es la distrofia de Duchenne? DMD
Distrofia más ʋ en la infancia.
1:3500 (1) recién nacidos ♂. Menor proporción ♀
Hereditaria ligada al cromosoma X
Ausencia de distrofina (gen de la distrofina, DMD)
Se expresa en músculo esquelético y cardíaco.
Cerebro, células de Swann, retina.
65% pacientes delección del exón del gen.
Rompe el marco de lectura del ARNm.
No rompe Distrofia muscular de Becker (DMB)
Portadoras del gen.
(1) Emery AE. Population frecuencies of inherited neuromuscular diseases

Manifestaciones
Afectación motora
Distribución de proximal a distal
Cintura pélvica Cintura escapular
~3 años
Retraso de la marcha, en puntillas... Pérdida ~ 10-13 años.
Maniobra de Gowers
Levantarse con apoyo de los brazos
Pseudohipertrofia gemelar
Escápula alada
DISTROFIAS: Debilidad muscular progresiva
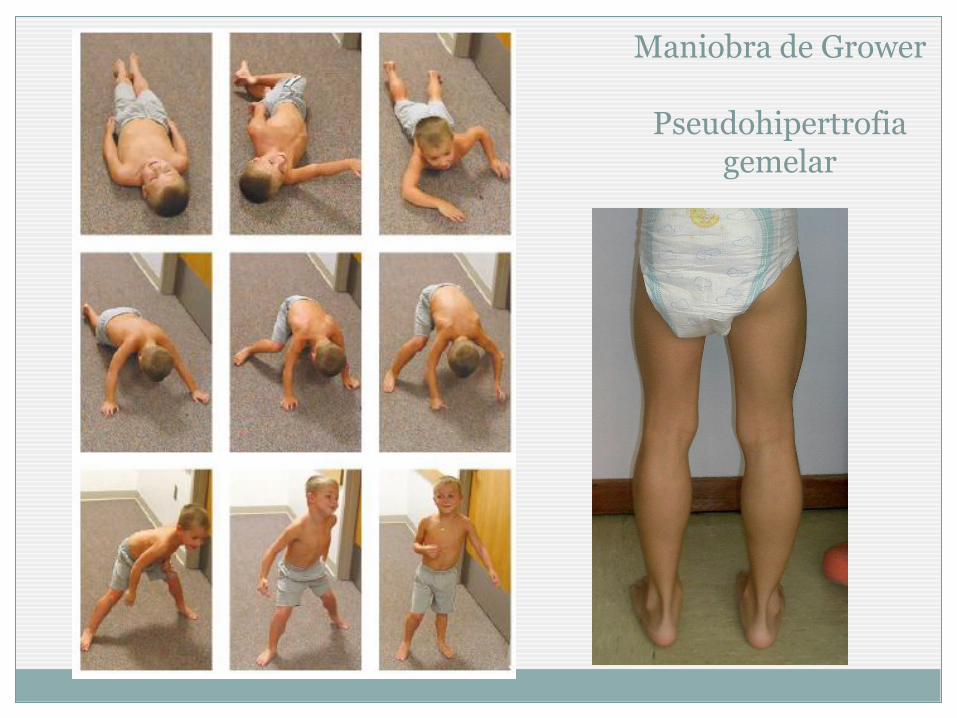
Maniobra de Grower
Pseudohipertrofia gemelar
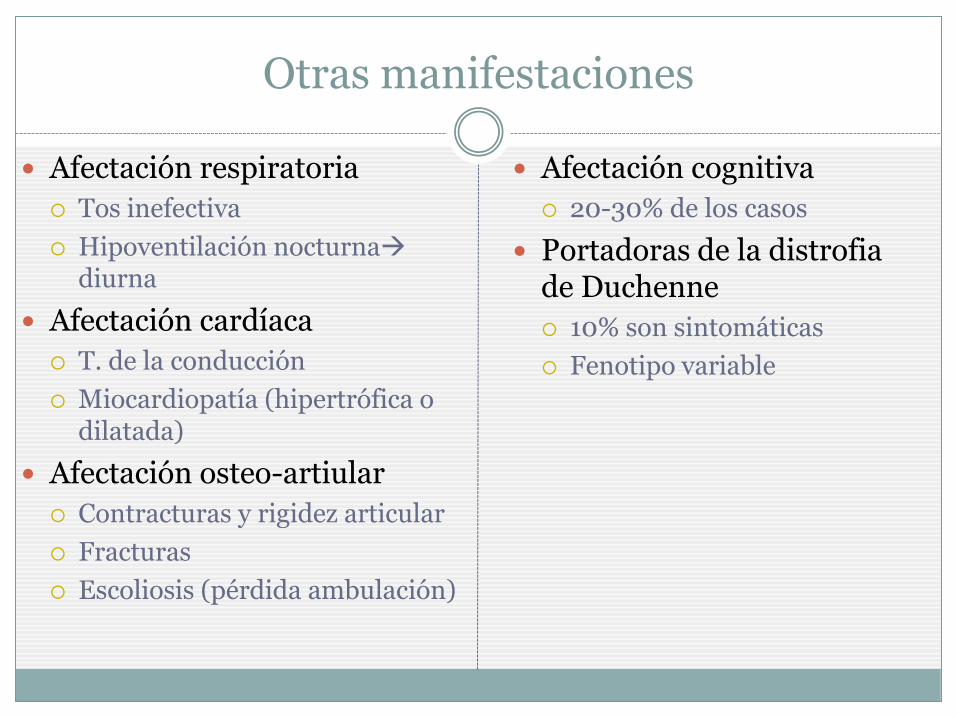
Otras manifestaciones
Afectación respiratoria
Tos inefectiva
Hipoventilación nocturna diurna
Afectación cardíaca
T. de la conducción
Miocardiopatía (hipertrófica o dilatada)
Afectación osteo-artiular
Contracturas y rigidez articular
Fracturas
Escoliosis (pérdida ambulación)
Afectación cognitiva
20-30% de los casos
Portadoras de la distrofia de Duchenne
10% son sintomáticas
Fenotipo variable

Ck elevada Maniobra Gowers
Marcha elavorada
Capaz de impulsarse por sí solo
Se va limitando función extremidades superiores
Hª familiar Marcha balanceada
No subir escaleras
Mantiene la postura
Dificultad para mantener postura
Incrementa daño respiratorio
Disfagia
Fase presintomática
(0 a 2 años)
Fase ambulatoria
temprana (3 a 4 años)
Fase ambulatoria tardía (5 a 8
años)
Fase no ambulatoria
temprana (9 a 11 años)
Fase no ambulatoria
tardía (mayores a 12 años)
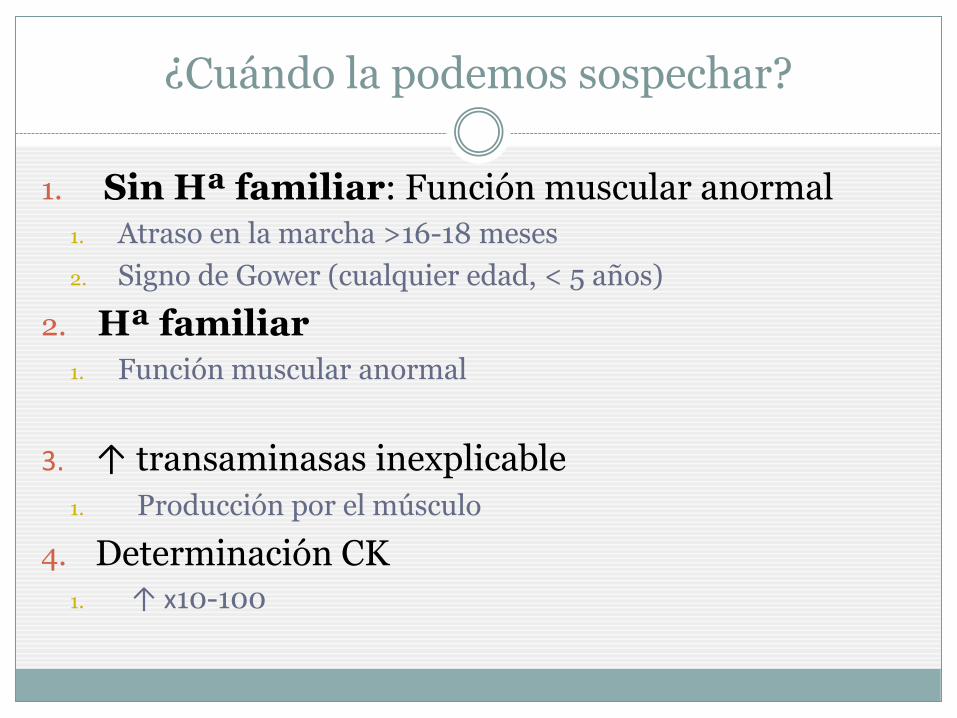
¿Cuándo la podemos sospechar?
1. Sin Hª familiar: Función muscular anormal
1. Atraso en la marcha >16-18 meses
2. Signo de Gower (cualquier edad, < 5 años)
2. Hª familiar
1. Función muscular anormal
3. ↑ transaminasas inexplicable
1. Producción por el músculo
4. Determinación CK 1. ↑ x10-100

¿Cómo lo diagnosticamos?
Biopsia muscular
Deficiencia distrofina
(+)
Estudio genético
MLPA (Multiplex Ligation-dependent Probe Amplification)
Detectar delecciones o duplicaciones de todos los exones
Permite estudio de portadoras
PCR
No cubre todo el gen
Secuenciar le gen DMD
Si el anterior es negativo
EMG: Determina dónde se encuentra la debilidad no discrimina el tipo de distrofia
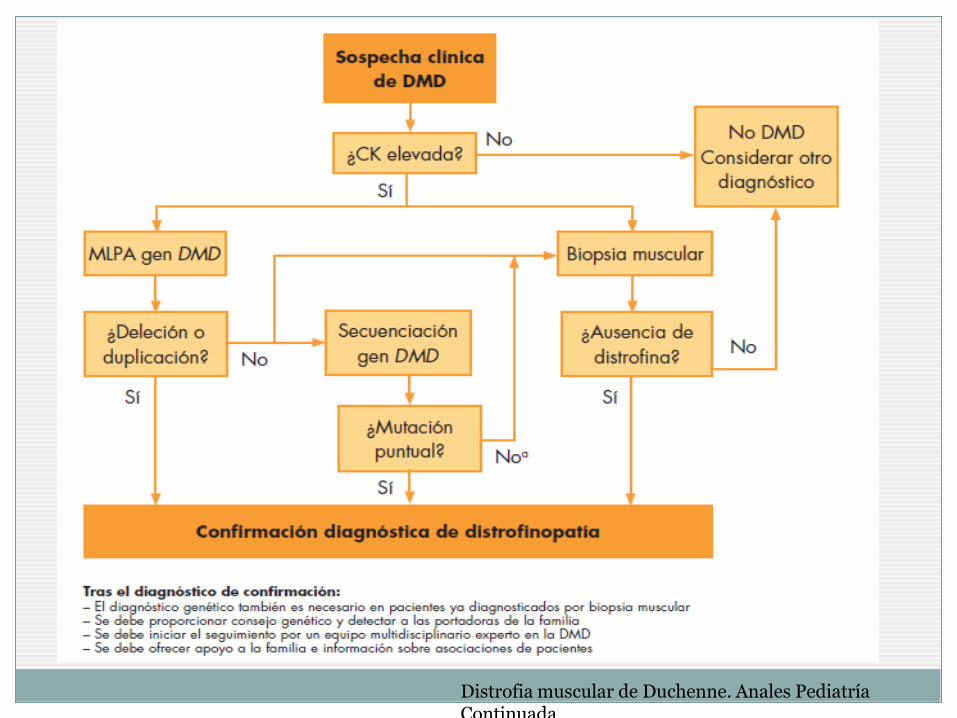
Distrofia muscular de Duchenne. Anales Pediatría Continuada

MANEJO FARMACOLÓGICO
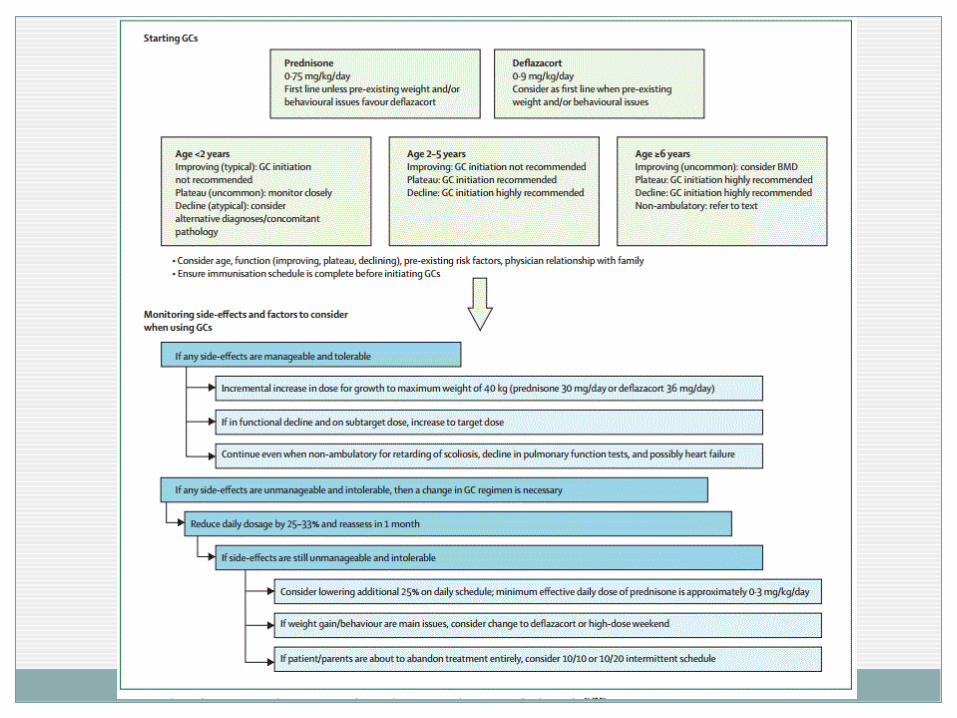
Corticoides Prednisona (0,75 mg/kg/día)/ Deflazacort (0,9 mg/kg/día) DIARIA
Estabilizar la función motora ( 6 meses a 2 años). Mejorar la función respiratoria y reducir escoliosis.
<0.3 mg/kg/día sin beneficio aparente.
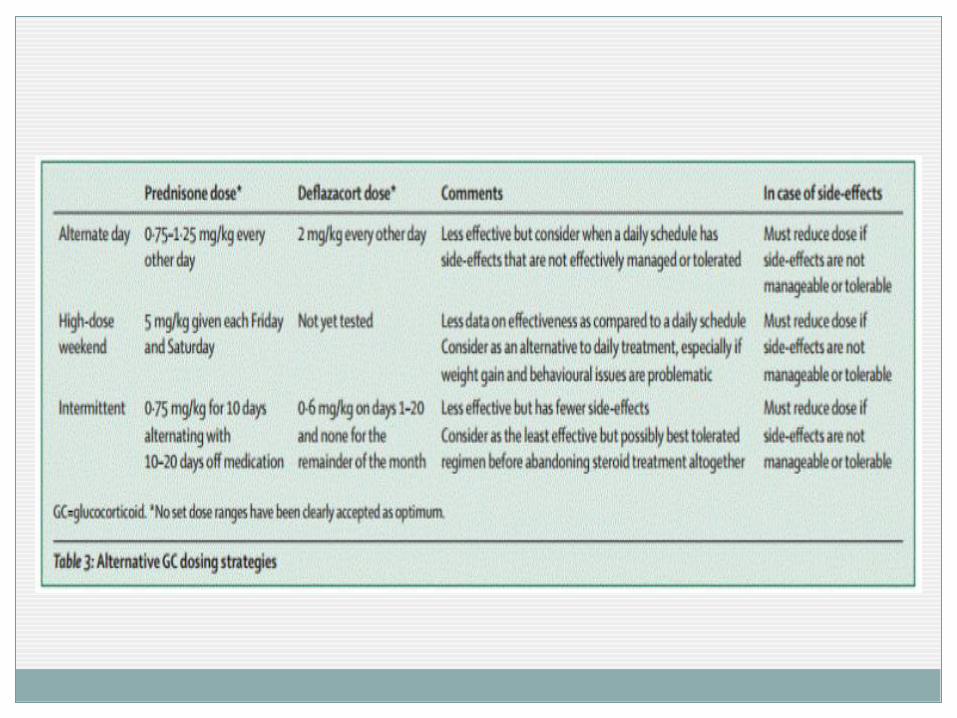
Otras pautas (minimizar los efectos secundarios) Uso alterno durante 10 días Pauta exclusiva los fines de semana ( 5mg/kg)
Inicio (discutido) Decisión individual Etapa plateau (4-6 años): antes de empeoramiento motor.
No <2 años : desarrollo motor
Mantenimiento: 0.3 mg/kg/día – 0.6 mg/kg/día
No diferencias “a priori” entre ellos

Efectos secundarios
Aumento de peso
Cambios estéticos (Cushingoide, hirsurtismo...)
Trastornos de conducta
Fracturas
Control de peso, talla, TA, glucemia, cuidado de salud ósea (suplementos de Ca y Vit D, densitometrías), revisión oftalmológica.
En caso de efectos secundarios no manejables retirada escalonada y planificada.

Otros fármacos y/o suplementos
Oxandrolona, toxina botulínica, creatinina NO recomendado, incluso perjudicial.
Suplementos como coenzima Q10, carnitina, aminoácidos, antioxidantes naturales u otros NO recomendados.
Se recomienda multivitamínico diario con vitamina D y minerales.

Fisioterapia y actividad física
Ejercicios de estiramiento muscular : mantener el rango de movimiento y simetría Mín. 4-6 veces/semana.
Tobillo, rodilla y cadera Ambas fases.
Miembros Superiores No ambulatoria.
Natación deporte de elección. Mantener actividad física mientras sea posible, siempre sin sobreesfuerzo.
Evitar ejercicio contra resitencia aumento del daño muscular.

Ortesis y cirugía ortopédica
Ortesis y Sillas (En primera etapa una manual ligera) Prevención de contracturas
Férulas antiequino nocturnas
Ortesis pie-tobillo-rodilla para prolongar la marcha
Cirugía (la función pulmonar la condiciona) Escoliosis
Ambulatoria • Ángulo de Cobb >20º
No ambulatoria • Ángulo de Cobb >40º
Contracturas Valoración individual

Manejo respiratorio
FVC: Predice la aparición de hipercapnia y determina supervivencia.
Seguimiento estrecho: Anualmente en fase ambulatoria Semestral tras pérdida de la marcha.
Determinar FVC, Saturación 02, flujo máximo de tos
Ventilación mecánica no invasiva de elección. Cuidado con el uso de oxigenoterapia riesgo de
hipercapnia.
Infecciones: Tos asistida manual, ATB si cultivo + ó saturación <95% Calendario vacunal completo
Neumococo, Influenza.

Manejo Cardíaco
ECG, Ecocardiograma
<10 años cada 2 años
>10 años o sintomático anualmente
Miocardiopatía:
IECAs > BB > diuréticos
Valorar anticoagulación ante disfunción severa.
Nutrición
Estado nutricional adecuado: objetivo esencial.
Monitorizar: talla/longitud brazo y peso.

Gastrointestinal: RGE:
IBPs o antagonistas receptor H2.
Estreñimiento:
Uso de laxantes, enemas, etc…
En impactación fecal se desaconseja técnica manual.
Deglución: Valoración clínica si:
Pérdida peso involuntaria >10%, ganancia ponderal limitada, tiempo de ingesta prolongado con fatiga excesiva, babeo, signos de disfagia, tos persistente, neumonía por broncoaspiración…
En fases finales, consensuar necesidad de gastrostomía.

Intervención cognitiva
Valorar remisión a logopedia, psicoterapia, adaptación curricular en función del estado del paciente.
Minimizar el aislamiento.
Anestesia y tratamiento del dolor
En caso de necesidad de cirugía, uso de anestésicos vía intravenosa por complicaciones de vía inhalada.
Manejo eficaz del dolor en función de la causa (terapia física, corrección postural, ortesis, fármacos...).
En casos extremos indicación quirúrgica.

Manejo psicosocial
Atención médica incompleta sin este abordaje del paciente y la familia.
Utilización de las mismas medidas y tratamientos que población general, poniendo énfasis en la prevención y abordaje precoz.
Incremento de alteraciones del neurodesarrollo con desórdenes del espectro autista, TDAH y trastornos obsesivo-compulsivos.
Presencia de ansiedad-depresión.
Exacerbación por déficit en flexibilidad mental, la regulación emocional y temperamento explosivo.
Abordaje individualizado y en edades tempranas.

Terapia en fase de investigación
Salto de exón (DRISAPERSEN (SALTO DEL EXON 51)) Actualmente suspendida en España Restablecer el marco de lectura ARN modificados que reconocen secuencias específicas e
interfieren en el proceso del ARNm ocultando determinados exones
Ensayos clínicos fase II/III
Ataluren ® (TRANSLARNA) Actualmente en España Compuesto con capacidad de hacer que se ignore el codón de
parada y se sintetice la distrofina Fase III de confirmación
Terapia génica y celular. Nuevos compuestos Idebedona, Sildenafilo, Inh de la miostatina....
Terapias España

Conclusiones
No existe en el momento un tratamiento curativo para estas enfermedades y solo se pueden tratar algunos de sus síntomas, por ello se requiere una aproximación multidisciplinaria enfocada en medidas preventivas e intervenciones activas para las alteraciones primarias y secundarias, para cambiar su historia natural y mejorar la función, la calidad de vida y la longevidad


Bibliografía
Bushby K; Finkel R; Birknkrant DJ; Case LE; Clemens PR, et al. DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy; part 1: diagnosis and pharmacological and psychosocial management. Lancet Neurol 2010;9: 77-93
Bushby K; Finkel R; Birknkrant DJ; Case LE; Clemens PR, et al. DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy; part 2: implementation of multidisciplinary care. Lancet Neurol 2010; 9: 177-89
Camacho Salas A. Distrofia muscular de Duchenne. An Pediatr Contin. 2014;12(2):47-44
Darras BT. Overview of muscular dystrophies (Beyond the basics). UptoDate Jan 2016
Darras BT. Clinical features and diagnosis of Duchenne and Becker muscular Distrophy. UptoDate Jan 2016
Viñas Pesqueira M. Revisión: Tratamiento en la distrofia muscular de Duchenne: fisioterapia respiratoria frente a nuevos avances. Fisioterapia. 2013;35 (1):32-39
Diagnostico y manejo de la Distrofia Muscular de Duchenne, una guía para familias. MDA,PPMD, TREAT-NMD, UPPMD. Traducción de Fanco G, Rojas R, Hasselkus G. www.upaduchenne.org

Gràcies!





























