Embed Size (px)
Citation preview

Límites nosológicos de la Epilepsia Ausencia InfantilDr. Rafael Camino León.
U. de Neuropediatría. H. U. Reina Sofía. Córdoba





Epilepsia ausencia infantil (EAI)
“La EAI se produce en los niños de edad escolar (con un
pico máximo de manifestaciones entre los 6 – 7 años), con
una fuerte predisposición genética en niños normales en
otros aspectos. Se da con mayor frecuencia en niñas que en
niños. Se caracteriza por ausencias muy frecuentes
(picnolepsia).
El EEG muestra un registro de PO simétricas, síncronas y
bilaterales, por lo general a 3 Hz, sobre una actividad de
fondo normal.
Durante la adolescencia evoluciona a menudo hacia una
epilepsia con crisis de Gran Mal (CTCG). De no ser así,
pueden remitir las ausencias o más raramente, persistir como
único tipo de crisis” (ICE, 1989)

Esta definición incluye las características clínicas y EEG
fundamentales de las ausencias en el niño, pero omite
otros rasgos que permiten diferenciar la EAI de otros SE
que cursan con ausencias, cómo síntomas asociados,
duración y respuesta al tratamiento
Nieto Barrera M, 2004
Críticas a la definición de la ILAE

Criterios obligatorios de Panayiotopoulos
Edad de inicio entre 4 a 10 años, con un pico entre 5 y 7 años.
Estado neurológico y desarrollo normal.
Crisis de inicio brusco, cortas de 4 a 20 segundos y varias (10 por día) en un mismo día, con afectación severa de la conciencia. Los automatismos son frecuentes, pero no tienen significación para el diagnóstico
Puntas rítmicas generalizadas o descargas de ondas con doble espiga a aproximadamente 3 Hz.
Panayiotopoulos CP 1994

Criterios de exclusión de Panayiotopoulos
Otro tipo de crisis previas o durante la etapa activa de ausencia, tales como CTCG o sacudidas mioclónicas.
Mioclonias masivas de miembros, mioclonias palpebrales, periorales. Sin embargo, leves mioclonias de ojos, cejas y párpados pueden observarse, especialmente en primeros 3 segundos de la crisis de ausencia
Sin alteración de conciencia, o muy leve durante la descarga de PO a 3 Hz.
EEG: breves PO a 3-4 Hz de menos de 4 segundos, descargas fragmentadas y múltiples puntas (más de 3) ictales.
Precipitantes visuales o sensoriales de crisis clínicas
Panayiotopoulos CP 1994

Panayiotopoulos CP 2008


Nosología
“Parte de la medicina que tiene por objeto
describir, diferenciar y clasificar las
enfermedades” (RAE)
Las diferentes clasificaciones de la ILAE
de crisis y síndromes epilépticos han ido
estableciendo distintas delimitaciones
nosológicas a la luz de los avances
concretos, utilizando criterios variados
(edad, etiología, genética, entre otros)

Ubicación nosológica de las
Mioclonias palpebrales con ausencias
Epilepsias fotosensibles
Epilepsias con ausencias
Epilepsias mioclónicas


Visión global del problema
Síndromes establecidos “oficiales”
• Criterios diagnósticos poco definidos
• Solapamiento entre diversos síndromes
Síndromes no considerados por la ILAE
• Dudas ante el pronóstico de los diferentes síndromes
• Clasificaciones y enfoques alternativos

Clasificaciones recientes en epilepsia
Clasificación de crisis
• ILAE 1981
• ILAE 2001, Eje 2
• ILAE 2010
• Clasificación semiológica, 1998
Clasificación de síndromes
• ILAE 1989
• ILAE 2001, Eje 3
• ILAE 2010

Clasificación Internacional de
Crisis Epilépticas ILAE 1981
1. Crisis parcial (focal, local)
A) Parcial simple
B) Crisis parcial compleja (con alteración de conciencia, lenguaje
o memoria)
2. Crisis generalizadas
A) Ausencias
B) Crisis mioclónicas
C) Crisis clónicas, D) Crisis tónicas y E) Crisis tónico-clónicas
F) Crisis atónicas
3. Crisis no clasificadas
Commission on Classification and Terminology, ILAE (1981).

Clasificación semiológica de Luders
1. Aura
— Aura somatosensorial
— Aura visual
— Aura auditiva
— Aura gustatoria
— Aura olfatoria
— Aura autonómica
— Aura abdominal
— Aura psíquica
2. Crisis dialéptica
— Crisis dialéptica típica
Lüders y cols. (1998).
3. Crisis motora
Motora simple
— Mioclónica
— Tónica
— Clónica
— Tónico-clónica
— Versiva
— Espasmo epiléptico
Motora compleja
— Hipermotora
— Gelástica
— Automotora
4. Crisis especiales
— Atónica
— Astática
— Hipomotora
— Acinética
— Mioclono negativo
— Crisis afásica
5. Evento paroxístico
Además se detalla:
Somatotopía
Evolución

Clasificación de CE. ILAE 2001, Eje 2
Generalizadas
• Tónico-clónicas
• Clónicas
• Ausencias típicas
• Ausencias atípicas
• Ausencias mioclónicas
• Tónicas
• Espasmos
• Mioclónicas
• Mioclonías palpebrales
• Mioclono negativo
• Atónicas
• Crisis reflejas en síndromes
generalizados
Focales
• Sensoriales
• Motoras
• Gelásticas
• Hemiclónicas
• Secundariamente generalizadas
• Crisis reflejas en síndromes
focales
1. Crisis autolimitadas
Engel (2001).

Clasificación Internacional de
síndromes epilépticos. ILAE 1989
Síndromes relacionados con localización (parcial, focal,
local)
Idiopáticos
Sintomáticos
Criptogénicos
Síndromes generalizados
Idiopáticos
Sintomáticos o criptogénicos: Epilepsia con ausencias mioclónicas
Sintomáticos
Epilepsias y síndromes de origen indeterminado, focal o
generalizado
Síndromes especiales

Epilepsias y S. epilépticos generalizados idiopáticos
• Convulsiones neonatales familiares benignas
• Convulsiones neonatales benignas
• Epilepsia mioclónica benigna de la infancia
• Epilepsia-ausencia de la infancia
• Epilepsia-ausencia juvenil
• Epilepsia mioclónica juvenil
• Epilepsia con CTC (gran mal) al despertar
• Otras epilepsias generalizadas idiopáticas no definidas
• Epilepsias con crisis reflejas
ILAE 1989

1. Epilepsias focales idiopáticas
2. Epilepsias focales familiares
3. Epilepsias focales sintomáticas o probablemente
sintomáticas
4. Epilepsias generalizadas idiopáticas
5. Epilepsias reflejas
6. Encefalopatías epilépticas
7. Epilepsias mioclónicas progresivas
8. Crisis que no requieren un diagnóstico de epilepsia
obligatoriamente
Clasificación de los S. epilépticos 2001. Eje 3
ILAE Commission Report. Epilepsia; 2.001

Epilepsias y S. epilépticos generalizados idiopáticos
• Epilepsia mioclónica benigna de la infancia
• Epilepsia con crisis mioclónico-astáticas
• Epilepsia-ausencia de la infancia
• Epilepsia con ausencias mioclónicas
• Epilepsias generalizadas idiopáticas con fenotipos variables
– Epilepsia-ausencia juvenil
– Epilepsia mioclónica juvenil
– Epilepsia sólo con CTCG
• Epilepsias con crisis reflejas
ILAE Commission Report. Epilepsia; 2.001

Revised terminology and concepts for organization
of seizures and epilepsies. ILAE 2010
Berg AT. Epilepsia, 2010: 51(4):676–685.

Clasificación de las CE. ILAE 2010
CE generalizadas (convulsivas o no convulsivas)
Tónico-clónicas
Ausencias Mioclónicas Clónicas TónicasAtónicas (astáticas)
CE focales
CE desconocidas
Espasmos epilépticos
Otros

CE generalizada
Es la que se origina en algún punto dentro de las
redes neuronales distribuidas bilateralmente y
se difunde rápidamente
Estas redes pueden incluir estructuras corticales y
subcorticales, pero no precisan afectar a las
totalidad de la corteza cerebral

Clasificación de las ausencias. ILAE 2010
Típicas AtípicasCon características
especiales
Ausencias mioclónicas
Mioclonias palpebrales

Síndromes electroclínicos de la ILAE 2010

Síndromes Electroclínicos y otras epilepsias
agrupadas por Especificidad del diagnóstico
• Síndromes electroclínicos según edad de comienzo
– Periodo neonatal
– Lactancia
– Infancia: EAI y EAM
– Adolescencia/adulto: EAJ
– Edad de inicio variable
• Constelaciones distintivas/Síndromes quirúrgicos
• Epilepsias no sindrómicas
– Epilepsias atribuidas a causas estructurales-metabólicas
– Epilepsias de causa desconocida
• Entidades con CE no diagnosticadas como epilepsia
– Crisis neonatales
– Crisis febrilesBerg AT. Epilepsia, 2010: 51(4):676–685.
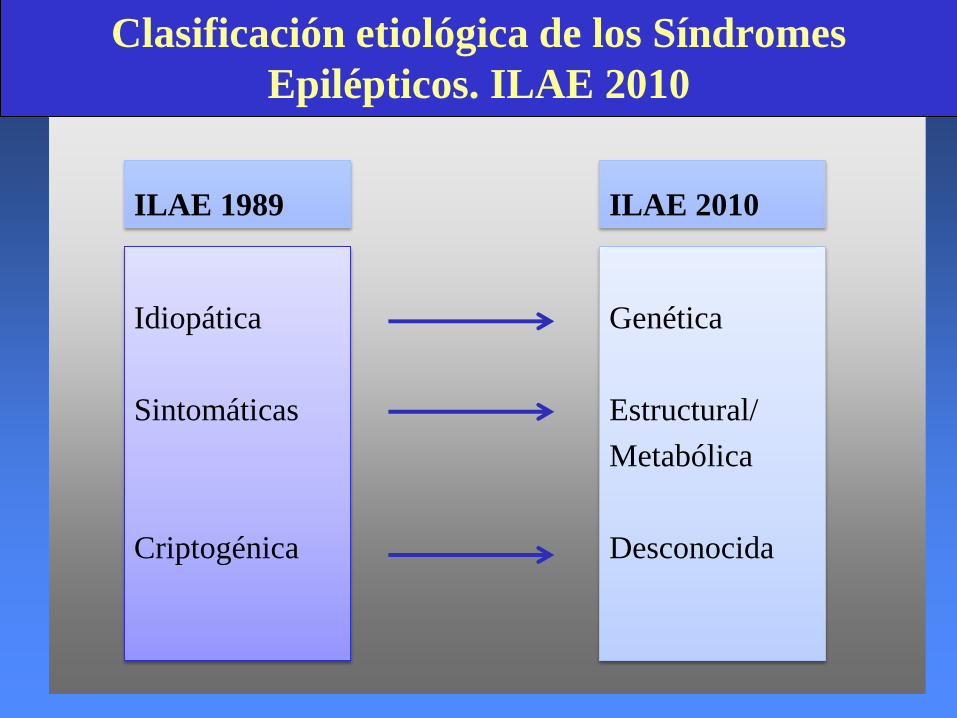
Clasificación etiológica de los Síndromes
Epilépticos. ILAE 2010
ILAE 1989
Idiopática
Sintomáticas
Criptogénica
ILAE 2010
Genética
Estructural/
Metabólica
Desconocida

Síndromes electroclínicos con ausencias
clasificados por edad de comienzo
En la infancia
Epilepsia ausencia de la
infancia
Epilepsia con ausencias
mioclónicas
En la adolescencia/edad
adulta
Epilepsia ausencia juvenil

Epilepsia ausencia infantil

Epilepsia ausencia infantil
Incidencia 2-8/100.000
Prevalencia Elevada: 2,6 – 5,7/1.000.
Representa el 8-15% de la Epilepsias y el 44% de la EGI, en la
edad escolar
Edad y sexo Comienzo habitual: 4-10 años (Pico: 6 – 7a)
Sexo. Predomina en mujeres (3:2)
AP CF en el 15%. En niños sin antecedentes de interés y con
neurodesarrollo previo normal.
AF CF y/o EGI en un 15-30%
El 75% de concordancia en gemelos monocigóticos. Los
familiares tienen con frecuencia crisis febriles o tónico-clónicas
Etiología La causa es genética, con gran heterogeneidad. Probable herencia
poligénica, encontrándose varios locus (cr 1p, 8q24, 5q31.1)
Alteraciones en el gen CAC- NA1H que codifica el canal de Ca++
tipo T activado por voltaje y genes receptores GABA podrían estar
implicadas.

Epilepsia ausencia infantil
Ausencias
típicas
Pérdida de conciencia con punta-onda a 3 Hz generalizada en el
EEG. Inicio y fin brusco. Muy breves. Alta frecuencia diaria.
Inducibles por hiperventilación
Pueden ser simples (9%) o complejas (91%)
Ausencias
complejas
Componente motor
- simétrico: 45.5%
- asimétrico: 5%
- atónico: 22.5%
Automatismos: 63.1%
Fenómenos autonómicos: 5-10%
Semiología
clínica
El 30-40% de los EAI presentan en la pubertad CTCG
Estado Mental normal, aunque presentan problemas cognitivos
- Disminución del tiempo de reacción
- Deterioro cognitivo transitorio
Trastornos conductuales, reactivos y iatrogénicos

Niveles de diagnóstico diferencial
TPE vs TPNE Tipo de CETipo de
epilepsia
Tipo de síndrome epiléptico
Entidad

EAI y DD con TPNE
Ensoñaciones Tics
Crisis psicógenas
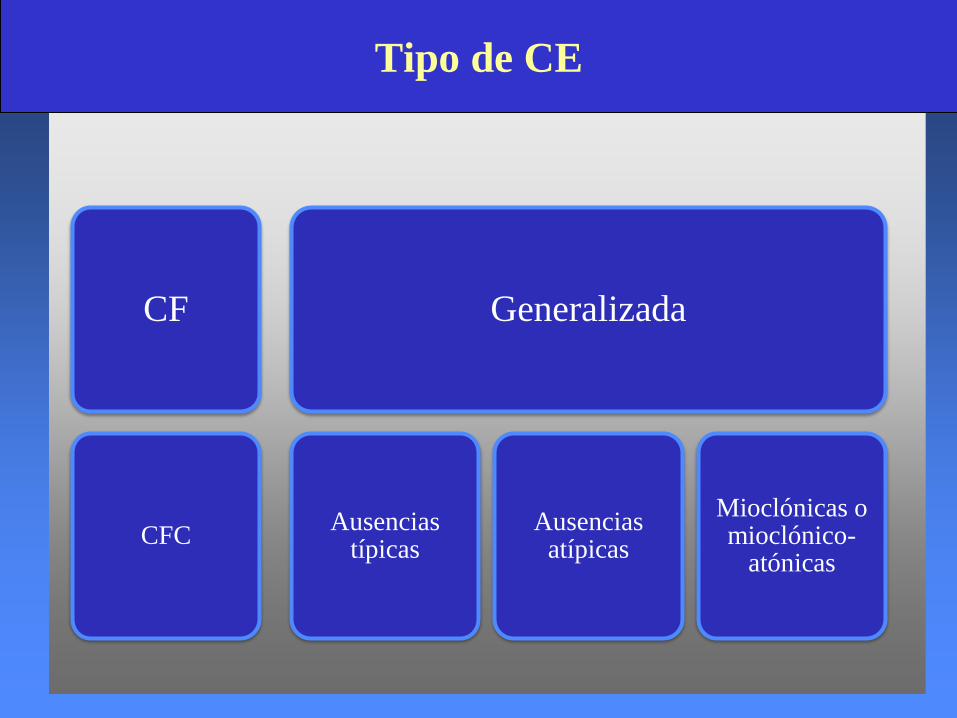
Tipo de CE
CF
CFC
Generalizada
Ausencias típicas
Ausencias atípicas
Mioclónicas o mioclónico-
atónicas

Tipo de Síndrome electroclínico
EAJ
EMJ
EAM
MPA
Ausencias sintomáticas
SLG
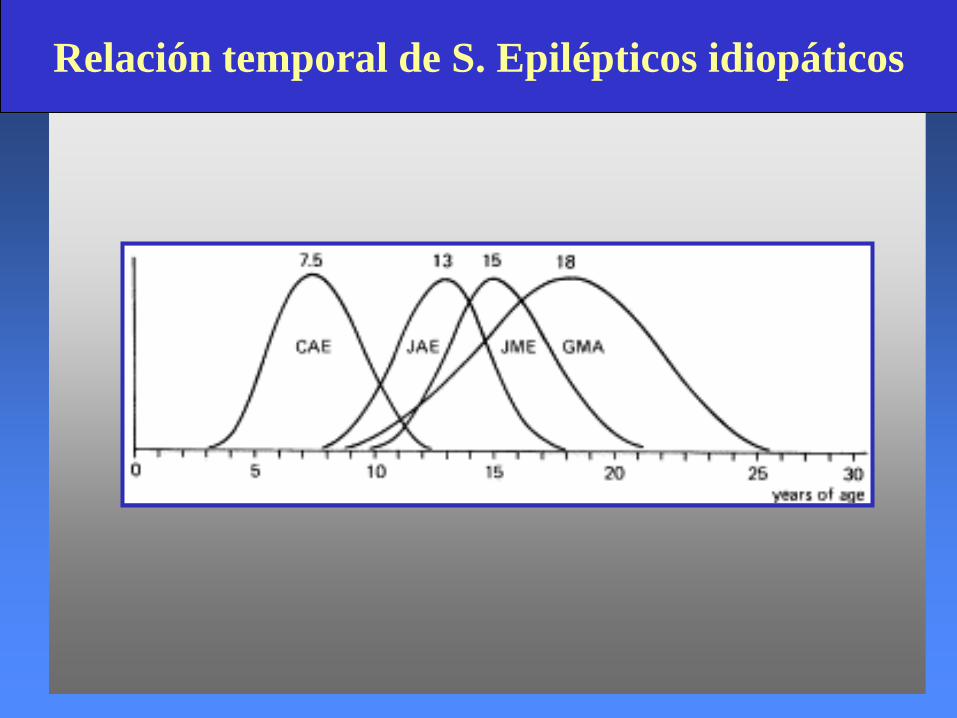
Relación temporal de S. Epilépticos idiopáticos

Diagnóstico de entidad
Tumor o LOC
Glut-1Trastornos genéticos
Cromosomopatías
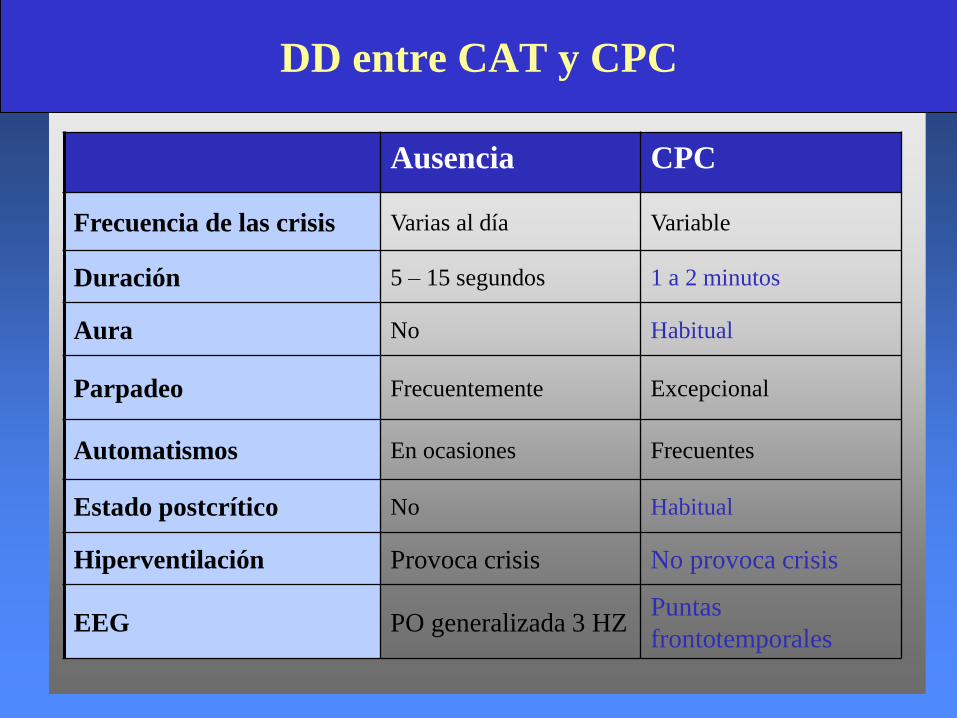
Ausencia CPC
Frecuencia de las crisis Varias al día Variable
Duración 5 – 15 segundos 1 a 2 minutos
Aura No Habitual
Parpadeo Frecuentemente Excepcional
Automatismos En ocasiones Frecuentes
Estado postcrítico No Habitual
Hiperventilación Provoca crisis No provoca crisis
EEG PO generalizada 3 HZPuntas
frontotemporales
DD entre CAT y CPC

A. típicas A. atípicas
Inicio y final Brusco Gradual
Pérdida de conciencia CompletaNo completa y más
prolongada
Cambios en tono Leve Moderado
Hiperventilación Provoca crisis No provoca crisis
EEG PO 3 Hz PO lenta (1,5 – 2,5 Hz)
Síndromes EAI y EAJ SLG y otras EGS
DD entre CAT y ausencias atípicas
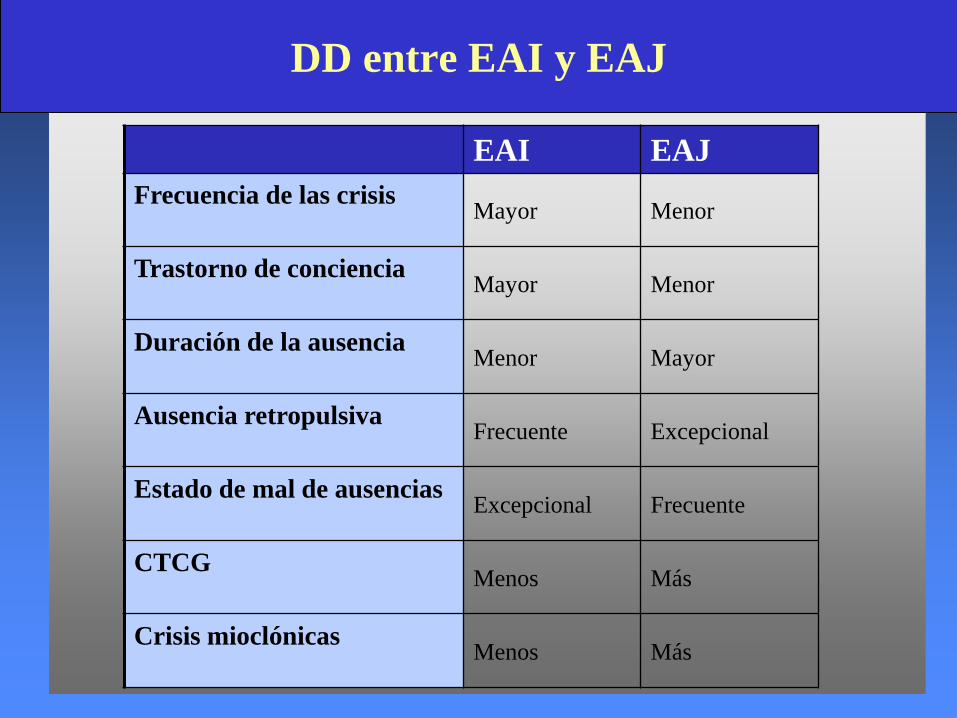
EAI EAJ
Frecuencia de las crisis Mayor Menor
Trastorno de conciencia Mayor Menor
Duración de la ausencia Menor Mayor
Ausencia retropulsiva Frecuente Excepcional
Estado de mal de ausencias Excepcional Frecuente
CTCGMenos Más
Crisis mioclónicas Menos Más
DD entre EAI y EAJ

Patrones EEG y ausencias
Patrón EEG Crisis Síndromes
PO a 3 Hz Ausencias
típicas
E. Con ausencias infantiles.
E. Con ausencias juveniles.
E. Con ausencias mioclónicas.
E. Mioclónico-atónica.
E. S. del lóbulo frontal con
ausencias.
PO a 2 Hz Ausencias
atípicas
Síndrome de Lennox-Gastaut.
E. Mioclónica severa.
Ausencias atípicas.
E. Parcial infantil atípica de
Aicardi.
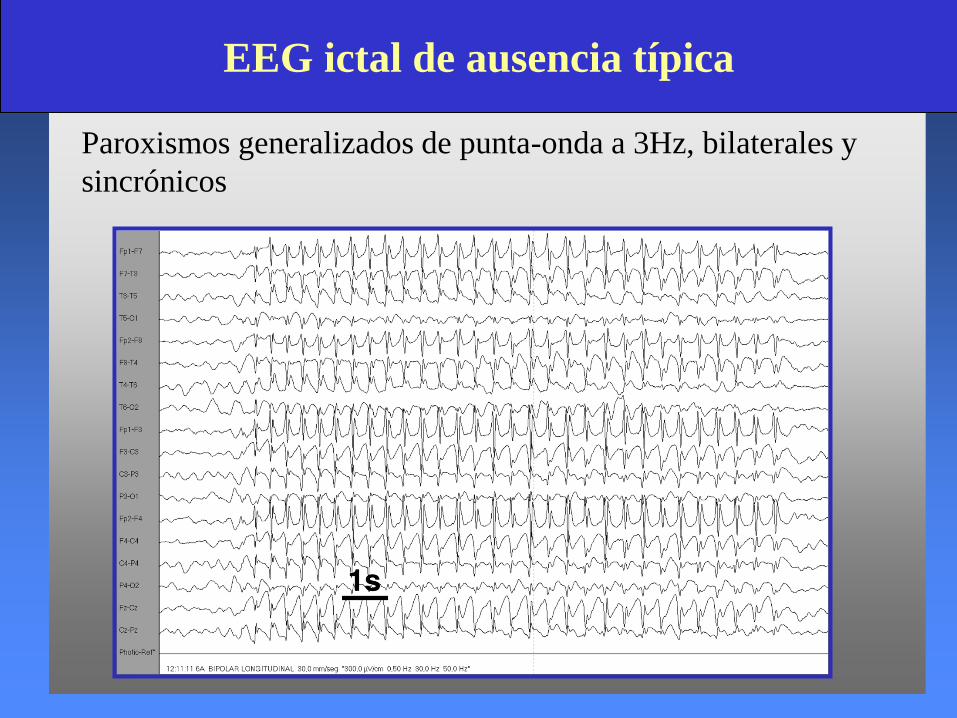
EEG ictal de ausencia típica
Paroxismos generalizados de punta-onda a 3Hz, bilaterales y
sincrónicos

EEG en la ausencia atípica. SLG

Tratamiento EAI. Guía SADE 2015

EAI. Pronóstico

Criterios predictivos en EAI
Favorables Desfavorables
Sexo mujer
Comienzo ausencias entre 4-9 a
CAT como único tipo de CE
Inteligencia normal
E. Neurológico normal
Tratamiento precoz y correcto
Respuesta rápida al tratamiento
EEG con salvas de ondas lentas
posteriores
Sexo varón
Comienzo ausencias después de 9 a
Asociación CTCG
Ausencias de larga duración
CI bajo
Daño cerebral previo
Pobre respuesta al tratamiento
Asimetría de descargas críticas
ELI positiva

Síndromes epilépticos idiopáticos generalizados
no incluidos en la ILAE
Mioclonias palpebrales con
ausencias
Mioclonia perioral con ausencias
Epilepsia generalizada
idiopática con ausencias fantasma
Epilepsia generalizada
idiopática con ausencias de la
primera infancia

Mioclonías palpebrales con ausencias

Mioclonías palpebrales con ausencias
(S. de Jeavons)
Frecuencia 13% de los pacientes con EGI con ausencias típicas.
Edad y sexo Inicio entre 2-14 años (pico a los 6-8 años).
Más en niñas
Antecedentes Sin antecedentes de interés y con neurodesarrollo
previo normal. Formas clínicas parecidas se han
descrito en pacientes con retraso cognitivo.
Puede existir agregación familiar.
Mioclonias
palpebrales
Sacudidas marcadas de los párpados, con desviación
de la mirada y retropulsión cefálica.
PUEDEN o NO acompañarse de una breve ausencia de
3-6 segundos de duración.
Las mioclonías se repiten frecuentemente a lo largo del
día de forma espontanea o autoinducida.
Desencadenantes: cierre palpebral, autoestimulación

Mioclonías palpebrales con ausencias
(S. de Jeavons)
Otras crisis Mioclonías de las extremidades.
Ausencias típicas espontáneas.
Estado epiléptico de ausencias palpebrales
(autoinducido con estímulos luminosos o por la
mañana al despertar).
CGTC en la evolución infrecuentes.
EEG Salvas de PPO generaliza a 3-6 Hz desencadenados
por el cierre palpebral, de 1-6 segundos de duración.
La hiperventilación aumenta las descargas.
Respuesta fotoparoxistica en todos
Pueden presentar, además, actividad epileptiforme con
la ausencia de fijación.

Mioclonías palpebrales con ausencias
(S. de Jeavons)
DD EGI que cursan con ausencias (EMJ, EAJ, CAE)
Tics faciales.
Evolución y
pronóstico
Las mioclonías palpebrales suelen persistir durante
toda la vida y suelen ser difíciles de controlar con
FAE.
Las ausencias y las CGTC responden mejor al
tratamiento farmacológico.
Tratamiento VPA – LTG – ESM – LEV
Medidas
higiénicas
Evitar la estimulación luminosa.
Usar gafas polarizadas.
Contemplar la TV a distancia

Mioclonías periorales con ausencias

Mioclonías periorales con ausencias
Frecuencia Alrededor del 9% de los pacientes con ausencias
típicas.
Edad y sexo Inicio entre 6-12 años (pico a los 10 años).
Más frecuente en niñas.
AF EGI de ausencias
AP Sin antecedentes de interés y con desarrollo previo
normal.
Crisis Ausencias típicas con mioclonias periorales
Ausencias Muy breves
Mioclonias
faciales
El paciente puede percibir contracción de los
músculos periorales con protrusión de los labios o
temblor en la mandíbula.

Mioclonías periorales con ausencias
Estado de ausencias Hasta un 50%.
EEG intercrítico PPO generalizada irregular a 4-7 Hz, muy breve (1-
2 segundos) y amplia
Puede existir focalidad. No fotosensibilidad
EEG crítico PPO rápidos, difusos, bilaterales y síncronos. No
fotosensibilidad
DD Con otras EGI que cursan con ausencias (EMJ, EAJ,
CAE). Puede confundirse con epilepsias focales por
los hallazgos del EEG y porque el paciente a veces
percibe las mioclonías de forma unilateral.
Evolución CTCG con frecuencia en la evolución
Pronóstico Persiste toda la vida
Tratamiento VPA – LTG – ESM – BZD. VPA + LTG

Epilepsia generalizada idiopática
con ausencias fantasma

Epilepsia generalizada idiopatica
con ausencias fantasma
Frecuencia 15% de los pacientes con EGI con ausencias.
Edad y sexo Inicio en la adolescencia en la mayoría de los casos.
Ambos sexos por igual
AP Sin antecedentes de interés y con desarrollo previo
normal.
Ausencias Ausencias sutiles de breve duración (3-4 s), que
interrumpen parcialmente la actividad del paciente, a
veces acompañadas con parpadeo.
Estado de
ausencias
Hasta un 50%
Pueden cursar con clínica sutil como confusión, dificultad
para comunicarse y lentitud mental
Otras crisis CGTC que suelen aparecer en la edad adulta y son
infrecuentes.

Epilepsia generalizada idiopatica
con ausencias fantasma
EEG
intercrítico
PPO generalizada irregular a 4-7 Hz, muy breve (1-2 s) y
amplia
Frecuente actividad rápida paroxística y polipuntas en los
registros EEG de sueño de estos pacientes
Puede existir focalidad. No fotosensibilidad
EEG crítico Trenes de polipuntas que preceden a la ausencia con la
descarga típica de puntaonda
DD Con otras EGI que cursan con ausencias (EMJ, EAJ). Puede
confundirse con epilepsias focales por los hallazgos del EEG
Evolución y
pronóstico
Muchos pacientes no sabían que tenían epilepsia hasta que
tuvieron una primera CGTC. Persiste toda la vida. No esta
claro si es necesario tratar al paciente que tan solo tiene
ausencias.
Tratamiento VPA y LTG

EGI con ausencias de de inicio precoz

EGI con ausencias de inicio precoz
Frecuencia Rara en relación con las ausencias de inicio en la infancia
Edad y sexo Inicio antes de los 3 años .
Más en varones.
AF Fuerte historia familiar de EGI y frecuentes EEG alterados
en miembros no afectados, especialmente las madres
AP Desarrollo previo normal
Ausencias Interrupción de la actividad y pérdida de conciencia difícil
de precisar por los cuidadores.
Menos frecuentes y menos severas que en niño mayor
Otras crisis CTCG frecuentes (2/3) y y a menudo son el primer tipo
de crisis
Sacudidas mioclónicas y C. mioclónico-atónicas en el 40%
Frecuente status de ausencias, que pueden afectar
cognitivamente

EGI con ausencias de inicio precoz
EEG interictal No específico, aunque puede estar lento
EEG ictal Descargas irregulares de PO a 2,5-3,5 Hz, sin final
brusco, de 2 a 10 segundos de duración
DD Epilepsia mioclónico-astática
Déficit de Glut-1
Pronóstico Menos favorable que el de la EAI, en control de las CE
y desarrollo neuropsicológico y de la conducta
Tratamiento VPA o combinación de VPA y ESM o LTG



Vamos finalizando

Clasificación de las epilepsias. CIE 10
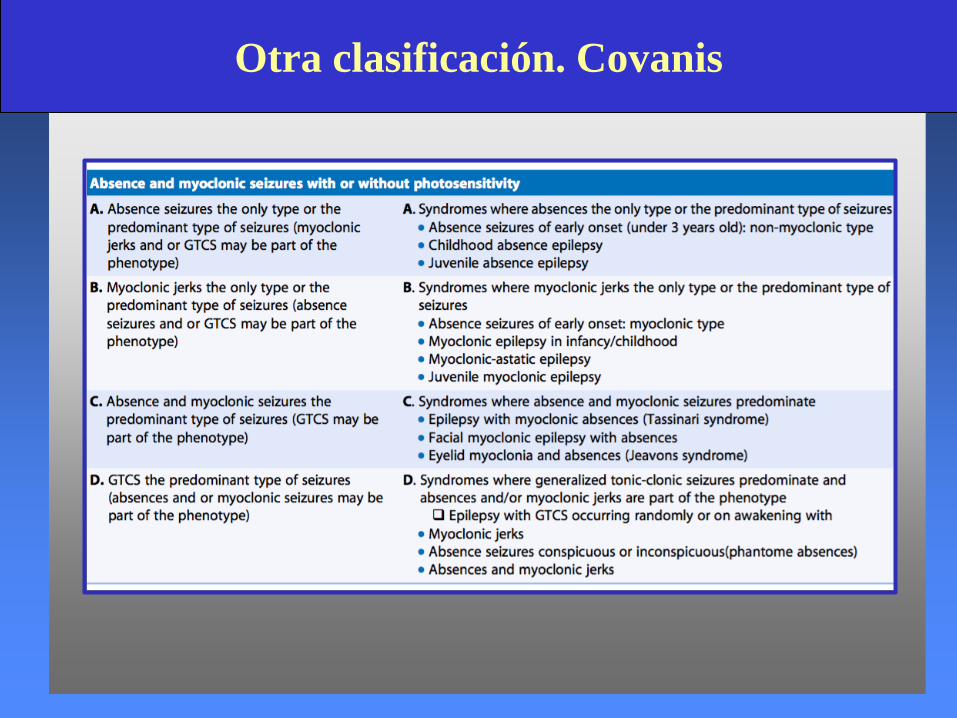
Otra clasificación. Covanis

Conclusiones
• Las epilepsias se han desarrollado históricamente desde perspectivas
anatómicas, neurofisiológicas y neuroquímicas.
• Recientemente desde perspectivas neurobiológicas desde sistema a
célula, desde modelos in vivo a estudios de neurotransmisores,
receptores y canales.
• Actualmente desde el estudio de moléculas y genes implicados en
interacción probable o posible con factores ambientales.
• Conocer los mecanismos cerebrales que subyacen a las crisis
epilépticas, así como los efectos de la genética, la edad y el entorno
sobre ellos nos va a permitir, probablemente, programar estrategias
terapéuticas mas adecuadas y…...
• Mejorar en la nosología de estos tratornos

Homenaje