Embed Size (px)
Citation preview

EVALUACIÓN DE LAS CARACTERÍSTICAS FÍSICAS DE VIDRIOS DEL SISTEMA GE-SB-TE, EN VOLUMEN Y COMO PELÍCULAS, CON EL OBJETO
DE SU APLICACIÓN A MEMORIAS DE CAMBIO DE FASE
TESIS DE INGENIERÍA INFORMÁTICA
FACULTAD DE INGENIERÍA
UNIVERSIDAD DE BUENOS AIRES
JAVIER ALEJANDRO ROCCA
DIRECTORA: DRA. BIBIANA ARCONDO
CO-DIRECTOR: DR. MARCELO FONTANA
DICIEMBRE DE 2012

2

Resumen
La tendencia actual de las aplicaciones de nivel empresarial a ser centradas en los datos (en vez de centradas en el cómputo) demanda la existencia de una memoria de estado sólido no volátil de alta densidad y rendimiento, de bajo costo, con tiempos de acceso entre la memoria principal y la secundaria. Por otra parte, el límite de integración alcanzado para las memorias no volátiles flash presenta la oportunidad para buscar tecnologías alternativas, que deberán ser superiores en cuanto a escalabilidad, costo por bit y rendimiento. A partir de ambas necesidades, se plantea el objetivo de desarrollar un tipo de dispositivo con las características mencionadas (storage-class memory), aplicable a un amplio rango de escenarios, entre sistemas portátiles y servidores de alto rendimiento. Entre las tecnologías candidatas a desplazar a las memorias flash de su posición dominante y ubicarse como una nueva capa en la jerarquía de memorias, la memoria de cambio de fase se postula como la más madura.Este trabajo tiene como objetivo evaluar la aplicación de aleaciones del sistema Ge-Sb-Te como materiales de cambio de fase. Los materiales son sintetizados y caracterizados estructural, térmica y eléctricamente. Con los resultados obtenidos, se desarrolla el modelado de una celda de memoria, por el método de los elementos finitos.
3

Índice de contenido1 Introducción a los sistemas de memoria............................................................................................8
1.1 Características principales y evolución histórica.......................................................................81.2 Limitaciones de las tecnologías actuales...................................................................................91.3 Tecnologías candidatas a la aplicación como memorias no volátiles de alto rendimiento......121.4 Estructura del presente trabajo.................................................................................................161.5 Referencias...............................................................................................................................16
2 Técnicas experimentales y desarrollo experimental para la síntesis y caracterización de los materiales estudiados..........................................................................................................................19
2.1 Síntesis de materiales amorfos.................................................................................................192.1.1 Melt Quenching................................................................................................................202.1.2 Melt Spinning...................................................................................................................212.1.3 Deposición física en fase vapor (Physical Vapour Deposition, PVD).............................22
2.2 Caracterización de los materiales obtenidos............................................................................262.2.1 Difracción de rayos X......................................................................................................262.2.2 Técnicas calorimétricas para el estudio de la transformación de cristalización...............32
2.2.2.1 Procesamiento de la señal calorimétrica..................................................................342.2.2.2 Características del equipamiento usado...................................................................37
2.2.3 Microscopía de fuerza atómica (AFM)............................................................................372.3 Referencias...............................................................................................................................41
3 Estabilidad térmica y cinética de cristalización en los sistemas Ge-Te y Ge-Sb-Te.......................433.1 Características estructurales de las aleaciones estudiadas as-quenched..................................433.2 Procesamiento de medidas en régimen de calentamiento continuo e isotérmicas...................44
3.2.1 Caracterización de los procesos de transformación ........................................................443.2.2 Identificación de los productos de cristalización.............................................................453.2.3 Determinación de parámetros calorimétricos...................................................................47
3.3 Modelización de la cinética de cristalización..........................................................................513.3.1 Modelo elegido................................................................................................................513.3.2 Determinación de diagramas Transformación-velocidad de calentamiento-Temperatura (THrT) y Transformación-Tiempo-Temperatura (TTT)...........................................................54
3.4 Análisis de la capacidad de amorfización para las composiciones estudiadas........................563.5 Referencias...............................................................................................................................58
4 Conductividad eléctrica en películas delgadas del sistema Ge-Sb-Te.............................................604.1 Características estructurales de las películas delgadas depositadas por ablación láser...........604.2 Mediciones a temperatura ambiente........................................................................................60
4.2.1 Variación de la resistencia a temperatura ambiente mediante tratamientos térmicos......604.2.2 Medición de la resistividad a temperatura ambiente con AFM........................................61
4.3 Mediciones durante el tratamiento térmico.............................................................................644.3.1 Dependencia de la resistencia con la temperatura............................................................644.3.2 Dependencia de la conductividad con la temperatura......................................................66
4.4 Estudio de la cristalización por DRX......................................................................................674.4.1 Identificación de los productos de cristalización.............................................................674.4.2 Cálculo del tamaño de grano............................................................................................68
4.5 Referencias...............................................................................................................................725 Simulación de celdas de memoria no volátil de cambio de fase en el sistema Ge-Sb-Te...............73
5.1 Consideraciones previas..........................................................................................................735.2 Descripción física del modelo..................................................................................................735.3 Aproximación por el Método de los Elementos Finitos..........................................................755.4 Resultados de las simulaciones................................................................................................79
4

5.5 Referencias...............................................................................................................................866 Conclusiones generales...................................................................................................................887 Apéndice I........................................................................................................................................89
Índice de figurasFigura 1.1..............................................................................................................................................9Figura 1.2............................................................................................................................................11Figura 1.3............................................................................................................................................11Figura 1.4............................................................................................................................................14Figura 1.5............................................................................................................................................15Figura 2.1............................................................................................................................................20Figura 2.2............................................................................................................................................21Figura 2.3............................................................................................................................................22Figura 2.4............................................................................................................................................24Figura 2.5............................................................................................................................................26Figura 2.6............................................................................................................................................27Figura 2.7............................................................................................................................................29Figura 2.8............................................................................................................................................30Figura 2.9............................................................................................................................................32Figura 2.10..........................................................................................................................................33Figura 2.11..........................................................................................................................................34Figura 2.12..........................................................................................................................................38Figura 2.13..........................................................................................................................................39Figura 2.14..........................................................................................................................................40Figura 3.1............................................................................................................................................43Figura 3.2............................................................................................................................................44Figura 3.3............................................................................................................................................45Figura 3.4............................................................................................................................................47Figura 3.5............................................................................................................................................48Figura 3.6............................................................................................................................................48Figura 3.7............................................................................................................................................50Figura 3.8............................................................................................................................................51Figura 3.9............................................................................................................................................56Figura 4.1............................................................................................................................................60Figura 4.2............................................................................................................................................62Figura 4.3............................................................................................................................................63Figura 4.4............................................................................................................................................64Figura 4.5............................................................................................................................................66Figura 4.6............................................................................................................................................68Figura 4.7............................................................................................................................................69Figura 4.8............................................................................................................................................69Figura 4.9............................................................................................................................................70Figura 4.10..........................................................................................................................................70Figura 5.1............................................................................................................................................74Figura 5.2............................................................................................................................................80Figura 5.3............................................................................................................................................81Figura 5.4............................................................................................................................................82Figura 5.5............................................................................................................................................83Figura 5.6............................................................................................................................................84Figura 5.7............................................................................................................................................85
5

Índice de tablasTabla 1.1.............................................................................................................................................12Tabla 3.1.............................................................................................................................................49Tabla 3.2.............................................................................................................................................50Tabla 3.3.............................................................................................................................................55Tabla 4.1.............................................................................................................................................61Tabla 4.2.............................................................................................................................................64Tabla 4.3.............................................................................................................................................66Tabla 4.4.............................................................................................................................................67Tabla 4.5.............................................................................................................................................71Tabla 5.1.............................................................................................................................................79Tabla 5.2.............................................................................................................................................79
Índice de ecuacionesEc 1.1..................................................................................................................................................10Ec 2.1..................................................................................................................................................27Ec 2.2..................................................................................................................................................30Ec 2.3..................................................................................................................................................30Ec 2.4..................................................................................................................................................31Ec 2.5..................................................................................................................................................34Ec 2.6..................................................................................................................................................35Ec 2.7..................................................................................................................................................35Ec 2.8..................................................................................................................................................35Ec 2.9..................................................................................................................................................35Ec 2.10................................................................................................................................................36Ec 2.11................................................................................................................................................36Ec 2.12................................................................................................................................................36Ec 2.13................................................................................................................................................36Ec 2.14................................................................................................................................................36Ec 2.15................................................................................................................................................36Ec 2.16................................................................................................................................................36Ec 2.17................................................................................................................................................36Ec 2.18................................................................................................................................................36Ec 3.1..................................................................................................................................................49Ec 3.2..................................................................................................................................................52Ec 3.3..................................................................................................................................................52Ec 3.4..................................................................................................................................................52Ec 3.5..................................................................................................................................................52Ec 3.6..................................................................................................................................................52Ec 3.7..................................................................................................................................................52Ec 3.8..................................................................................................................................................53Ec 3.9..................................................................................................................................................53Ec 3.10................................................................................................................................................53Ec 3.11................................................................................................................................................53Ec 3.12................................................................................................................................................53Ec 3.13................................................................................................................................................57Ec 3.14................................................................................................................................................57Ec 4.1..................................................................................................................................................61Ec 4.2..................................................................................................................................................61
6

Ec 4.3..................................................................................................................................................62Ec 4.4..................................................................................................................................................62Ec 4.5..................................................................................................................................................62Ec 4.6..................................................................................................................................................63Ec 4.7..................................................................................................................................................63Ec 4.8..................................................................................................................................................63Ec 4.9..................................................................................................................................................65Ec 4.10................................................................................................................................................65Ec 4.11................................................................................................................................................65Ec 4.12................................................................................................................................................65Ec 4.13................................................................................................................................................66Ec 4.14................................................................................................................................................68Ec 5.1..................................................................................................................................................74Ec 5.2..................................................................................................................................................74Ec 5.3..................................................................................................................................................74Ec 5.4..................................................................................................................................................75Ec 5.5..................................................................................................................................................75Ec 5.6..................................................................................................................................................75Ec 5.7..................................................................................................................................................75Ec 5.8..................................................................................................................................................75Ec 5.9..................................................................................................................................................76Ec 5.10................................................................................................................................................76Ec 5.11................................................................................................................................................76Ec 5.12................................................................................................................................................76Ec 5.13................................................................................................................................................76Ec 5.14................................................................................................................................................76Ec 5.15................................................................................................................................................76Ec 5.16................................................................................................................................................76Ec 5.17................................................................................................................................................76Ec 5.18................................................................................................................................................76Ec 5.19................................................................................................................................................77Ec 5.20................................................................................................................................................77Ec 5.21................................................................................................................................................77Ec 5.22................................................................................................................................................77Ec 5.23................................................................................................................................................77Ec 5.24................................................................................................................................................77Ec 5.25................................................................................................................................................77Ec 5.26................................................................................................................................................78Ec 5.27................................................................................................................................................78Ec 5.28................................................................................................................................................78Ec 5.29................................................................................................................................................78Ec 5.30................................................................................................................................................78Ec 5.31................................................................................................................................................78Ec 5.32................................................................................................................................................78
7

1 Introducción a los sistemas de memoria
1.1 Características principales y evolución histórica
Las computadoras digitales convencionales desarrolladas en nuestros días siguen la estructura y el funcionamiento generales planteadas por el modelo presentado por John von Neumann en 1945, para el diseño de la EDVAC (Electronic Discrete Variable Computer) [von Neumann, 1993]. La principal novedad que propuso la máquina de von Neumann es el programa almacenado: el programa se representa en una forma tal que puede guardarse en memoria junto con los datos. Puede crearse o modificarse cargando los valores adecuados en una zona de memoria; de allí el computador consigue las instrucciones a ejecutar. El modelo consta de cinco componentes:
• Una memoria principal que almacena datos e instrucciones.• Una unidad aritmético-lógica capaz de operar sobre los datos.• Una unidad de control que interpreta las instrucciones en memoria y provoca su ejecución.• Una unidad de entrada y salida.
Las restricciones al diseño de la memoria de un computador están dadas para la capacidad, la velocidad de acceso y el costo, según las siguientes afirmaciones:
• Alcanzando cierta capacidad, probablemente se desarrollarán aplicaciones que la utilicen.• La velocidad de acceso debe ser tal que no deje al procesador a la espera de instrucciones u
operandos.• El costo debe ser razonable respecto de los otros componentes.
Existe una relación de compromiso entre las tres características, en un momento dado, para el espectro de tecnologías posibles en un sistema de memorias [Stallings, 2006]:
• A mayor velocidad de acceso, mayor será el costo por bit.• A mayor capacidad, menor será el costo por bit.• A mayor capacidad, menor será la velocidad de acceso.
Si bien la capacidad de las memorias ha aumentado a igual ritmo que la velocidad de los procesadores manteniendo costos constantes, no sucede lo mismo con su velocidad de acceso. Si se pretendiera construir un sistema con un solo tipo de memorias, el uso de la tecnología más rápida disponible para la capacidad que se requiere resultaría prohibitivo en costos, mientras las tecnologías de bajo costo implicarían un rendimiento inaceptable. La organización jerárquica de distintos tipos de memorias (ver figura 1.1, de [Stallings, 2006]) busca acortar la brecha de velocidades y exhibir al procesador el comportamiento de una memoria única, rápida y grande.A medida que se desciende por los niveles de la jerarquía, se encuentra: disminución de costo por bit, aumento en la capacidad y aumento en el tiempo de acceso. Para que esta organización resulte exitosa también debería sumarse la disminución de la frecuencia de acceso del procesador al nivel de memoria en cuestión. Esta condición resulta válida por la propiedad conocida como principio de localidad de las referencias. Al ejecutar un programa, existe una tendencia a la iteración sobre una sección tanto del código como de los datos; es decir, cuando se hace referencia a una ubicación de memoria, muy probablemente se repita la referencia en un plazo reducido, lo que se conoce como localidad temporal. La mayor probabilidad de acceder a ubicaciones cercanas de memoria ocurre debido a la tendencia de almacenar datos relacionados en zonas contiguas y es conocida como localidad espacial [Murdocca, 2002].En un primer grupo de la jerarquía, se encuentran los registros del procesador, la memoria caché y la memoria principal, habitualmente volátiles y de tecnología semiconductora. Los registros del
8

procesador, con una velocidad similar a la unidad de proceso y alto consumo energético, se ubican en nivel más alto, como el tipo de memoria más rápida, más costosa y de menor capacidad. El sistema de memoria caché consiste en una memoria asociativa que se ubica entre la memoria principal, a fin de salvar la brecha entre su velocidad de acceso y la velocidad del procesador. Típicamente los tiempos de acceso crecen con un factor 10 en estos tres niveles.El segundo grupo, de memoria secundaria, corresponde al almacenamiento persistente de datos (no volátil) en discos magnéticos (conocidos como discos rígidos) y ópticos (variantes de Compact Disc y Digital Versatile Disc), donde los tiempos de acceso crecen con un factor 100.000 respecto del nivel anterior. Técnicas de buffer de disco (como buffer caché) se utilizan para mejorar las velocidades de acceso.Por último, el grupo de almacenamiento fuera de línea se utiliza para datos persistentes y masivos que se escriben una sola vez y vuelven a accederse con baja frecuencia. Se caracteriza por un bajo costo tanto por bit almacenado, como en términos de consumo de energía.
Figura 1.1: Jerarquía de memorias [Stallings, 2006].
1.2 Limitaciones de las tecnologías actuales
En su publicación de 1965 [Moore, 1998], Gordon Moore predecía, a partir de la tendencia observada, que el número de dispositivos integrado en una oblea semiconductora de área fija (y, por consiguiente, a costo constante) se duplicaría cada 12 meses. Esta afirmación, posteriormente denominada Ley de Moore y convertida en la fuerza impulsora de la industria electrónica, se verificaría en los primeros 10 años y luego se ajustaría a períodos de 18 a 24 meses, manteniendo su
9

validez, como en el caso de la integración de las memorias RAM dinámicas y los microprococesadores, entre 1972 y 1997 [Schaller, 1997]. En general, la miniaturización optimiza la operación en otros aspectos: los transitores funcionan más rápido y consumen menos potencia; la integración de funciones más complejas mejora la confiabilidad de los sistemas [Moore, 2003].El trabajo de Dennard [Dennard, 1974] postula una ley complementaria a la de Moore, que describe más formalmente la relación de los parámetros de diseño de los dispositivos para reducir la escala, a la vez que se mejora el rendimiento general: aumenta la velocidad de conmutación y disminuye la potencia disipada [Bohr, 2007]. Plantea el cambio de escala a campo eléctrico constante, como resultado de la reducción conjunta de la tensión y los tamaños característicos (feature sizes), que garantiza mantener constante la densidad de potencia. Considerando la densidad de potencia dinámica P/A de la ecuación 1.1 (donde Ceff es la capacidad debida a la conmutación por ciclo, f, la frecuencia del reloj y A, el área), esto es posible tomando un factor de escala α<1 para Ceff y Vdd,
1α para f, y α2 para A [Azizi, 2010].
Ec 1.1
Históricamente, ambas leyes se verificaron en simultáneo. Cuando la miniaturización deja de implicar mejoras en el rendimiento, dejan de justificarse los esfuerzos en este sentido, pues también se discontinúa el beneficio económico que predice la ley de Moore [Burr, 2008].La propuesta de Dennard encuentra su límite por debajo del tamaño característico de 130 nm, donde las corrientes de fuga comienzan a ser una apreciable fuente de disipación de potencia (estática), creciendo en forma exponencial con el decrecimiento de la tensión umbral de los transistores Vth
(dependiente del espesor del sustrato semiconductor). En consecuencia, la tensión de operación Vdd
(en relación lineal con Vth) no se ha reducido significativamente en las generaciones de procesadores desde 130 hasta 32 nm. Esta restricción imposibilita mantener constante la densidad de potencia:
cuando no es posible escalar Vdd y se escala f con un factor entre 1 y1α , la densidad de potencia
se incrementa entre 1α y
1
α2
. La publicación International Technology Roadmap for
Semiconductors de 2001 [ITRS-PIDS, 2001] esperaba un valor de 0,4 V para Vdd en dispositivos de alto rendimiento en 2016, mientras en la edición de 2011 [ITRS-PIDS, 2011] es de 0,57 V en 2026.A pesar de la tendencia a la reducción de la tensión de operación (ver figura 1.2), se ha observado, en el mismo período, un sostenido aumento de las potencias disipadas, tanto activa como de fuga (ver figura 1.3). Éstas, por los motivos ya mencionados, se encuentran muy próximas hacia el final del período. Cabe destacar que, desde principios del siglo XXI, se ha alcanzado el límite de potencia en que puede refrigerarse por convección forzada de aire.Las reglas de Dennard para el escalado a densidad de potencia constante parten del supuesto de reducir la escala manteniendo un diseño fijo. Sin embargo, esa no es la única forma en que ha avanzado la industria. Para lograr funcionalidad y complejidad mayores, los diseños han requerido aumentar el área del circuito integrado (die size) o la densidad de potencia. Por lo tanto, ésta resulta otra causa del aumento de la potencia consumida por los procesadores.Por ambas causas, la potencia consumida ha cobrado mayor importancia como restricción para el diseño de microprocesadores, no sólo en soluciones sobre ambientes portátiles, sino también para sistemas de escritorio y hasta para servidores con requerimientos de alto rendimiento. Se han desarrollado técnicas que adaptan dinámicamente la tensión de alimentación o la frecuencia del reloj, según la carga de trabajo generada por las aplicaciones en el procesador. Sin embargo, la potencia disipada debida a las corrientes de fuga resulta comparable con la potencia dinámica y
10
PA=C eff V dd
2 fA
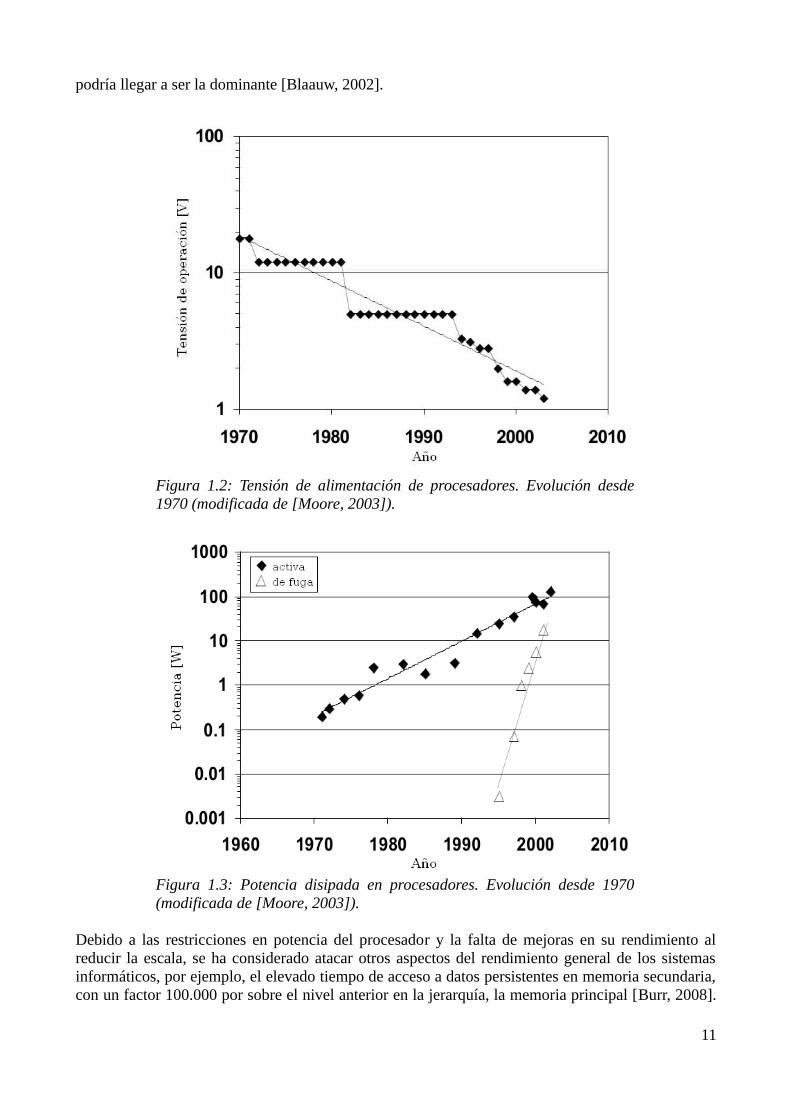
podría llegar a ser la dominante [Blaauw, 2002].
Figura 1.2: Tensión de alimentación de procesadores. Evolución desde 1970 (modificada de [Moore, 2003]).
Debido a las restricciones en potencia del procesador y la falta de mejoras en su rendimiento al reducir la escala, se ha considerado atacar otros aspectos del rendimiento general de los sistemas informáticos, por ejemplo, el elevado tiempo de acceso a datos persistentes en memoria secundaria, con un factor 100.000 por sobre el nivel anterior en la jerarquía, la memoria principal [Burr, 2008].
11
Figura 1.3: Potencia disipada en procesadores. Evolución desde 1970 (modificada de [Moore, 2003]).

El aumento de 35 millones de veces en la densidad superficial de los discos rígidos magnéticos, con la consiguiente reducción de costo por bit almacenado, entre 1956 y 2003 [Grochowski, 2003], alentó el uso de arreglos de múltiples discos (Redundant Array of Inexpensive Disks, RAID) para compensar la discrepancia de tiempos de acceso, debida principalmente a los tiempos de latencia de los discos magnéticos [Patterson, 2004]. Este método llevaría al uso de cantidades prohibitivas de discos para grandes instalaciones, tanto por el consumo energético, como por la complejidad de su mantenimiento.Teniendo en cuenta que las aplicaciones de nivel empresarial han adquirido la tendencia a ser centradas en los datos, en vez de centradas en el cómputo, la existencia de una memoria de estado sólido no volátil de alta densidad y rendimiento, de bajo costo, con tiempos de acceso entre la memoria principal y el disco rígido, podría representar una mejora significativa en el rendimiento general de los sistemas. Por otra parte, el límite de integración para las memorias no volátiles flash presenta la oportunidad para buscar tecnologías alternativas, que deberán ser superiores en cuanto a escalabilidad, costo por bit y rendimiento (considerando tiempos de acceso, ancho de banda de lectura/escritura, tiempos de retención, cantidad de ciclos de escritura y consumo energético). Ambas necesidades convergen en el objetivo de desarrollar un tipo de dispositivo con las características mencionadas (denominado storage-class memory), beneficioso en un amplio rango de escenarios, entre sistemas portátiles y servidores de alto rendimiento. En la tabla 1.1, se resumen algunas especificaciones esperadas:
• La variante NAND flash tiene una arquitectura que optimiza el acceso masivo a datos aunque el acceso aleatorio puede llegar a 10 μs, mientras en la variante NOR flash se logra un acceso aleatorio menor a 100 ns [Mikolajick, 2005] y pueden leerse hasta 100 MiB/s.
• No se requieren mejoras en el tiempo de retención de datos.• Las tecnologías flash no superan los 106 ciclos de escritura [Mikolajick, 2005; ITRS-PIDS,
2011], cantidad que debería aumentarse en memorias no volátiles a utilizar como caché de la memoria secundaria: para tecnologías de cambio de fase se espera entre 109 y 1013 ciclos, y para aquellas de conmutación magnética o ferroeléctrica, hasta 1016 ciclos [Burr, 2008; ITRS-PIDS, 2001; ITRS-PIDS, 2011].
Tiempo de acceso aleatorio 50 – 1.000 ns [Burr, 2008]
Tasa de transferencia de datos 100 MiB/s [Burr, 2008]
Tiempo de retención > 10 años [Burr, 2008; ITRS-PIDS, 2001; ITRS-PIDS, 2011]
Ciclos de escritura 109 – 1016 [Burr, 2008; ITRS-PIDS, 2001; ITRS-PIDS, 2011]
Potencia consumida en actividad 100 mW [Burr, 2008]
Potencia consumida por inactividad (stand-by) 1 mW [Burr, 2008]
Costo << 5 US$/GiB [Burr, 2008]
Tabla 1.1: Especificaciones esperadas para tecnologías alternativas a las flash.
1.3 Tecnologías candidatas a la aplicación como memorias no volátiles de alto rendimiento
Las memorias flash de tipo floating gate presentan dificultades para su escalabilidad, pues junto con la miniaturización [ITRS-PIDS, 2011]:
• Cada celda debe mantener una cantidad de electrones con la que pueda sensarse
12

confiablemente su estado, a pesar de la fluctuación estadística.• Debe prevernirse la interferencia entre celdas vecinas que se aproximan.• Deben conservarse los tiempos de retención y cantidad de ciclos de escritura.
Entre las tecnologías de memoria por almacenamiento de cargas, surgen las basadas en capas que atrapan cargas (charge trapping layers), como SONOS (Silicon-Oxide-Nitride-Oxide-Silicon), que atacan efectivamente el problema de la reducción de escala, pero enfrentan un compromiso entre rendimiento de escritura/borrado y retención de datos [Mikolajick, 2005]. A estas variantes, se suman dos estrategias de diseño para aumentar la densidad de las memorias: i) las celdas multinivel, en las cuales se distinguen 2n estados para almacenar n bits, y ii) la integración vertical o tridimensional, es decir, apilar múltiples capas de memoria sobre un mismo transistor [Burr, 2008]. Otros tipos de memorias que funcionan por conmutación magnética o ferroeléctrica también afrontan dificultades de escalabilidad [Meijer, 2008].Entre las tecnologías candidatas a desplazar a las memorias flash de su posición dominante actual en el mercado y consolidarse por su aplicación como un nuevo caché en la jerarquía de memorias (situándose entre la principal y la secundaria), se destacan aquellas cuyo principio de funcionamiento consiste en identificar estados de resistividad eléctrica bien diferenciados entre los que el material sensible de la celda de memoria puede transitar: i) memoria por cambio de fase (Phase Change Memory, PCM), en calcogenuros, ii) memoria por metalización programable de celdas (Programmable Metalization Cell, PMC o Conductive Bridge), en electrolitos sólidos [Kozicki, 2005; Waser, 2007], y iii) memoria por cambio de resistencia inducida por pulsos eléctricos, en óxidos de metales de transición [Baek, 2004; Beck, 2000; Liu, 2000].La memoria de cambio de fase se postula como la más madura de las tecnologías emergentes. Si bien, los principios físicos que sustentan su funcionamiento se estudian desde la década de los '60, no es hasta principios del siglo XXI, con el estado de la situación ya planteado para las tecnologías de memoria, que se retomó el desarrollo de los materiales de cambio de fase en vista de esta aplicación [Ielmini, 2011]. El primer antecedente relacionado se trató del cambio irreversible en la conductividad de una sal de MoS2, inducido por calor, luz o aplicación de un campo eléctrico, que fue informado en 1923; pasó desapercibido debido a la falta de técnicas de caracterización modernas y de visión a su aplicación tecnológica, cuando aún no habían sido desarrollados los primeros computadores electrónicos. En 1962, un trabajo de Pearson et al. presentaba un comportamiento novedoso de las curvas I-V en vidrios de As-Te-I y As-Te-Br: se observa en la figura 1.4, una región de alta (1) y otra de baja (2) conductividad entre las cuales podía transitar reversiblemente el material por la aplicación de pulsos eléctricos adecuados. En el mismo, no se buscaba una explicación al mecanismo ni una aplicación práctica. En contraste, el trabajo comenzado por Ovshinsky en 1960, para el desarrollo de dispositivos basados en efectos de conmutación por aplicación de campo eléctrico (threshold switching effect y memory switching effect) en calcogenuros (compuestos químicos con al menos un calcógeno, o sea, un elemento del grupo VI A: S, Se o Te) queda plasmado en patentes, consideradas más amplias y a la vez exhaustivas que las de Dewald, Northover y Pearson. Su publicación de 1968 [Ovshinsky, 1968] resulta fundacional en el tema y sus esfuerzos posteriores para impulsar los materiales de cambio de fase tanto en la industria del almacenamiento óptico como de las memorias semiconductoras, lo impondrían como pionero. Entre las mayores desventajas de los prototipos de la década de los '70 (como el de Neale et al.), se encuentra la elevada potencia de operación de los dispositivos, que resulta proporcional al volumen del material de cambio de fase. Como consecuencia del escalado del tamaño característico de los dispositivos por debajo de los 180 nm, resurge fuertemente el interés académico e industrial por los materiales de cambio de fase (en auge desde los '90 en el almacenamiento óptico) como memorias electrónicas no volátiles [Lam, 2008].
13

El principio para almacenar información en materiales de cambio de fase se basa en el cambio de las propiedades electrónicas (u ópticas) que ocurren por el reordenamiento de los átomos en la transición entre las fases cristalina y amorfa. Una porción limitada de material es sometida a controlados pulsos eléctricos (o de radiación láser), con potencia y duración adecuadas para una transición de fase en una escala de tiempo del orden de los nanosegundos. Partiendo de un volumen cristalino confinado, un pulso corto de alta corriente (o intensidad lumínica) eleva la temperatura por encima de la temperatura de liquidus del material, fundiéndolo. Debido al gradiente de temperatura que se establece con el material circundante, se alcanzan velocidades de enfriamiento suficientemente altas para amorfizar el material (ver sección 2.1). Un pulso de menor potencia, capaz de elevar la temperatura por encima de la temperatura de transición vítrea del material aplicado por un tiempo algo mayor, permite cristalizar nuevamente el volumen amorfo. El proceso de cristalización suele ser el más lento y, por ello resulta crítico en la evaluación de una aleación para estas aplicaciones [Asokan, 2011].El estado de una celda de memoria (o almacenamiento) es sensado con pulsos de baja corriente (o intensidad) tales que no lo modifiquen. Para que el mismo sea confiable, el contraste de las propiedades debe encontrarse típicamente como una variación en tres órdenes de magnitud para la conductividad eléctrica, y un cambio del 30% para la reflectividad óptica (dependiendo de la longitud de onda del láser y el espesor de la capa de material sensible).En resumen, las propiedades deseadas para un material de cambio de fase resultan [Wuttig, 2007]:
• Alta velocidad de transición de fase, inducida por estímulos en el orden de los nanosegundos.
• Alta estabilidad de la fase amorfa, para temperaturas superiores a la ambiente en algunas decenas de grados.
• Composición estable a través de los ciclos.• Cambio significativo en conductividad eléctrica (o índice de refracción/coeficiente de
absorción) entre estados.
La conductividad eléctrica del amorfo depende fuertemente de la magnitud del campo eléctrico aplicado. Un estado, antes de la tensión umbral, se caracteriza por su baja conductividad, separada en tres regiones: lineal, exponencial y superexponencial (ver figura 1.5). Pasada la tensión umbral Vth, alcanza otro estado (threshold switching), en el cual la conductividad exhibe un crecimiento tal
14
Figura 1.4: Curvas I-V en vidrios de As-Te-I y As-Te-Br (modificada de [Lam, 2008]).
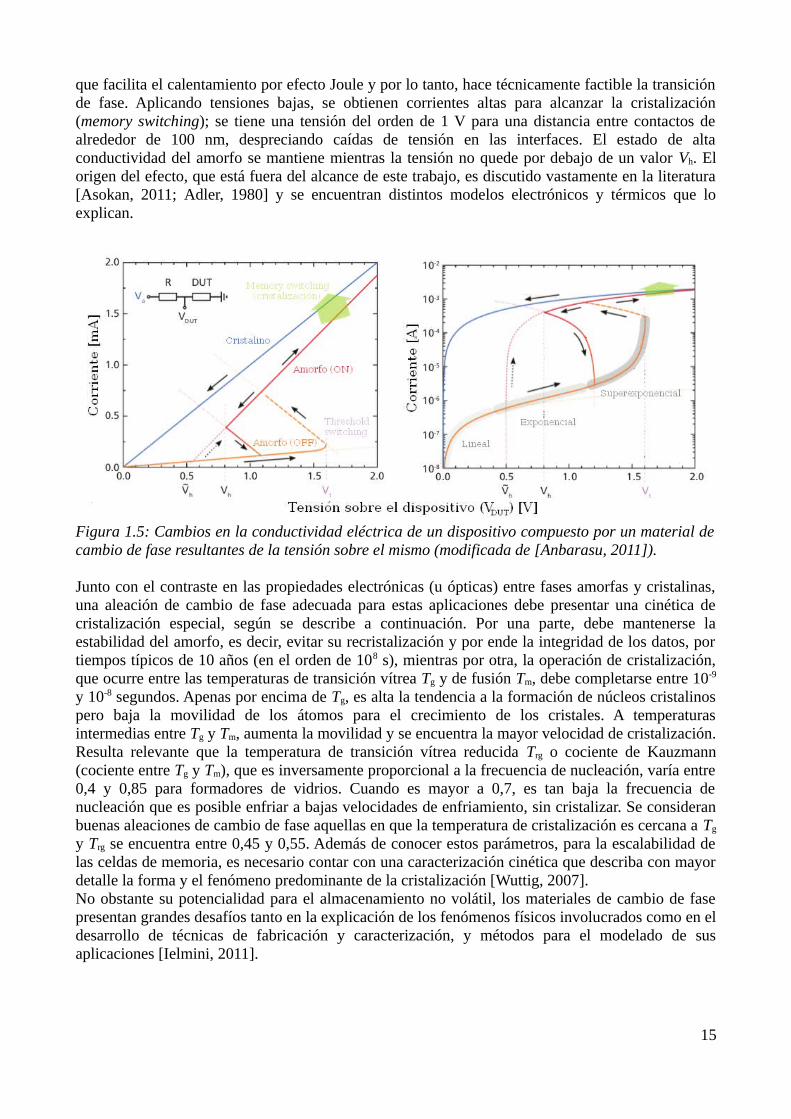
que facilita el calentamiento por efecto Joule y por lo tanto, hace técnicamente factible la transición de fase. Aplicando tensiones bajas, se obtienen corrientes altas para alcanzar la cristalización (memory switching); se tiene una tensión del orden de 1 V para una distancia entre contactos de alrededor de 100 nm, despreciando caídas de tensión en las interfaces. El estado de alta conductividad del amorfo se mantiene mientras la tensión no quede por debajo de un valor Vh. El origen del efecto, que está fuera del alcance de este trabajo, es discutido vastamente en la literatura [Asokan, 2011; Adler, 1980] y se encuentran distintos modelos electrónicos y térmicos que lo explican.
Junto con el contraste en las propiedades electrónicas (u ópticas) entre fases amorfas y cristalinas, una aleación de cambio de fase adecuada para estas aplicaciones debe presentar una cinética de cristalización especial, según se describe a continuación. Por una parte, debe mantenerse la estabilidad del amorfo, es decir, evitar su recristalización y por ende la integridad de los datos, por tiempos típicos de 10 años (en el orden de 108 s), mientras por otra, la operación de cristalización, que ocurre entre las temperaturas de transición vítrea Tg y de fusión Tm, debe completarse entre 10-9
y 10-8 segundos. Apenas por encima de Tg, es alta la tendencia a la formación de núcleos cristalinos pero baja la movilidad de los átomos para el crecimiento de los cristales. A temperaturas intermedias entre Tg y Tm, aumenta la movilidad y se encuentra la mayor velocidad de cristalización. Resulta relevante que la temperatura de transición vítrea reducida Trg o cociente de Kauzmann (cociente entre Tg y Tm), que es inversamente proporcional a la frecuencia de nucleación, varía entre 0,4 y 0,85 para formadores de vidrios. Cuando es mayor a 0,7, es tan baja la frecuencia de nucleación que es posible enfriar a bajas velocidades de enfriamiento, sin cristalizar. Se consideran buenas aleaciones de cambio de fase aquellas en que la temperatura de cristalización es cercana a Tg
y Trg se encuentra entre 0,45 y 0,55. Además de conocer estos parámetros, para la escalabilidad de las celdas de memoria, es necesario contar con una caracterización cinética que describa con mayor detalle la forma y el fenómeno predominante de la cristalización [Wuttig, 2007].No obstante su potencialidad para el almacenamiento no volátil, los materiales de cambio de fase presentan grandes desafíos tanto en la explicación de los fenómenos físicos involucrados como en el desarrollo de técnicas de fabricación y caracterización, y métodos para el modelado de sus aplicaciones [Ielmini, 2011].
15
Figura 1.5: Cambios en la conductividad eléctrica de un dispositivo compuesto por un material de cambio de fase resultantes de la tensión sobre el mismo (modificada de [Anbarasu, 2011]).

1.4 Estructura del presente trabajo
A continuación, el capítulo 2 se dedica a la presentación de técnicas de síntesis y caracterización de los materiales estudiados. El capítulo 3 trata la caracterización de sus propiedades térmicas y el capítulo 4, de sus propiedades eléctricas. Con los resultados obtenidos, el capítulo 5 plantea el modelado de una celda de memoria por el método de los elementos finitos.
1.5 Referencias
[Adler, 1980] Adler, D., Shur, M. S., Silver, M., Ovshinsky, S. R. (1980). Threshold switching in chalcogenide‐glass thin films. Journal of Applied Physics, 51(6), 3289-3309. doi:10.1063/1.328036
[Anbarasu, 2011] Anbarasu, M., Wuttig, M. (2011). Understanding the Structure and Properties of Phase Change Materials for Data Storage Applications. Journal of the Indian Institute of Science, 91(2), 259-274. Recuperado de http://journal.library.iisc.ernet.in/vol201102/JIISc-9102-WUTTIG.pdf
[Asokan, 2011] Asokan, S., Lakshmi, K. P. (2011). Electrical Switching and Other Properties of Chalcogenide Glasses. Journal of the Indian Institute of Science, 91(2), 319-330. Recuperado de http://journal.library.iisc.ernet.in/vol201102/JIISc-9102-ASOKAN.pdf
[Azizi, 2010] Azizi, O. J. (2010). Design and optimization of processors for energy efficiency: A joint architecture-circuit approach. Stanford, CA. Presentada como tesis Ph.D., Stanford University.
[Baek, 2004] Baek, I. G., Lee, M. S., Seo, S., Lee, M. J., Seo, D. H., Suh, D.-S., Park, J. C., Park, S. O., Kim, H. S., Yoo, I. K., Chung, U.-In., Moon, J.T. (2004). Highly scalable nonvolatile resistive memory using simple binary oxide driven by asymmetric unipolar voltage pulses. IEDM Technical Digest, Electron Devices Meeting 2004, 587-590. doi:10.1109/IEDM.2004.1419228
[Beck, 2000] Beck, A., Bednorz, J. G., Gerber, Ch., Rossel, C., Widmer, D. (2000). Reproducible switching effect in thin oxide films for memory applications. Applied Physics Letters, 77(1), 139-141. doi:10.1063/1.126902
[Blaauw, 2002] Blaauw, D., Martin, S., Mudge, T., Flautner, K. (2002). Leakage Current Reduction in VLSI Systems.Journal of Circuits, Systems and Computers, 11(6), 621. doi: 10.1142/S0218126602000665
[Bohr, 2007] Bohr, M. (2007). A 30 Year Retrospective on Dennard's MOSFET Scaling Paper. IEEE Solid-State Circuits Newsletter, 12(1), 11-13. doi:10.1109/N-SSC.2007.4785534
[Burr, 2008] Burr, G. W., Kurdi, B. N., Scott, J. C., Lam, C. H., Gopalakrishnan, K., Shenoy, R. S. (2008). Overview of candidate device technologies for storage-class memory. IBM Journal of Research and Development, 52(4.5), 449-464. doi:10.1147/rd.524.0449
[Dennard, 1974] Dennard, R.H., Gaensslen, F.H., Rideout, V.L., Bassous, E., LeBlanc, A.R. (1974). Design of ion-implanted MOSFET's with very small physical dimensions. IEEE Journal of Solid-State Circuits, 9(5), 256-268. doi:10.1109/JSSC.1974.1050511
[Grochowski, 2003] Grochowski, E., Halem, R. D. (2003). Technological impact of magnetic hard disk drives on storage systems. IBM Systems Journal, 42(2), 338-346. doi:10.1147/sj.422.0338
16

[Ielmini, 2011] Ielmini, D., Lacaita, A. L. (2011). Phase change materials in non-volatile storage. Materials Today, 14(12), 600-607. doi:10.1016/S1369-7021(11)70301-7[ITRS-PIDS, 2001] ITRS, Equipo PIDS (2001). Process integration, devices, and structure and emerging devices. En International Technology Roadmap for Semiconductors (ed. 2001), recuperado de: http://www.itrs.net/Links/2001ITRS/PIDS.pdf
[ITRS-PIDS, 2011] ITRS, Equipo PIDS (2011). Process integration, devices, and structure and emerging devices. En International Technology Roadmap for Semiconductors (ed. 2011), recuperado de: http://www.itrs.net/Links/2011ITRS/2011Chapters/2011PIDS.pdf y http://www.itrs.net/Links/2011ITRS/2011Tables/PIDS_2011Tables.xlsx
[Kozicki, 2005] Kozicki, M., Park, M., Mitkova, M. (2005). Nanoscale memory elements based on solid-state electrolytes. IEEE Transactions on Nanotechnology, 4(3), 331-338. doi:10.1109/TNANO.2005.846936
[Lam, 2008] Lam, C. H. (2008). History of Phase Change Memories. En Raoux, S., Wuttig, M. (Eds.), Phase Change Materials: Science and Applications (p. 1). Boston, MA: Springer US.
[Liu, 2000] Liu, S., Wu, N., Ignatiev, A. (2000). Electric-pulse-induced reversible resistance change effect in magnetoresistive films. Applied Physics Letters, 76(19), 2749-2751. doi:10.1063/1.126464
[Meijer, 2008] Meijer, G. I. (2008). Who Wins the Nonvolatile Memory Race? Science, 319(5870), 1625-1626. doi:10.1126/science.1153909
[Mikolajick, 2005] Mikolajick, T., Pinnow, C.-U. (2005). An Introduction to Nonvolatile Memory Technology. En Zschech, E.; Whelan, C., Mikolajick, T. (Eds.), Materials for Information Technology: Devices, Interconnects and Packaging (1a. ed., p. 111). Londres: Springer-Verlag.
[Moore, 1998] Moore, G.E. (1998). Cramming More Components Onto Integrated Circuits. Proceedings of the IEEE, 86(1), 82-85. doi:10.1109/JPROC.1998.658762
[Moore, 2003] Moore, G.E. (2003). No exponential is forever: but "Forever" can be delayed! Solid-State Circuits Conference, 1, 20-23. doi:10.1109/ISSCC.2003.1234194
[Murdocca, 2002] Murdocca, M., Heuring, V. (2002). Memoria. En Principios de arquitectura de computadoras (1a. ed., p. 243). Buenos Aires: Pearson Education.
[Ovshinsky, 1968] Ovshinsky, S. (1968). Reversible Electrical Switching Phenomena in Disordered Structures. Physical Review Letters, 21, 1450–1453. doi:10.1103/PhysRevLett.21.1450
[Patterson, 2004] Patterson D. A. (2004). Latency lags bandwith. Communications of the ACM, 47(10), 71-75. doi:10.1145/1022594.1022596
[Schaller, 1997] Schaller, R.R. (1997). Moore's law: past, present and future. IEEE Spectrum, 34(6), 52-59. doi:10.1109/6.591665
[Stallings, 2006] Stallings, W. (2006). Memoria caché. En Organización y arquitectura de computadores (7a. ed., p. 103). Madrid: Pearson Educación.
17

[von Neumann, 1993] von Neumann, J. (1993 ). First draft of a report on the EDVAC. IEEE Annals of the History of Computing , 15(4), 27-75. doi:10.1109/85.238389
[Waser, 2007] Waser, R., Aono M. (2007). Nanoionics-based resistive switching memories. Nature Materials, 6(11), 833-840. doi:10.1038/nmat2023[Wuttig, 2007] Wuttig, M., Yamada, N. (2007). Phase-change materials for rewriteable data storage. Nature Materials, 6(11), 824-832. doi:10.1038/nmat2009
18

2 Técnicas experimentales y desarrollo experimental para la síntesis y caracterización de los materiales estudiados
2.1 Síntesis de materiales amorfos
Se puede definir un material amorfo como aquel sólido que no tiene la periodicidad propia de un
cristal, su estructura es desordenada y no presenta un orden de largo alcance [Fontana, 1998]. La
obtención de un sólido amorfo es posible a través de técnicas que busquen: i) retener a temperatura
ambiente el estado desordenado del líquido o el gas, ii) destruir la estructura del cristal, o iii)
directamente producir la estructura desordenada mediente reacciones químicas adecuadas. Dentro
del primer grupo describiremos técnicas de síntesis de materiales amorfos por enfriamiento rápido
desde el líquido (melt quenching, melt spinning) o deposición desde la fase vapor (ablación láser)
[Feltz, 1993]. Todo proceso de fabricación de materiales amorfos debe presentar restricciones
cinéticas que impidan que los átomos alcancen la situación de equilibrio termodinámico y la
distribución periódica espacial de largo alcance [Ureña, 2002].
La obtención de aleaciones amorfas desde el líquido dependerá de la velocidad de enfriamiento
(q=dT/dt). La transformación para la formación del sólido cristalino no es un proceso instantáneo:
comienza a una temperatura inferior a la de fusión Tm (líquido subenfriado) y no lo hace
simultáneamente en todo el volumen. La cristalización ocurre en dos fases:
• La formación de núcleos o centros de cristalización (agrupamientos de pocos átomos),
caracterizada por la frecuencia de nucleación I.
• El crecimiento de los cristales a partir de los núcleos, caracterizado por la velocidad de
crecimiento de los cristales u.
La velocidad del proceso completo quedará determinada tanto por la frecuencia de nucleación como
por la velocidad de crecimiento. La cristalización requiere del tiempo necesario para que los átomos
difundan y ocupen sus posiciones en la estructura cristalina. Si se alcanzan grados de
subenfriamiento suficientemente altos para suprimir los procesos de nucleación y crecimiento, el
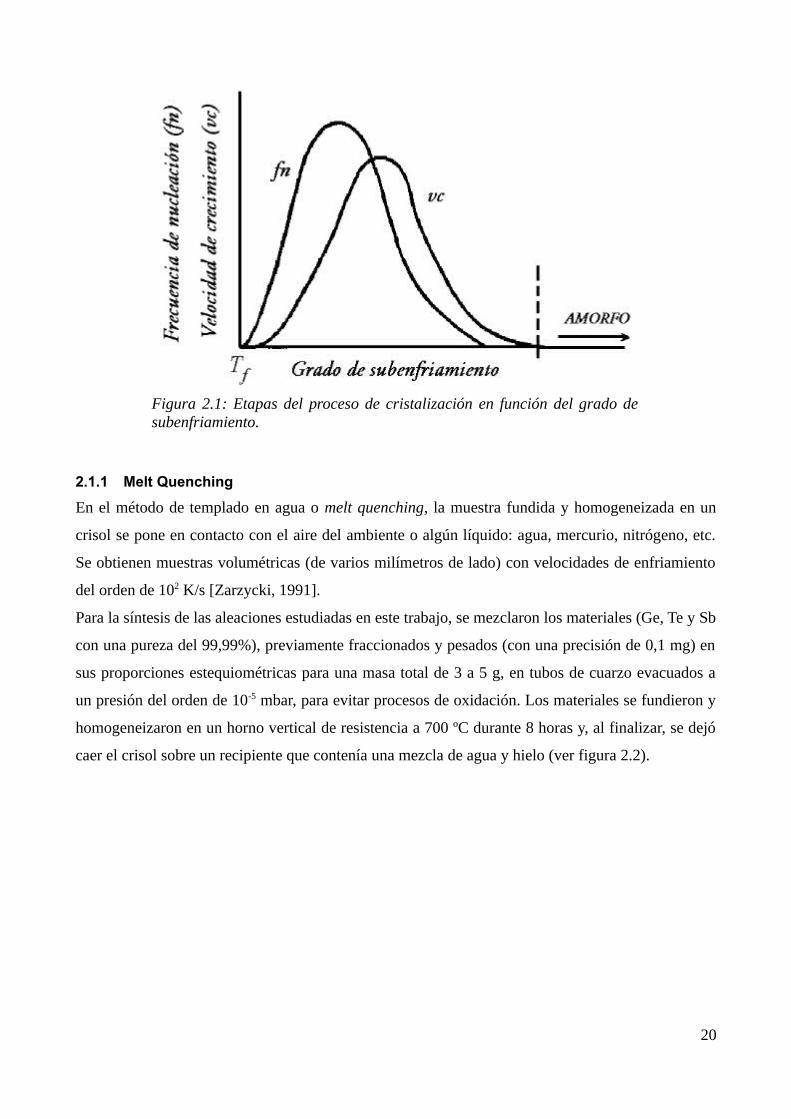
líquido subenfriado se congela obteniéndose un sólido amorfo (ver figura 2.1).
19
2.1.1 Melt Quenching
En el método de templado en agua o melt quenching, la muestra fundida y homogeneizada en un
crisol se pone en contacto con el aire del ambiente o algún líquido: agua, mercurio, nitrógeno, etc.
Se obtienen muestras volumétricas (de varios milímetros de lado) con velocidades de enfriamiento
del orden de 102 K/s [Zarzycki, 1991].
Para la síntesis de las aleaciones estudiadas en este trabajo, se mezclaron los materiales (Ge, Te y Sb
con una pureza del 99,99%), previamente fraccionados y pesados (con una precisión de 0,1 mg) en
sus proporciones estequiométricas para una masa total de 3 a 5 g, en tubos de cuarzo evacuados a
un presión del orden de 10-5 mbar, para evitar procesos de oxidación. Los materiales se fundieron y
homogeneizaron en un horno vertical de resistencia a 700 ºC durante 8 horas y, al finalizar, se dejó
caer el crisol sobre un recipiente que contenía una mezcla de agua y hielo (ver figura 2.2).
20
Figura 2.1: Etapas del proceso de cristalización en función del grado de subenfriamiento.

2.1.2 Melt Spinning
Para alcanzar mayores velocidades de enfriamiento, la muestra fundida se pone en contacto con
sólidos de alta conductividad térmica como el cobre, buscando reducir el espesor de la misma, de
modo que resulte más rápida la extracción de calor. La técnica de splat cooling consiste en aplicar
una presión suficiente sobre la muestra, para que ésta sea eyectada a través de un orificio en el
crisol, hacia un plato de cobre curvo (ver figura 2.3(a)). El material amorfo se obtiene como
escamas de pocos micrómetros, alcanzando velocidades de enfriamiento de 105 a 109 K/s. La
variante de splat cooling con martillo/pistón y yunque se basa en atrapar la gota de material
fundido, entre dos platos metálicos: uno fijo y el otro accionado por una fotocelda que sensa la
caída de la gota (ver figura 2.3(b)). Se obtienen muestras de espesor uniforme, aunque la velocidad
de enfriamiento no supera los 105 K/s. Otra variante, denominada roller quenching melt spinning,
consiste en dejar caer la gota entre dos cilindros rotantes, obteniendo escamas (ver figura 2.3(c)).
El dispositivo de Chen y Miller [Chen, 1976] mantiene un flujo de material fundido sobre la
superficie convexa de un cilindro hueco que es centrifugado para obtener cintas continuas (ver
figura 2.3(d)). La técnica fue mejorada con el uso de la superficie exterior de una rueda para obtener
cintas más anchas a velocidades de enfriamiento entre 106 y 108 K/s y se conoce como free jet melt
spinning (ver figura 2.3(e)) [Feltz, 1993; Zarzycki, 1991]. Entre las variables que influyen en el
proceso de obtención de cintas, se destacan: diámetro del orificio de salida del crisol, presión de
eyección, velocidad de la rueda, atmósfera circundante, posición y ángulo del crisol respecto de la
rueda y rugosidad superficial de la rueda [Silveyra, 2012].
21
Figura 2.2: Foto (izquierda) y esquema (derecha) del dispositivo utilizado en este trabajo para la síntesis de amorfos por melt quenching.

Para este trabajo, se utilizó un horno de inducción de alta frecuencia (870 kHz – 1,25 MHz, 12 kW
de potencia máxima) para fundir lingotes de 0,3 a 0,8 g de las aleaciones. Los mismos se colocaron
rodeados por una camisa de grafito dentro de un tubo de cuarzo, que se ubica en el centro de una
bobina por la que circula una corriente alterna de alta frecuencia. El campo magnético alterno
induce corrientes parásitas en la camisa de grafito, que se calienta por efecto Joule y funde el
material que contiene. Los crisoles usados presentan forma cónica en su extremo inferior con un
orificio de diámetro tal que la tensión superficial no permita que el líquido fluya por propio peso.
Aplicando una sobrepresión de argón de hasta 250 mm de Hg, la aleación fundida es eyectada sobre
la superficie de una rueda de cobre que gira aproximadamente a 20 m/s.
2.1.3 Deposición física en fase vapor (Physical Vapour Deposition, PVD)
En este grupo de técnicas, el material de partida (blanco) se encuentra en estado sólido y es
sometido a un calentamiento hasta su evaporación, ya sea por efecto Joule, bombardeo intenso de
electrones o iones de alta energía, incidencia de un haz de láser, etc. El material en forma de vapor
22
Figura 2.3: Esquemas de técnicas de templado rápido: a) splat cooling, b) splat cooling con martillo y yunque, c) rollet quenching melt spinning, d) centrifugado de material fundido, e) free jet melt spinning (modificado de [Feltz, 1993; Zarzycki, 1991])

es eyectado de la superficie del blanco y finalmente se condensa sobre la superficie del substrato
sobre la cual crece una película delgada [Piarristeguy, 2005]. En general, aquellos sistemas que
muestran tendencia a la amorfización por enfriamiento desde el líquido, también permiten
solidificar amorfos con estas técnicas, manteniendo prácticamente constante la composición
química de los compuestos [Zarzycki, 1991]. Entre las técnicas más importantes de PVD se
encuentran: la evaporación térmica, la pulverización catódica o sputtering, la ablación láser (Pulsed
Laser Deposition, PLD), y la implantación iónica [Piarristeguy, 2005].
La remoción de material causada por pulsos de láser de corta duración y alta intensidad es a
menudo referida como ablación por pulsos de láser. Este método permite suprimir la disipación de
la energía de excitación más allá del volumen que es ablacionado (removido) durante el pulso,
evitando la segregación de los componentes del blanco. Esta condición se verifica si el espesor de la
capa ablacionada por pulso es del orden de la longitud de penetración térmica o de la longitud de
penetración óptica (el que resulte mayor). En muchos materiales, puede ser satisfecha con radiación
láser ultravioleta y pulsos del orden de los nanosegundos.
En los materiales irradiados con pulsos de láser breves de alta intensidad se observan dos tipos de
interacción según la energía por unidad de área incidente, denominada fluencia Φ. Con fluencias
menores a determinado umbral (Φu), la radiación absorbida produce un aumento de temperatura que
puede llegar a fundir el material. Se observan cambios en la morfología y microestructura
superficial, la generación de defectos y la disminución de la concentración de uno o más
componentes del material. Cuando la fluencia supera el umbral, el proceso observado se caracteriza
por la expulsión de fase líquida, vaporización y formación de una pluma de plasma compuesto por
las especies ionizadas [Golmar, 2009].
Existen varios modelos [Srinivasan, 1983; Srinivasan, 1986; Sutcliffe, 1986] que explican la
interacción radiación-blanco y la extracción de material del interior del blanco, como procesos
térmicos, fotofísicos o fotoquímicos. Uno de ellos [Srinivasan, 1983] supone un proceso
fotoquímico donde la absorción de la radiación ultravioleta produce la ruptura de las ligaduras
químicas en el sólido, lo cual aumenta el volumen respecto de aquel que ocupaban inicialmente,
generando la expulsión de fragmentos del blanco. Debido a la interacción entre la radiación y el
material expulsado, se produce la disociación de sus agregados más grandes y de la especie
molecular y, luego, su fotoionización, resultando la formación de un plasma. El plasma se calienta
al absorber la radiación ya sea mediante la dispersión inelástica con electrones libres presentes en el
mismo, o bien por los electrones ligados a un átomo, que absorben fotones (efecto bremsstrahlung
inverso). El material expulsado y calentado se concentra en un volumen pequeño rodeado del vacío
en la cámara de ablación; el gradiente de presión provoca su expansión de manera tal que el flujo de
23

material adquiere una distribución angular estrecha y perpendicular a la superficie del blanco, para
depositarse sobre un sustrato ubicado frente a él. En este régimen puede evitarse el daño en el
blanco y su segregación en componentes distintos; la concentración relativa de las especies dentro
de la pluma prácticamente se conserva como en el material blanco sin alterarse por los sucesivos
pulsos. En consecuencia, resulta una técnica muy adecuada para la deposición de películas delgadas
de composición compleja si se pretende controlar con precisión su estequiometría.
La figura 2.4 muestra el esquema típico de un dispositivo para deposición de películas por ablación
láser. Está compuesto por un láser pulsado de alta potencia como fuente de energía para ablacionar
el material, una cámara conectada a un sistema de vacío (o preparada para trabajar en atmósferas
controladas), donde se ubican blanco y sustrato. Un sistema óptico de lentes y espejos se utiliza para
enfoncar y dirigir el haz láser hacia el blanco. Según las características de la película a depositar,
deberá controlarse la temperatura del sustrato.
Algunos de los parámetros involucrados en la deposición por PLD que determinan las propiedades
de las películas [Chrisey, 1994] se enumeran a continuación.
• Longitud de onda del láser: establece la efectividad de absorción de la energía del láser por
24
Figura 2.4: Esquema de un dispositivo para deposición por ablación láser.

parte del blanco. En la región del espectro electromagnético comprendida entre 200 y 400
nm, la mayoría de los materiales utilizados presentan una absorción adecuada.
• Densidad de energía del láser o fluencia: permite controlar la estequiometría y la calidad de
la película depositada. La densidad de energía umbral, a partir de la cual las películas
obtenidas reproducen la estequiometría del blanco, es característica del material que lo
compone y se encuentra generalmente entre 0,6 y 0,8 J/cm2. Experimentalmente puede
controlarse variando la potencia del láser o el tamaño del área de impacto (spot), a través del
sistema óptico que enfoca el haz.
• Densidad de potencia: Cuando se encuentra entre 105 y 108 W/cm2, la temperatura de la
superficie que está siendo evaporada permanece constante, alcanzando el régimen de
evaporación cuasi-estacionario del blanco.
• Distancia blanco–substrato: Para la técnica de PLD, se encuentra entre 30 y 150 mm, aunque
podría ser mayor (alrededor de 200 mm), para extender el área de uniformidad en el espesor
de la película. La optimización de esta distancia depende de la fluencia del haz de láser, la
presión de la cámara y la morfología del blanco, entre otros. Cuando la deposición se realiza
en vacío, el efecto de esta distancia se refleja en el ancho angular del flujo expulsado.
Cuando es menor que la longitud de la pluma, no existe una diferencia marcada en el
tamaño de las partículas y su densidad.
• Distribución angular del material ablacionado: El haz del láser se enfoca en una región
reducida del blanco y distribución de material en la pluma posee un máximo en la dirección
de propagación. Para evitar la deposición de películas de espesor uniforme solamente en un
rango angular estrecho, es necesario mover la pluma respecto de la superficie del sustrato.
En este trabajo se utilizó un láser de Nd:YAG (neodymium-doped yttrium aluminium garnet) que
consiste en un cristal de óxido de itrio y aluminio (Y3Al5O12), dopado con iones de Nd3+ como
medio activo. El equipo emite pulsos de 5 ns de duración, 10 Hz de frecuencia de repetición,
energía entre 45 y 70 mJ. Se configuró el sistema óptico del mismo para emplear longitudes de onda
de 266 y 355 nm. El sistema de vacío está formado por una bomba mecánica en serie con una
difusora que se conecta a la cámara de ablación, para alcanzar una presión del orden de 10-5 mbar.
Se usó una configuración on-axis (sobre el eje), donde el sustrato se ubica perpendicular a la pluma.
Como blanco y sustrato se encuentran fijos a la cámara, un espejo del sistema óptico se monta sobre
una plataforma móvil que le confiere un movimiento oscilatorio en una dimensión para que el haz
barra mayor superficie del blanco y no incida permanentemente en la misma región del blanco (ver
25

figura 2.5).
2.2 Caracterización de los materiales obtenidos
2.2.1 Difracción de rayos X
Se considera a un cristal como un arreglo tridimensional de átomos ordenados que forman una serie
de planos paralelos separados por una distancia d. Por ser las distancias de los centros de dispersión
(átomos) del mismo orden de magnitud que la longitud de onda de la radiación incidente en el caso
de los rayos X, ocurren fenómenos de interferencia.
Cuando un haz de rayos X incide sobre una muestra cristalina, estos son reflejados por los planos
sucesivos del cristal. Se encontrará un máximo de intensidad (interferencia constructiva) en la
radiación dispersada cuando la diferencia de caminos entre dos rayos reflejados en distintos planos
sea un múltiplo entero de la longitud de onda λ. Esta condición, expresada en la ecuación 2.1, donde
n es el orden difracción y θ, el ángulo entre el la dirección de incidencia y el plano cristalino
(ángulo de Bragg), se conoce como ley de Bragg y se intrerpreta geométricamente según la figura
2.6.
26
Figura 2.5: Foto del sistema de ablación láser utilizado en este trabajo.

Ec 2.1
Entre los tres métodos que existen para determinar los máximos de difracción, el de Debye-Scherrer
o método de polvos, a diferencia de los otros dos, prescinde de la fuerte restricción de utilizar
muestras monocristalinas. Éste consiste en reducir a polvo muy fino la muestra policristalina e
incidir sobre ella con radiación monocromática. Los granos poseen dimensiones mucho mayores a
la escala atómica y, por ello, son capaces de sostener la difracción. Los ejes cristalinos de los granos
están orientados al azar y siempre existirán casos en los que el ángulo de incidencia coincidirá con
el de Bragg: el diagrama de difracción producido es el que se produciría por la combinación de
diagramas de difracción para todas las orientaciones posibles de un cristal único. Esta afirmación es
válida cuando el tamaño de las partículas es suficientemente pequeño en comparación con el
volumen irradiado, de modo que todas las familias de planos estén en condiciones de difractar
[Conde Garrido, 2007; Golmar, 2009; Ureña, 2002; Vignolo, 2005].
El difractómetro es un instrumento que consiste en: una fuente de rayos X, un mecanismo que
detecte la radiación dispersada por la muestra, un goniómetro, el hardware y software para control
de movimiento de las partes móviles y adquisición de datos. En la geometría de focalización de
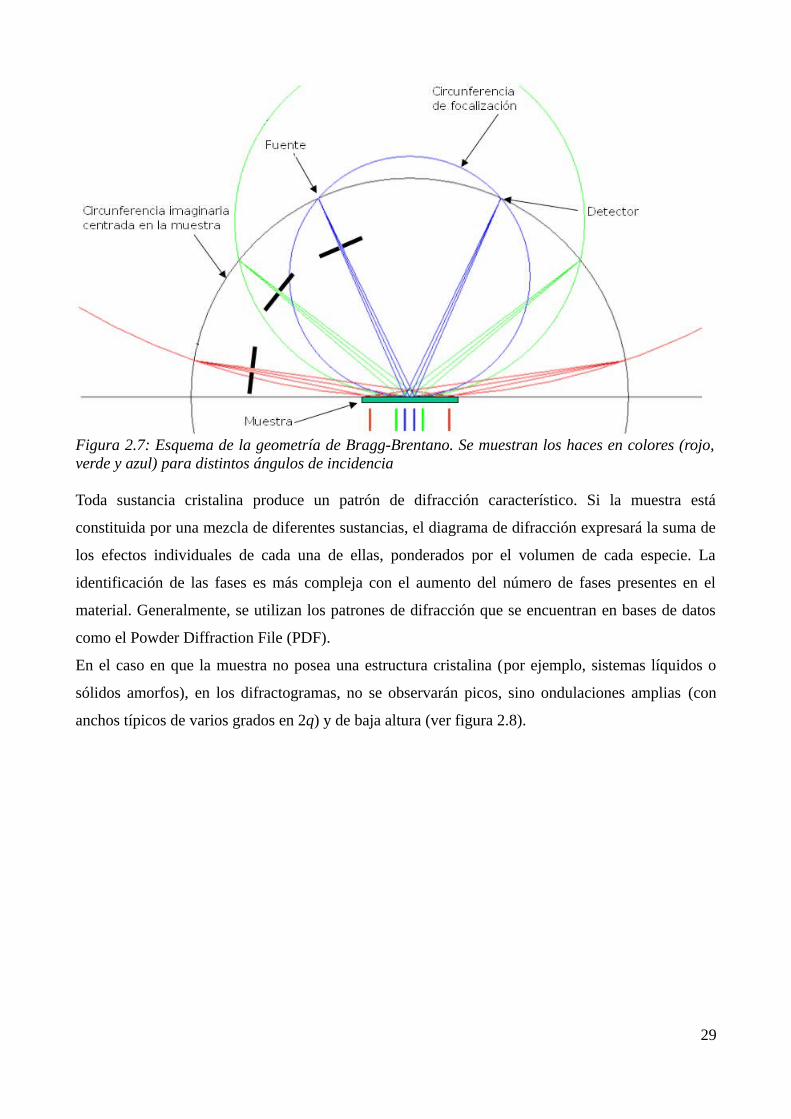
Bragg-Brentano se trabaja por reflexión: la fuente de rayos X y el detector se mueven en una
27
n λ=2d senθ
Figura 2.6: Esquema de la reflexión de rayos X, con longitud de onda λ, incidentes en una red cristalina, donde d es la distancia entre planos cristalinos y θ, el ángulo entre el la dirección de incidencia y el plano cristalino (ángulo de Bragg).

circunferencia imaginaria centrada en la muestra; los haces incidentes y difractados forman el
mismo ángulo θ con la superficie plana de la muestra y son coplanares con la normal a la misma. En
esta geometría, hay por lo menos una restricción geométrica importante: que la distancia entre el
foco de la fuente y la muestra sea igual a la distancia entre la muestra y el foco del detector (ver
figura 2.7). Idealmente, la superficie de la muestra debería seguir la curvatura de la circunferencia
de enfoque, pero, lo habitual es que la muestra sea plana y no muy extensa. Esta geometría admite
dos configuraciones posibles a modo de mantener igual ángulo de incidencia y de reflexión θ:
• Theta - 2 Theta: La fuente de rayos X está fija. La muestra gira a una velocidad ω y el
detector a una velocidad 2ω.
• Theta - Theta: La muestra está fija. La fuente y el detector giran a igual velocidad ω.
Se llama diagrama de difracción o difractograma a la representación gráfica de la intensidad en
función del ángulo de barrido 2θ (ángulo formado entre las direcciones de incidencia y reflexión).
Las posiciones de los picos están determinadas por la simetría y dimensiones de la celda unidad en
el cristal. La ley de Bragg asume dos condiciones ideales: cristales perfectos infinitos, y haz
incidente compuesto de radiación estrictamente monocromática y rayos perfectamente paralelos
[Cullity, 1956]. Como consecuencia, cada pico tiene un ancho (típicamente de unas décimas de
grados en el ángulo 2q) que depende de la muestra y del instrumental, a partir del cual puede
estimarse el tamaño de grano de las muestras. La intensidad de los picos está determinada por el
arreglo de átomos dentro de la celda y por factores instrumentales.
28
Toda sustancia cristalina produce un patrón de difracción característico. Si la muestra está
constituida por una mezcla de diferentes sustancias, el diagrama de difracción expresará la suma de
los efectos individuales de cada una de ellas, ponderados por el volumen de cada especie. La
identificación de las fases es más compleja con el aumento del número de fases presentes en el
material. Generalmente, se utilizan los patrones de difracción que se encuentran en bases de datos
como el Powder Diffraction File (PDF).
En el caso en que la muestra no posea una estructura cristalina (por ejemplo, sistemas líquidos o
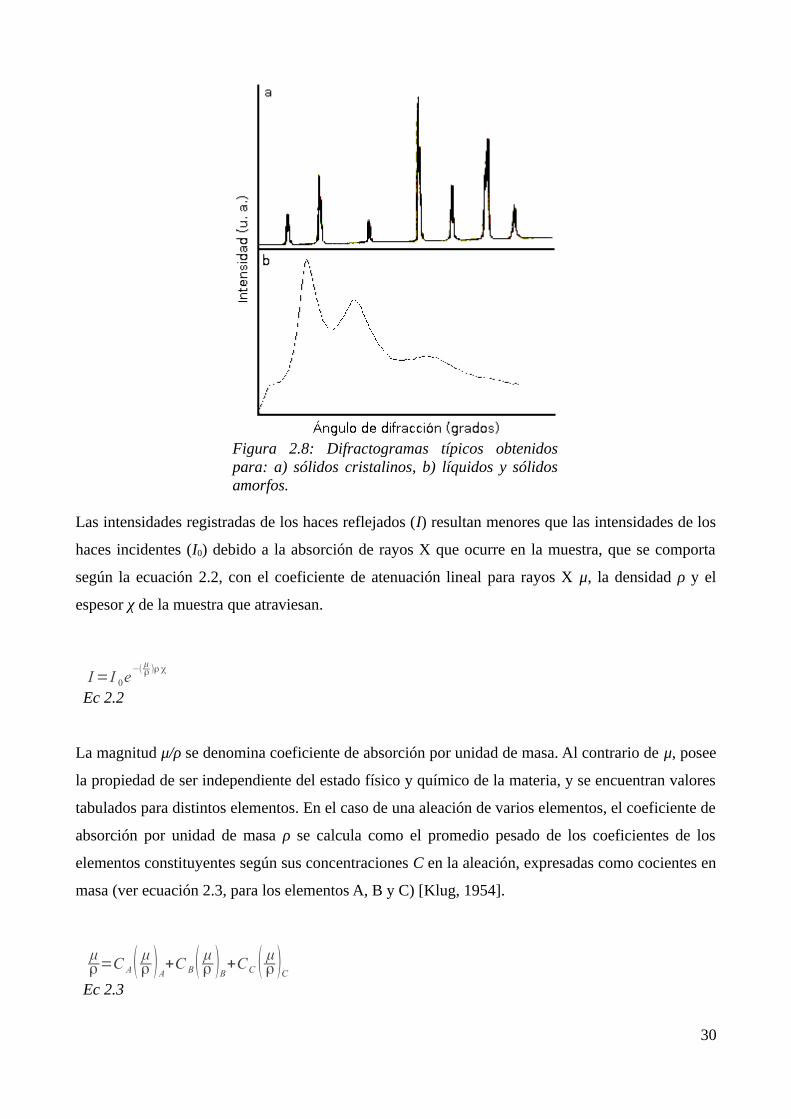
sólidos amorfos), en los difractogramas, no se observarán picos, sino ondulaciones amplias (con
anchos típicos de varios grados en 2q) y de baja altura (ver figura 2.8).
29
Figura 2.7: Esquema de la geometría de Bragg-Brentano. Se muestran los haces en colores (rojo, verde y azul) para distintos ángulos de incidencia
Las intensidades registradas de los haces reflejados (I) resultan menores que las intensidades de los
haces incidentes (I0) debido a la absorción de rayos X que ocurre en la muestra, que se comporta
según la ecuación 2.2, con el coeficiente de atenuación lineal para rayos X μ, la densidad ρ y el
espesor χ de la muestra que atraviesan.
Ec 2.2
La magnitud μ/ρ se denomina coeficiente de absorción por unidad de masa. Al contrario de μ, posee
la propiedad de ser independiente del estado físico y químico de la materia, y se encuentran valores
tabulados para distintos elementos. En el caso de una aleación de varios elementos, el coeficiente de
absorción por unidad de masa ρ se calcula como el promedio pesado de los coeficientes de los
elementos constituyentes según sus concentraciones C en la aleación, expresadas como cocientes en
masa (ver ecuación 2.3, para los elementos A, B y C) [Klug, 1954].
Ec 2.3
30
I=I 0e−(
μρ )ρ χ
μρ=C A( μρ )A+C B( μρ )B+CC ( μρ )C
Figura 2.8: Difractogramas típicos obtenidos para: a) sólidos cristalinos, b) líquidos y sólidos amorfos.

A partir de los difractogramas obtenidos para una muestra depositada como película delgada sobre
un sustrato y para el mismo sustrato por separado, es posible restar del primero la señal debida al
segundo. La contribución del sustrato puede calcularse aplicando la ecuación 2.2 al difractograma
del sustrato solo. Es decir, se considera la atenuación en el camino recorrido por los rayos X en la
muestra, con el espesor atravesado χ por los mismos según la ecuación 2.4, donde h, es el espesor
medio de la muestra. Luego, puede restarse al difractograma del arreglo película-sustrato y obtener
el de la película.
Ec 2.4
Para realizar la caracterización estructural por rayos X, se utilizó un difractómetro Rigaku con
goniómetros horizontal y vertical y ranuras tipo soller, montados sobre un equipo generador de
rayos X Philips PW 1730, utilizando radiación Kα de cobre (con longitud de onda λ = 1,5418 Å),
trabajando bajo la geometría de Bragg-Brentano anteriormente mencionada (ver figura 2.9).
31
χ=2h
sen( 2θ2 )

2.2.2 Técnicas calorimétricas para el estudio de la transformación de cristalización
Cuando un material experimenta un cambio de estado físico o participa de una reacción química,
ocurre una absorción o un desprendimiento de calor. Pueden medirse las entalpías involucradas en
estos procesos y, cuando los mismos son activados térmicamente, es posible analizar características
de los mecanismos de las transformaciones que tienen lugar en el material [Ureña, 2002].
Las técnicas de análisis calorimétrico se basan en la interacción térmica de la muestra estudiada con
su entorno, resultante de cómo se programe la temperatura de éste. Las medidas calorimétricas
suelen realizarse manteniendo constante la temperatura (experiencias isotérmicas) o variándola
linealmente con el tiempo (experiencias de calentamiento continuo). Las experiencias isotérmicas
suponen mayor precisión por contar con una modelización más simple y no presentan problemas de
gradientes térmicos, histéresis o retrasos térmicos, aunque implican mayor lentitud y difícil elección
de la temperatura adecuada para que se pueda registrar el proceso térmico buscado [Fontana, 1998].
Entre las diferentes técnicas calorimétricas, la calorimetría diferencial de barrido (Differential
Scanning Calorimeter, DSC) es ampliamente utilizada en el estudio de la cinética de cristalización y
la relajación estructural de materiales amorfos. Se basa en la comparación de la evolución térmica
de la muestra en estudio con una referencia inerte, en un rango de temperaturas. Existen dos tipos
de calorímetros diferenciales de barrido: de compensación de potencia y de flujo de calor. En los del
32
Figura 2.9: Foto del difractómetro con goniómetro vertical utilizado.

primer tipo se determinan las entalpías de los procesos, midiendo la diferencia de flujo calórico
necesaria para mantener una muestra y la referencia inerte a la misma temperatura.
En el calorímetro, la muestra y la referencia se ubican en celdas que cuentan con calefactores y
termorresistencias de platino que pueden controlarse y sensarse independientemente (ver figura
2.10). Para mantener simultáneamente la misma temperatura en ambos crisoles, el sistema cuenta
con dos lazos de control: i) el que controla la temperatura media, para que las temperaturas de la
muestra y de la referencia varíen a la velocidad programada, y ii) el que asegura que si se produce
una diferencia de temperatura entre la muestra y la referencia (debido a alguna reacción exotérmica
o endotérmica de la muestra), se modifique la potencia de entrada al elemento calefactor
correspondiente a fin de anular esta diferencia, es decir, suministra o absorbe energía hasta que la
diferencia de temperatura sea menor que el valor umbral del equipo (0,01 K).
La diferencia de potencia entregada a los calefactores resulta proporcional a las diferencias de
calores intercambiados por la muestra y por la referencia, es decir, proporcional a la diferencia de
entalpía del proceso dH/dt. La señal que registrará el equipo es la diferencia de potencia dQ/dt,
conjuntamente con la temperatura media del sistema. Entonces, en una curva de DSC típica para
régimen isotérmico se representa dH/dt en función del tiempo, t, y en el caso de calentamiento
continuo se representa dH/dt en función de la temperatura media, T, del sistema.
La señal calorimétrica se produce cuando un cambio energético en la muestra en estudio tiende a
provocar una diferencia de temperatura entre la misma y la referencia. En caso de no existir ningún
cambio en la muestra, la línea registrada resulta paralela al eje de tiempos o de temperatura,
recibiendo el nombre de línea base. Si la muestra experimenta un cambio energético, la línea
cambiará de forma, dependiendo de si el proceso es endotérmico, exotérmico o un cambio de calor
específico. En el caso de tener un proceso endotérmico (dH/dt positivo), ocurrirá una tendencia al
retraso en la temperatura de la muestra frente a la referencia, produciendo un dQ/dt positivo, ya que
el DSC deberá entregar energía a la muestra para mantener su temperatura igual a la de la
33
Figura 2.10: Esquema de las celdas portamuestra (M) y de referencia (R)
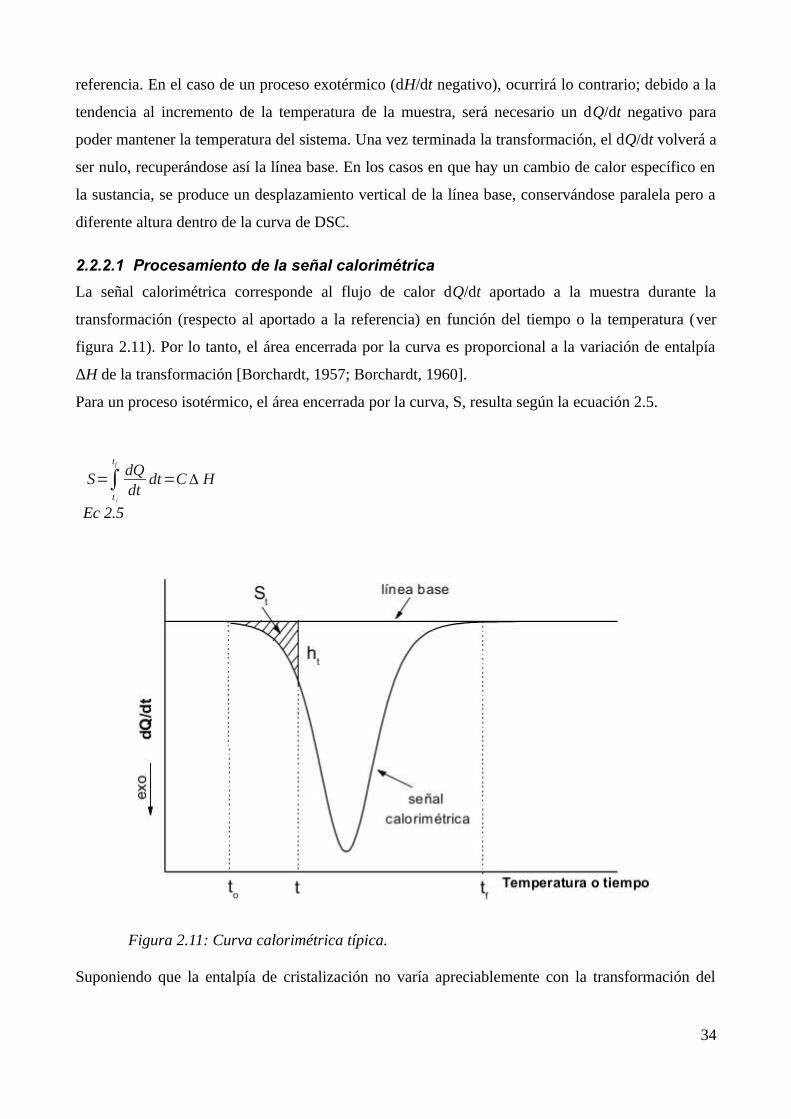
referencia. En el caso de un proceso exotérmico (dH/dt negativo), ocurrirá lo contrario; debido a la
tendencia al incremento de la temperatura de la muestra, será necesario un dQ/dt negativo para
poder mantener la temperatura del sistema. Una vez terminada la transformación, el dQ/dt volverá a
ser nulo, recuperándose así la línea base. En los casos en que hay un cambio de calor específico en
la sustancia, se produce un desplazamiento vertical de la línea base, conservándose paralela pero a
diferente altura dentro de la curva de DSC.
2.2.2.1 Procesamiento de la señal calorimétrica
La señal calorimétrica corresponde al flujo de calor dQ/dt aportado a la muestra durante la
transformación (respecto al aportado a la referencia) en función del tiempo o la temperatura (ver
figura 2.11). Por lo tanto, el área encerrada por la curva es proporcional a la variación de entalpía
ΔH de la transformación [Borchardt, 1957; Borchardt, 1960].
Para un proceso isotérmico, el área encerrada por la curva, S, resulta según la ecuación 2.5.
Ec 2.5
Suponiendo que la entalpía de cristalización no varía apreciablemente con la transformación del
34
S=∫t i
tf
dQdt
dt=CΔ H
Figura 2.11: Curva calorimétrica típica.

amorfo, la fracción cristalizada de material x puede evaluarse a partir del cociente entre la entalpía
parcial a tiempo t (∆Ht) y la entalpía total del proceso (∆H), proporcionales a St (el área parcial a
tiempo t) y S, respectivamente (ecuación 2.6). Derivando, se obtiene la velocidad de transformación
dx/dt (ecuación 2.7), siendo ht la altura de la curva experimental respecto de la línea base a tiempo t.
Ec 2.6
Ec 2.7
Para un proceso de calentamiento continuo, con velocidad de calentamiento β, la señal calorimétrica será dQ/dt en función de la temperatura T. Análogamente, obtenemos las expresiones para la fracción cristalizada x y la velocidad de transformación dx/dt según las ecuaciones 2.8 y 2.9, donde S es el área encerrada por la curva, ST, el área parcial a temperatura T y hT, la altura de la curva respecto de la línea base a temperatura T.
Ec 2.8
Ec 2.9
Los parámetros cinéticos de una reacción pueden calcularse a partir de expresar la velocidad de reacción dx/dt según la ecuación cinética fundamental (ecuación 2.10), donde x es la fracción transformada, K(T) la constante de velocidad, y f(x) una función que depende únicamente de la fracción transformada y se comporta según el mecanismo de reacción. En general, esto es válido en procesos de cristalización activados térmicamente, donde la ley de Arrhenius es utilizada para describir la constante de velocidad K(T) (ecuación 2.11) [Chen, 1978; Henderson, 1979; Šesták,1971], con el factor preexponencial k0, la energía de activación aparente del proceso Ea, la temperatura T y la constante de los gases R.
35
x=Δ H t
Δ H=
St
S=
∫ti
tdQdt
dt
∫ti
tf
dQdt
dt
dxdt
=ddt
(∫ti
tdQdt
dt
S)= dQ
dt ∣t
S=
h(t )S
x=∫t i
tdQdt
dt
∫t i
tf
dQdt
dt
=
∫T (ti )
T (t )dQdt
dTβ
∫T (t i)
T (tf )
dQdt
dTβ
=
1β∫
T i
TdQdt
dT
1β∫
T i
T f
dQdt
dT
=ST
S
dxdt
=
ddt
ST
S=
ddT ( dT
dtST)
S=
ddT
(βST )
S=β
ddT∫T i
TdQdt
dT
S=β
dQdt ∣T
S=βhT
S

Ec 2.10
Ec 2.11
Para determinar K(T), el método de Kissinger [Kissinger, 1957] parte de la hipótesis de que en el máximo de la curva calorimétrica ocurre la máxima velocidad de transformación (ecuación 2.12). Desarrollando la ecuación 2.13 (ver ecuaciones 2.14, 2.15, 2.16, y 2.17) e imponiendo la condición de máximo se llega a la expresión de la ecuación 2.18. Representando el primer miembro de la ecuación ecuación 2.18 en función de 1/Tp con los datos obtenidos para diferentes velocidades de calentamiento, y si se ajusta con una función lineal por el método de cuadrados mínimos, se obtiene la pendiente -Ea/R.
Ec 2.12
Ec 2.13
Ec 2.14
Ec 2.15
Ec 2.16
Ec 2.17
Ec 2.18
36
dxdt
=K (T ) f (x )
K (T )=k0 e−
E a
RT
d2 x
dt 2 ∣T=T p ,t=t p , x= xp
=0
dx2
dt2 =K (T )df (x )
dt+
dK (T )
dtf (x)
df (x)dt
=df (x )
dxdxdt
=f ' (x )dxdt
dK (T )
dt=
dK (T )
dTdTdt
=βEa
RT2 k 0e−
Ea
RT =βEa
RT 2 K (T )
dx2
dt 2 =K (T ) f ' ( x)dxdt
+βEa
RT 2 K (T ) f (x )=dxdt [K (T ) f ' (x )+β
E a
RT 2 ]
βEa
RT p2 =−k0 e
−E a
RT p f '(x p)
ln(β
T p2 )=ln(− k0 R
Ea
f ' (x p)e−
E a
RT p)=−Ea
RT p
+ln(−k0 REa
f '(x p))

2.2.2.2 Características del equipamiento usado
En este trabajo, se ha utilizado un calorímetro diferencial de barrido Perkin-Elmer modelo DSC7,
que funciona por compensación de potencia, cuyas prestaciones se enumeran a continuación:
a) Rango de temperaturas: 323 K a 1000 K.
b) Velocidad de calentamiento: de 2,5 K/min a 100 K/min.
c) Sensibilidad en la medida calorimétrica: depende de la calibración realizada, puede llegar a 0,1
%. La máxima sensibilidad del instrumento es de 35 μW.
d) Precisión en la medida en temperaturas: depende de la calibración realizada, puede llegar a 0,2
K.
La calibración de temperatura se realizó utilizando los puntos de fusión del indio (429,6 K) y del
zinc (692,7 K), y la calibración en áreas mediante el pico de fusión del indio (28,47 J/g). La
calibración realizada permite alcanzar, en experiencias a distintas velocidades y con diferentes
cantidades de muestra, una precisión de 1 K en temperaturas y de un 5 % en la medida de la
entalpía.
Durante un programa de calentamiento continuo existe un gradiente de temperatura entre el horno y
la muestra, y también dentro de la muestra misma. La magnitud del gradiente de temperatura se
incrementa con el aumento de la masa de muestra y con la velocidad de calentamiento. Para evitar
estos retrasos térmicos, es importante trabajar con muestras de masa pequeña (en el orden de los 5
mg) dispuestas con poco espacio entre partículas y un buen contacto térmico con la cazuela donde
se empaquetan. También debe existir buen contacto térmico entre el sensor en la celda y la
superficie de la cazuela.
2.2.3 Microscopía de fuerza atómica (AFM)
El principio de funcionamiento de la microscopía de fuerza atómica (Atomic Force Microscopy,
AFM) consiste en medir las fuerzas de interacción entre la superficie de una muestra y una punta
afilada con un radio de curvatura en el orden de los nanómetros sujeta al extremo de un fleje
elástico (cantilever). Cuando la punta se pone en la proximidad de la superficie de una muestra, las
interacciones de Van der Waals entre la punta y la muestra deflectan el cantilever de acuerdo con la
ley de Hooke: una atracción de largo alcance debida a la interacción dipolo-dipolo y una repulsión
de corto alcance debida al principio de exclusión de Pauli. Generalmente, estas fuerzas de
interacción tienen una componente normal a la superficie de la muestra (provoca la deflexión del
cantilever) y una componente lateral, coplanar a la misma (provoca la torsión del cantilever)
37

[Golmar, 2009; Mironov, 2004; Piarristeguy, 2005].
Los métodos de adquisición de imágenes por AFM, sean topográficas o relativas a propiedades
locales de la muestra, se clasifican en modos de contacto (cuasiestáticos) y modos de no-contacto y
semi-contacto (oscilatorios). En los primeros, el extremo de la punta está en contacto directo con la
superficie y la fuerza de atracción o repulsión entre los átomos de la punta y la muestra se
contrarresta con la fuerza elástica producida por la deflexión o torsión del cantilever. En
consecuencia, los cantilevers usados tienen baja rigidez para lograr alta sensibilidad y evitar
excesiva interacción que dañe la muestra. Las pequeñas deflexiones y torsiones del cantilever son
registradas por un sistema óptico compuesto de un diodo láser y un fotodetector. El sistema se
alinea de manera tal que el haz láser sea enfocado sobre el cantilever y el haz reflejado incida sobre
el centro de cuatro secciones sensadas por fotodiodos. Las variaciones en las intensidades
percibidas por cada fotodiodo permiten medir deflexión y torsión del cantilever (figuras 2.12 y
2.13).
38
Figura 2.12: Esquema del sistema óptico para detectar la deformación del cantilever (modificado de [Mironov, 2004]).
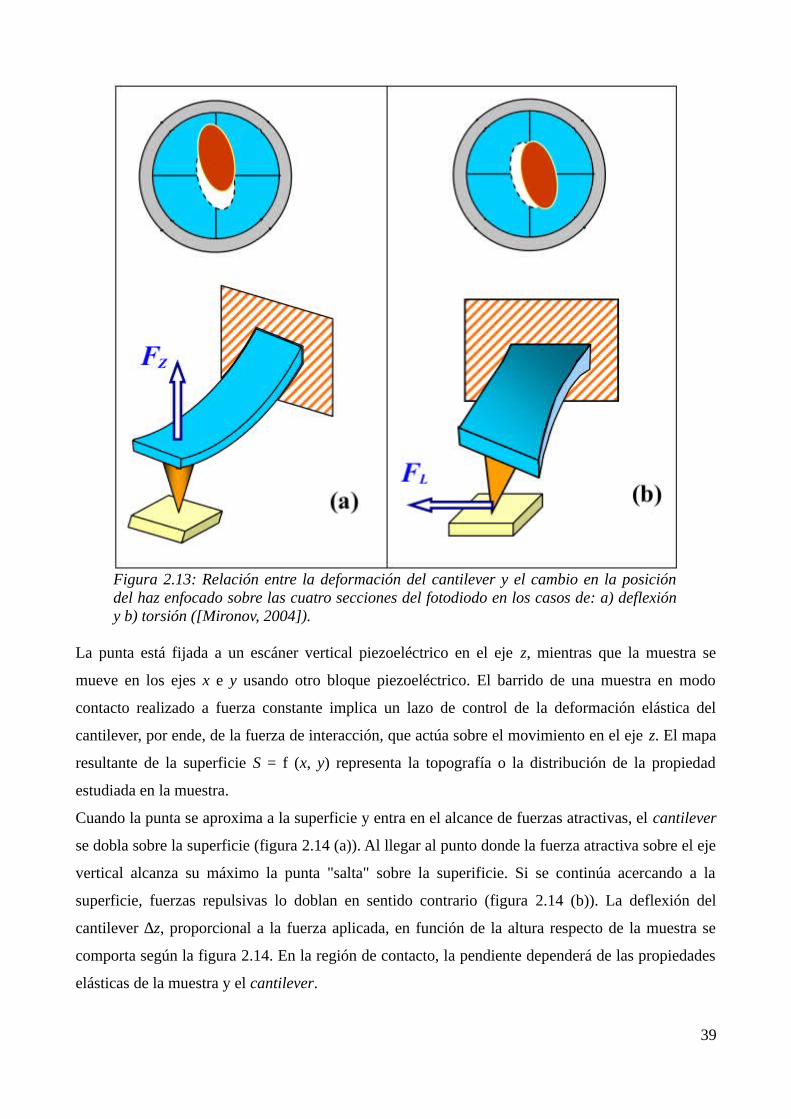
La punta está fijada a un escáner vertical piezoeléctrico en el eje z, mientras que la muestra se
mueve en los ejes x e y usando otro bloque piezoeléctrico. El barrido de una muestra en modo
contacto realizado a fuerza constante implica un lazo de control de la deformación elástica del
cantilever, por ende, de la fuerza de interacción, que actúa sobre el movimiento en el eje z. El mapa
resultante de la superficie S = f (x, y) representa la topografía o la distribución de la propiedad
estudiada en la muestra.
Cuando la punta se aproxima a la superficie y entra en el alcance de fuerzas atractivas, el cantilever
se dobla sobre la superficie (figura 2.14 (a)). Al llegar al punto donde la fuerza atractiva sobre el eje
vertical alcanza su máximo la punta "salta" sobre la superificie. Si se continúa acercando a la
superficie, fuerzas repulsivas lo doblan en sentido contrario (figura 2.14 (b)). La deflexión del
cantilever ∆z, proporcional a la fuerza aplicada, en función de la altura respecto de la muestra se
comporta según la figura 2.14. En la región de contacto, la pendiente dependerá de las propiedades
elásticas de la muestra y el cantilever.
39
Figura 2.13: Relación entre la deformación del cantilever y el cambio en la posición del haz enfocado sobre las cuatro secciones del fotodiodo en los casos de: a) deflexión y b) torsión ([Mironov, 2004]).
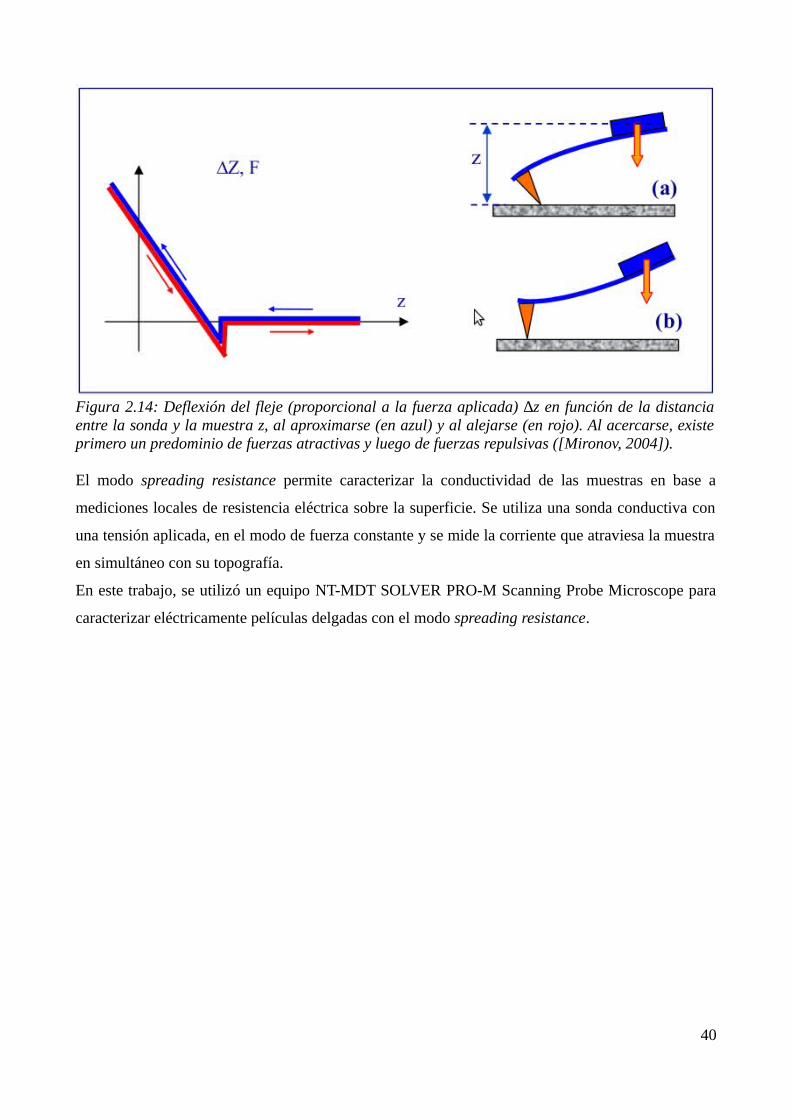
El modo spreading resistance permite caracterizar la conductividad de las muestras en base a
mediciones locales de resistencia eléctrica sobre la superficie. Se utiliza una sonda conductiva con
una tensión aplicada, en el modo de fuerza constante y se mide la corriente que atraviesa la muestra
en simultáneo con su topografía.
En este trabajo, se utilizó un equipo NT-MDT SOLVER PRO-M Scanning Probe Microscope para
caracterizar eléctricamente películas delgadas con el modo spreading resistance.
40
Figura 2.14: Deflexión del fleje (proporcional a la fuerza aplicada) ∆z en función de la distancia entre la sonda y la muestra z, al aproximarse (en azul) y al alejarse (en rojo). Al acercarse, existe primero un predominio de fuerzas atractivas y luego de fuerzas repulsivas ([Mironov, 2004]).

2.3 Referencias
[Borchardt, 1957] Borchardt, H. J., Daniels, F. (1957). The Application of Differential Thermal Analysis to the Study of Reaction Kinetics. Journal of the American Chemical Society, 79(1), 41-46. doi:10.1021/ja01558a009
[Borchardt, 1960] Borchardt, H. J. (1960). Initial reaction rates from DTA. Journal of Inorganic and Nuclear Chemistry, 12(3–4), 252–254. doi:10.1016/0022-1902(60)80369-7
[Chen, 1978] Chen, H. S. (1978). A method for evaluating viscosities of metallic glasses from the rates of thermal transformations. Journal of Non-Crystalline Solids, 27(2), 257–263. doi:10.1016/0022-3093(78)90128-X
[Chen, 1976] Chen, H. S., Miller C. E. (1976). Centrifugal spinning of metallic glass filaments. Materials Research Bulletin, 11(1), 49-54. doi:10.1016/0025-5408(76)90213-0
[Chrisey, 1994] Chrisey, D., Hubler, G. (1994). Pulsed Laser Deposition of Thin Films. New York: John Wiley & Sons, Inc.
[Conde Garrido, 2007] Conde Garrido, J. M. (2007). Vidrios Calcogenuros: Aplicación a electrodos selectivos de iones (ISE). Buenos Aires. Presentada como Tesis de Licenciatura en Ciencias Físicas, Universidad de Buenos Aires.
[Cullity, 1956] Cullity, B. D. (1956). Elements of X-ray diffraction. Reading, Massachusetts: Addison-Wesley Publishing Company, Inc.
[Feltz, 1993] Feltz, A. (1993). Amorphous Inorganic Materials and Glasses. Weinheim: VCH.
[Fontana, 1998] Fontana, M. (1998). Cinética de cristalización de aleaciones amorfas base Te y base Mg. Buenos Aires. Presentada como Tesis de Doctorado en Ciencias Físicas, Universidad de Buenos Aires.
[Golmar, 2009] Golmar, F. (2009). Nuevos Dispositivos para Aplicaciones de Espintrónica. Buenos Aires. Presentada como Tesis de Doctorado en Ingeniería, Universidad de Buenos Aires.
[Henderson, 1979] Henderson, D. W. (1979). Thermal analysis of non-isothermal crystallization kinetics in glass forming liquids. Journal of Non-Crystalline Solids, 30(3), 301–315. doi:10.1016/0022-3093(79)90169-8
[Kissinger, 1957] Kissinger, H. E. (1957). Reaction Kinetics in Differential Thermal Analysis. Analytical Chemistry, 29(11), 1702–1706. doi:10.1021/ac60131a045
[Klug, 1954] Klug, H. P., Alexander, L. E. (1954). X-ray diffraction procedures for polycrystalline and Amorphous Materials. New York: John Wiley & Sons, Inc.
[Mironov, 2004] Mironov, V. L. (2004). Fundamentals of Scanning Probe Microscopy. The Russian Academy of Sciences, Institute of Physics of Microstructures Nizhniy Novgorod.
[Piarristeguy, 2005] Piarristeguy, A. (2005). Vidrios calcogenuros: aplicación a la construcción de baterías. Buenos Aires. Presentada como Tesis de Doctorado en Ingeniería, Universidad de Buenos
41

Aires.
[Šesták, 1971] Šesták, J. (1971). Thermal Analysis Proceedings third ICTA, 2.
[Silveyra, 2012] Silveyra, J. (2012). Desarrollo de materiales magnéticos para transformadores. Buenos Aires. Presentada como Tesis de Doctorado en Ingeniería, Universidad de Buenos Aires.
[Srinivasan, 1983] Srinivasan, R. (1983). Kinetics of the ablative photodecomposition of organic polymers in the far ultraviolet (193 nm). Journal of Vacuum Science & Technology B: Microelectronics and Nanometer Structures, 1(4), 923-926. doi:10.1116/1.582712
[Srinivasan, 1986] Srinivasan, V., Smrtic, M. A., Babu, S. V. (1986). Excimer laser etching of polymers. Journal of Applied Physics, 59(11), 3861-3867. doi:10.1063/1.336728
[Sutcliffe, 1986] Sutcliffe, E., Srinivasan, R. (1986). Dynamics of UV laser ablation of organic polymer surfaces. Journal of Applied Physics, 60(9), 3315-3322. doi:10.1063/1.337698
[Ureña, 2002] Ureña, M. A. (2002). Vidrios calcogenuros conductores superiónicos. Buenos Aires. Presentada como Tesis de Doctorado en Ingeniería, Universidad de Buenos Aires.
[Vignolo, 2005] Vignolo, M. F. (2005). Sensores Químicos de Película Delgada. Buenos Aires. Presentada como Tesis de Doctorado en Ciencias Físicas, Universidad de Buenos Aires.
[Zarzycki, 1991] Zarzycki, J. (1991). Special Methods of Obtanining Glasses and Amorphous Materials. En J. Zarzycki (Ed.), Materials Science and Technology: A Comprehensive Treatment (Vol. 9). Weinheim: VCH.
42
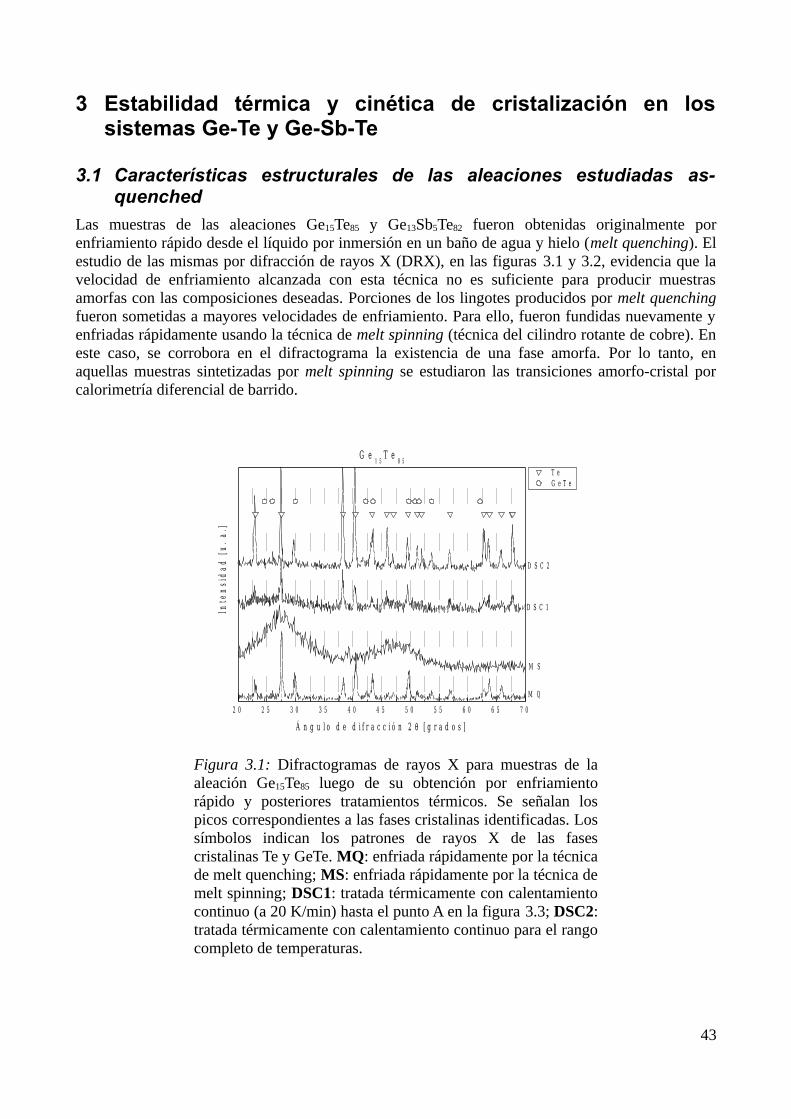
3 Estabilidad térmica y cinética de cristalización en los sistemas Ge-Te y Ge-Sb-Te
3.1 Características estructurales de las aleaciones estudiadas as-quenched
Las muestras de las aleaciones Ge15Te85 y Ge13Sb5Te82 fueron obtenidas originalmente por enfriamiento rápido desde el líquido por inmersión en un baño de agua y hielo (melt quenching). El estudio de las mismas por difracción de rayos X (DRX), en las figuras 3.1 y 3.2, evidencia que la velocidad de enfriamiento alcanzada con esta técnica no es suficiente para producir muestras amorfas con las composiciones deseadas. Porciones de los lingotes producidos por melt quenching fueron sometidas a mayores velocidades de enfriamiento. Para ello, fueron fundidas nuevamente y enfriadas rápidamente usando la técnica de melt spinning (técnica del cilindro rotante de cobre). En este caso, se corrobora en el difractograma la existencia de una fase amorfa. Por lo tanto, en aquellas muestras sintetizadas por melt spinning se estudiaron las transiciones amorfo-cristal por calorimetría diferencial de barrido.
43
Figura 3.1: Difractogramas de rayos X para muestras de la aleación Ge15Te85 luego de su obtención por enfriamiento rápido y posteriores tratamientos térmicos. Se señalan los picos correspondientes a las fases cristalinas identificadas. Los símbolos indican los patrones de rayos X de las fases cristalinas Te y GeTe. MQ: enfriada rápidamente por la técnica de melt quenching; MS: enfriada rápidamente por la técnica de melt spinning; DSC1: tratada térmicamente con calentamiento continuo (a 20 K/min) hasta el punto A en la figura 3.3; DSC2: tratada térmicamente con calentamiento continuo para el rango completo de temperaturas.
2 0 2 5 3 0 3 5 4 0 4 5 5 0 5 5 6 0 6 5 7 0
Inte
nsi
da
d [
u.
a.]
G e1 5
T e8 5
Á n g u l o d e d i f r a c c i ó n 2 θ [ g r a d o s ]
T e G e T e
D S C 2
D S C 1
M S
M Q
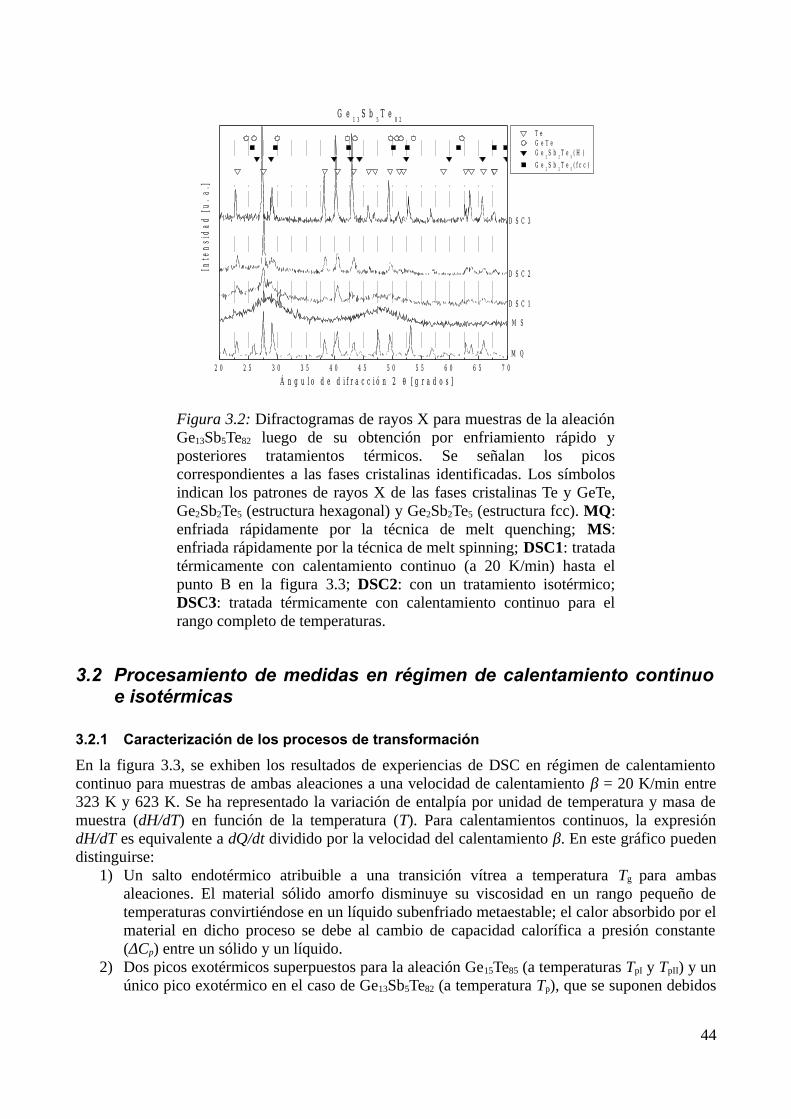
3.2 Procesamiento de medidas en régimen de calentamiento continuo e isotérmicas
3.2.1 Caracterización de los procesos de transformación
En la figura 3.3, se exhiben los resultados de experiencias de DSC en régimen de calentamiento continuo para muestras de ambas aleaciones a una velocidad de calentamiento β = 20 K/min entre 323 K y 623 K. Se ha representado la variación de entalpía por unidad de temperatura y masa de muestra (dH/dT) en función de la temperatura (T). Para calentamientos continuos, la expresión dH/dT es equivalente a dQ/dt dividido por la velocidad del calentamiento β. En este gráfico pueden distinguirse:
1) Un salto endotérmico atribuible a una transición vítrea a temperatura Tg para ambas aleaciones. El material sólido amorfo disminuye su viscosidad en un rango pequeño de temperaturas convirtiéndose en un líquido subenfriado metaestable; el calor absorbido por el material en dicho proceso se debe al cambio de capacidad calorífica a presión constante (ΔCp) entre un sólido y un líquido.
2) Dos picos exotérmicos superpuestos para la aleación Ge15Te85 (a temperaturas TpI y TpII) y un único pico exotérmico en el caso de Ge13Sb5Te82 (a temperatura Tp), que se suponen debidos
44
Figura 3.2: Difractogramas de rayos X para muestras de la aleación Ge13Sb5Te82 luego de su obtención por enfriamiento rápido y posteriores tratamientos térmicos. Se señalan los picos correspondientes a las fases cristalinas identificadas. Los símbolos indican los patrones de rayos X de las fases cristalinas Te y GeTe, Ge2Sb2Te5 (estructura hexagonal) y Ge2Sb2Te5 (estructura fcc). MQ: enfriada rápidamente por la técnica de melt quenching; MS: enfriada rápidamente por la técnica de melt spinning; DSC1: tratada térmicamente con calentamiento continuo (a 20 K/min) hasta el punto B en la figura 3.3; DSC2: con un tratamiento isotérmico; DSC3: tratada térmicamente con calentamiento continuo para el rango completo de temperaturas.
2 0 2 5 3 0 3 5 4 0 4 5 5 0 5 5 6 0 6 5 7 0
Á n g u l o d e d i f r a c c i ó n 2 θ [ g r a d o s ]
Inte
nsi
da
d [
u.
a.]
T e G e T e G e
2S b
2T e
5( H )
G e2
S b2
T e5
( f c c )
D S C 3
D S C 2
D S C 1
M S
G e1 3
S b5
T e8 2
M Q
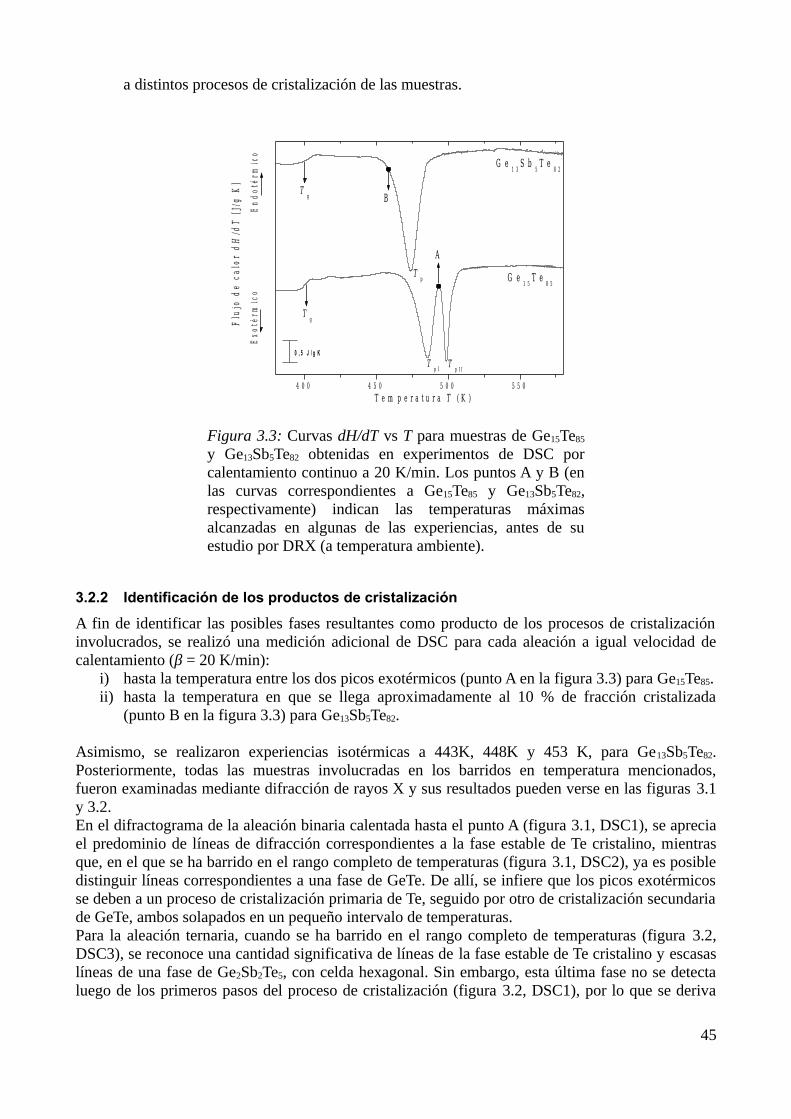
a distintos procesos de cristalización de las muestras.
3.2.2 Identificación de los productos de cristalización
A fin de identificar las posibles fases resultantes como producto de los procesos de cristalización involucrados, se realizó una medición adicional de DSC para cada aleación a igual velocidad de calentamiento (β = 20 K/min):
i) hasta la temperatura entre los dos picos exotérmicos (punto A en la figura 3.3) para Ge15Te85.ii) hasta la temperatura en que se llega aproximadamente al 10 % de fracción cristalizada
(punto B en la figura 3.3) para Ge13Sb5Te82.
Asimismo, se realizaron experiencias isotérmicas a 443K, 448K y 453 K, para Ge13Sb5Te82. Posteriormente, todas las muestras involucradas en los barridos en temperatura mencionados, fueron examinadas mediante difracción de rayos X y sus resultados pueden verse en las figuras 3.1 y 3.2.En el difractograma de la aleación binaria calentada hasta el punto A (figura 3.1, DSC1), se aprecia el predominio de líneas de difracción correspondientes a la fase estable de Te cristalino, mientras que, en el que se ha barrido en el rango completo de temperaturas (figura 3.1, DSC2), ya es posible distinguir líneas correspondientes a una fase de GeTe. De allí, se infiere que los picos exotérmicos se deben a un proceso de cristalización primaria de Te, seguido por otro de cristalización secundaria de GeTe, ambos solapados en un pequeño intervalo de temperaturas. Para la aleación ternaria, cuando se ha barrido en el rango completo de temperaturas (figura 3.2, DSC3), se reconoce una cantidad significativa de líneas de la fase estable de Te cristalino y escasas líneas de una fase de Ge2Sb2Te5, con celda hexagonal. Sin embargo, esta última fase no se detecta luego de los primeros pasos del proceso de cristalización (figura 3.2, DSC1), por lo que se deriva
45
Figura 3.3: Curvas dH/dT vs T para muestras de Ge15Te85
y Ge13Sb5Te82 obtenidas en experimentos de DSC por calentamiento continuo a 20 K/min. Los puntos A y B (en las curvas correspondientes a Ge15Te85 y Ge13Sb5Te82, respectivamente) indican las temperaturas máximas alcanzadas en algunas de las experiencias, antes de su estudio por DRX (a temperatura ambiente).
4 0 0 4 5 0 5 0 0 5 5 0
Tp I
Tp I I
Tp
A
BT
g
Tg
En
do
térm
ico
Ex
oté
rmic
o
Flu
jo d
e c
alo
r d
H/d
T [
J/g
K]
G e1 5
T e8 5
G e1 3
S b5
T e8 2
T e m p e r a t u r a T ( K )
0 , 5 J / g K

que ambas fases no cristalizan en forma simultánea. La micrografía óptica en la figura 3.4 (obtenida después de un tratamiento térmico hasta el punto B) sugiere una estructura que consiste en partículas esféricas de cristales de Te rodeados por una matriz amorfa. Difractogramas posteriores a los tratamientos isotérmicos (figura 3.2, DSC2) muestran sólo una incipiente línea de difracción debido a la fase ternaria, es decir que la cristalización principal en el proceso es la de cristales de Te, en acuerdo con los resultados de calentamiento continuo.
3.2.3 Determinación de parámetros calorimétricos
En las figuras 3.5 y 3.6, se exhiben las curvas dH/dT vs T para muestras de las aleaciones binaria y ternaria respectivamente, obtenidas por calentamientos continuos a velocidad β, constante, de 5 K/min, 10 K/min, 20 K/min, 40 K/min y 80 K/min entre 323 K y 623 K. Si bien las formas de las curvas son similares y estaríamos siempre en presencia de los procesos analizados previamente para el caso de 20 K/min, se advierte que a medida que aumenta β, las transformaciones se desplazan a mayores temperaturas. Esta situación resulta característica de los procesos activados térmicamente [Christian, 1975].En la tabla 3.1, se resume la dependencia con la velocidad de calentamiento de los siguientes parámetros calorimétricos: temperatura de transcición vítrea (Tg), variación de calor específico a presión constante de la transición vítrea (Δcp), temperatura de onset de la cristalización (Tx), temperaturas de picos exotérmicos de cristalización (Tp, TpI, TpII) y variación de entalpía de transformación, en este caso cristalización (ΔHc).
46
Figura 3.4: Micrografía óptica de una muestra de Ge13Sb5Te82 después de un tratamiento térmico hasta el punto B en la figura 3.3 (aumento óptico: 1000x)
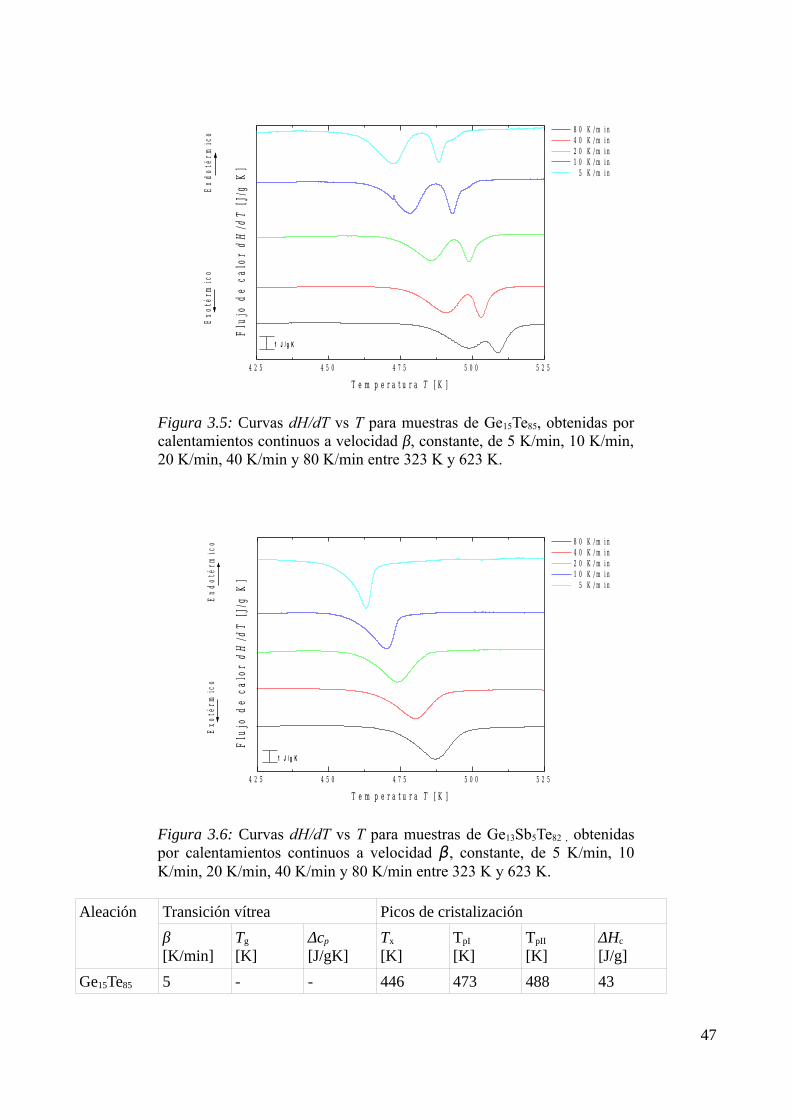
Aleación Transición vítrea Picos de cristalización
β[K/min]
Tg
[K]Δcp
[J/gK]Tx
[K]TpI
[K]TpII
[K]ΔHc
[J/g]
Ge15Te85 5 - - 446 473 488 43
47
Figura 3.5: Curvas dH/dT vs T para muestras de Ge15Te85, obtenidas por calentamientos continuos a velocidad β, constante, de 5 K/min, 10 K/min, 20 K/min, 40 K/min y 80 K/min entre 323 K y 623 K.
4 2 5 4 5 0 4 7 5 5 0 0 5 2 5
8 0 K / m i n 4 0 K / m i n 2 0 K / m i n 1 0 K / m i n 5 K / m i n
Flu
jo d
e c
alo
r d
H/d
T [
J/g
K]
T e m p e r a t u r a T [ K ]
1 J / g K
En
do
térm
ico
Ex
oté
rmic
o
Figura 3.6: Curvas dH/dT vs T para muestras de Ge13Sb5Te82 , obtenidas por calentamientos continuos a velocidad β, constante, de 5 K/min, 10 K/min, 20 K/min, 40 K/min y 80 K/min entre 323 K y 623 K.
4 2 5 4 5 0 4 7 5 5 0 0 5 2 5
1 J / g K
8 0 K / m i n 4 0 K / m i n 2 0 K / m i n 1 0 K / m i n 5 K / m i n
Flu
jo d
e c
alo
r d
H/d
T [
J/g
K]
T e m p e r a t u r a T [ K ]
En
do
térm
ico
Ex
oté
rmic
o
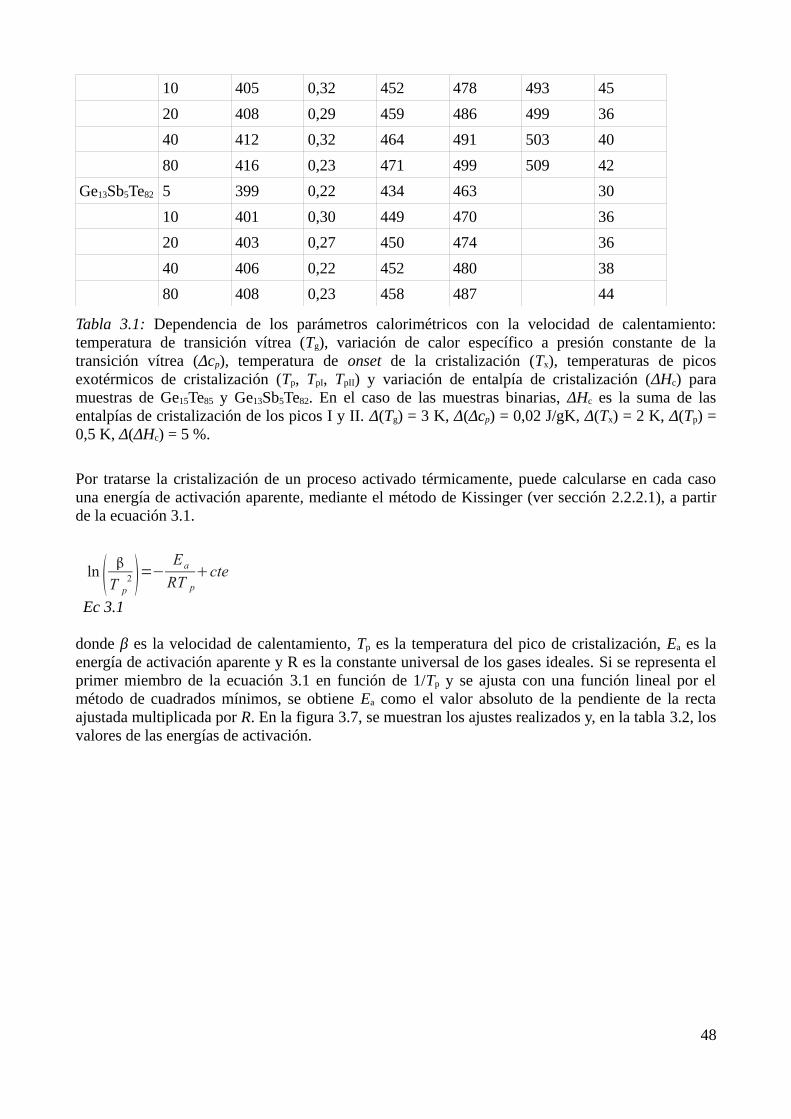
10 405 0,32 452 478 493 45
20 408 0,29 459 486 499 36
40 412 0,32 464 491 503 40
80 416 0,23 471 499 509 42
Ge13Sb5Te82 5 399 0,22 434 463 30
10 401 0,30 449 470 36
20 403 0,27 450 474 36
40 406 0,22 452 480 38
80 408 0,23 458 487 44
Tabla 3.1: Dependencia de los parámetros calorimétricos con la velocidad de calentamiento: temperatura de transición vítrea (Tg), variación de calor específico a presión constante de la transición vítrea (Δcp), temperatura de onset de la cristalización (Tx), temperaturas de picos exotérmicos de cristalización (Tp, TpI, TpII) y variación de entalpía de cristalización (ΔHc) para muestras de Ge15Te85 y Ge13Sb5Te82. En el caso de las muestras binarias, ΔHc es la suma de las entalpías de cristalización de los picos I y II. Δ(Tg) = 3 K, Δ(Δcp) = 0,02 J/gK, Δ(Tx) = 2 K, Δ(Tp) = 0,5 K, Δ(ΔHc) = 5 %.
Por tratarse la cristalización de un proceso activado térmicamente, puede calcularse en cada caso una energía de activación aparente, mediante el método de Kissinger (ver sección 2.2.2.1), a partir de la ecuación 3.1.
Ec 3.1
donde β es la velocidad de calentamiento, Tp es la temperatura del pico de cristalización, Ea es la energía de activación aparente y R es la constante universal de los gases ideales. Si se representa el primer miembro de la ecuación 3.1 en función de 1/Tp y se ajusta con una función lineal por el método de cuadrados mínimos, se obtiene Ea como el valor absoluto de la pendiente de la recta ajustada multiplicada por R. En la figura 3.7, se muestran los ajustes realizados y, en la tabla 3.2, los valores de las energías de activación.
48
ln
T p2 =−
Ea
RT p
cte
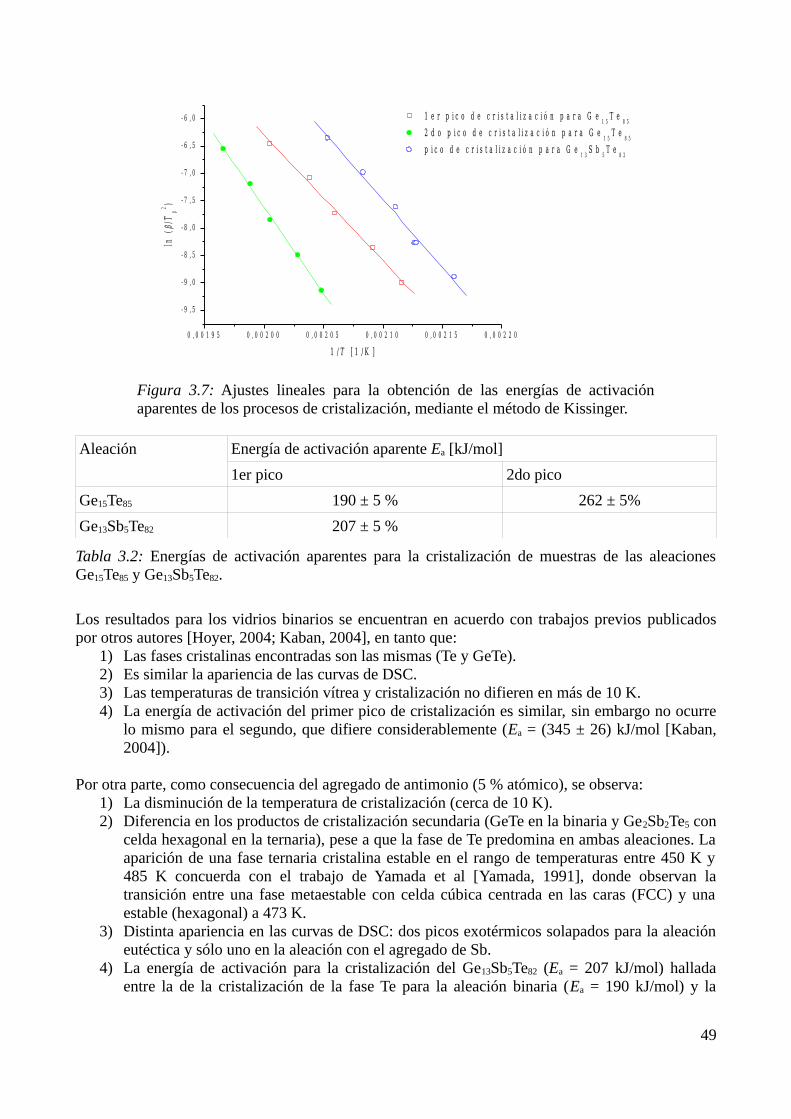
Aleación Energía de activación aparente Ea [kJ/mol]
1er pico 2do pico
Ge15Te85 190 ± 5 % 262 ± 5%
Ge13Sb5Te82 207 ± 5 %
Tabla 3.2: Energías de activación aparentes para la cristalización de muestras de las aleaciones Ge15Te85 y Ge13Sb5Te82.
Los resultados para los vidrios binarios se encuentran en acuerdo con trabajos previos publicados por otros autores [Hoyer, 2004; Kaban, 2004], en tanto que:
1) Las fases cristalinas encontradas son las mismas (Te y GeTe).2) Es similar la apariencia de las curvas de DSC.3) Las temperaturas de transición vítrea y cristalización no difieren en más de 10 K.4) La energía de activación del primer pico de cristalización es similar, sin embargo no ocurre
lo mismo para el segundo, que difiere considerablemente (Ea = (345 ± 26) kJ/mol [Kaban,2004]).
Por otra parte, como consecuencia del agregado de antimonio (5 % atómico), se observa:1) La disminución de la temperatura de cristalización (cerca de 10 K).2) Diferencia en los productos de cristalización secundaria (GeTe en la binaria y Ge2Sb2Te5 con
celda hexagonal en la ternaria), pese a que la fase de Te predomina en ambas aleaciones. La aparición de una fase ternaria cristalina estable en el rango de temperaturas entre 450 K y 485 K concuerda con el trabajo de Yamada et al [Yamada, 1991], donde observan la transición entre una fase metaestable con celda cúbica centrada en las caras (FCC) y una estable (hexagonal) a 473 K.
3) Distinta apariencia en las curvas de DSC: dos picos exotérmicos solapados para la aleación eutéctica y sólo uno en la aleación con el agregado de Sb.
4) La energía de activación para la cristalización del Ge13Sb5Te82 (Ea = 207 kJ/mol) hallada entre la de la cristalización de la fase Te para la aleación binaria (Ea = 190 kJ/mol) y la
49
Figura 3.7: Ajustes lineales para la obtención de las energías de activación aparentes de los procesos de cristalización, mediante el método de Kissinger.
0 , 0 0 1 9 5 0 , 0 0 2 0 0 0 , 0 0 2 0 5 0 , 0 0 2 1 0 0 , 0 0 2 1 5 0 , 0 0 2 2 0
- 9 , 5
- 9 , 0
- 8 , 5
- 8 , 0
- 7 , 5
- 7 , 0
- 6 , 5
- 6 , 0
ln (
β/T
p
2)
1 / T [ 1 / K ]
1 e r p i c o d e c r i s t a l i z a c i ó n p a r a G e1 5
T e8 5
2 d o p i c o d e c r i s t a l i z a c i ó n p a r a G e1 5
T e8 5
p i c o d e c r i s t a l i z a c i ó n p a r a G e1 3
S b5
T e8 2
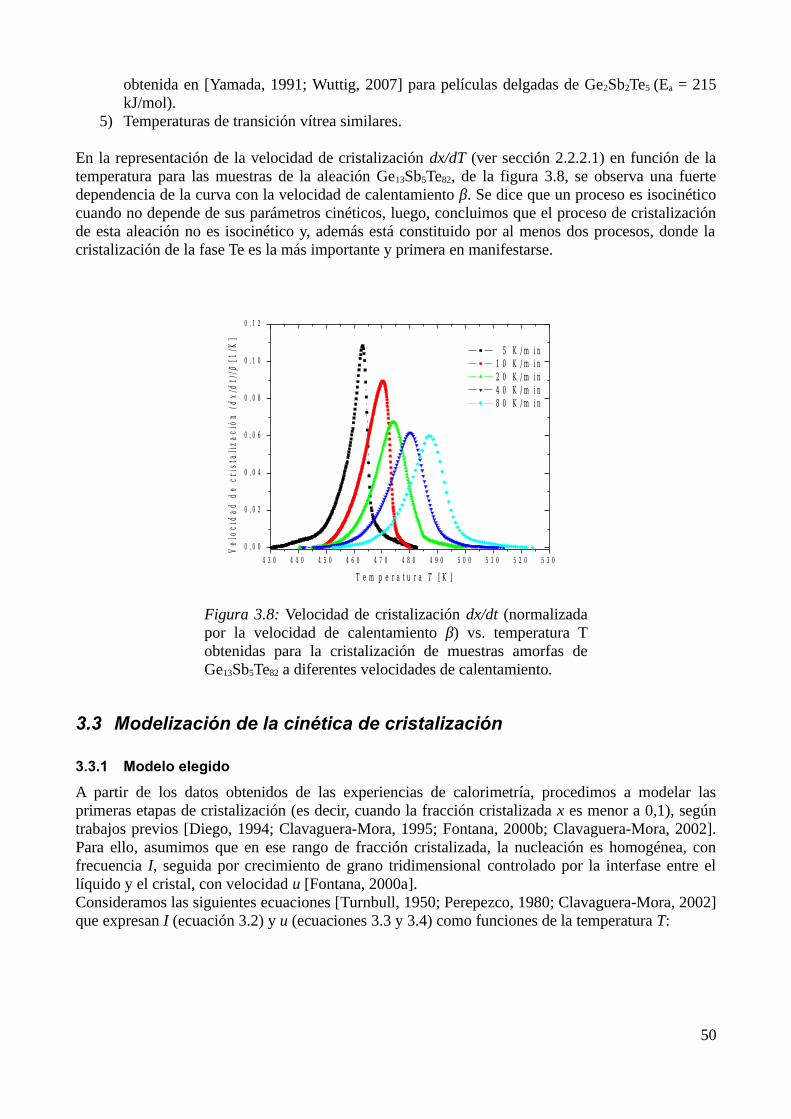
obtenida en [Yamada, 1991; Wuttig, 2007] para películas delgadas de Ge2Sb2Te5 (Ea = 215 kJ/mol).
5) Temperaturas de transición vítrea similares.
En la representación de la velocidad de cristalización dx/dT (ver sección 2.2.2.1) en función de la temperatura para las muestras de la aleación Ge13Sb5Te82, de la figura 3.8, se observa una fuerte dependencia de la curva con la velocidad de calentamiento β. Se dice que un proceso es isocinético cuando no depende de sus parámetros cinéticos, luego, concluimos que el proceso de cristalización de esta aleación no es isocinético y, además está constituido por al menos dos procesos, donde la cristalización de la fase Te es la más importante y primera en manifestarse.
3.3 Modelización de la cinética de cristalización
3.3.1 Modelo elegido
A partir de los datos obtenidos de las experiencias de calorimetría, procedimos a modelar las primeras etapas de cristalización (es decir, cuando la fracción cristalizada x es menor a 0,1), según trabajos previos [Diego, 1994; Clavaguera-Mora, 1995; Fontana, 2000b; Clavaguera-Mora, 2002]. Para ello, asumimos que en ese rango de fracción cristalizada, la nucleación es homogénea, con frecuencia I, seguida por crecimiento de grano tridimensional controlado por la interfase entre el líquido y el cristal, con velocidad u [Fontana, 2000a].Consideramos las siguientes ecuaciones [Turnbull, 1950; Perepezco, 1980; Clavaguera-Mora, 2002] que expresan I (ecuación 3.2) y u (ecuaciones 3.3 y 3.4) como funciones de la temperatura T:
50
Figura 3.8: Velocidad de cristalización dx/dt (normalizada por la velocidad de calentamiento β) vs. temperatura T obtenidas para la cristalización de muestras amorfas de Ge13Sb5Te82 a diferentes velocidades de calentamiento.
4 3 0 4 4 0 4 5 0 4 6 0 4 7 0 4 8 0 4 9 0 5 0 0 5 1 0 5 2 0 5 3 0
0 , 0 0
0 , 0 2
0 , 0 4
0 , 0 6
0 , 0 8
0 , 1 0
0 , 1 2
Ve
loci
da
d d
e c
rist
ali
zaci
ón
(d
x/d
t)/β
[1
/K]
T e m p e r a t u r a T [ K ]
5 K / m i n 1 0 K / m i n 2 0 K / m i n 4 0 K / m i n 8 0 K / m i n

Ec 3.2
Ec 3.3
Ec 3.4
donde Nv es el número de átomos por unidad de volumen; a0, la distancia media entre dos átomos; R, la constante universal de los gases ideales; k, la constante de Boltzmann; σ, la energía interfasial molar entre el núcleo y el líquido; L, el espesor interfasial medio, en unidades de a0; λ, el producto entre la fracción de sitios superficiales donde se agregan preferentemente los átomos y el largo de la interfase, en unidades de a0. La viscosidad η viene dada por la expresión de Vogel-Fulcher [Diego,1994; Clavaguera-Mora, 1995], en la ecuación 3.5, donde η0, A y T0 son constantes a determinar.
Ec 3.5
ΔG* es la energía libre de Gibbs de formación de un núcleo de tamaño crítico [Hrubý, 1972; Biswas, 2006] y viene dada por la ecuación 3.6.
Ec 3.6
A partir de la ecuación 3.7, donde ΔCp es la diferencia de capacidad calorífica a presión constante entre el líquido y el cristal y ΔSm la entropía de fusión, si se admite que Γ se mantiene constante, ΔG es la diferencia de energía libre de Gibbs entre el líquido subenfriado y el cristal y está dada por la ecuación 3.8 [Perepezco, 1980; Fontana, 2000b], donde Tr es la temperatura reducida (ecuación 3.9, siendo Tm la temperatura de fusión).
Ec 3.7
51
I T =[ 4
RT 1/2 N v b
] exp−G *RT
u T =a0b [ 1−exp−G
RT ]
b=kT
3 L2a03
=0 exp AT−T 0
G *=16 3
3G 2
=C p
Sm

Ec 3.8
Ec 3.9
Se calculan las curvas Transformación-Tiempo-Temperatura (TTT) y Transformación-velocidad de calentamiento-Temperatura (ThrT), según proponen trabajos de otros autores [Diego, 1994; Clavaguera-Mora, 1995; Clavaguera-Mora, 2002; Ureña, 2002].
Para un tratamiento térmico a temperatura T constante (condiciones isotérmicas), la fracción cristalizada después de un tiempo t está dada por la ecuación 3.10.
Ec 3.10
Las curvas TTT se obtienen graficando T vs t para valores elegidos de x, a partir de los datos experimentales. Con las expresiones de I(T) y u(T) asumidas y algunos parámetros conocidos o estimados, se ajusta el resto de los parámetros involucrados.
De forma análoga, para un tratamiento térmico a velocidad de calentamiento constante β (condiciones de calentamiento continuo), la fracción cristalizada al alcanzar una temperatura T está dada por la ecuación 3.11, donde Vng(T', T) es el volumen a la temperatura T de un núcleo formado a la temperatura T' y se expresa en la ecuación 3.12.
Ec 3.11
Ec 3.12
Las curvas THrT se obtienen graficando T vs β para valores elegidos de x, a partir de los datos experimentales. Con las expresiones de I(T) y u(T) asumidas y algunos parámetros conocidos o estimados, se ajusta el resto de los parámetros involucrados.
52
G= H m[1−T r1−− T r ln T r]
T r=TT m
x T , t =1−exp−3I T u T
3 t 4
x T ,=1−exp 1∫0
T
I T ' V ng T ' ,T dT '
V ng T ' ,T =43 [ 1
∫T '
T
u T ' ' dT ' ' ]3

3.3.2 Determinación de diagramas Transformación-velocidad de calentamiento-Temperatura (THrT) y Transformación-Tiempo-Temperatura (TTT)
Asumiendo la fase Te como único producto de las primeras etapas de cristalización tanto de la aleación Ge15Te85 como de la aleación Ge13Sb5Te82, junto con las suposiciones del modelo elegido, calculamos las curvas THrT y TTT. Se cuenta con datos experimentales en condiciones de calentamiento continuo e isotérmicas para la aleación ternaria y sólo de calentamiento continuo para la aleación binaria.Los parámetros ΔHm, σ, a0, L2/λ, Tm y Γ se admiten iguales para ambas aleaciones. Para ΔHm, σ, a0, L2/λ se toman los valores presentados por trabajos previos que estudiaron la cristalización de la fase Te en las aleaciones Ga20Te80 y otras del sistema Ga-Sn-Te [Clavaguera, 1998; Fontana, 2000b]. Tm
se toma como la temperatura de fusión del eutéctico binario y Γ se estima a partir de ΔCp, ΔHm y Tm.Siendo cercanas ambas composiciones, así como sus temperaturas de transición vítrea y de fusión, consideramos iguales los parámetros de viscosidad η(Tm), A y T0 para ambas aleaciones. Sí habrá diferencias en el parámetro Nv/L2 por tratarse de distintas composiciones.Imponiendo además η(Tg) ~ 1013 P, valor de viscocidad que habitualmente se considera para una transición líquido subenfriado-sólido amorfo, se ajustaron los parámetros Nv/L2, η(Tm), A y T0 con los datos experimentales isotérmicos y de calentamiento continuo de las curvas correspondientes a x = 0,1. Los parámetros ajustados junto con los estimados se resumen en la tabla 3.3.Las curvas THrT y TTT calculadas para x = 0,1 y x = 10-6 se muestran en la figura 3.9. Exhiben un ajuste adecuado con los valores experimentales.
Parámetros estimados
ΔHm [kJ/mol] 17,0
Tm [K] 648
Γ 1,2
σ 0,33 ΔHm
a0 [m] 2,8.10-10
L2/λ 10
Parámetros ajustados
Aleación Ge15Te85 Ge13Sb5Te82
Nv/L2 [1/m3] 1,27.1017 ± 10 % 1,16.1019 ± 10 %
η(Tm) [P] 0,010
A [K] 1250,0 ± 5 %
T0 [K] 370 ± 4
Tabla 3.3: Valores de los parámetros estimados y ajustados al modelar los primeros pasos de cristalización de las aleaciones Ge15Te85 y Ge13Sb5Te82.
53
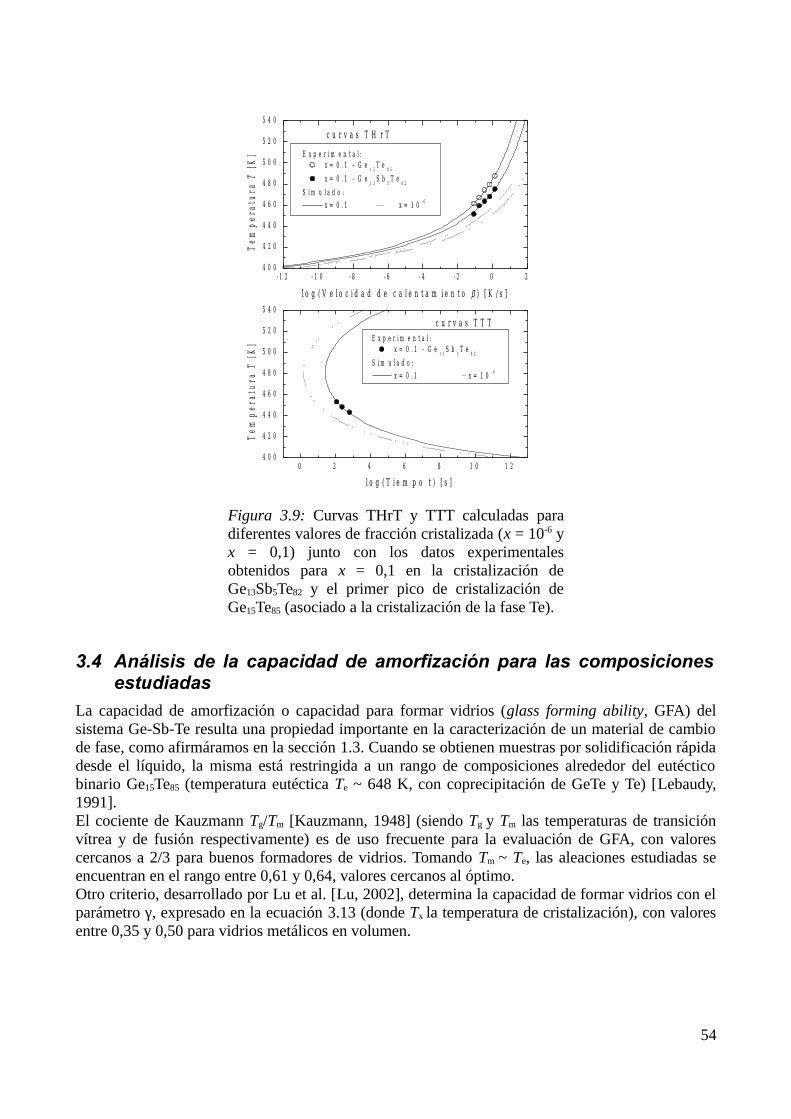
3.4 Análisis de la capacidad de amorfización para las composiciones estudiadas
La capacidad de amorfización o capacidad para formar vidrios (glass forming ability, GFA) del sistema Ge-Sb-Te resulta una propiedad importante en la caracterización de un material de cambio de fase, como afirmáramos en la sección 1.3. Cuando se obtienen muestras por solidificación rápida desde el líquido, la misma está restringida a un rango de composiciones alrededor del eutéctico binario Ge15Te85 (temperatura eutéctica Te ~ 648 K, con coprecipitación de GeTe y Te) [Lebaudy,1991].El cociente de Kauzmann Tg/Tm [Kauzmann, 1948] (siendo Tg y Tm las temperaturas de transición vítrea y de fusión respectivamente) es de uso frecuente para la evaluación de GFA, con valores cercanos a 2/3 para buenos formadores de vidrios. Tomando Tm ~ Te, las aleaciones estudiadas se encuentran en el rango entre 0,61 y 0,64, valores cercanos al óptimo.Otro criterio, desarrollado por Lu et al. [Lu, 2002], determina la capacidad de formar vidrios con el parámetro γ, expresado en la ecuación 3.13 (donde Tx la temperatura de cristalización), con valores entre 0,35 y 0,50 para vidrios metálicos en volumen.
54
Figura 3.9: Curvas THrT y TTT calculadas para diferentes valores de fracción cristalizada (x = 10-6 y x = 0,1) junto con los datos experimentales obtenidos para x = 0,1 en la cristalización de Ge13Sb5Te82 y el primer pico de cristalización de Ge15Te85 (asociado a la cristalización de la fase Te).
4 0 0
4 2 0
4 4 0
4 6 0
4 8 0
5 0 0
5 2 0
5 4 0
0 2 4 6 8 1 0 1 2
4 0 0
4 2 0
4 4 0
4 6 0
4 8 0
5 0 0
5 2 0
5 4 0
- 1 2 - 1 0 - 8 - 6 - 4 - 2 0 2
c u r v a s T T T
l o g ( T i e m p o t ) [ s ]
Te
mp
era
tura
T [
K] E x p e r i m e n t a l :
x = 0 . 1 - G e1 3
S b5
T e8 2
S i m u l a d o :
x = 0 . 1 x = 1 0 - 6
c u r v a s T H r T
l o g ( V e l o c i d a d d e c a l e n t a m i e n t o β ) [ K / s ]
Te
mp
era
tura
T [
K] E x p e r i m e n t a l :
x = 0 . 1 - G e1 5
T e8 5
x = 0 . 1 - G e1 3
S b5
T e8 2
S i m u l a d o :
x = 0 . 1 x = 1 0 - 6

Ec 3.13
Para las aleaciones estudiadas, resulta en el rango entre 0,41 y 0,44. Ambos criterios evidencian buena GFA.La estabilidad del amorfo se analiza con el parámetro de Hrubý Kgl [Hrubý, 1972; Biswas, 2006], expresado en la ecuación 3.14. Mayor será la estabilidad y presentará mayor dificultad para cristalizar cuando es calentado, conforme aumenta su valor. También es indicio de mejor GFA: para Kgl ~ 0,1 es muy difícil preparar vidrios, mientras que para Kgl ~ 0,5 se obtienen por templado al aire [Majid, 1982].
Ec 3.14
En el caso de las muestras estudiadas en este trabajo, resulta entre 0,24 y 0,31 para la aleación binaria; entre 0,24 y 0,26 para la ternaria. Por ello, puede considerarse la aleación binaria apenas más estable.
Los resultados de este capítulo fueron parcialmente publicados en [Rocca, 2009].
55
=T x
T gT m
K gl=T x−T g
T m−T x

3.5 Referencias
[Biswas, 2006] Biswas, K., Venkataraman, S., Zhang, W. Y., Ram, S., Eckert, J. (2006). Glass-forming ability and fragility parameter of amorphous Fe67Co9.5Nd3Dy0.5B20. Journal of Applied Physics, 100(2). doi:10.1063/1.2214333
[Christian, 1975] Christian, J.W. (1975). Theory of Transformations in Metals and Alloys, Oxford: Pergamon.
[Clavaguera-Mora, 1995] Clavaguera-Mora, M. T. (1995). Glassy materials: thermodynamic and kinetic quantities. Journal of Alloys and Compounds, 220(1-2), 197-205. doi:10.1016/0925-8388(94)06034-7
[Clavaguera-Mora, 2002] Clavaguera-Mora, M. T., Clavaguera, N., Crespo, D., Pradell, T. (2002). Crystallisation kinetics and microstructure development in metallic systems. Progress in Materials Science, 47(6), 559–619. doi:10.1016/S0079-6425(00)00021-9
[Clavaguera, 1998] Clavaguera, N., Clavaguera-Mora, M. T., Fontana, M. (1998). Accuracy in the experimental calorimetric study of the crystallization kinetics and predictive transformation diagrams: Application to a Ga–Te amorphous alloy. Journal of Materials Research, 13(3), 744-753. doi:10.1557/JMR.1998.0094
[Diego, 1994] Diego J. A., Clavaguera-Mora, M. T., Clavaguera, N. (1994). Thermodynamic, kinetic and structural mechanisms controlling the formation of nanocrystalline Nd-Fe-B materials. Materials Science and Engineering: A, 179–180, 526–530. doi:10.1016/0921-5093(94)90260-7
[Fontana, 2000a] Fontana, M., Arcondo, B., Clavaguera-Mora, M. T., Clavaguera, N. (2000). Crystallization kinetics driven by two simultaneous modes of crystal growth. Philosophical Magazine Part B, 80(10), 1833-1856. doi:10.1080/13642810008216509
[Fontana, 2000b] Fontana, M., Arcondo B., Clavaguera-Mora, M. T., Clavaguera, N., Greneche, J. M. (2000). Crystallization kinetics and structural aspects of TeGaSn amorphous alloys. Journal of Applied Physics, 88(6), 3276-3284. doi:10.1063/1.1288691
[Hoyer, 2004] Hoyer W., Kaban, I., Jóvári, P., Dost, E. (2004). Crystallization behavior and structure of amorphous Ge15Te85 and Ge20Te80 alloys. Journal of Non-Crystalline Solids, 338-340, 565-568. doi: 10.1016/j.jnoncrysol.2004.03.043
[Hrubý, 1972] Hrubý, A. (1972). Evaluation of glass-forming tendency by means of DTA. Czechoslovak Journal of Physics, 22(11), 1187-1193. doi:10.1007/BF01690134
[Kaban, 2004] Kaban, I., Dost, E., Hoyer W. (2004). Thermodynamic and structural investigations of heat-treated amorphous Ge-Te alloys. Journal of Alloys and Compounds, 379(1-2), 166-170. doi: 10.1016/j.jallcom.2004.02.033
[Kauzmann, 1948] Kauzmann, W. (1948). The Nature of the Glassy State and the Behavior of Liquids at Low Temperatures. Chemical Reviews, 43(2), 219-256. doi:10.1021/cr60135a002
56

[Lebaudy, 1991] Lebaudy, P., Saiter, J. M., Grenet, J., Belhadji, M., Vautier, C. (1991). Identification of amorphous zones in the GeTeSb system. Materials Science and Engineering: A, 132, 273–276. doi:10.1016/0921-5093(91)90384-Y
[Lu, 2002] Lu, Z.P., Liu, C.T. (2002). A new glass-forming ability criterion for bulk metallic glasses. Acta Materialia, 50(13), 3501–3512. doi:10.1016/S1359-6454(02)00166-0
[Majid, 1982] Majid, C.A. (1982). Thermal analysis of chalcogenide glasses of the system (As2Se3)1-x:(Tl2Se)x. Internal Report IC/82/131. Trieste, Italia: International Centre for Theoretical Physics.
[Perepezco, 1980] Perepezco, J.H. (1980). En R. Mehrabian, B.H. Kear, M. Cohen (Eds.), Rapid Solidification Processing Principles and Technologies (p. 56). Baton Rouge, LA: Claitor’s.
[Rocca, 2009] Rocca, J., Erazú, M., Fontana, M., Arcondo, B. (2009). Crystallization process on amorphous GeTeSb samples near to eutectic point Ge15Te85. Journal of Non-Crystalline Solids, 355 (37–42), 2068–2073. doi:10.1016/j.jnoncrysol.2008.10.020
[Turnbull, 1950] Turnbull, D. (1950). Formation of Crystal Nuclei in Liquid Metals. Journal of Applied Physics, 21(10), 1022-1028. doi:10.1063/1.1699435
[Ureña, 2002] Ureña, A., Fontana M., Arcondo, B., Clavaguera-Mora, M. T., Clavaguera, N. (2002). Influence of Cu addition in the crystallization of the superionic glass (Ge25Se75)75Ag25. Journal of Non-Crystalline Solids, 304(1-3), 306-314. doi:10.1016/S0022-3093(02)01039-6
[Wuttig, 2007] Wuttig, M., Lüsebrink, D., Wamwangi, D., Wełnic, W., Gilleßen, M., Dronskowski, R. (2007). The role of vacancies and local distortions in the design of new phase-change materials. Nature Materials, 6, 122-128. doi:10.1038/nmat1807
[Yamada, 1991] Yamada, N., Ohno, E., Nishiuchi, K., Akahira, N., Takao M. (1991). Rapid-phase transitions of GeTe-Sb2Te3 pseudobinary amorphous thin films for an optical disk memory. Journal of Applied Physics, 69(5), 2849-2856. doi:10.1063/1.348620
57
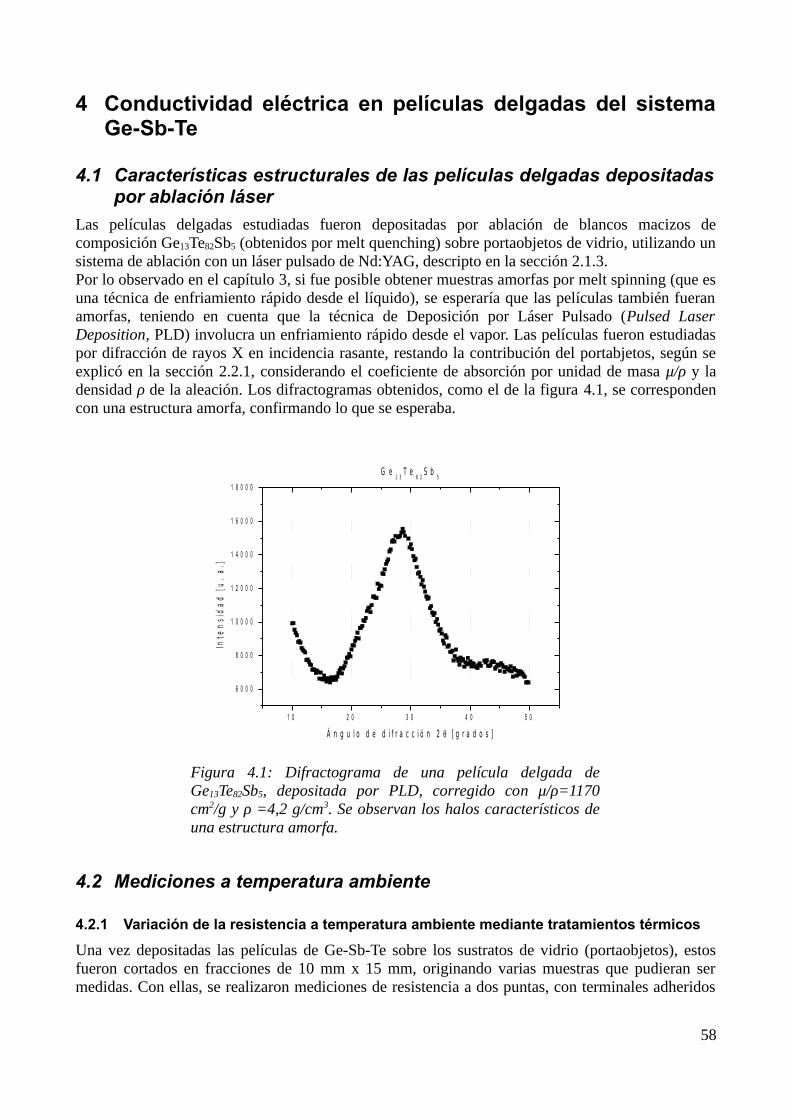
4 Conductividad eléctrica en películas delgadas del sistema Ge-Sb-Te
4.1 Características estructurales de las películas delgadas depositadas por ablación láser
Las películas delgadas estudiadas fueron depositadas por ablación de blancos macizos de composición Ge13Te82Sb5 (obtenidos por melt quenching) sobre portaobjetos de vidrio, utilizando un sistema de ablación con un láser pulsado de Nd:YAG, descripto en la sección 2.1.3.Por lo observado en el capítulo 3, si fue posible obtener muestras amorfas por melt spinning (que es una técnica de enfriamiento rápido desde el líquido), se esperaría que las películas también fueran amorfas, teniendo en cuenta que la técnica de Deposición por Láser Pulsado (Pulsed Laser Deposition, PLD) involucra un enfriamiento rápido desde el vapor. Las películas fueron estudiadas por difracción de rayos X en incidencia rasante, restando la contribución del portabjetos, según se explicó en la sección 2.2.1, considerando el coeficiente de absorción por unidad de masa μ/ρ y la densidad ρ de la aleación. Los difractogramas obtenidos, como el de la figura 4.1, se corresponden con una estructura amorfa, confirmando lo que se esperaba.
4.2 Mediciones a temperatura ambiente
4.2.1 Variación de la resistencia a temperatura ambiente mediante tratamientos térmicos
Una vez depositadas las películas de Ge-Sb-Te sobre los sustratos de vidrio (portaobjetos), estos fueron cortados en fracciones de 10 mm x 15 mm, originando varias muestras que pudieran ser medidas. Con ellas, se realizaron mediciones de resistencia a dos puntas, con terminales adheridos
58
Figura 4.1: Difractograma de una película delgada de Ge13Te82Sb5, depositada por PLD, corregido con μ/ρ=1170 cm2/g y ρ =4,2 g/cm3. Se observan los halos característicos de una estructura amorfa.
1 0 2 0 3 0 4 0 5 0
6 0 0 0
8 0 0 0
1 0 0 0 0
1 2 0 0 0
1 4 0 0 0
1 6 0 0 0
1 8 0 0 0
G e1 3
T e8 2
S b5
Inte
nsi
da
d [
u.
a.]
Á n g u l o d e d i f r a c c i ó n 2 θ [ g r a d o s ]

con pintura conductora de plata (con un secado mínimo de cuatro horas) y dispuestos en forma coplanar sobre la película.En primer lugar, la resistencia de cada muestra fue medida a temperatura ambiente. Luego de someterlas a un tratamiento isotérmico (cerca de 473 K durante una hora y media, previo calentamiento continuo a aproximadamente 4 K/min desde temperatura ambiente) sin haber retirado los contactos de plata, se repitió la medición a temperatura ambiente. En la tabla 4.1, se resumen los resultados de las mediciones sobre una muestra a la que se realizó un tratamiento térmico como el que se ha descripto. Se observa una disminución de cinco órdenes de magnitud en la resistencia medida, y por tanto, en la resistividad. Debido al conocimiento de las transformaciones sufridas por muestras en volumen de la aleación en el rango de temperaturas estudiado, atribuimos la variación significativa de la resistividad o conductividad a la transición amorfo-cristal, siendo el estado amorfo el de baja conductividad en apreciable contraste con la mayor conductividad en presencia de fases cristalizadas.
R ± ΔR [Ω]
Antes del tratamiento térmico (9,1 ± 0,2).108
Después del tratamiento térmico (1,5 ± 0,1).104
Tabla 4.1: Resistencia eléctrica medida para una película delgada de Ge13Te82Sb5, a temperatura ambiente, antes (con la película amorfa, como fue depositada) y después de un tratamiento térmico
(por el cual se cristaliza). El error expresado proviene de la fluctuación en la medición de la corriente para la tensión que impone la fuente del electrómetro utilizado.
4.2.2 Medición de la resistividad a temperatura ambiente con AFM
Utilizando un microscopio de fuerza atómica en modo spreading resistance, según se describe en la sección 2.2.3, se midió la resistencia de la película con una configuración distinta a la empleada en la sección 4.2.1. Esta vez, la película de material calcogenuro fue depositada por PLD sobre una película delgada de oro previamente adherida a un vidrio portaobjetos, por sputtering DC. Así, la sonda del microscopio que explora la superficie de la muestra se conectó a una fuente de tensión de manera de establecer una diferencia de potencial V respecto del contacto de oro ubicado entre la muestra y el sustrato de vidrio, y medir la corriente I que circula a través de la muestra.Simultáneamente con la medición de corriente a través de la película al explorar su superficie, se obtuvo un mapa topográfico de la misma, midiendo la altura h en función de la posición (x, y) de la punta durante el barrido. Adaptando la definición de resistividad ρ, según la ecuación 4.1, para un cuerpo de sección s y longitud l, y considerando que el volumen de la muestra no estará involucrado en su totalidad en la conducción eléctrica para la geometría estudiada, llegamos a la ecuación 4.2, donde s' es la sección efectiva de la película por la que circula corriente (ver figura 4.2).
Ec 4.1
Ec 4.2
59
ρ=R( sl )
=VI s '
h

Como se verá a continuación, el problema de la conducción eléctrica entre dos discos paralelos, cuando se encuentran separados por una capa infinita de material, presenta ecuaciones análogas a las de un capacitor electrostático, cuando se consideran efectos de borde. Por lo tanto, ambos fenómenos físicos pueden reducirse a un problema geométrico.En primer lugar, si consideraremos un capacitor de discos paralelos de radio r, separados por una distancia d, despreciando efectos de borde, su capacitancia C0 sigue la ecuación 4.3.
Ec 4.3
Al considerar los efectos de borde [Nishiyama, 1993], la capacitancia C se corrige con un factor de capacitancia normalizada Cn, que es función de b (cociente entre d y r). Cn tiene valores calculados por métodos numéricos para distintos b y una expresión empírica según la ecuación 4.4.
Ec 4.4
Para dos discos paralelos de radio r, separados una distancia 2h por un material con una constante dieléctrica ε, según el esquema de la figura 4.3, su capacitancia sigue la ecuación 4.5.
Ec 4.5
60
C0= sd
= r 2
d
Cn=11,298 b0,8670,01≤b≤1,0
Cn=11,298 b0,9821,0≤b≤10,0
C=C nbC0=Cns
2h
Figura 4.2: Medición de resistencia de películas delgadas por AFM en el modo spreading resistance. Las líneas representan las direcciones del vector densidad de corriente. Se denomina s a la sección de la punta AFM y s', a la sección promedio por la que circula corriente en la película.

Figura 4.3: Esquema de un capacitor de discos paralelos separados por una distancia 2h.
La capacitancia C es equivalente a la de dos capacitores en serie con el doble de capacitancia, a saber: entre uno de los discos y la equipotencial a distancia h, y entre la misma y el otro disco, también a distancia h. Dicha capacitancia viene expresada en la ecuación 4.6, donde Cn resulta un
factor de corrección del parámetrosh
.
Ec 4.6
La geometría del capacitor de capacitancia C1/2 es análoga a la de la punta de la sonda AFM con radio de curvatura característico r a una distancia h del sustrato de la película. Por lo tanto reescribimos la ecuación 4.2, con el factor de corrección conocido, en las ecuaciones 4.7 y 4.8.
Ec 4.7
Ec 4.8
Para la superficie de película estudiada (en cada medición, menor a 7 μm por 7μm), se ajustaron las imágenes topográficas para eliminar ruido instrumental y obtener la altura de la película de calcogenuro respecto de la base de oro. Se calculó la resistividad como el cociente entre la resistencia y la altura h para cada punto medido, multiplicado por la sección efectiva correspondiente a la muestra barrida con determinada punta de sonda AFM, usando un valor medio de h y el valor conocido del radio de curvatura característico r de la sonda, para el cálculo de b. Para las mediciones realizadas, se obtuvo un valor medio de 77,4 Ωm y un desvío estándar de 25,1 Ωm. En la tabla 4.2, se resumen los valores utilizados y obtenidos.
61
h
h
V
-V
0
C1/2=2Cns
2h=Cn
sh
=VI C n
sh = V
I hCn s =
VI h
s '
s '=C n s=C n r2
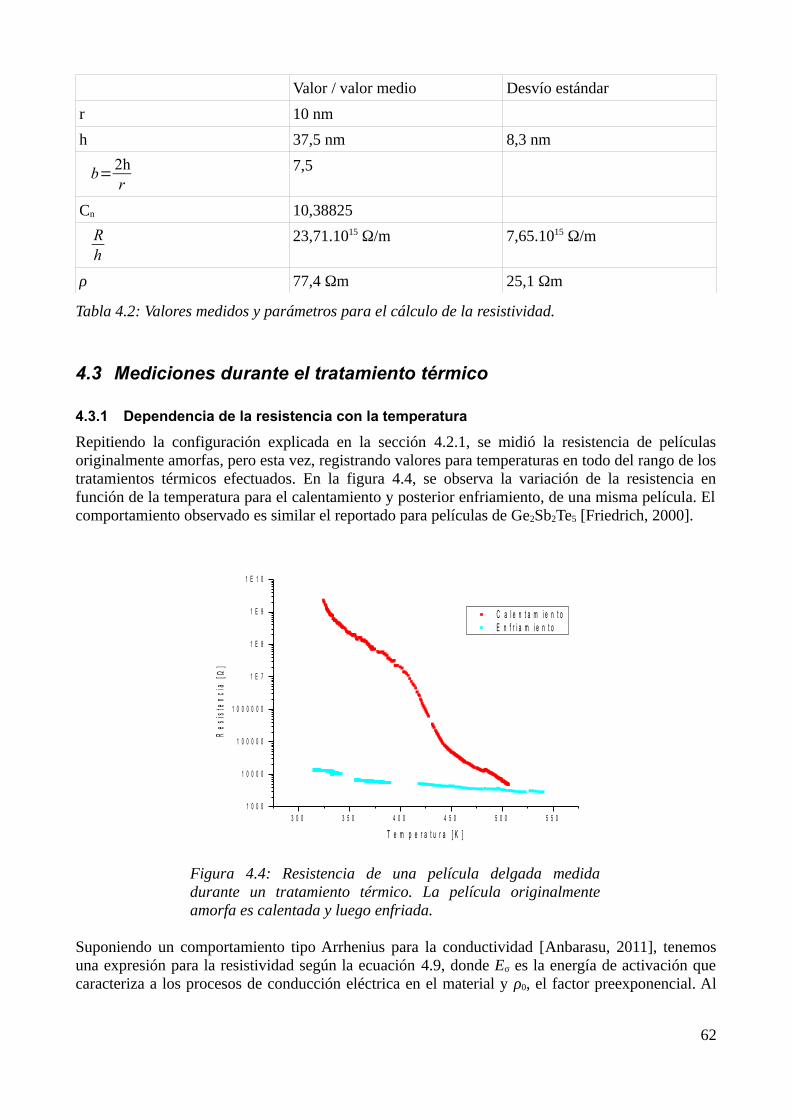
Valor / valor medio Desvío estándar
r 10 nm
h 37,5 nm 8,3 nm
b=2hr
7,5
Cn 10,38825
Rh
23,71.1015 Ω/m 7,65.1015 Ω/m
ρ 77,4 Ωm 25,1 Ωm
Tabla 4.2: Valores medidos y parámetros para el cálculo de la resistividad.
4.3 Mediciones durante el tratamiento térmico
4.3.1 Dependencia de la resistencia con la temperatura
Repitiendo la configuración explicada en la sección 4.2.1, se midió la resistencia de películas originalmente amorfas, pero esta vez, registrando valores para temperaturas en todo del rango de los tratamientos térmicos efectuados. En la figura 4.4, se observa la variación de la resistencia en función de la temperatura para el calentamiento y posterior enfriamiento, de una misma película. El comportamiento observado es similar el reportado para películas de Ge2Sb2Te5 [Friedrich, 2000].
Suponiendo un comportamiento tipo Arrhenius para la conductividad [Anbarasu, 2011], tenemos una expresión para la resistividad según la ecuación 4.9, donde Eσ es la energía de activación que caracteriza a los procesos de conducción eléctrica en el material y ρ0, el factor preexponencial. Al
62
Figura 4.4: Resistencia de una película delgada medida durante un tratamiento térmico. La película originalmente amorfa es calentada y luego enfriada.
3 0 0 3 5 0 4 0 0 4 5 0 5 0 0 5 5 01 0 0 0
1 0 0 0 0
1 0 0 0 0 0
1 0 0 0 0 0 0
1 E 7
1 E 8
1 E 9
1 E 1 0
C a l e n t a m i e n t o E n f r i a m i e n t o
Re
sist
en
cia
[Ω
]
T e m p e r a t u r a [ K ]

combinarse con la ecuación 4.1, llegamos a la ecuación 4.10.
Ec 4.9
Ec 4.10
Es posible independizarse de los parámetros geométricos de las muestras medidas si utilizamos el resultado de resistividad a temperatura ambiente obtenido en la sección 4.2.2, en la ecuación 4.11, donde RRT y ρRT son la resistencia y la resistividad a temperatura ambiente, respectivamente.
Dividiendo cada valor medido de resistencia de la película R porls
, se obtiene su
correspondiente de resistividad del material ρ a igual temperatura.
Ec 4.11
Buscamos ajustar la resistividad del material como función de la temperatura, según la ecuación 4.9. Para ello, representando los datos obtenidos como el ln ρ en función de 1/kT (con k la constante de Maxwell-Boltzmann y T la temperatura de la película), se obtienen energías de activación Eσ de los procesos de conducción eléctrica en el material como las pendientes de los ajustes lineales y el logaritmo natural de los factores preexponenciales como las ordenadas al origen (ver ecuación 4.12).
Ec 4.12
Los datos experimentales y sus ajustes se exhiben en la figura 4.5, y se resumen en la tabla 4.3. En la curva de calentamiento, se observa que la resistencia muestra tres regiones diferentes con comportamiento del tipo Arrhenius.
63
T =0 eE
kT
R=( ls )ρ0 e
Eσ
kT
ls=
RRT
RT
ln(ρ)=ln (ρ0)+Eσ
1kT
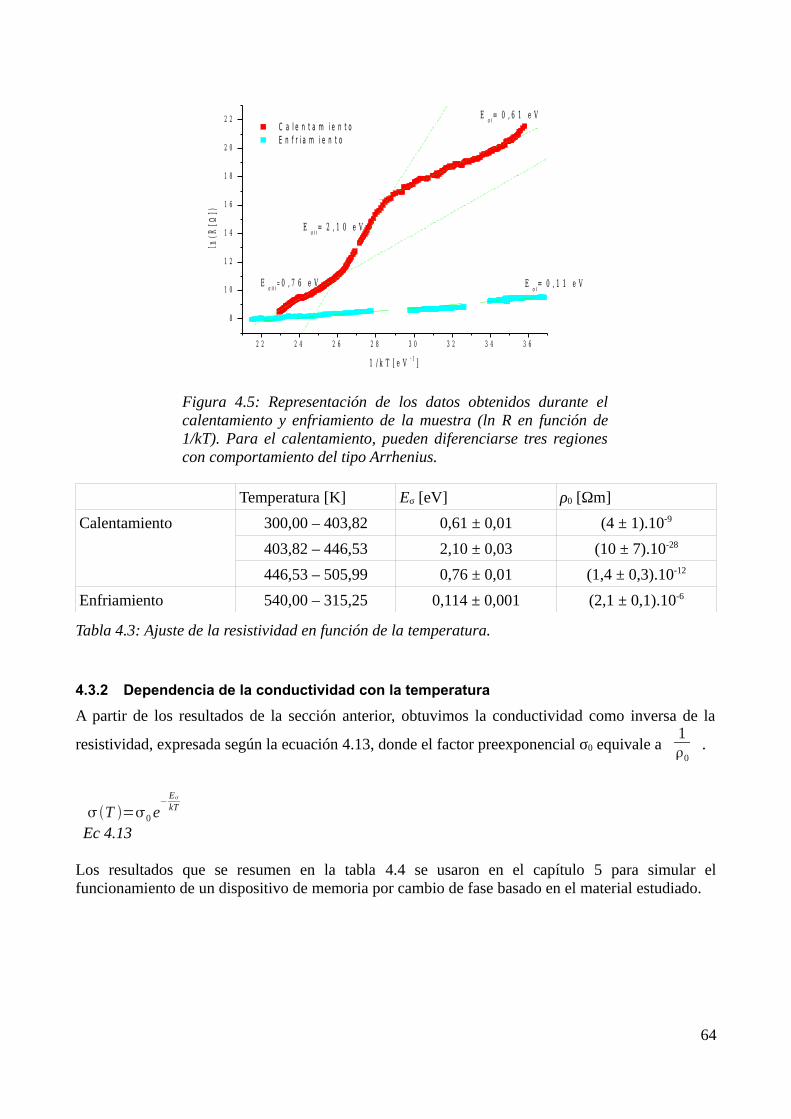
Temperatura [K] Eσ [eV] ρ0 [Ωm]
Calentamiento 300,00 – 403,82 0,61 ± 0,01 (4 ± 1).10-9
403,82 – 446,53 2,10 ± 0,03 (10 ± 7).10-28
446,53 – 505,99 0,76 ± 0,01 (1,4 ± 0,3).10-12
Enfriamiento 540,00 – 315,25 0,114 ± 0,001 (2,1 ± 0,1).10-6
Tabla 4.3: Ajuste de la resistividad en función de la temperatura.
4.3.2 Dependencia de la conductividad con la temperatura
A partir de los resultados de la sección anterior, obtuvimos la conductividad como inversa de la
resistividad, expresada según la ecuación 4.13, donde el factor preexponencial σ0 equivale a10
.
Ec 4.13
Los resultados que se resumen en la tabla 4.4 se usaron en el capítulo 5 para simular el funcionamiento de un dispositivo de memoria por cambio de fase basado en el material estudiado.
64
T =0 e−
E
kT
Figura 4.5: Representación de los datos obtenidos durante el calentamiento y enfriamiento de la muestra (ln R en función de 1/kT). Para el calentamiento, pueden diferenciarse tres regiones con comportamiento del tipo Arrhenius.
2 2 2 4 2 6 2 8 3 0 3 2 3 4 3 6
8
1 0
1 2
1 4
1 6
1 8
2 0
2 2
Eσ Ι
= 0 , 1 1 e VEσ Ι Ι Ι
= 0 , 7 6 e V
Eσ Ι Ι
= 2 , 1 0 e V
Eσ Ι
= 0 , 6 1 e V C a l e n t a m i e n t o E n f r i a m i e n t o
ln(R
[Ω])
1 / k T [ e V - 1 ]

Temperatura [K] Eσ [eV] σ0 [(Ωm)-1]
Calentamiento 300,00 – 403,82 0,61 ± 0,01 (2,4 ± 0,6).108
403,82 – 446,53 2,10 ± 0,03 (1,0 ± 0,7).1027
446,53 – 505,99 0,76 ± 0,01 (7 ± 1).1011
Enfriamiento 540,00 – 315,25 0,114 ± 0,001 (4,8 ± 0,2).105
Tabla 4.4: Ajuste de la conductividad en función de la temperatura.
A temperatura ambiente, se observa un contraste de cinco órdenes de magnitud en la conductividad, atribuible a la cristalización del material. En este caso, se supera el contraste mínimo de tres órdenes de magnitud esperable para el sensado confiable del estado de una celda de memoria (ver sección 1.3).
4.4 Estudio de la cristalización por DRX
4.4.1 Identificación de los productos de cristalización
Las películas tratadas térmicamente fueron nuevamente analizadas por difracción de rayos X. En el difractograma de la figura 4.6, puede observarse la coincidencia de los dos picos más intensos con líneas de difración correspondientes a la fase estable de Ge2Sb2Te5 de celda hexagonal y el pico que les sigue en intensidad en coincidencia con una línea de la fase metaestable de Ge2Sb2Te5 de celda FCC. La cristalización de la fase hexagonal se encuentra en acuerdo con lo obtenido en el capítulo 3 para muestras en volumen. Sin embargo, la aparición de una fase metaestable (en coincidencia con [Yamada, 1991]) sólo fue identificada en las experiencias con películas presentadas en este capítulo y no se observa el predominio de picos correspondientes a la cristalización de Te, que se presentaba en las muestras masivas. Teniendo en cuenta que se trata de muestras de igual composición, este resultado es destacable: o bien, existirían diferencias estructurales de los amorfos debidas a la técnica de síntesis y/o geometría de las muestras, o bien, ésta última influiría en la cristalización y sus productos.
65
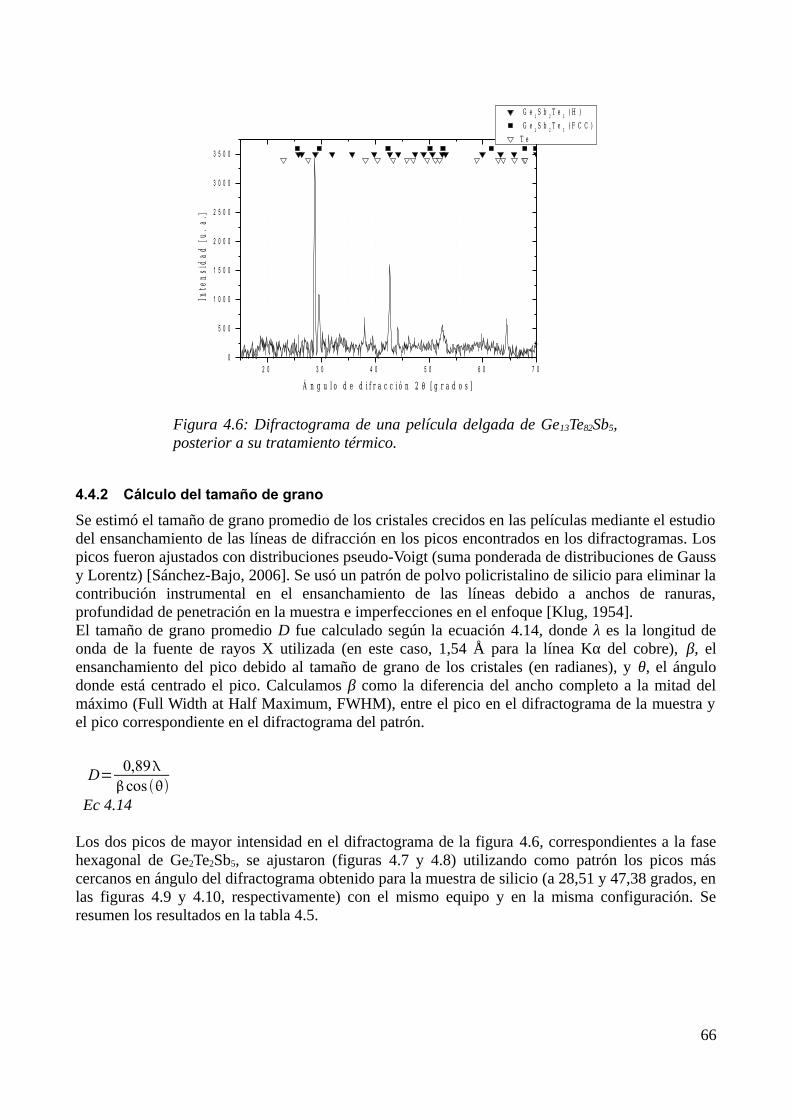
4.4.2 Cálculo del tamaño de grano
Se estimó el tamaño de grano promedio de los cristales crecidos en las películas mediante el estudio del ensanchamiento de las líneas de difracción en los picos encontrados en los difractogramas. Los picos fueron ajustados con distribuciones pseudo-Voigt (suma ponderada de distribuciones de Gauss y Lorentz) [Sánchez-Bajo, 2006]. Se usó un patrón de polvo policristalino de silicio para eliminar la contribución instrumental en el ensanchamiento de las líneas debido a anchos de ranuras, profundidad de penetración en la muestra e imperfecciones en el enfoque [Klug, 1954].El tamaño de grano promedio D fue calculado según la ecuación 4.14, donde λ es la longitud de onda de la fuente de rayos X utilizada (en este caso, 1,54 Å para la línea Kα del cobre), β, el ensanchamiento del pico debido al tamaño de grano de los cristales (en radianes), y θ, el ángulo donde está centrado el pico. Calculamos β como la diferencia del ancho completo a la mitad del máximo (Full Width at Half Maximum, FWHM), entre el pico en el difractograma de la muestra y el pico correspondiente en el difractograma del patrón.
Ec 4.14
Los dos picos de mayor intensidad en el difractograma de la figura 4.6, correspondientes a la fase hexagonal de Ge2Te2Sb5, se ajustaron (figuras 4.7 y 4.8) utilizando como patrón los picos más cercanos en ángulo del difractograma obtenido para la muestra de silicio (a 28,51 y 47,38 grados, en las figuras 4.9 y 4.10, respectivamente) con el mismo equipo y en la misma configuración. Se resumen los resultados en la tabla 4.5.
66
D=0,89cos
Figura 4.6: Difractograma de una película delgada de Ge13Te82Sb5, posterior a su tratamiento térmico.
2 0 3 0 4 0 5 0 6 0 7 00
5 0 0
1 0 0 0
1 5 0 0
2 0 0 0
2 5 0 0
3 0 0 0
3 5 0 0
G e2
S b2
T e5
( H )
G e2
S b2
T e5
( F C C )
T e
Inte
nsi
da
d [
u.
a.]
Á n g u l o d e d i f r a c c i ó n 2 θ [ g r a d o s ]
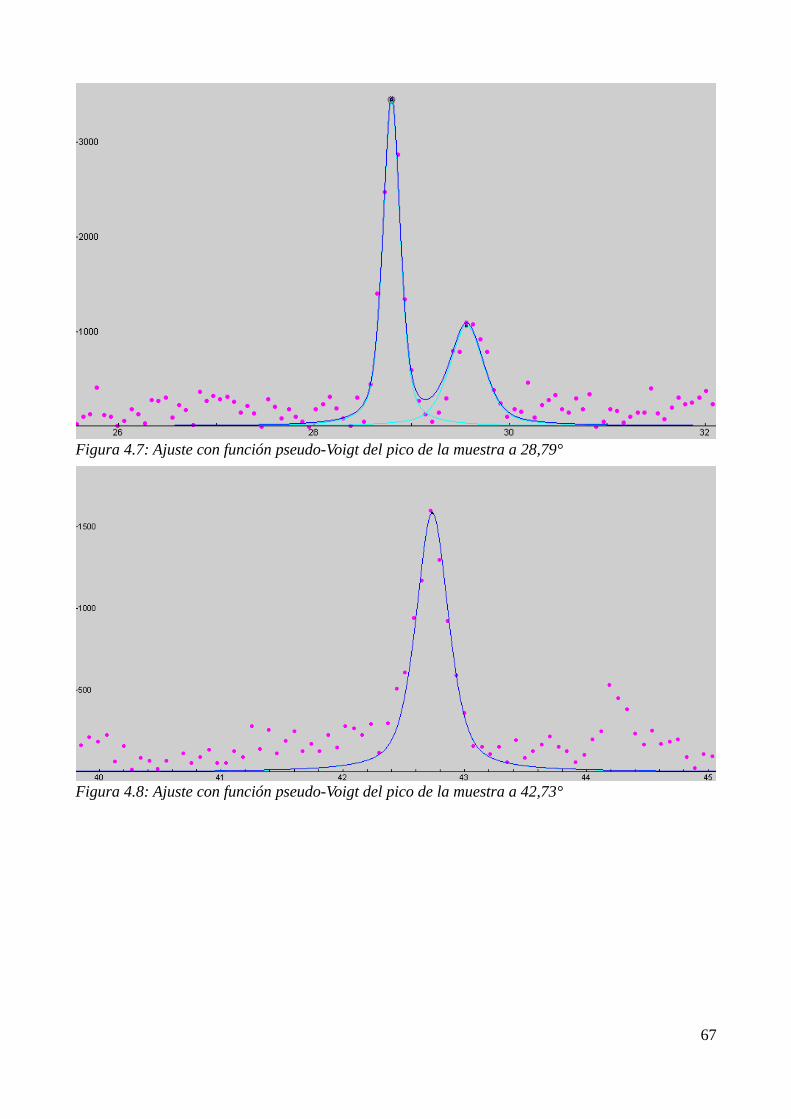
Figura 4.7: Ajuste con función pseudo-Voigt del pico de la muestra a 28,79°
Figura 4.8: Ajuste con función pseudo-Voigt del pico de la muestra a 42,73°
67
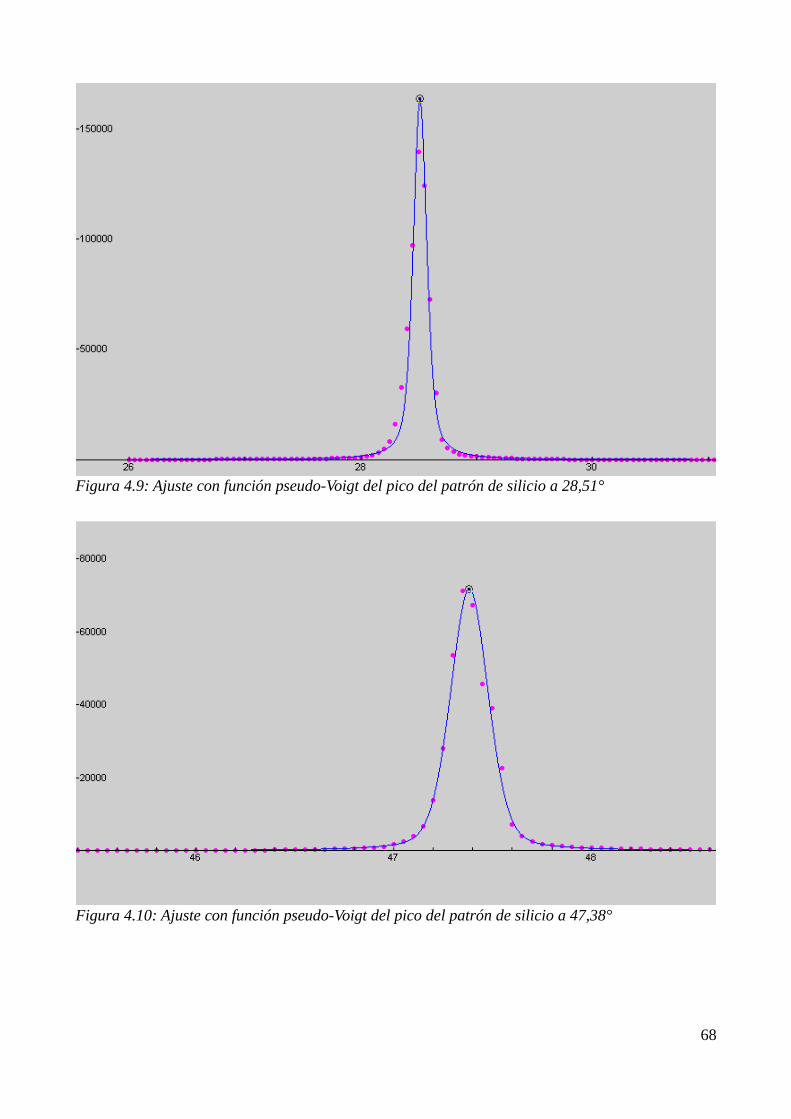
Figura 4.9: Ajuste con función pseudo-Voigt del pico del patrón de silicio a 28,51°
Figura 4.10: Ajuste con función pseudo-Voigt del pico del patrón de silicio a 47,38°
68
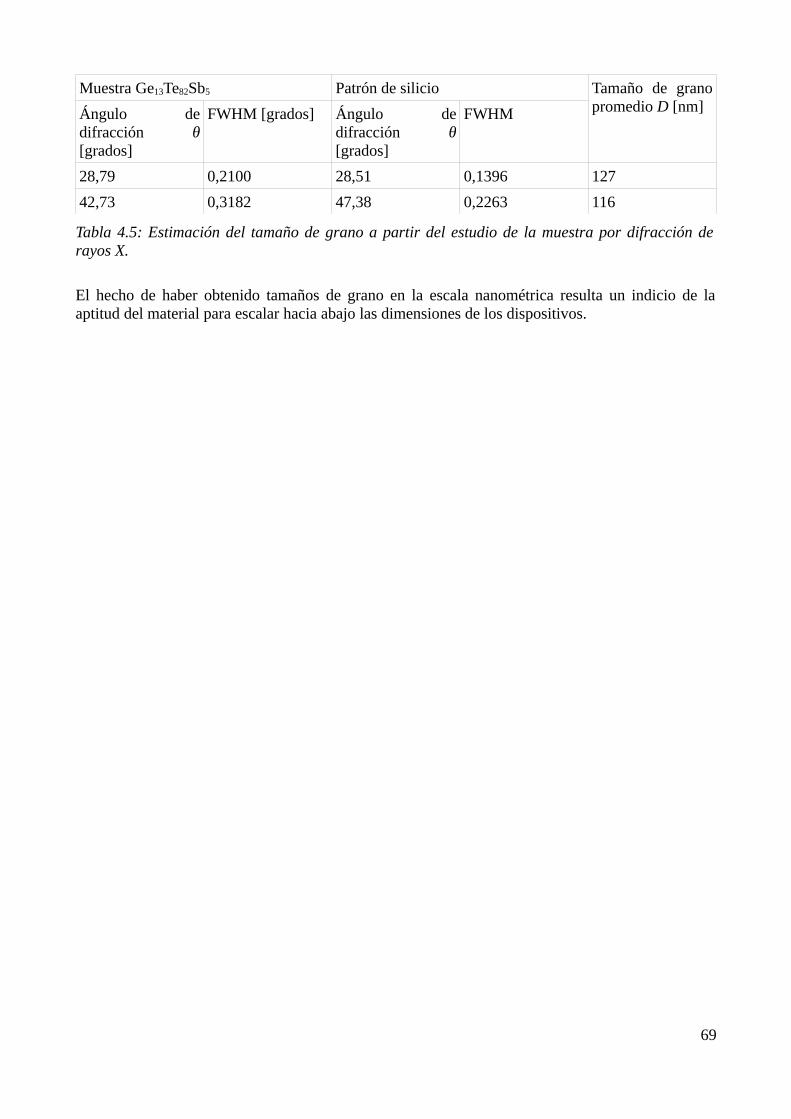
Muestra Ge13Te82Sb5 Patrón de silicio Tamaño de grano promedio D [nm]Ángulo de
difracción θ [grados]
FWHM [grados] Ángulo de difracción θ [grados]
FWHM
28,79 0,2100 28,51 0,1396 127
42,73 0,3182 47,38 0,2263 116
Tabla 4.5: Estimación del tamaño de grano a partir del estudio de la muestra por difracción de rayos X.
El hecho de haber obtenido tamaños de grano en la escala nanométrica resulta un indicio de la aptitud del material para escalar hacia abajo las dimensiones de los dispositivos.
69

4.5 Referencias
[Anbarasu, 2011] Anbarasu, M., Wuttig, M. (2011). Understanding the Structure and Properties of Phase Change Materials for Data Storage Applications. Journal of the Indian Institute of Science, 91(2), 259-274. Recuperado de http://journal.library.iisc.ernet.in/vol201102/JIISc-9102-WUTTIG.pdf
[Friedrich, 2000] Friedrich, I., Weidenhof, V., Njoroge, W., Franz, P., Wuttig, M. (2000). Structural transformations of Ge2Sb2Te5 films studied by electrical resistance measurements. Journal of Applied Physics, 87(9), 4130-4134. doi:10.1063/1.373041
[Klug, 1954] Klug, H. P., Alexander, L. E. (1954). X-ray diffraction procedures for polycrystalline and Amorphous Materials. New York: John Wiley & Sons, Inc.
[Nishiyama, 1993] Nishiyama, H., Nakamura, M. (1993). Capacitance of Disk Capacitors. IEEE Transactions on components, hybrids, and manufacturing technology, 16(3), 360-366. doi:10.1109/33.232065
[Sánchez-Bajo, 2006] Sánchez-Bajo, F., Ortiz, A.L. (2006). Novel analytical model for the determination of grain size distributions in nanocrystalline materials with low lattice microstrains by X-ray diffractometry. Acta Materialia, 54(1), 1-10. doi:10.1016/j.actamat.2005.08.018
[Yamada, 1991] Yamada, N., Ohno, E., Nishiuchi, K., Akahira, N., Takao M. (1991). Rapid-phase transitions of GeTe-Sb2Te3 pseudobinary amorphous thin films for an optical disk memory. Journal of Applied Physics, 69(5), 2849-2856. doi:10.1063/1.348620
70

5 Simulación de celdas de memoria no volátil de cambio de fase en el sistema Ge-Sb-Te
5.1 Consideraciones previas
En la sección 1.2, nos hemos referido a las limitaciones que presentan las tecnologías de memorias no volátiles basadas en almacenamiento de cargas en cuanto a velocidades de programación, consumo eléctrico, retención de la información y cantidad de ciclos de escritura/borrado.Por encontrarse la tecnología de cambio de fase entre las candidatas a superar estas limitaciones, el presente capítulo propone un modelo con el cual puede simularse el funcionamiento de una celda de memoria no volátil basada en la tecnología mencionada. Para tal fin, son parametrizables factores geométricos y propiedades físicas dependientes de los materiales a estudiar (conductividad térmica, conductividad eléctrica, calor específico).Para las simulaciones realizadas, se utilizaron las aleaciones del sistema Ge-Sb-Te estudiadas en capítulos previos.
5.2 Descripción física del modelo
En la figura 5.1, se muestra el esquema bidimensional propuesto para la celda de memoria no volátil por cambio de fase. Está formada por capas de tres materiales: un material dieléctrico, un conductor metálico y un vidrio calcogenuro como material sensible, aquel que guardará el estado de la memoria. Se asume una simetría azimutal y se utilizan las coordenadas cilíndricas ρ y z. Los tamaños de las capas son dados por las coordenadas radiales ρ1, ρ2, ρ3 y ρ4 y los espesores definidos por L1, L2, L3, L4 y L5.Hemos utilizado vidrios calcogenuros del sistema Ge-Sb-Te con dos composiciones correspondientes a dos zonas diferentes del diagrama de equilibrio de fases del mismo:
a) Ge13Sb5Te82 (en porcentaje atómico), cercano al eutéctico binario Ge15Te85.b) Ge2Sb2Te5, en el sistema seudobinario GeTe-Sb2Te3.
En la composición Ge2Sb2Te5 se reconocen propiedades ideales para su empleo en celdas de memoria no volátil: un salto abrupto de la resistividad con el cambio de fase, rápida cinética de cristalización y muy buena reversibilidad entre las fases amorfa y cristalina. Las aleaciones con composiciones cercanas al eutéctico binario Ge15Te85 exhiben el deseado contraste de sus propiedades eléctricas con el cambio de fase, pero los tiempos de cristalización resultan elevados para su aplicación tecnológica [Wuttig, 2007].
71

El modelo considera el efecto térmico generado en el material sensible debido a la aplicación de un pulso eléctrico entre las capas del conductor metálico. También tiene en cuenta la dependencia con la temperatura de las propiedades eléctricas del calcogenuro, como su conductividad. En consecuencia, se trata de un problema transitorio acoplado: la distribución espacial de temperaturas en la celda, que evoluciona en el tiempo, depende de la conductividad eléctrica pues el calor es generado por efecto Joule y transmitido por conducción, mientras la conductividad eléctrica depende fuertemente de la temperatura. Se desprecian las resistencias eléctrica y térmica en los bordes de la celda.Cuando se establece una diferencia de potencial V=V 2−V 1 entre las placas de conductor metálico, el potencial eléctrico V dentro de la celda puede calcularse resolviendo la ecuación de Laplace (ecuación 5.1) [Li, 2009], con las condiciones de contorno dadas por la ecuación 5.2, donde σ es la conductividad eléctrica dependiente de la temperatura, z1=L1 y
z5=L1L2L3L4L5 .
Ec 5.1
Ec 5.2
La densidad de corriente j puede determinarse usando la Ley de Ohm (ecuación 5.3).
Ec 5.3
La distribución espacial de temperaturas en la celda T , z y su dependencia con el tiempo se obtienen resolviendo la ecuación de conducción de calor estándar (ecuación 5.4), donde δ es la densidad, Cp el calor específico, k la conductividad térmica, Qv el calor Joule por unidad de volumen y por unidad de tiempo (ecuación 5.5), y t el tiempo.
72
Figura 5.1: Estructura de la celda simulada.
0= ∇⋅ ∇ V
V , z =V 2 03 ∧ z=z1
V 1 03 ∧ z=z5
j=− ∇ V

Ec 5.4
Ec 5.5
Las condiciones de contorno y las condiciones iniciales de la ecuación 5.4 se expresan en las ecuaciones 5.6 y 5.7, respectivamente, siendo T0 la temperatura ambiente.
Ec 5.6
Ec 5.7
5.3 Aproximación por el Método de los Elementos Finitos
El Método de los Elementos Finitos (Finite Element Method, FEM) [Bathe, 1996] se ha utilizado para resolver el sistema de ecuaciones acopladas, formado por las ecuaciones 5.1 y 5.4, con las condiciones de contorno dadas por las ecuaciones 5.2, 5.6 y 5.7. En primer lugar, se procede a la división del dominio en un conjunto de subdominios discretos. Como se ha mencionado, se trata de un problema bidimensional, con las coordenadas ρ y z, por ello, utilizamos elementos rectangulares con nodos en sus cuatro esquinas. Dadas las magnitudes potencial eléctrico V (según la ecuación 5.1) y temperatura T (según la ecuación 5.4), sus valores en los nodos de cada elemento se denotan V1, V2, V3, V4 y T1, T2, T3 y T4, respectivamente. Para cada elemento pueden expresarse las variables potencial eléctrico y temperatura según las ecuaciones 5.8 y 5.9 donde: V es el vector que indica el potencial eléctrico en los nodos (ecuación 5.10), T el que indica la temperatura en los nodos (ecuación 5.11) y H el vector de las funciones de interpolación (ecuación 5.12); en las ecuaciones 5.13, 5.14, 5.15, 5.16 se indican las formas empleadas para las funciones de interpolación y en la ecuación 5.17, el rango de valores posibles para las variables u y v) [Bathe, 1996]. Es decir, el potencial eléctrico o la temperatura en un punto cualquiera de un elemento se determina con los valores en sus nodos mediante las funciones de interpolación h1, h2, h3 y h4, que son funciones lineales de las coordenadas. La derivada temporal de la temperatura viene dada por la ecuación 5.18.
Ec 5.8
73
Cp∂T∂ t
− ∇⋅k ∇ T=Qv
Qv=∣ ∇ V∣2
T , z , t =T 0 z=0∧∀∧∀ tT 0 z=z5∧∀∧∀ tT 0 =4∧∀ z∧∀ t
T , z , t =T 0 t=0∧∀∧∀ z
V (u , v )=∑i=1
4
h i(u , v )⋅V i=H⋅V

Ec 5.9
Ec 5.10
Ec 5.11
Ec 5.12
Ec 5.13
Ec 5.14
Ec 5.15
Ec 5.16
Ec 5.17
Ec 5.18
Aplicando el método de Galerkin [Bathe, 1996] para la ecuación 5.1, se obtiene la ecuación 5.19.
74
T (u , v)=∑i=1
4
hi(u , v )⋅T i=H⋅V
V=[V 1
V 2
V 3
V 4]
T=[T 1
T 2
T 3
T 4]
H=[h1(u ,v ) h2(u , v) h3(u , v ) h4(u , v)]
h1u , v =141u1v
h2u , v =141−u1v
h3u , v=141−u1−v
h4u , v =141u 1−v
−1u , v1
∂T∂ t
=H∂T∂ t
=H⋅T

Ec 5.19
Reescribiendo la ecuación 5.19 con coordenadas cilíndricas e integrando por partes, se obtienen las ecuaciones 5.20 y 5.21, respectivamente.
Ec 5.20
Ec 5.21
En consecuencia, si la conductividad eléctrica es conocida, el vector de potencial eléctrico en los nodos de todos los elementos es obtenido usando las condiciones de contorno en la ecuación 5.2 y la ecuación 5.22.
Ec 5.22
Ec 5.23
En forma análoga, usando el método de Galerkin [Bathe, 1996] para la ecuación 5.4, se obtiene la ecuación 5.24.
Ec 5.24
La ecuación 5.5 (calor por unidad de volumen y por unidad de tiempo, generado por efecto Joule) también puede expresarse como la ecuación 5.25. Reescribiendo la ecuación 5.24 con coordenadas cilíndricas, integrando por partes en las derivadas espaciales, se obtienen las ecuaciones 5.26 y 5.27, respectivamente.
Ec 5.25
75
∫
HT⋅∇⋅ ∇ V d=0
∫
HT⋅[ ∂∂
∂H⋅V
∂ ∂∂ z
∂H⋅V
∂ z ]d=0
∫∂ HT
∂
∂H∂
∂ HT
∂ z
∂H∂ z ⋅V d=0
K v⋅V =0
K v= ∑∀ elemento
∫ ∂HT
∂
∂ H∂
∂HT
∂ z
∂ H∂ z d
∫
HT⋅C p
∂T∂ t
− ∇⋅k ∇ T −Q vd=0
Qv=∣ ∂H∂, z
⋅V∣2

Ec 5.26
Ec 5.27
Luego, el vector de temperatura en los nodos de todos los elementos satisface la ecuación 5.28. Las ecuaciones 5.29, 5.30 y 5.31 corresponden a las matrices y los vectores involucrados en la ecuación 5.28.
Ec 5.28
Ec 5.29
Ec 5.30
Ec 5.31
Las condiciones iniciales y de contorno para la ecuación 5.28 están dadas por las ecuaciones 5.6 y 5.7. Las ecuaciones 5.22, 5.23 y 5.31 muestran que el problema está acoplado: la temperatura T se obtiene de la ecuación 5.28 conociendo F, F es determinado conociendo el potencial eléctrico de los nodos y la conductividad eléctrica σ(T); para ello, debe conocerse la temperatura.
T en la ecuación 5.28 puede determinarse mediante el método alfa [Bathe, 1996]. T t t
sigue la ecuación 5.32, donde T para el tiempo t se denota T(t) y T para el tiempo t t , con 01 .
Ec 5.32
Las simulaciones se realizaron con la condición adicional de mantener constante en el tiempo la corriente que circula por la celda. Es por ello que las condiciones de contorno relativas al potencial eléctrico (ecuación 5.2) varían con el tiempo.El código fuente desarrollado se encuentra en el Apéndice I.
76
∫
HT⋅C p H⋅
∂T∂ t
−[ ∂∂ k ∂H⋅T
∂ ∂∂ z k
∂H⋅T
∂ z ]−Qvd=0
T t t=T t t T t
t
∫Ω [HT
⋅ρδC p H⋅T−(∂HT
∂ρ⋅kρ
∂ H∂ρ
+∂HT
∂ z⋅k ρ
∂H∂ z )⋅T−HT
⋅ρσ∣ ∂ H∂(ρ , z)
⋅V∣2
]dΩ=0
Cq⋅T+ Kq⋅T=F
F= ∑∀ elemento
∫HT
⋅∣ ∂ H∂ , z
⋅V∣2
d
Cq= ∑∀ elemento
∫
HT⋅C p H d
K q= ∑∀ elemento
∫ ∂HT
∂⋅k
∂ H∂
∂ HT
∂ z⋅k
∂H∂ z d
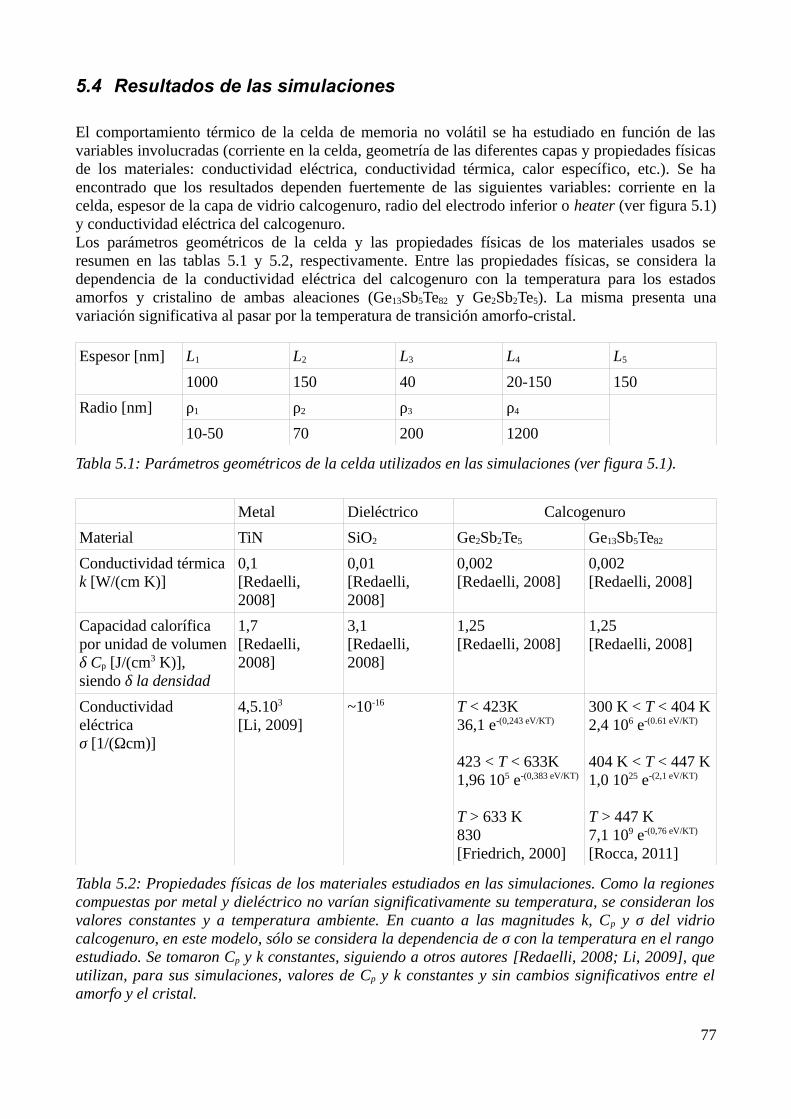
5.4 Resultados de las simulaciones
El comportamiento térmico de la celda de memoria no volátil se ha estudiado en función de las variables involucradas (corriente en la celda, geometría de las diferentes capas y propiedades físicas de los materiales: conductividad eléctrica, conductividad térmica, calor específico, etc.). Se ha encontrado que los resultados dependen fuertemente de las siguientes variables: corriente en la celda, espesor de la capa de vidrio calcogenuro, radio del electrodo inferior o heater (ver figura 5.1) y conductividad eléctrica del calcogenuro.Los parámetros geométricos de la celda y las propiedades físicas de los materiales usados se resumen en las tablas 5.1 y 5.2, respectivamente. Entre las propiedades físicas, se considera la dependencia de la conductividad eléctrica del calcogenuro con la temperatura para los estados amorfos y cristalino de ambas aleaciones (Ge13Sb5Te82 y Ge2Sb2Te5). La misma presenta una variación significativa al pasar por la temperatura de transición amorfo-cristal.
Espesor [nm] L1 L2 L3 L4 L5
1000 150 40 20-150 150
Radio [nm] ρ1 ρ2 ρ3 ρ4
10-50 70 200 1200
Tabla 5.1: Parámetros geométricos de la celda utilizados en las simulaciones (ver figura 5.1).
Metal Dieléctrico Calcogenuro
Material TiN SiO2 Ge2Sb2Te5 Ge13Sb5Te82
Conductividad térmica k [W/(cm K)]
0,1[Redaelli, 2008]
0,01[Redaelli, 2008]
0,002[Redaelli, 2008]
0,002[Redaelli, 2008]
Capacidad calorífica por unidad de volumen δ Cp [J/(cm3 K)], siendo δ la densidad
1,7[Redaelli, 2008]
3,1[Redaelli, 2008]
1,25[Redaelli, 2008]
1,25[Redaelli, 2008]
Conductividad eléctricaσ [1/(Ωcm)]
4,5.103
[Li, 2009]~10-16 T < 423K
36,1 e-(0,243 eV/KT)
423 < T < 633K1,96 105 e-(0,383 eV/KT)
T > 633 K 830[Friedrich, 2000]
300 K < T < 404 K2,4 106 e-(0.61 eV/KT)
404 K < T < 447 K1,0 1025 e-(2,1 eV/KT)
T > 447 K7,1 109 e-(0,76 eV/KT)
[Rocca, 2011]
Tabla 5.2: Propiedades físicas de los materiales estudiados en las simulaciones. Como la regiones compuestas por metal y dieléctrico no varían significativamente su temperatura, se consideran los valores constantes y a temperatura ambiente. En cuanto a las magnitudes k, Cp y σ del vidrio calcogenuro, en este modelo, sólo se considera la dependencia de σ con la temperatura en el rango estudiado. Se tomaron Cp y k constantes, siguiendo a otros autores [Redaelli, 2008; Li, 2009], que utilizan, para sus simulaciones, valores de Cp y k constantes y sin cambios significativos entre el amorfo y el cristal.
77
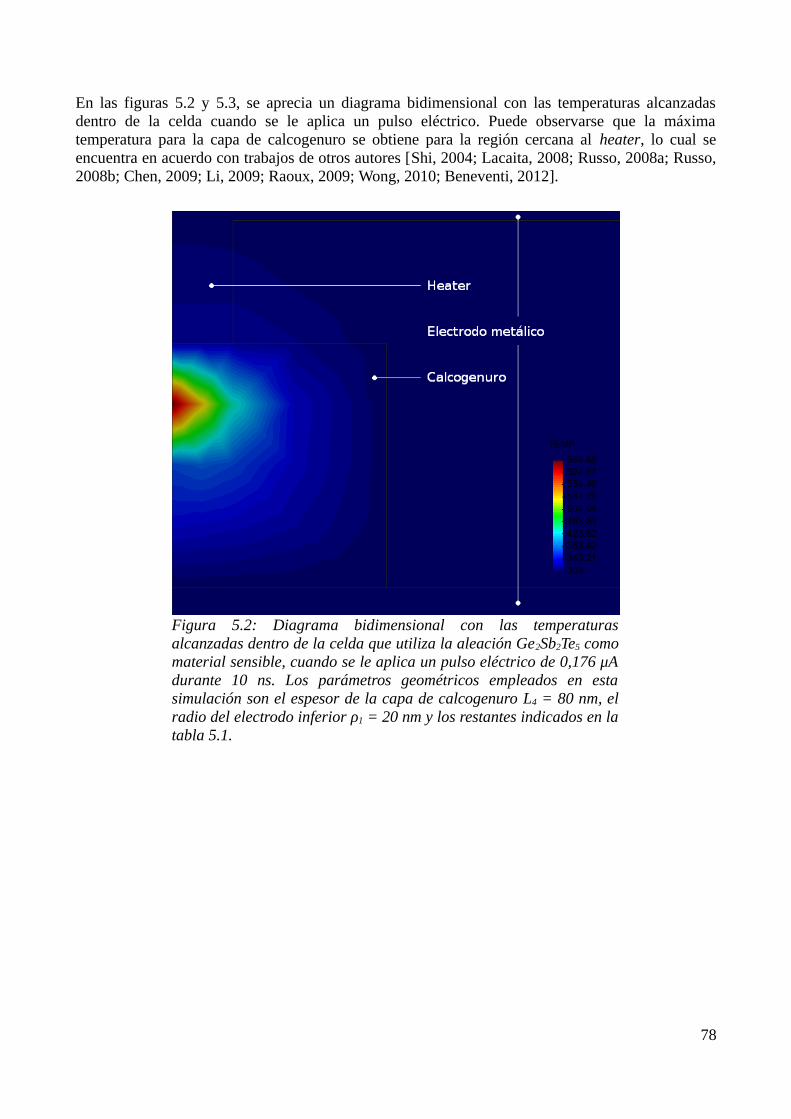
En las figuras 5.2 y 5.3, se aprecia un diagrama bidimensional con las temperaturas alcanzadas dentro de la celda cuando se le aplica un pulso eléctrico. Puede observarse que la máxima temperatura para la capa de calcogenuro se obtiene para la región cercana al heater, lo cual se encuentra en acuerdo con trabajos de otros autores [Shi, 2004; Lacaita, 2008; Russo, 2008a; Russo,2008b; Chen, 2009; Li, 2009; Raoux, 2009; Wong, 2010; Beneventi, 2012].
78
Figura 5.2: Diagrama bidimensional con las temperaturas alcanzadas dentro de la celda que utiliza la aleación Ge2Sb2Te5 como material sensible, cuando se le aplica un pulso eléctrico de 0,176 μA durante 10 ns. Los parámetros geométricos empleados en esta simulación son el espesor de la capa de calcogenuro L4 = 80 nm, el radio del electrodo inferior ρ1 = 20 nm y los restantes indicados en la tabla 5.1.
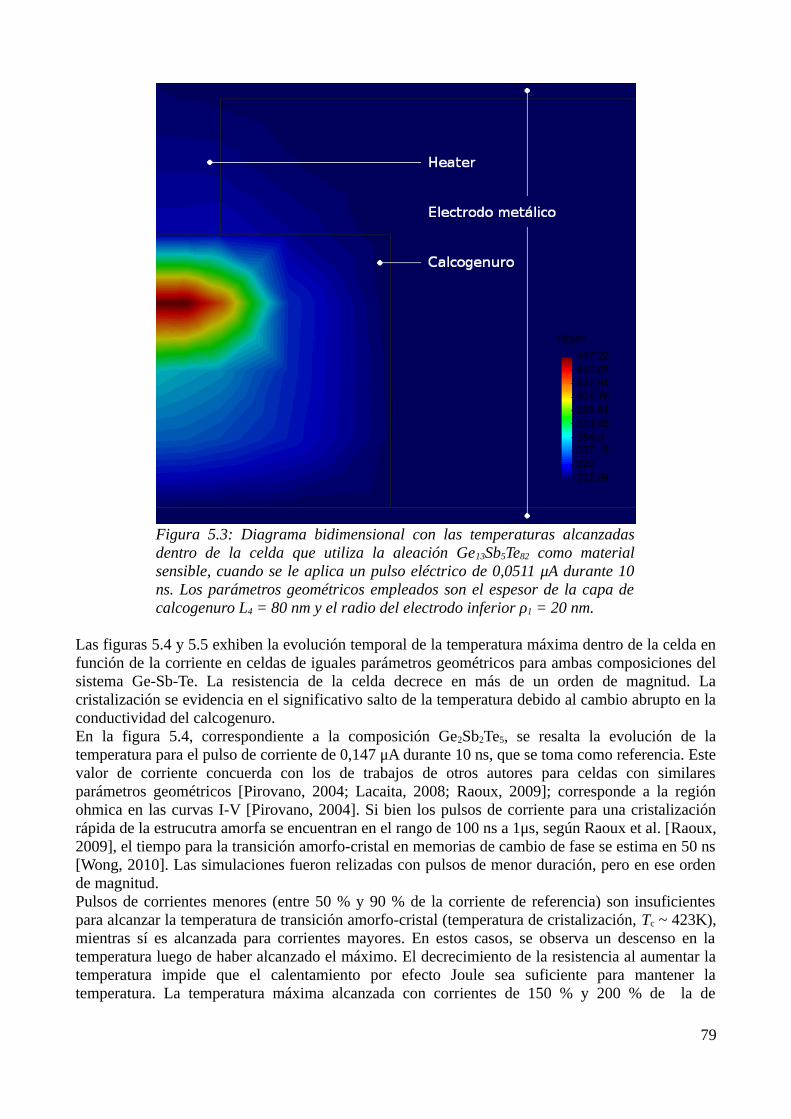
Las figuras 5.4 y 5.5 exhiben la evolución temporal de la temperatura máxima dentro de la celda en función de la corriente en celdas de iguales parámetros geométricos para ambas composiciones del sistema Ge-Sb-Te. La resistencia de la celda decrece en más de un orden de magnitud. La cristalización se evidencia en el significativo salto de la temperatura debido al cambio abrupto en la conductividad del calcogenuro.En la figura 5.4, correspondiente a la composición Ge2Sb2Te5, se resalta la evolución de la temperatura para el pulso de corriente de 0,147 μA durante 10 ns, que se toma como referencia. Este valor de corriente concuerda con los de trabajos de otros autores para celdas con similares parámetros geométricos [Pirovano, 2004; Lacaita, 2008; Raoux, 2009]; corresponde a la región ohmica en las curvas I-V [Pirovano, 2004]. Si bien los pulsos de corriente para una cristalización rápida de la estrucutra amorfa se encuentran en el rango de 100 ns a 1μs, según Raoux et al. [Raoux,2009], el tiempo para la transición amorfo-cristal en memorias de cambio de fase se estima en 50 ns [Wong, 2010]. Las simulaciones fueron relizadas con pulsos de menor duración, pero en ese orden de magnitud. Pulsos de corrientes menores (entre 50 % y 90 % de la corriente de referencia) son insuficientes para alcanzar la temperatura de transición amorfo-cristal (temperatura de cristalización, Tc ~ 423K), mientras sí es alcanzada para corrientes mayores. En estos casos, se observa un descenso en la temperatura luego de haber alcanzado el máximo. El decrecimiento de la resistencia al aumentar la temperatura impide que el calentamiento por efecto Joule sea suficiente para mantener la temperatura. La temperatura máxima alcanzada con corrientes de 150 % y 200 % de la de
79
Figura 5.3: Diagrama bidimensional con las temperaturas alcanzadas dentro de la celda que utiliza la aleación Ge13Sb5Te82 como material sensible, cuando se le aplica un pulso eléctrico de 0,0511 μA durante 10 ns. Los parámetros geométricos empleados son el espesor de la capa de calcogenuro L4 = 80 nm y el radio del electrodo inferior ρ1 = 20 nm.
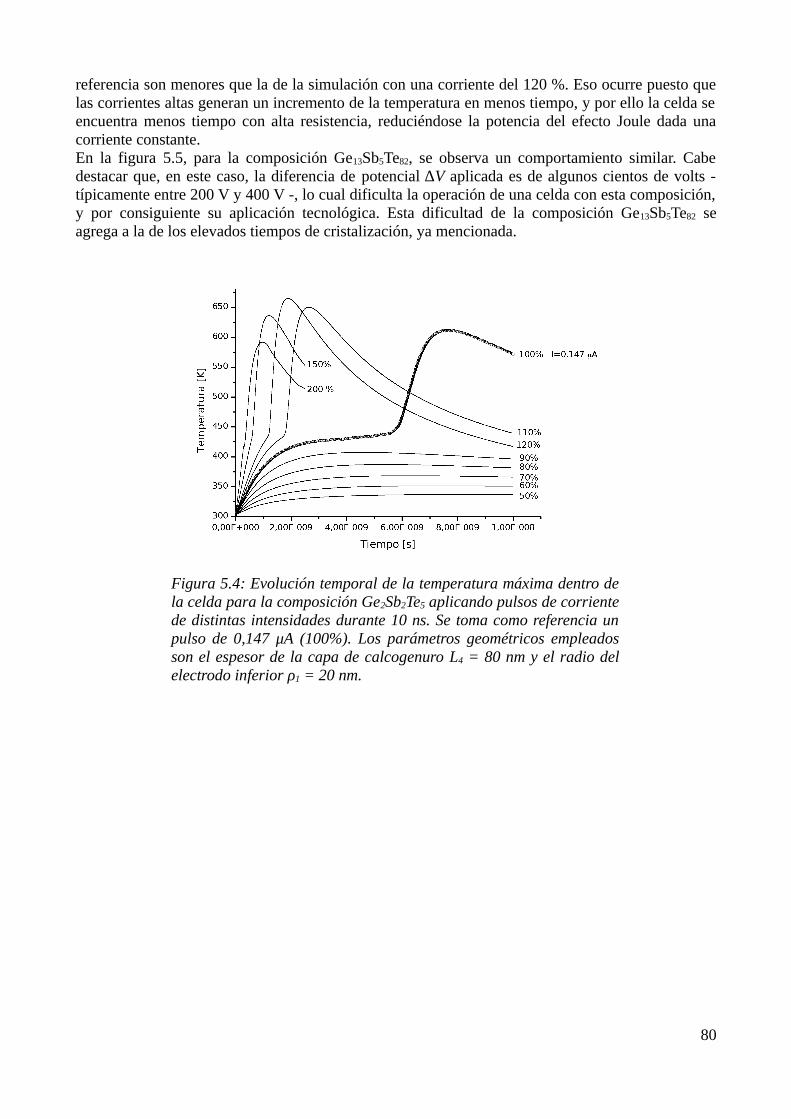
referencia son menores que la de la simulación con una corriente del 120 %. Eso ocurre puesto que las corrientes altas generan un incremento de la temperatura en menos tiempo, y por ello la celda se encuentra menos tiempo con alta resistencia, reduciéndose la potencia del efecto Joule dada una corriente constante.En la figura 5.5, para la composición Ge13Sb5Te82, se observa un comportamiento similar. Cabe destacar que, en este caso, la diferencia de potencial ΔV aplicada es de algunos cientos de volts - típicamente entre 200 V y 400 V -, lo cual dificulta la operación de una celda con esta composición, y por consiguiente su aplicación tecnológica. Esta dificultad de la composición Ge13Sb5Te82 se agrega a la de los elevados tiempos de cristalización, ya mencionada.
Figura 5.4: Evolución temporal de la temperatura máxima dentro de la celda para la composición Ge2Sb2Te5 aplicando pulsos de corriente de distintas intensidades durante 10 ns. Se toma como referencia un pulso de 0,147 μA (100%). Los parámetros geométricos empleados son el espesor de la capa de calcogenuro L4 = 80 nm y el radio del electrodo inferior ρ1 = 20 nm.
80
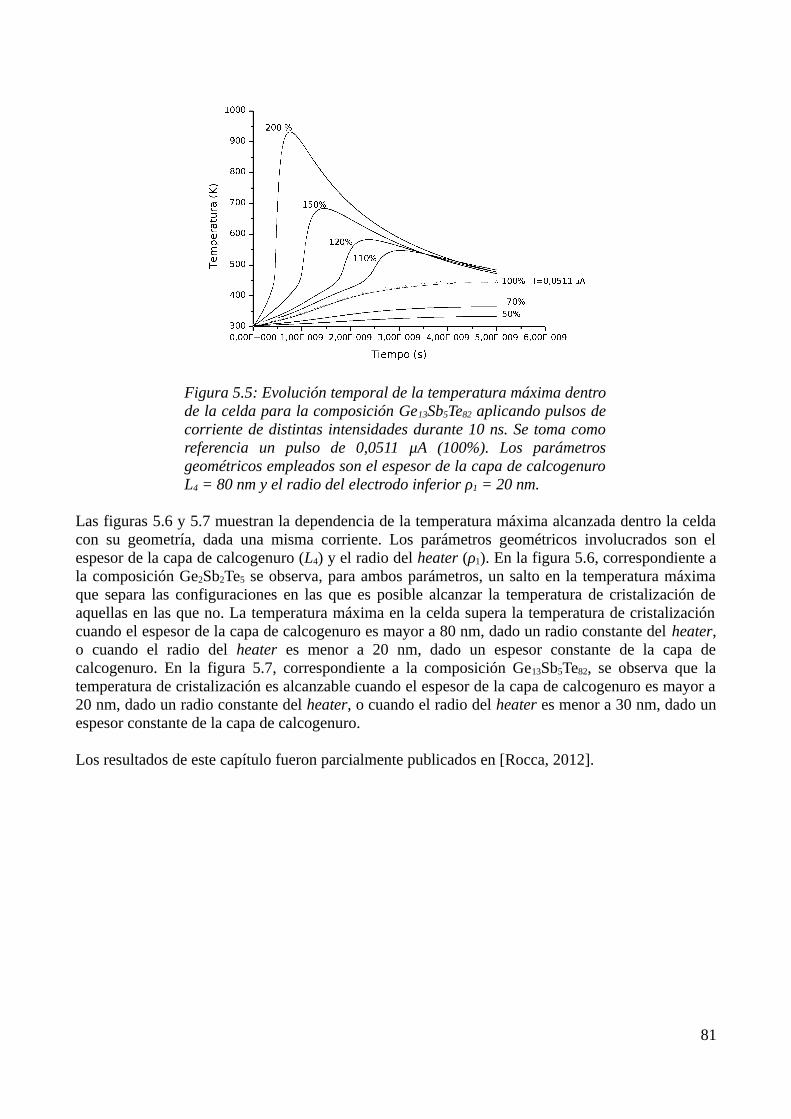
Figura 5.5: Evolución temporal de la temperatura máxima dentro de la celda para la composición Ge13Sb5Te82 aplicando pulsos de corriente de distintas intensidades durante 10 ns. Se toma como referencia un pulso de 0,0511 μA (100%). Los parámetros geométricos empleados son el espesor de la capa de calcogenuro L4 = 80 nm y el radio del electrodo inferior ρ1 = 20 nm.
Las figuras 5.6 y 5.7 muestran la dependencia de la temperatura máxima alcanzada dentro la celda con su geometría, dada una misma corriente. Los parámetros geométricos involucrados son el espesor de la capa de calcogenuro (L4) y el radio del heater (ρ1). En la figura 5.6, correspondiente a la composición Ge2Sb2Te5 se observa, para ambos parámetros, un salto en la temperatura máxima que separa las configuraciones en las que es posible alcanzar la temperatura de cristalización de aquellas en las que no. La temperatura máxima en la celda supera la temperatura de cristalización cuando el espesor de la capa de calcogenuro es mayor a 80 nm, dado un radio constante del heater, o cuando el radio del heater es menor a 20 nm, dado un espesor constante de la capa de calcogenuro. En la figura 5.7, correspondiente a la composición Ge13Sb5Te82, se observa que la temperatura de cristalización es alcanzable cuando el espesor de la capa de calcogenuro es mayor a 20 nm, dado un radio constante del heater, o cuando el radio del heater es menor a 30 nm, dado un espesor constante de la capa de calcogenuro.
Los resultados de este capítulo fueron parcialmente publicados en [Rocca, 2012].
81
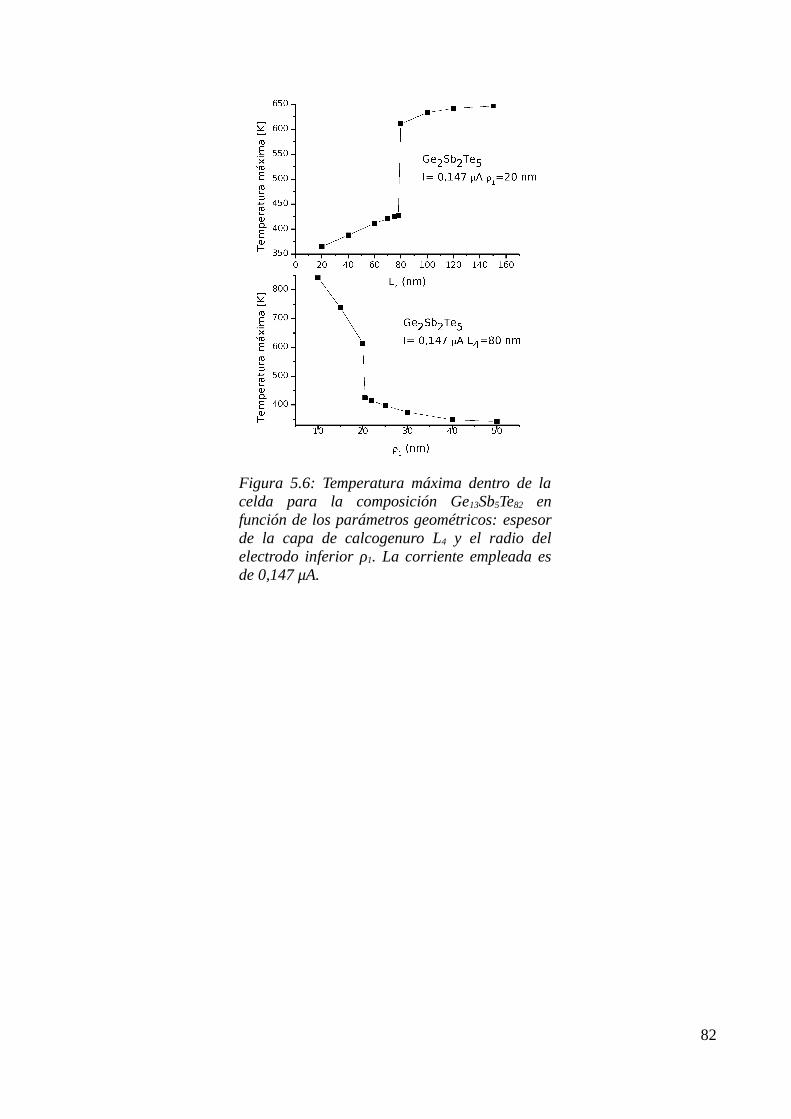
Figura 5.6: Temperatura máxima dentro de la celda para la composición Ge13Sb5Te82 en función de los parámetros geométricos: espesor de la capa de calcogenuro L4 y el radio del electrodo inferior ρ1. La corriente empleada es de 0,147 μA.
82
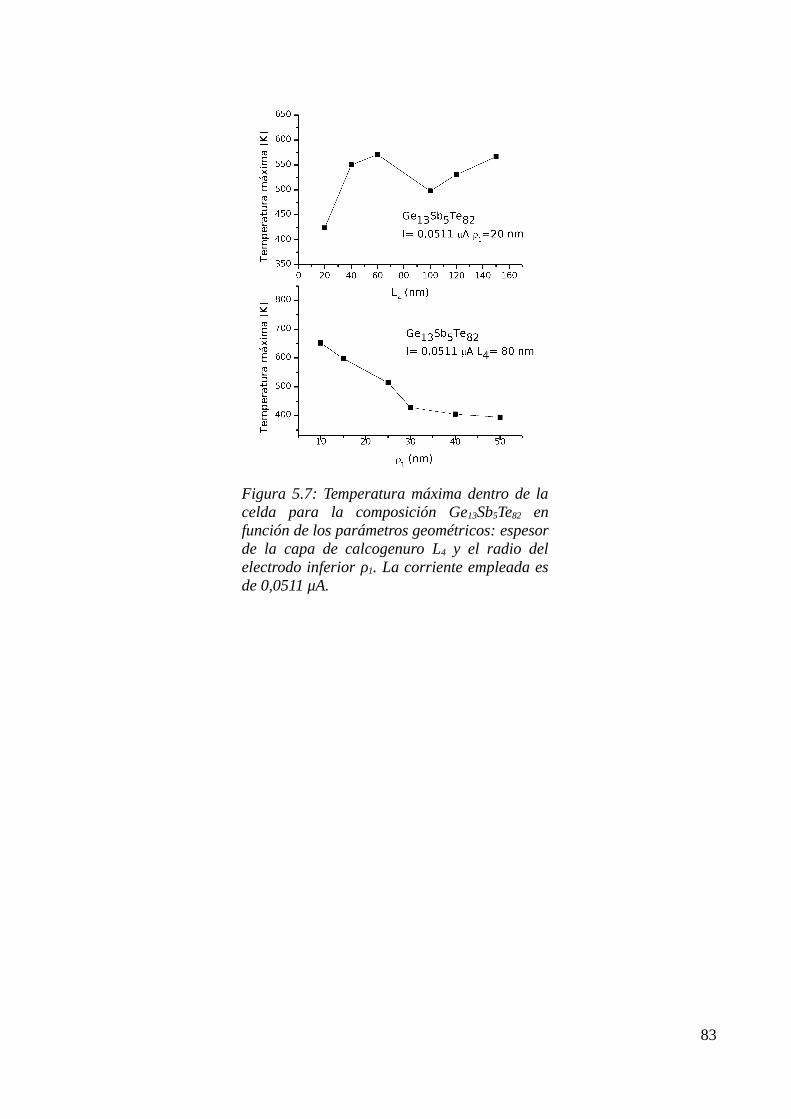
83
Figura 5.7: Temperatura máxima dentro de la celda para la composición Ge13Sb5Te82 en función de los parámetros geométricos: espesor de la capa de calcogenuro L4 y el radio del electrodo inferior ρ1. La corriente empleada es de 0,0511 μA.

5.5 Referencias
[Bathe, 1996] Bathe, K. J. (1996). Finite Elements Procedures. New Jersey: Prentice Hall.
[Beneventi, 2012] Beneventi, G. B., Perniola, L., Hubert, Q., Gliere, A., Larcher, L., Pavan, P., De Salvo, B. (2012) Assessment of Self-Induced Joule-Heating Effect in the I – V Readout Region of Polycrystalline Ge2Sb2Te5 Phase-Change Memory. IEEE Transactions on Electron Devices, 59(1), 188-196. doi:10.1109/TED.2011.2170840
[Chen, 2009] Chen, I. R., Pop, E. (2009). Compact thermal model for vertical nanowire phase-change memory cells. IEEE Transactions on Electron Devices, 56(7), 1523-1528. doi:10.1109/TED.2009.2021364
[Friedrich, 2000] Friedrich, I., Weidenhof, V., Njoroge, W., Franz, P., Wuttig, M. (2000). Structural transformations of Ge2Sb2Te5 films studied by electrical resistance measurements. Journal of Applied Physics, 87(9), 4130-4134. doi:10.1063/1.373041
[Lacaita, 2008] Lacaita A. L., Ielmini, D., Mantegazza, D. (2008). Status and challenges of phase change memory modeling. Solid-State Electronics, 52(9), 1443–1451. doi:10.1016/j.sse.2008.04.020
[Li, 2009] Li, Y., Hwang, C.,Li, T., Cheng, H. (2009). The geometric effect and programming current reduction in cylindrical-shaped phase change memory. Nanotechnology, 20, 285701. doi:10.1088/0957-4484/20/28/285701
[Pirovano, 2004] Pirovano, A., Lacaita, A. L., Benvenuti, A., Pellizzer, F., Bez, R. (2004). Electronic switching in phase-change memories. IEEE Transactions on Electron Devices, 51(3), 452-459. doi:10.1109/TED.2003.823243
[Raoux, 2009] Raoux, S., Wuttig, M. (Eds.). (2009). Phase Change Materials, Science and Applications, New York: Springer.
[Redaelli, 2008] Redaelli, A., Pirovano, A., Benvenuti, A., Lacaita, A. L. (2008). Threshold switching and phase transition numerical models for phase change memory simulations. Journal of Applied Physics, 103(11). doi:10.1063/1.2931951
[Rocca, 2011] Rocca, J.A., Fontana, M., Arcondo, B., Molina Ruiz, M., Rodríguez Viejo, J. (2011). Amorphous-crystal transition of Ge-Sb-Te thin films using resistivity measurements. Humboldt Kolleg - International Conference on Physics (HK2011). La Plata, Argentina.
[Rocca, 2012] Rocca, J., Fontana, M., Arcondo, B. (2012). Simulation of non-volatile memory cell using chalcogenide glasses. Journal of Alloys and Compounds, 536(1), S516–S521. doi:10.1016/j.jallcom.2012.01.052
[Russo, 2008a] Russo, U., Ielmini, D., Redaelli, A., Lacaita, A. L. (2008). Modeling of Programming and Read Performance in Phase-Change Memories - Part I: Cell Optimization and Scaling. IEEE Transactions on Electron Devices, 55(2), 506-514. doi:10.1109/TED.2007.911630
[Russo, 2008b]Russo, U., Ielmini, D., Redaelli, A., Lacaita, A. L. (2008). Modeling of
84

Programming and Read Performance in Phase-Change Memories - Part II: Program Disturb and Mixed-Scaling Approach. IEEE Transactions on Electron Devices, 55(2), 515-522. doi:10.1109/TED.2007.913573
[Shi, 2004] Shi, L. P., Chong, T. C., Li, J. M., Koh, D., Zhao, R., Yang, H., Tan, P., Wei, X., Song, W. (2004). Thermal modelling and simulation of non-volatile and non-rotating phase change memory cell. Non-Volatile Memory Technology Symposium, 2004. 83-87. doi:10.1109/NVMT.2004.1380811
[Wong, 2010] Wong, H.P., Raoux, S., Kim, S., Liang, J., Reifenberg, J.P., Rajendran, B., Asheghi, M., Goodson, K.E. (2010). Phase Change Memory. Proceedings of the IEEE, 98(12), 2201-2227. doi:10.1109/JPROC.2010.2070050
[Wuttig, 2007] Wuttig, M., Yamada, N. (2007). Phase-change materials for rewriteable data storage. Nature Materials, 6, 824-832. doi:10.1038/nmat2009
85

6 Conclusiones generales
En este trabajo se han estudiado aleaciones del sistema Ge-Sb-Te, cuyo mecanismo de cambio de fase amorfo-cristal, de probada aptitud para el funcionamiento de dispositivos de almacenamiento óptico de información, tiene un fuerte potencial de aplicación en el ámbito de la memorias electrónicas no volátiles. Se eligió la composición Ge13Sb5Te82, que ha sido poco explorada para el almacenamiento electrónico, y se encuentra cercana al eutéctico binario Ge-Te (Ge15Te85) con un pequeño agregado de Sb (5 % at.). Se compararon algunos aspectos de esta aleación con la vastamente estudiada composición Ge2Sb2Te5.
Muestras en volumen de la aleación en estudio fueron caracterizadas por técnicas calorimétricas. Los resultados obtenidos presentan como novedad, a diferencia de los reportes de otros autores, en la aleación Ge2Sb2Te5, la cristalización de la fase cristalina estable Ge2Sb2Te5, de estructura hexagonal. Ésta es acompañada de la fase estable de Te, como principal producto de cristalización. Aunque ambas aparecen en un mismo pico exotérmico, se atribuyen altos tiempos de cristalización (en el orden de los μs) al hecho de que la aleación no cristalice en una única fase.Si bien la temperatura de transición vítrea reducida Trg se encuentra apenas por encima del rango ideal para materiales de cambio de fase (0,45 – 0,55), éste no es un parámetro para descartar su aplicación. Los resultados relacionados con la estabilidad del amorfo de la aleación, hacen suponer que no debería presentar problemas en cuanto a retención de datos para el almacenamiento no volátil.Los primeros pasos de cristalización en las curvas TTT-THrT fueron ajustados con un modelo de nucleación homogénea y crecimiento de grano tridimensional controlado por la interfase entre el líquido y el cristal.
Para las muestras conformadas como películas delgadas, se encontraron simultáneamente las fases hexagonal estable y cúbica centrada en las caras (FCC) metaestable de Ge2Sb2Te5 como productos de cristalización. Esta diferencia sugiere que debe estudiarse más detalladamente la cinética de cristalización para muestras obtenidas de esta forma, pues para el rango de temperaturas de operación de un dispositivo de memoria, podrían lograrse cristalizaciones rápidas por la obtención de un único producto de cristalización.El tamaño de grano estimado supone una aleación apta para el escalado de dispositivos que la utlicen como material sensible. El contraste en la conductividad eléctrica resultó adecuado para lo que se espera de un material de cambio de fase.
El modelado por elementos finitos de una de celda de memoria permitió, a partir de las magnitudes de ciertas propiedades térmicas y eléctricas en las aleaciones Ge2Sb2Te5 y Ge13Sb5Te82, comparar el desempeño que tendrían dispositivos fabricados con dichas aleaciones. Las simulaciones resultaron favorables para la aleación Ge2Sb2Te5. Los resultados obtenidos en simulaciones para la aleación Ge13Sb5Te82 ponen en duda su factibilidad tecnológica, por sus requerimientos en términos de consumo energético.
Si bien el presente trabajo se centró en el estudio de una determinada aleación, sienta un marco de trabajo que puede extenderse para la evaluación de otras composiciones de vidrios para memorias de cambio de fase. El mismo admite tanto añadir o mejorar el uso de las técnicas de caracterización que alimentan el modelo, como refinar el modelo en sí mismo y también su implementación algorítmica.
86

7 Apéndice I
Código fuente MATLAB• cálculo principal: transitorio_memoria.m• configuración de datos físicos y geométricos, condiciones de contorno e iniciales:
F_G_CC_CI.m, ConductividadElectrica.m• funciones auxiliares al cálculo: b.m, h.m, der.m, des.m
transitorio_memoria.m
% Problema de calculo de Potencial y Temperaturas %% %% Se usan los las funciones en los archivos %% B.m deR.m deS.m H.m sigmac.m %% %% Se usan coordendas cilindricas %% El isoparametrico tiene nombrados los nodos de la sig. forma: %% 1 3 osea h1 h3 %% 2 4 h2 h4 %
F_G_CC_CI; % Cargamos datos físicos y geometricos, condiciones de contorno e iniciales
% Inicializamos a cero las matrices del programa (tanto parte electrica como termica (T)) %
FgT =zeros(gof, 1);Kg = zeros(gof, gof);KgT = zeros(gof, gof);CgT = zeros(gof, gof);
FT = zeros(4,1);K = zeros(4,4);KT = zeros(4,4);CT = zeros(4,4);Corrheater=0;Corrheater2=0;
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
% Calculo de las temperaturas en regimen transitorio %% AQUI COMIENZA EL PROGRAMA %
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
% REGIMEN TRANSITORIO DE TEMPERATURA, usando el metodo alfa con alfa = 0 %
% Condicion inicial para la temperatura, en el vector Tvie %
Tvie = ones(1, gof)*Tin;
Tviemax=max(Tvie);Tmax=0;Tc = 0;
87

% AQUI EMPIEZA UN LOOP EN EL TIEMPO %
for it=1:4000; %%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%% % Condiciones de contorno %% Potecial en Volt %% 0: libre %% distinto de 0: trabado en ese valor %% Electricas %CC(1) = v1; CC(2) = v1; CC(3) = v1; CC(4) = v1; CC(5) = v1;CC(6) = v1; CC(7) = v1;CC(81) = v2; CC(82) = v2; CC(83) = v2; CC(84) = v2; CC(85) = v2;CC(86) = v2; CC(87) = v2;%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
% ESTE INDICE ES PARA EL TIEMPO % tact=dtau*it;% tiempo actual %
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
% PARTE ELECTRICA
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
for i=1:totalElementos;% Nos paramos en un elemento i %
n1=Ele(i,1);n2=Ele(i,2);n3=Ele(i,3);n4=Ele(i,4); ro=[Coor(n1,1) Coor(n2,1) Coor(n3,1) Coor(n4,1)]; ze=[Coor(n1,2) Coor(n2,2) Coor(n3,2) Coor(n4,2)]; Tcond=[Tvie(1,n1) Tvie(1,n2) Tvie(1,n3) Tvie(1,n4)];
% Matriz K %
for l1=1:nG; for l2=1:nG; J=[deR(r(l1),s(l2))*ro' deR(r(l1),s(l2))*ze'; deS(r(l1),s(l2))*ro' deS(r(l1),s(l2))*ze']; % jacobiano de los elementos evaluado en los puntos de Gauss Legendre % erre=H(r(l1),s(l2))*ro'; if Material(i) == 2 Tc=H(r(l1),s(l2))*Tcond'; end K=w(l1)*w(l2)*erre*(B(r(l1),s(l2)))'*inv(J)'*inv(J)*B(r(l1),s(l2))*ConductividadElectrica(Material(i),Tc)*det(J)+K;% Aqui la matriz K de 4x4 es calculada como un producto de matrices % endend
% ensamble de las matrices %
for j=1:4
88
for l=1:4 jg=M(i,j); lg=M(i,l); Kg(jg,lg)=Kg(jg,lg)+K(j,l); endendK=0;end
% Condiciones de contorno %for i1=1:gof; if CC(i1)>0 for j1=1:gof; if j1==i1 Kg(i1,j1)=1; else Kg(i1,j1)=0; end end endend
% solucion %
V=inv(Kg)*CC';
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
for j=1:gof;Solu(j,1)=Coor(j,1); Solu(j,2)=Coor(j,2);Solu(j,3)=V(j);end
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
% PARTE TERMICA %
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
for i=1:(eletot(1));% Nos paramos en un elemento i %
n1=Ele(i,1);n2=Ele(i,2);n3=Ele(i,3);n4=Ele(i,4); ro=[Coor(n1,1) Coor(n2,1) Coor(n3,1) Coor(n4,1)]; ze=[Coor(n1,2) Coor(n2,2) Coor(n3,2) Coor(n4,2)]; Tcond=[Tvie(1,n1) Tvie(1,n2) Tvie(1,n3) Tvie(1,n4)];
% Matrices KT, CT y vector FT (evalua el calor por efecto Joule) del elemento i%
for l1=1:nG; for l2=1:nG; % jacobiano de los elementos evaluado en los puntos de Gauss Legendre %
89

J=[deR(r(l1),s(l2))*ro' deR(r(l1),s(l2))*ze'; deS(r(l1),s(l2))*ro' deS(r(l1),s(l2))*ze']; erre=H(r(l1),s(l2))*ro';
KT=w(l1)*w(l2)*erre*(B(r(l1),s(l2)))'*inv(J)'*inv(J)*B(r(l1),s(l2))*ConductividadTermica(Material(i))*det(J)+KT; % Aqui la matriz KT de 4x4 es calculada como un producto de matrices % CT=w(l1)*w(l2)*erre*(H(r(l1),s(l2)))'*H(r(l1),s(l2))*CapacidadCalorifica(Material(i))*det(J)+CT; % Densidad de corriente/sigma %
densiJ=-1*inv(J)*(B(r(l1),s(l2)))*[V(n1) V(n2) V(n3) V(n4)]'; if Material(i) == 2 Tc=H(r(l1),s(l2))*Tcond'; end
FT=w(l1)*w(l2)*erre*H(r(l1),s(l2))'*densiJ'*densiJ*ConductividadElectrica(Material(i),Tc)*det(J)+FT; endend
% ensamble de la matrices KgT, CgT y vector FgT %
for j=1:4 jg=M(i,j); FgT(jg,1)=FgT(jg,1)+FT(j,1); for l=1:4 lg=M(i,l); KgT(jg,lg)=KgT(jg,lg)+KT(j,l); CgT(jg,lg)=CgT(jg,lg)+CT(j,l); endendKT=0;FT=0;CT=0;end
% condiciones de contorno %
for i1=1:gof; if CCT(i1)>0 FgT(i1,1)=0; for j1=1:gof; if j1==i1 KgT(i1,j1)=1; CgT(i1,j1)=dtau; else KgT(i1,j1)=0; CgT(i1,j1)=0; end end endend
% Solucion %
90
T=Tvie'+inv(CgT/dtau)*(-KgT*Tvie'+FgT+CCT');
Tvie=T';
fprintf('tiempo: %e, %f, Tmax : %f \n', tact, it, max(T))
SoluT(it,1)= tact;SoluT(it,2:gof+1)= T';
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%Itotal=0;
% calculo de la corriente del elemento integrado sobre la coordenada roI(i)=Itotal % for i=43:49; % suma sobre los nodos del heater % if Material(i) == 1 n1=Ele(i,1); n2=Ele(i,2); n3=Ele(i,3); n4=Ele(i,4); ro=[Coor(n1,1) Coor(n2,1) Coor(n3,1) Coor(n4,1)]; ze=[Coor(n1,2) Coor(n2,2) Coor(n3,2) Coor(n4,2)]; Tcond=[Tvie(1,n1) Tvie(1,n2) Tvie(1,n3) Tvie(1,n4)]; l2=2; for l1=1:nG; J1=[deR(r(l1),s(l2))*ro' deR(r(l1),s(l2))*ze'; deS(r(l1),s(l2))*ro' deS(r(l1),s(l2))*ze']; erre1=H(r(l1),s(l2))*ro';
densiJ1=-ConductividadElectrica(Material(i),Tc) *inv(J1)* (B(r(l1),s(l2)))* [V(n1) V(n2) V(n3) V(n4)]';
% Corriente del elemento integral sobre ro %
Itotal=Itotal+w(l1)*erre1*densiJ1(2)*deR(r(l1),s(l2))*ro';
end %cerrando l1% % Luego de integrar en ro, o sea en l1, se asigna a la corriente del elemento en cuestion %
I(i)=Itotal; end Corrheater=Itotal;end
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%
Itotal=0;
% calculo de la corriente del elemento integrado sobre la coordenada roI(i)=Itotal;% for i=50:56; % suma sobre los nodos del heater 2da hilera% if Material(i) == 1
91

n1=Ele(i,1); n2=Ele(i,2); n3=Ele(i,3); n4=Ele(i,4); ro=[Coor(n1,1) Coor(n2,1) Coor(n3,1) Coor(n4,1)]; ze=[Coor(n1,2) Coor(n2,2) Coor(n3,2) Coor(n4,2)]; Tcond=[Tvie(1,n1) Tvie(1,n2) Tvie(1,n3) Tvie(1,n4)]; l2=2; for l1=1:nG; J1=[deR(r(l1),s(l2))*ro' deR(r(l1),s(l2))*ze'; deS(r(l1),s(l2))*ro' deS(r(l1),s(l2))*ze']; erre1=H(r(l1),s(l2))*ro';
densiJ1=-ConductividadElectrica(Material(i),Tc) *inv(J1)* (B(r(l1),s(l2)))* [V(n1) V(n2) V(n3) V(n4)]';
% Corriente del elemento integral sobre ro %
Itotal=Itotal+w(l1)*erre1*densiJ1(2)*deR(r(l1),s(l2))*ro';
end %cerrando l1% % Luego de integrar en ro, o sea en l1, se asigna a la corriente del elemento en cuestion %
I(i)=Itotal; end Corrheater2=Itotal;
end
fprintf('corriente heater %e y %e potencial V1 %f \n', Corrheater, Corrheater2,v1);
% reasignacion del valor de v1 %
if it == 1 Corrienteini=(Corrheater+Corrheater2)/2;end
if it > 1 Corrientenueva=(Corrheater+Corrheater2)/2; v1=v2+(v1-v2)*Corrienteini/Corrientenueva;end
%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%%% end% Aqui cierra el loop del tiempo it %
save solucion.txt SoluT /ascii
F_G_CC_CI.m (ejemplo)
92
clear;
% DATOS FISICOS Y GEOMETRICOS%
% Conductividad Electrica en 1/Ohm cm %% En el archivo ConductividadElectrica.m %
% Conductividad Termica en W/cm K %k1 = 0.1; % Heater %k2 = 0.002; % Calcogenuro %k3 = 0.01; % Silicon Oxide %ConductividadTermica = [k1; k2; k3];
% Capacidad calorifica por unidad de volumem (ro*Cp) en J/cm3 K %c1 = 1.7; % Heater %c2 = 1.25; % Calcogenuro %c3 = 3.1; % Silicon Oxide %CapacidadCalorifica = [c1; c2; c3];
% Factor de escala (en cm) %FactorEscala = 1e-6; % equivale a 10 nm %
% Coordenadas de los nodos %x1=0;x2=1;x3=2;x4=4;x5=7;x6=10;x7=20;x8=120;X = [x1 x2 x3 x4 x5 x6 x7 x8];
y1=0;y2=7.5;y3=15;y4=17;y5=19;y6=21;y7=23;y8=25;y9=27;y10=34.5;y11=42;y12=142;Y=[y1 y2 y3 y4 y5 y6 y7 y8 y9 y10 y11 y12];
% Paso del Tiempo caracteristico del transitorio %dtau = (c2/k3)*FactorEscala^2/50;
% Coordenadas r, z de los nodos (en cm) %Coor =[];for j=Y for i=X Coor = [Coor; i j]; endend
Coor = FactorEscala*Coor;
93

%Grados de libertad totalesgof=length(X)*length(Y);
% CONDICIONES DE CONTORNO %% Potecial en Volt %v1=30; v2=1;% Temperatura ambiente %TAmb=303;
% 0: libre %% distinto de 0: trabado en ese valor %% Electricas %CC = zeros(1, gof);CC(1) = v1; CC(2) = v1; CC(3) = v1; CC(4) = v1; CC(5) = v1;CC(6) = v1; CC(7) = v1;CC(81) = v2; CC(82) = v2; CC(83) = v2; CC(84) = v2; CC(85) = v2;CC(86) = v2; CC(87) = v2;
% Termicas % CCT=zeros(1, gof);CCT(1) = TAmb; CCT(2) = TAmb; CCT(3) = TAmb; CCT(4) = TAmb; CCT(5) = TAmb;CCT(6) = TAmb; CCT(7) = TAmb; CCT(8) = TAmb; CCT(16) = TAmb; CCT(24) = TAmb;CCT(32) = TAmb; CCT(40) = TAmb; CCT(48) = TAmb; CCT(56) = TAmb; CCT(64) = TAmb;CCT(72) = TAmb; CCT(80) = TAmb; CCT(88) = TAmb; CCT(89) = TAmb; CCT(90) = TAmb;CCT(91) = TAmb; CCT(92) = TAmb; CCT(93) = TAmb; CCT(94) = TAmb; CCT(95) = TAmb;CCT(96) = TAmb;
% Vector de materiales por elemento %Material = [1; 1; 1; 1; 1; 1; 3; 1; 1; 1; 1; 1; 1; 3; 2; 2; 2; 2; 3; 3; 3; 2; 2; 2; 2; 3; 3; 3; 2; 2; 2; 2; 3; 3; 3; 2; 2; 2; 2; 3; 3; 3; 1; 1; 3; 3; 3; 3; 3; 1; 1; 3; 3; 3; 3; 3; 1; 1; 1; 1; 1; 1; 3; 1; 1; 1; 1; 1; 1; 3; 3; 3; 3; 3; 3; 3; 3];
% CONDICION INICIAL %% Temperatura inicial (a t=0) %Tin = TAmb;
% Nodos de los elementos %Ele =[];for j=1:length(Y)-1 for i=1:length(X)-1 nodo1 = (j-1)*length(X)+i; nodo2 = j*length(X)+i; nodo3 = nodo1+1; nodo4 = nodo2+1; Ele = [Ele; nodo1 nodo2 nodo3 nodo4]; endend
eletot=size(Ele);totalElementos = eletot(1);
94

% matriz de ensamble M %
M = Ele;
% puntos de Gauss Legendre %% 3x3 %r=[-.774596669241483 0 .774596669241483]; s=[-.774596669241483 0 .774596669241483];% (funciones de peso) % w=[0.555555555555556 0.888888888888889 0.555555555555556];
nG=length(r);
ConductividadElectrica.m (ejemplo)
% Funcion que devuelve la conductividad térmica de un elemento %
function [ConductividadElectrica] = ConductividadElectrica(material,T) switch material case 1, ConductividadElectrica = 4500; case 2, if T < 423 Ea = 0.243; %eV rho0 = 1/36.1; %ohm cm elseif T < 633 Ea = 0.383; %eV rho0 = 1/1.96e5; %ohm cm else Ea = 0; %eV rho0 = 1/830; %ohm cm end k = 8.617343e-5; % eV/K kT = k*T; ConductividadElectrica = (1/rho0)*exp(-Ea/kT); case 3, ConductividadElectrica = 1e-8; otherwise, ConductividadElectrica = 0; end
b.m
% Funcion para calcular la matriz B %
function [B] = B(r1,s1) B=[-(1+s1)/4 -(1-s1)/4 (1+s1)/4 (1-s1)/4; (1-r1)/4 -(1-r1)/4 (1+r1)/4 -(1+r1)/4];
95

h.m
% Funcion que se usa para calcular la matriz H %
function [H] = H(r1,s1) H=(1/4)*[(1-r1)*(1+s1) (1-r1)*(1-s1) (1+r1)*(1+s1) (1+r1)*(1-s1)];
der.m
% Funcion que se usa para calcular el jacobiano de los elementos evaluado en los puntos de Gauss Legendre %
function [deR] = deR(r1,s1) deR=(1/4)*[-(1+s1) -(1-s1) (1+s1) (1-s1)];
des.m
% Funcion que se usa para calcular el jacobiano de los elementos evaluado en los puntos de Gauss Legendre %
function [deS] = deS(r1,s1) deS=(1/4)*[(1-r1) -(1-r1) (1+r1) -(1+r1)];
96





























