Embed Size (px)
Citation preview

24
artíCulo de revisión
Escleromixedema
RESUMEN
El escleromixedema, también conocido como liquen generalizado mixedematoso, es una mucinosis rara, crónica e incapacitante. Se distingue por una erupción papular generalizada y esclerodermoide con gammapatía monoclonal en 83% de los casos y sin enfermedad tiroidea, que afecta a adultos sin distinción de género. Se conocen tres subtipos clinicopatológicos de mucinosis: localizada, generali-zada (escleromixedema) y atípica. La infiltración de mucina en el escleromixedema tiene diversas manifestaciones en otros órganos y sistemas, por lo que se considera una enfermedad sistémica. En la piel se distingue por un depósito excesivo de mucina en el tejido conectivo, mismo que puede estimular la síntesis de colágeno y glucosaminogli-canos, que engrosan a las fibras de colágeno y, en consecuencia, se forman pápulas liquenoides de consistencia dura. El curso grave de la enfermedad requiere un tratamiento agresivo y la mayoría de los casos necesita un tratamiento de mantenimiento de largo plazo. Se han prescrito esteroides, retinoides alquilantes, inmunosupresores y trasplante de médula ósea, entre otros.
Palabras clave: escleromixedema, diagnóstico, tratamiento.
Diana Elizabeth Medina-Castillo1
José Ángel Martinez-Muñoz2
Nestor Caballero-Hernández3
Rosa María Ortiz-Guerrero3
Carolina Paola Ortiz-Valdés3
Carlos Brani Rodríguez-Baron3
Andrea Talavera-Díaz3
Miguel Alejandro Azuara-Varela3
1 Dermatóloga, Hospital General Regional núm. 220 Gral. José Vicente Villada, IMSS, Toluca, México.2 Dermatólogo y dermatopatólogo. Práctica privada.3 Estudiantes de pregrado de cuarto año de la Licen-ciatura de Médico Cirujano, Facultad de Médicina, UAEMex, Toluca, México.
Correspondencia: Dra. Diana Elizabeth Medina Castillo Comonfort 100, planta baja 8 52140 Metepec, Estado de México [email protected]
Este artículo debe citarse comoMedina-Castillo DE, Martinez-Muñoz JA, Caballero-Hernández N, Ortiz-Guerrero RM y col. Esclero-mixedema. Dermatol Rev Mex 2016;60:24-36.
Recibido: 11 de agosto 2015
Aceptado: 14 de octubre 2015
Scleromyxedema
ABSTRACT
Scleromyxedema, also known as myxedematosus lichen, is a rare, chronic, disabling disease, characterized by generalized papular and esclerodermoid rash with monoclonal gammopathy and absence of thyroid disease, which usually affects adults regardless of gender. Three clinicopathological mucinosis types are known: localized, generalized and atypical. Scleromyxedema is a generalized mucinosis and may have cutaneous manifestations and affect other organs; thus, it is considered a systemic disease. It is characterized by excessive mucin in connective tissue, which can stimulate the synthesis of collagen and glycosaminoglycans, that thicken the collagen fibers and thus causing lichenoid papules are formed tissue hardening. The severe course of the disease requires aggressive treatment and in most cases long-term maintenance is necessary. There have been prescribed: steroids, alkylating retinoids, immunosuppressive therapy and bone marrow transplantation, among others.
Key words: scleromyxedema, diagnosis, treatment.
Dermatol Rev Mex 2016;60:24-36.Dermatología
R e v i s t a m e x i c a n a

25
Medina-Castillo DE y col. Escleromixedema
ANTECEDENTES
En 1954, Arndt y Gottron definieron el mixede-ma, que fue redefinido por Rongioletti y Rebora en 2001 como una enfermedad caracterizada por una erupción papular generalizada1 y es-clerodermoide, acompañada de gammapatía monoclonal, en su mayor parte inmunoglobulina lambda o kappa en suero, la piel, la médula ósea, y ausencia de trastorno tiroideo.2 Puede ir seguido de otras manifestaciones sistémicas, que más adelante describiremos.
El escleromixedema afecta el grupo etario entre 30 y 80 años; sin embargo, cuando afecta a niños, ocurre después de infecciones generales o locales, sobre todo por estreptococcias. Se considera que es consecuencia de un mecanis-mo autoinmunitario inducido por la infección estreptocócica, que aumenta los mucopolisacá-ridos y el diagnóstico histológico se establece al demostrar un aumento considerable de muco-polisacáridos ácidos.3
Fisiopatología
El escleromixedema pertenece al grupo de las mucinosis cutáneas; conjunto heterogéneo de enfermedades en las que la mucina se acumula de manera focal o difusa en la dermis o los folí-culos pilosos. La razón por la que la mucina se acumula de manera anómala en la piel no está bien definida aún. Se sugiere que factores séri-cos, por ejemplo inmunoglobulinas, citocinas o ambas, podrían inducir la sobrerregulación de la síntesis de los glucosaminoglucanos.4 Varios estudios demostraron que el suero de los pacientes con escleromixedema estimulado in vitro genera la proliferación de fibroblastos, lo que resulta en producción de ácido hialurónico y de prostaglandinas E.5
Las mucinosis cutáneas se dividen en dos gru-pos: específicas, denominadas “primarias”, que
se manifiestan con lesiones clínicas en las que el depósito de mucina es el signo histológico distintivo, y mucinosis “secundarias”, enferme-dades cutáneas en las que el depósito de mucina es un signo histológico adicional. Las mucinosis primarias pueden ser de origen inflamatorio, degenerativo, hamartomatoso o neoplásico. En las mucinosis secundarias, los depósitos se encuentran en las lesiones específicas de lupus eritematoso, dermatomiositis y esclerodermia o en el curso de otras dermatosis, por ejemplo, granuloma anular y enfermedad de Degos.6,7
Algunos autores sugieren que las paraproteínas actúan como autoanticuerpos que estimulan la proliferación de fibrobastos y la producción de mucina in vitro.
También se describen erupciones generalizadas tras la exposición intensa a UVB y casos aislados de escleromixedema asociado con neoplasias sólidas, pero esta relación tampoco está bien establecida.8
Manifestaciones clínicas
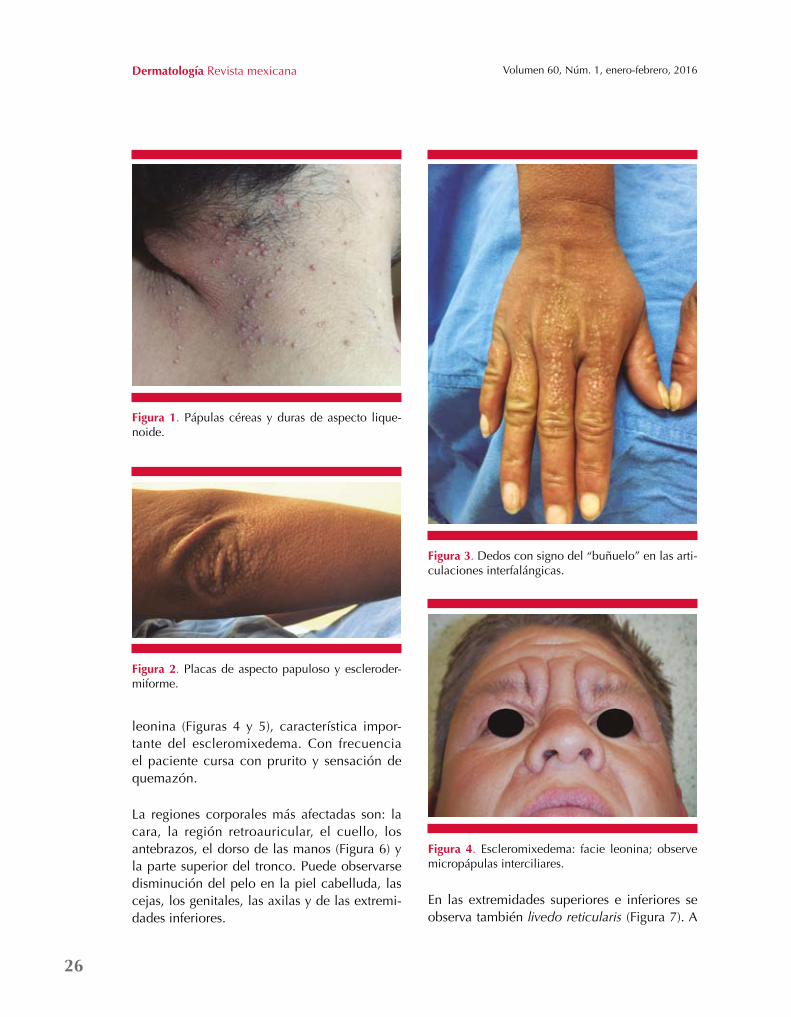
Los datos clínicos se distinguen por eritema difuso, pápulas liquenoides de consistencia dura y aspecto céreo que pueden estar aisladas o confluir en placas, miden entre 1 y 3 mm de diámetro, lo que a la palpación denota un estado esclerodermiforme (Figura 1) con disminución severa de la elasticidad cutánea (Figura 2), así como disminución importante de las amplitudes articulares, lo que puede generar que la piel tenga un aspecto brillante y edematoso.
Cuando hay engrosamiento en la piel de las articulaciones interfalángicas aparecerá el signo del “buñuelo”, descrito como una depresión central o fóvea cutánea, en la que la afectación difusa de los dedos también da un aspecto de esclerodactilia (Figura 3). La acumulación de mucina en la zona glabelar resulta en la facie
DermatologíaR e v i s t a m e x i c a n a
26
Volumen 60, Núm. 1, enero-febrero, 2016
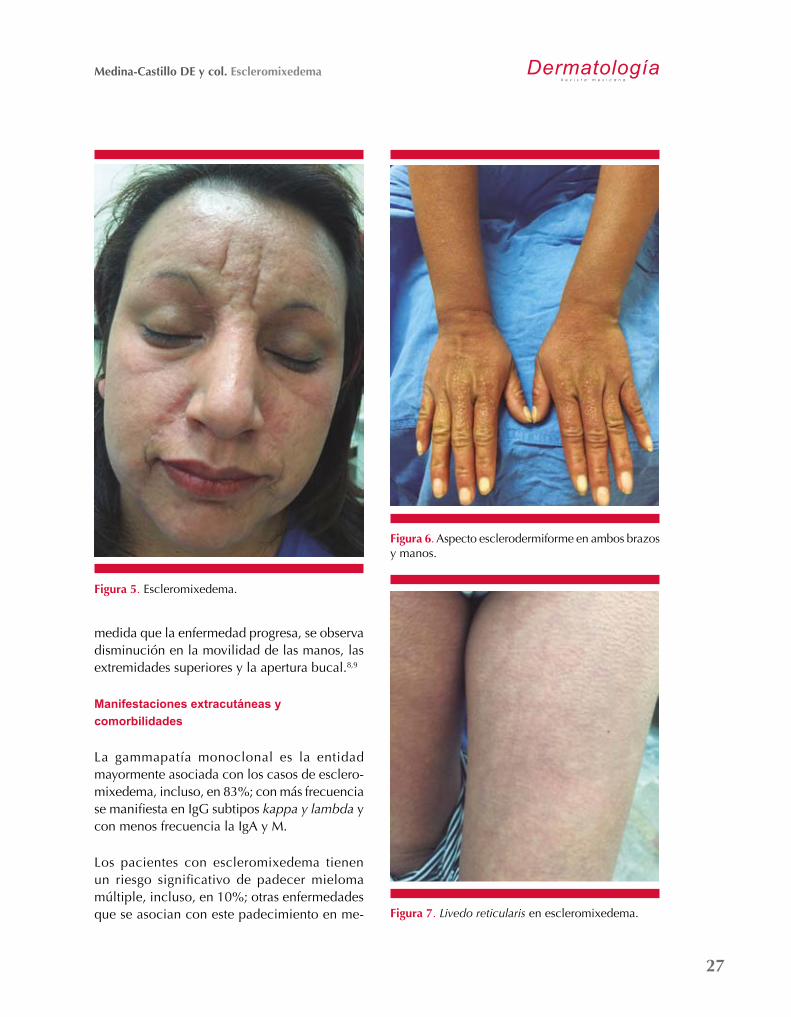
leonina (Figuras 4 y 5), característica impor-tante del escleromixedema. Con frecuencia el paciente cursa con prurito y sensación de quemazón.
La regiones corporales más afectadas son: la cara, la región retroauricular, el cuello, los antebrazos, el dorso de las manos (Figura 6) y la parte superior del tronco. Puede observarse disminución del pelo en la piel cabelluda, las cejas, los genitales, las axilas y de las extremi-dades inferiores.
En las extremidades superiores e inferiores se observa también livedo reticularis (Figura 7). A
Figura 1. Pápulas céreas y duras de aspecto lique-noide.
Figura 3. Dedos con signo del “buñuelo” en las arti-culaciones interfalángicas.
Figura 2. Placas de aspecto papuloso y escleroder-miforme.
Figura 4. Escleromixedema: facie leonina; observe micropápulas interciliares.
Dermatología Revista mexicana
27
Medina-Castillo DE y col. Escleromixedema
Figura 7. Livedo reticularis en escleromixedema.
Figura 5. Escleromixedema.
Figura 6. Aspecto esclerodermiforme en ambos brazos y manos.
medida que la enfermedad progresa, se observa disminución en la movilidad de las manos, las extremidades superiores y la apertura bucal.8,9
Manifestaciones extracutáneas y
comorbilidades
La gammapatía monoclonal es la entidad mayormente asociada con los casos de esclero-mixedema, incluso, en 83%; con más frecuencia se manifiesta en IgG subtipos kappa y lambda y con menos frecuencia la IgA y M.
Los pacientes con escleromixedema tienen un riesgo significativo de padecer mieloma múltiple, incluso, en 10%; otras enfermedades que se asocian con este padecimiento en me-
DermatologíaR e v i s t a m e x i c a n a

28
Volumen 60, Núm. 1, enero-febrero, 2016
nor porcentaje son la macroglobulinemia de Waldenström y los linfomas de Hodgkin y no Hodgkin.10
La disfagia es la manifestación extracutánea más común del escleromixedema.11 La alteración esofágica puede evaluarse por motilidad gas-trointestinal superior disminuida, regurgitación e incluso peristalsis abolida. La disfonía es el resultado de la afección de la laringe con dismi-nución de la movilidad de las cuerdas vocales.
El escleromixedema en el área reumatológica tiene importancia debido a su parecido con en-fermedades del tejido conjuntivo, especialmente la esclerosis sistémica progresiva, dermatomio-sitis o artritis reumatoide.
Las áreas de superposición clínica entre estas dos entidades incluyen la esclerodactilia, fenómeno de Raynaud, rigidez generalizada de la piel, disfagia, afectación pulmonar y microstomía o disminución de la apertura bucal. La biopsia de piel distingue entre la esclerosis sistémica pro-gresiva y escleromixedema, pero en ocasiones pueden estar mal organizados los depósitos de mucina y eso dificulta su diagnóstico, lo que puede causar un diagnóstico incorrecto.12 La miopatía también ocurre en el escleromixedema. En una serie de casos se informa debilidad proxi-mal en 5 de 19 pacientes, generalmente ocurre varios meses o años después del inicio de las lesiones en la piel. En términos histológicos, se trata de una miopatía vacuolar, también pueden observarse infiltrados inflamatorios linfocitarios intersticiales y causar confusión con polimiositis (más comunes en esta última).13
Respecto de la artropatía, aunque no es común, se describe una poliartritis erosiva que afecta las manos y se asemeja a la quiroartropatía diabética, sobre todo si las contracturas afectan los extremos proximales de las articulaciones interfalángicas.14
La afección neurológica más frecuente es el síndrome del túnel carpiano, se cree que se debe al depósito de mucopolisacáridos en el túnel carpiano o a un efecto directo en el nervio mediano.15 Otra manifestación neurológica es el “síndrome dermatoneuro”, constituido por pró-dromo gripal con exacerbación cutánea seguido por fiebre alta, convulsiones y coma. Los resul-tados de las pruebas de diagnóstico neurológico de rutina con frecuencia son negativos, incluida la resonancia magnética.16,17
En otros órganos, como el corazón y los pul-mones, la mucina se identifica en la adventicia alrededor de los vasos. Si hay afectación pulmo-nar, la mayoría de los pacientes comúnmente tienen disnea obstructiva o restrictiva.
Las manifestaciones cardiacas incluyen: enfer-medad cardiaca aterosclerótica, infarto agudo de miocardio, bloqueo cardiaco y derrame peri-cárdico. Otros autores reportan manifestaciones cardiacas por depósito de mucina en una válvula.
Algunos pacientes con enfermedad renal crónica muestran hallazgos dermatológicos similares a escleromixedema. Un estudio18 describió 15 pacientes en diálisis que posteriormente padecieron una enfermedad cutánea similar a escleromixedema con lesiones limitadas a las extremidades y el tronco. La dermopatía nefro-génica fibrosante o fibrosis sistémica nefrogénica tiene características dermatológicas similares a las del escleromixedema. Un mecanismo posible es que en los pacientes con insuficiencia renal, el suero contiene pequeños fragmentos que son sumamente angiogénicos e inflamatorios y éste sea un estímulo para que los fibroblastos produzcan fibras colágenas engrosadas; esto, a su vez, puede conducir a mayor producción de ácido hialurónico y su depósito en la dermis.18
Kucher comparó nueve pacientes con dermo-patía nefrogénica fibrosante con otros siete
Dermatología Revista mexicana

29
Medina-Castillo DE y col. Escleromixedema
Figura 8. Panorámica: se observa la dermis superficial con aspecto mixoide, dermis media con espacios entre las fibras de colágena.
pacientes con escleromixedema. Encontró que la expresión de procolágeno I estaba aumentada en pacientes con dermopatía nefrogénica fibrosante comparados con los sujetos con escleromixede-ma. En estudios con inmunohistoquímica CD34, el factor XIIIa, CD31, actina de músculo liso, CD68 y hierro coloidal se expresaron de manera similar en dermopatía nefrogénica fibrosante y escleromixedema.19
Las complicaciones del escleromixedema son numerosas, el inicio de estas manifestaciones puede ser agudo.
Las complicaciones neurológicas incluyen alteraciones de la conducta, encefalopatía, convulsiones, coma, psicosis, eventos cere-brovasculares, trastornos del sistema nervioso periférico. Otros hallazgos pueden ser pérdida de memoria, vértigo, problemas de la marcha y disartria. Si bien estos déficits neurológicos generalmente son reversibles, pueden persistir a pesar del tratamiento eficaz del padecimiento de la piel. Por ejemplo, Nieves y colaboradores informaron síntomas visuales persistentes en un paciente cuyas lesiones cutáneas habían desaparecido con melfalán y plasmaféresis.17 Kaufman sugirió que el depósito de mucina en el cerebro puede ser parcialmente responsable de los síntomas neurológicos de los pacientes; sin embargo, esto no está demostrado en las biopsias y los estudios patológicos realiza-dos.20
Diagnóstico
El diagnóstico de escleromixedema es clínico e histopatológico. Las manifestaciones clínicas son orientadoras de la enfermedad; la manifestación clínica típica es: erupción cutánea en la cabeza, el cuello y las extremidades superiores.21
Bolognia indicó el seguimiento de cuatro crite-rios diagnósticos:
1. Erupción generalizada papular y esclero-dermiforme.
2. Hallazgos microscópicos: depósitos de mucina, proliferación de fibroblastos y fibrosis.
3. Gammapatía monoclonal.
4. Ausencia de enfermedad tiroidea.
Entre los hallazgos histopatológicos está el depósito de mucina que suele distribuirse en la dermis superior y media, compuesto por glucosaminoglucanos, el ácido hialurónico es el principal; hay mayor cantidad de fibras de co-lágeno dispuestas a mayor distancia entre ellas, disminución cuantitativa de fibras elásticas y adelgazamiento de la epidermis (Figuras 8 y 9).
Los depósitos son compuestos por glucosami-noglucanos, principalmente ácido hialurónico, que se evidencia con la tinción azul alcián y hierro coloidal, entre otras (Figura 10). En las lesiones más incipientes es frecuente observar proliferación de fibroblastos estrellados o fusi-formes. Además, puede encontrarse aumento de las fibras de colágeno con mayor distancia entre ellas, disminución del número de fibras elásticas
DermatologíaR e v i s t a m e x i c a n a

30
Volumen 60, Núm. 1, enero-febrero, 2016
y adelgazamiento de la epidermis. También es habitual observar atrofia de los folículos pilosos e infiltrado perivascular superficial de tipo linfo-plasmocitario, así como esclerosis en las lesiones más evolucionadas.
Clasificación clínico-patológica
Según la clasificación de Montgomery y Un-derwood realizada en 1953 y posteriormente
revisada por Rongioletti,22 las mucinosis pueden clasificarse en:
1) Escleromixedema o liquen mixedematoso generalizado
2) Liquen mixedematoso localizado
a) mucinosis papular*
b) mucinosis papular persistente acral*
c) mucinosis papular de la infancia
d) mucinosis nodular
3) Formas atípicas o intermedias
* también se manifiestan en pacientes con el virus de la inmunodeficiencia humana.
Diagnóstico diferencial
En términos clínicos e histopatológicos, el escle-romixedema debe diferenciarse de enfermedades generales, como escleredema, esclerodermia sis-témica progresiva, fibrosis nefrogénica y fascitis eosinofílica (Cuadro 1).6,22
Tratamiento
Debido a la rareza de la enfermedad, no se ha podido determinar la superioridad de un trata-miento.20
Desai y James categorizaron los diversos trata-mientos en tres líneas terapéuticas (Cuadro 2).23
Aunque melfalán es un fármaco de primea línea en el tratamiento del escleromixedema, su prescripción está limitada debido a su toxi-cidad, aproximadamente 30% de las muertes secundarias a malignidad hematológica y complicaciones sépticas se deben a su adminis-tración.24 De 17 pacientes tratados con melfalán, en 12 hubo remisión parcial de los síntomas cutáneos; sin embargo, sólo 8 de éstos tuvieron mejoría temporal y 10 tuvieron complicaciones
Figura 10. Tinción especial para mucopolisacáridos, tinción positiva de hierro coloidal.
Figura 9. Dermis superficial con aspecto mixoide con infiltrado de linfocitos y fibroblastos (infiltrado característico del padecimiento).
Dermatología Revista mexicana

31
Medina-Castillo DE y col. Escleromixedema
mortales secundarias a la enfermedad y al tra-tamiento. Se observó que melfalán no produce efectos tóxicos a corto plazo; sin embargo, dos
pacientes padecieron leucemia mielomonocítica aguda a 9.5 y 10 años de tratamiento con este fármaco, por lo que su administración requiere
Cuadro 1. Diagnóstico diferencial clínico e histológico de escleromixedema6
Diagnóstico diferencial clínico e histológico
Escleromixedema Esclerodermiasistémica progresiva
Escleredema Fascitis eosinofílica Fibrosis nefrogénicasistémica
Morfología Pápulas céreas Esclerosis Induración einfiltracion
Induración,aspecto leñoso
Placas induradasy nodulares
Topografía Cara, cuello, extremidades superiores y
manos
Inicia en los dedos, afecta las manos, la cara y el cuello
Cuello, espalda y cara
Extremidades y tronco
Extremidades y tronco
Alteraciones sistémicas, Raynaud
Poco habitual Universal No Ocasional Ocasional
Capilaroscopia Normal Capilares engro-sados
Normal Normal Normal
Anticuerpos antinucleares
Poco común positivos
Positivos Negativos Poco común Negativos
Enfermedadneurológica
Crisis convulsivas, demencia, coma
Rara No Síndrome del túnel carpiano
Neuropatía periférica
Clínica asociada Gammapatía monoclonal
Gammapatía monoclonal, infecciones y diabetes mellitus
Morfea, neoplasias sólidas y
hematológicas
Enfermedad renal crónica,
trasplante renal y exposición a
gadolinio
Histopatología mucina Sí No No Sí Sí
Fibrosis Dermoepidérmica Dermo/hipodérmica Epidérmica/dérmica
Epidérmica/dérmica Epidérmica/dérmica
Fibroblastos einflamación
Sí, perivascular NoNo
NoSí, eosinófilos
NoSí, eosinófilos
SíNo
Cuadro 2. Líneas de tratamiento contra escleromixedema23
Primera línea Segunda línea Tercera línea
MelfalánCorticoesteroides sistémicosPlasmaféresis Inmunoglobulina IV
Isotretinoína Talidomida Corticoesteroides tópicos e intralesionalesAcitretina
Trasplante autólogo de células madre he-matopoyéticas
Bortezomib CiclofosfamidaCiclosporina Metotrexato Clorambucilo Interferón alfaFotoféresis extracorporalPsoralenos con radiación ultravioletaRadiación Vincristina, idarrubicina, dexametasona talidomida
DermatologíaR e v i s t a m e x i c a n a

32
Volumen 60, Núm. 1, enero-febrero, 2016
supervisión estrecha en la cuenta de plaquetas y leucocitos cada tres semanas, así como el ajuste o retiro de las dosis de melfalán.25
Los corticoesteroides sistémicos han tenido resultados variables, con reportes de respuesta clínica con la administración de dosis altas de dexametasona en tres pacientes y con curación de la paraproteinemia en uno.26,27 Con la admi-nistración de prednisona se reportó remisión parcial de los síntomas en tres casos: el primero con dosis de 0.3 mg/kg/día dividida en cuatro dosis al día durante una semana a dosis de re-ducción durante tres semanas, con mejoría en las primeras cuatro semanas de tratamiento.28 Otro caso tratado con prednisona oral tuvo alivio de los síntomas y permaneció en remisión durante 24 meses después del tratamiento.29 La predni-sona a dosis de 60 mg/día dio resultados a dos, seis y nueve meses cuando comenzó la dosis de reducción del esteroide; el paciente permaneció en remisión durante los 10 meses siguientes al tratamiento.30
El tratamiento con plasmaféresis fue curativo en dos casos en combinación con melfalán en uno, con remisión parcial de los síntomas neuroló-gicos31 y en combinación con ciclofosfamida y metilprednisolona en el otro; en otro de los casos, el tratamiento con plasmaféresis aceleró el progreso de la enfermedad.32
La inmunoglobulina intravenosa es otro trata-miento de elección contra escleromixedema. Blum y colaboradores evaluaron la eficacia de inmunoglobulina intravenosa como tratamiento inicial y encontraron que 8 de 10 pacientes recibieron tratamiento con inmunoglobulina intravenosa, con 100% de remisión completa o parcial con una dosis inicial de 2 g/kg dividida en dosis de 2-5 días durante seis meses.33 En otro estudio, después del tratamiento con inmuno-globulina intravenosa durante seis meses hubo alivio de los síntomas cutáneos del paciente; en el examen histológico de la piel no se encontró
incremento de los fibroblastos ni de los depósitos de mucina.34 Sin embargo, ambos estudios se limitaron a la vigilancia a corto plazo de la remi-sión de los síntomas de los pacientes. Asimismo, en un reporte de tres casos se confirmó la eficacia del tratamiento con inmunoglobulina intraveno-sa; en dos de ellos hubo remisión completa de la enfermedad a tres y dos años después de la sus-pensión del tratamiento; sin embargo, en el otro caso el tratamiento se mantuvo después de dos años de iniciado debido a que no hubo remisión después de completado el tratamiento inicial. En un estudio se administró inmunoglobulina intravenosa a 12 pacientes (6 como tratamiento inicial, 6 como segunda a quinta línea) a dosis de 2 g/kg. La inmunoglobulina intravenosa se combinó en una ocasión con esteroides orales y en otra con lenalidomida; cuatro pacientes (tres con inmunoglobulina intravenosa como monoterapia y una con lenalidomida) lograron la remisión clínica completa, la remisión parcial se observó en nueve pacientes, las manifestacio-nes extracutáneas se aliviaron en dos pacientes y hubo reducción en las concentraciones de paraproteína en un paciente. No hubo efectos secundarios que causaran la interrupción del tratamiento.35
La isotretinoína pertenece a la segunda línea de tratamiento; hay reportes de pacientes tratados con este fármaco a dosis de 60 mg diarios duran-te seis meses que tuvieron mejoría.36 Otro estudio menciona alivio de los síntomas cutáneos aso-ciados con miositis como síntoma extracutáneo con la administración de isotretinoína a dosis de 40 mg dos veces al día.37 Se observó remisión parcial de las manifestaciones cutáneas en un paciente tratado con isotretinoína durante 10 meses; sin embargo, la afección articular tuvo solamente disminución ligera.
El tratamiento con talidomida fue exitoso en tres pacientes con escleromixedema resistente en quienes hubo alivio parcial de las lesiones cutáneas y mejoría de la movilidad articular a
Dermatología Revista mexicana

33
Medina-Castillo DE y col. Escleromixedema
dos meses de iniciado el tratamiento con reduc-ción de las concentraciones de paraproteína a los cuatro meses.38 En otro estudio, un paciente tuvo mejoría considerable después de 10 meses de iniciado el régimen con talidomida, pero du-rante el mismo el paciente padeció neuropatía periférica leve secundaria al tratamiento.39 En una paciente con tratamiento inicial de 50 mg/día durante seis meses se observó notable alivio de los síntomas cutáneos; la dosis de talidomida se incrementó gradualmente durante un año hasta la administración de 400 mg/día, dosis que se mantuvo por cuatro meses con posterior reducción de la dosis a 300 mg/día durante dos meses hasta llegar a 200 mg/día; dosis que mantiene desde entonces hasta el momento del informe del caso. Los efectos teratogénicos de la talidomida son bien conocidos; sin embargo, otros efectos colaterales incluyen: sedación, náusea, cefalea, fiebre, neutropenia, prurito, exantema cutáneo, xerostomía, edema y neutro-penia que pueden limitar su administración; no obstante, la limitante de mayor importancia en su administración a largo plazo es la polineuropatía axonal sensitiva.40
En un estudio en el que se trató a siete pacientes con dosis altas de melfalán (180 mg/m2 vía intra-venosa) y trasplante autólogo de células madre hematopoyéticas se observó remisión completa en cinco pacientes y remisión parcial en los dos restantes. Se observó remisión completa de las manifestaciones gastrointestinales de seis pacientes; dos pacientes con manifestaciones pulmonares tuvieron remisión parcial de las mismas; seis pacientes tuvieron disminución de las concentraciones de paraproteína.41 En otro estudio, un paciente fue tratado con dosis altas de melfalán, busulfán y trasplante autólogo de células madre hematopoyéticas con remisión parcial de las lesiones cutáneas en los primeros días posteriores al trasplante y se observó re-misión completa de las lesiones cutáneas y las manifestaciones gastrointestinales, musculoes-
queléticas y neurológicas en los primeros seis meses posteriores al trasplante y la paraproteína era apenas detectable.42 Otro paciente con escle-romixedema grave que atentaba contra su vida se trató con quimioterapia (BCNU, etopósido, citarabina y melfalán) y trasplante autólogo de células madre hematopoyéticas con remisión completa seis meses posteriores al trasplante y permaneció asintomático después de tres años sin otro tratamiento, aunque las concentraciones de paraproteína aún son detectables. En cinco pacientes tratados con ciclofosfamida (105 g/m2/día durante dos días) y factor estimulante de colonias de granulocitos seguido de trasplante autólogo de células hematopoyéticas se observó remisión completa de las concentraciones de pa-raproteína en dos pacientes y remisión parcial en dos; la remisión completa de las lesiones cutáneas se observó 100 días posteriores al trasplante, tres pacientes tuvieron remisión completa de las ma-nifestaciones gastrointestinales y respiratorias; sin embargo, tres pacientes tuvieron recidiva cutánea a los 14, 37 y 5 meses posteriores al trasplante, dos de ellos tuvieron remisión con el tratamiento con inmunoglobulina intravenosa.43,44
Otros tratamientos comunicados son: PUVA, bortezomib y cloroquinas en informes de casos; sin embargo, aún no hay un tratamiento defini-tivo contra esta enfermedad.45
COMENTARIOS
El escleromixedema es una enfermedad poco frecuente que causa afectación sistémica con predominio en la edad adulta.
Es una mucinosis generalizada cuya fisiopato-logía se desconoce, pero debido a que afecta a otros órganos y sistemas, debe tratarse de manera multidisciplinaria.
Es importante que el dermatólogo y el médico en general, como apunta el Dr. Lifshitz, estén al ace-
DermatologíaR e v i s t a m e x i c a n a

34
Volumen 60, Núm. 1, enero-febrero, 2016
cho de comorbilidades, establezcan estrategias para jerarquizarlas y prevengan interacciones entre los tratamientos.46
REFERENCIAS
1. Zarragan EZF, Guevara CRM, López IMM, Quintanal RMJ. Escleromixedema: Informe de un caso clínico. Dermatolo-gía CMQ 2013;11:253-256.
2. Vidarte OG, Rivitti E. Escleromixedema: tratamiento con cloroquina. Dermatologia Peruana 1997;7:71-73.
3. Coto-Borges R. Escleromixedema. Rev Biomed 1998;9:46-47.
4. Gómez AS, Figueras NI, Jucglá S. Escleromixedema. Piel 2013;29:96-100.
5. Yaron M, Yaron I, Brenner S. Lichen myxedematosus (scleromyxedema) serum stimulates hyaluronic acid and prostaglandin production by human fibroblasts. Rheuma-tol 1985;12:171-175.
6. Boin F, Hummers LK. Scleroderma-like fibrosing disorders. Rheum Dis Clin North Am 2008;34:199-220.
7. Maruani A, Matos M, de Muret A, Lorette G. Sclé-romyxœdème. Presse Med 2006;35:1401-1402.
8. Serdar ZA, Yasar SP, Erfan GT, Gunes P. Generalized papular sclerodermoid eruption: scleromyxedema. Indian J Derma-tol Venerol Leprol 2010;76:592.
9. Le Moigne M, Mazereeuw-Hautir J, Astudillo L, D’Incan M, et al. Caractéristiques cliniques et évolutives du scléromyxœdème: étude rétrospective multicentrique. Annales Dermatologie Vénéréologie 2010;137:782-788.
10. Cokonis Geoegakis CD, Falasca G, Geoargakis A, Heymann WR. Scleromyxedema. Clinic Dermatol 2006;24:493-497.
11. Pomman JJ, Rudner EJ. Scleromyxedema revised. Int J Dermatol 2003;42:31-35.
12. Rongioletti F. Lichen myxedematosus (papular mucinosis): New concepts and perspectives for an old disease. Semin Cutan Med Surg 2006;25:100-104.
13. Launay D, Hatron PY, Delaporte E, Hachulla E, et al. Scle-romyxedema (lichen myxedematosus) associated with dermatomyositis. Br J Dermatol 2001;144:359-362.
14. Gabriel SE, Perry HO, Oleson GB, Bowles CA. Scleromyxe-dema: a scleroderma-like disorder with systemic manifes-tations. Medicine 1988;67:58-65.
15. Breguen JR, Dobbs MR, Terhune MH, Maragos WF. The neurologic complications of scleromyxedema. Medicine 2001;80:313-319.
16. River Y, Levy I, Gilead L, Orbach H, Almong Y. Fever, con-vulsions and coma in scleromyxedema: A “dermato-neuro syndrome”. Neurology 1996;46:1778-1779.
17. Nieves DS, Bondi TT, Wallmark J, Raps EC, Seycora JT. Scleromyxedema: Successful treatment of cutaneous and neurologic symptoms. Cutis 2000;65:89-92.
18. Neudecker BA, Stern R, Mark LA, Steinberg S. Scleromyxe-dema-like lesions of patients in renal failure contain hyalu-ronan: a possible pathophysiological mechanism. J Cutan Pathol 2005;32:612-615.
19. Kucher C, Xu X, Pasha T, Elenitsas R. Histopathologic comparison of nephrogenic fibrosing dermopathy and scleromyxedema. J Cutan Pathol 2005;32:484-490.
20. Kaufman D, Truhan AP, Roegnick HH Jr. Scleromyxedema: Systemic manifestations and cosmetic improvement from dermabrasion. Cutis 1987;39:321-324.
21. Dinneen AM, Dicken CH. Scleromyxedema. J Am Acad Dermatol 1995;33:37-43.
22. Rongioletti F, Merlo G, Cinotti E, Fausti V, et al. Scleromyxe-dema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol 2013;69:66-72.
23. Desai A, James W. Lichen myxedematosus. In: Lebwohl M, Heymann W, Berth-Jones J, Coulson I, editors. Treatments of skin disease: comprehensive therapeutic strategies. 2nd ed. Philadelphia: Elsevier Science, 2006;343-344.
24. Helm F, Helm TN: Iatrogenic mylemonocytic leukemia following melphalan treatment of scleromyxedema. Cutis 1987;39:219-223.
25. Harris RB, Perry HO, Kyle RA, Winklemann RK. Treatment of scleromyxedema with melphalan. Arch Dermatol 1979;115:295-299.
26. Kreuter A, Altmeyer P: High-dose dexamethasone in scleromyxedema: report of 2 additional cases. J Am Acad Dermatol 2005;53:739-740.
27. Horn KB, Horn MA, Swan J, Singhal S, Guitart J. A complete and durable clinical response to high-dose dexamethasone in a patient with scleromyxedema. J Am Acad Dermatol 2004;5:S120-S123.
28. Lin Yc, Wang HC, Shen Jl. Scleromyxedema: An experience using treatment with systemic corticosteroid and review of the published work. J Dermatol 2006;33:207-210.
29. Rayson D, Lust JA, Duncan A, Su WP. Scleromyxedema: a complete response to prednisone. Mayo Clin Proc 1999;74:481-484.
30. Trinidade Neto PBD, Sales ADO, Silva ACDO, Nunes JC. Scleromyxedema: a case treated with oral prednisone. An Bras Dermatol 2006;81:55-58.
31. Keong CH, Asaka Y, Fukuro S, Miyamoto C, et al. Successful treatment of scleromyxedema with plasmapheresis and immunosuppression. J Am Dermatol 1990;22:842-844.
32. Westheim AI, Lookingbill DP. Plasmapheresis in a patient with scleromyxedema. Arch Dermatol 1987;123:786-789.
33. Blum M, Wigley FM, Hummers LK. Scleromyxedema: a case series highlighting long-term outcomes of treatment with intravenous immunoglobulin (IV IG). Medicine 2008;87:10-20.
34. Caudill L, Howell E. Scleromyxedema: a case clinically and histologically responsive to intravenous immunoglobulin. J Clin Aesthet Dermatol 2014;7:45-47.
Dermatología Revista mexicana

35
Medina-Castillo DE y col. Escleromixedema
35. Righi A, Schiavon F, Jablonska S, Doria A, et al. Intravenous immunoglobulins control scleromyxedema. Ann Rheum Dis 2002;61:59-61.
36. Hisler BM, Savoy LB, Hashimoto K. Improvement of scle-romyxedema associated with isotretinoid therapy. J Am Acad Dermatol 1991;24:854-857.
37. Lominska-Lasota K, Rosen-Uzelac G, Reichi W, Bauer R. Scleromyxedema therapy with isotretinoid. Z Hautkr 1988;63:137-138, 141.
38. Sansbury JC, Cocuroccia B, Jorizzo Jl, Gubinelli, et al. Treatment of recalcitrant slceromyxedema with thalido-mide in 3 patients. J Am Acad Dermatol 2004;51:126-131.
39. Jacob SE, Fien S, Kerdel FA. Scleromyxedema, a positive effect with thalidomide. Dermatology 2006;213:150-152.
40. Martins A, Paiva-Lopes MJ, Tavares-Belo R, Rodrigues JC. Scleromyxedema—thalidomide therapy. J Eur Acad Der-matol Venereol 2008;22:622-624.
41. Donato ML, Feasel AM, Weber DM, Prieto VG, et al. Scle-romyxedema: role of high-dose melphalan with autologous stem cell transplantation. Blood 2006;107:463-466.
42. Cheng T, Gnanakumar V, Hegedus C, Stewart DA. Complete and durable remission in a patient with life threatening scleromyxedema treated with high-dose melphalan and BU with auto-SCT. Bone Marrow Transplantation 2008;42:215-217.
43. Illa I, De la Torre C, Rojas-García R, Altes A, et al. Steady remission of scleromyxedema 3 years after autologous stem cell transplantation: an in vivo and in vitro study. Blood 2006;108:773-774.
44. Lacy MQ, Hogan WJ, Gertz MA, et al. Successful treatment of scleromyxedema with autologous peripheral blood stem cell transplantation. Arch Dermatol 2005;141:1277-1282.
45. Salas-Alanis JC, Martinez-Jaramillo B, Gomez- Flores M, Ocampo Candiani J. Scleromyxedema, a therapeutic di-lemma. Indian J Dermatol 2015;2:215.
46. Lifshitz A. Sobre la asociación de enfermedades. Gac Méd Méx 2015;151:149.
DermatologíaR e v i s t a m e x i c a n a
EVALUACIÓN
1. ¿Cómo se define el escleromixedema?a) mucinosis localizada que afecta las
extremidades inferioresb) mucinosis atípica con esclerodermia
sistémica progresivac) mucinosis generalizada con esclerosis
múltipled) mucinosis generalizada escleroder-
moide con gammapatía monoclonale) todas las anteriores
2. ¿A qué grupo de pacientes afecta el escle-romixedema?a) mujeres en edad pediátricab) hombres en senectudc) adultos sin distinción de género d) mujeres embarazadase) ninguna de las anteriores
3. Mencione tres enfermedades relacionadas con el subtipo de mucinosis secundarias lu-pus eritematoso sistémico, esclerodermia sistémica progresiva, dermatomiositis
a) esclerosis múltiple, esclerosis lateral, miastenia gravis
b) artritis reumatoide, fibromialgia, ami-loidosis
c) sólo a y cd) todas las anteriores
4. De las hipótesis acerca de la fisiopatología del escleromixedema, responda falso o ver-dadero a las siguientes aseveraciones:a) disminución de la vigilancia inmuno-
lógica F V b) mayor producción de glucosaminogli-
canos F Vc) proliferación de fibroblastos F Vd) respuesta anómala de linfocitos F V
5. ¿Cuál es el aspecto clínico de un paciente con escleromixedema?a) facies leonina, vasculitis de las extremi-
dades inferiores y nódulos generaliza-dos

36
Volumen 60, Núm. 1, enero-febrero, 2016Dermatología Revista mexicana
b) facies acartonada “de pajarito”, escle-rosis cutánea, calcinosis, disminución de la amplitud articular
c) facies leonina, pápulas liquenoides y collarete escamoso periférico
d) facies pálida, macroglosia, piel elásti-ca e hiperlaxitud articular
c) facies leonina, pápulas generalizadas duras y céreas, aspecto escleroder-moide de la piel, disminución de la amplitud articular
6. ¿Cuál es la enfermedad asociada con más frecuencia en el escleromixedema?a) linfomas cutáneos de células T y linfo-
mas cutáneos de células Bb) esclerosis lateral amiotrófica c) vasculitis linfocítica agudad) amiloidosis sistémicae) gammapatía monoclonal Ig G subtipo
kappa-lambda
7. Mencione la afección extracutánea o sisté-mica mas común en escleromixedemaa) cardiacab) pulmonarc) esofágicad) neurológicae) muscular
8. ¿Cuál es la afección neurológica más
común en escleromixedema?a) síndrome “dermatoneuro”b) síndrome del túnel carpianoc) síndrome CREST
d) síndrome de Sjögrene) síndrome de Cushing
9. Mencione la tríada histológica característi-ca del escleromixedemaa) acantosis, papilomatosis y microabs-
cesos de neutrófilosb) atrofia, fibras colágenas engrosadas e
infiltrado perivascular linfocíticoc) acantosis, infiltrado lliquenoide en
banda, ausencia de granulosad) depósitos de mucina, proliferación de
fibroblastos y fibrosise) todas las anteriores
10. Es el medicamento de primera línea en el tratamiento del escleromixedemaa) melfalánb) acitretinac) isotretinoínad) adapalenoe) metoxaleno
11. Mencione otros medicamentos que se pres-criben en el tratamiento del escleromixede-ma a) inmunoglobulina IV, plasmaféresis, ta-
lidomida, esteroides sistémicosb) rituximab, adalimumab, infliximab,
etanerceptc) bortezomib, anakrina, leflunamidad) esteroides sistémicos, esteroides tópi-
cos, cloroquinas, talidomida, rituxi-mab
e) todos los anteriores
El Consejo Mexicano de Dermatología, A.C. otorgará dos puntos con validez para la recertificación a quienes envíen correctamente contestadas las evaluaciones que aparecen en cada número de Dermatología Revista Mexicana.
El lector deberá enviar todas las evaluaciones de 2016, una por una o todas juntas, a la siguiente dirección:
Dermatología Revista MexicanaJosé Martí 55, colonia Escandón, CP 11800, México, DF.
Fecha límite de recepción de evaluaciones: 31 de enero de 2017.





























