Embed Size (px)
Citation preview

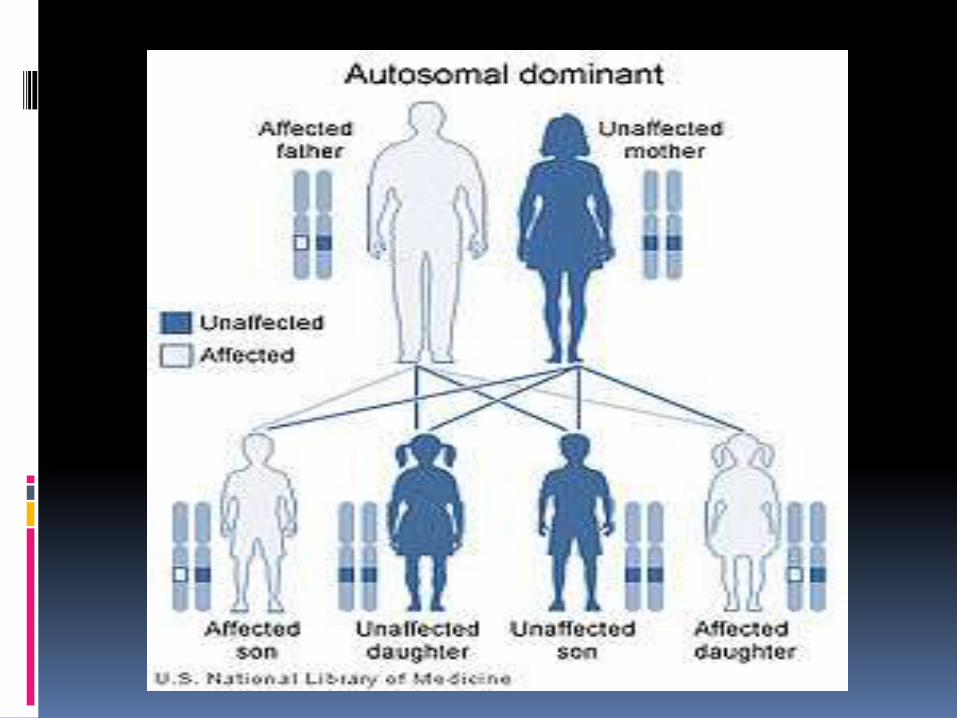
FACOMATOSIS KARINA ALVARADO

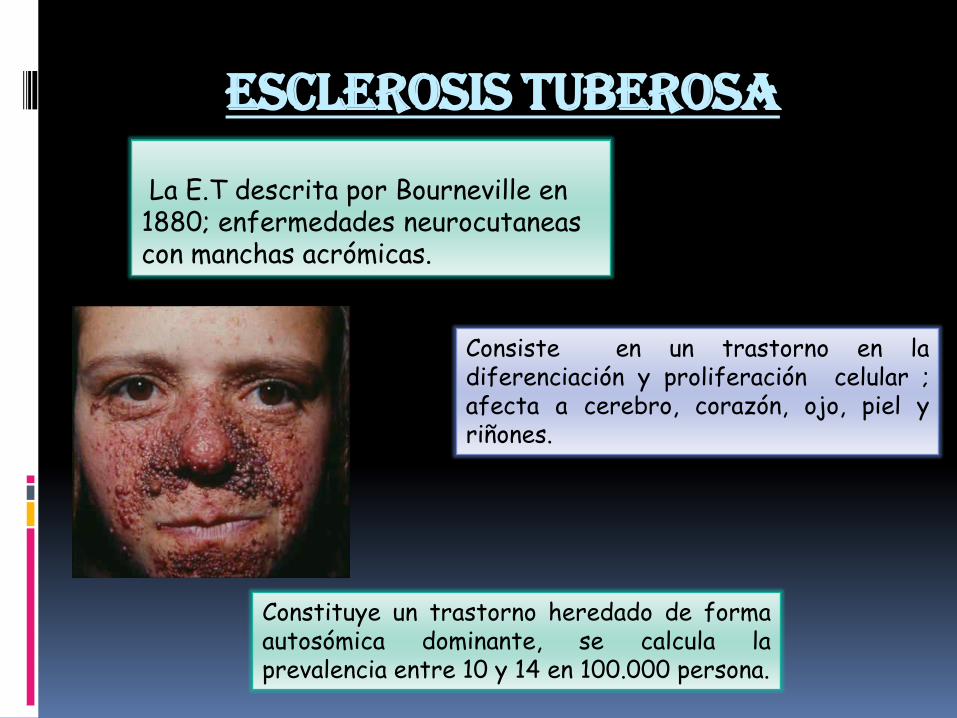
Esclerosis tuberosa
La E.T descrita por Bourneville en 1880; enfermedades neurocutaneascon manchas acrómicas.
Consiste en un trastorno en ladiferenciación y proliferación celular ;afecta a cerebro, corazón, ojo, piel yriñones.
Constituye un trastorno heredado de formaautosómica dominante, se calcula laprevalencia entre 10 y 14 en 100.000 persona.
etiopatogenia
Se hereda con carácter autosómicodominante y penetrancia variable, debidoa mutaciones en los genes TSC1 (9q34) oTSC2 (16p13). Tales mutacionesacontecen de forma espontánea en pocomás del 50% de los casos.
Las proteínas afectas que derivan demutaciones en los genes TSC1 y TSC2 sedenominan, respectivamente, hamartina ytuberina, y actúan como represorestumorales..

Cuadro clínico
Retraso psicomotor
Epilepsia
Deterioro cognitivo
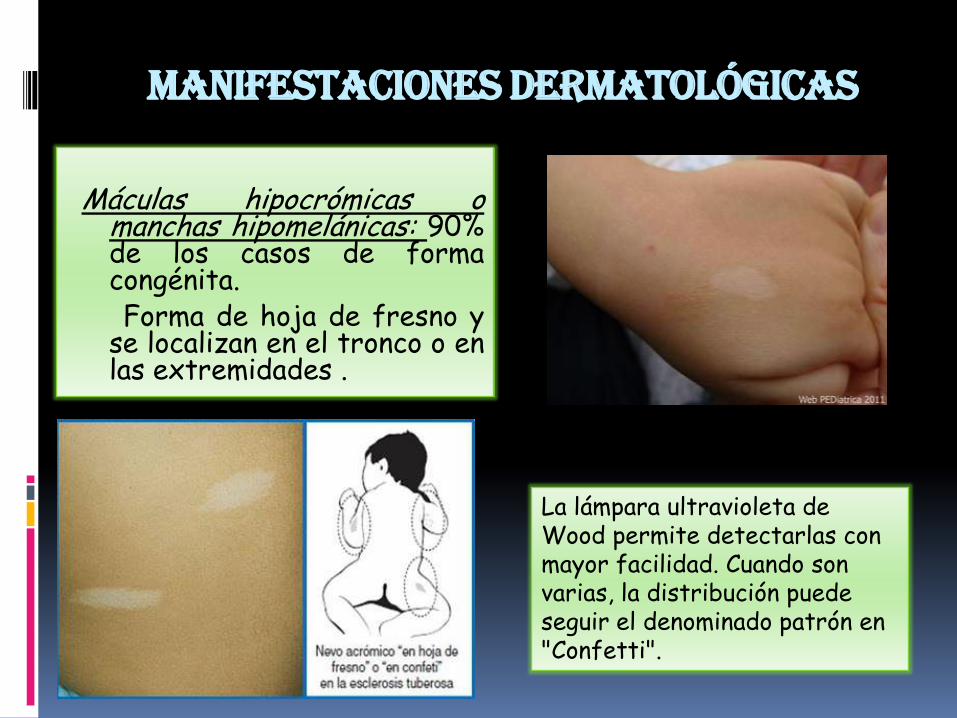
Manifestaciones dermatológicas
Máculas hipocrómicas omanchas hipomelánicas: 90%de los casos de formacongénita.Forma de hoja de fresno y
se localizan en el tronco o enlas extremidades .
La lámpara ultravioleta de Wood permite detectarlas con mayor facilidad. Cuando son varias, la distribución puede seguir el denominado patrón en "Confetti".
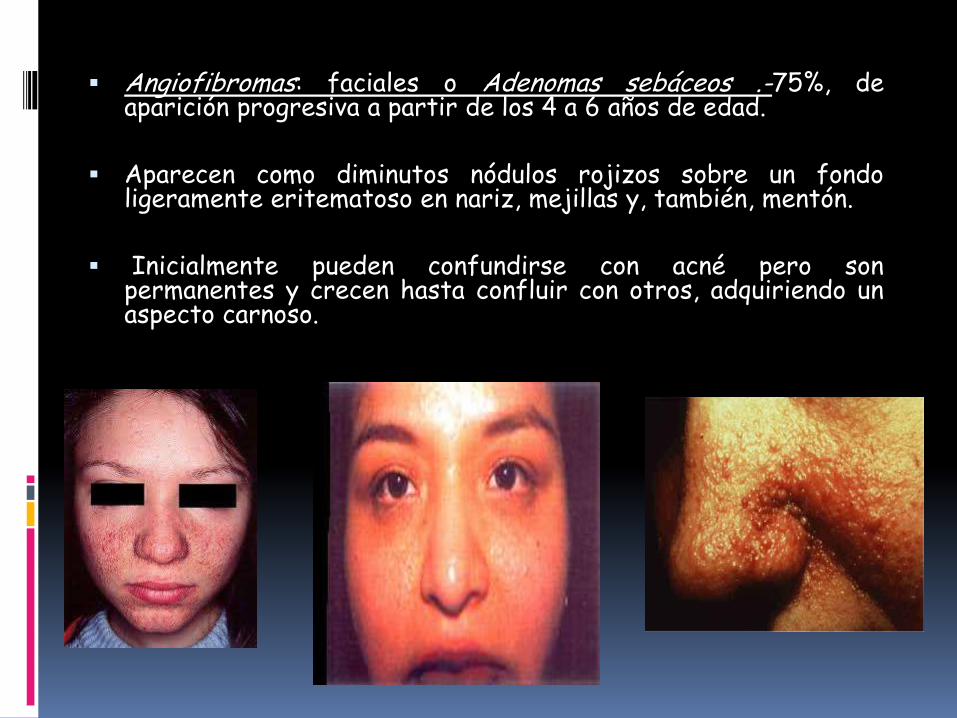
Angiofibromas: faciales o Adenomas sebáceos .-75%, deaparición progresiva a partir de los 4 a 6 años de edad.
Aparecen como diminutos nódulos rojizos sobre un fondoligeramente eritematoso en nariz, mejillas y, también, mentón.
Inicialmente pueden confundirse con acné pero sonpermanentes y crecen hasta confluir con otros, adquiriendo unaspecto carnoso.
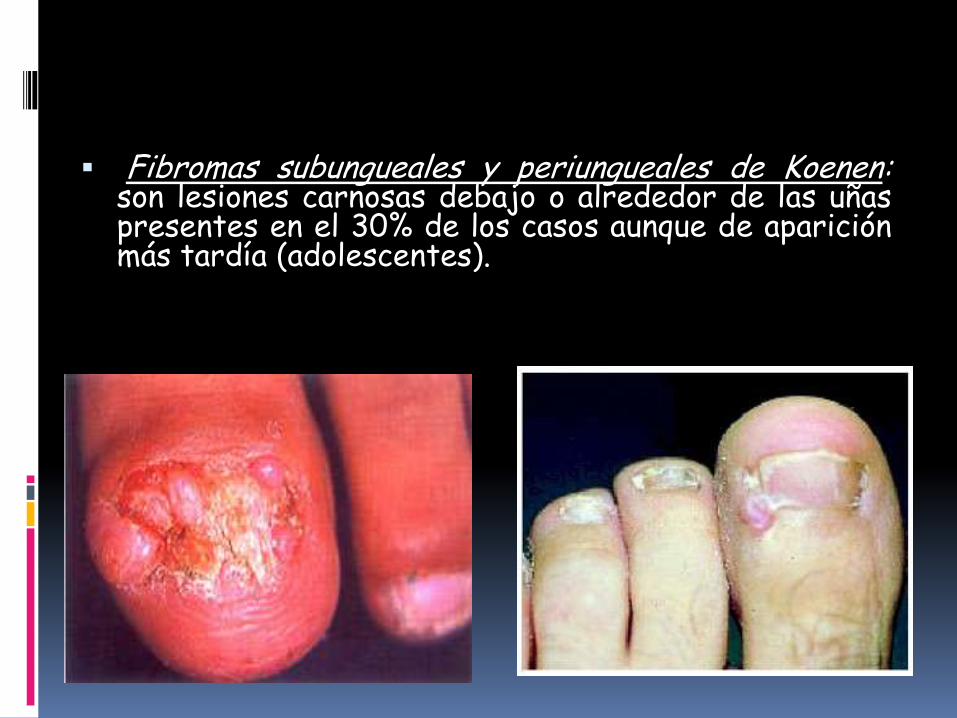
Fibromas subungueales y periungueales de Koenen:son lesiones carnosas debajo o alrededor de las uñaspresentes en el 30% de los casos aunque de apariciónmás tardía (adolescentes).

Placa de Chagrin o Nevus deltejido conectivo: 65% de loscasos. Es una lesión rugosaelevada con consistencia de pielde naranja, en la regiónlumbosacra.
Parche lijoso: es una lesiónligeramente elevada, de bordesirregulares , se localiza enespalda o en flancos. Del 20 al35% en adolescentes.
Placas fibrosas: formaciones decolor rosado y prominentes,localizadas en frente y mejillas ,aparición en la infancia..

Manifestaciones neurologicas
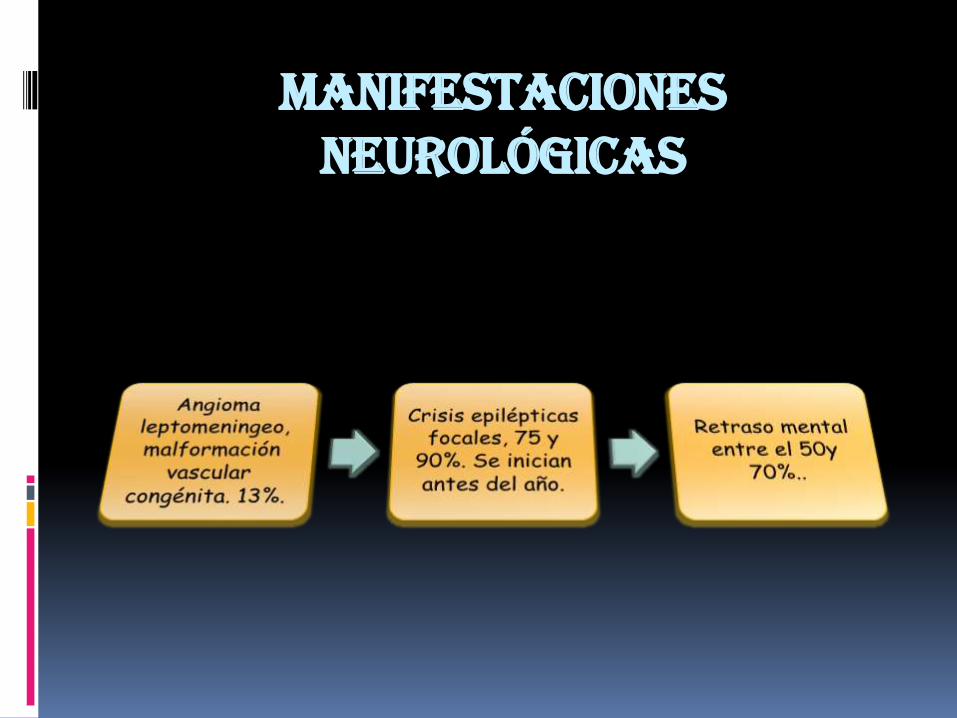
Triada clínica de Vogt: epilepsia, retraso mental y angiofibromas, se
encuentra en un tercio de los casos y el 6% nopresenta estas manifestaciones.
Las crisis epilépticas se presentan en el 80 al90% de los casos.
En menores de 1 año da importanciadiagnostica.
En niños mayores y adultos hay crisis parcialessimples o complejas

Retraso mental en el 60% de los casos…
Los trastornos de conducta y el autismoestán presentes en pctes. Con esclerosistuberosa habitualmente asociado con laepilepsia y el retraso mental…

Del 6 al 14% astrocitomas de célulasgigantes, en las dos primeras décadas de lavida…
Aunque poco frecuente, la oclusión súbita delagujero de Monro, hipertensión intracraneal.
La afectación neurológica esta dada por:
Crisis epilépticas
Retraso mental
Trastornos de la conducta
Astrocitomas cerebrales.
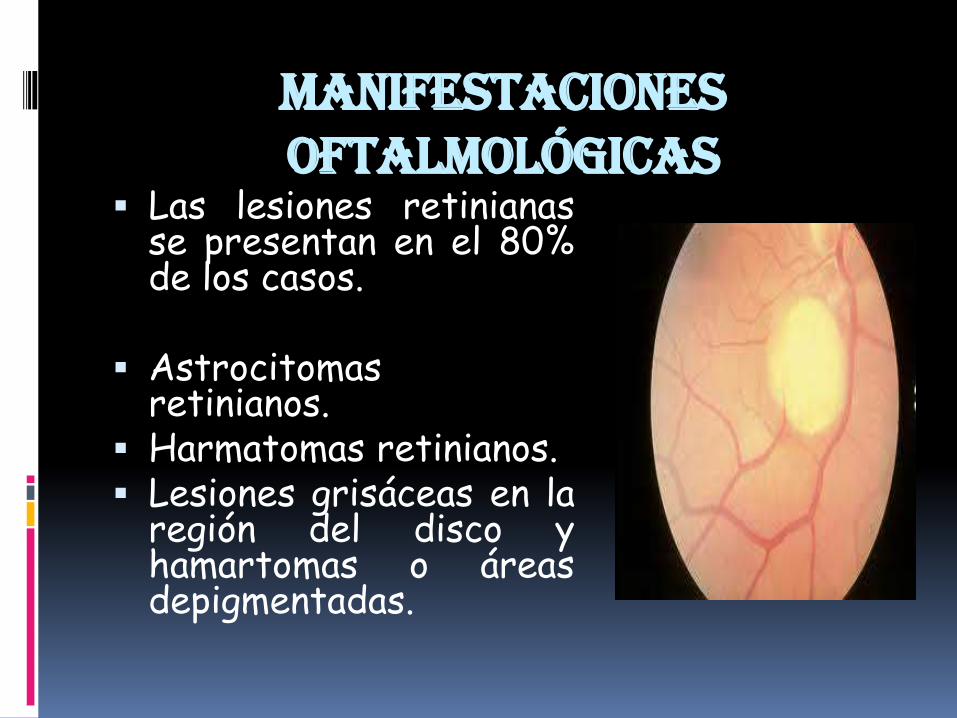
Manifestaciones oftalmológicas
Las lesiones retinianasse presentan en el 80%de los casos.
Astrocitomasretinianos.
Harmatomas retinianos. Lesiones grisáceas en la
región del disco yhamartomas o áreasdepigmentadas.

Manifestaciones renales
Los angiomiolipomas renales constituidospor musculo liso, tejido adiposo yelementos vasculares…. Entre el 50 al 80%de los casos.
edades jóvenes.Afectación bilateralAsintomáticos Tendencia a crecer
-Se presenta dolor abdominal o en el flanco
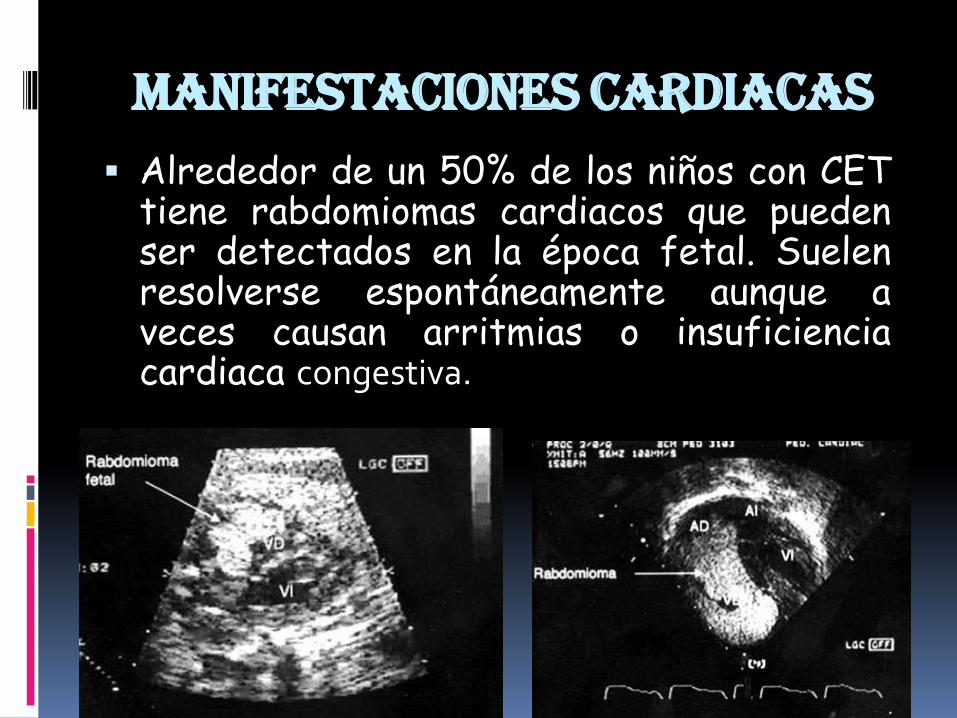
Manifestaciones cardiacas
Alrededor de un 50% de los niños con CETtiene rabdomiomas cardiacos que puedenser detectados en la época fetal. Suelenresolverse espontáneamente aunque aveces causan arritmias o insuficienciacardiaca congestiva.
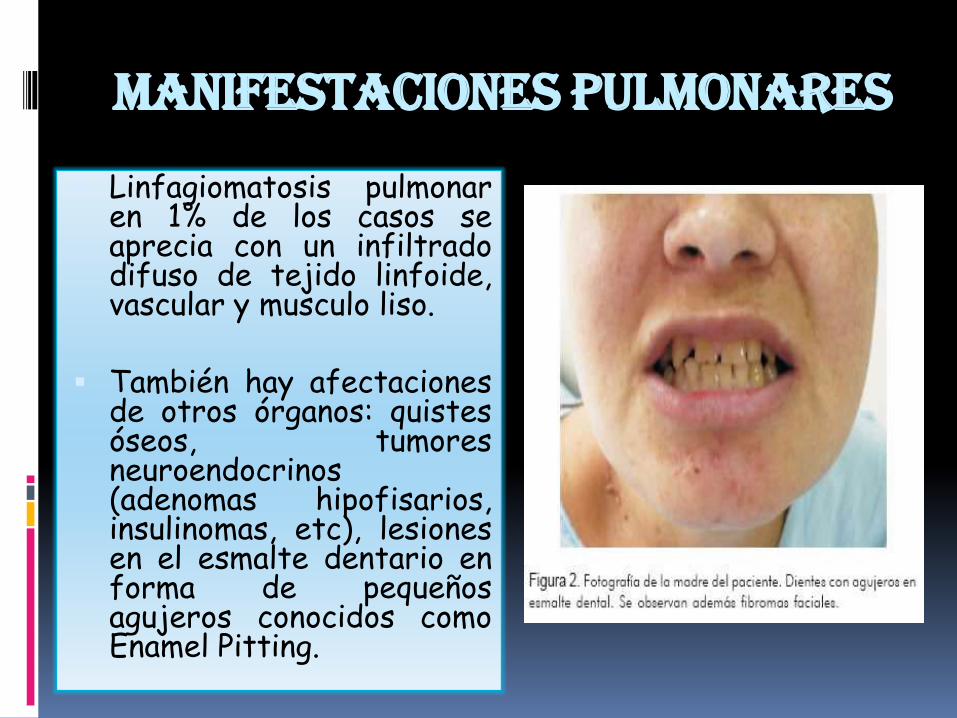
Manifestaciones pulmonares
Linfagiomatosis pulmonaren 1% de los casos seaprecia con un infiltradodifuso de tejido linfoide,vascular y musculo liso.
También hay afectacionesde otros órganos: quistesóseos, tumoresneuroendocrinos(adenomas hipofisarios,insulinomas, etc), lesionesen el esmalte dentario enforma de pequeñosagujeros conocidos comoEnamel Pitting.
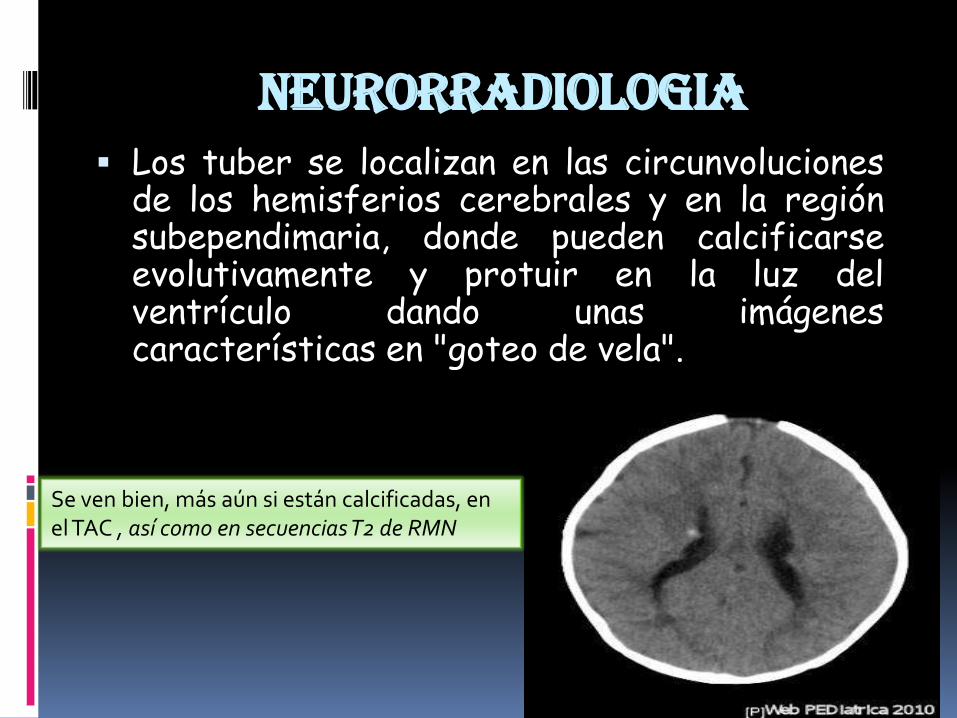
neurorradiologia Los tuber se localizan en las circunvoluciones
de los hemisferios cerebrales y en la regiónsubependimaria, donde pueden calcificarseevolutivamente y protuir en la luz delventrículo dando unas imágenescaracterísticas en "goteo de vela".
Se ven bien, más aún si están calcificadas, en el TAC , así como en secuencias T2 de RMN

neuropatologia
Lesiones cerebrales en 3 categorías:
o Tuberosidades corticales tambiénllamadas “Escleromas”.
o Nódulos gliales subependimarios
o Astrocitomas subependimarios decélulas gigantes.
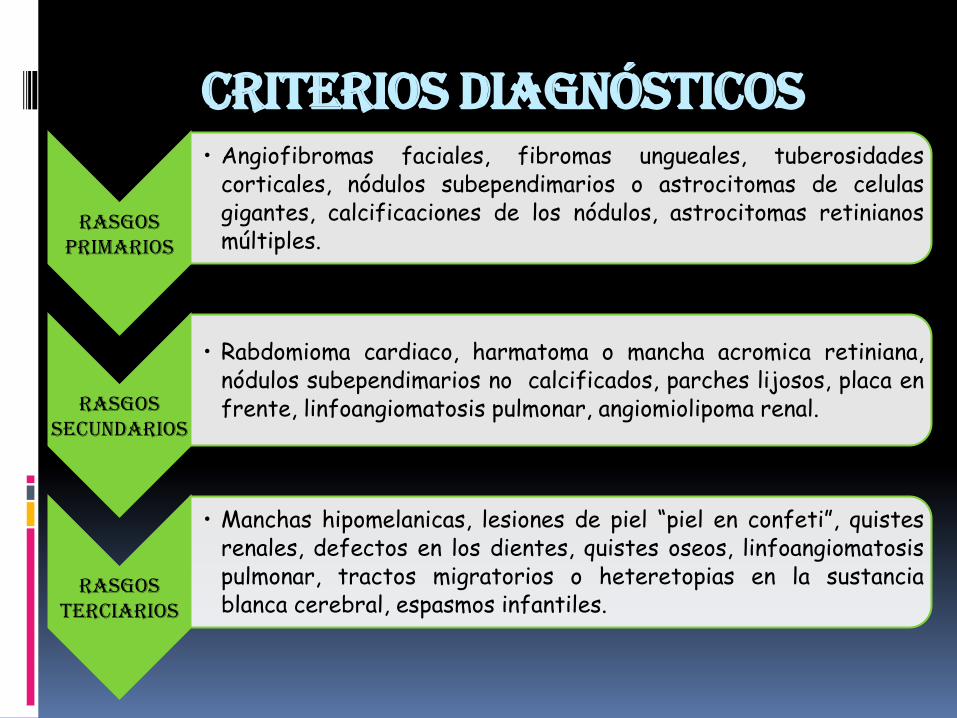
Criterios Diagnósticos
Rasgos primarios
• Angiofibromas faciales, fibromas ungueales, tuberosidadescorticales, nódulos subependimarios o astrocitomas de celulasgigantes, calcificaciones de los nódulos, astrocitomas retinianosmúltiples.
Rasgos secundarios
• Rabdomioma cardiaco, harmatoma o mancha acromica retiniana,nódulos subependimarios no calcificados, parches lijosos, placa enfrente, linfoangiomatosis pulmonar, angiomiolipoma renal.
Rasgos terciarios
• Manchas hipomelanicas, lesiones de piel “piel en confeti”, quistesrenales, defectos en los dientes, quistes oseos, linfoangiomatosispulmonar, tractos migratorios o heteretopias en la sustanciablanca cerebral, espasmos infantiles.

Síndrome de sturge weber
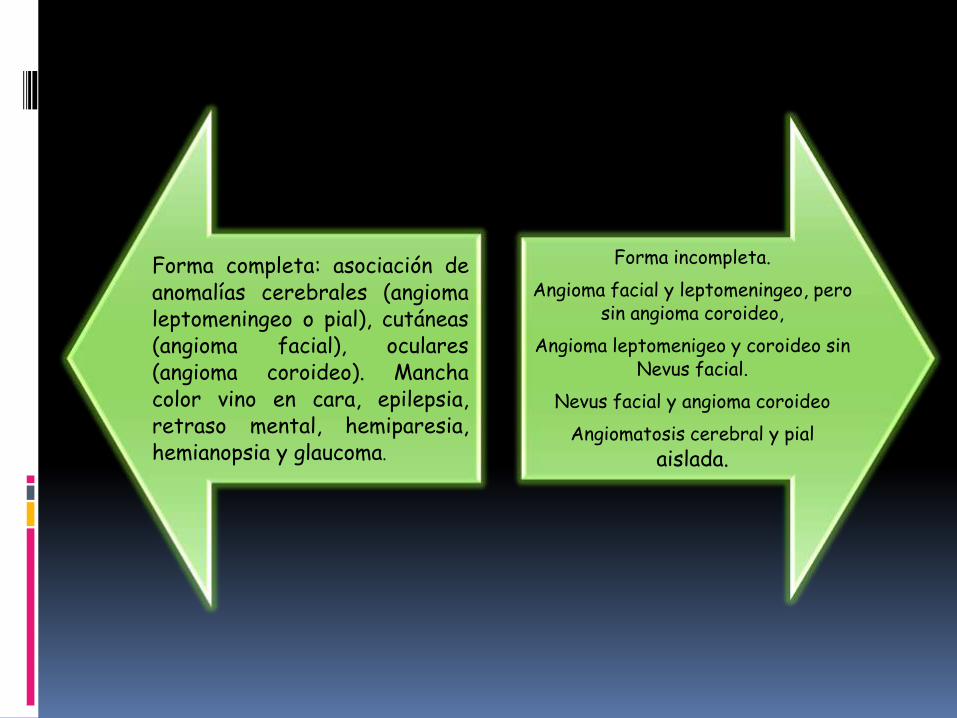
Forma completa: asociación deanomalías cerebrales (angiomaleptomeningeo o pial), cutáneas(angioma facial), oculares(angioma coroideo). Manchacolor vino en cara, epilepsia,retraso mental, hemiparesia,hemianopsia y glaucoma.
Forma incompleta.
Angioma facial y leptomeningeo, pero sin angioma coroideo,
Angioma leptomenigeo y coroideo sin Nevus facial.
Nevus facial y angioma coroideo
Angiomatosis cerebral y pial
aislada.
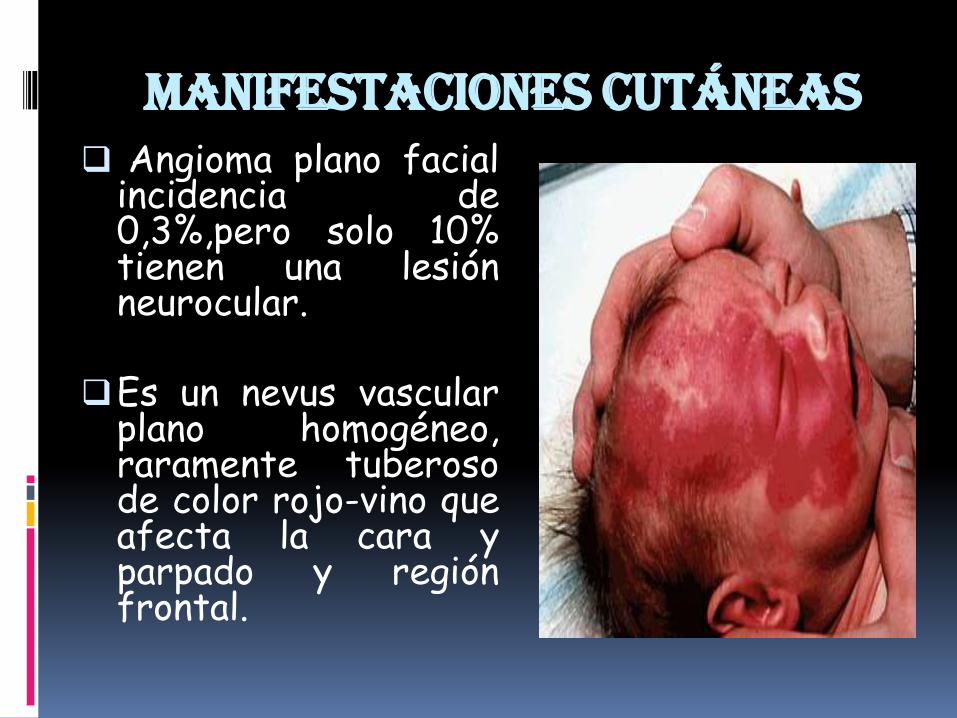
Manifestaciones cutáneas Angioma plano facial
incidencia de0,3%,pero solo 10%tienen una lesiónneurocular.
Es un nevus vascularplano homogéneo,raramente tuberosode color rojo-vino queafecta la cara yparpado y regiónfrontal.
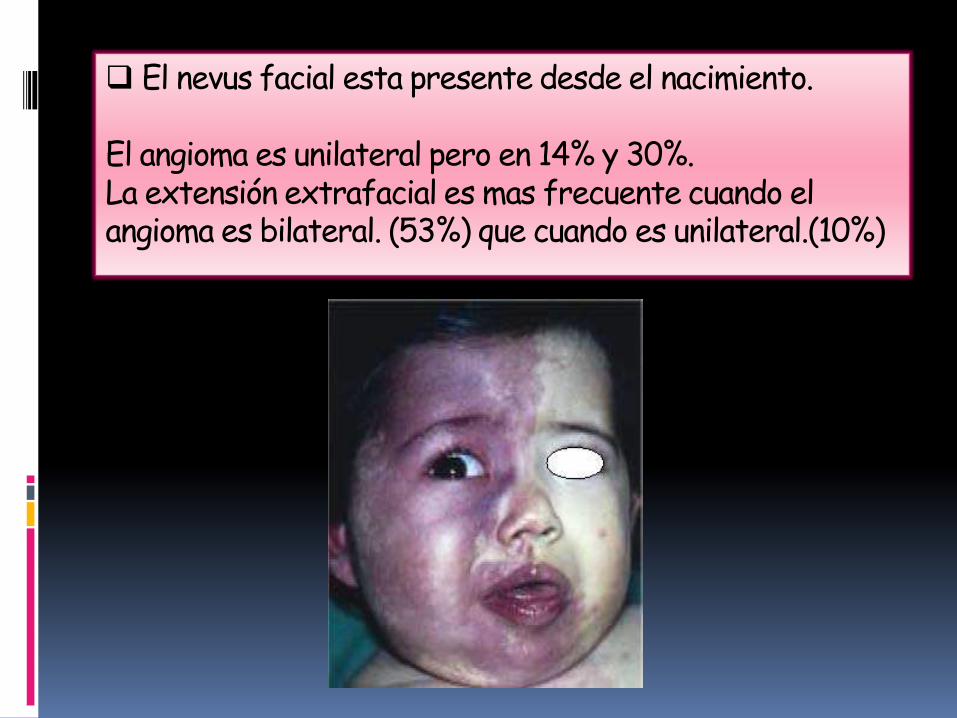
El nevus facial esta presente desde el nacimiento.
El angioma es unilateral pero en 14% y 30%.La extensión extrafacial es mas frecuente cuando el angioma es bilateral. (53%) que cuando es unilateral.(10%)

Afectación ocular
El angioma ocular aparece en el 30%, afecta a la coroides y laesclerótica ocular. Se demuestra una elevación anaranjadalocalizada en el polo posterior del ojo.
El angioma coroide produce glaucoma entre el 25% y 60% de los casos., puede ser congénito o aparecer con la edad y producir BUFTALMO.
Hay dolor retroorbitario,, deterioro de la visión, heterocromia del iris, angioma orbitario y dilatación de los vasos retinianos
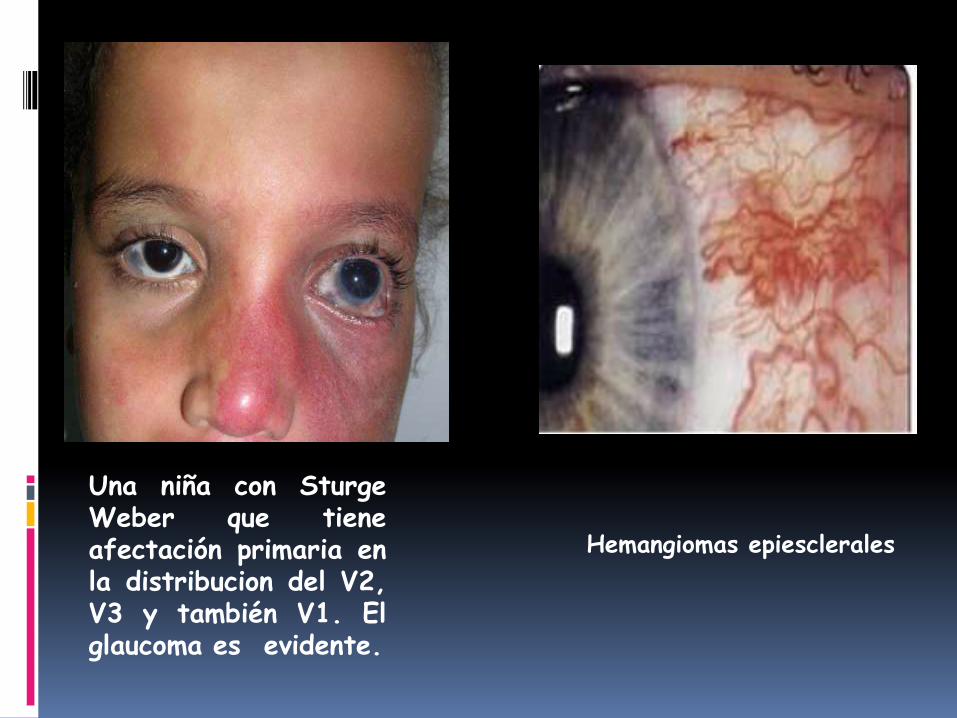
Una niña con SturgeWeber que tieneafectación primaria enla distribucion del V2,V3 y también V1. Elglaucoma es evidente.
Hemangiomas epiesclerales
Manifestaciones neurológicas

diagnostico
Clínica
Los hallazgos tomográficos característicosen el SSW son las calcificacionesgiriformes, serpenteantes, “en vías detren”,
En la región occipito-parietal, la atrofiahemisférica cerebral progresiva y elaumento del tamaño del plexo coroideoipsilateral.)
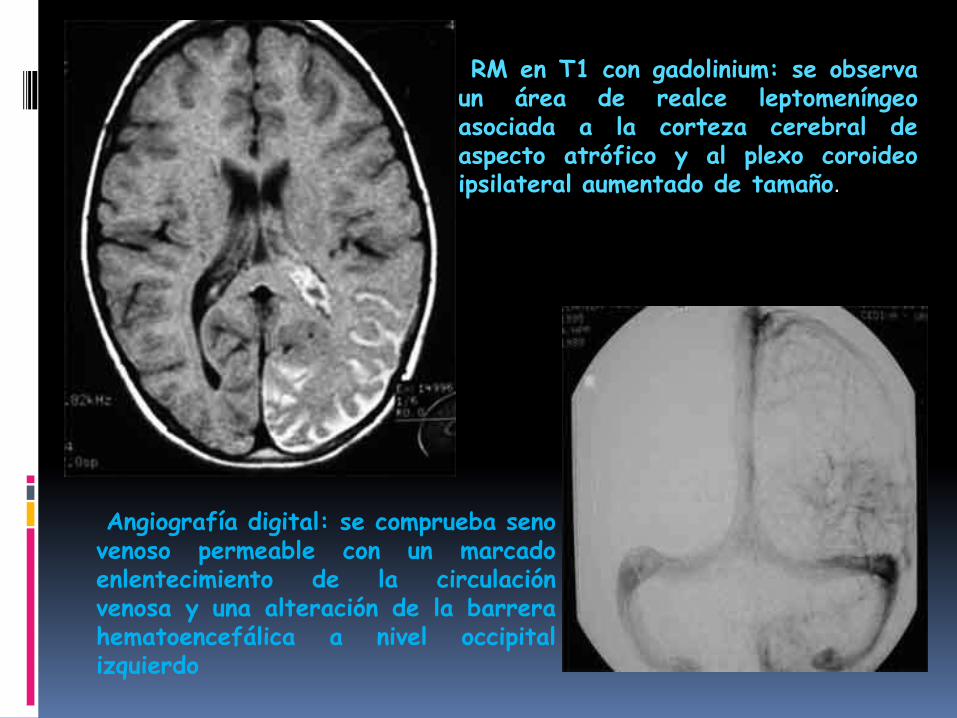
RM en T1 con gadolinium: se observaun área de realce leptomeníngeoasociada a la corteza cerebral deaspecto atrófico y al plexo coroideoipsilateral aumentado de tamaño.
Angiografía digital: se comprueba senovenoso permeable con un marcadoenlentecimiento de la circulaciónvenosa y una alteración de la barrerahematoencefálica a nivel occipitalizquierdo

tratamiento
Control de las crisis epilépticas.
Epilepsia refractaria indicada lalobectomia e incluso la hemisfe-rectomia. Para la resección del áreaafectada.


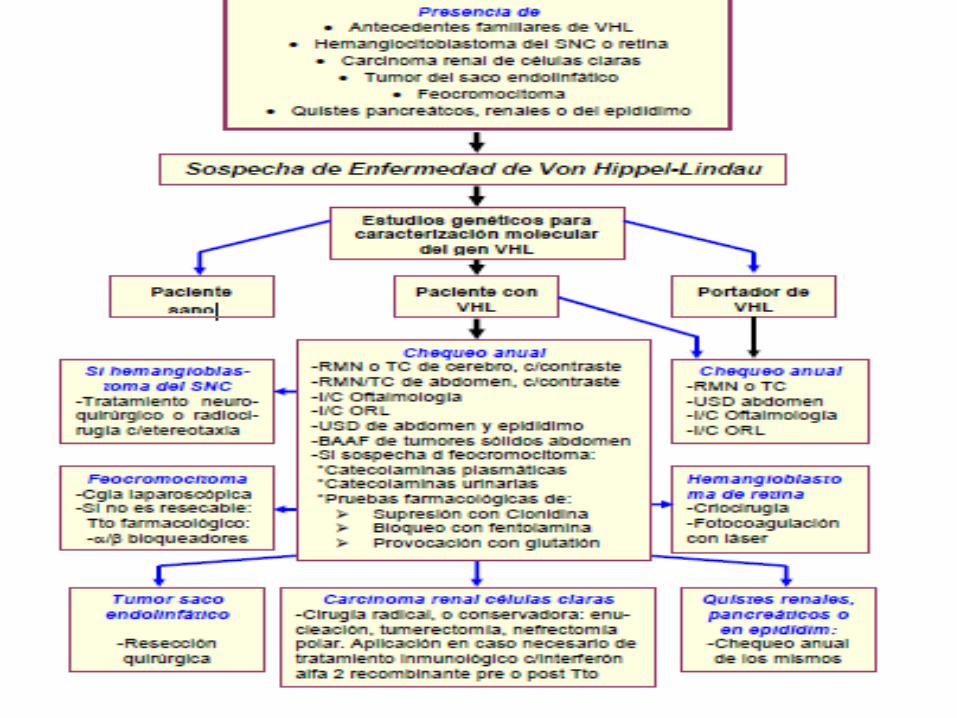
El Síndrome de von Hippel-Lindau, produce tumores en la región ocular, sistema nervioso central, saco endolinfático, riñones, páncreas, glándulas suprarrenales, hígado y epidídimo.
Se presentan 3 en cada 100.000 individuos.
La enfermedad fue descrita por dos gruposindependientes, dirigidos por Eugen vonHippel (en 1894) y Arvid Lindau (en 1926).trastorno neoplásico hereditario autosómicodominante, predisposición a desarrollar tumores.

etiología
Está relacionada con una mutación genéticaen la síntesis del gen supresor tumoral devon Hippel-Lindau (VHL), situado enel cromosoma 3.
Casi todas las personas que tienen unamutación en el gen VHL expresarán síntomasrelacionados con la enfermedad para la edadde la adolescencia..
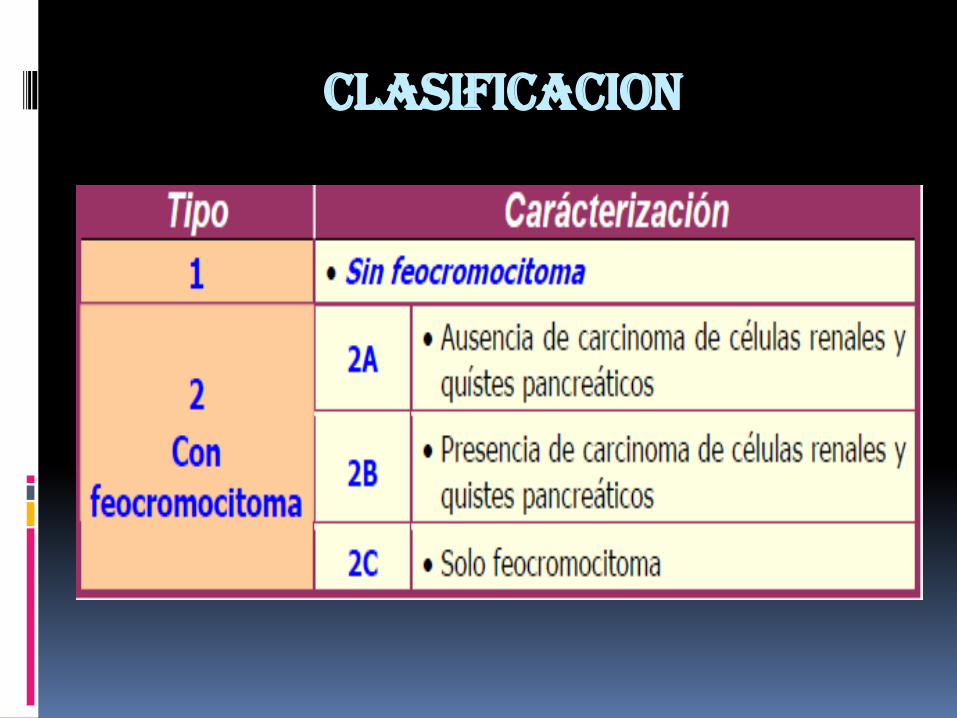
CLASIFICACION

CUADRO CLINICO Se observa la proliferación de tumores, de los cuales lo más esencial es su
localización en el cerebro, la médula espinal, la retina y los riñones, y del mismomodo se multiplican más rápidamente en un paciente joven que en la poblaciónde edad más avanzada.
Rasgos
Los rasgos del VHL son:
Angiomatosis- pequeños nódulos de capilares en la retina y otros órganos.
Hemangioblastomas - tumores del sistema nervioso central (especialmente enel cerebelo, tronco del encéfalo y médula espinal).
Feocromocitoma - tumores de la médula adrenal que frecuentementeproducen catecolaminas
Carcinoma de células renales- tumores malignos del riñón.
Páncreas - quistes y tumores del páncreas, que pueden ser tumoresneuroendocrinos.

diagnostico
El diagnóstico clínico de la enfermedad de von Hippel-Lindau (VHL) seestablece en:
Un individuo sin antecedentes familiares de VHL que presenta dos o máslesiones características (por ejemplo, dos o más hemangioblastomas de laretina o el cerebro o un hemangioblastoma.
Un individuo con un historial familiar positivo del síndrome de VHL, en losque uno o más de las siguientes manifestaciones de la enfermedad estápresente: angioma retiniano, espinal o hemangioblastoma cerebeloso,feocromocitoma, múltiples quistes pancreáticos, epidídimo o citoadenomas,múltiples quistes renales, o carcinoma de células renales antes de la edad de30 años.
Las neurofibromatosis:afectan al desarrollo ycrecimiento de los tejidos delas células neurales(nerviosas).
Estos trastornosocasionan tumores que crecenen los nervios y producen otrasanormalidades tales comocambios en la piel ydeformidades en los huesos.
Las neurofibromatosis ocurren enambos sexos, todas las razas ygrupos étnicos.
Se transmiten a la descendenciade forma autosómica dominante.
neurofibromatosis tipo 1 (NF1) yexiste una alteración en elcromosoma 17.
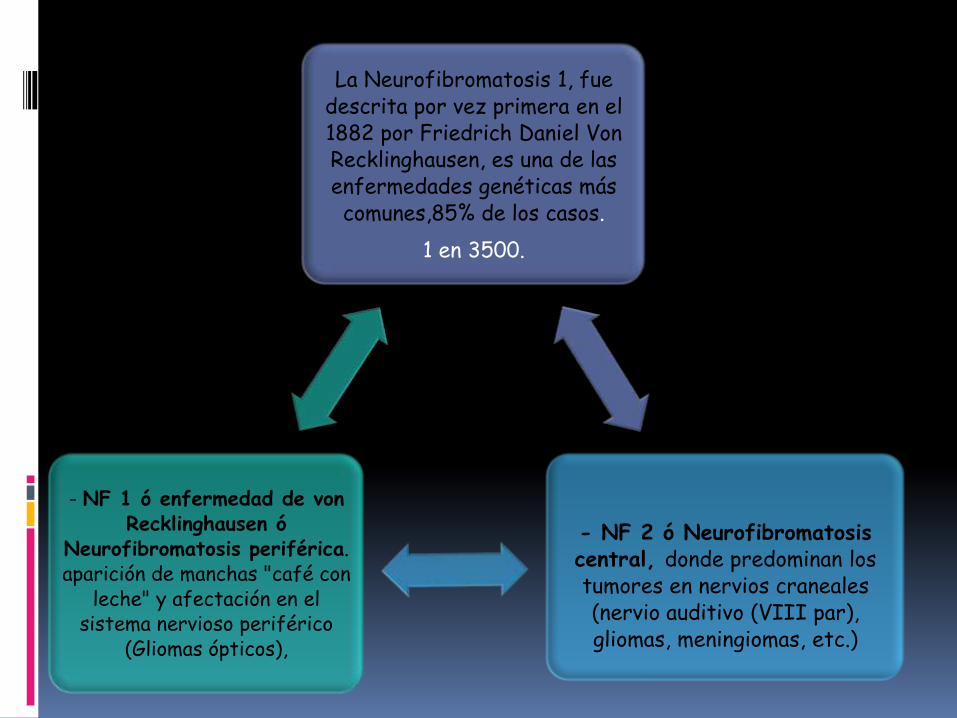
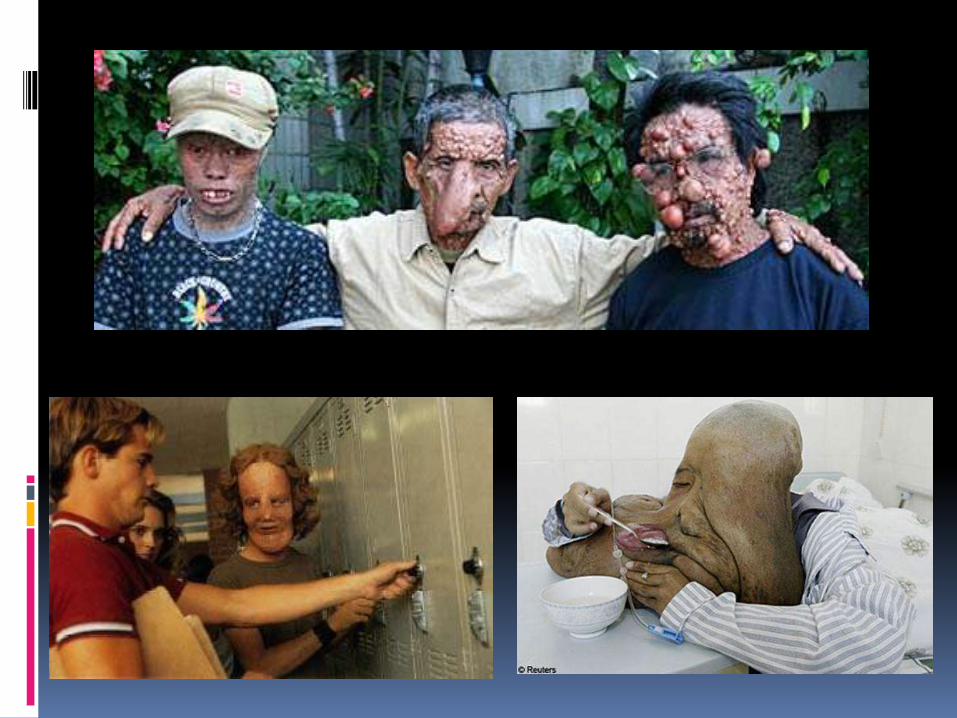
La Neurofibromatosis 1, fue descrita por vez primera en el 1882 por Friedrich Daniel Von Recklinghausen, es una de las enfermedades genéticas más comunes,85% de los casos.
1 en 3500.
- NF 2 ó Neurofibromatosiscentral, donde predominan los tumores en nervios craneales (nervio auditivo (VIII par), gliomas, meningiomas, etc.)
- NF 1 ó enfermedad de von Recklinghausen ó
Neurofibromatosis periférica. aparición de manchas "café con
leche" y afectación en el sistema nervioso periférico
(Gliomas ópticos),
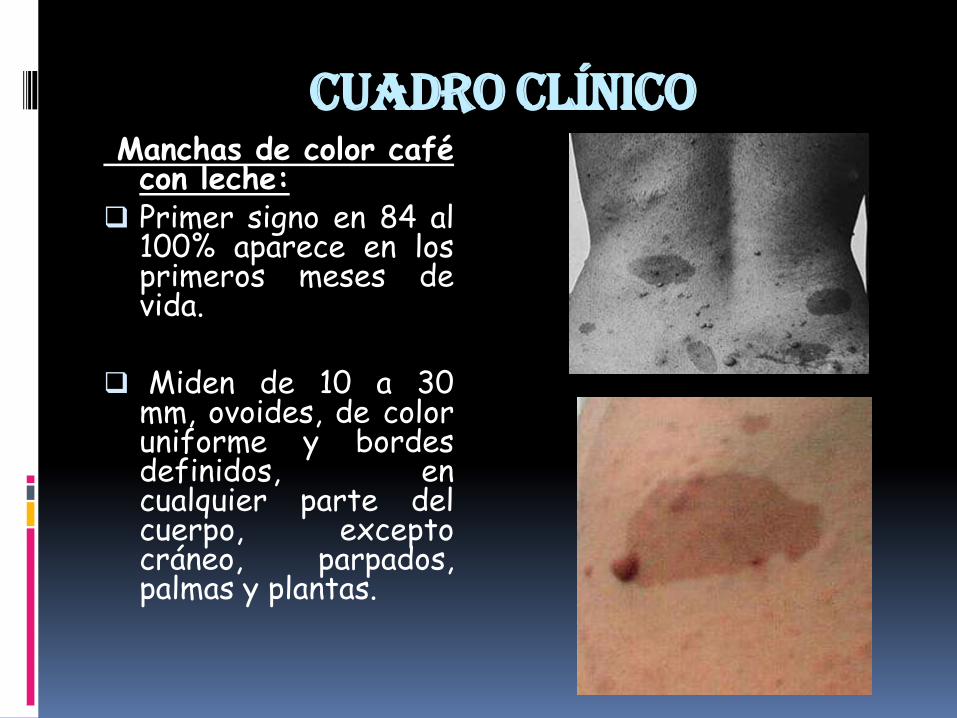
Cuadro clínicoManchas de color cafécon leche:
Primer signo en 84 al100% aparece en losprimeros meses devida.
Miden de 10 a 30mm, ovoides, de coloruniforme y bordesdefinidos, encualquier parte delcuerpo, exceptocráneo, parpados,palmas y plantas.
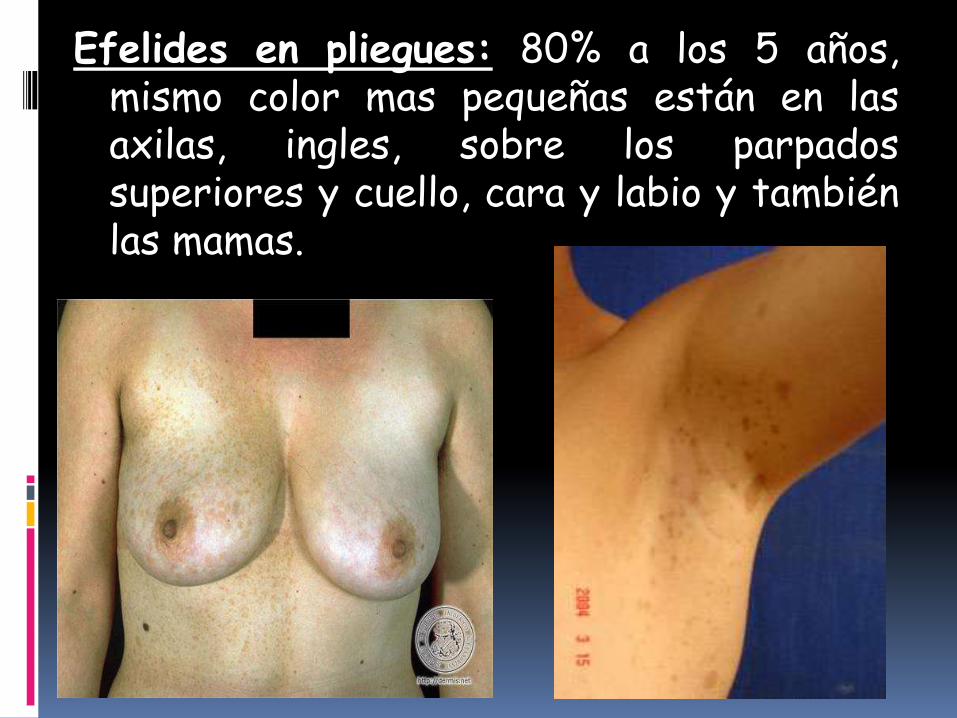
Efelides en pliegues: 80% a los 5 años,mismo color mas pequeñas están en lasaxilas, ingles, sobre los parpadossuperiores y cuello, cara y labio y tambiénlas mamas.
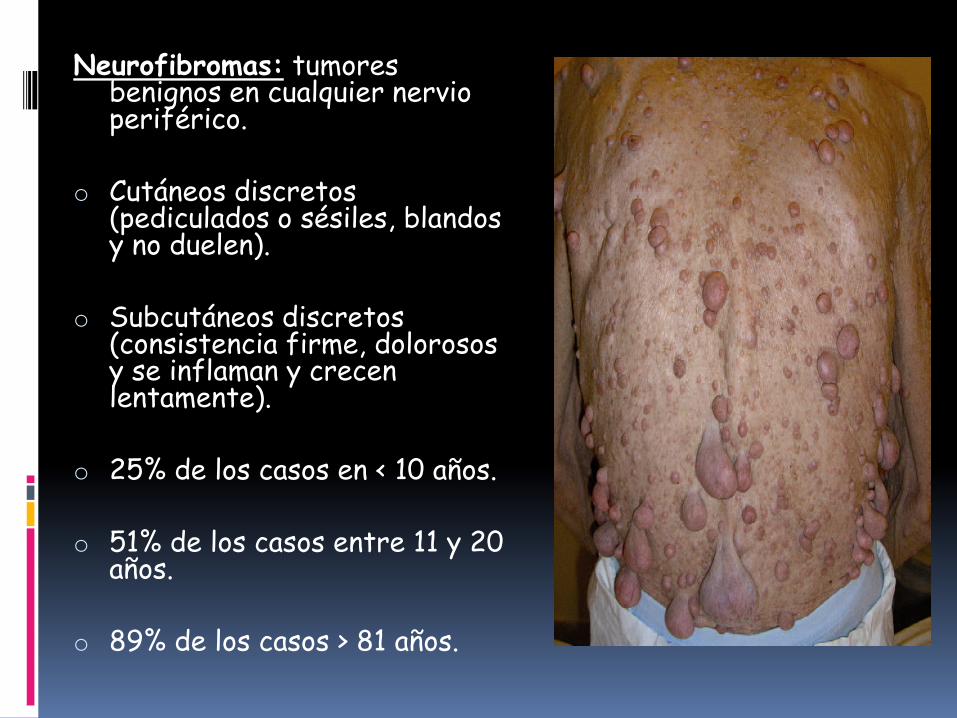
Neurofibromas: tumores benignos en cualquier nervio periférico.
o Cutáneos discretos (pediculados o sésiles, blandos y no duelen).
o Subcutáneos discretos (consistencia firme, dolorosos y se inflaman y crecen lentamente).
o 25% de los casos en < 10 años.
o 51% de los casos entre 11 y 20 años.
o 89% de los casos > 81 años.
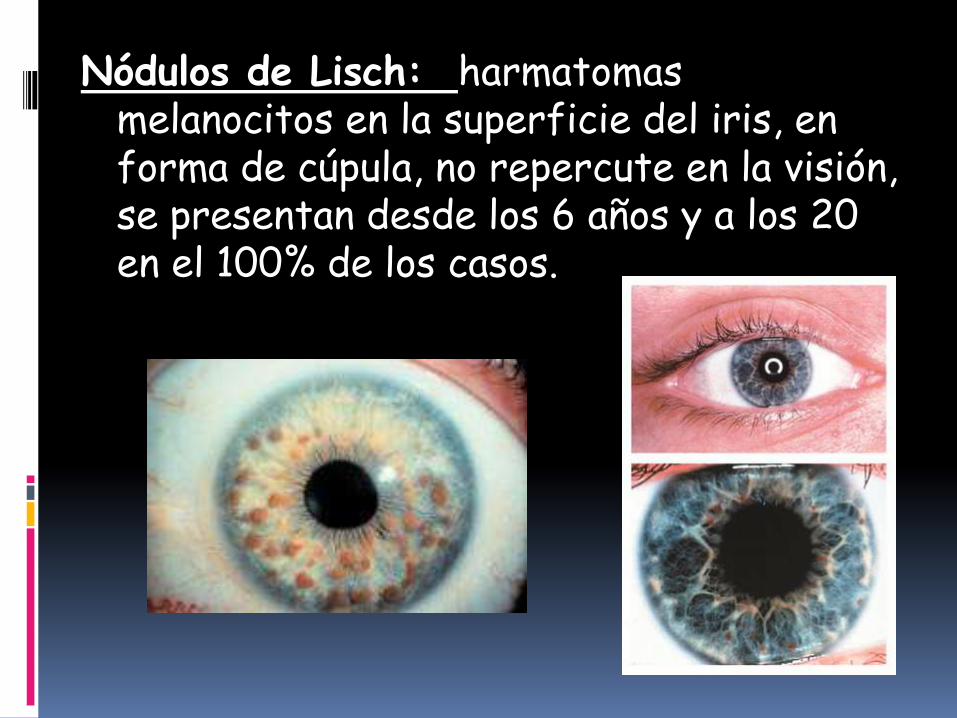
Nódulos de Lisch: harmatomasmelanocitos en la superficie del iris, en forma de cúpula, no repercute en la visión, se presentan desde los 6 años y a los 20 en el 100% de los casos.
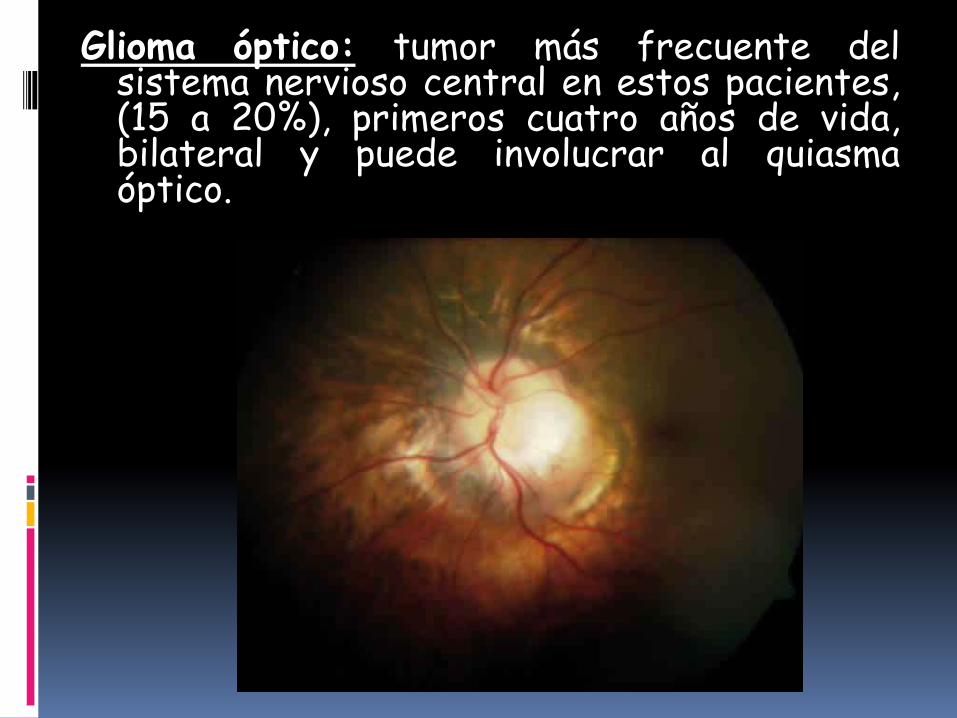
Glioma óptico: tumor más frecuente delsistema nervioso central en estos pacientes,(15 a 20%), primeros cuatro años de vida,bilateral y puede involucrar al quiasmaóptico.
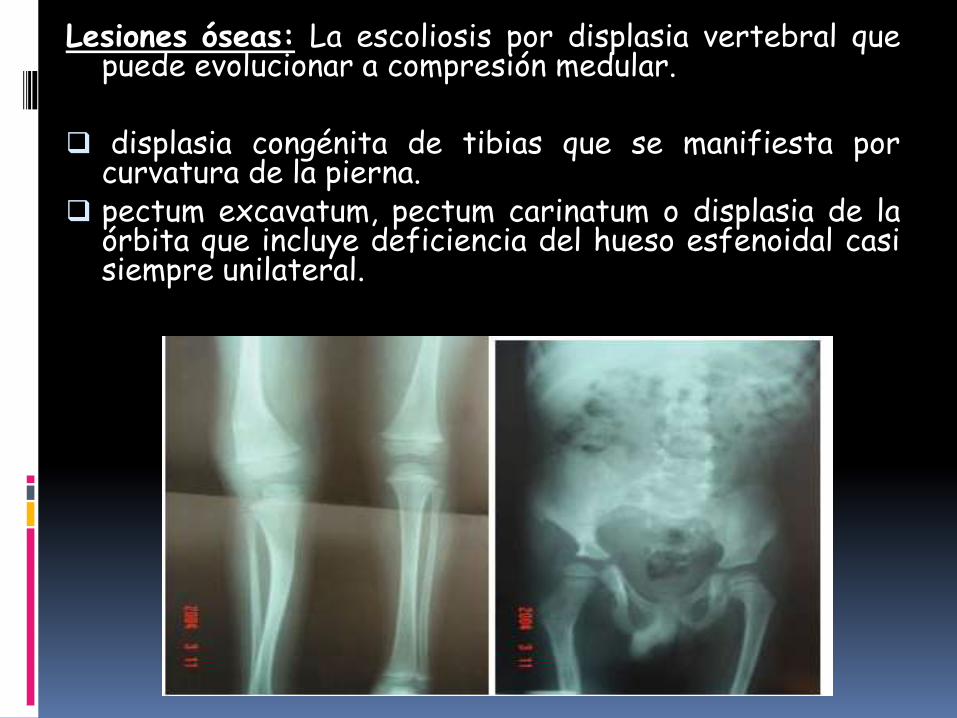
Lesiones óseas: La escoliosis por displasia vertebral quepuede evolucionar a compresión medular.
displasia congénita de tibias que se manifiesta porcurvatura de la pierna.
pectum excavatum, pectum carinatum o displasia de laórbita que incluye deficiencia del hueso esfenoidal casisiempre unilateral.

Criterios diagnósticosDos o más de los siguientes criterios:
Seis o más manchas café con leche mayores de 5 mm de diámetro en pre-púberes o con más de 15mm en mayores.
Dos o más neurofibromas de cualquier tipo o uno o más plexiformes
Efélides axilares o inguinales
Un tumor de la vía óptica
Dos o más nódulos de Lisch (hamartomas de iris)
Una lesión ósea distintiva (displasia del ala del esfenoides, adelgazamiento de la corteza de los huesos largos (con o sin pseudoartrosis)
Un pariente en primer grado con NF TI (padre, hermano o hijo)

tratamiento Sobrevida promedio de 59 años debido a
tumores malignos, vasculopatíaespecialmente enfermedad cerebrovasculare infarto.
Todos los parientes en primer gradodeberán revisarse para buscar lesionesdérmicas y oftalmológicas.
El consejo a los familiares y al pacientedebe basarse en pronóstico, riesgo genéticopara futuros casos y apoyo psicosocial.

Las neurofibromatosis(NF) forman un grupo de
3
trastornos relacionados, pero geneticamente
distintos del
sistema nervioso.
Se heredan de forma autosomica
dominante, y que causan tumores de las células de la
neuroglia.
Su prevalencia es igual para ambos sexos y no tienen
predileccion por ningunaraza o grupo étnico.
Afecta a 1 de cada 50.000

En la clasificacion de las NF, diferenciamos:
Neurofibromatosis tipo1 o enfermedad de Von Recklinghausen (NF1)
Neurofibromatosis tipo 2 (NF2)
Schwanomatosis
En 1822 fue descrito por primera vez un caso de NF2 por un cirujano escocés llamado Wishart y, a finales del siglo XIX.
Son el resultado de mutaciones puntuales o de delecciones o grandes reordenamientos del gen NF2 localizado en el brazo largo del cromosoma 22.
Esto produce como resultado la disminución de la producción de la proteína merlina o schwanomina, que sirve como un supresor tumoral.
La edad de inicio de hallazgos en pacientes con NF2 suele ser de entre 18 – 24 años, aunque el rango va desde el nacimiento hasta los 70 años.
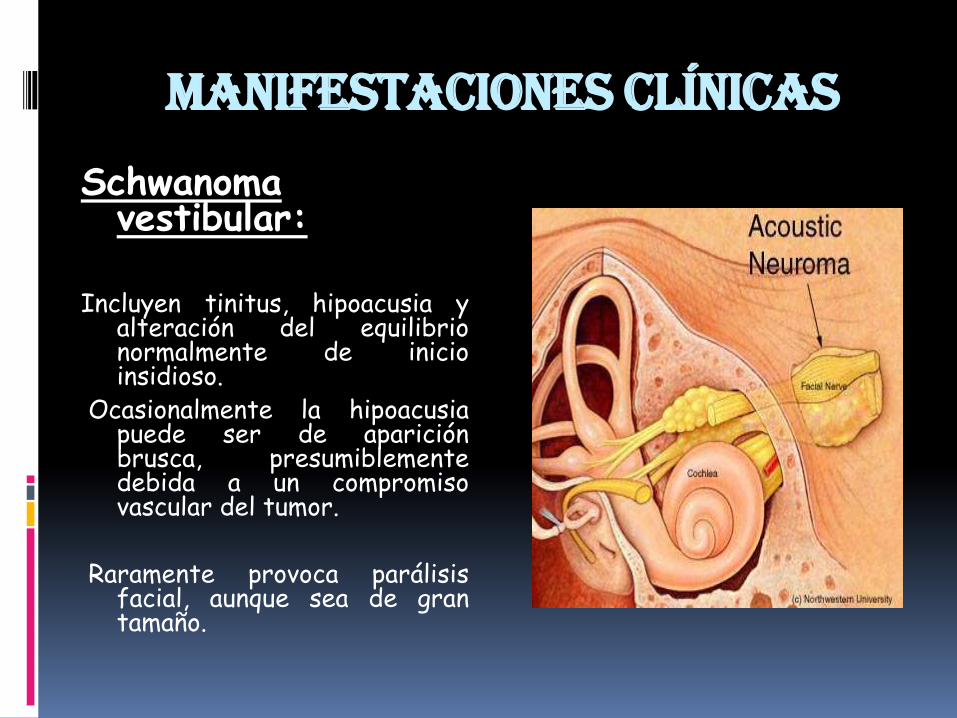
Manifestaciones clínicas
Schwanomavestibular:
Incluyen tinitus, hipoacusia yalteración del equilibrionormalmente de inicioinsidioso.
Ocasionalmente la hipoacusiapuede ser de apariciónbrusca, presumiblementedebida a un compromisovascular del tumor.
Raramente provoca parálisisfacial, aunque sea de grantamaño.
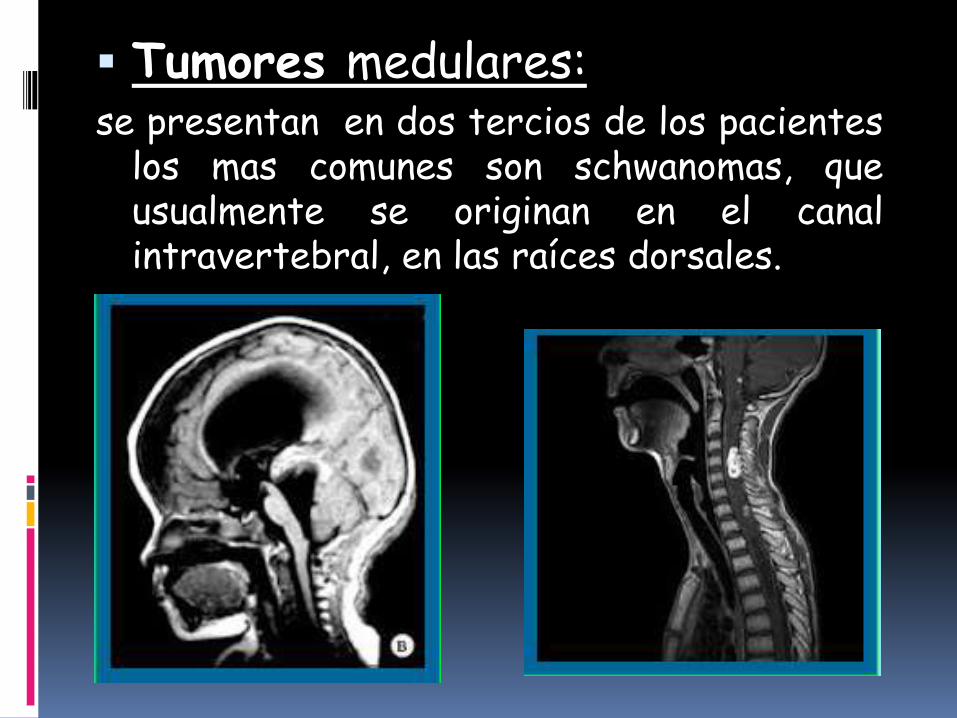
Tumores medulares:se presentan en dos tercios de los pacientes
los mas comunes son schwanomas, queusualmente se originan en el canalintravertebral, en las raíces dorsales.

Meningiomas:
La mayoría de ellos intracraneales y principalmente supratentoriales.
Meningiomas en la orbita provocaran perdida de visión porcompresión del nervio óptico y los que se originen en la base delcráneo pueden causar neuropatıa, compresión cerebral ehidrocefalia.
Afectación ocular:
Una opacidad subcapsular posterior, aunque raramente evolucionahasta una catarata significativa.
Dichas opacidades corneales suelen aparecer antes del inicio de los síntomas del neurinoma vestibular y pueden observarse en niños.
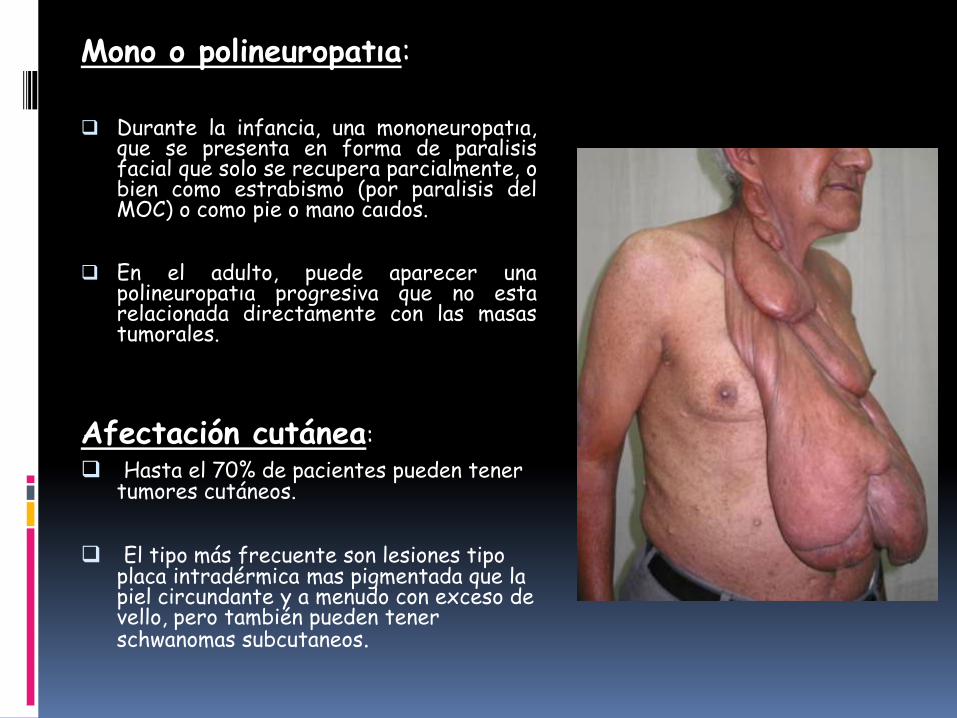
Mono o polineuropatıa:
Durante la infancia, una mononeuropatıa,que se presenta en forma de paralisisfacial que solo se recupera parcialmente, obien como estrabismo (por paralisis delMOC) o como pie o mano caıdos.
En el adulto, puede aparecer unapolineuropatıa progresiva que no estarelacionada directamente con las masastumorales.
Afectación cutánea:
Hasta el 70% de pacientes pueden tener tumores cutáneos.
El tipo más frecuente son lesiones tipo placa intradérmica mas pigmentada que la piel circundante y a menudo con exceso de vello, pero también pueden tener schwanomas subcutaneos.

diagnostico1. Schwanomas bilaterales del VIII
par craneal diagnosticados en la RM o el TC (biopsia no necesaria)
2. Familiar de primer grado con NF2 y:
a) schwanoma unilateral del VIIIpc, de inicio temprano (edad o30 años)
b) dos de los siguientes:
Meningioma
Glioma
Schwanoma
Menores con opacidad lenticular subcapsular posterior (catarata cortical juvenil)
3. Schwanoma de VIII par craneal unilateral diagnosticado por TAC o RNM de inicio precoz (detectado en paciente menor de 30 años) y 2 de los siguientes:a) Meningioma b) Glioma
c) Schwanoma d) Catarata cortical juvenil.
4. Meningiomas multiples (42) y:a) schwanoma del VIIIpcunilateralb) 2 de los siguientes:GliomaSchwanomaCatarata cortical juvenil

tratamiento La radioterapia externa convencional esta
contraindicada por transformación maligna de lostumores.
Una alternativa a la cirugía es la radiocirugíaestereotactica en pacientes seleccionados (contumores muy agresivos o para aquellos pacientes quese niegan a una intervención quirúrgica).
La edad de mortalidad es variable en pacientes conNF2 y muchos pacientes tienen un ciclo de vidarelativamente normal.