Embed Size (px)
Citation preview

Facu l tad de C ienc ias
Desarrollo de nuevos métodos de vacunación basados en
la anafilotoxina C5a y la proteína extra A de la
fibronectina y péptidos sintéticos inhibidores de las
células T reguladoras
Francesc Rudilla Salvador
2011


F a c u l t a d d e C i e n c i a s
Desarrollo de nuevos métodos de vacunación basados en la
anafilotoxina C5a y la proteína extra A de la fibronectina y
péptidos sintéticos inhibidores de las células T reguladoras
Memoria presentada por D. Francesc Rudilla Salvador para aspirar al grado
de Doctor por la Universidad de Navarra
El presente trabajo ha sido realizado bajo mi dirección en el Departamento de Hepatología y Terapia Génica y autorizo su presentación ante el Tribunal que lo ha de juzgar.
Pamplona, 17 de Junio de 2011
Dr. Juan José Lasarte Sagastibelza


A mi mujer Sílvia
y a mi hijo Biel.
“Caminant sempre junts”


AGRADECIMIENTOS


Han sido muchas las personas que me han ayudado y apoyado antes y durante
la realización de esta tesis. El espacio para darles las más sinceras gracias se
hace corto. A todas ellas quiero darles las gracias y espero de todo corazón no
dejarme a nadie.
A la Clínica Universidad de Navarra por la formación especializada en
Inmunología que he recibido.
Por la formación académica que en ellas he adquirido, quiero expresar
mi más profundo y sincero agradecimiento a la Universidad de Navarra, a la
Universitat de Girona y a la Universitat Autònoma de Barcelona.
Al Dr. Jesús Prieto, por permitirme formar parte del equipo de la
División de Terapia Génica y Hepatología y por el gran entusiasmo que
muestra en los proyectos. Muchas gracias por su apoyo.
A mi director de tesis, el Dr. Juan José Lasarte, creador de este proyecto.
Gracias por confiar en mí y darme la oportunidad de trabajar con un equipo
humano extraordinario. Gracias por tu dedicación e ilusión en el trabajo, tus
brillantes ideas, tu optimismo y por tener siempre en cuenta mis opiniones.
A los Dres. Francisco Borrás y Pablo Sarobe, por todas las buenas ideas
que aportáis y porque siempre se puede contar con vuestra inestimable ayuda y
objetividad científica, muchas gracias.
Quiero expresar, también, mi gratitud y mi consideración más sincera a
la Dra Noelia Casares y al Dr Javier Dotor porque cuando más lo he necesitado
me han proporcionado una dosis de esperanza e ilusión. Muchas Gracias Noe,
muchas gracias Dotor.
Mi gratitud también para la Dra Laura Arribillaga y la Dra Maika
Durántez, por su estímulo y ayuda desinteresada.
Para Cristina Mansilla, Marta Martínez, Teresa Lozano y Lorea
Villanueva por ser unas excelentes compañeras de trabajo. Muchas gracias por
vuestro apoyo.

A todos los peptídicos, tengo la gran suerte de poder contar con vosotros
siempre que lo he necesitado, por eso esta tesis también es en parte vuestra.
Muchas gracias.
Al resto de mis compañeros del departamento y, en especial, a aquellos
con los que comparto planta gracias por hacer agradable el simple hecho de
estar. Gracias al Dr José Ignacio Riezu, por tener siempre una sonrisa. Muchas
gracias Edurne por tu ayuda en el trabajo realizado.
Quiero agradecer al Dr Rubén Pío por proporcionarme ratones C5aR ko
y al Dr Daniel Ajona por su entusiasmo y apoyo en el proyecto.
A la Dra Claude Leclerc, y a Catherine por su colaboración con los
ensayos con ratones TLR4 ko.
Al equipo del animalario, por ser un apoyo imprescindible para la
manipulación de los ratones.
Han sido prácticamente cinco años de trabajo tres de los cuales han sido
especialmente duros, por tener que combinar el trabajo como biólogo residente
(guardias incluidas) con la tesis doctoral y vivir lejos de la familia. Quiero
agradecer profundamente a mis padres Núria y Francisco por el apoyo que
constituyen en mi vida y por animarme siempre a mirar hacia delante sin
perder el presente. A mis hermanas Núria y Laia por el cariño que siempre me
han transmitido y por hacerme saber que siempre tendré cerca a mis hermanas.
A mis amigos y compañeros de viaje, gracias por ayudarme a crecer
como persona.
Mis últimas palabras de agradecimiento son para ti, Sílvia. Cada día
recibo mucho de ti, me apoyas incondicionalmente en todo aquello que me
propongo realizar y sin tu apoyo este proyecto no hubiese terminado. Muchas
gracias por ayudarme en todo aquello que has podido y gracias por permitirme
crecer a tu lado.
A todas aquellas personas que, aunque no he nombrado, han sido muy
importantes y necesarias. Muchas y muchísimas gracias.

ABREVIATURAS


Aa: aminoácido
AMPc: adenosín monofosfato cíclico
APC: célula presentadora de antígeno (del inglés antigen presenting cell)
as: antisentido (aplicado a los cebadores)
CFSE: carboxifluorescein succinimidil ester
CML: cultivo mixto leucocitario
CpG: regiones de DNA viral o bacteriano ricas en pares de citosina y guanina enlazados por fosfatos
Cpm: cuentas por minuto
DAF: factor acelerador de la degradación
DC: célula dendrítica (del inglés dendritic cell)
DIDS: ácido 4,4'-diisotiocianostilbeno-2,2'-disulfónico
DNA: ácido desoxirribonucléico (del inglés deoxyribonucleic acid)
DTc: determinante T citotóxico
DTh: determinante T helper
EDA: dominio extra A de la fibronectina (del inglés, extradomain-A fibronectin)
FBS: suero fetal bovino (del inglés foetal bovine serum)
FITC: isotiocianato de fluoresceína
FPLC: cromatografía liquida de proteína (del inglés Fast protein liquid chromatography)
GM-CSF: factor estimulador de colonias de granulocitos y macrófagos (del inglés, granulocyte-macrophage colony stimulatory factor)
HPPTLP: péptido señal de la pre-pro-tripsina humana (del inglés human preprotrypsin leader peptide)
Ig: inmunoglobulina
IL: interleucina
i.p.: intraperitoneal
IPTG: isopropil β-D-1-tiogalactopiranosido
i.t.: intratumoral
i.v.: intravenosa
LB: linfocitos B
LPS: lipopolisacárido

LTc: linfocito T citotóxico
LTh: linfocito T helper
MC: medio completo
MHC: complejo principal de histocompatibilidad (del inglés major histocompatibility complex)
MyD88: factor de diferenciación mieloide 88 (del inglés myeloid differentiation factor 88)
NF-кB: factor de transcripción nuclear kappa de los linfocitos B (del inglés, nuclear factor kappa-light-chain-enhancer of activated B cells)
NK: célula asesina natural (del inglés natural killer)
NKT: linfocitos T asesinos naturales (del inglés natural killer T lymphocyte)
PAMP: patrones de moléculas asociados a patógenos (del inglés pathogen-associated molecular patterns)
PBS: tampón fosfato salino (del inglés phosphate buffered saline)
PCR: reacción en cadena de la polimerasa (del inglés polymerase chain reaction)
PE: ficoeritrina (del inglés phycoerythrin)
poly(I:C): ácido poliinosínico:policitidílico
PRR: receptores de patrones de reconocimiento (del inglés pattern recognition receptors)
RNA: ácido ribonucléico (del inglés ribonucleic acid)
s: sentido (aplicado a los cebadores)
TCR: receptor de células T (del inglés T cell receptor)
TLR: receptores tipo peaje (del inglés toll-like receptors)
TNF: factor de necrosis tumoral (del inglés tumor necrosis factor)
Treg: células T reguladoras

ÍNDICE


INTRODUCCIÓN................................................................................................................................................. 1
1. Sistema inmunitario ......................................................................................................................................... 3
1.1. Inmunidad innata.................................................................................................................................... 3
1.1.1. Receptores Toll-like (TLR) y vías de señalización.......................................................................... 5
1.1.2. Sistema del complemento. Generalidades ...................................................................................... 9
1.1.2.1. Activación del complemento ................................................................................................... 10
1.1.2.1.1. Vía clásica del complemento ............................................................................................. 10
1.1.2.1.2. Vía de las lectinas................................................................................................................ 11
1.1.2.1.3. Vía alternativa o sistema properdina ............................................................................... 12
1.1.2.1.4. Rutas alternativas de la activación del complemento.................................................... 12
1.1.2.2. Funciones efectoras del complemento ................................................................................... 13
1.1.3. Interacción del complemento con los receptores Toll like (TLR)................................................. 16
1.2. Inmunidad adaptativa o adquirida .................................................................................................... 20
1.2.1. Respuesta humoral........................................................................................................................... 20
1.2.2. Respuesta celular.............................................................................................................................. 21
1.2.2.1. Linfocitos Th............................................................................................................................... 21
1.2.2.2. Linfocitos T citotóxicos ............................................................................................................. 22
1.2.2.3. Linfocitos T reguladores CD4+CD25+.................................................................................... 24
1.3. Activación de una respuesta inmunitaria .......................................................................................... 25
1.3.1. Presentación antigénica .................................................................................................................. 25
1.3.2. Células dendríticas .......................................................................................................................... 27
1.3.3. Señalización del calcio en la activación linfocitaria .................................................................... 30
2. Estrategias de vacunación .............................................................................................................................. 31
2.1. DNA desnudo ......................................................................................................................................... 35
2.2. Proteínas recombinantes de fusión basadas en el dominio extra A de la fibronectina (EDA). 36
2.2.1. El domino extra A de la fibronectina (EDA) ................................................................................ 37
2.3. Péptidos inhibidores de las células T reguladoras........................................................................... 39
2.4. Anafilotoxinas. Generalidades ............................................................................................................ 40
2.4.1. Efecto biológico de C5a................................................................................................................... 40
2.4.2. Receptores del fragmento C5a ....................................................................................................... 45
2.4.2.1. Receptor C5aR (CD88) .............................................................................................................. 46
2.4.2.2. Receptor C5L2 (GPR77) ............................................................................................................ 47
2.5. Adyuvantes.............................................................................................................................................. 48

OBJETIVOS........................................................................................................................................................... 51
MATERIAL Y MÉTODOS ............................................................................................................................... 55
2. Células............................................................................................................................................................... 57
2. Ratones.............................................................................................................................................................. 58
3. Antígenos.......................................................................................................................................................... 58
3.1. Péptidos.................................................................................................................................................... 58
3.1.1. Síntesis de péptidos......................................................................................................................... 58
3.1.2. Péptidos comerciales....................................................................................................................... 61
3.1.3. Análisis de interacción biomolecular............................................................................................ 61
3.2. Plásmidos de expresión......................................................................................................................... 61
3.2.1. Construcción .................................................................................................................................... 61
3.2.2. Transfección en células adherentes ............................................................................................... 63
3.2.2.1. Western blot ............................................................................................................................... 64
3.3. Proteínas recombinantes de fusión ..................................................................................................... 65
3.3.1. Construcción .................................................................................................................................... 65
3.3.2. Purificación....................................................................................................................................... 67
3.3.2.1. Purificación por afinidad.......................................................................................................... 68
3.3.2.2. Intercambio iónico..................................................................................................................... 69
3.3.2.3. Replegado y detoxificado ......................................................................................................... 69
3.3.3. Análisis de la pureza....................................................................................................................... 71
3.3.3.1. Geles SDS-PAGE y tinción con Azul de Coomassie ............................................................. 71
3.3.4. Ensayos de actividad....................................................................................................................... 72
3.3.3.1. Análisis de activación de monocitos ....................................................................................... 72
3.3.4.2. Análisis de maduración de células dendríticas ..................................................................... 72
3.3.4.2.1. Diferenciación DC ............................................................................................................... 72
3.3.4.2.2. Análisis in vitro de la maduración de las células dendríticas......................................... 73
3.3.4.2.2.1. Expresión de marcadores de maduración .................................................................. 74
3.3.4.2.2.2. Producción de citocinas ................................................................................................ 74
3.3.4.3. Presentación antigénica ............................................................................................................ 74
3.3.4.4. Migración celular....................................................................................................................... 75
4. Estudio in vitro de los intercambiadores de iones cloruro, calcio y potasio en la activación linfocitaria ........................................................................................................................................................ 75
4.1. Análisis de la expresión de mRNA por PCR quantitativa .............................................................. 75
4.2. Purificación de células T reguladoras................................................................................................. 76
4.3. Ensayos de proliferación celular.......................................................................................................... 76

4.4. Cinéticas de calcio intracelular ............................................................................................................ 76
5. Estudio de la apoptosis linfocitaria in vitro ............................................................................................... 77
5.1. Técnica de Tunel..................................................................................................................................... 77
6. Estudio de la respuesta inmunitaria en ratones......................................................................................... 78
6.1. Inmunización .......................................................................................................................................... 78
6.1.1. Plásmido ........................................................................................................................................... 78
6.1.2. Proteína............................................................................................................................................. 78
6.2. Estudio in vitro e in vivo de las respuestas inmunitarias inducidas............................................. 78
6.2.1. Aislamiento de esplenocitos........................................................................................................... 78
6.2.2. ELISPOT ........................................................................................................................................... 79
6.2.3. In vivo killing ..................................................................................................................................... 79
6.2.4. Inducción de la respuesta inmunitaria en ausencia de señalización C5aR.............................. 80
6.2.5. Depleción in vivo de células NK1.1 ............................................................................................... 80
6.3. Estudios in vivo de protección tumoral .............................................................................................. 81
6.3.1. Modelo tumoral ................................................................................................................................ 81
6.3.2. Estudios de protección de tumores ................................................................................................ 81
6.3.3. Estudios de tratamiento de tumores .............................................................................................. 82
7. Estadística ......................................................................................................................................................... 82
RESULTADOS ..................................................................................................................................................... 83
1. Implicación de los intercambiadores de Cl- y canales de Ca2+ en la activación celular de los linfocitos T efectores y T reguladores (CD4+CD25+FOXP3+) ................................................................ 85
1.1. Efecto de la inhibición de los intercambiadores de aniones cloruro sobre la acción de las células T reguladoras (CD4+CD25+FOXP3+)................................................................................. 85
1.2. Efecto de la inhibición de los intercambiadores de aniones cloruro sobre la expresión de mRNAs en los linfocitos T................................................................................................................ 90
1.3. Efecto de la inhibición de los intercambiadores de aniones cloruro sobre el flujo de entrada de calcio en los linfocitos ................................................................................................................... 92
2. Desarrollo de péptidos inhibidores del intercambiador cloruro/bicarbonato AE2............................. 97
2.1. Síntesis de los péptidos y ensayos funcionales de selección de los péptidos inhibidores ...... 97
2.2. Ensayos de unión del péptido AE2-17 al bucle 3 del intercambiador AE2 .................................. 99
2.3. Efecto del péptido p17AE2 en la proliferación celular .................................................................. 102
2.4. Efecto del péptido p17AE2 en la apoptosis celular......................................................................... 106
3. Estudio del potencial inmunomodulador de las anafilotoxinas para el desarrollo de vacunas...... 107
3.1. Cribado con plásmidos de expresión................................................................................................ 107
3.2. Utilización del fragmento C5a como adyuvante de vacunación .................................................. 110
3.2.1. Purificación de las proteínas recombinantes de fusión ............................................................. 110
3.2.1.1. Purificación de EDA-SIINFEKL.............................................................................................. 113

3.2.1.2. Purificación de C5a-EDA-SIINFEKL ..................................................................................... 113
3.2.1.3. Purificación de EDA-SIINFEKL-C5a y SIINFEKLC5a......................................................... 115
3.2.2. Inducción de la producción de TNF-α por la línea THP-1........................................................ 117
3.2.3. Maduración de células dendríticas. ............................................................................................. 118
3.2.3.1. Estudio de la expresión de marcadores de superficie en las células dendríticas ............. 118
3.2.3.2. Inducción de citocinas y quimiocinas por las células dendríticas...................................... 120
3.2.4. Análisis in vitro de la quimiotaxis inducida por C5a ................................................................ 121
3.2.5. Presentación antigénica de EDA-SIINFEKL, EDA-SIINFEKL-C5a y SIINFEKL-C5a .......... 124
3.2.6. Inducción de la respuesta inmunitaria frente a SIINFEKL ...................................................... 125
3.2.7. Efecto de las proteínas recombinantes sobre la población de las células T reguladoras en la inducción de la respuesta inmunitaria ........................................................................................ 128
3.2.8. Inducción de la respuesta inmunitaria en ausencia de señalización TLR4............................ 130
3.2.9. Inducción de la respuesta inmunitaria en ausencia de señalización C5aR............................ 132
3.2.10. Inducción de la respuesta inmunitaria NK. ............................................................................. 134
3.2.11. Inducción de protección antitumoral y tratamiento de tumores establecidos .................... 137
DISCUSIÓN........................................................................................................................................................ 141
1. Estudio del desarrollo de inhibidores de la acción de las células T reguladoras .............................. 143
2. Estudio del efecto adyuvante de las proteínas del complemento......................................................... 147
CONCLUSIONES ............................................................................................................................................. 155
BIBLIOGRAFÍA................................................................................................................................................. 159
ANEXOS ............................................................................................................................................................... 179

INTRODUCCIÓN


Introducción
3
1. Sistema inmunitario
El sistema inmunitario está compuesto de muchos tipos celulares
interdependientes que en conjunto protegen al organismo contra bacterias,
parásitos, hongos, infecciones virales y frente al crecimiento tumoral. Muchos
de estos tipos celulares tienen funciones especializadas, pueden engullir
bacterias y parásitos o eliminar células tumorales o células propias del
organismo infectadas por virus. Por lo tanto, es un sistema que discrimina entre
lo propio y lo ajeno, permitiendo actuar de forma específica. Cuando esta
tolerancia inmunológica se altera, es decir, cuando falla la no respuesta a lo
propio y a lo no dañino, se generan respuestas frente a estructuras propias
dando lugar a procesos autoinmunes o bien procesos de hipersensibilidad
donde el sistema inmunitario responde de forma exagerada a estructuras
foráneas no patógenas, produciendo daño al organismo.
La respuesta inmunitaria está formada por dos tipos principales de
respuesta en función de la naturaleza de las estructuras de reconocimiento del
patógeno. Estas estructuras pueden ser genéricas (respuesta innata) o bien
específicas, utilizando receptores generados al azar que tienen un repertorio de
reconocimiento prácticamente ilimitado (respuesta adaptativa).
La interacción entre ambas formas de respuesta es esencial para una
respuesta inmunológica eficaz. Los mecanismos innatos son fundamentales
para la iniciación de las respuestas adaptativas y además permiten controlar el
tipo de respuesta inducida, por otra parte la respuesta innata también es
reclutada en la fase efectora de la respuesta adaptativa. Es importante destacar
que las interacciones entre ambas respuestas son muchas, complejas y
bidireccionales, formando parte de una única respuesta global de defensa.
1.1. Inmunidad innata
La inmunidad innata constituye la primera línea de defensa contra las
infecciones. Comprende toda una serie de mecanismos de defensa constitutivos
y listos para movilizarse cuando se produzca una infección, preparados para
responder con gran rapidez. Se caracteriza por ser una respuesta no antígeno

Introducción
4
específica que reacciona inicialmente del mismo modo frente a una amplia
variedad de infecciones. Es una respuesta que no genera memoria
inmunológica, es decir, que en encuentros posteriores con el mismo patógeno
no se produce una respuesta mejorada y por lo tanto no es una respuesta
inmunitaria duradera.
Los mecanismos defensivos de la respuesta innata están formados por
barreras físicas como la piel, las mucosas, el ácido estomacal, el reflejo de la tos
entre otros y barreras químicas como la fiebre, la inflamación y el sistema del
complemento. El sistema del complemento constituye el mayor componente
humoral de la respuesta innata y actúa como mediador de varios mecanismos
de la respuesta adaptativa, entre ellos la citotoxicidad mediada por anticuerpos.
Además, la respuesta innata tiene un componente celular importante
cuyo mecanismo defensivo es la fagocitosis y la citotoxicidad celular. Las
poblaciones celulares responsables del desarrollo de una respuesta inmunitaria
innata son las células fagocíticas (macrófagos, neutrófilos, células dendríticas),
mastocitos, basófilos, eosinófilos y las células citotóxicas naturales o células NK
(del inglés natural killer). Las células de la respuesta innata también son
importantes mediadores en la activación del sistema inmune adaptativo. Un
ejemplo es la secreción de interferón gamma (IFN-γ) por parte de las células NK
(Mandelboim et al. 1998; Arase et al. 1996; Young and Hardy 1995); otro
ejemplo lo encontramos en las células dendríticas que presentan antígenos a las
células T, uno de los tipos celulares clave del sistema inmunitario adaptativo
(Guermonprez et al. 2002). Los determinantes reconocidos por los componentes
de la respuesta innata (no específica) difieren de los reconocidos por la
respuesta adaptativa (específica). Mientras que los componentes de la respuesta
adaptativa reconocen y reaccionan frente a un patógeno en particular, los
componentes del sistema inmunitario innato reconocen amplios patrones
moleculares que se encuentran en grupos de patógenos relacionados,
careciendo de un alto grado de especificidad. Los patrones moleculares
comunes reconocidos por el sistema inmunitario innato han sido llamados
patrones moleculares asociados a patógenos (PAMP, del inglés “pathogen

Introducción
5
associated molecular patterns”), y sus receptores se llaman receptores de patrones
de reconocimiento (PRR, del inglés pattern recognition receptors) (Medzhitov
2001; Janeway and Medzhitov 2002).
1.1.1. Receptores Toll-like (TLR) y vías de señalización
Los PRR se pueden dividir en tres clases dependiendo de su ubicación:
secretables, de membrana o citosólicos. Los PRR transmembrana incluyen
varios tipos de receptores, entre ellos destacamos la familia de los receptores
TLR (del inglés Toll-like receptors).
Los TLR son glicoproteínas transmembrana tipo I formadas por un
dominio extracelular N terminal rico en leucinas, contiene entre 15-30 LRR (del
inglés leucine-rich repeats) que media el reconocimiento de los PAMPs, un
dominio transmembrana y el dominio intracelular C terminal, conocido como
TIR (del inglés Toll/Interleukin-1 receptor) que está implicado en la transducción
de la señalización. El número de TLRs puede diferir en función de la especie, se
han identificado entre 10 y 12 TLRs funcionales en humanos y ratones
respectivamente. Los TLRs 1-9 están conservados en ambas especies. Los
PAMPs reconocidos por los TLR son lípidos, lipoproteínas, proteínas y ácidos
nucleicos derivados de una amplia gama de microbios, como bacterias, virus,
parásitos y hongos. Se localizan principalmente en células presentadoras de
antígeno profesionales (células mononucleares, dendríticas y macrófagos) y en
diferentes células epiteliales (Takeda et al. 2003; Akira and Takeda 2004).
Los TLRs se clasifican en dos tipos en función de su localización y sus
respectivos ligandos PAMPs. Por un lado, los TLR 1, 2, 4, 5 y 6 reconocen
principalmente productos bacterianos (lipoproteínas, lípidos y proteínas), por
lo que se expresan en la membrana. Y por otro lado, los TLR 3, 7, 8 y 9 que se
localizan en los endosomas, retículo endoplasmático, lisosomas y
endolisosomas donde reconocen ácidos nucleicos virales (Tabla 1).
Está descrita la existencia de una comunicación cruzada entre los TLRs y
las moléculas adaptadoras, lo que aumenta la especificidad y amplía el patrón
de PAMPs reconocidos por un mismo receptor (Latz et al. 2003; Martin et al.
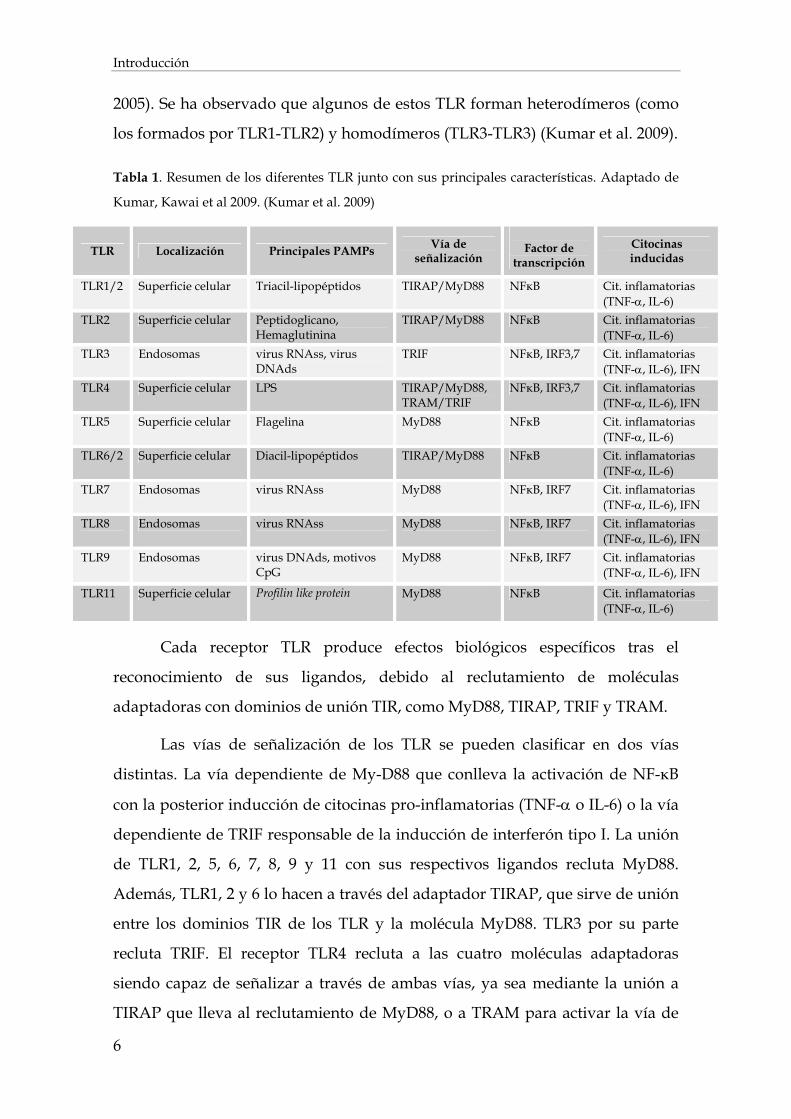
Introducción
6
2005). Se ha observado que algunos de estos TLR forman heterodímeros (como
los formados por TLR1-TLR2) y homodímeros (TLR3-TLR3) (Kumar et al. 2009).
Tabla 1. Resumen de los diferentes TLR junto con sus principales características. Adaptado de
Kumar, Kawai et al 2009. (Kumar et al. 2009)
TLR Localización Principales PAMPs Vía de señalización
Factor de
transcripción
Citocinas inducidas
TLR1/2 Superficie celular Triacil-lipopéptidos TIRAP/MyD88 NFкB Cit. inflamatorias (TNF-, IL-6)
TLR2 Superficie celular Peptidoglicano, Hemaglutinina
TIRAP/MyD88 NFкB Cit. inflamatorias (TNF-, IL-6)
TLR3 Endosomas virus RNAss, virus DNAds
TRIF NFкB, IRF3,7 Cit. inflamatorias (TNF-, IL-6), IFN
TLR4 Superficie celular LPS TIRAP/MyD88, TRAM/TRIF
NFкB, IRF3,7 Cit. inflamatorias (TNF-, IL-6), IFN
TLR5 Superficie celular Flagelina MyD88 NFкB Cit. inflamatorias (TNF-, IL-6)
TLR6/2 Superficie celular Diacil-lipopéptidos TIRAP/MyD88 NFкB Cit. inflamatorias (TNF-, IL-6)
TLR7 Endosomas virus RNAss MyD88 NFкB, IRF7 Cit. inflamatorias (TNF-, IL-6), IFN
TLR8 Endosomas virus RNAss MyD88 NFкB, IRF7 Cit. inflamatorias (TNF-, IL-6), IFN
TLR9 Endosomas virus DNAds, motivos CpG
MyD88 NFкB, IRF7 Cit. inflamatorias (TNF-, IL-6), IFN
TLR11 Superficie celular Profilin like protein MyD88 NFкB Cit. inflamatorias (TNF-, IL-6)
Cada receptor TLR produce efectos biológicos específicos tras el
reconocimiento de sus ligandos, debido al reclutamiento de moléculas
adaptadoras con dominios de unión TIR, como MyD88, TIRAP, TRIF y TRAM.
Las vías de señalización de los TLR se pueden clasificar en dos vías
distintas. La vía dependiente de My-D88 que conlleva la activación de NF-κB
con la posterior inducción de citocinas pro-inflamatorias (TNF- o IL-6) o la vía
dependiente de TRIF responsable de la inducción de interferón tipo I. La unión
de TLR1, 2, 5, 6, 7, 8, 9 y 11 con sus respectivos ligandos recluta MyD88.
Además, TLR1, 2 y 6 lo hacen a través del adaptador TIRAP, que sirve de unión
entre los dominios TIR de los TLR y la molécula MyD88. TLR3 por su parte
recluta TRIF. El receptor TLR4 recluta a las cuatro moléculas adaptadoras
siendo capaz de señalizar a través de ambas vías, ya sea mediante la unión a
TIRAP que lleva al reclutamiento de MyD88, o a TRAM para activar la vía de
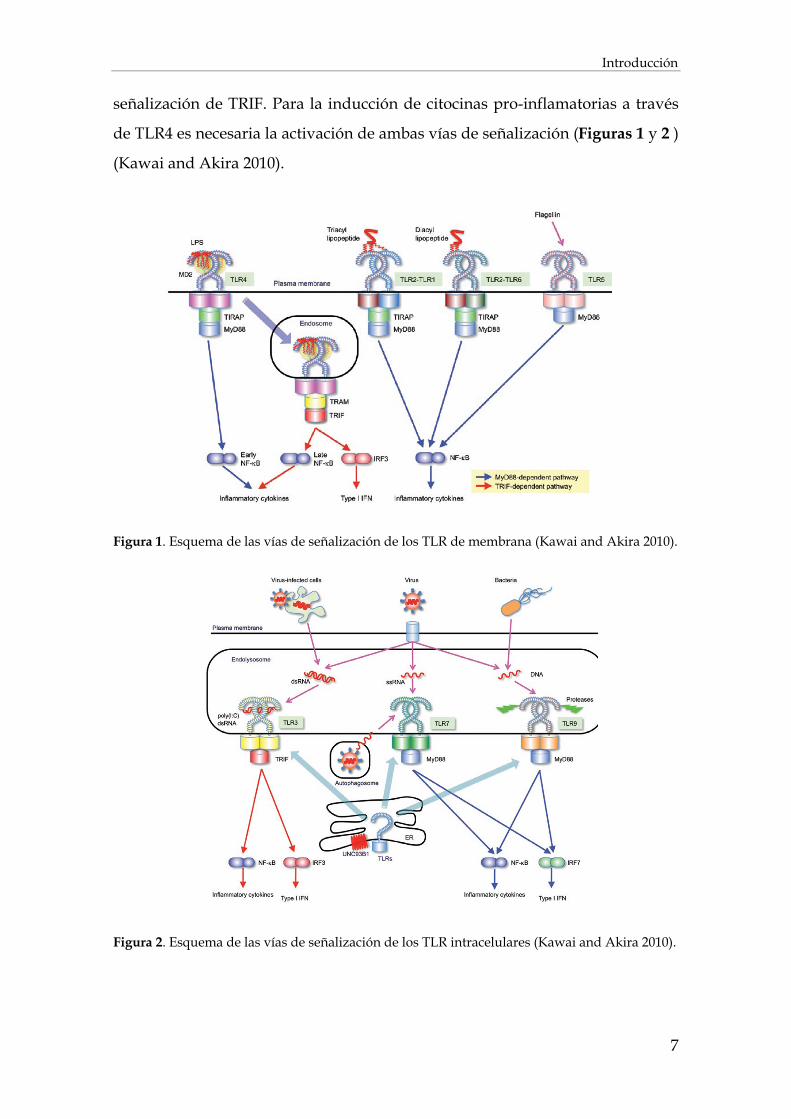
Introducción
7
señalización de TRIF. Para la inducción de citocinas pro-inflamatorias a través
de TLR4 es necesaria la activación de ambas vías de señalización (Figuras 1 y 2 )
(Kawai and Akira 2010).
Figura 1. Esquema de las vías de señalización de los TLR de membrana (Kawai and Akira 2010).
Figura 2. Esquema de las vías de señalización de los TLR intracelulares (Kawai and Akira 2010).

Introducción
8
Hay una creciente evidencia de que los receptores TLR juegan un papel
clave en la mediación de las respuestas sistémicas, a la invasión de patógenos
durante la sepsis, así como también en otras enfermedades inflamatorias y
autoinmunes. En consecuencia, se ha sugerido que el bloqueo de la función de
los TLR puede, en el futuro, traer nuevas posibilidades terapéuticas con el
objetivo de suprimir estados de inflamación. Algunos de los inhibidores
estudiados son variantes de las moléculas adaptadoras, ubiquitin ligasas,
deubiquitinasas, micro RNAs y reguladores transcripcionales (O'Neill and
Bowie 2007; Kawai and Akira 2010).
Por otra parte, la señalización de los TLR podría utilizarse para potenciar
el efecto terapéutico de las vacunas, puesto que la activación de la señalización
TLR provoca una activación rápida de la inmunidad innata mediante la
maduración de las células dendríticas (DC), producción de citocinas pro-
inflamatorias y citocinas antivirales, así como la expresión de moléculas co
estimuladoras y receptores de quimiocinas (Aderem and Ulevitch 2000; Kaisho
and Akira 2002; Werling and Jungi 2003). Además, está descrita la importancia
de la señalización TLR en células T efectoras y en células T reguladoras,
asociándose dichas vías a la diferenciación de las células T memoria (Pasare
and Medzhitov 2004; Salem et al. 2005), polarización de la respuesta Th1
(Schnare et al. 2001), pérdida temporal de la función supresora de las células T
reguladoras (reduciendo la expresión de Foxp3) (Hackl et al. 2011; Liu et al.
2006; Sutmuller et al. 2006), la generación de células Th17 o el bloqueo de la
conversión de las células T reguladoras promoviendo la respuesta autoinmune
(Chen et al. 2007; Abdollahi-Roodsaz et al. 2008). En definitiva, los TLR
permiten modular la respuesta celular T a través de la respuesta innata y
directamente por su acción en la respuesta adaptativa, permitiendo la inducción
de una inmunidad adaptativa eficaz. En este sentido se pueden considerar
receptores adyuvantes.

Introducción
9
1.1.2. Sistema del complemento. Generalidades
El complemento es un conjunto de proteínas termolábiles, la gran
mayoría plasmáticas y algunas de membrana, cuya función principal es
potenciar la inflamación, la fagocitosis y la lisis de los microorganismos. El
hepatocito es el principal productor de factores del complemento, también los
macrófagos activados en el foco inflamatorio, lo que es de gran importancia
porque así se garantiza la presencia de estos factores del complemento en el
foco inflamatorio. Las citocinas inflamatorias (IL-1, IL-6 y TNFα) y el IFNγ,
incrementan la síntesis de algunos factores del complemento en el hígado.
La gran mayoría de los factores del complemento son enzimas
proteolíticas. Cuando una de estas enzimas actúa sobre su sustrato, este se
escinde en dos fragmentos. Estos fragmentos se nombran igual que al sustrato
pero añadiéndoles una letra minúscula. Por ejemplo, C3 se escinde en dos
fragmentos, al mayor se le llama C3b y al menor C3a. Los dos fragmentos
resultantes tienen actividad biológica, si bien dichas acciones son distintas para
cada uno de ellos. Son un ejemplo, algunos pequeños péptidos con actividad de
anafilotoxinas, liberados durante la activación del complemento. El más potente
es el C5a, seguido por el C3a. Por lo tanto, durante la activación del
complemento tienen lugar una serie de reacciones en cascada, en cada reacción
se genera un producto activo, que además de determinar que la cadena prosiga
hasta la reacción siguiente, puede tener funciones biológicas en la defensa
frente a microorganismos.
El complemento es activado por tres vías distintas, la clásica, la de las
lectinas (MBL) y la alternativa (o properdina) (Vergani 1986) (Figura 3). Las tres
tienen en común la activación del componente C3 y por lo tanto confluyen en
una vía común de ataque a membrana, pero difieren en la naturaleza del
reconocimiento. La vía clásica es activada por anticuerpos liberados tras una
respuesta humoral. La vía de las lectinas es activada tras el reconocimiento e
interacción de patrones asociados a patógenos (PAMP´s) por proteínas lectinas.
Y por último, la vía alternativa se diferencia del resto debido a una activación
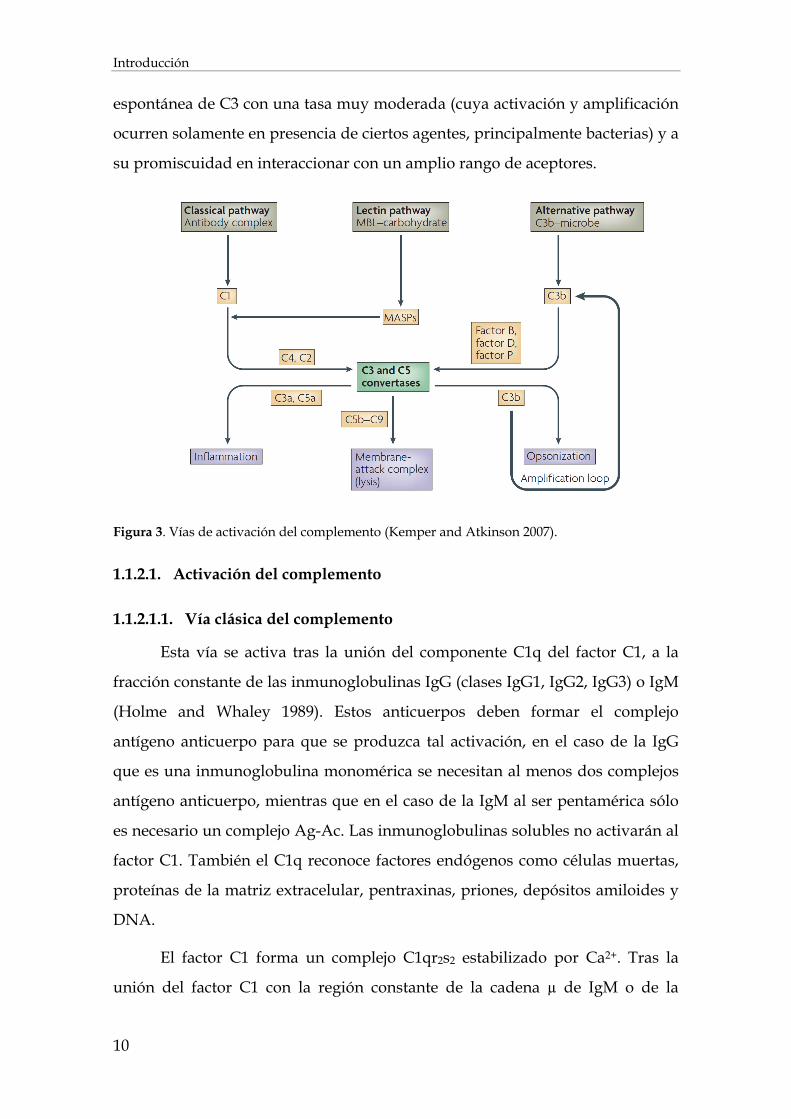
Introducción
10
espontánea de C3 con una tasa muy moderada (cuya activación y amplificación
ocurren solamente en presencia de ciertos agentes, principalmente bacterias) y a
su promiscuidad en interaccionar con un amplio rango de aceptores.
Figura 3. Vías de activación del complemento (Kemper and Atkinson 2007).
1.1.2.1. Activación del complemento
1.1.2.1.1. Vía clásica del complemento
Esta vía se activa tras la unión del componente C1q del factor C1, a la
fracción constante de las inmunoglobulinas IgG (clases IgG1, IgG2, IgG3) o IgM
(Holme and Whaley 1989). Estos anticuerpos deben formar el complejo
antígeno anticuerpo para que se produzca tal activación, en el caso de la IgG
que es una inmunoglobulina monomérica se necesitan al menos dos complejos
antígeno anticuerpo, mientras que en el caso de la IgM al ser pentamérica sólo
es necesario un complejo Ag-Ac. Las inmunoglobulinas solubles no activarán al
factor C1. También el C1q reconoce factores endógenos como células muertas,
proteínas de la matriz extracelular, pentraxinas, priones, depósitos amiloides y
DNA.
El factor C1 forma un complejo C1qr2s2 estabilizado por Ca2+. Tras la
unión del factor C1 con la región constante de la cadena µ de IgM o de la

Introducción
11
cadena γ de IgG, tendrá lugar una activación en secuencia de la actividad
proteolítica de los componentes C1r y C1s. C1r activa a C1s. Por acción de C1s,
con actividad serín proteasa, se escinde el fragmento C4, liberándose un
pequeño fragmento C4a y un C4b. C4a tiene una actividad anafilotoxina leve, y
C4b tiene actividad opsonina. En presencia de Mg2+, C2 forma un complejo con
C4b y se transforma en un nuevo sustrato para C1s, formándose el complejo
C4b2a con actividad C3 convertasa para escindir C3. La escisión de C3 implica
que se agrega C3b al complejo C4b2a formando la C5 convertasa (C4b2a3b). La
degradación de C4b2a es inducida por una proteína de unión a C4 (C4bp) o a
un receptor de C3b (C1R) en presencia del factor I.
C5 se escinde en C5a y C5b por la acción de la C5a convertasa. C5a es la
anafilotoxina más potente. En este momento empieza la segunda fase de
activación del complemento que consiste en el ensamblaje de los componentes
terminales. C6 se une a C5b y luego a C7, se insertan en la membrana
plasmática y posteriormente se une C8, causando una penetración en la
membrana plasmática y la inducción de la polimerización de C9 en membrana.
El resultado final es la formación del complejo de ataque a membrana (CAM) o
componente terminal del complemento (CTC), con la generación de un poro de
100 Å en la membrana plasmática que causa la osmólisis celular (Morgan 1989;
Bhakdi et al. 1990).
1.1.2.1.2. Vía de las lectinas
Es una vía análoga a la clásica que se activa por proteínas homólogas a
C1q: ficolinas y lectina de unión a manosa (MBL). Su activación es
independiente de anticuerpos. Estas proteínas reconocen específicamente
carbohidratos en la superficie de patógenos o células apoptóticas, como la
manosa y la N-acetil-glucosamina (GlcNac). Tras su unión con los
carbohidratos, la proteína de unión a manosa activa al complejo C1qrs. Otras
proteínas, las serín proteasas asociadas a MBL (MASP-1 y MASP-2) funcionan
de manera análoga a C1r y C1s, hidrolizando los componentes C4 y C2 para
formar la C3 convertasa (C4bC2a), que es común para ambas vías, la clásica y la

Introducción
12
de la lectina. Los fragmentos C3b generados presentan actividad opsónica,
estimulando la opsonización para el aclaramiento de microorganismos y
residuos celulares resultantes de la apoptosis.
1.1.2.1.3. Vía alternativa o sistema properdina
La vía alternativa se inicia por la interacción del componente C3 con
polisacáridos, su activación es independiente de anticuerpos, presenta un
mecanismo de acción distinto a la vía clásica y a la lectina. Constituye una
primera línea de defensa en la respuesta innata. Es la vía más primitiva.
Constituye un estado de activación permanente del componente C3, se
inicia tras la hidrólisis espontánea de C3 y en presencia de niveles bajos de C3.
En ausencia de microorganismos, la cantidad de C3b producida es inactivada
por el Factor H. En presencia de microorganismos, C3 se une a la superficie
invasora evadiendo la acción del Factor H, se forma un complejo con el Factor
B, el cual se fragmenta por acción del factor D en presencia de Mg2+. El
complejo C3bBb es altamente inestable y la vía alternativa no continúa sin la
presencia de una proteína circulante llamada properdina, que permite
estabilizar el complejo C3bBb. Se forma la C3 convertasa de la vía alternativa
(compuesta por C3bBb), que actuará enzimáticamente sobre moléculas de C3,
amplificando la cascada. Parte de este C3b se puede unir a la C3 convertasa y
formar la C5 convertasa de la vía alternativa (C3bBb3b) que activará a C6,
convergiendo en los mismos pasos no enzimáticos y de ensamblaje finales de la
vía clásica.
1.1.2.1.4. Rutas alternativas de la activación del complemento
Además de los tres principales mecanismos de activación del
complemento, hay varias rutas accesorias que permiten activar el sistema del
complemento en respuesta a varios estados.
La vía de “la lectina ligadora de manosa” (MBL) puede ser activada
directamente por la unión de lectina de unión a manosa (MBL) a
inmunoglobulina IgM que interacciona con antígenos isquémicos de células

Introducción
13
endoteliales en situación de daño por isquemia-reperfusión (McMullen et al.
2006).
En ausencia de C2 y C4 pero en presencia de componentes de la vía
alternativa es posible que complejos antígeno anticuerpo o oligonucleótidos
activen a C1 o lectina de unión a manosa (MBL) respectivamente (Selander et al.
2006).
Está descrito que las anafilotoxinas se pueden generar por la acción de
ciertas proteasas extrínsecas, el componente C3 puede ser degradado y activado
por proteasas como la trombina o la calicreína (Markiewski et al. 2007; Amara et
al. 2008), además el componente C5 puede degradarse por la trombina sin la
necesidad de la acción de C3 (Huber-Lang et al. 2006). Por lo tanto existe un
nexo entre el sistema del complemento y la cascada de coagulación. Asimismo,
las anafilotoxinas C3a y C5a se pueden generar directamente de los
componentes C3 y C5 respectivamente por proteasas que se presentan en
procesos alérgicos, en mecanismos que involucran la generación de radicales
libres y en la activación de la calicreína debido a la presencia de fibras de
amianto y de sílice (Maruo et al. 1997; Governa et al. 2000; Governa et al. 2005)
1.1.2.2. Funciones efectoras del complemento
El sistema de complemento funciona como una herramienta de vigilancia
inmunológica. En un ambiente sano, hay una actividad constitutiva y ligera del
complemento, asegurándose un sondeo que es llevado a cabo por la presencia
de proteínas solubles que reconocen estructuras propias y proteínas de
membrana reguladoras del complemento, de modo que se previene la
amplificación de la señal de activación del complemento. Un ejemplo de estas
proteínas de membrana es el factor DAF (factor acelerador de la degradación)
que está ampliamente distribuído y cuya función es inhibir la unión y facilitar la
disociación de C4b y C2a. De este modo, estos factores reguladores pueden
prevenir la lisis de las células autólogas vivas. En presencia de células
apoptóticas o de microorganismos bacterianos se produce un descenso de estos
factores reguladores, al mismo tiempo que las proteínas que reconocen los

Introducción
14
patrones moleculares asociados a patógenos (PAMPs) amplifican la activación
del complemento. Tiene lugar la opsonización de la célula apoptótica o del
microorganismo por fragmentos del complemento y la posterior fagocitosis o la
formación del complejo de ataque a membrana que conlleva la lisis celular, la
señalización pro-inflamatoria mediada por la liberación de los fragmentos del
complemento con actividad biológica llamados anafilotoxinas, y además, se
induce una estimulación downstream de la respuesta inmune.
Por lo tanto tiene lugar una acción de reconocimiento, activación y
regulación del sistema del complemento, condicionada a la presencia de células
sanas, apoptóticas o microorganismos bacterianos.
Las proteínas reguladoras del complemento pueden ser solubles o de
membrana y ayudan al control del sistema de ataque del complemento
ajustando su severidad, propagación y destino en la célula diana. Ejercen su
acción en distintos puntos, tanto en la vía clásica como en la alternativa o de las
lectinas, centrándose fundamentalmente en la activación de C3.
El sistema del complemento tiene una diversidad de funciones biológicas
defensivas. Estas funciones se llevan a cabo por diferentes fracciones activas del
complemento que interaccionan con sus respectivos receptores de membrana.
Las acciones anafilotóxicas, quimiotáxicas y opsonizantes del
complemento lo convierten en un factor fundamental en la potenciación de la
inflamación, fenómeno básico en la defensa frente a la infección y a los procesos
tumorales. Las principales acciones del complemento son las siguientes:
Acción lítica: la lisis se produce como consecuencia de la formación del
CAM (complejo de ataque a la membrana), este complejo puede lisar bacterias
gram-negativas, parásitos, virus encapsulados, eritrocitos y células nucleadas.
Las bacterias gram positivas son bastante resistentes a la acción lítica del
complemento. Se conoce como citotoxicidad dependiente de complemento.
Respuesta inflamatoria. Acción anafilotóxica: como consecuencia de la
fragmentación de distintos componentes del complemento se generan unos
fragmentos activos llamados anafilotoxinas, estos son el C3a, C4a y C5a. Estas

Introducción
15
anafilotoxinas se unen a sus respectivos receptores induciendo la liberación de
mediadores de la inflamación. Estas sustancias aumentan la vasodilatación y la
permeabilidad de los vasos sanguíneos. Así mismo, inducen la extravasación de
los leucocitos al endotelio. C3a y C5a son potentes quimiotácticos para
neutrófilos, monocitos y macrófagos hacia el lugar de activación del
complemento. Causan una fuerte señalización inflamatoria a través de sus
receptores acoplados a proteínas G (Ames et al. 1996; Monk et al. 2007). C5a
también co-regula la fagocitosis mediada por inmunocomplejos, modulando la
expresión de los receptores de activación FcγRI/II y de inhibición FcγRIIB
(Shushakova et al. 2002; Kumar et al. 2006). La única regulación endógena de
C3a y C5a, es la pérdida de un residuo de arginina terminal mediada por la
actividad carboxipeptidasa, dando lugar a C5a desArg y C3a desArg (Bokisch
and Muller-Eberhard 1970). Estos productos son activos pero con un diferente
espectro de acción (Jalili et al. ; Ratajczak et al. 2006; MacLaren et al. 2008).
Opsonización: C3b es la principal opsonina del complemento. Los
antígenos recubiertos con C3b se unen a receptores específicos en células
fagocíticas facilitando la fagocitosis.
La neutralización de virus: C3b induce la agregación de partículas virales
formando una capa gruesa que bloquea la fijación de los virus a la célula. Este
agregado puede ser fagocitado mediante la interacción de receptores del
complemento y C3b en las células fagocíticas.
Eliminación de inmunocomplejos: Los inmunocomplejos (complejos
antígeno-anticuerpo circulantes) pueden ser eliminados de la circulación si el
complejo se une a C3b. Los eritrocitos tienen receptores del complemento que
interactúan con los complejos inmunes cubiertos por C3b y los lleva al hígado y
al bazo para su destrucción.
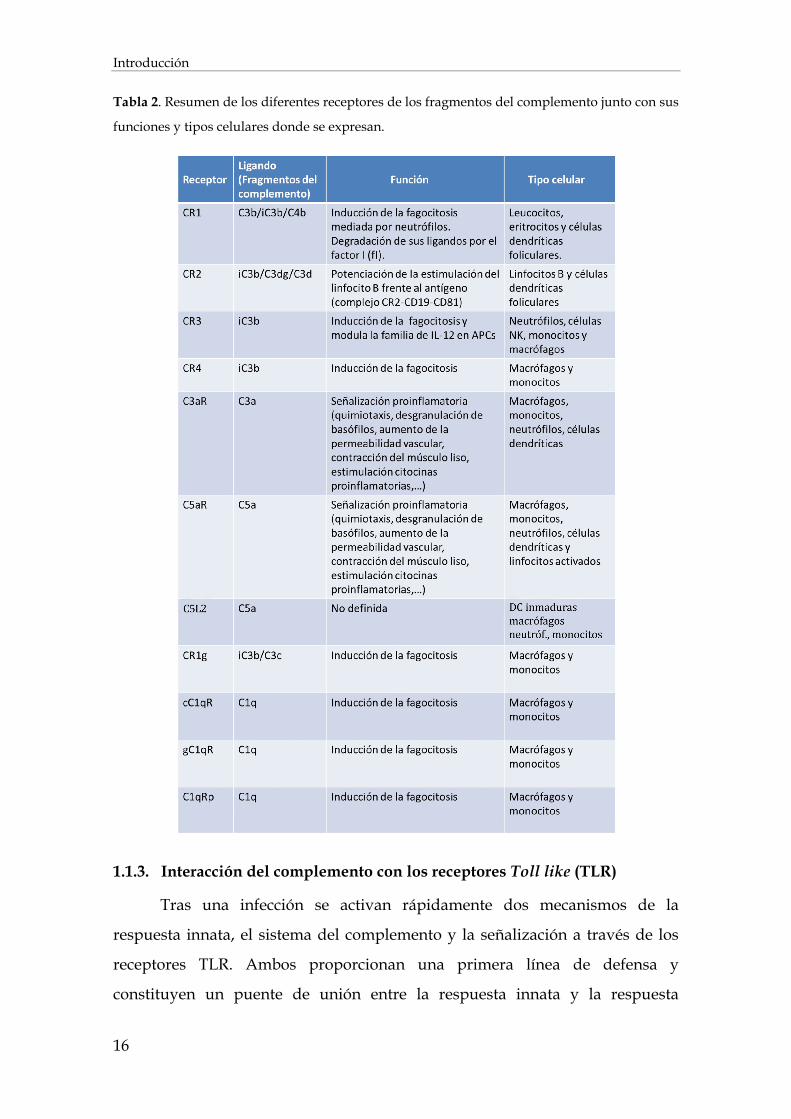
A continuación, mostramos una tabla resumen (Tabla 2) que muestra las
principales funciones efectoras del complemento, en función de la interacción
entre el fragmento del complemento y su receptor.
Introducción
16
Tabla 2. Resumen de los diferentes receptores de los fragmentos del complemento junto con sus
funciones y tipos celulares donde se expresan.
1.1.3. Interacción del complemento con los receptores Toll like (TLR)
Tras una infección se activan rápidamente dos mecanismos de la
respuesta innata, el sistema del complemento y la señalización a través de los
receptores TLR. Ambos proporcionan una primera línea de defensa y
constituyen un puente de unión entre la respuesta innata y la respuesta

Introducción
17
humoral y celular (Dunkelberger and Song 2010; Bhakdi et al. 1990). Coordinan
la respuesta innata en cooperación bidireccional, potenciando la respuesta
innata con acciones sinérgicas o bien regulando un exceso de respuesta
mediante acciones antagónicas (Hajishengallis and Lambris 2010).
La producción de algunas citocinas pro-inflamatorias, inducidas in vivo
por la señalización de los receptores TLR (principalmente TLR4, TLR2, TLR6 y
TLR9), están reguladas por el complemento a través de los receptores C5aR y
C3aR. Las vías de señalización de estos receptores TLR convergen con la
señalización de las anafilotoxinas C3a y C5a a nivel de MAP quinasas,
especialmente ERK1/2 y JNK (Zhang et al. 2007). La interacción de ambos
sistemas, se observa cuando al inhibir la vía de señalización de C5a, se confiere
protección frente a la sepsis que es inducida por altas dosis de LPS (Guo et al.
2004). Recíprocamente, la activación de los receptores TLR induce la expresión
de componentes del complemento y de sus receptores (Kaczorowski et al. 2010).
Como demostraron Zhang et al, los ratones knockout para el factor
acelerador de la degradación DAF, cuando son tratados con LPS, zymosan o
CpG, presentan aumentados los niveles en suero de citocinas pro-inflamatorias
como el factor de necrosis tumoral (TNFα), las interleucinas 1β (IL-1β) y 6 (IL-6)
pero también tienen disminuidos los niveles de la interleucina 12 (IL-12) en
comparación con ratones wild-type. Este fenotipo es mediado por las
anafilotoxinas C5a (en el caso del receptor TLR4) y C3a (en el caso del receptor
TLR9). También demostraron que tras la activación con LPS, estos ratones
knockout para DAF presentan mayor inducción del factor de transcripción NF-
кB y se incrementa la fosforilación de MAP quinasas como ERK y JNK. La
inmunización de estos ratones con distintos antígenos y en combinación con el
adyuvante completo de Freund (CFA) presenta una mayor respuesta T efectora
y de memoria. Además, estos ratones muestran susceptibilidad a procesos
autoinmunes y de inflamación (Zhang et al. 2007). Este es un ejemplo en el que
el complemento y los receptores TLR, promueven la inflamación y modulan la
inmunidad adaptativa.

Introducción
18
Sin embargo otros estudios afirman que el fragmento C5a inhibe la
señalización del TLR4. Según el estudio de Hawlisch et al, el fragmento C5a
regula negativamente la señalización mediada por TLR4 y por la molécula
CD40, inhibiendo la síntesis de citocinas inducidas por la familia de la citocina
IL-12 (IL-12, IL-23 e IL-27) en macrófagos activados. Esta disminución se
traduce en una respuesta Th1 disminuida in vitro e in vivo, observándose una
mayor respuesta Th1 en ratones deficientes del receptor C5aR, confiriéndose
una protección contra la infección por Leishmania major (Hawlisch et al. 2005).
Sin embargo, C5a no inhibe la producción de IL-12 mediada por TLR y por la
interacción entre la molécula CD40 y CD154 en células dendríticas humanas y
murinas. De hecho, en las células dendríticas, la activación de los receptores
C5aR y C3aR promueve la producción de las citocinas IL-12 y IL-23. Además,
los ratones deficientes en ambos receptores presentan un déficit en la
estimulación de la respuesta T in vivo e in vitro. La activación de los receptores
C5aR y C3aR da lugar a la activación de las proteínas ERK1/2 y así, mientras en
macrófagos se induce la inhibición de la producción de IL-12, en las células
dendríticas se produce el efecto opuesto. Una explicación del efecto producido
por el factor C5a al receptor TLR4 en la producción de IL-12 en las células
dendríticas, es la capacidad de C5aR para inhibir la señalización de la proteína
quinasa A (PKA) dependiente de AMPc, incrementando los niveles de TNFα y
disminuyendo la producción de IL-10 (Peng et al. 2009).
El complemento también puede modular la acción de los ligandos para
TLR9, como por ejemplo los oligodeoxinucleótidos CpG. Los CpG inducen la
maduración de las células presentadoras de antígeno (APC) a través de su
receptor TLR9. La inhibición del componente C3 del sistema del complemento
inhibe la acción de los CpG en las células dendríticas, influyendo
negativamente en la señalización TLR9. Es un ejemplo de una
inmunomodulación de la función CpG dependiente del complemento y del
receptor TLR9 (Mangsbo et al. 2009).
El bloqueo de la molécula CD14, co-receptor de TLR4 y TLR2, inhibe
algunas actividades del complemento, esto indica la existencia de un cierto
Introducción
19
grado de cooperación entre el complemento y la molécula CD14 (Lappegard et
al. 2009). Esta cooperación se podría atribuir a las interacciones extracelulares
entre la molécula CD14 y los receptores del complemento o bien a fragmentos
activos del complemento que podrían activar directamente la señalización de
los receptores TLR.
Hay evidencias de que la señalización de los TLR también puede afectar
a la expresión de los receptores del complemento y por lo tanto a su actividad.
Está descrito que la inducción de IL-6 a través del TLR4 induce un aumento de
la expresión de los receptores C5aR y C3aR (Koleva et al. 2002).
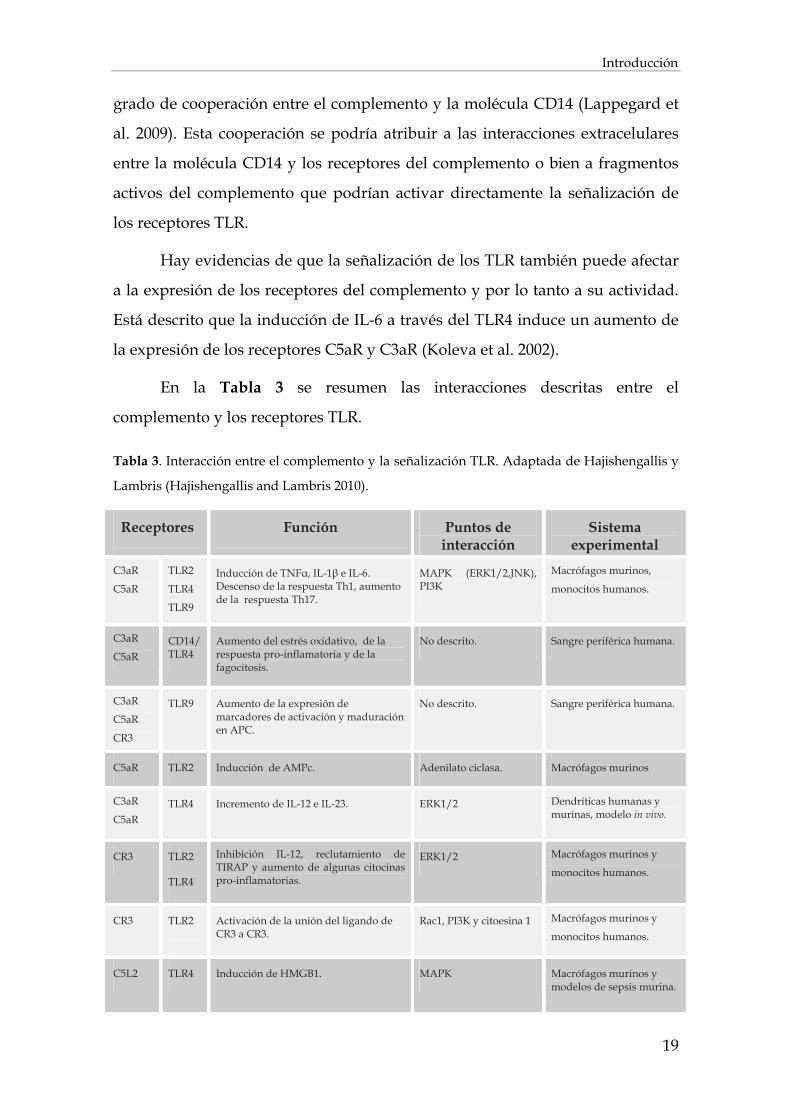
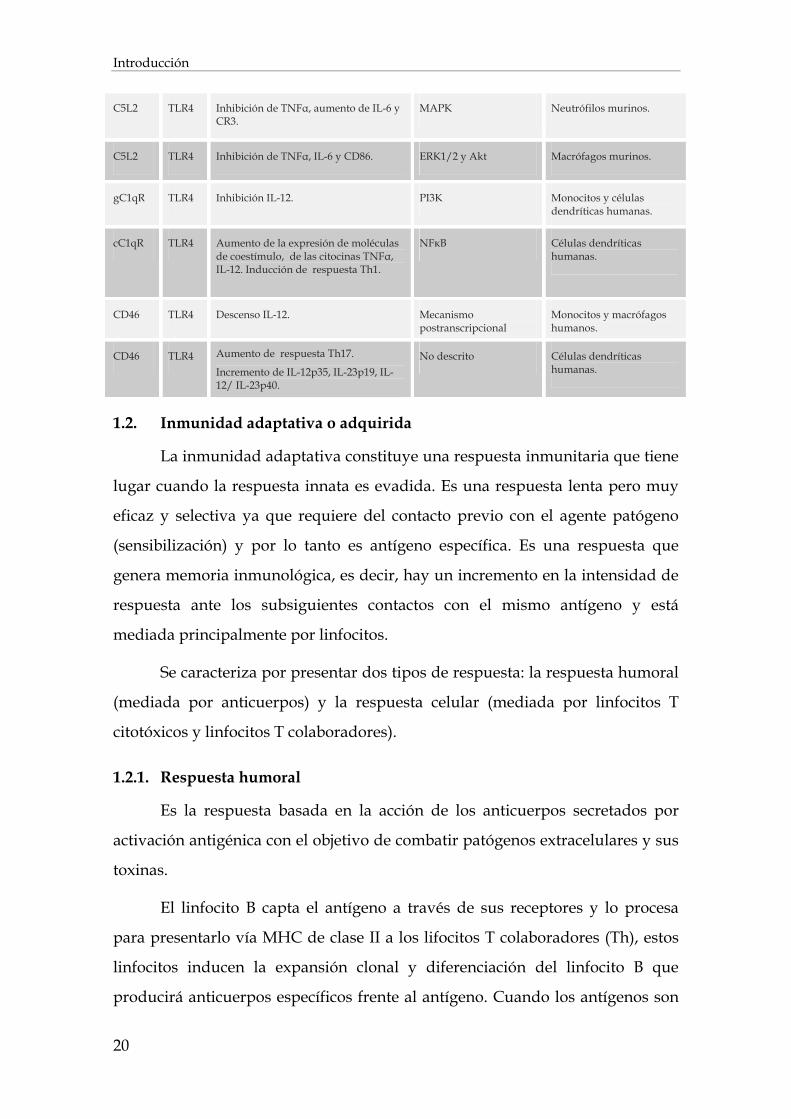
En la Tabla 3 se resumen las interacciones descritas entre el
complemento y los receptores TLR.
Tabla 3. Interacción entre el complemento y la señalización TLR. Adaptada de Hajishengallis y
Lambris (Hajishengallis and Lambris 2010).
Receptores
Función Puntos de interacción
Sistema experimental
C3aR
C5aR
TLR2
TLR4
TLR9
Inducción de TNFα, IL-1β e IL-6. Descenso de la respuesta Th1, aumento de la respuesta Th17.
MAPK (ERK1/2,JNK), PI3K
Macrófagos murinos,
monocitos humanos.
C3aR
C5aR CD14/TLR4
Aumento del estrés oxidativo, de la respuesta pro-inflamatoria y de la fagocitosis.
No descrito. Sangre periférica humana.
C3aR
C5aR
CR3
TLR9 Aumento de la expresión de marcadores de activación y maduración en APC.
No descrito. Sangre periférica humana.
C5aR TLR2 Inducción de AMPc. Adenilato ciclasa. Macrófagos murinos
C3aR
C5aR TLR4 Incremento de IL-12 e IL-23. ERK1/2 Dendríticas humanas y
murinas, modelo in vivo.
CR3 TLR2
TLR4
Inhibición IL-12, reclutamiento de TIRAP y aumento de algunas citocinas pro-inflamatorias.
ERK1/2 Macrófagos murinos y
monocitos humanos.
CR3 TLR2
Activación de la unión del ligando de CR3 a CR3.
Rac1, PI3K y citoesina 1 Macrófagos murinos y
monocitos humanos.
C5L2 TLR4 Inducción de HMGB1. MAPK Macrófagos murinos y modelos de sepsis murina.
Introducción
20
C5L2 TLR4 Inhibición de TNFα, aumento de IL-6 y CR3.
MAPK Neutrófilos murinos.
C5L2 TLR4 Inhibición de TNFα, IL-6 y CD86. ERK1/2 y Akt Macrófagos murinos.
gC1qR TLR4 Inhibición IL-12. PI3K Monocitos y células dendríticas humanas.
cC1qR TLR4 Aumento de la expresión de moléculas de coestímulo, de las citocinas TNFα, IL-12. Inducción de respuesta Th1.
NFкB Células dendríticas humanas.
CD46 TLR4 Descenso IL-12. Mecanismo postranscripcional
Monocitos y macrófagos humanos.
CD46 TLR4 Aumento de respuesta Th17.
Incremento de IL-12p35, IL-23p19, IL-12/ IL-23p40.
No descrito Células dendríticas humanas.
1.2. Inmunidad adaptativa o adquirida
La inmunidad adaptativa constituye una respuesta inmunitaria que tiene
lugar cuando la respuesta innata es evadida. Es una respuesta lenta pero muy
eficaz y selectiva ya que requiere del contacto previo con el agente patógeno
(sensibilización) y por lo tanto es antígeno específica. Es una respuesta que
genera memoria inmunológica, es decir, hay un incremento en la intensidad de
respuesta ante los subsiguientes contactos con el mismo antígeno y está
mediada principalmente por linfocitos.
Se caracteriza por presentar dos tipos de respuesta: la respuesta humoral
(mediada por anticuerpos) y la respuesta celular (mediada por linfocitos T
citotóxicos y linfocitos T colaboradores).
1.2.1. Respuesta humoral
Es la respuesta basada en la acción de los anticuerpos secretados por
activación antigénica con el objetivo de combatir patógenos extracelulares y sus
toxinas.
El linfocito B capta el antígeno a través de sus receptores y lo procesa
para presentarlo vía MHC de clase II a los lifocitos T colaboradores (Th), estos
linfocitos inducen la expansión clonal y diferenciación del linfocito B que
producirá anticuerpos específicos frente al antígeno. Cuando los antígenos son

Introducción
21
de naturaleza no proteica, no se requiere de la presencia de los linfocitos Th
para la activación de las células B. Estos anticuerpos pueden tener efecto
neutralizante del patógeno directamente o indirectamente a través de células
fagocíticas, activación del complemento o por unión de las células NK a la
fracción Fc de la inmunoglobulina.
1.2.2. Respuesta celular
Los linfocitos T son los responsables de la respuesta inmunitaria celular
permitiendo la destrucción de las células infectadas o células tumorales. Esta
respuesta presenta una fase de activación (expansión de las células T
citotóxicas), una fase efectora (encuentro y eliminación de la célula infectada o
tumoral) y una fase de contracción (apoptosis y memoria).
Los linfocitos T después de un proceso de selección tímica, donde forman
un repertorio de linfocitos T con un receptor clonal (TCR) que les permite
reconocer determinantes antigénicos presentados por moléculas MHC, migran
a los órganos linfoides secundarios donde tendrá lugar la presentación
antigénica mediante la interacción entre su receptor TCR (del inglés T Cell
Receptor) y el complejo MHC-antígeno (expresado en la superfície de las APC)
(Germain et al. 1988; McMichael 1979; Townsend and Bodmer 1989).
Dentro de esta población celular encontramos varias subpoblaciones,
entre las que destacan los linfocitos T CD4+ o colaboradores (Th) , los linfocitos
CD8+ o citotóxicos (Tc), los linfocitos T CD4+CD25 + reguladores (Treg), los
linfocitos Th17 y las NKT (del inglés natural killer T lymphocyte).
1.2.2.1. Linfocitos Th
Los linfocitos T colaboradores o helper (Th) fueron descritos por primera
vez por Mossman y Coffman, observaron que las células T podían dividirse en
dos subgrupos que llamaron las células Th1 y Th2 en función de su patrón de
producción de citocinas, interferón-γ, TNF- e IL-2 (células Th1) o interleucina-
4 (IL-4), IL-5, IL-6, IL-10 e IL-13 (células Th2) (Mosmann and Coffman 1989;
Cherwinski et al. 1987). Pero la relevancia entre la distinción de ambos subtipos

Introducción
22
fue descrita por Heinzel et al. (Heinzel et al. 1989), observaron que en ratones
una respuesta predominantemente Th1 permitía superar la infección por
Leishmania major mientras que una respuesta tipo Th2 no era capaz de combatir
dicha infección.
Estos linfocitos Th no presentan actividad citotóxica, pero controlan la
respuesta inmunitaria dirigiendo a otras células para que realicen esas tareas,
tienen efectos protectores y promotores de la función de las células B, T CD8+ y
macrófagos. Los linfocitos Th1 están asociados a respuestas inmunitarias
mediadas por linfocitos T citotóxicos, macrófagos y neutrófilos. Los linfocitos
Th2 están destinados a ayudar a las células B en la producción de anticuerpos y
a activar a los eosinófilos. Además, los linfocitos Th pueden tener un papel
antiviral directo a través de las citocinas que producen.
1.2.2.2. Linfocitos T citotóxicos
Los linfocitos T CD8+ (Tc) tienen su capacidad de actuación restringida
por las moléculas MHC-I por lo tanto se encargan de combatir las infecciones
intracelulares destruyendo las células infectadas, de eliminar células tumorales
o células dañadas por otras causas. El principal mecanismo efector utilizado es
la inducción de la apoptosis segregando una serie de moléculas inductoras de la
apoptosis (FasL, perforina, granzimas) (Harty et al. 2000; Pardoll 1993;
Zinkernagel and Althage 1977). Son los principales causantes del rechazo de
tejidos y órganos transplantados (Bothwell 1999; Onoe et al. 1997), estando
también implicados en fenómenos de autoinmunidad (Billet et al. 2006).
En virtud de un conjunto definido de receptores de homing como la
molécula CD62L y receptores de quimiocinas, como el receptor CCR7, los
linfocitos T CD8+, del mismo modo que los linfocitos T CD4+, circulan por el
torrente circulatorio hasta llegar a los órganos linfoides secundarios, donde
tiene lugar la presentación antigénica por las células dendríticas (DC) que
actúan como células presentadoras de antígeno profesionales. Los linfocitos Tc
tras este encuentro se expanden, proliferan y se diferencian a células efectoras.

Introducción
23
La activación del linfocito Tc tiene unos controles muy estrictos y, por lo
general, requiere una señal de activación muy fuerte por parte del complejo
MHC-I/DTc y de señales adicionales proporcionadas por las células T
colaboradoras (Bourgeois et al. 2002; Freeman et al. 1993; Linsley and Ledbetter
1993). Sin embargo, no siempre la ayuda CD4 es indispensable, existiendo
varios modelos de activación de linfocitos CD8 en donde las células CD4 no
participan (Bachmann et al. 1998; Lasarte et al. 1995).
Los linfocitos T CD8+ ejercen su función antiviral mediante mecanismos
no citolíticos con la producción de citocinas (IFN-, TNF- o IL-2) y a través de
mecanismos citolíticos que convergen en la inducción de apoptosis en la célula
diana por activación de caspasas. Uno de los mecanismos consiste en la
liberación de gránulos que contienen la molécula formadora de poros perforina
y la serín esterasa granzima B. El otro mecanismo implica la expresión de Fas
ligando (CD95L) y su interacción con la molécula Fas (CD95) expresada en la
superficie de la célula diana.
La gran mayoría de células efectoras tras cumplir con su cometido
entrarán en apoptosis celular, el resto formarán un pool de células memoria.
Estos linfocitos memoria tienen una vida larga pudiendo circular durante meses
o años y están preparados para desencadenar una respuesta inmunitaria rápida
y potente si se produce una nueva exposición con el mismo patógeno (Ahmed
and Gray 1996). De hecho, éste es el principal objetivo de las vacunas, generar
linfocitos memoria (T y B) de manera que el organismo responda de manera
rápida y eficaz frente al patógeno activo (Ahmed and Gray 1996).
Está descrito que se requiere la presencia de los linfocitos T CD4+ para la
generación de estos linfocitos T CD8+ memoria en respuestas inmunitarias
contra virus y bacterias (Shedlock and Shen 2003), así como en la erradicación
de tumores (Marzo et al. 2000), aunque las células T CD8+ memoria podrían
generarse en su ausencia, siendo menos eficaces que las generadas con su
ayuda (Janssen et al. 2003; Shedlock and Shen 2003). La población de los
linfocitos T CD8+ memoria se regula mediante mecanismos homeostáticos para

Introducción
24
mantener a lo largo del tiempo una representación de la misma.
Fundamentalmente dos citocinas son las responsables de la homeostasis de esta
población celular: la IL-7, que es esencial para su supervivencia, y la IL-15, que
se encarga principalmente de su proliferación para mantener una población
mínima de reserva (Surh and Sprent 2008).
1.2.2.3. Linfocitos T reguladores CD4+CD25+
A principios de los años 70 se describió por primera vez la presencia de
unos linfocitos T capaces de suprimir las respuestas inmunitarias. En 1995,
Sakaguchi y col. (Sakaguchi et al. 1995) encontraron que una población
minoritaria de células CD4+ (10%) que co-expresaban la cadena alfa del
receptor para la interleuquina 2 (CD25) era crucial para el control de las células
autorreactivas y la autoinmunidad in vivo. Desde entonces, muchos grupos han
demostrado que esta subpoblación de células CD4+CD25+, también conocidas
como células Treg o linfocitos Treg, son inmunosupresoras (Takahashi et al.
1998; Thornton and Shevach 1998). Estas células fueron identificadas primero
en ratones pero después han sido ampliamente caracterizadas en humanos
(Dieckmann et al. 2001; Jonuleit et al. 2001; Levings et al. 2001). Actualmente, la
existencia de una subpoblación inmunosupresora específica es aceptada
ampliamente por la comunidad científica y se busca la forma de manipular su
actividad para su uso clínico. La principal cuestión es cómo podría modularse
su actividad.
Los linfocitos Treg son esenciales para la protección frente a las
enfermedades autoinmunes y para la prevención del rechazo a los transplantes;
por ello, la posibilidad de potenciar su actividad tiene un gran potencial en el
tratamiento de las enfermedades autoinmunes y en los transplantes de órganos.
Sin embargo, debido a que los tumores expresan autoantígenos, los linfocitos
Treg pueden ser capaces de inhibir la activación de respuestas inmunitarias
frente al cáncer.
Varios grupos, han demostrado que la simple eliminación de las células
CD4+CD25+FOXP3+ (Treg) por la administración in vivo de anticuerpos

Introducción
25
deplecionantes facilita la inducción de inmunidad antitumoral y la protección
frente al desarrollo del cáncer (Casares et al. 2003; Onizuka et al. 1999; Shimizu
et al. 1999; Steitz et al. 2001; Sutmuller et al. 2001). Así, se cree que las células
Treg están continuamente frenando la activación de los linfocitos T efectores
para evitar procesos de autoinmunidad, pero dificultando a su vez la correcta
activación de una respuesta antitumoral cuando ésta es necesaria.
1.3. Activación de una respuesta inmunitaria
1.3.1. Presentación antigénica
Para que tenga lugar una activación específica de la respuesta
inmunitaria frente a un antígeno, es necesario que se de la presentación
antigénica (Mellman 2005), un proceso de reconocimiento molecular entre el
péptido expuesto por moléculas de MHC (complejo mayor de
histocompatibilidad) en las APC y el receptor TCR o BCR de linfocitos T y B
respectivamente. Para ello, el antígeno debe ser previamente capturado por la
APC y procesado hasta ser convertido en péptido. Además, se requieren señales
de coestímulo (interacción de factores solubles y proteínas de membrana) para
una activación eficaz del linfocito y por lo tanto de la respuesta inmunitaria
(Koch et al. 1996; Vazeux et al. 1992; Zhang et al. 1999; Zuckerman et al. 1998).
Esta interacción entre la APC y el linfocito se conoce como sinapsis
inmunológica.
Existen dos vías principales de presentación antigénica, la vía clase I y la
vía clase II, en función del origen y la vía de entrada del antígeno (Jensen 2007;
Villadangos and Schnorrer 2007) (Figura 4).
La vía clase I consiste en la presentación de antígenos endógenos
procedentes de proteínas propias, proteínas intracelulares derivadas de un
patógeno que ha infectado la célula o proteínas inducidas durante el desarrollo
de un tumor que son procesadas por el proteosoma. Tras la degradación de la
proteína en el proteosoma, los fragmentos peptídicos generados (determinantes
T citotóxicos) son transportados mediante un transportador dependiente de
ATP llamado TAP (del inglés, transporter associated protein) desde el citoplasma

Introducción
26
hasta el retículo endoplasmático, donde se unen a las moléculas MHC de clase
I. El complejo MHC-I/DTc pasa a través del Golgi antes de llegar a la superficie
celular, donde será presentado a los linfocitos CD8+ citotóxicos. Todas las
células nucleadas pueden presentar determinantes antigénicos mediante esta
ruta. En estos casos, la presentación de antígenos puede ocurrir en ausencia de
otras señales inmunoestimuladoras (como moléculas de coestímulo situadas
sobre las APC o señales emitidas por los LTh), por lo que este proceso puede
terminar en tolerancia.
La vía clase II es propia de las APC y consiste en la captación e
internalización en endosomas de los antígenos exógenos. Los antígenos se
procesan en el lisosoma por la acción de enzimas proteolíticas y los péptidos
resultantes del procesamiento antigénico se unen a moléculas MHC de clase II.
El complejo MHC-II/determinante T helper es transportado a la superficie
celular para ser presentado a los linfocitos T colaboradores (Th).
Ciertos antígenos exógenos a pesar de ser fagocitados por las APC
profesionales, son capaces de salir de los fagosomas y entrar en el citosol. De
esta forma son procesados por la ruta citosólica mediante mecanismos que
pueden ser tanto dependientes como independientes del proteasoma. Así, los
antígenos son presentados a través del MHC-I de la APC al LTc (Gil-Torregrosa
et al. 1998; Heath and Carbone 2001; Rock and Shen 2005). Este proceso se
conoce como cross-presentation (presentación cruzada).

Introducción
27
Figura 4. Esquema del procesamiento y presentación de los péptidos (Heath and Carbone 2001).
1.3.2. Células dendríticas
Las DC son células que derivan de las células madre hematopoyéticas de
médula ósea y han sido identificadas como las APC más eficaces. Actúan de
enlace entre la inmunidad innata y adaptativa permitiendo el inicio y la
modulación de la activación de ambas respuestas. Su papel principal es la
captura, procesamiento y presentación de antígenos a través del MHC a los
linfocitos T y, de este modo, activación de la respuesta inmunitaria mediante la
producción de citocinas y la inducción de la activación de los linfocitos.
Constituye una población celular heterogénea que controla tanto la
inmunidad como la tolerancia inmunológica. En sangre se distinguen dos tipos
de DC, en función de sus características fenotípicas (expresión diferencial de
marcadores de superficie) y funcionales (patrón diferencial de citocinas). Estas
células dendríticas son las DC mieloides y las DC plasmacitoides.
Las DC convencionales o mieloides (DCm), provienen de la línea
mieloide y su función principal es la presentación antigénica. Migran a los
tejidos periféricos, donde interceptan y capturan los patógenos, antes de su
migración a los ganglios linfáticos para activar la respuesta T (Belmonte et al.

Introducción
28
2007). Se caracterizan por su capacidad para producir citocinas como IL-12 y
TNF-α, con el objetivo de promover una respuesta de tipo Th1 y desarrollo de
linfocitos T citotóxicos. La segunda subpoblación de DC, de origen linfoide, se
denomina DC plasmacitoide (DCp). Se distinguen de las DCm por no expresar
CD11c y se caracterizan por su capacidad para producir grandes cantidades de
IFN-γ en respuesta a infecciones virales (Cella et al. 1999; Siegal et al. 1999; Sun
et al. 1998), cumpliendo de esta forma un importante papel en la respuesta
inmunitaria innata. Al contrario de las DCm, migran directamente de la sangre
a los tejidos linfáticos secundarios. El IFN de tipo I inducido por las DCp puede
activar las células NK y las células T CD8+, regular la producción de IL-12
mediada por las DCm, e intervenir en la diferenciación de células B en células
plasmáticas (Liu 2005). Otra función de las DCp es el procesamiento de
antígenos y su presentación a los linfocitos T. Sin embargo, las DCm son más
eficaces en esta función (Krug et al. 2003).
La capacidad de las DC para activar la repuesta inmunitaria depende de
su estado de maduración. Se encuentran ampliamente distribuidas como células
inmaduras en todos los tejidos, especialmente en los que interactúan con el
medio ambiente (epitelios de la piel y mucosas). La internalización de antígenos
extraños puede desencadenar su maduración y la migración desde los tejidos
periféricos a los órganos linfoides donde tendrá lugar la activación linfocitaria.
En estado inmaduro, poseen una gran capacidad fagocítica y expresan
un amplio espectro de receptores (TLR, receptores de quemoquinas entre otros).
Sin embargo, el nivel de expresión en la superficie celular de moléculas de
MHC, coestímulo y adhesión es muy bajo. La principal función de estas células
consiste en detectar la presencia de agentes extraños, capturarlos y
transportarlos a los nódulos linfoides. Si la DC reconoce señales de peligro sufre
un proceso de maduración que activa el proceso de migración a los nódulos
linfoides. Durante la migración, las DC maduras, presentan un cambio en su
morfología, aumentan su movilidad y se vuelven muy eficaces en la
presentación antigénica y en la estimulación de los linfocitos T vírgenes. Los
procesos de maduración y migración están íntimamente ligados y la completa
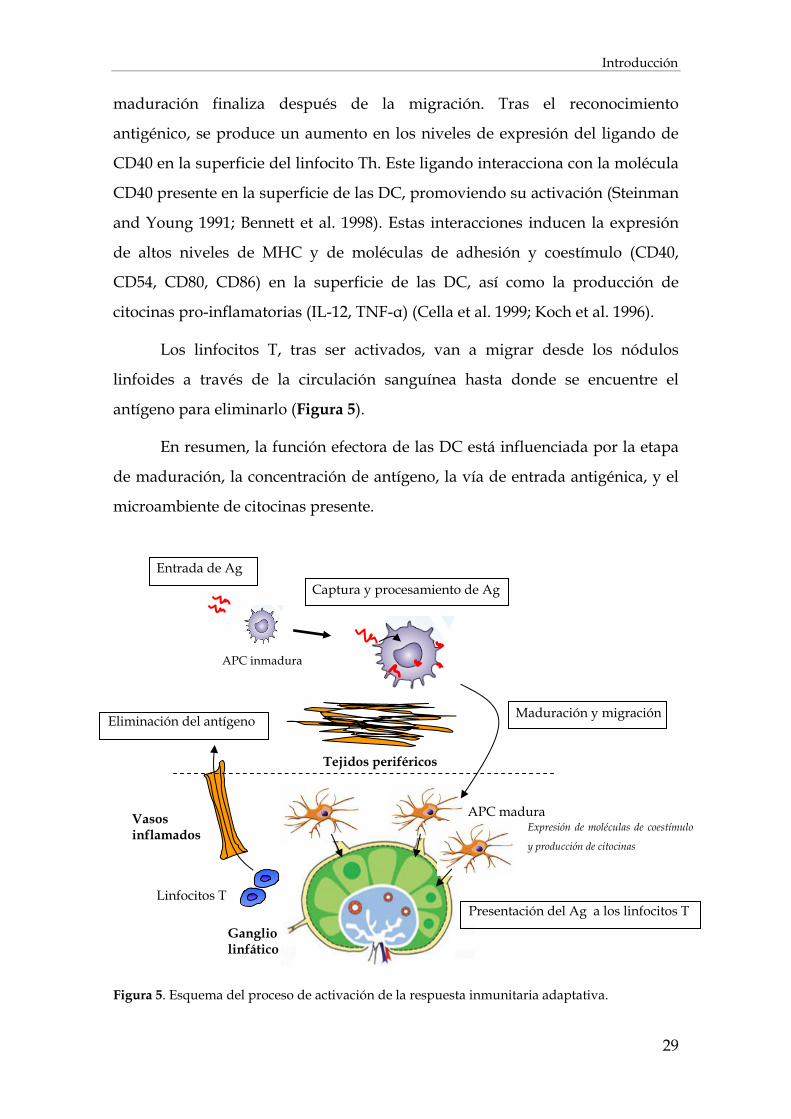
Introducción
29
maduración finaliza después de la migración. Tras el reconocimiento
antigénico, se produce un aumento en los niveles de expresión del ligando de
CD40 en la superficie del linfocito Th. Este ligando interacciona con la molécula
CD40 presente en la superficie de las DC, promoviendo su activación (Steinman
and Young 1991; Bennett et al. 1998). Estas interacciones inducen la expresión
de altos niveles de MHC y de moléculas de adhesión y coestímulo (CD40,
CD54, CD80, CD86) en la superficie de las DC, así como la producción de
citocinas pro-inflamatorias (IL-12, TNF-α) (Cella et al. 1999; Koch et al. 1996).
Los linfocitos T, tras ser activados, van a migrar desde los nódulos
linfoides a través de la circulación sanguínea hasta donde se encuentre el
antígeno para eliminarlo (Figura 5).
En resumen, la función efectora de las DC está influenciada por la etapa
de maduración, la concentración de antígeno, la vía de entrada antigénica, y el
microambiente de citocinas presente.
Figura 5. Esquema del proceso de activación de la respuesta inmunitaria adaptativa.
Maduración y migración
APC madura
Presentación del Ag a los linfocitos T
Tejidos periféricos
Entrada de Ag
Expresión de moléculas de coestímulo
y producción de citocinas
APC inmadura
Captura y procesamiento de Ag
Linfocitos T
Eliminación del antígeno
Ganglio linfático
Vasos inflamados

Introducción
30
1.3.3. Señalización del calcio en la activación linfocitaria
Los iones de Ca2+ funcionan como segundos mensajeros en todas las
células eucariotas, incluyendo células del sistema inmunitario. La entrada de
Ca2+ al interior celular es la señal crucial para la correcta activación de los
linfocitos, la realización de sus funciones efectoras, de diferenciación, la correcta
sinapsis entre células dendríticas y células T, así como, la exocitosis en células T
citotóxicas.
Un influjo de Ca2+ en el citosol ocurre después de la unión a
inmunorreceptores de superficie celular, por ejemplo la unión al BCR (B cell
receptor) o al TCR (T cell receptor) (van Leeuwen and Samelson 1999). En los
linfocitos el principal mecanismo de incremento de las concentraciones celulares
de calcio ocurre tras el vaciado de los depósitos intracelulares, mecanismo
conocido como SOCE (store operated calcium entry), con la participación de la
activación de los canales de liberación de calcio CRAC situados en la membrana
plasmática (Feske 2007) (Figura 6).
Figura 6. Mecanismo SOCE (store operated calcium entry) de Stefan Feske (Feske 2007).
Cuando tiene lugar el reconocimiento antigénico a través del TCR, los
reservorios de calcio en el retículo endoplasmático se vacían. Esto produce
activación de CRAC generándose un incremento de calcio citosólico, que a su
vez, permitirá la activación de enzimas dependientes de calcio, como la

Introducción
31
calcineurina, factores de transcripción como NFAT (nuclear factor of activated T
cells) o el NFκB (nuclear factor κB). Este mecanismo es necesario para la
activación y expresión génica de citocinas por los linfocitos T y B. En ausencia
del influjo de calcio, la activación celular, proliferación y funciones efectoras
estarían comprometidas (Feske et al. 2001; Partiseti et al. 1994).
Varios mecanismos influencian en la forma, duración y fuerza de la señal
de Ca2+, entre otros están: la despolarización de la membrana plasmática que
reduce la fuerza de entrada de calcio, los canales Kv1.3 que favorecen la
hiperpolarización de la membrana plasmática y las bombas de Ca2+
dependientes de ATP que permiten la entrada de Ca2+ al RE inhibiendo
indirectamente a los CRAC (Cahalan et al. 2001; Chandy et al. 2004). Por este
motivo cualquier alteración en alguno de los mecanismos que afecten a la
entrada de Ca2+ a la célula puede tener gran repercusión en la activación
linfocitaria.
2. Estrategias de vacunación
La activación de los linfocitos T está dirigida por células presentadoras
de antígeno profesionales conocidas como células dendríticas (DCs). Estas
células juegan un rol fundamental en la iniciación de la respuesta inmune
celular contra patógenos. Las DCs actúan como centinelas en tejidos periféricos,
linfa y órganos linfoides secundarios, todos ellos lugares que se encuentran
constantemente expuestos a patógenos, cumpliendo la función estratégica de
captura y presentación de antígenos propios y ajenos a linfocito T. Los
antígenos son procesados hasta fragmentos peptídicos que se presentan unidos
a moléculas MHC de clase I y II, para la activación de linfocitos T CD8+ o
CD4+, respectivamente, necesarios para dar inicio a la inmunidad celular contra
el agente patógeno. Este proceso de captación de antígeno, degradación y carga,
se denomina presentación antigénica. Sin embargo, en ausencia de
estimulación, las células dendríticas periféricas presentan los antígenos de
modo ineficaz. Las señales exógenas provenientes de los patógenos (tales como
el LPS, DNA, CpG, RNA), o las señales endógenas activadas en los procesos

Introducción
32
inflamatorios (en conjunto denominadas señales de peligro), inducen a las
células dendríticas para que inicien un proceso de desarrollo denominado
maduración, que transforma a las células dendríticas. De este modo, las células
dendríticas se convierten en las APC más potentes y las únicas capaces de
activar a los linfocitos T no activados e iniciar la respuesta inmunológica.
El reconocimiento de estos “patrones moleculares asociados a
patógenos” (PAMPs) por medio de receptores específicos en la DC, así como,
citocinas pro-inflamatorias (señales endógenas), produce una serie de cambios
que conducen a la maduración de esta célula. Mientras las DCs inmaduras son
tolerogénicas, las maduras son, por el contrario, inmunogénicas.
Se han observado al menos seis tipos de receptores de superficie capaces
de desencadenar la maduración de células dendríticas: (i) receptores tipo peaje
“toll-like receptors” (TLR), (ii) receptores de citocinas, (iii) moléculas de la
familia de receptores TNF (TNF-R), (iv) FcR, (v) sensores de muerte celular y
(vi) receptores del complemento. Algunos de los estímulos más eficaces de
maduración están mediados por interacciones TLR (TLR1-10) con sus
respectivos ligandos (Latz et al. 2003).
El reconocimiento de ligandos por los TLR activa la vía del NF-kB,
conduciendo a la iniciación de la respuesta inmune adaptativa por la
producción de citocinas inflamatorias tales como la interleucina IL-1, IL-8, TNF-
alfa, IL-12 y la inducción de moléculas de co-estimulación, tales como la CD80,
CD86 y CD40. En nuestro laboratorio hemos mostrado que el dominio extra A
de la fibronectina (EDA), es un ligando natural de la molécula TLR4 y que su
unión a un antígenio viral o tumoral puede aumentar su inmunogenicidad,
convirtiendo a EDA en un candidato para el desarrollo de vacunas (Aranda et
al. 2011; Mansilla et al. 2009). La combinación de varias vías de activación de las
DC podría derivar en el desarrollo de una estrategia de vacunación más eficaz.
En este sentido, las proteínas del complemento podrían constituir una
herramienta interesante.

Introducción
33
Actualmente, una estrategia utilizada en la vacunación frente a infección
es la utilización del fragmento C3d como molécula adyuvante en la vacunación.
C3d conjugado a un antígeno mejora la respuesta inmune frente al antígeno.
C3d se une al receptor del complemento 2 (CR2) que se encuentra en la
superficie de las células dendríticas foliculares, las células B y células T,
estimulando la presentación antigénica por las células dendríticas foliculares y
ayudando a mantener la memoria inmunológica de las células B. En las células
B, la interacción C3d y CR2 reagrupa algunas moléculas como CD19,
desencadenando la activación y proliferación celular. Por otra parte, la unión
simultánea de C3d, CR2 y del anticuerpo unido al antígeno de superficie, activa
otra vía de señalización sinérgica. También está descrito que C3d sin presencia
de CR2 induce la producción de anticuerpos, indicando un mecanismo de
acción CR2 independiente (Dempsey et al. 1996; Watanabe et al. 2003; Bower et
al. 2004; Wang et al. 2004; Kolla et al. 2007). Esta estrategia también es utilizada
para la vacunación frente al crecimiento tumoral con resultados muy parecidos.
Xu Gl et al. han diseñado una vacuna de DNA que expresa la región
extracelular murina del VEGFR-2 y 3 copias del fragmento C3d (C3d3). Estos
autores describen que con esta proteína de fusión se puede inhibir de manera
significativa el crecimiento del tumor y que la administración de C3d como
adyuvante molecular aumenta los niveles de anticuerpos contra el VEGFR-2,
proporcionando una alternativa terapéutica a las terapias frente al cáncer (Xu et
al. 2010).
Otro fragmento del complemento descrito como posible vacuna es el
C5a. Un ejemplo es la fusión entre anticuerpos dirigidos al antígeno tumoral
HER/neu con el fragmento C5a y C5a desArg, que induce una mayor muerte
celular en el tumor que aquellos anticuerpos sin la fusión con el fragmento C5a
(Fuenmayor et al. 2010).
Por otra parte la inmunosupresión ejercida por las células T reguladoras
es un mecanismo de regulación de la respuesta inmunitaria, constituyendo una
diana perfecta para bloquear con el fin de potenciar la respuesta inmunitaria.
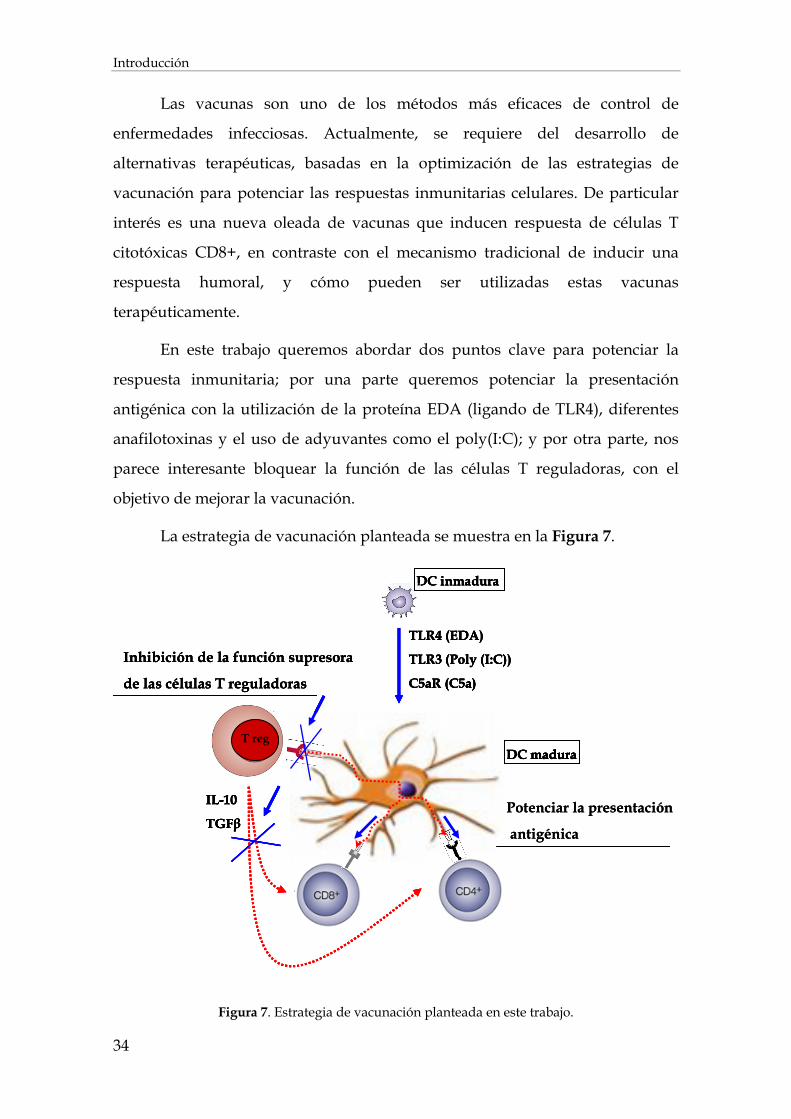
Introducción
34
Las vacunas son uno de los métodos más eficaces de control de
enfermedades infecciosas. Actualmente, se requiere del desarrollo de
alternativas terapéuticas, basadas en la optimización de las estrategias de
vacunación para potenciar las respuestas inmunitarias celulares. De particular
interés es una nueva oleada de vacunas que inducen respuesta de células T
citotóxicas CD8+, en contraste con el mecanismo tradicional de inducir una
respuesta humoral, y cómo pueden ser utilizadas estas vacunas
terapéuticamente.
En este trabajo queremos abordar dos puntos clave para potenciar la
respuesta inmunitaria; por una parte queremos potenciar la presentación
antigénica con la utilización de la proteína EDA (ligando de TLR4), diferentes
anafilotoxinas y el uso de adyuvantes como el poly(I:C); y por otra parte, nos
parece interesante bloquear la función de las células T reguladoras, con el
objetivo de mejorar la vacunación.
La estrategia de vacunación planteada se muestra en la Figura 7.
Potenciar la presentación
antigénica
TLR4 (EDA)
TLR3 (Poly (I:C))
C5aR (C5a)
DC inmadura
DC madura
IL-10
TGFβ
T reg
Inhibición de la función supresora
de las células T reguladoras
Potenciar la presentación
antigénica
TLR4 (EDA)
TLR3 (Poly (I:C))
C5aR (C5a)
DC inmadura
DC madura
IL-10
TGFβ
T reg
Inhibición de la función supresora
de las células T reguladoras
TLR4 (EDA)
TLR3 (Poly (I:C))
C5aR (C5a)
DC inmadura
DC madura
IL-10
TGFβ
T reg
TLR4 (EDA)
TLR3 (Poly (I:C))
C5aR (C5a)
DC inmadura
DC maduraDC madura
IL-10
TGFβ
T reg
Inhibición de la función supresora
de las células T reguladoras
Figura 7. Estrategia de vacunación planteada en este trabajo.

Introducción
35
Como se refleja en la Tabla 4, se pueden utilizar distintas combinaciones
de una gran variedad de inmunógenos, administrados en diferentes vehículos y
utilizando distintos adyuvantes.
Tabla 4. Estrategias de inducción de respuestas inmunitarias celulares.
-Péptidos-Lipop éptidos-Proteínas-ADN desnudo-ARNm-Virus recomb. -Bacts recomb.
Ant ígenos
Vehículos
-Salino-Emulsiones, aceites-Liposomas-Virosomas-Virus like particles-Nanopartículas-Proteínas-Anticuerpos-Células dendríticas
Adyuvantes
-IFA, CFA, ISCOMS...-Citoquinas, quemoquinas.-Mol éculas-anti 4-1BB, anti -CD40, anti-CD28…-Estímulos maduración de células dendríticas.-Ligandos de TLR.-Estrategiasde Prime-boost-Inhibidores de Treg-Polietilenimina (PEI)
-Péptidos-Proteínas-ADN desnudo-ARN mensajero -Virus recomb.-Bacts recomb.-...
Antígenos
Vehículos
-Salino-Emulsiones-Liposomas-Virosomas-Virus like particles-Nanopartículas-Proteínas-Anticuerpos-Células dendr
Vehículos
-Salino-Liposomas-Virosomas-Virus like particles-Nanopartículas-Proteínas-Anticuerpos-Células dendríticas-...
Adyuvantes
-IFA, CFA, ISCOMS, ...-Citoquinas, quemoquinas.-Moléculas coestimuladoras:anti-4-1BB, anti-CD40, anti-CD28, …
-Estímulos maduración de células dendríticas
-Ligandos de TLR-Estrategias de prime-boost-Inhibidores de Treg-Polietilenimina (PEI)-…-
-Péptidos-Lipop éptidos-Proteínas-ADN desnudo-ARNm-Virus recomb. -Bacts recomb.
Ant ígenos
Vehículos
-Salino-Emulsiones, aceites-Liposomas-Virosomas-Virus like particles-Nanopartículas-Proteínas-Anticuerpos-Células dendríticas
Vehículos
-Salino-Emulsiones, aceites-Liposomas-Virosomas-Virus like particles-Nanopartículas-Proteínas-Anticuerpos-Células dendríticas
Adyuvantes
-IFA, CFA, ISCOMS...-Citoquinas, quemoquinas.-Mol éculas-anti 4-1BB, anti -CD40, anti-CD28…-Estímulos maduración de células dendríticas.-Ligandos de TLR.-Estrategiasde Prime-boost-Inhibidores de Treg-Polietilenimina (PEI)
-Péptidos-Proteínas-ADN desnudo-ARN mensajero -Virus recomb.-Bacts recomb.-...
Antígenos
Vehículos
-Salino-Emulsiones-Liposomas-Virosomas-Virus like particles-Nanopartículas-Proteínas-Anticuerpos-Células dendr
Vehículos
-Salino-Liposomas-Virosomas-Virus like particles-Nanopartículas-Proteínas-Anticuerpos-Células dendríticas-...
Adyuvantes
-IFA, CFA, ISCOMS, ...-Citoquinas, quemoquinas.-Moléculas coestimuladoras:anti-4-1BB, anti-CD40, anti-CD28, …
-Estímulos maduración de células dendríticas
-Ligandos de TLR-Estrategias de prime-boost-Inhibidores de Treg-Polietilenimina (PEI)-…-
A continuación se enumeran las diferentes estrategias utilizadas en este
trabajo.
2.1. DNA desnudo
Son plásmidos que codifican para un antígeno tumoral o viral. Las
células que han sido transfectadas expresan esa proteína y, una vez procesada,
los determinantes antigénicos T citotóxicos (DTc) y los determinantes
antigénicos T colaboradores (DTh) son presentados a los linfocitos T para la
inducción de una respuesta inmunitaria. Además, estas células transfectadas
expresan el antígeno con las modificaciones translacionales y conformacionales
nativas, por lo tanto estimulan la producción de anticuerpos de alta
especificidad para el antígeno. Su acción imita lo que ocurre en un proceso
infeccioso o tumoral. Estos plásmidos pueden llevar genes que codifican las
secuencias de proteínas completas (Ulmer et al. 1993), o secuencias cortas, que
codifican únicamente las regiones de las proteínas con interés desde el punto de
vista inmunológico, que corresponden a los DTc y/o DTh (Ishioka et al. 1999;
Depla et al. 2008; Durantez et al. 2009). A pesar de que la cantidad de proteína
sintetizada a partir del vector plasmídico es relativamente pequeña, es
suficiente para desencadenar una respuesta inmunitaria eficaz, debido en parte
a las características inmunopotenciadoras intrínsecas del DNA, como las

Introducción
36
secuencias CpG (ligando de TLR9); (Klinman et al. 1996; Corr et al. 1997). La
existencia de dichas secuencias induce la producción de diferentes citocinas
pro-inflamatorias, como IFN-γ. Esto hace que una de las propiedades generales
de la vacunación con DNA desnudo sea la generación de respuestas
inmunitarias tipo Th1. Esta estrategia de vacunación presenta la eficacia de las
vacunas vivas convencionales con la seguridad de las vacunas inactivadas,
aunque existen algunos riesgos asociados como la posible integración del
plásmido en el genoma de la célula transfectada, inducción de anticuerpos
contra el DNA del plásmido y la inducción de procesos autoinmunes, entre
otros riesgos potenciales.
2.2. Proteínas recombinantes de fusión basadas en el dominio extra A de la
fibronectina (EDA)
El desarrollo de vectores o métodos de transporte y captación de
antígenos en las DC in vivo, constituye una estrategia alternativa en el diseño de
nuevas vacunas para potenciar la respuesta inmunitaria frente a tumores o
agentes infecciosos.
Un ejemplo es la incorporación de ligandos para los PRRs en las vacunas.
Con ello se promueve por un lado, la activación de la respuesta innata con la
producción de citocinas pro-inflamatorias y el aumento de moléculas de
coestímulo que permite inducir una respuesta adaptativa efectiva, y por otro
lado se induce el transporte y captación del antígeno a la DC, así como la
maduración de la DC (Kaisho and Akira 2002; Guy 2007; Werling and Jungi
2003). Varios grupos ya han contrastado esta hipótesis y han descrito que la
unión covalente de un antígeno a ligandos de TLR (por ejemplo, de TLR5 como
la flagelina (Cuadros et al. 2004) o de TLR9 como los CpGs (Tighe et al. 2000)) o
a otros receptores de las DC (como complejos peptídicos de las chaperonas a
través de los receptores Hsp (Delneste et al. 2002; Binder and Srivastava 2005)),
péptidos derivados de las modulinas de Staphylococcus epidermidis (Durantez et
al. 2010), proteínas de fusión recombinantes basadas en la adenilato ciclasa de
Bordetella pertussis que se unen al complejo CD11b/CD18 (Fayolle et al. 1999;

Introducción
37
Guermonprez et al. 2001), o anticuerpos monoclonales específicos de antígenos
superficiales de las DC (Bonifaz et al. 2004; Sancho et al. 2008) son capaces de
dirigir antígenos a las DC in vivo, de fomentar su presentación y, por tanto, de
favorecer la iniciación de una respuesta inmunitaria específica frente a esos
antígenos.
Buscando el mismo objetivo, y aprovechando su unión al receptor TLR4
y su capacidad de inducir citocinas pro-inflamatorias, en esta tesis hemos
seleccionado el domino extra A de la fibronectina (Okamura et al. 2001).
2.2.1. El domino extra A de la fibronectina (EDA)
La fibronectina es una glicoproteína presente en la matriz extracelular
(en su forma insoluble) y en los fluidos corporales (en su forma soluble) de los
vertebrados. Para ser funcional, forma dímeros consistentes en 2 subunidades
similares unidas por dos puentes disulfuro. Cada monómero es una molécula
larga y flexible con tres tipos de dominio homólogos repetidos conocidos como
I, II, y III. La fibronectina juega un papel muy importante en el mantenimiento
de la morfología normal de la célula, así como en la adhesión celular,
migración, diferenciación y proliferación (Kaspar et al. 2006). La forma soluble
incrementa la coagulación y la fagocitosis (Engvall and Ruoslahti 1977;
Bohnsack et al. 1985).
Esta proteína presenta distintas isoformas generadas por splicing
alternativo del RNAm. En humanos se han detectado hasta 20 de estas
isoformas. Estos splicing pueden ocurrir en tres regiones: el dominio extra A
(EDA), el dominio extra B (EDB) y la región variable IIICS. Las dos primeras
regiones o se incluyen o se excluyen totalmente. Sin embargo la tercera región
tiene varios sitios de splicing, pudiendo dar lugar a 5 variantes distintas (Figura
8).

Introducción
38
Figura 8. Representación de los dominios de la fibronectina (dominios extra A, B y IIICS).
El dominio EDA se encuentra elevado durante el desarrollo embrionario,
y disminuye sustancialmente tras el nacimiento y con la edad (Hynes 1990). Sin
embargo se ha descrito un aumento en su producción en circunstancias
concretas, como reparación tisular, fibrosis, angiogénesis, artritis reumatoide o
inflamación. Así, se ha empezado a estudiar el papel de EDA como marcador
en diversas enfermedades como tumores sólidos y metástasis (Rybak et al. 2007;
Villa et al. 2008), artritis reumatoide (Przybysz et al. 2009) o fibrosis (Hackl et al.
2010).
Por otro lado se ha descrito que tanto EDA como la fibronectina que
contiene EDA inducen una respuesta inmunitaria parecida a la observada con
LPS, en la que se observa una activación de TLR4, la inducción del factor de
transcripción NF-kB, la inducción de genes codificantes para citocinas pro-
inflamatorias, la liberación de proteoglicanos y la producción de las
metaloproteinasas de matriz 1, 2 y 9 (Saito et al. 1999). Todos estos resultados
sugieren que la producción local de EDA en respuesta a daño tisular u otros
daños podría inducir el reclutamiento de DC al sitio del daño y la posterior
maduración de estas DC.
Con el fin de dirigir el antígeno a las DC y así facilitar la inducción de
respuestas celulares in vivo, en trabajos anteriores hemos unido covalentemente
esta proteína a diferentes DTc bien caracterizados o a proteínas con varios
determinantes, tanto DTc como DTh (Lasarte et al. 2007; Mansilla et al. 2009;
Aranda et al. 2011) (Figura 9).

Introducción
39
TLR4
Célula T
MHC-IMHC-II CD54
CD80CD86
1. Unión2. Activación de la señalización TLR
3. Maduración de CD
4. Presentación antigénica
5. Activación célula TCD Madura
Nódulo linfoide
Citoquinas
Tejido
CD Inmadura
EDA-DTc
Figura 9. Esquema del proceso de activación de la respuesta inmunitaria adaptativa a través de
las proteínas de fusión EDA-DTc o EDA-proteína.
En este trabajo, con la idea de potenciar el efecto inmunomodulador de
EDA y aprovechar las propiedades pro-inflamatorias de las anafilotoxinas,
hemos construido proteínas de fusión donde añadimos covalentemente el
fragmento del complemento C5a al constructo EDA-DTc con el objetivo de
mejorar la respuesta celular in vivo. Posteriormente hemos estudiado su
potencial como adyuvante de vacunación.
2.3. Péptidos inhibidores de las células T reguladoras
La activación del sistema inmunitario es el resultado de un proceso muy
complejo de interacciones entre moléculas inmunoestimuladoras y moléculas
inmunosupresoras. Estas últimas pueden afectar enormemente a la
inmunogenicidad de una vacuna. Por este motivo, otro de los abordajes en el
desarrollo de vacunas radica en el diseño de inhibidores de los mediadores de
inmunosupresión. La inhibición de la función de las células Treguladoras
CD4+CD25+ en pacientes con cáncer es un paso esencial para mejorar la eficacia
de las terapias antitumorales, especialmente los enfoques basados en
inmunoterapia (Colombo and Piconese 2007).
Nuestro grupo ha desarrollado un péptido inhibidor de la molécula
FOXP3 con capacidad para reducir la función supresora de las células Treg
(Casares et al. 2010). En este trabajo pretendemos demostrar que la inhibición
de canales de Cl- mediante péptidos frente al intercambiador AE2, puede afectar
a la función supresora de las Treg, sin que las células T efectoras vean

Introducción
40
comprometidas sus funciones efectoras, nos basamos en los resultados descritos
por el grupo del Dr Medina que observan que ratones knockout para AE2
presentan un reducido número de Treg y elevado número de linfocitos T CD8+
(Salas et al. 2008).
2.4. Anafilotoxinas. Generalidades
Como se ha comentado anteriormente, durante la activación del sistema
del complemento se generan unos pequeños péptidos con actividad de
anafilotoxinas. El más potente es el fragmento C5a, seguido de C3a y con muy
poca actividad el fragmento C4a. Sus principales funciones son la activación de
células mieloides, quimiotaxis de los PMN, degranulación de mastocitos
(contracción de la musculatura lisa e incremento de la permeabilidad capilar) y
la potenciación de la vasodilatación que permite acelerar el paso del patógeno a
los ganglios con lo que se iniciará la respuesta adaptativa. Las propiedades pro-
inflamatorias de las anafilotoxinas, nos motivaron al estudio de su potencial
como adyuvantes de vacunas. A continuación, nos referiremos principalmente
al fragmento C5a.
2.4.1. Efecto biológico de C5a
C5a es una glicoproteína de 74 aminoácidos (≈ 11 kDa) con una
estructura terciaria característica formada por un haz de cuatro hélices α, es una
molécula bioactiva potente que puede actuar sobre una amplia variedad de
tipos celulares. Expresa una alta afinidad por los receptores transmembrana
C5aR/CD88 y C5L2 (anteriormente conocido como GPR77 o DST11).
La interacción del fragmento C5a con su receptor C5aR sobre los
neutrófilos, macrófagos y las células endoteliales da lugar a una serie de
eventos que inducen una respuesta inflamatoria localizada y contenida. Estos
eventos son estrictamente regulados y locales limitándose a una pequeña área
del tejido. C5a induce distintos tipos de respuesta en función de los diferentes
tipos celulares. Induce la vasodilatación y aumento del flujo sanguíneo a nivel
local, la contracción del músculo liso, edema, el aumento de la temperatura en

Introducción
41
el tejido y la acumulación de neutrófilos en los tejidos extravasculares (Marder
et al. 1985; Schumacher et al. 1991). Además, C5a tiene importantes funciones
de protección inmunológica al actuar en fagocitos como los neutrófilos y
macrófagos, potenciando la respuesta fagocítica cuando entran en contacto con
los PAMPs (patrones moleculares asociados a patógenos) y aumentando el
ensamblaje del sistema enzimático NADPH oxidasa, cuya actividad constituye
una de las fuentes endógenas más importantes de especies reactivas de oxígeno
en el organismo. C5a también induce quimiotaxis, los fagocitos se acumulan en
los sitios donde se da la producción local de C5a. En conjunto, C5a confiere una
mayor protección contra la presencia local de agentes infecciosos.
Otro efecto que hemos mencionado anteriormente es la capacidad que
tiene el fragmento C5a para conferir resistencia a la apoptosis tanto en
neutrófilos como en linfocitos T CD4+ (Lalli et al. 2008; Strainic et al. 2008;
Perianayagam et al. 2002).
Cuando los neutrófilos son sometidos a una situación de estrés como la
incubación in vitro a 37 ºC durante un tiempo prolongado se induce la apoptosis
celular pero si son expuestos a C5a o a otros mediadores como el factor
estimulante de colonias (G-CSF) se vuelven resistentes a la apoptosis. C5a activa
la vía de la fosfatidilinositol 3-quinasa (PI3K). El bloqueo de esta vía con el
inhibidor de PI3K elimina la resistencia de los neutrófilos a la apoptosis tras la
exposición a C5a (Perianayagam et al. 2002). Por lo tanto, C5a prolonga la vida
de los neutrófilos en los sitios de inflamación mejorando la protección frente a
agentes infecciosos. Sin embargo, la apoptosis de los neutrófilos es un
mecanismo importante para su eliminación y por lo tanto su regulación y en
definitiva la regulación de la respuesta inmunitaria. Si el mecanismo de la
apoptosis se ve disminuido, la vida útil de los neutrófilos se prolonga
implicando una respuesta inflamatoria exagerada.
Las células endoteliales también se ven influenciadas por la acción de la
anafilotoxina C5a. Estas células tienen un papel clave en la respuesta
inflamatoria, son activadas por los mediadores de la inflamación y tras su

Introducción
42
activación expresan moléculas en sus membranas que favorecen la adhesión de
los leucocitos y su posterior activación para la transmigración a tejidos
extravasculares. C5a induce la expresión de moléculas de adhesión como la P-
selectina en la membrana de las células endoteliales, así como también induce la
liberación del factor Von Willebrand (Schumacher et al. 1991). En la superficie
celular de los neutrófilos la P-selectina interactúa con su ligando, el glicolípido
P-selectina 1 (PSGL1), para iniciar el rodamiento de los leucocitos como los
neutrófilos. Este es la primera de una serie de interacciones adhesivas que
tienen lugar antes de que se produzca la transmigración de los leucocitos en los
tejidos extravasculares.
C5a también mejora la función de los agonistas de las células
endoteliales, como el LPS, causando aumento en la producción de la
interleucina IL-8 y de interleucina IL-6. Ambas citocinas pertenecen a la familia
de las quimiocinas y por lo tanto con funciones quimiotácticas (Riedemann et
al. 2002; Laudes et al. 2002; Hopken et al. 1996; Strieter et al. 1992). C5a también
puede inducir la expresión de su propio receptor C5aR en las células
endoteliales (Laudes et al. 2002).
En la Figura 10 mostramos un ejemplo de la señalización mediada por
C5a en los neutrófilos.
Figura 10. Señalización de C5a en neutrófilos. Peter A. Ward (Ward 2004).

Introducción
43
La unión C5a con su receptor da lugar a la activación de la enzima
adenilato ciclasa que cataliza la conversión de ATP a AMPc. Posteriormente
tiene lugar la activación de la fosfolipasa C (PLC), el AMPc activa la
fosfoquinasa A (PKA), que tiene algunas acciones reguladoras, inhibiendo las
moléculas efectoras de señalización mitogénica RAF1 y B-RAF. La fosfolipasa C
a su vez activa la proteína quinasa C y ésta activa fuertemente a RAF1 y B-RAF.
A continuación, tiene lugar la activación de MEK1, que induce la activación de
la MAP quinasa ERK1/2, esta última causa la fosforilación de las subunidades
citosólicas de la NADPH oxidasa. El resultado final de estos eventos es el
ensamblaje de la NADPH oxidasa.
El sistema de complemento puede modular la respuesta celular T
durante las fases de inducción, efectora y de contracción. Algunos datos
sugieren efectos pleomórficos y complejos del complemento en la producción
de citocinas clave para la diferenciación y función de las células T, actuando no
sólo en células T vírgenes sino también localmente en la sinapsis inmunológica
entre las células dendríticas y células T determinando el resultado de dicha
interacción. Es la señalización mediada por los receptores C3aR y C5aR la que
proporciona una señal de coestimulación que permite aumentar la activación y
la supervivencia de las células T.
Los ratones deficientes en el componente DAF, tras ser inmunizados,
presentan una mayor respuesta celular Th1 con una mayor secreción de
interleuquina IL-2 y de IFNγ y un descenso de los niveles de interleuquina IL-
10 (Lalli et al. 2008). También presentan mayores efectos adversos en patologías
autoinmunes como la esclerosis múltiple y la encefalomielitis autoinmune
experimental (Li et al. 2009). Además, muestran una mejora de la respuesta de
células T en un modelo de infección por la coriomeningitis linfocítica (LCMV)
(Fang et al. 2007). Mientras el mecanismo subyacente que contribuye a este
fenotipo queda por determinar. Una hipótesis es que, en ausencia de DAF, la
activación del complemento en las células presentadoras de antígeno y en las
células T se aumenta debido a una mayor producción local de anafilotoxinas.

Introducción
44
Otro efecto causado por fragmentos activados del complemento y
mencionado anteriormente es la inducción de la expansión de las células T por
inhibición de la apoptosis (Lalli et al. 2008; Strainic et al. 2008).
Se ha descrito que las células dendríticas deficientes en los receptores
C3aR o C5aR pierden su capacidad para inducir una potente respuesta celular
T, ya que ambos receptores contribuyen en la regulación de la maduración de
las células dendríticas y en la captación antigénica (Li et al. 2008; Peng et al.
2009).
A través de la interacción del fragmento C5a y su receptor C5aR
expresado en la membrana de las células dendríticas tiene lugar una
modulación directa de la activación de las células dendríticas como células
presentadoras de antígeno y activadoras de las células T. Los ratones deficientes
en el receptor C5aR o tratados con antagonistas de C5aR, presentan un fenotipo
de célula dendrítica menos activo con un aumento de la expresión de la citocina
inmunosupresora IL-10 y una disminución de la producción de la citocina pro-
inflamatoria IL-12p70 en respuesta a la estimulación con LPS. También se
observa un descenso en la expresión de marcadores de maduración como las
moléculas MHC de clase II y B7.2 y una capacidad reducida de estimular
células T aloespecíficas. Por otra parte, la estimulación del receptor C5aR media
la inhibición de la producción de AMPc y de la actividad de la proteína quinasa
A participando en la activación de la señalización NFкB y PI3K/AKT. Por lo
tanto, C5a actúa directamente sobre su receptor C5aR expresado en dendríticas
potenciando la capacidad de las células dendríticas para estimular la respuesta
celular T (Peng et al. 2009).
El receptor C5aR es necesario para una respuesta antiviral CD8+ (Kim et
al. 2004). Se ha demostrado que los ratones tratados con antagonistas del
receptor C5aR producen una menor respuesta de las células T CD8+ antígeno
específico frente a la infección por influenza tipo A (Kim et al. 2004). Esto
concuerda con la conclusión de que los ratones con deficiencia del factor DAF
presentan una mayor respuesta de las células T CD8+ antígeno específico tanto

Introducción
45
naїve como de memoria en respuesta a la infección por LCMV dependiendo de
la presencia de los receptores C3aR o C5aR (Fang et al. 2007).
Los efectos del complemento en las células T CD4+ vienen determinados
en función del entorno en el que se da la señalización. Un ejemplo es el caso del
asma alérgica, donde los componentes C3a y C5a respectivamente se
consideran mediadores pro-inflamatorios en el proceso de asma alérgica ya que
permiten el reclutamiento de células inflamatorias, inducen edema y la bronco
constricción. Sin embargo, una deficiencia en C5 produce una mayor
susceptibilidad en ratones a padecer asma experimental. Esto es debido a una
mayor sensibilización de las células Th2 en la primera exposición al alérgeno
inhalado en ausencia de señalización C5aR y mediado por las células
dendríticas. Por otro lado se ha descrito que un bloqueo de la señalización del
receptor C5aR durante el proceso inflamatorio suprime la sintomatología del
asma. Un tratamiento frente al asma mediante bloqueo del receptor C5aR
puede ser beneficioso pero a su vez podría inducir una sensibilización a nuevos
antígenos (Lambrecht 2006).
Algunos estudios describen que el sistema del complemento,
indirectamente y a través del componente C5a puede sinergizar con los
receptores TLR-4 para inducir una respuesta celular de tipo Th17, este podría
ser uno de los mecanismos que explicarían la inducción de procesos
autoinmunes por el complemento (Fang et al. 2009).
Por lo tanto, podemos concluir que el sistema del complemento modula
la intensidad y el fenotipo de la respuesta celular T.
2.4.2. Receptores del fragmento C5a
Hay descrito dos receptores para la anafilotoxina C5a, el receptor clásico
C5aR (CD88) que forma parte de la superfamilia de los receptores acoplados a
proteína G (Figura 11) y un receptor no acoplado a proteína G y descrito
posteriormente llamado C5L2 (GPR77). El receptor C5aR tiene una elevada
afinidad para C5a sin embargo esta afinidad es del orden de 10 a 100 veces
menor para el producto C5a desArg. Por otra parte, el receptor C5L2

Introducción
46
interacciona con C5a con una afinidad parecida a C5aR pero unas 20 veces
mayor con el producto C5a desArg. Asimismo, C5L2 también interacciona con
C3a y C3a desArg (Cain and Monk 2002; Scola et al. 2007).
2.4.2.1. Receptor C5aR (CD88)
El receptor C5aR se localiza principalmente en células mieloides,
incluyendo neutrófilos, monocitos, células dendríticas, macrófagos y
eosinófilos. Pero también se ha demostrado que se expresa en las células del
parénquima de varios órganos sólidos, incluyendo hepatocitos humanos,
células del epitelio alveolar de bronquios y pulmón, en músculo liso vascular de
pulmón y en las células endoteliales (Wetsel 1995). Los neutrófilos son las
células con una mayor densidad de expresión de C5aR, y por este motivo son
las células más sensibles al efecto de la anafilotoxina C5a.
Últimamente se ha descrito la expresión de C5aR en los linfocitos T
CD4+. La señalización C5aR en los linfocitos T parece ser importante para
inducir una correcta expansión de las células T efectoras, limitando la apoptosis
inducida por activación antigénica regulando la expresión de Bcl-2 y Fas (CD95)
(Lalli et al. 2008; Strainic et al. 2008). También hay trabajos que describen la
expresión de C5aR en células B.
A través de la interacción con el receptor C5aR (Figura 11), la
anafilotoxina C5a induce toda la señalización pro-inflamatoria descrita.

Introducción
47
Figura 11. Estructura molecular e interacción entre C5a y su receptor C5aR humano. Peter
A.Ward (Ward 2004).
2.4.2.2. Receptor C5L2 (GPR77)
Los estudios de bloqueo del receptor C5L2 así como en ratones knockout
han sugerido que el receptor C5L2 es importante en la regulación de la función
de C5a y C5a desArg, pudiendo actuar como receptor señuelo para la
señalización de C5a permitiendo la eliminación en el ambiente extracelular de
los fragmentos C5a (Okinaga et al. 2003; Scola et al. 2007).
Otros estudios indican una función de señalización para C5a distinta a la
mediada por el receptor C5aR (Gao et al. 2005; Gerard et al. 2005).
Así mismo, está descrito que pacientes que sobreviven a la sepsis
presentan mayores niveles de expresión del receptor C5L2 que aquellos que no
sobreviven a la sepsis (Huber-Lang et al. 2005). Este es un ejemplo de función
de señuelo del receptor C5L2. Sin embargo otros estudios de sepsis posicionan
este receptor C5L2 como un receptor que promueve la inflamación en situación
de sepsis ya que parece ser necesario para la inducción de la proteína HMGB1
(High mobility group box 1) a través del cual C5L2 sinergiza con C5aR para llevar
a cabo un proceso de inflamación letal durante la sepsis. Además, se postula
que C5L2 podría actuar como co-receptor en la activación del receptor TLR4
(Rittirsch et al. 2008). También está descrita la interacción de C5L2 con los

Introducción
48
fragmentos C3a y C3a desArg mediando la síntesis de triglicéridos en los
adipocitos (Kalant et al. 2005).
Podemos concluir que el receptor C5L2 presenta ambas funciones,
reguladoras como receptor señuelo de C5a y pro-inflamatorias sinergizando
con el receptor C5aR.
2.5. Adyuvantes
Las proteínas y los péptidos presentan en general una baja
inmunogenicidad, por lo que es necesario combinarlos con diversos adyuvantes
para inducir una respuesta inmunitaria más fuerte (Kim et al. 1998). Muchas de
las propiedades inmunopotenciadoras de los adyuvantes radican en su
capacidad de interactuar con las APC.
Como adyuvante hemos seleccionado el poly(I:C) (ácido
poliinosínico:policitidílico), que es un análogo sintético del RNA de doble
cadena capaz de unirse al TLR3, un receptor para RNA de doble cadena que
está expresado en células NK, macrófagos, DC (Banchereau and Steinman 1998)
y células T CD4+ (Gelman et al. 2004). Los mecanismos por los que el poly(I:C)
es capaz de mejorar las respuestas celulares no están bien definidos, aunque
parece ser que son parcialmente dependientes de las células NK, pero no NKT.
Esta molécula está asociada con una rápida activación de las células NK, que
contribuyen a la rápida inducción de citocinas pro-inflamatorias como IFN tipo
I, TNF-, IFN-, IL-12 e IL-15, y al aumento de la proliferación de las células T
CD8 (Salem et al. 2005). Además, el poly(I:C) puede madurar eficazmente las
DC (que también contribuyen a la producción de citocinas pro-inflamatorias) al
unirse a ellas a través de su TLR3 (Schwarz et al. 2003), y mejora las respuestas
T CD8 memoria y antitumorales (Salem et al. 2005; Zhang et al. 1998).
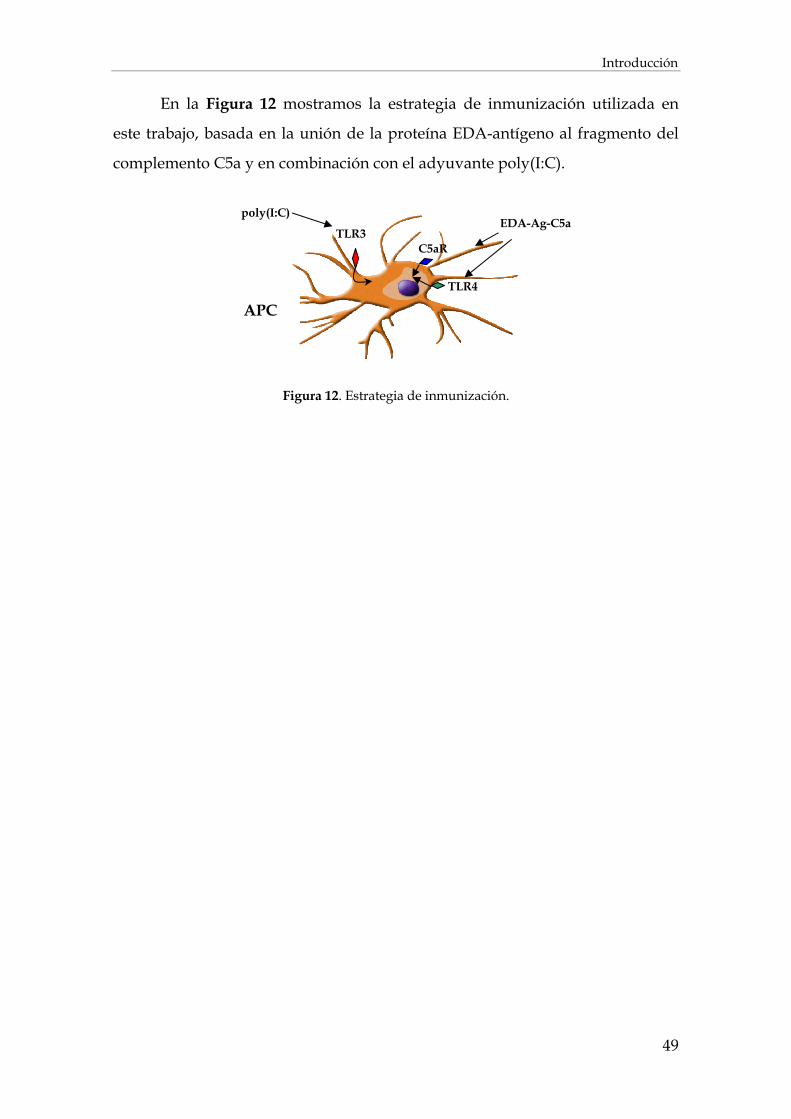
Introducción
49
En la Figura 12 mostramos la estrategia de inmunización utilizada en
este trabajo, basada en la unión de la proteína EDA-antígeno al fragmento del
complemento C5a y en combinación con el adyuvante poly(I:C).
Figura 12. Estrategia de inmunización.
TLR3
poly(I:C)
TLR4
EDA-Ag-C5a C5aR
APC


OBJETIVOS


Objetivos
53
Una de las características del sistema inmunitario es su sofisticación en el
control de la activación de una respuesta inmunitaria celular T. Así, existen una
serie de células y moléculas inmunopotenciadoras que pueden a su vez ser
controladas por células y moléculas inmunosupresoras. Las células dendríticas
son las células presentadoras de antígeno encargadas de la activación de los LT,
mientras que las células T reguladoras (Treg) presentan una actividad
inmunosupresora que, aunque es esencial para la protección frente a las
enfermedades autoinmunes, puede dificultar la activación de una respuesta
antiviral o antitumoral. Por lo tanto, planteamos dos estrategias para la
optimización de las estrategias de vacunación. La primera de ellas es el
desarrollo de inhibidores de las células T reguladoras que hacen de freno del
sistema inmunitario y la segunda es el desarrollo de moléculas que faciliten la
captura del antígeno por las células dendríticas y por tanto su
inmunogenicidad.
En este trabajo nos planteamos el abordaje de estas dos estrategias con
los siguientes objetivos:
1. Conocer la implicación de los canales de Cl- y Ca2+ en la activación
celular de los linfocitos T, así como en la función supresora de las células T
reguladoras. Diseñar péptidos inhibidores de los canales de Cl- AE2, a fin de
revertir la función supresora de las células T reguladoras.
2. Estudiar el potencial inmunomodulador in vivo de proteínas de fusión
formadas por las anafilotoxinas derivadas del complemento y el dominio EDA
de la fibronectina, unidos a un antígeno.


MATERIAL Y MÉTODOS


Material y métodos
57
2. Células
Todas las líneas celulares descritas en este apartado se cultivaron en estufas
incubadoras estándar a 37 ºC y con un 5% de CO2.
Las células tumorales E.G7-OVA (ATCC CRL-2113) derivan de la transfección de
la línea celular de timoma EL-4 con el DNA complementario de la proteína
ovoalbúmina (OVA) de la clara de huevo del pollo. Presentan la restricción H-2b y se
cultivan en medio RPMI-1640 (BioWhittaker, Bélgica) suplementado con 10% de
suero bovino fetal (FBS) (Invitrogen, Scotland, UK), antibióticos (penicilina 100
U/mL y estreptomicina 100 g/mL) y 510-5 M 2-mercaptoetanol. A partir de ahora
nos referiremos a esta formulación como medio completo (MC). El medio fue
suplementado con 400 μg/mL del antibiótico geneticina (G418, Invitrogen) para
seleccionar únicamente las células transfectadas con el plásmido que expresa la
proteína ovoalbúmina. Estas células se usaron en experimentos in vivo de protección
y tratamiento de tumores con EDA-SIINFEKL, EDA-SIINFEKL-C5a y SIINFEKL-
C5a.
Las células THP-1 (ATCC TIB-202), derivadas de monocitos humanos, se
cultivan en MC y se utilizaron para estudiar la capacidad de las proteínas EDA-
SIINFEKL, EDA-SIINFEKL-C5a y SIINFEKL-C5a para inducir la producción de
citocinas pro-inflamatorias.
La línea celular B3Z (cedida por la Dra. C. Leclerc, Institute Pasteur, Paris,
Francia) proviene de un hibridoma de células T CD8+ específico de SIINFEKL. Se
utilizaron para estudiar la presentación antigénica de EDA-SIINFEKL, EDA-
SIINFEKL-C5a y SIINFEKL-C5a.
Las células CTLL-2 (ATCC TIB-214) son una línea de células T citotóxicas de
ratón. Son dependientes de IL-2, por lo que se cultivan en MC suplementado con
esta citocina a una concentración de 10 U/mL. Se utilizaron para estudiar la
presentación antigénica de EDA-SIINFEKL, EDA-SIINFEKL-C5a y SIINFEKL-C5a.
Las células YAC-1 (ATCC TIB-160) (Manassas, UA) se utilizaron para estudiar la
actividad de las células NK.

Material y métodos
58
Las células HEK293 (Invitrogen, CA, EE.UU.) derivan de riñón embrionario
humano y están transfectadas establemente con la región E1 del adenovirus tipo 5
humano (Ad5). Se cultivaron en medio DMEM (BioWhittaker, Bélgica)
suplementado con 10% de suero bovino fetal (FBS) (Invitrogen, Scotland, UK),
antibióticos (penicilina 100 U/ml y estreptomicina 100 g/ml), 2 mM glutamina y
510-5 M 2-mercaptoetanol. Se utilizaron para comprobar la expresión de las formas
secretables de los plásmidos de expresión.
2. Ratones
Se utilizaron ratones de distintas cepas en función del antígeno inoculado en
la inmunización o del ensayo realizado. Todos ellos se mantuvieron con los
cuidados y condiciones adecuadas, siguiendo las normas institucionales requeridas
para la experimentación con animales.
Hembras de 6 semanas de edad de la cepa BALB/c (Harlan, Barcelona),
restricción H-2d.
Hembras de 6 semanas de edad de la cepa C57BL/6 (Harlan), restricción H-2b.
Ratones hembras TLR4-KO con fondo C57BL/6. Fueron cedidas por el Dr. M.
Chingard (Institute Pasteur, Paris, Francia).
Ratones macho C5aR-KO con fondo C57BL/6. Fueron cedidos por Rubén Pío
(Centro de Investigación Médica Aplicada, Pamplona, España).
3. Antígenos
3.1. Péptidos
3.1.1. Síntesis de péptidos
La síntesis de péptidos se realizó en un sintetizador manual múltiple de 9
pocillos diseñado en nuestro laboratorio (Borras-Cuesta et al. 1991), así como en uno
automático (Apex 396 Aapptec LLC; Louisville, KY, EE.UU.). En ambos casos se usó
el método de síntesis en fase sólida de Merrifield (Merrifield 1964), utilizando como
grupo protector temporal el fluorenilmetoxicarbonil (Fmoc). Los péptidos se

Material y métodos
59
sintetizaron unidos a una resina de poliestireno-divinilbenceno (p-Alkoxybenzyl
Alcohol resin, Sigma Chemical Co., St Louis, EE.UU.). El primer aminoácido estaba
unido directamente a la resina, cuyo grado de sustitución era diferente en función de
cual fuese ese aminoácido (en nuestros péptidos varió entre 0.5-0.8 mEq/g). Dicha
resina se colocó en los pocillos del sintetizador y los solventes (que generalmente
fueron dimetilformamida), se eliminaron mediante filtración y con ayuda de una
bomba de vacío. La síntesis se realizó desde el extremo carboxi terminal hasta el
extremo amino terminal, añadiendo aminoácidos acetilados (en el sintetizador
automático) o ésteres activos de los distintos aminoácidos (en el sintetizador
manual), con el extremo amino terminal protegido por el grupo temporal base lábil
Fmoc y las cadenas laterales protegidas por grupos ácidos lábiles (Bachem, Suiza).
En este proceso de síntesis peptídica, la adición de cada aminoácido a la
cadena peptídica supone tres pasos: la escisión del grupo protector Fmoc del último
aminoácido, la adición de un aminoácido activado y finalmente la monitorización de
la eficacia de unión mediante el ensayo de Kaiser o test de ninhidrina, que detecta
grupos amino libres. Este proceso de escisión del grupo Fmoc, adición de
aminoácido y monitorización, se repite tantas veces como aminoácidos forman la
secuencia del péptido.
Tras la desprotección del último aminoácido, el péptido es escindido del
soporte sólido mediante un tratamiento ácido, que permite cortar selectivamente el
péptido del agente de unión a la resina, manteniendo estable el enlace peptídico.
Además, el medio ácido también escinde los grupos protectores de las cadenas
laterales de los aminoácidos.
Al final de la síntesis, los péptidos desecados se disolvieron en agua Milli-Q
para su liofilización (Benchtop, The Virtis Company Inc., NY, EE.UU.). Una vez
liofilizados, los péptidos se conservaron a -20 ºC.
Los péptidos sintetizados se analizaron mediante cromatografía líquida de
alta presión (HPLC), utilizando el sistema Agilent G1311A (Agilent Technologies,
Santa Clara, CA, EE.UU.) y una columna Zorbax Eclipse XDB-C18, 5 µm, 4,6x150

Material y métodos
60
mm (Agilent Technologies). Sólo se utilizaron aquellos péptidos con una pureza
superior al 80%.
Los péptidos se disolvieron en PBS (Invitrogen, Paisley, Inglaterra) o medio
RPMI-1640 (BioWhittaker, Bélgica). Si la solubilidad del péptido no era muy alta, se
disolvió en dimetilsulfóxido (nunca superando el 20% p/v) y se llevó al volumen
final con PBS o RPMI-1640.
En la Tabla 5 se muestran las secuencias aminoacídicas de los diferentes
péptidos sintetizados frente al intercambiador AE2, diseñados en función de
posibles interacciones entre aminoácidos afines, utilizando matrices de
complementariedad y teniendo en cuenta el volumen de cada aminoácido.
Asimismo, nos basamos en la estructura del intercambiador AE2 y en la presencia
de bucles extracelulares que están implicados en su función.
Tabla 5. Secuencia aminoacídica de los diferentes péptidos sintetizados frente AE2.
Abreviatura péptido Proteína Secuencia
Mouse AE2-13 Bucle 3 del intercambiador AE2 KKKAGKKKKKKFFWA
Mouse AE2-14 Bucle 3 del intercambiador AE2 DSEAGSSSSSNMTVA
Mouse AE2-15 Bucle 3 del intercambiador AE2 RRRAGRRRRRRFFWA
Mouse AE2-16 Bucle 3 del intercambiador AE2 RRRAGRRRRRRFFVA
Mouse AE2-17 Bucle 3 del intercambiador AE2 KKKKKKFFWAFFILF
Mouse AE2-18 Bucle 3 del intercambiador AE2 SSSSSNMTVAFFILF
Mouse AE2-19 Bucle 3 del intercambiador AE2 KFFWAFFILFGKKKK
Mouse AE2-20 Bucle 3 del intercambiador AE2 NMTVAFFILFGRRRR
Mouse AE2-21 Bucle 3 del intercambiador AE2 FFILFGKKKKAKGKK
Mouse AE2-22 Bucle 3 del intercambiador AE2 FFILFGRRRRARGRR
Mouse AE2-23 Bucle 3 del intercambiador AE2 KKKKAKGKKGKKEGE
Mouse AE2-24 Bucle 3 del intercambiador AE2 RRRRARGRRGRRDGD
Mouse AE2-25 Bucle 4 del intercambiador AE2 KKFLFKELKFGKGLK
Mouse AE2-26 Bucle 4 del intercambiador AE2 RRFLFRELRFGRGLR
Mouse AE2-27 Bucle 4 del intercambiador AE2 KELKFGKGLKFFAGK
Mouse AE2-28 Bucle 4 del intercambiador AE2 RELRFGRGLRFFAGR
Mouse AE2-29 Bucle 4 del intercambiador AE2 GKGLKFFAGKEEGWL
Mouse AE2-30 Bucle 4 del intercambiador AE2 FFAGKEEGWLIDGLG

Material y métodos
61
Mouse AE2-31 Bucle 1 del intercambiador AE2 DFDKLIGL
Mouse AE2-32 Bucle 1 del intercambiador AE2 DTDKLIGL
Mouse AE2-33 Bucle 1 del intercambiador AE2 ETEKLIGL
3.1.2. Péptidos comerciales
El péptido SIINFEKL, determinante T citotóxico de la proteína ovoalbúmina
(OVA) que corresponde a la secuencia 257-264, fue sintetizado por la empresa
Neomps (Estrasburgo, Francia). Su pureza fue analizada por HPLC y fue superior al
90%.
3.1.3. Análisis de interacción biomolecular
Se confirmó la interacción del péptido AE2-17 con AE2, por el método de
resonancia de plasmones utilizando el ProteOn XPR36™ Protein Interaction Array
System (Biorad). El bucle 3 extracelular de AE2 (secuencia
DSEAGSSSSSNMTWATTILVPDNSSASGQSGQEKPR) biotinilado se inmovilizó a la
superficie de un chip NLC funcionalizado con neutravidina (Biorad). Como
controles negativos usamos PBS y un péptido biotinilado del factor de transcripción
NFAT. El péptido (5 μM) fue inyectado con PBS Tween 20 0.005% pH 7.4 a un flujo
de 50 μL/min. Los resultados obtenidos son normalizados con la señal obtenida en
la interacción con el péptido biotinilado de NFAT.
3.2. Plásmidos de expresión
3.2.1. Construcción
Los plásmidos pSHuttle-EDA-SIINFEKL, pSHuttle-C3d-EDA-SIINFEKL,
pSHuttle-C5a-EDA-SIINFEKL, pSHuttle-C3a-EDA-SIINFEKL y pSHuttle-C4a-EDA-
SIINFEKL que expresan versiones secretables de EDA-SIINFEKL, C3d-EDA-
SIINFEKL, C5a-EDA-SIINFEKL, C3a-EDA-SIINFEKL y C4a-EDA-SIINFEKL
respectivamente, se construyeron previo clonaje de los insertos diseñados en el
vector TOPOTA (Invitrogen, Carlsbad, CA, USA). Los insertos se sintetizaron por
PCR añadiendo la secuencia del péptido señal de la pre-pro-tripsina (HPPTLP) en el
extremo N terminal.

Material y métodos
62
Inicialmente, se amplificaron por transcripción inversa-PCR o RT-PCR los
distintos fragmentos del complemento utilizando cebadores específicos (Tabla 6) y
partiendo de muestras de RNA de hepatocito de ratón tratado con ConA. Las
muestras de RNA se extrajeron según el método Chomczynski y Sacchi
(Chomczynski and Sacchi 1987), partiendo de tejido hepático homogeneizado y
lisado en Ultraspec (Biotech, Houston, TX, EE.UU.) empleando Ultra Turrax Driver
T.25 (Jankel and Kunkel, Ika Labortechnik, Staufen,Alemania).
Paralelamente amplificamos por PCR la secuencia EDASIINFEKL utilizando
cebadores específicos y partiendo del plásmido pET20b-EDA-SIINFEKL que expresa
la secuencia EDASIINFEKL (Tabla 6).
Los productos amplificados por PCR se clonaron en el vector pCR2.1-TOPO
(Invitrogen, Carlsbad, CA, USA), utilizando el kit de clonaje TOPO TA (Invitrogen)
(Tabla 8).
Posteriormente se subclonó la secuencia EDASIINFEKL del vector pCR2.1-
TOPO en el plásmido de expresión pShuttle (Clontech, BD Biosciences, NJ, EE.UU.)
(Tabla 8). Para ello, se realizó la digestión del vector con las enzimas de restricción
NheI y Afl II (BioLabs) para extraer la secuencia EDASIINFEKL y poder hacer la
ligación de este inserto con el vector pShuttle previamente digerido con las mismas
enzimas NheI y Afl II (BioLabs).
Los distintos fragmentos del complemento clonados en el vector TOPOTA se
subclonaron en el plásmido pShuttle-EDASIINFEKL, para ello se realizó la digestión
de los plásmidos pCR2.1-TOPO y el plásmido pShuttle-EDASIINFEKL con la
enzima de restricción Kpn1 (New England BioLabs), posteriormente se llevó a cabo
la ligación de los insertos en el plásmido digerido pShuttle-EDASIINFEKL.
Todas las secuencias obtenidas fueron verificadas por secuenciación de DNA,
por el servicio de secuenciación del Centro de Investigación Médica Aplicada
(CIMA).
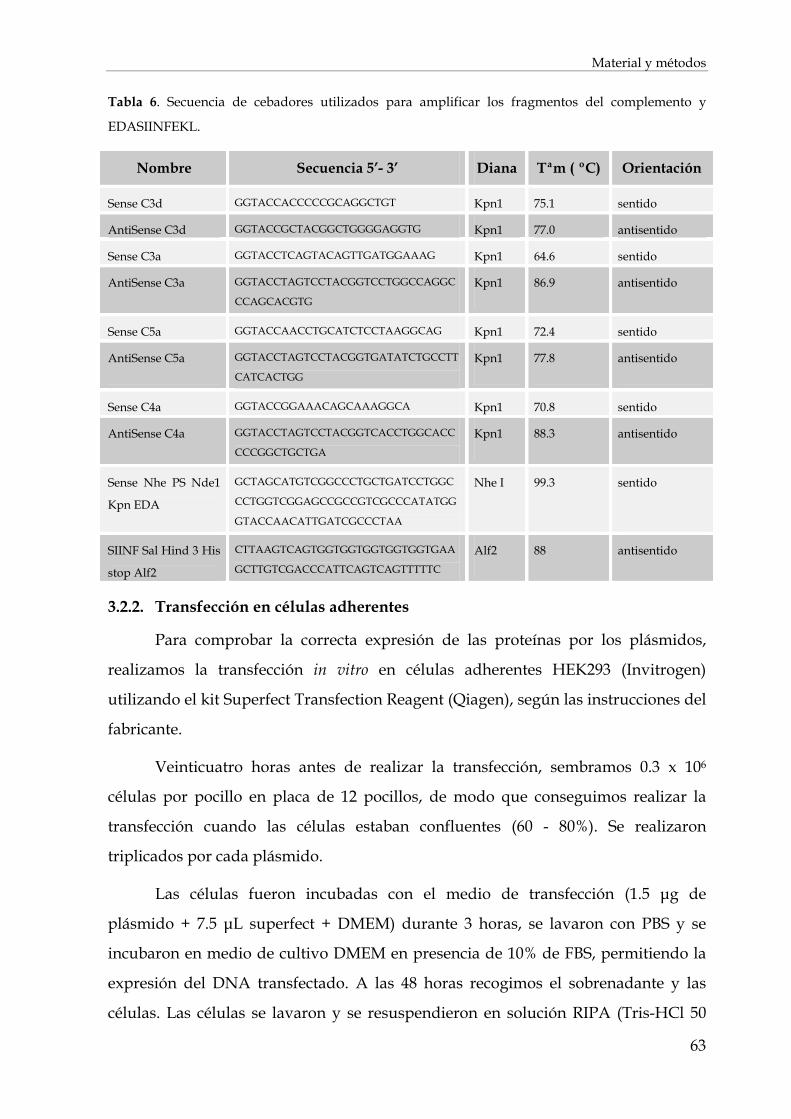
Material y métodos
63
Tabla 6. Secuencia de cebadores utilizados para amplificar los fragmentos del complemento y
EDASIINFEKL.
Nombre Secuencia 5’- 3’ Diana Tªm ( ºC) Orientación
Sense C3d GGTACCACCCCCGCAGGCTGT Kpn1 75.1 sentido
AntiSense C3d GGTACCGCTACGGCTGGGGAGGTG Kpn1 77.0 antisentido
Sense C3a GGTACCTCAGTACAGTTGATGGAAAG Kpn1 64.6 sentido
AntiSense C3a GGTACCTAGTCCTACGGTCCTGGCCAGGC
CCAGCACGTG
Kpn1 86.9 antisentido
Sense C5a GGTACCAACCTGCATCTCCTAAGGCAG Kpn1 72.4 sentido
AntiSense C5a GGTACCTAGTCCTACGGTGATATCTGCCTT
CATCACTGG
Kpn1 77.8 antisentido
Sense C4a GGTACCGGAAACAGCAAAGGCA Kpn1 70.8 sentido
AntiSense C4a GGTACCTAGTCCTACGGTCACCTGGCACC
CCCGGCTGCTGA
Kpn1 88.3 antisentido
Sense Nhe PS Nde1
Kpn EDA
GCTAGCATGTCGGCCCTGCTGATCCTGGC
CCTGGTCGGAGCCGCCGTCGCCCATATGG
GTACCAACATTGATCGCCCTAA
Nhe I 99.3 sentido
SIINF Sal Hind 3 His
stop Alf2
CTTAAGTCAGTGGTGGTGGTGGTGGTGAA
GCTTGTCGACCCATTCAGTCAGTTTTTC
Alf2 88 antisentido
3.2.2. Transfección en células adherentes
Para comprobar la correcta expresión de las proteínas por los plásmidos,
realizamos la transfección in vitro en células adherentes HEK293 (Invitrogen)
utilizando el kit Superfect Transfection Reagent (Qiagen), según las instrucciones del
fabricante.
Veinticuatro horas antes de realizar la transfección, sembramos 0.3 x 106
células por pocillo en placa de 12 pocillos, de modo que conseguimos realizar la
transfección cuando las células estaban confluentes (60 - 80%). Se realizaron
triplicados por cada plásmido.
Las células fueron incubadas con el medio de transfección (1.5 μg de
plásmido + 7.5 μL superfect + DMEM) durante 3 horas, se lavaron con PBS y se
incubaron en medio de cultivo DMEM en presencia de 10% de FBS, permitiendo la
expresión del DNA transfectado. A las 48 horas recogimos el sobrenadante y las
células. Las células se lavaron y se resuspendieron en solución RIPA (Tris-HCl 50

Material y métodos
64
mM pH 7.4, Igepal CA-630 0.5%, EDTA 5 mM, NaCl 250 mM, NaF 50 mM, Na3VO4
0.1 mM) suplementado con PMSF 1 mM (SIGMA) y una dilución 1:100 de inhibidor
de proteasas (SIGMA). Las células se incubaron durante 30 minutos a 4ºC,
posteriormente fueron sometidas a un proceso de sonicación y finalmente se
centrifugaron durante 10 minutos a 13000 rpm y 4ºC y se recogió los sobrenadantes
para cuantificar las proteínas por el método BCA y congelarlas a -20ºC.
3.2.2.1. Western blot
Para comprobar la expresión de los plásmidos en las células HEK293 se
realizó la detección de las proteínas secretables mediante la técnica de Western blot.
Los extractos celulares se sometieron a electroforesis en un gel de acrilamida-
SDS al 15% (véase el apartado 3.3.3.1). Posteriormente se realizó la transferencia de
las proteínas separadas en la electroforesis en un soporte de nitrocelulosa (Bio-Rad,
Alemania) a partir del gel de acrilamida. El tampón de transferencia utilizado fue
Tris-HCl 0.25 M, glicina 1.92 M y metanol al 20%(v/v), pH 8.3. La transferencia se
realizó a un voltaje fijo de 100V y a una intensidad variable de 20- 30 mA durante 40
minutos.
Posteriormente realizamos el inmunoblot que consiste en cuatro pasos. El
primero es bloquear la membrana de nitrocelulosa (PBS con 5% de leche y 0.05%
Tween-20) durante 1 hora a temperatura ambiente, permitiendo el bloqueo de las
uniones inespecíficas. Posteriormente incubamos el anticuerpo primario anti-EDA
(dilución 1/2000 en PBS 1X con 5% leche) durante 2 horas, seguido de 5 lavados de
10 minutos con PBS Tween-20 0.05% (v/v) para eliminar el exceso de anticuerpo. El
siguiente paso es la incubación del anticuerpo secundario unido a peroxidasa anti-
rabbit IgG HRP-linked (Cell Signaling) durante 1 hora. Tras 5 lavados de 10 minutos
con PBS y Tween-20 0.05% (v/v) para eliminar el exceso de anticuerpo, se incuba la
membrana durante 5 minutos con la solución que contiene los sustratos de la
reacción enzimática (ECL PLUS Western-Blot KIT, Amersham) y se pone en contacto
con la película fotográfica. Después de una exposición se procede al revelado
utilizando el revelador Curix60, AGFA.

Material y métodos
65
3.3. Proteínas recombinantes de fusión
3.3.1. Construcción
Para la obtención de la proteína C5a-EDA-SIINFEKL, el inserto C5a-EDA-
SIINFEKL del plásmido pShuttle-C5a-EDA-SIINFEKL fue subclonado en el
plásmido pET20b (Novagen, Darmstadt, Alemania), que permite la expresión de
proteínas de fusión con una cola de 6 histidinas en el extremo C-terminal (Tabla 8).
Ambos plásmidos fueron digeridos con las enzimas de restricción NdeI y HindIII.
Para obtener la proteína EDA-SIINFEKL-C5a, inicialmente se amplificó la
secuencia de C5a flanqueada con la enzima SalI partiendo del clon C5a-pCR2.1-
TOPO que previamente habíamos clonado. Para realizar esta amplificación
utilizamos cebadores que contienen la secuencia de SalI, así como una secuencia
espaciadora (GS(G4S)2GS) en el cebador antisentido (Tabla 7). Esta secuencia
espaciadora permite mayor flexibilidad en el plegado de la proteína. Posteriormente
se realizó la digestión del vector pShuttle EDA-SIINFEKL con la enzima SalI para
realizar la clonación. Una vez confirmada la clonación, el fragmento EDA-
SIINFEKL-C5a se subclonó en el plásmido pET20b.
La proteína SIINFEKL-C5a se obtuvo amplificando la secuencia
SIINFEKL-C5a, utilizando el vector C5a pCR2.1-TOPO obtenido previamente y la
secuencia SIINFEKL presente en el cebador sentido. Utilizando como cebador
antisentido un fragmento de la secuencia de C5a (Tabla 7). El inserto amplificado
SIINFEKL-C5a se clonó en el vector TOPO TA, para posteriormente insertar el
fragmento SIINFEKL-C5a en el vector pShuttle EDA-SIINFEKL que previamente
digerimos con los mismos enzimas de restricción NdeI y AflII para extraer la
secuencia EDA-SIINFEKL y sustituirla por la secuencia SIINFEKL-C5a. Una vez
obtuvimos el vector pShuttle SIINFEKL-C5a el inserto se subclonó en el vector de
expresión pET20b.
Para construir la proteína EDA-SIINFEKL se amplificó el cDNA resultante de
la RT-PCR de muestras de RNA de hepatocito de ratones tratados con ConA,
utilizando los cebadores EDAs y EDA-SIINFEKLas (Tabla 7). Los productos
resultantes se clonaron en pCR2.1 TOPO usando el mismo kit de clonado. Estos
Material y métodos
66
plásmidos fueron digeridos con NdeI y NotI, los fragmentos obtenidos fueron
subclonados en el plásmido pET20b.
Todos los constructos fueron verificados por secuenciación de DNA.
Una vez obtenidos y confirmados todos los plásmidos, fueron transfectados
en bacterias BL21(DE3) para la expresión de la proteína recombinante.
Tabla 7. Secuencias de los cebadores utilizados para la construcción de las diferentes proteínas de
fusión.
Nombre Secuencia 5’- 3’ Diana Tªm ( ºC) Orientación
NdeI SIINFEKLC5a CATATGCAGCTTGAGAGTATAATCAACTT
TGAAAAACTGAATGGAACCTGCATCTCCT
AAGGCAG
NdeI 85.2 sentido
C5a Stop AflII CTTAAGTCATATATCTGCCTTCATCACTGG AflII 67.0 antisentido
Sense SalI C5a GTCGACGGGTCGAACCTGCATCTCCTAAG
GCAG
SalI 82.5 sentido
Antisense C5a linker
XbaiI SalI
GTCGACTCTAGACGACCCTGATCCTCCTCC
TCCTGATCCTCCTCCTCCCGACCCGATATCT
GCCTTCATCACTGG
SalI 94.1 antisentido
EDAs CCATATGAACATTGATCGCCCTAAAGGAC
T
KpnI 85 sentido
EDASIINFEKLas AGCGGCCGCCCATTCAGTCAGTTTTTCAA
AGTTGATTATACTCTCAAGCTGTGTGGACT
GGATTCCAATCAGGGG
AlfI 93 antisentido
Tabla 8. Mapa de los vectores utilizados.
Nombre Características Mapa
pCR 2.1-TOPO
Vector de clonaje para productos de PCR por
el método TA.
Plásmido lineal con los extremos activados
para la unión covalente de la Topoisomerasa I.
Contiene múltiples lugares de restricción para
facilitar el clonaje.
Posee resistencia a ampicilina y a kanamicina
para su selección en bacterias.

Material y métodos
67
Nombre Características Mapa
pShuttle2
Vector de expresión con promotor de CMV,
que permite niveles altos de expresión y de
manera constitutiva del gen de interés.
Expresión en gran variedad de células
eucariotas.
Posee resistencia a kanamicina para su
selección en bacterias.
Nombre Características Mapa
pET20b
Vector de expresión en células procariotas.
Contiene múltiples lugares de restricción.
Contine el Tag C-Terminal con His. Para
facilitar la purificación de la proteína en
resinas de níquel (purificación por afinidad).
Posee resistencia a ampicilina para su
selección en bacterias.
3.3.2. Purificación
Las bacterias transformadas se crecieron a 37ºC en medio LB. Cuando el
cultivo alcanzó una densidad óptica de 0.5, se indujo la expresión de las proteínas
agregando al medio 4 mL/L de IPTG (SIGMA, Alemania) a una concentración de
100 mM (400 μM, concentración final de IPTG). En el caso de las proteínas
purificadas de fracción soluble se hizo una incubación de 16 horas a temperatura
ambiente en agitación. En el caso de las obtenidas a partir de cuerpos de inclusión la
incubación fue de 3 horas a 37 ºC. Posteriormente se centrifugó el cultivo a 8000
rpm. El precipitado se resuspendió en 8 mL de Tris-HCl 0.1 N.
A la hora de lisar las bacterias, se trató el extracto bacteriano con anti-
proteasas (Complete Protease Inhibitor Cocktail de Roche, Alemania) y 1.2 L/L de
lisozima (Novagen). Se incubó durante 15 minutos a 30 ºC, se pasó por la prensa de
French a 2000 psi de presión y se centrifugó el extracto a 11000 rpm. Las proteínas de
fracción soluble se purificaron a partir del sobrenadante y las proteínas de cuerpos

Material y métodos
68
de inclusión del precipitado, que se resuspendió en una solución de urea a una
concentración 8 M, a la que se la añadió HEPES-Na a una concentración de 20 mM.
En todo el proceso de purificación de las proteínas de fracción soluble se
utilizó el PBS como base de las soluciones requeridas en las diferentes técnicas. Sin
embargo, en la purificación de las proteínas extraídas de cuerpos de inclusión, la
base de todas las soluciones fue urea 8 M, HEPES-Na 20 mM.
A la hora de purificar las proteínas se utilizaron diversas técnicas. Todas estas
técnicas, a excepción del isoelectroenfoque y el detoxificado manual, se realizaron en
una plataforma FPLC Äkta (Pharmacia, GE Healthcare, EE.UU.).
Esta plataforma cuenta con dos bombas para la inyección de soluciones.
Además tiene un sistema de inyección de muestra que permite cargar desde 100 μL
hasta 50 mL. La plataforma cuenta con diferentes columnas intercambiables,
dependiendo del tipo de purificación que se vaya a realizar. Este equipo tiene
además un lector de UV (280 nm) que monitoriza a cada momento la absorbancia de
la muestra a la salida de la columna. Finalmente el equipo cuenta con un recolector
de muestras.
El aparato se puede obstruir fácilmente, por lo que todas las soluciones y las
muestras que se utilizan en las purificaciones fueron previamente filtradas con un
filtro Millex de 0.8 m de tamaño de poro (Millipore, MA, EE.UU.) en el caso de las
muestras, o un filtro Express PLUS de tamaño de poro 0.22 m (Millipore) para las
soluciones.
Las diferentes técnicas utilizadas fueron:
3.3.2.1. Purificación por afinidad
Dado que las proteínas diseñadas poseen una cola de histidinas en su
extremo C-terminal, utilizamos las columnas Histrap HP (GE Healthcare) para su
purificación por cromatografía de afinidad.
Las columnas Histrap HP (GE Healthcare) están empaquetadas con una
resina de Ni2+ (Ni Sepharose High Performance), el níquel es un metal con gran
afinidad por el aminoácido histidina, al igual que por el imidazol. Por lo tanto las

Material y métodos
69
proteínas con una cola de histidinas son capaces de quedarse unidas a la columna
Histrap HP. El imidazol se utiliza para competir en la unión de la proteína a la
columna, en presencia de imidazol en la solución, la unión níquel-proteína está
desfavorecida. A mayor concentración de imidazol, menor cantidad de proteína se
une al níquel. Con el fin de evitar uniones inespecíficas de proteínas en las
columnas, las muestras se inyectaron con una pequeña cantidad de imidazol, en un
rango entre 0 y 100 mM de imidazol en función de la afinidad de la proteína por el
níquel. La proteína se recuperó mediante la elución con una solución con 500 mM
imidazol.
3.3.2.2. Intercambio iónico
Esta técnica permite separar y por lo tanto purificar proteínas en función de
su carga neta. Las proteínas dependiendo del pH en el que se encuentran tienen
diferente carga, por lo que se unen con diferente afinidad a una columna cargada.
Esta columna puede contener aniones o cationes. El pI de la proteína a purificar es lo
que determina el tipo de intercambio: si el pH utilizado es mayor que el pI se realiza
un intercambio aniónico y si es menor, uno catiónico. Por otro lado, la diferencia
entre el pH y el pI debe ser como mínimo de 1.5-2 unidades.
Para eluir las proteínas retenidas en la columna se sube la fuerza iónica de la
fase móvil, aumentando la concentración de sal (NaCl), de esta forma se eluyen
primero las proteínas más débilmente retenidas y cuando la fuerza iónica sea mayor
saldrán las proteínas más cargadas y por lo tanto más retenidas.
En nuestro caso, realizamos un intercambio aniónico para purificar EDA-
SIINFEKL y un intercambio catiónico para purificar SIINFEKL-C5a. Para purificar la
proteína SIINFEKLC5a recogimos la fracción no retenida en columna. En ambos
casos usamos la columna HI TRAP DEAE FF (GE Healthcare).
Para eluir la muestra se utilizó una solución con NaCl 0.5 M.
3.3.2.3. Replegado y detoxificado
Las proteínas extraídas a partir de cuerpos de inclusión deben ser replegadas
para recuperar su actividad biológica.

Material y métodos
70
El replegado se realiza en columna cromatográfica mediante la realización de
un gradiente decreciente en concentración de urea. Las columnas utilizadas fueron
HiTrap™ Desalting Column (GE Healthcare). El fundamento de esta técnica, es que
la proteína avanza a través de una columna equilibrada con el tampón de replegado
consistente en 50 mM Tris-HCl con 1 mM glutatión reducido y 1 mM glutatión
oxidado, en la que previamente se ha hecho un gradiente decreciente de urea (desde
8 M a 0 M) en la cabeza de la columna. Al atravesar el gradiente, la proteína va
pasando lentamente de una solución de urea, a una solución Tris, favoreciendo el
replegado de la proteína.
Con estas columnas se realizó además del replegado de la proteína el
desalado de la misma. La proteína procedente de fracción soluble, EDA-SIINFEKL,
se desaló utilizando las columnas PD-10 (GE Healthcare).
En los casos en los que se necesitó concentrar la proteína antes de proceder al
siguiente paso de purificación, se utilizaron tubos de concentración por
centrifugación Amicon Ultra 4-5000 MWCO (Millipore).
Para finalizar la purificación de las proteínas, se procedió al detoxificado.
Para ello se emplearon las columnas Endotrap (Profos, Alemania) siguiendo las
instrucciones del fabricante. Para comprobar que realmente se habían eliminado las
endotoxinas de las proteínas se realizó un ensayo con el kit Quantitative
Chromogenic Limulus Amebocyte Lysate (Cambrex, IA, EE.UU.). Se consideró un
nivel de endotoxinas aceptable cuando las muestras no superaron 0.1 unidades de
endotoxina por μg de proteína.
Dependiendo de las características de cada proteína se utilizó un protocolo
distinto. En la Tabla 9 se resumen los diferentes protocolos.

Material y métodos
71
Tabla 9. Resumen de los protocolos seguidos para la purificación de las diferentes proteínas.
Proteína Tipo de purificación Soluciones utilizadas BA: PBS + 20 mM imidazol (pH 7,2)
Afinidad BB: PBS + 500 mM imidazol (pH 7,2) BA: Tris + 20 mM imidazol + 0,1 M NaCl (pH 8,28)
Intercambio aniónico BB: Tris + 20 mM imidazol + 0,5 M NaCl (pH 8,28)
EDA-SIINFEKL Fracción soluble
Desalado y detoxificado
BA: PBS + 60 mM imidazol (pH 7,2) Afinidad BB: PBS + 500 mM imidazol (pH 7,2)
Replegado 50 mM Tris-HCl + 1 mM glutatión oxidado + 1 mM glutatión reducido
C5a-EDA-SIINFEKL Cuerpos de inclusión
Detoxificado
BA: Urea + 60 mM imidazol (pH 7,2) Afinidad
BB: Urea + 500 mM imidazol (pH 7,2)
Replegado 50 mM Tris-HCl + 1 mM glutatión oxidado + 1 mM glutatión reducido
EDA-SIINFEKL-C5a Cuerpos de inclusión
Detoxificado
BA: Urea + 60 mM imidazol (pH 7,2) 8 M urea-20 mM HEPES + 0.4 % TritonX + 0.4% Tween 20
Afinidad
BB: Urea + 500 mM imidazol (pH 7,2)
BA: Urea + 20 mM Hepes (pH 5)
Intercambio catiónico BB: Urea + 20 mM Hepes + 0.5 M NaCl (pH 8)
Replegado 50 mM Tris-HCl + 1 mM glutatión oxidado + 1 mM glutatión reducido
SIINFEKL-C5a Cuerpos de inclusión
Detoxificado
El rendimiento de cada una de las producciones dependió de la proteína,
aunque en términos generales osciló entre 1-10 mg por litro de cultivo bacteriano.
3.3.3. Análisis de la pureza
Después de cada paso de purificación y también al final de la misma se
realizaron análisis de la pureza de la proteína mediante SDS-PAGE y tinción con
Azul de Coomassie.
3.3.3.1. Geles SDS-PAGE y tinción con Azul de Coomassie
Las muestras de proteínas se sometieron a electroforesis en un gel de
acrilamida-SDS al 15%, en el que también se cargó un marcador de peso molecular,
el RainbowTM Coloured Protein Molecular Weight Marker (Amersham Biosciences,
Uppsala, Suecia). El tampón utilizado fue Tris-HCl 25 mM, glicina 192 mM, SDS

Material y métodos
72
0.1%, pH 8.3. La electroforesis se realizó a un voltaje fijo de 90 V y a una intensidad
variable de 20-30 mA, durante 2 horas aproximadamente.
A continuación se lavó el gel 3 veces con H2O destilada, y se procedió a su
tinción con Bio-Safe Coomassie (Bio-Rad) durante 30 minutos. Una vez transcurrido
este tiempo se lavó el gel con H2O destilada. Los geles teñidos fueron desecados en
un desecador (Bio-Rad).
3.3.4. Ensayos de actividad
3.3.3.1. Análisis de activación de monocitos
Para comprobar que las proteínas purificadas eran capaces de activar a
células TLR4+ para la producción de citocinas pro-inflamatorias, se utilizó la línea
celular THP-1. Se incubaron las células THP-1 (2x105 células/pocillo) con las
diferentes proteínas a una concentración de 1 μM, 0.1 g/mL de LPS (Sigma) como
control positivo o MC como control negativo. A las 16 horas de cultivo, se
recogieron los sobrenadantes y se midió la producción de TNF-α humano mediante
un ensayo de ELISA comercial (OptEIA ELISA Set, BD Biosciences), de acuerdo con
las instrucciones del fabricante.
3.3.4.2. Análisis de maduración de células dendríticas
3.3.4.2.1. Diferenciación DC
Para la generación de DC de ratón a partir de precursores de médula ósea, se
extrajeron las médulas del fémur y la tibia, y se lisaron los eritrocitos utilizando una
solución de lisis ACK (NH4Cl 0,15 M, KHCO3 10 mM, Na2EDTA 0,1 mM) durante 1
minuto. A continuación, se realizó un lavado con RPMI 1640 y se procedió a
eliminar los linfocitos y granulocitos mediante lisis por complemento. Para ello, las
células se incubaron con una mezcla de anticuerpos frente a las distintas poblaciones
celulares y complemento de conejo:
Anticuerpo anti-CD4. Se obtuvo a partir del hibridoma GK1.5 (ATCC). El
anticuerpo así obtenido se añadió a una concentración de 100 μg/mL.

Material y métodos
73
Anticuerpo anti-CD8. Se obtuvo a partir del hibridoma de rata H35.17.2
(ATCC)(Pierres et al. 1982). También se utilizó a 100 μg/mL.
Anticuerpo Ly-6G/Gr-1 (BD Biosciences). Se usó a 10 μL/mL. Este anticuerpo
se utilizó para eliminar los neutrófilos y granulocitos.
Anticuerpo anti-CD45R/B220 (BD Biosciences). Empleado a 50 μL/mL. Este
anticuerpo se utilizó para eliminar los linfocitos B
Complemento de conejo (SIGMA). Se utilizó a 50 μg/mL.
Esta mezcla se incubó en una estufa a 37˚C durante 50 minutos, agitando cada
20 minutos. Tras la depleción se realizó un lavado, y las células que quedan se
cultivaron a una concentración de 106 células/mL en placas de 12 o 96 pocillos
(Iwaki, Japón) en MC suplementado con 20 ng/mL de GM-CSF e IL-4 (ambos de
Peprotech, Londres, Reino Unido) en estufas incubadoras estándar a 37 ºC y con un
5% de CO2. Los días 2 y 5 se reemplazaron dos terceras partes del medio por MC
fresco suplementado con las citocinas en la misma concentración. A día 6 se
añadieron los estímulos de maduración, 0.1 μg/mL de lipopolisacárido (LPS de
Escherichia coli 0111:B4; SIGMA) ó 500 nM de las proteínas.
Pasadas 24 horas, se recogieron las células para analizar por citometría de
flujo el nivel de expresión de marcadores de maduración, medir la producción de
citoquinas, o hacer ensayos de presentación antigénica.
3.3.4.2.2. Análisis in vitro de la maduración de las células dendríticas
Las células dendríticas se crecieron a partir de células de médula ósea. Tal y
como está descrito en el apartado anterior (3.3.4.2.1.). Las células dendríticas
resultantes se cultivaron en presencia o ausencia de las proteínas EDA-SIINFEKL
(500 nM), EDA-SIINFEKL- C5a (1 μM), SIINFEKL- C5a (1 μM), LPS (0.1 μg/ml)
como control positivo y medio de cultivo como control negativo. Tras 24 h de cultivo
se analizó la expresión de moléculas de coestímulo por citometría de flujo y la
producción de citocinas mediante el uso de una matriz comercial que contenía
anticuerpos (Raybio mouse citokine antibody array (RayBiotech)), de acuerdo con
las instrucciones del fabricante.

Material y métodos
74
3.3.4.2.2.1. Expresión de marcadores de maduración
Al tratarse de una población homogénea donde la mayoría de las células son
CD11c+, se realizó un marcaje simple con el marcador que se quiere analizar.
Los anticuerpos que se utilizaron fueron: anti-CD40 (clon 3/23), anti-CD54
(clon 3E2), anti-CD80 (clon 16-10A1), anti-CD86 (clon GL1), anti-I-Ab (clon AF6-
120.1) anti-H-2Kb (clon AF6-88.5), anti-CD11c (clon HL3) marcados en verde con
fluorescein isothiocyanate (FITC) (todos de BD Biosciences). Además se incubaron
las células con un isotipo marcado con FITC (también de BD Biosciences) como
control para eliminar la unión inespecífica de los anticuerpos. El marcaje se realizó a
4ºC en PBS con 2% de FBS. Tras 15 minutos, las células se lavaron y se analizó la
expresión de los distintos marcadores de superficie mediante citometría de flujo
(BD, FACSCalibur TM).
3.3.4.2.2.2. Producción de citocinas
Transcurridas 24h de incubación con los diferentes estímulos se recogió el
sobrenadante y se midieron distintas citoquinas mediante un Array comercial
(RayBiotech) de acuerdo con las instrucciones del fabricante. Los resultados se
expresaron como unidades relativas cuantificando la señal por densitometria.
3.3.4.3. Presentación antigénica
Para estudiar la capacidad de la proteína EDA-SIINFEKL-C5a de ser
procesada y presentada por la vía de señalización del MHC de clase I, quisimos
estudiar si la expresión de TLR4 y C5aR en las DC podía favorecer la presentación
de EDA-SIINFEKL-C5a en mayor medida que con la proteína EDA-SIINFEKL. Se
incubaron las células B3Z (específicas de SIINFEKL) a una concentración de 105
células por pocillo con diferentes concentraciones de EDA-SIINFEKL y EDA-
SIINFEKL-C5a en presencia de 105 esplenocitos por pocillo de ratones C57/BL6. La
activación de las células B3Z se midió mediante la producción de la citocina IL-2,
para lo que se utilizó la línea celular CTLL-2, cuyo crecimiento es dependiente de
esta citocina. Para realizar el ensayo, se cultivaron 50 L de los sobrenadantes con
3000-5000 células CTLL-2 por pocillo, diluidos hasta un volumen final de 100 L.

Material y métodos
75
Tras 24 horas de cultivo, se añadieron 0.5 Ci (25 Ci/mmol) de timidina tritiada
(Amersham) por pocillo y 20 horas después se cosecharon las células, con un
cosechador (Harvester, Filtermate 196, Packard). La timidina incorporada al DNA se
cuantificó en un contador de centelleo (Topcount, Packard) (Lai et al. 1987).
3.3.4.4. Migración celular
Para estudiar el efecto quimiotáctico directo de las proteínas de fusión o por
la activación de la liberación de agentes quimiotácticos, realizamos ensayos de
transmigración celular en transwell. Se usaron cámaras transwell con filtro de poro
de 5 µm de diámetro (Costar, Corning Incorporated). En la cámara superior se
añadieron esplenocitos de ratón (106) C57BL/6 en medio libre de suero (100 µL). En
la parte inferior se añadieron 50 µM de la proteína en un volumen final de 500 µL de
medio sin suero o bien 500 μL de sobrenadante de esplenocitos de ratón (106)
C57BL/6 (Harlan) incubados con 50 µM de proteína en medio libre de suero durante
3 horas. Tras 3 horas de incubación a 37 ºC, retiramos la cámara superior donde se
situaban las células que no habían migrado. Se añadió 105 bolitas CalibriteTM3 (BD,
Biosciences) por pocillo en un volumen de 50 µL. Se recogieron las células y tras
realizar tres lavados con PBS, se procedió a la cuantificación celular mediante
citometría de flujo (BD, FACSCalibur™) adquiriendo 4000 bolitas Calibrite por
muestra. Los esplenocitos se marcaron con anti-Ly6G FITC y anti-CD11b PE (BD
Biosciences) para diferenciar los macrófagos (Ly6G- CD11b+). También se cuantificó
por ELISA (BD, Biosciences), la expresión de la quimiocina MIP-1α presente en el
sobrenadante utilizado como quimiotáctico.
4. Estudio in vitro de los intercambiadores de iones cloruro, calcio y
potasio en la activación linfocitaria
4.1. Análisis de la expresión de mRNA por PCR quantitativa
La extracción de RNA de las células T efectoras CD4+CD25- (Tefect) y de las
células T reguladoras CD4+CD25+ (Treg), se realizó usando una solución de lisis
para la purificación de ácidos nucleicos (Applied Biosystems) y el sistema ABI
PRISM 6100 nucleic acid Prepstation (Applied Biosystems). Las muestras fueron

Material y métodos
76
sometidas a un tratamiento con DNasa, transcripción reversa y PCR cuantitativa a
tiempo real utilizando cebadores específicos para diferentes moléculas. Los valores
de mRNA se representan por la fórmula 2ΔCt donde ACt indica la diferencia en el
umbral del ciclo entre el gen control (beta-actina) y el gen diana.
4.2. Purificación de células T reguladoras
La purificación de las células Tefect y de las células Treg se realizó partiendo
de esplenocitos de ratón BALB/c utilizando un kit de separación de células Treg
(Milteny Biotec, Bergish Gladbach, Germany) de acuerdo con las instrucciones del
fabricante. Se confirmó una pureza de las poblaciones purificadas superior al 95%
por citometría de flujo.
4.3. Ensayos de proliferación celular
Cultivamos esplenocitos CD4+CD25- (Tefect) de ratones hembras BALBc 105
cels/pocillo con 0.5 μg/mL de anticuerpo anti mouse CD3 (BD Pharmigen, San
Diego CA), con los distintos inhibidores de canales de Cl- , péptidos frente AE2 y las
proteínas recombinantes en presencia o ausencia de células T reguladoras
CD4+CD25+ (104/pocillo). La proliferación celular se midió por incorporación de 0.5
μCi/pocillo de timidina tritiada (Amersham, Pharmacia) al transcurrir tres días y
tras 16 horas, se procedió al cosechado de las placas de cultivo utilizando un
cosechador (Filtermate 96 harvester; Packard Instrument, Meriden, CT). El contaje
de la radioactividad incorporada al DNA celular (como medida de proliferación
celular) se realizó mediante un contador de centelleo (Topcount; Packard
Instrument). Para inducir un cultivo mixto leucocitario (CML) también usamos
células dendríticas 103 cels /pocillo de ratones C57BL/6. Dependiendo del ensayo se
utilizó 100 U/mL de la citocina IL-2 (Roche) para inducir la proliferación celular de
las células Treg.
4.4. Cinéticas de calcio intracelular
Medimos la cinética del aumento del Ca2+ intracelular por citometría de flujo
y por un lector de luminiscencia en microplacas NOVOstar (BMG Labtech,
Alemania). Las células (Tefect o Treg) fueron marcadas con el quelante fluorescente

Material y métodos
77
de Ca2+ Fluo-4 AM (Invitrogen), siguiendo las instrucciones del fabricante. Tras la
adquisición de la línea base, las células fueron activadas con distintos estímulos
según el ensayo, en presencia o ausencia de DIDS (100 μM). Entre los estímulos
destacamos la concanavalina A (40 μg/mL), la tapsigargina (0.5 μg/mL) o el anti-
CD3 + IL-2 (0.5 μg/mL y 100 U/mL respectivamente). El análisis del flujo de
entrada Ca2+ intracelular para las muestras medidas por citometría de flujo, se
realizó con el programa Flow jo (Tree Star, EE.UU.). Las muestras medidas por el
lector de luminiscencia se analizaron con el software de análisis del mismo lector.
5. Estudio de la apoptosis linfocitaria in vitro
5.1. Técnica de Tunel
Para detectar la rotura del DNA celular inducido por el péptido p17AE2 en
las células Treg, hemos utilizado el kit, In situ cell death detection POD (Roche
Molecular Biochemicals). Con este objetivo, purificamos las células T efectoras
(CD4+CD25-) y las células T reguladoras (CD4+CD25+) partiendo de esplenocitos
de ratones BALB/c. Posteriormente incubamos las dos subpoblaciones celulares
overnight con o sin el péptido p17AE2 (50 μg/mL), utilizando como péptido control
el péptido P301, que comprende los aminoácidos 301-315 de la proteína gp120 del
VIH-1, y en presencia de 0.5 μg/mL de anti CD3 (BD Pharmigen, San Diego CA) y
100 U/mL de IL-2 (Roche Molecular Biochemicals, Alemania). Tras la incubación,
añadimos 103 células en un porta para cada condición, con su control positivo y
negativo. Adherimos las células por citospin y las fijamos con una solución de
fijación (4% paraformaldehído en PBS, pH 7.4), tras hacer el lavado con PBS en
cubeta, incubamos los portas con la solución de bloqueo (3% H2O2 en metanol)
durante 10 minutos a 15-25ºC. Seguido de un lavado con PBS incubamos los portas
con una solución de permeabilización (0.1% triton X-100 en 0.1% de citrato sódico)
durante 2 minutos a 4ºC, posteriormente realizamos dos lavados con PBS y
añadimos la mezcla de reacción Tunel (marcaje de las roturas de DNA con
transferasa terminal desoxinucleotídica), los controles positivos previamente fueron
tratados con DNasa I (3 U/mL en 50 mM Tris-HCl, pH 7.5, 10 mM MgCl2 1 mg/mL
BSA, durante 10 minutos a 15-25 °C) y a los controles negativos añadimos sólo la

Material y métodos
78
solución de marcado (sin la enzima transferasa terminal), dejamos 60 minutos a 37
ºC en estufa húmeda y en oscuridad. La reacción se detuvo con tres lavados con PBS
y analizamos las muestras por microscopía de fluorescencia (IRE2; Leica, equipado
con una cámara DC300F; Leica), previo marcaje con DAPI (marcaje de núcleo celular
de color azul). Las células en apoptosis y por lo tanto con roturas en el DNA se
marcaron de color verde. Calculamos el porcentaje de apoptosis celular tras la
cuantificación de núcleos apoptóticos (marcaje Tunel) respecto al total de núcleos
celulares (marcaje DAPI). Para realizar este análisis, utilizamos el software ImageJ
(NIH, Bethesda, EE.UU.).
6. Estudio de la respuesta inmunitaria en ratones
6.1. Inmunización
6.1.1. Plásmido
Los ratones C57BL/6 recibieron una inyección intramuscular de 50 L de
cardiotoxina (10 nM) en el músculo tibial anterior. Cuatro días después recibieron
una inyección de 100 g de pShuttle-EDA-SIINFEKL, pShuttle-C3d-EDA-SIINFEKL,
pShuttle-C5a-EDA-SIINFEKL, pShuttle-C3a-EDA-SIINFEKL o pShuttle-C4a-EDA-
SIINFEKL en el mismo lugar. Dos semanas después recibieron una segunda dosis
del plásmido correspondiente.
6.1.2. Proteína
Las diferentes proteínas recombinantes se administraron por vía intravenosa
con o sin el adyuvante poly(I:C) (50 g/ratón) en un volumen final de 200 L de
solución salina. La cantidad de proteína administrada por ratón fue de 3 nmol, en
algunos ensayos fue de 1.5 nmol (como se indica en el apartado de resultados).
6.2. Estudio in vitro e in vivo de las respuestas inmunitarias inducidas
6.2.1. Aislamiento de esplenocitos
Entre 6 y 15 días después de la última inmunización, se sacrificaron los
animales y se extrajeron los bazos. A continuación, estos órganos se

Material y métodos
79
homogeneizaron con la base del émbolo de una jeringa o utilizando 2 portas
esmerilados, y los eritrocitos se lisaron utilizando solución de lisis ACK (1 mL/bazo)
durante 1 minuto. Tras un lavado con RPMI 1640, las células se resuspendieron en
medio de cultivo y se utilizaron en los diferentes ensayos que se citan a
continuación.
6.2.2. ELISPOT
La cuantificación de células productoras de IFN-γ se midió mediante un
ensayo de ELISPOT utilizando un kit comercial (BD-Biosciences.). Las placas
(Multiscreen HTS, Millipore) se incubaron con el anticuerpo purificado anti-IFN-γ (5
μg/mL) durante 16 horas a 4 ºC. Al día siguiente, las placas se bloquearon con MC
durante 2 horas y, a continuación, se cultivaron en MC entre 5x105 y 106 esplenocitos
a 37 ºC y 5% de CO2, en ausencia o presencia del péptido SIINFEKL (1 g/mL) para
la estimulación de la respuesta celular T, o utilizando células YAC-1 irradiadas (3000
células por pocillo) como estímulo de la respuesta mediada por las células NK. Un
día después, las placas se lavaron y se incubaron con el anticuerpo biotinilado anti-
IFN-γ (2 μg/mL). Tras 2 horas, se lavaron y se incubaron con una dilución 1:100 de
estreptavidina-peroxidasa. Una hora más tarde, se lavaron las placas y se revelaron
con el sustrato AEC (BD Biosciences). La reacción colorimétrica se paró con agua
destilada, y el número de puntos se cuantificó utilizando un contador de ELISPOT
(CTL, Aale, Alemania).
6.2.3. In vivo killing
Los ensayos de lisis in vivo o in vivo killing consistieron en inyectar
esplenocitos de ratones no inmunizados a ratones previamente inmunizados (tras 7
días de la inmunización). Estos esplenocitos se extrajeron de ratones, de la misma
cepa que los inmunizados, el día anterior a la realización del ensayo. La mitad de los
esplenocitos se incubó con el DTc SIINFEKL (10 g/mL) durante 30 minutos a 37ºC,
la otra mitad se incubó en las mismas condiciones, pero sin DTc. Posteriormente se
marcaron los esplenocitos, durante 5 minutos a temperatura ambiente y en
oscuridad, con 2,5 μM de CFSE (Invitrogen, Eugene, Oregon, EE.UU.) en el caso de
los esplenocitos incubados con DTc y 0, 25 μM CFSE en el caso de los incubados sin

Material y métodos
80
DTc. A los ratones inmunizados se les administró, por vía intravenosa, una mezcla
de 1x107 de ambos tipos de esplenocitos marcados en una proporción 1:1. A las 16
horas se sacrificaron los ratones y se extrajeron los bazos, que se homogenizaron
como se ha descrito en el apartado 6.2.1. Los esplenocitos de cada uno de los ratones
se analizaron por citometría de flujo para cuantificar la lisis de aquellos esplenocitos
pulsados con el DTc. Así, a mayor eficacia de la inmunización, habría una mayor
lisis de los esplenocitos marcados con 2,5 μM CFSE y el DTc. Sin embargo, los
esplenocitos marcados únicamente con 0,25 μM CFSE, no se lisarían. Como control
negativo, se usaron ratones a los que también se les inyectó la mezcla de
esplenocitos marcados, pero que previamente no habían sido inmunizados, por lo
que no se observaría lisis en ninguna de las dos poblaciones celulares. El porcentaje
de lisis específica se calculó según la siguiente fórmula:
6.2.4. Inducción de la respuesta inmunitaria en ausencia de señalización C5aR
Se inmunizaron por vía intravenosa ratones C57BL/6 con 1.5 nmol de las
proteínas EDA-SIINFEKL, EDA-SIINFEKL-C5a y SIINFEKL-C5a suplementado con
50 g de poly(I:C) por ratón en combinación con 100 g de anticuerpo bloqueante
para el receptor C5aR, clon 20/70 (AbD Serotec, Oxford, UK). Siete días después de
las inmunizaciones, los ratones fueron sacrificados y la respuesta de células T
específicas para el péptido SIINFEKL se midió in vivo.
6.2.5. Depleción in vivo de células NK1.1
Se inmunizaron por vía intravenosa ratones C57BL/6 con 3 nmol de las
proteínas EDA-SIINFEKL, EDA-SIINFEKL-C5a y SIINFEKL-C5a suplementado con
50 g de poly(I:C) por ratón. En algunos casos los ratones recibieron un tratamiento
de depleción de células NK1.1 que consistió en la administración de 200 g de
anticuerpo anti-NK1.1, clon PK136 (BioXCell, West Lebanon, NH.) o del
% Lisis específica = 100 - x 100
% esplenocitos CFSE alto (ratón inmunizado)
% esplenocitos CFSE bajo (ratón inmunizado)
% esplenocitos CFSE alto (ratón no inmunizado)
% esplenocitos CFSE bajo (ratón no inmunizado)

Material y métodos
81
correspondiente isotipo control a los días -1, 0 y 3 de la inmunización. La eficiencia
de la depleción de las células NK1.1 se analizó por citometría de flujo y en todos los
casos fue superior al 95%. Para estudiar el efecto de la actividad NK se realizó el
ensayo de in vivo killing a día 7 tras la inmunización. Además, se llevó a cabo un
ensayo de ELISPOT para medir la cantidad de células productoras de IFN-γ,
utilizando como células diana, la línea celular YAC-1 (3000 por pocillo) sensitiva a
las células NK.
6.3. Estudios in vivo de protección tumoral
6.3.1. Modelo tumoral
Se estudió la protección conferida por la inmunización frente al crecimiento
de una línea tumoral de timoma (E.G7-OVA) establemente transfectada con un
plásmido que codifica para la proteína OVA, capaz de desarrollar tumores en
ratones C57BL/6.
6.3.2. Estudios de protección de tumores
Los ratones se inmunizaron por vía venosa con 3 nmol de EDA-SIINFEKL,
EDA-SIINFEKL-C5a, SIINFEKL-C5a o SIINFEKL junto con el adyuvante poly(I:C)
(50 μg/ratón), a día 0. Siete días después de la inmunización se inocularon, por vía
subcutánea (en el flanco derecho de la espalda), las células E.G7-OVA (5 x 105
células/ratón) en un volumen final de 200 μL en los ratones que previamente habían
sido tratados. En todos los experimentos se incluyó también un grupo de ratones
que no recibió ningún tratamiento, y que permitió observar que el tumor se había
establecido correctamente.
Una semana después se comenzó a tomar la medida de los tumores y se
estudió la progresión del volumen tumoral: V= (longitud x anchura2)/2, así como la
supervivencia de los animales. Los animales fueron sacrificados cuando el volumen
tumoral alcanzó un diámetro de 20 mm.

Material y métodos
82
6.3.3. Estudios de tratamiento de tumores
Se inocularon por vía subcutánea (en la base de la espalda), 5 x 105 células
E.G7-OVA por ratón en un volumen de 200 L. Una vez que los tumores alcanzaron
un diámetro de 6 mm se trataron los ratones con las diferentes proteínas; EDA-
SIINFEKL, EDA-SIINFEKL-C5a y SIINFEKL-C5a (3 nmol/ratón) junto con el
adyuvante poly(I:C) (50 μg/mL) por vía intratumoral. Se monitorizó el crecimiento
tumoral dos veces por semana con un calibre. El tamaño tumoral medio se expresó
en milímetros cúbicos, usando la formula V= (longitud x anchura2)/2. Al igual que
en el estudio de protección, los ratones fueron sacrificados cuando el volumen
tumoral alcanzó un diámetro de 20 mm.
7. Estadística
La normalidad se estudió con un test Shapiro-Wilk. Los análisis estadísticos
de los resultados se realizaron utilizando test paramétricos (t de Student y ANOVA
de una vía) y no paramétricos (Kruskal-Wallis y U de Mann-Whitney). La
supervivencia de los ratones en los experimentos de tumor se analizó mediante el
método Kaplan-Meier y el test log-rank. Se consideraron significativos aquellos
valores de p < 0.05. Los análisis estadísticos se realizaron con la ayuda del programa
SPSS v.013 para Windows y el programa GraphPad Prism 5.03.

RESULTADOS


Resultados
85
1. Implicación de los intercambiadores de Cl- y canales de Ca2+ en
la activación celular de los linfocitos T efectores y T reguladores
(CD4+CD25+FOXP3+)
1.1. Efecto de la inhibición de los intercambiadores de aniones cloruro
sobre la acción de las células T reguladoras (CD4+CD25+FOXP3+)
Partiendo de la hipótesis de que la inhibición de los intercambiadores de
aniones cloruro puede afectar al flujo de entrada de calcio y por lo tanto a la
activación de los linfocitos (Wang et al. 2006), quisimos ver como afectaba un
bloqueante inespecífico de los intercambiadores de aniones cloruro en la
activación y proliferación celular. Así mismo, quisimos estudiar si la función
supresora de las células T reguladoras podría ser revertida con bloqueantes de
los intercambiadores de aniones cloruro, ya que está descrito que ratones Knock-
out para AE2 (intercambiador de aniones cloruro/bicarbonato) presentan un
elevado número de linfocitos T CD8+, un descenso de células T reguladoras
CD4+CD25+FOXP3+ y un patrón de expresión de anticuerpos autoinmunes
(Salas et al. 2008).
En nuestra primera aproximación a este estudio utilizamos el DIDS
(Ácido 4,4'-Diisotiocianostilbeno-2,2'-Disulfónico), por ser un inhibidor de la
conductancia aniónica, incluyendo el transporte de aniones cloruro mediados
por el intercambiador de Cl-/HCO3- AE1-3 y otros intercambiadores de HCO3-
como los intercambiadores de Na+/HCO3- (NBCe1, NBCe2, NBCn1, NDCBE,
NCBE) (Romero et al. 2004).
Se incubaron esplenocitos de ratones BALB/c (105 cel/pocillo) junto con
células dendríticas de ratones C57BL/6 (106 cel/primeros pocillos) a distintas
diluciones, en presencia o ausencia de células T reguladoras (104 cel/pocillo)
obtenidas de ratones C57BL/6 en presencia o ausencia de DIDS (50 μM). Tras
72 horas de cultivo, la proliferación celular se midió por incorporación de
timidina tritiada empleando un contador de centelleo.
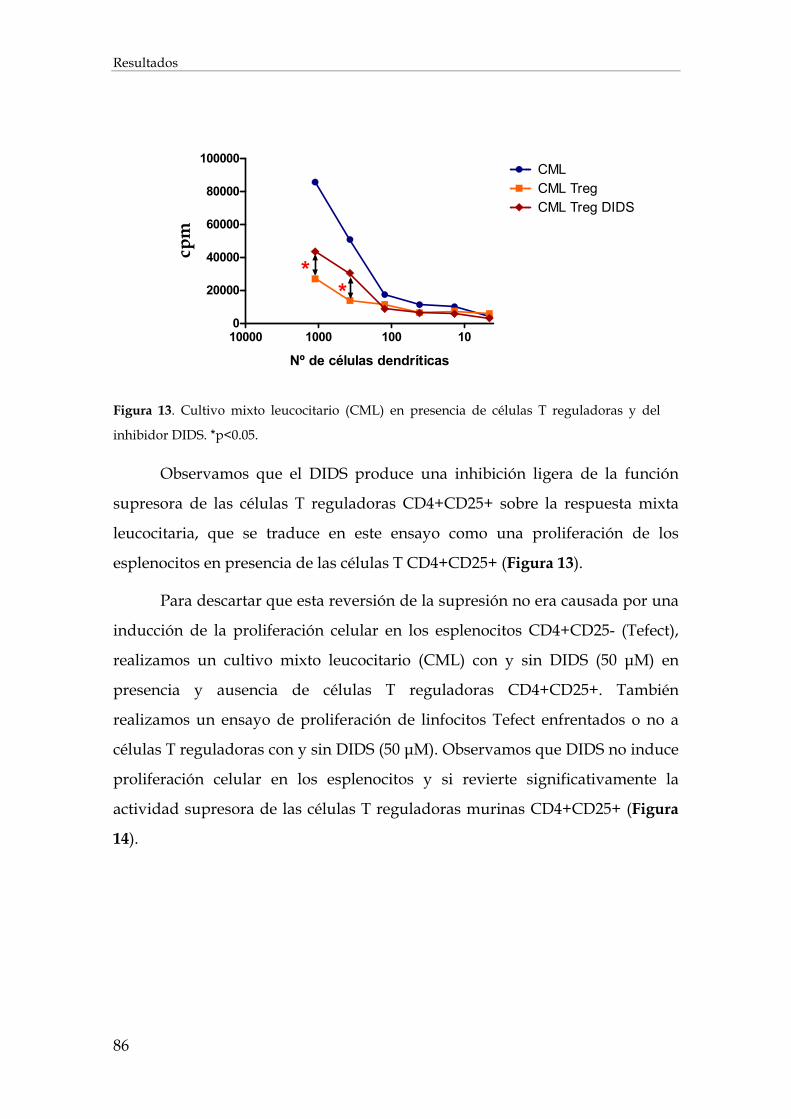
Resultados
86
101001000100000
20000
40000
60000
80000
100000CMLCML TregCML Treg DIDS
**
Nº de células dendríticas
cpm
Figura 13. Cultivo mixto leucocitario (CML) en presencia de células T reguladoras y del
inhibidor DIDS. *p<0.05.
Observamos que el DIDS produce una inhibición ligera de la función
supresora de las células T reguladoras CD4+CD25+ sobre la respuesta mixta
leucocitaria, que se traduce en este ensayo como una proliferación de los
esplenocitos en presencia de las células T CD4+CD25+ (Figura 13).
Para descartar que esta reversión de la supresión no era causada por una
inducción de la proliferación celular en los esplenocitos CD4+CD25- (Tefect),
realizamos un cultivo mixto leucocitario (CML) con y sin DIDS (50 μM) en
presencia y ausencia de células T reguladoras CD4+CD25+. También
realizamos un ensayo de proliferación de linfocitos Tefect enfrentados o no a
células T reguladoras con y sin DIDS (50 μM). Observamos que DIDS no induce
proliferación celular en los esplenocitos y si revierte significativamente la
actividad supresora de las células T reguladoras murinas CD4+CD25+ (Figura
14).
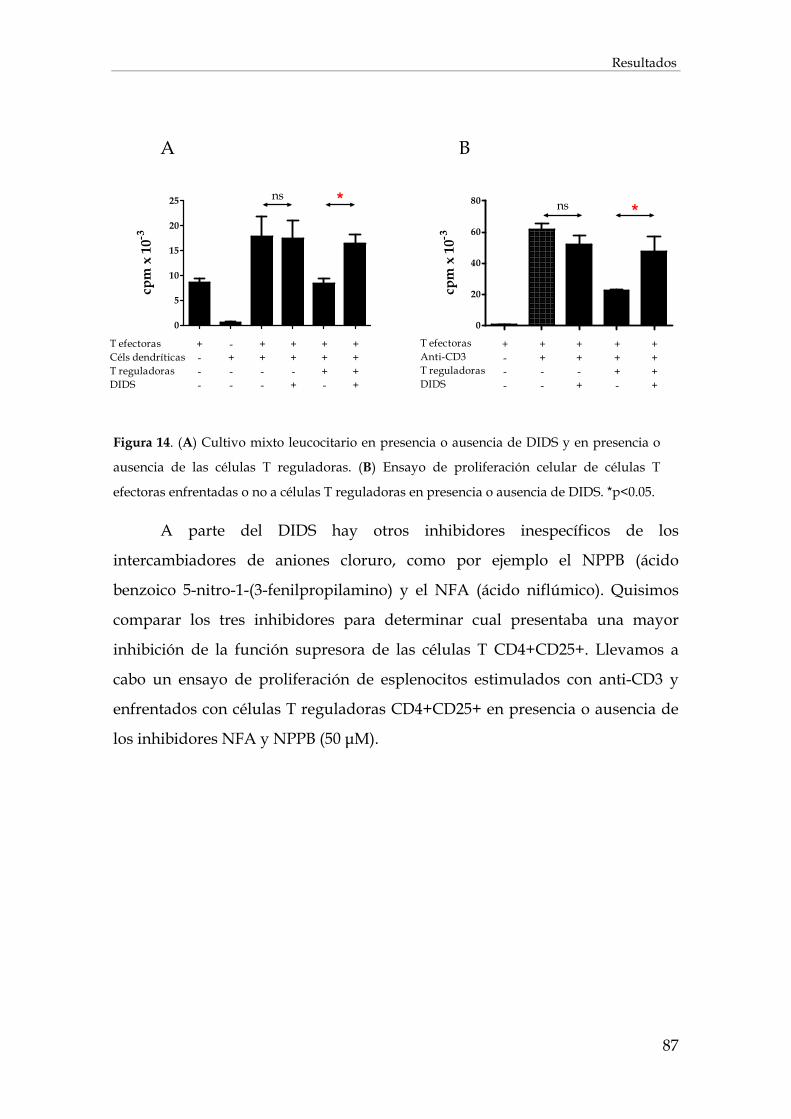
Resultados
87
A B
Figura 14. (A) Cultivo mixto leucocitario en presencia o ausencia de DIDS y en presencia o
ausencia de las células T reguladoras. (B) Ensayo de proliferación celular de células T
efectoras enfrentadas o no a células T reguladoras en presencia o ausencia de DIDS. *p<0.05.
A parte del DIDS hay otros inhibidores inespecíficos de los
intercambiadores de aniones cloruro, como por ejemplo el NPPB (ácido
benzoico 5-nitro-1-(3-fenilpropilamino) y el NFA (ácido niflúmico). Quisimos
comparar los tres inhibidores para determinar cual presentaba una mayor
inhibición de la función supresora de las células T CD4+CD25+. Llevamos a
cabo un ensayo de proliferación de esplenocitos estimulados con anti-CD3 y
enfrentados con células T reguladoras CD4+CD25+ en presencia o ausencia de
los inhibidores NFA y NPPB (50 μM).
0
20
40
60
80*
T efectorasAnti-CD3T reguladorasDIDS
++++
+++-
++-+
++--
+---
ns
cpm
x 1
0-3
0
5
10
15
20
25
T efectorasCéls dendríticasT reguladorasDIDS
*
+---
-+--
++--
++-+
+++-
++++
ns
cpm
x 1
0-3
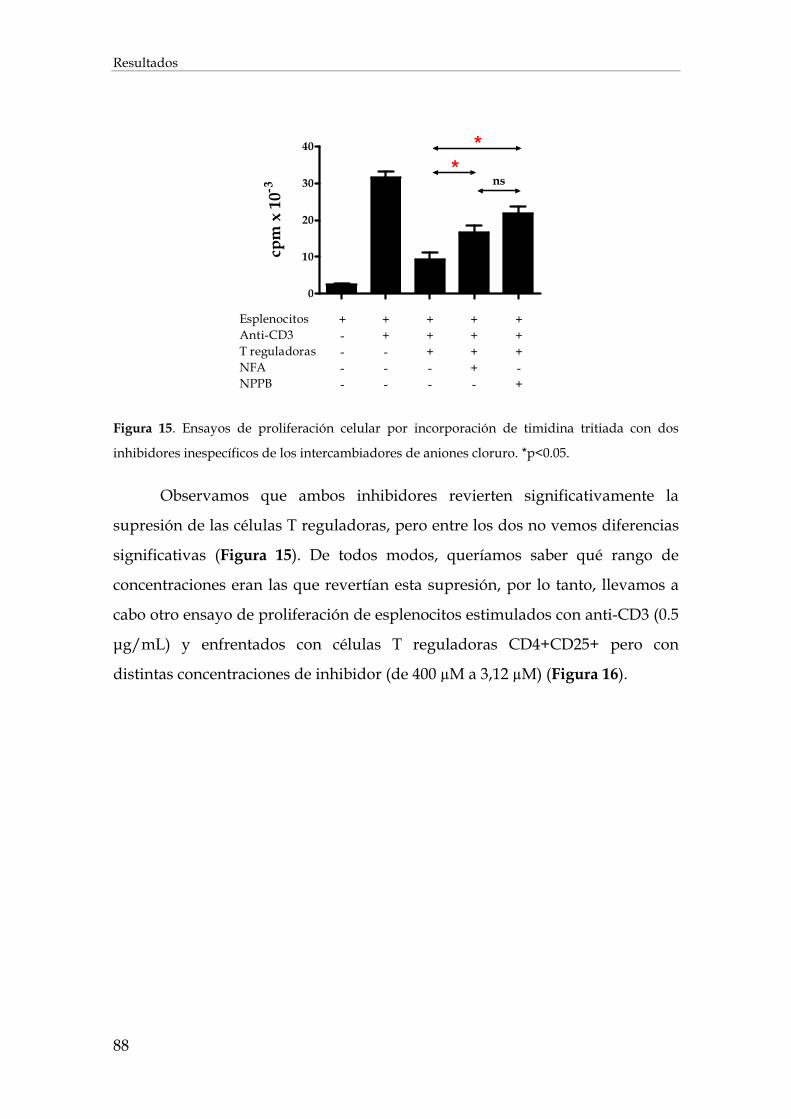
Resultados
88
Figura 15. Ensayos de proliferación celular por incorporación de timidina tritiada con dos
inhibidores inespecíficos de los intercambiadores de aniones cloruro. *p<0.05.
Observamos que ambos inhibidores revierten significativamente la
supresión de las células T reguladoras, pero entre los dos no vemos diferencias
significativas (Figura 15). De todos modos, queríamos saber qué rango de
concentraciones eran las que revertían esta supresión, por lo tanto, llevamos a
cabo otro ensayo de proliferación de esplenocitos estimulados con anti-CD3 (0.5
μg/mL) y enfrentados con células T reguladoras CD4+CD25+ pero con
distintas concentraciones de inhibidor (de 400 µM a 3,12 µM) (Figura 16).
0
10
20
30
40
EsplenocitosAnti-CD3T reguladorasNFANPPB
+++-+
++++-
+++--
++---
+----
**
ns
cpm
x 1
0-3
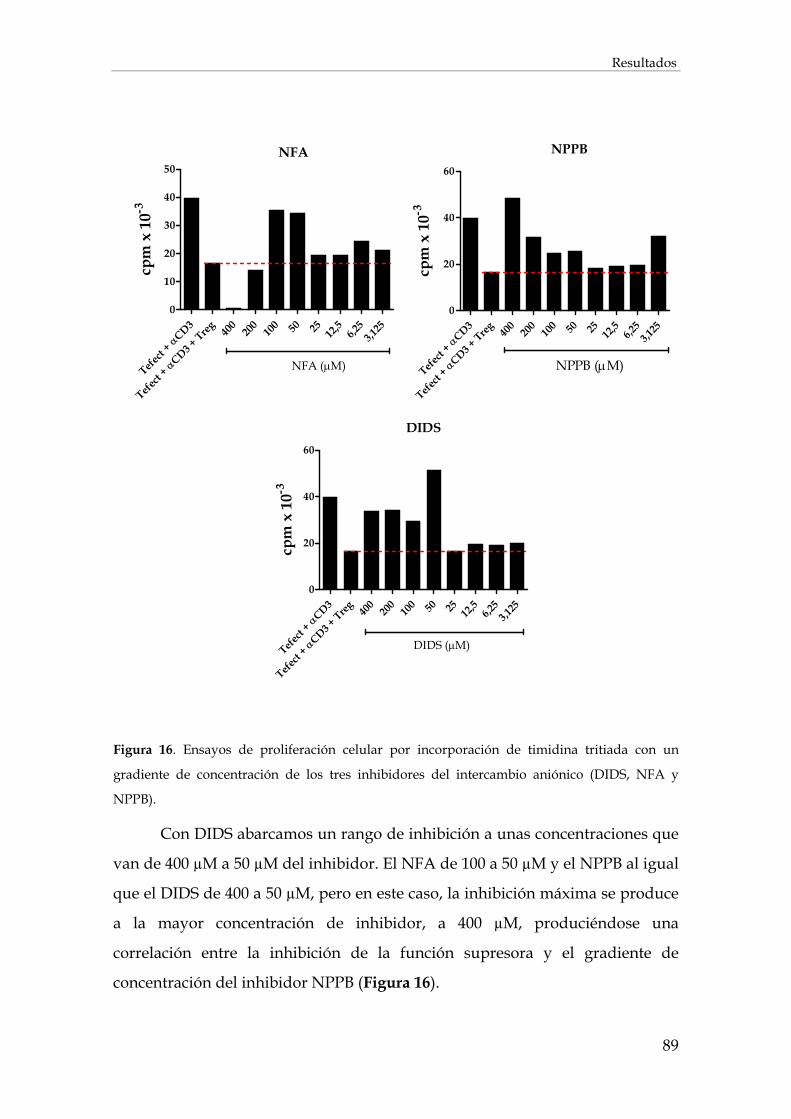
Resultados
89
NFA
CD3
Tefec
t +
CD3 + T
reg
Tefec
t +
400
200
100 50 25 12
,56,2
53,1
250
10
20
30
40
50
NFA (M)
cpm
x 1
0-3 NPPB
CD3
Tefec
t +
CD3 + T
reg
Tefec
t +
400
200
100 50 25 12
,56,2
53,1
250
20
40
60
NPPB (M)
cpm
x 1
0-3 DIDS
CD3
Tefec
t +
CD3 + T
reg
Tefec
t +
400
200
100 50 25 12
,56,2
53,1
250
20
40
60
DIDS (M)
cpm
x 1
0-3
Figura 16. Ensayos de proliferación celular por incorporación de timidina tritiada con un
gradiente de concentración de los tres inhibidores del intercambio aniónico (DIDS, NFA y
NPPB).
Con DIDS abarcamos un rango de inhibición a unas concentraciones que
van de 400 µM a 50 µM del inhibidor. El NFA de 100 a 50 µM y el NPPB al igual
que el DIDS de 400 a 50 µM, pero en este caso, la inhibición máxima se produce
a la mayor concentración de inhibidor, a 400 µM, produciéndose una
correlación entre la inhibición de la función supresora y el gradiente de
concentración del inhibidor NPPB (Figura 16).

Resultados
90
1.2. Efecto de la inhibición de los intercambiadores de aniones cloruro
sobre la expresión de mRNAs en los linfocitos T
Incubamos esplenocitos CD4+CD25- (Tefect) en presencia y ausencia de
células T reguladoras con y sin DIDS (50 µM), tras 72 horas de incubación
medimos la cantidad de mRNA de interleucina IL-2, interleucina IL-10, CTLA-4
y Foxp3 como medidas de activación y proliferación celular de las células T
efectoras y de actividad supresora de las células T reguladoras. Podemos
observar que en los esplenocitos estimulados con anti-CD3 (0.5 μg/mL), cuando
se cocultivan con las células CD4+CD25+, el mRNA de la interleucina IL-2
desciende, siendo un reflejo de la supresión en la proliferación celular ejercida
por la población Treg. Sin embargo, este descenso se revierte con la presencia en
el cocultivo del inhibidor DIDS a una concentración de 50 μM. También
observamos el efecto supresor de las Treg en el aumento de mRNA de la
interleucina IL-10 en el cocultivo, que a su vez, desciende por el efecto de DIDS
(50 μM) (Figura 17).
Con respecto a CTLA-4 y Foxp3 mRNA, como era de esperar se observa
un aumento de sus niveles en el cocultivo en presencia de Treg, ya que son las
células Treg las que expresan mayores niveles de ambas moléculas. El inhibidor
DIDS no produce descenso de sus valores (Figura 17).
En conclusión, podemos decir que tras 72 h con 50 µM DIDS se restaura
la activación y proliferación de las células T efectoras y la producción de IL-2,
en presencia de células T reguladoras CD4+CD25+, además de un descenso de
los niveles de producción de la citocina inmunosupresora IL-10.
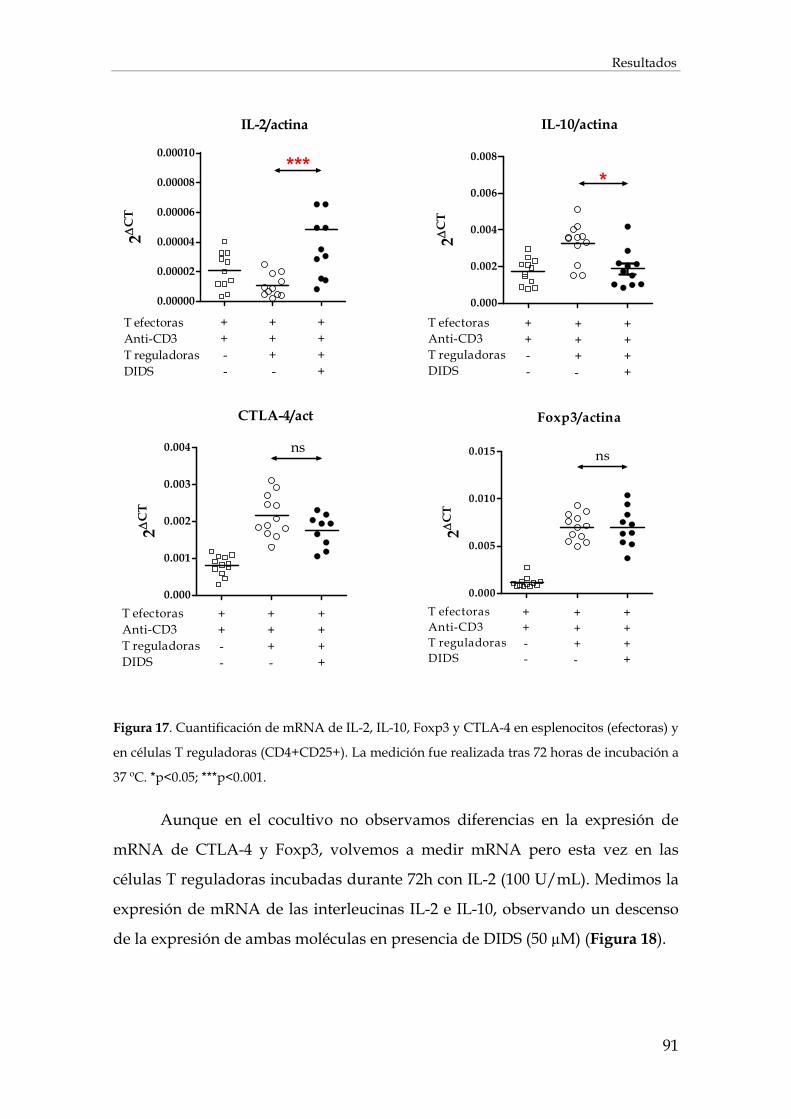
Resultados
91
Figura 17. Cuantificación de mRNA de IL-2, IL-10, Foxp3 y CTLA-4 en esplenocitos (efectoras) y
en células T reguladoras (CD4+CD25+). La medición fue realizada tras 72 horas de incubación a
37 ºC. *p<0.05; ***p<0.001.
Aunque en el cocultivo no observamos diferencias en la expresión de
mRNA de CTLA-4 y Foxp3, volvemos a medir mRNA pero esta vez en las
células T reguladoras incubadas durante 72h con IL-2 (100 U/mL). Medimos la
expresión de mRNA de las interleucinas IL-2 e IL-10, observando un descenso
de la expresión de ambas moléculas en presencia de DIDS (50 µM) (Figura 18).
IL-2/actina
0.00000
0.00002
0.00004
0.00006
0.00008
0.00010
T efectorasAnti-CD3T reguladorasDIDS
++ - -
++++
+++ -
***
2C
T IL-10/actina
0.000
0.002
0.004
0.006
0.008
*
T efectorasAnti-CD3T reguladorasDIDS
++--
++++
+++-
2C
T
CTLA-4/act
0.000
0.001
0.002
0.003
0.004
T efectorasAnti-CD3T reguladorasDIDS
++--
++++
+++-
ns
2C
T
Foxp3/actina
0.000
0.005
0.010
0.015
T efectorasAnti-CD3T reguladorasDIDS
++--
++++
+++-
ns2
CT
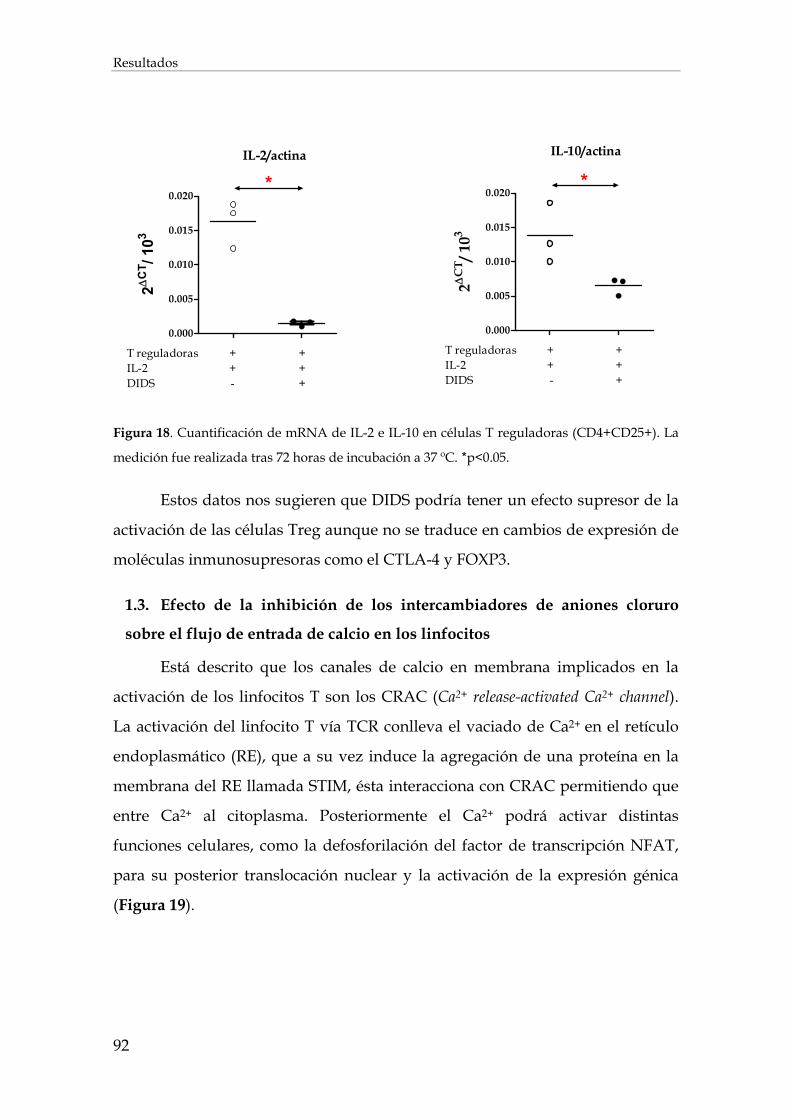
Resultados
92
Figura 18. Cuantificación de mRNA de IL-2 e IL-10 en células T reguladoras (CD4+CD25+). La
medición fue realizada tras 72 horas de incubación a 37 ºC. *p<0.05.
Estos datos nos sugieren que DIDS podría tener un efecto supresor de la
activación de las células Treg aunque no se traduce en cambios de expresión de
moléculas inmunosupresoras como el CTLA-4 y FOXP3.
1.3. Efecto de la inhibición de los intercambiadores de aniones cloruro
sobre el flujo de entrada de calcio en los linfocitos
Está descrito que los canales de calcio en membrana implicados en la
activación de los linfocitos T son los CRAC (Ca2+ release-activated Ca2+ channel).
La activación del linfocito T vía TCR conlleva el vaciado de Ca2+ en el retículo
endoplasmático (RE), que a su vez induce la agregación de una proteína en la
membrana del RE llamada STIM, ésta interacciona con CRAC permitiendo que
entre Ca2+ al citoplasma. Posteriormente el Ca2+ podrá activar distintas
funciones celulares, como la defosforilación del factor de transcripción NFAT,
para su posterior translocación nuclear y la activación de la expresión génica
(Figura 19).
IL-2/actina
0.000
0.005
0.010
0.015
0.020
T reguladorasIL-2DIDS
++-
+++
*
2C
T/ 1
03 IL-10/actina
0.000
0.005
0.010
0.015
0.020*
T reguladorasIL-2DIDS
++ -
+++
2C
T/ 1
03

Resultados
93
Figura 19. Señalización del flujo de entrada de calcio en la activación celular. (Winslow et al.
2003).
Se ha descrito que el potencial de membrana podría afectar a la
activación de CRAC. Por este motivo, quisimos comprobar si un bloqueante de
los intercambiadores de aniones cloruro, podría afectar a los canales de calcio
en la membrana de los esplenocitos y detectar diferencias entre las células T
efectoras y las células T reguladoras.
Estudiamos de forma basal las diferencias entre células T efectoras
(CD4+CD25-) o células T reguladoras (CD4+CD25+) en la entrada de calcio tras
la estimulación con un mitógeno como la concanavalina A. Observamos que la
afluencia de Ca2+ en las células Treg era muy inferior a la que encontrábamos en
las células T efectoras tras el mismo estímulo (Figura 20). Esto nos hace pensar
que las células T reguladoras presentan mecanismos de activación vía
señalización de Ca2+ distintos o con diferencias al resto de los linfocitos.
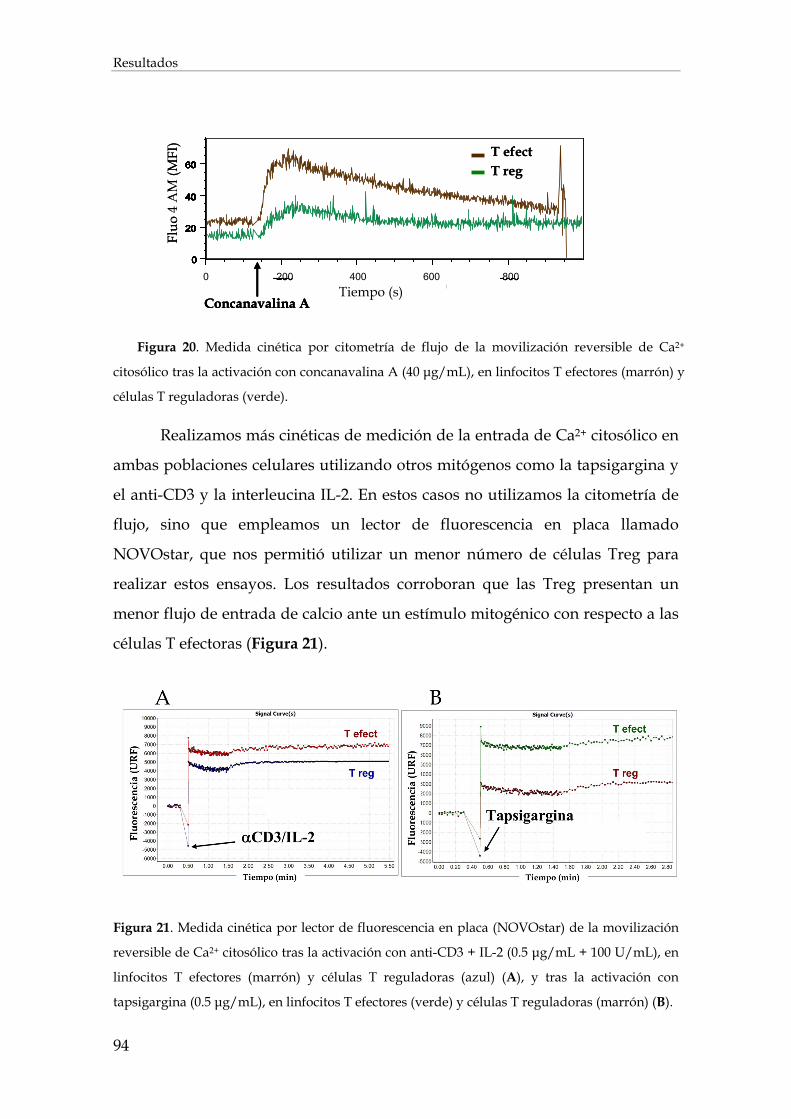
Resultados
94
Figura 20. Medida cinética por citometría de flujo de la movilización reversible de Ca2+
citosólico tras la activación con concanavalina A (40 μg/mL), en linfocitos T efectores (marrón) y
células T reguladoras (verde).
Realizamos más cinéticas de medición de la entrada de Ca2+ citosólico en
ambas poblaciones celulares utilizando otros mitógenos como la tapsigargina y
el anti-CD3 y la interleucina IL-2. En estos casos no utilizamos la citometría de
flujo, sino que empleamos un lector de fluorescencia en placa llamado
NOVOstar, que nos permitió utilizar un menor número de células Treg para
realizar estos ensayos. Los resultados corroboran que las Treg presentan un
menor flujo de entrada de calcio ante un estímulo mitogénico con respecto a las
células T efectoras (Figura 21).
Figura 21. Medida cinética por lector de fluorescencia en placa (NOVOstar) de la movilización
reversible de Ca2+ citosólico tras la activación con anti-CD3 + IL-2 (0.5 μg/mL + 100 U/mL), en
linfocitos T efectores (marrón) y células T reguladoras (azul) (A), y tras la activación con
tapsigargina (0.5 μg/mL), en linfocitos T efectores (verde) y células T reguladoras (marrón) (B).
00 200 400 600 800Time: Time (1024.00 sec.)
0
20
40
60
FL1-H
Efect.Treg
0 200 400 600 800Time: Time (1024.00 sec.)
0
20
40
60
FL1-H
EfectTreg
0 200 400 600 800Time: Time (1024.00 sec.)
0
20
40
60
FL1-H
0 200 400 600 800Time: Time (1024.00 sec.)
0
20
40
60
FL1-H
Efect.Treg
0 200 400 600 800Time: Time (1024.00 sec.)
0
20
40
60
FL1-H
0 200 400 600 800Time: Time (1024.00 sec.)
0
20
40
60
FL1-H
EfectTreg
Concanavalina ATiempo (s)
0 200 400 600 800
T regT efect
Flu
o4
AM
(MFI
)
00 200 400 600 800Time: Time (1024.00 sec.)
0
20
40
60
FL1-H
Efect.Treg
0 200 400 600 800Time: Time (1024.00 sec.)
0
20
40
60
FL1-H
EfectTreg
0 200 400 600 800Time: Time (1024.00 sec.)
0
20
40
60
FL1-H
0 200 400 600 800Time: Time (1024.00 sec.)
0
20
40
60
FL1-H
Efect.Treg
0 200 400 600 800Time: Time (1024.00 sec.)
0
20
40
60
FL1-H
0 200 400 600 800Time: Time (1024.00 sec.)
0
20
40
60
FL1-H
EfectTreg
Concanavalina ATiempo (s)
0 200 400 600 800
T regT efect
00 200 400 600 800Time: Time (1024.00 sec.)
0
20
40
60
FL1-H
Efect.Treg
0 200 400 600 800Time: Time (1024.00 sec.)
0
20
40
60
FL1-H
EfectTreg
0 200 400 600 800Time: Time (1024.00 sec.)
0
20
40
60
FL1-H
0 200 400 600 800Time: Time (1024.00 sec.)
0
20
40
60
FL1-H
Efect.Treg
0 200 400 600 800Time: Time (1024.00 sec.)
0
20
40
60
FL1-H
0 200 400 600 800Time: Time (1024.00 sec.)
0
20
40
60
FL1-H
EfectTreg
Concanavalina ATiempo (s)
0 200 400 600 800
T regT efectT regT efectT regT efect
Flu
o4
AM
(MFI
)

Resultados
95
Quisimos entonces estudiar el efecto de la adición de DIDS a los cultivos.
Incubamos las células T con DIDS (100 μM) durante 4 horas antes de realizar la
medición del flujo de Ca2+. Observamos que el DIDS no modificaba
significativamente las curvas de entrada de calcio ni en células T efectoras
(CD4+CD25-) ni en Treg (CD4+CD25+) (Panel A y B, Figura 22). Quisimos
estudiar el efecto del DIDS en células T que ya habían recibido un estímulo
previo de activación. Para ello, incubamos las células T efectoras con anti-CD3
durante 24h y medimos la cinética de Ca2+ tras la estimulación con
concanavalina A. En este caso, observamos que la adición del DIDS aumentó la
entrada de Ca2+ en las células T efectoras (Panel C, Figura 22). Lo mismo
ocurría si realizábamos un co-cultivo de células T efectoras y células T
reguladoras (Panel E, Figura 22). Sin embargo, cuando incubábamos las células
T reguladoras con anti-CD3 o IL-2 como estímulo, no veíamos cambios
significativos con DIDS (Panel D y F, Figura 22).
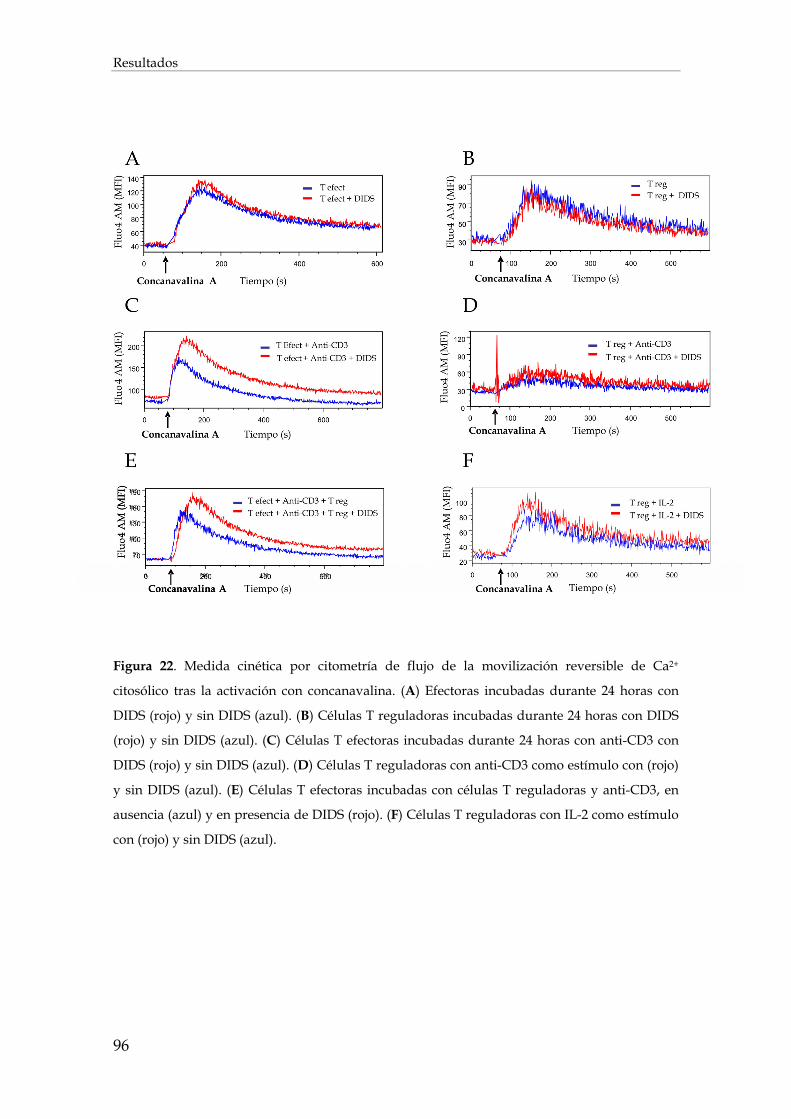
Resultados
96
Figura 22. Medida cinética por citometría de flujo de la movilización reversible de Ca2+
citosólico tras la activación con concanavalina. (A) Efectoras incubadas durante 24 horas con
DIDS (rojo) y sin DIDS (azul). (B) Células T reguladoras incubadas durante 24 horas con DIDS
(rojo) y sin DIDS (azul). (C) Células T efectoras incubadas durante 24 horas con anti-CD3 con
DIDS (rojo) y sin DIDS (azul). (D) Células T reguladoras con anti-CD3 como estímulo con (rojo)
y sin DIDS (azul). (E) Células T efectoras incubadas con células T reguladoras y anti-CD3, en
ausencia (azul) y en presencia de DIDS (rojo). (F) Células T reguladoras con IL-2 como estímulo
con (rojo) y sin DIDS (azul).

Resultados
97
2. Desarrollo de péptidos inhibidores del intercambiador
cloruro/bicarbonato AE2
2.1. Síntesis de los péptidos y ensayos funcionales de selección de los
péptidos inhibidores
Tras los resultados obtenidos con el inhibidor DIDS y teniendo en cuenta
el fenotipo inmunitario que tienen los ratones knockout para AE2 (incremento de
linfocitos CD8+ y descenso de células T reguladoras CD4+CD25+) hemos
diseñado unos péptidos que potencialmente podrían unirse a los bucles
extracelulares de AE2, con el fin de bloquear estos canales de forma específica.
Con esto queremos obtener unos péptidos que nos permitan revertir la función
supresora de las células T reguladoras, de una manera similar al DIDS.
Basados en la estructura del intercambiador AE2 y la presencia de bucles
extracelulares que están implicados en su función (Stewart et al. 2007),
diseñamos secuencias de aminoácidos que podrían interactuar con secuencias
de AE2. El diseño de estos péptidos se realizó basándonos en posibles
interacciones entre aminoácidos afines, utilizando matrices de
complementariedad y teniendo en cuenta el volumen de cada aminoácido. Así,
sintetizamos 20 péptidos que fueron probados en su capacidad de bloquear la
acción inmunosupresora de las células Treg in vitro (véase en el apartado 3.1.1.
de material y métodos).
Aislamos linfocitos T efectores CD4+CD25- y linfocitos Treg
CD4+CD25+FOXP3+ a partir de esplenocitos de ratones BALB/c. Tras la
purificación, se analizaron las células por citometría de flujo, obteniéndose una
pureza superior al 95% en todos los ensayos. Los linfocitos T efectores fueron
cultivados en ausencia o presencia del estímulo mitogénico anti-CD3 y en
ausencia o presencia de células Treg y de los péptidos sintetizados. A los 3 días
de cultivo, se añadieron 0,5 µCi/pocillo de timidina tritiada y tras 16 horas, se
procedió al cosechado de las placas y a la realización del contaje de la
radiactividad incorporada al DNA celular (como medida de la proliferación
celular) mediante un contador de centelleo. Los resultados de este ensayo se
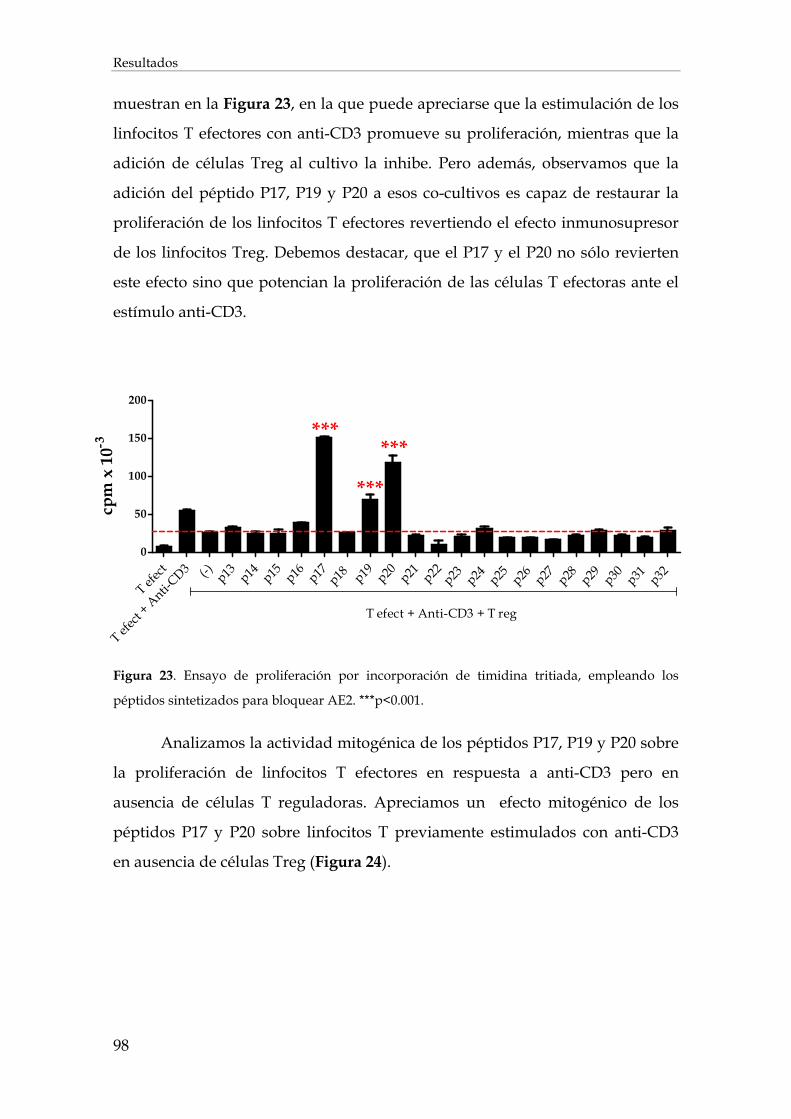
Resultados
98
muestran en la Figura 23, en la que puede apreciarse que la estimulación de los
linfocitos T efectores con anti-CD3 promueve su proliferación, mientras que la
adición de células Treg al cultivo la inhibe. Pero además, observamos que la
adición del péptido P17, P19 y P20 a esos co-cultivos es capaz de restaurar la
proliferación de los linfocitos T efectores revertiendo el efecto inmunosupresor
de los linfocitos Treg. Debemos destacar, que el P17 y el P20 no sólo revierten
este efecto sino que potencian la proliferación de las células T efectoras ante el
estímulo anti-CD3.
Figura 23. Ensayo de proliferación por incorporación de timidina tritiada, empleando los
péptidos sintetizados para bloquear AE2. ***p<0.001.
Analizamos la actividad mitogénica de los péptidos P17, P19 y P20 sobre
la proliferación de linfocitos T efectores en respuesta a anti-CD3 pero en
ausencia de células T reguladoras. Apreciamos un efecto mitogénico de los
péptidos P17 y P20 sobre linfocitos T previamente estimulados con anti-CD3
en ausencia de células Treg (Figura 24).
T efec
t
T efec
t + A
nti-CD3 (-) p13 p14 p15 p16 p17 p18
p19 p20 p21 p22
p23
p
24 p
25 p
26 p
27
p28
p
29
p30
p
31
p32
0
50
100
150
200
T efect + Anti-CD3 + T reg
***
***
***
cpm
x 1
0-3
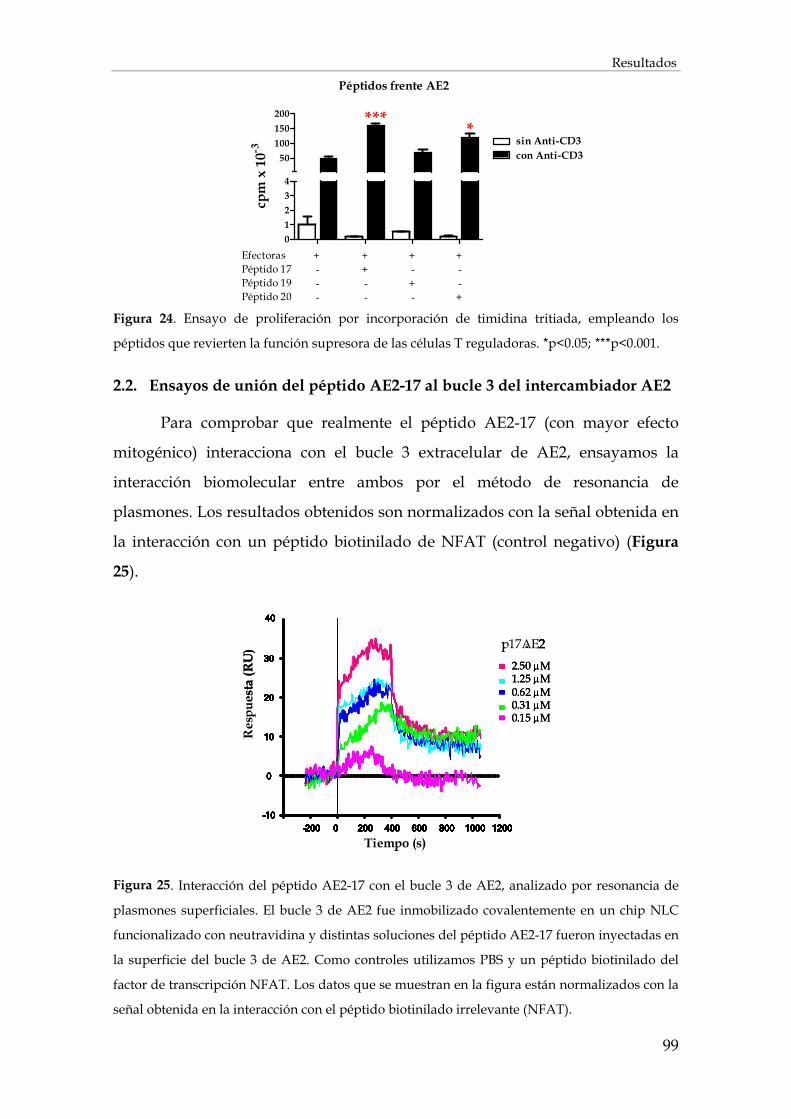
Resultados
99
Figura 24. Ensayo de proliferación por incorporación de timidina tritiada, empleando los
péptidos que revierten la función supresora de las células T reguladoras. *p<0.05; ***p<0.001.
2.2. Ensayos de unión del péptido AE2-17 al bucle 3 del intercambiador AE2
Para comprobar que realmente el péptido AE2-17 (con mayor efecto
mitogénico) interacciona con el bucle 3 extracelular de AE2, ensayamos la
interacción biomolecular entre ambos por el método de resonancia de
plasmones. Los resultados obtenidos son normalizados con la señal obtenida en
la interacción con un péptido biotinilado de NFAT (control negativo) (Figura
25).
Figura 25. Interacción del péptido AE2-17 con el bucle 3 de AE2, analizado por resonancia de
plasmones superficiales. El bucle 3 de AE2 fue inmobilizado covalentemente en un chip NLC
funcionalizado con neutravidina y distintas soluciones del péptido AE2-17 fueron inyectadas en
la superficie del bucle 3 de AE2. Como controles utilizamos PBS y un péptido biotinilado del
factor de transcripción NFAT. Los datos que se muestran en la figura están normalizados con la
señal obtenida en la interacción con el péptido biotinilado irrelevante (NFAT).
Tiempo (s)
-10
0
10
20
30
-200 0 200 400 600 800 1000 1200
40
-10
0
10
20
30
-200 0 200 400 600 800 1000 1200
40
-10
0
10
20
30
-200 0 200 400 600 800 1000 1200
40
-10
0
10
20
30
-200 0 200 400 600 800 1000 1200
40
p17AE2--
2.50 µM1.25 µM0.62 µM0.31 µM0.15 µM
Res
pue
sta
(RU
)
Tiempo (s)
-10
0
10
20
30
-200 0 200 400 600 800 1000 1200
40
-10
0
10
20
30
-200 0 200 400 600 800 1000 1200
40
-10
0
10
20
30
-200 0 200 400 600 800 1000 1200
40
-10
0
10
20
30
-200 0 200 400 600 800 1000 1200
40
p17AE2--
2.50 µM1.25 µM0.62 µM0.31 µM0.15 µM
Tiempo (s)
-10
0
10
20
30
-200 0 200 400 600 800 1000 1200
40
-10
0
10
20
30
-200 0 200 400 600 800 1000 1200
40
-10
0
10
20
30
-200 0 200 400 600 800 1000 1200
40
-10
0
10
20
30
-200 0 200 400 600 800 1000 1200
40
p17AE2--p17AE2--
2.50 µM1.25 µM0.62 µM0.31 µM0.15 µM
Res
pue
sta
(RU
)
Péptidos frente AE2
0
1
2
3
4
50
100
150
200
con Anti-CD3sin Anti-CD3
EfectorasPéptido 17Péptido 19Péptido 20
++ - -
+ -+ -
+ - - -
+ - -+
****
cpm
x 1
0-3

Resultados
100
Se confirmó la interacción del péptido AE2-17 con el bucle 3 extracelular
de AE2 de forma dosis dependiente.
Con estos resultados nos centramos en el péptido P17 (desde ahora
p17AE2), para estudiar con más detalle su efecto inhibidor de las células T
reguladoras y su efecto estimulador de las células T efectoras.
Posteriormente quisimos ver si hay interacción entre el péptido p17AE2
y la vía de señalización del Ca2+.
Se ha descrito que la tapsigargina, un inhibidor de las bombas de calcio
intracelulares, inhibe la entrada del Ca2+ citoplasmático al retículo
endoplasmático. Esto provoca el aumento de la concentración de Ca2+
citoplasmático que finalmente activa los canales de CRAC y la entrada masiva
de calcio extracelular.
Empleamos la tapsigargina a distintas concentraciones (1.5 y 2.5 μg/mL)
como un activador de los canales CRAC, en un ensayo de proliferación de
linfocitos T efectores y de linfocitos Treg. Los resultados de este experimento
mostraron que en ambas poblaciones celulares, estimuladas con anti-CD3 (0.5
μg/mL) e IL-2 (100 U/mL), la tapsigargina a 2.5 μg/mL aumenta la
proliferación celular (Panel A y B, Figura 26). Además, se realizó un co-cultivo
de células T efectoras con células Treg en presencia de tapsigargina (1.5 μg/mL
) para determinar si la función supresora de la proliferación ejercida por las
Treg podría verse alterada. Paralelamente, se realizó un ensayo de proliferación
de linfocitos T efectores co-cultivados con las células Treg y tapsigargina (1.5
μg/mL) en presencia o ausencia del péptido p17AE2 (100 μg/mL). Observamos
que el uso de la tapsigargina a 1.5 μg/mL (una concentración que no efecta a la
proliferación celular de ambas poblaciones), potencia el efecto supresor de la
proliferación de las células T reguladoras CD4+CD25+ (Panel C, Figura 26). Así
mismo, es importante destacar que la adición del péptido p17AE2 a estos
cultivos fue capaz de bloquear el efecto de la tapsigargina, revertiendo la
supresión de la proliferación ejercida por las Treg (Panel D, Figura 26).
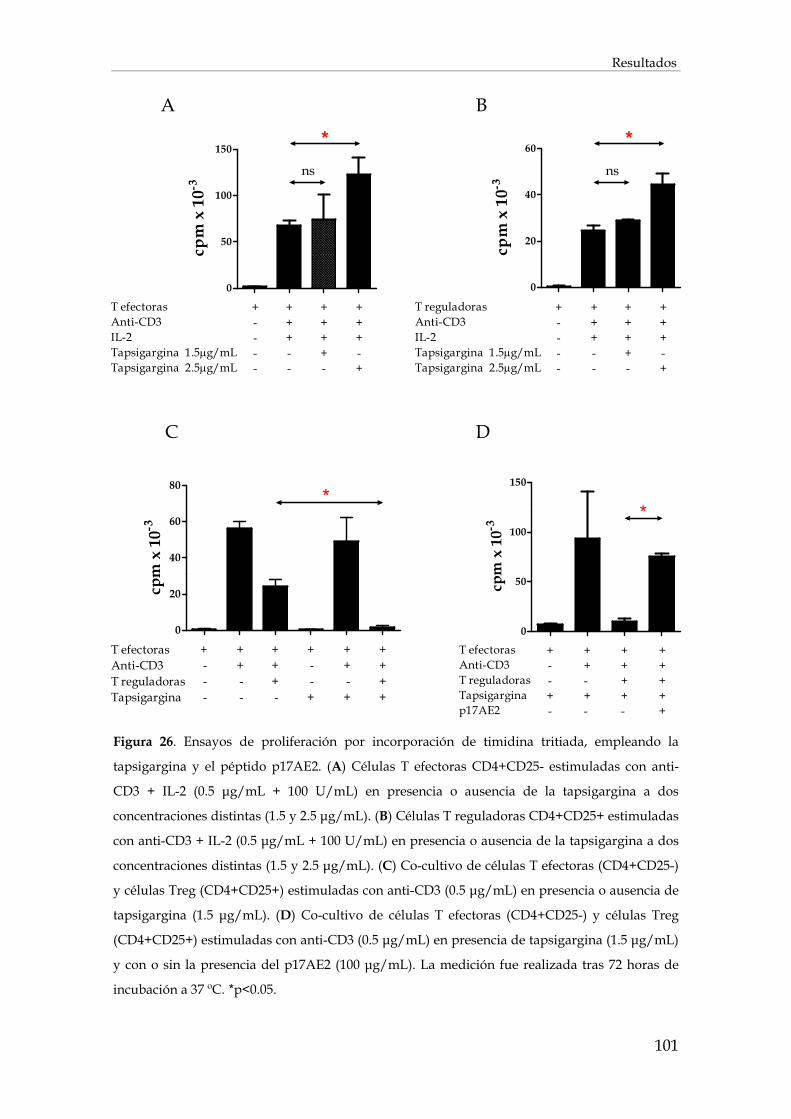
Resultados
101
A B
C D
Figura 26. Ensayos de proliferación por incorporación de timidina tritiada, empleando la
tapsigargina y el péptido p17AE2. (A) Células T efectoras CD4+CD25- estimuladas con anti-
CD3 + IL-2 (0.5 μg/mL + 100 U/mL) en presencia o ausencia de la tapsigargina a dos
concentraciones distintas (1.5 y 2.5 μg/mL). (B) Células T reguladoras CD4+CD25+ estimuladas
con anti-CD3 + IL-2 (0.5 μg/mL + 100 U/mL) en presencia o ausencia de la tapsigargina a dos
concentraciones distintas (1.5 y 2.5 μg/mL). (C) Co-cultivo de células T efectoras (CD4+CD25-)
y células Treg (CD4+CD25+) estimuladas con anti-CD3 (0.5 μg/mL) en presencia o ausencia de
tapsigargina (1.5 μg/mL). (D) Co-cultivo de células T efectoras (CD4+CD25-) y células Treg
(CD4+CD25+) estimuladas con anti-CD3 (0.5 μg/mL) en presencia de tapsigargina (1.5 μg/mL)
y con o sin la presencia del p17AE2 (100 μg/mL). La medición fue realizada tras 72 horas de
incubación a 37 ºC. *p<0.05.
0
50
100
150
T efectorasAnti-CD3IL-2Tapsigargina 1.5g/mLTapsigargina 2.5g/mL
+++--
+++-+
+----
*
ns
++++-
cpm
x 1
0-3
0
20
40
60
T reguladorasAnti-CD3IL-2Tapsigargina 1.5g/mLTapsigargina 2.5g/mL
+++--
+++-+
+----
++++-
*
ns
cpm
x 1
0-3
0
50
100
150
T efectorasAnti-CD3T reguladorasTapsigarginap17AE2
++++-
+++++
++-+-
+--+-
*cp
m x
10-3
0
20
40
60
80
T efectorasAnti-CD3T reguladorasTapsigargina
*
+ - - -
++ - -
+++ -
+ - -+
++ -+
++++
cpm
x 1
0-3
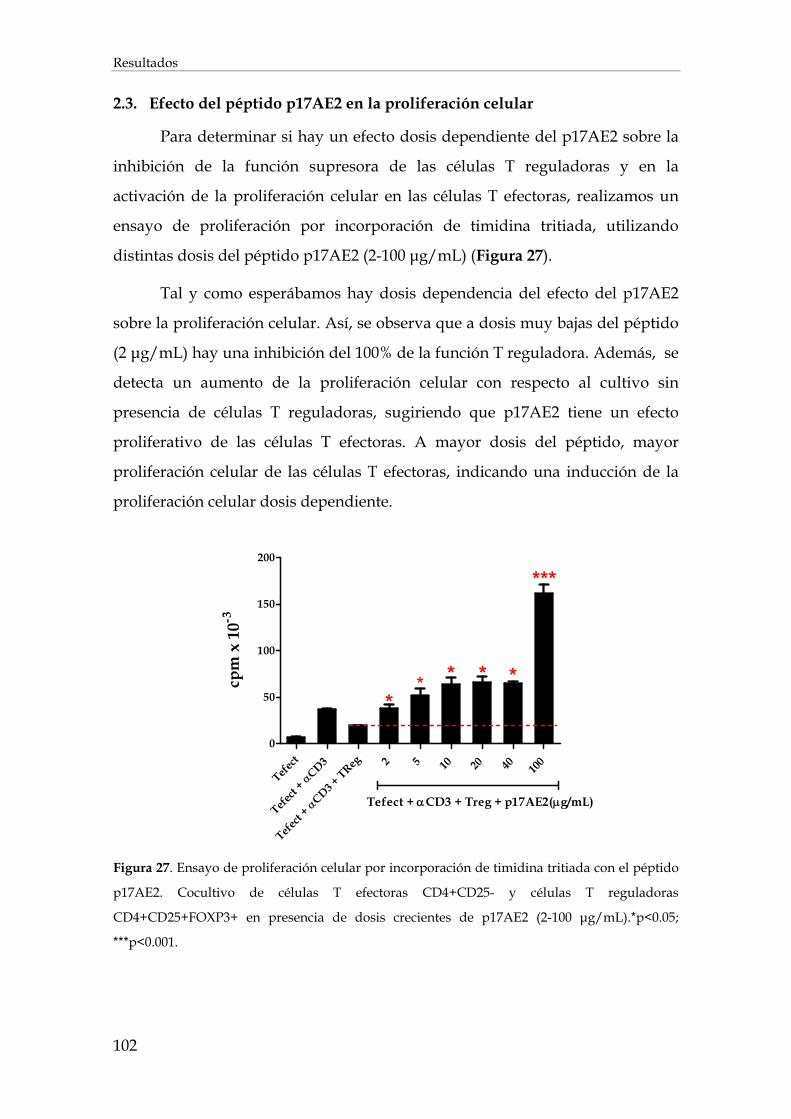
Resultados
102
2.3. Efecto del péptido p17AE2 en la proliferación celular
Para determinar si hay un efecto dosis dependiente del p17AE2 sobre la
inhibición de la función supresora de las células T reguladoras y en la
activación de la proliferación celular en las células T efectoras, realizamos un
ensayo de proliferación por incorporación de timidina tritiada, utilizando
distintas dosis del péptido p17AE2 (2-100 μg/mL) (Figura 27).
Tal y como esperábamos hay dosis dependencia del efecto del p17AE2
sobre la proliferación celular. Así, se observa que a dosis muy bajas del péptido
(2 μg/mL) hay una inhibición del 100% de la función T reguladora. Además, se
detecta un aumento de la proliferación celular con respecto al cultivo sin
presencia de células T reguladoras, sugiriendo que p17AE2 tiene un efecto
proliferativo de las células T efectoras. A mayor dosis del péptido, mayor
proliferación celular de las células T efectoras, indicando una inducción de la
proliferación celular dosis dependiente.
Figura 27. Ensayo de proliferación celular por incorporación de timidina tritiada con el péptido
p17AE2. Cocultivo de células T efectoras CD4+CD25- y células T reguladoras
CD4+CD25+FOXP3+ en presencia de dosis crecientes de p17AE2 (2-100 μg/mL).*p<0.05;
***p<0.001.
Tefec
tCD3
Tefec
t +
CD3 + T
Reg
Tefec
t +
2 5 10
20
40
100
0
50
100
150
200
Tefect + CD3 + Treg + p17AE2(g/mL)
***
** **
*
cpm
x 1
0-3
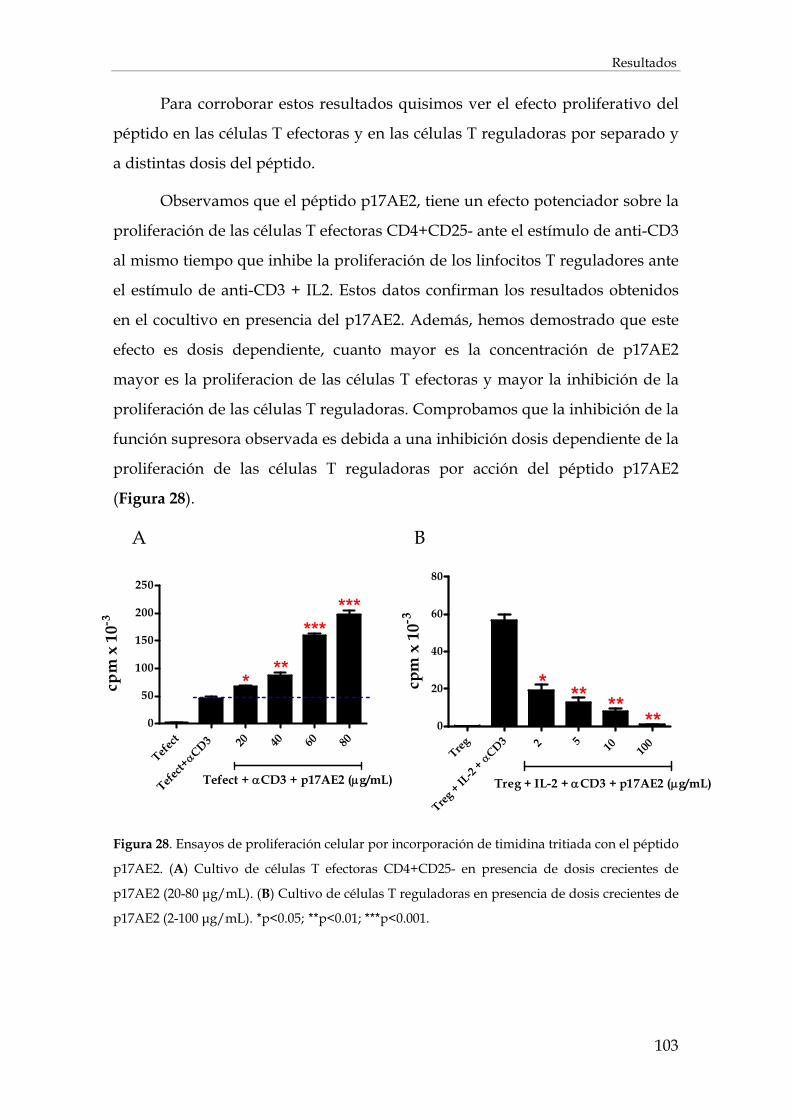
Resultados
103
Para corroborar estos resultados quisimos ver el efecto proliferativo del
péptido en las células T efectoras y en las células T reguladoras por separado y
a distintas dosis del péptido.
Observamos que el péptido p17AE2, tiene un efecto potenciador sobre la
proliferación de las células T efectoras CD4+CD25- ante el estímulo de anti-CD3
al mismo tiempo que inhibe la proliferación de los linfocitos T reguladores ante
el estímulo de anti-CD3 + IL2. Estos datos confirman los resultados obtenidos
en el cocultivo en presencia del p17AE2. Además, hemos demostrado que este
efecto es dosis dependiente, cuanto mayor es la concentración de p17AE2
mayor es la proliferacion de las células T efectoras y mayor la inhibición de la
proliferación de las células T reguladoras. Comprobamos que la inhibición de la
función supresora observada es debida a una inhibición dosis dependiente de la
proliferación de las células T reguladoras por acción del péptido p17AE2
(Figura 28).
A B
Figura 28. Ensayos de proliferación celular por incorporación de timidina tritiada con el péptido
p17AE2. (A) Cultivo de células T efectoras CD4+CD25- en presencia de dosis crecientes de
p17AE2 (20-80 μg/mL). (B) Cultivo de células T reguladoras en presencia de dosis crecientes de
p17AE2 (2-100 μg/mL). *p<0.05; **p<0.01; ***p<0.001.
Tefec
tCD3
Tefec
t+
20 40 60 800
50
100
150
200
250
Tefect + CD3 + p17AE2 (g/mL)
******
***cp
m x
10-3
Treg
CD3
Treg +
IL-2
+ 2 5 10
10
0 0
20
40
60
80
Treg + IL-2 + CD3 + p17AE2 (g/mL)
******
*cpm
x 1
0-3

Resultados
104
Al igual que en el caso de los inhibidores del intercambio aniónico,
quisimos comprobar si el péptido p17AE2 era capaz de activar la producción de
interleucina IL-2 en las células T efectoras estimuladas como medida de
activación y proliferación celular. Al mismo tiempo estudiamos si el p17AE2
reduce la producción de interleucina IL-10 por parte de las células T
reguladoras estimuladas, como medida de inhibición de la función supresora
de las T reguladoras. Para ello, incubamos las respectivas células estimuladas
con anti-CD3 + IL-2 (0.5 μg/mL y 100 U/mL respectivamente) con el péptido
p17AE2 (40 μg/mL) durante 48 horas a 37ºC de incubación, recogimos el
sobrenadante y por ELISA medimos la producción de IL-2 en las T efectoras y
IL-10 en las T reguladoras. A su vez, hicimos una medición de la expresión de
mRNA a las 8 horas de incubación a 37ºC, de las interleucinas IL-2 y IL-10 para
las células efectoras y T reguladoras respectivamente.
Tal como se puede apreciar en la Figura 29 (Panel A y B), el péptido
p17AE2 es capaz de aumentar la producción de interleucina IL-2 por parte de
las células T efectoras, de manera muy significativa cuando son estimuladas con
anti-CD3 + IL-2. Mientras que en las células T reguladoras se observa que el
péptido p17AE2, induce un descenso significativo de la expresión de la
interleucina IL-10, tras ser estimuladas con el mismo estímulo anti-CD3 + IL-2
(Panel D, Figura 29). Este descenso no se observa cuando cuantificamos mRNA
de IL-10 tras 8 horas de incubación de las células Treg con el péptido p17AE2
(Panel C, Figura 29).
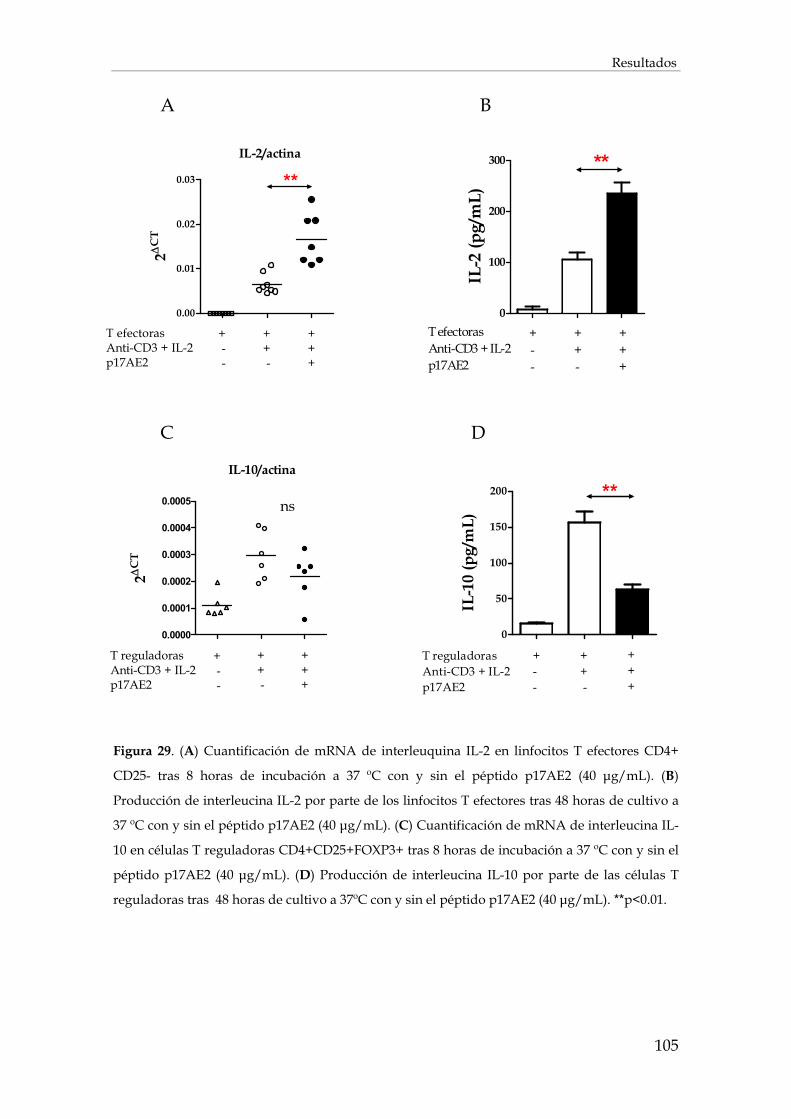
Resultados
105
A B
C D
Figura 29. (A) Cuantificación de mRNA de interleuquina IL-2 en linfocitos T efectores CD4+
CD25- tras 8 horas de incubación a 37 ºC con y sin el péptido p17AE2 (40 μg/mL). (B)
Producción de interleucina IL-2 por parte de los linfocitos T efectores tras 48 horas de cultivo a
37 ºC con y sin el péptido p17AE2 (40 μg/mL). (C) Cuantificación de mRNA de interleucina IL-
10 en células T reguladoras CD4+CD25+FOXP3+ tras 8 horas de incubación a 37 ºC con y sin el
péptido p17AE2 (40 μg/mL). (D) Producción de interleucina IL-10 por parte de las células T
reguladoras tras 48 horas de cultivo a 37ºC con y sin el péptido p17AE2 (40 μg/mL). **p<0.01.
0
100
200
300 **
T efectorasAnti-CD3 + IL-2p17AE2
+--
+++
++-
IL-2
(pg/
mL)
IL-2/actina
0.00
0.01
0.02
0.03
T efectorasAnti-CD3 + IL-2p17AE2
+ - -
++ -
+++
**2
CT
0.0000
0.0001
0.0002
0.0003
0.0004
0.0005
T reguladorasAnti-CD3 + IL-2p17AE2
+++
+ - -
++ -
IL-10/actina
ns
2C
T
0
50
100
150
200 **
T reguladorasAnti-CD3 + IL-2p17AE2
+--
+++
++ -
IL-1
0 (p
g/m
L)
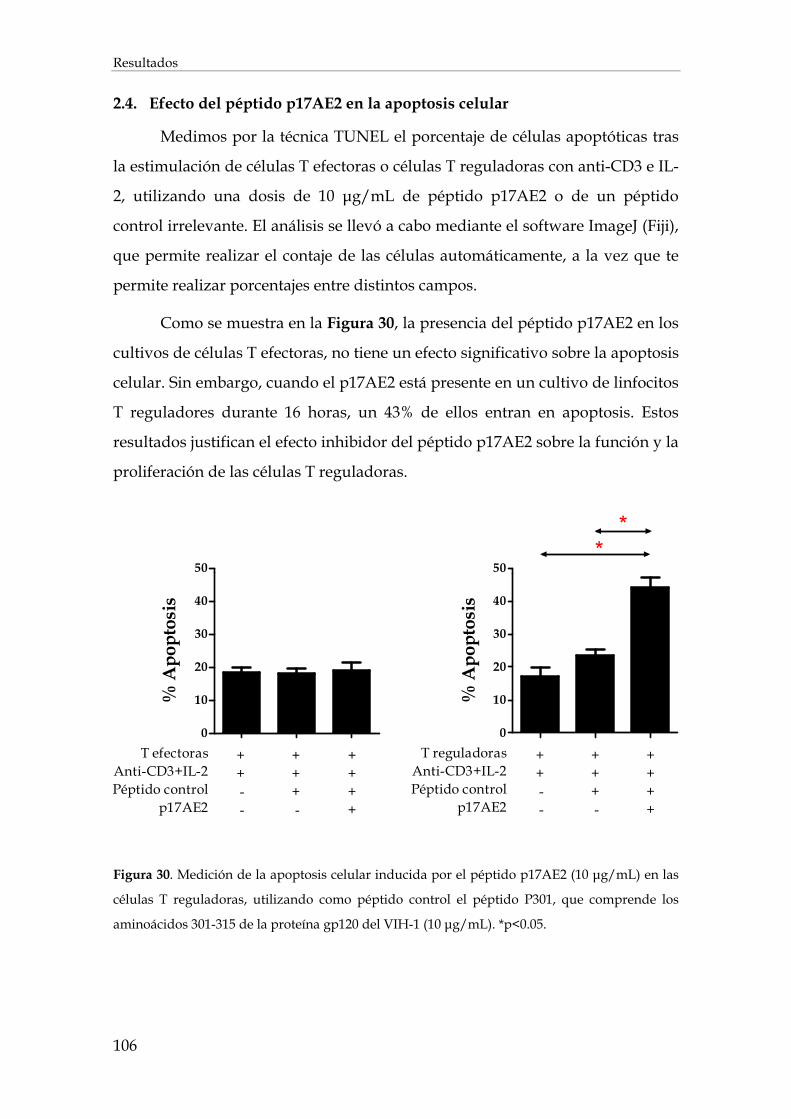
Resultados
106
2.4. Efecto del péptido p17AE2 en la apoptosis celular
Medimos por la técnica TUNEL el porcentaje de células apoptóticas tras
la estimulación de células T efectoras o células T reguladoras con anti-CD3 e IL-
2, utilizando una dosis de 10 μg/mL de péptido p17AE2 o de un péptido
control irrelevante. El análisis se llevó a cabo mediante el software ImageJ (Fiji),
que permite realizar el contaje de las células automáticamente, a la vez que te
permite realizar porcentajes entre distintos campos.
Como se muestra en la Figura 30, la presencia del péptido p17AE2 en los
cultivos de células T efectoras, no tiene un efecto significativo sobre la apoptosis
celular. Sin embargo, cuando el p17AE2 está presente en un cultivo de linfocitos
T reguladores durante 16 horas, un 43% de ellos entran en apoptosis. Estos
resultados justifican el efecto inhibidor del péptido p17AE2 sobre la función y la
proliferación de las células T reguladoras.
Figura 30. Medición de la apoptosis celular inducida por el péptido p17AE2 (10 μg/mL) en las
células T reguladoras, utilizando como péptido control el péptido P301, que comprende los
aminoácidos 301-315 de la proteína gp120 del VIH-1 (10 μg/mL). *p<0.05.
0
10
20
30
40
50
T reguladorasAnti-CD3+IL-2Péptido control
p17AE2
++++
+++-
++--
**
% A
popt
osis
0
10
20
30
40
50
T efectorasAnti-CD3+IL-2Péptido control
p17AE2
++++
+++-
++--
% A
popt
osis

Resultados
107
3. Estudio del potencial inmunomodulador de las anafilotoxinas
para el desarrollo de vacunas
3.1. Cribado con plásmidos de expresión
En un estudio previo realizado por nuestro grupo, demostramos que la
inmunización de ratones con DNA desnudo que codifica para una proteína de
fusión entre EDA y un antígeno, induce una respuesta de células T específicas
para el antígeno superior a la inducida por un plásmido que codifica el
antígeno solo (Mansilla et al. 2009). Utilizando el mismo sistema y para evaluar
la capacidad adyuvante de las anafilotoxinas para mejorar la inmunogenicidad
de la proteína EDA-SIINFEKL, construimos plásmidos que codifican versiones
secretables de EDA-SIINFEKL, C3d-EDA-SIINFEKL, C3a-EDA-SIINFEKL, C4a-
EDA-SIINFEKL y C5a-EDA-SIINFEKL. Los constructos diseñados fueron los
siguientes:
Figura 31. Esquema de las construcciones pC5a-EDA-SIINFEKL (A), pC4a-EDA-SIINFEKL (B),
pC3d-EDA-SIINFEKL (C), pC3a-EDA-SIINFEKL (D), pEDA-SIINFEKL (E).
La expresión genética de estos plásmidos se lleva a cabo bajo la
influencia de un promotor de la región inmediata temprana de Citomegalovirus
(CMV/IE), el cual permite altos niveles de transcripción de manera constitutiva
en una gran variedad de células eucariotas.
Tal y como se detalla en el punto 3.2.1 del apartado de material y
métodos, inicialmente se procedió a la realización del constructo péptido señal-
EDA-SIIINFEKL para posteriormente insertar los distintos fragmentos del
complemento. Se utilizaron cebadores específicos para introducir en el extremo
N-terminal de la proteína una secuencia que codificase para el péptido señal de

Resultados
108
la pre-pro-tripsina humana (HPPTLP) cuyo papel era promover que las
proteínas que se transcribiesen a partir de estos plásmidos fueran secretadas al
exterior. Todas las secuencias obtenidas en los distintos pasos de clonaje fueron
confirmadas mediante secuenciación del DNA. Posteriormente, se procedió a la
transformación de los plásmidos de expresión en bacterias BL21, para
posteriormente purificar los plásmidos.
Una vez confirmada la obtención de los distintos plásmidos de expresión
quisimos confirmar que expresaban las proteínas recombinantes diseñadas.
Para ello, transfectamos células HEK293 y mediante western blot de extracto
celular utilizando anticuerpos anti-EDA (Figura 32), fuimos capaces de detectar
la expresión de todas las proteínas con la excepción de EDA-SIINFEKL.
A: EDA-SIINFEKL (14 KDa)B: C3a-EDA-SIINFEKL (32 KDa)C: C3d-EDA-SIINFEKL (48,7 KDa)D: C4a-EDA-SIINFEKL (54 KDa)E: C5a-EDA-SIINFEKL (25.8 KDa)
A B C D E
A: EDA-SIINFEKL (14 KDa)B: C3a-EDA-SIINFEKL (32 KDa)C: C3d-EDA-SIINFEKL (48,7 KDa)D: C4a-EDA-SIINFEKL (54 KDa)E: C5a-EDA-SIINFEKL (25.8 KDa)
A B C D EA B C D E
ABCDE
A: EDA-SIINFEKL (14 KDa)B: C3a-EDA-SIINFEKL (32 KDa)C: C3d-EDA-SIINFEKL (48,7 KDa)D: C4a-EDA-SIINFEKL (54 KDa)E: C5a-EDA-SIINFEKL (25.8 KDa)
A B C D E
A: EDA-SIINFEKL (14 KDa)B: C3a-EDA-SIINFEKL (32 KDa)C: C3d-EDA-SIINFEKL (48,7 KDa)D: C4a-EDA-SIINFEKL (54 KDa)E: C5a-EDA-SIINFEKL (25.8 KDa)
A B C D EA B C D E
ABCDE
Figura 32. Análisis de expresión por Western blot del extracto celular de células HEK293,
transfectadas con los plásmidos de expresión diseñados.
Una vez demostrada la expresión de las proteínas in vitro, procedimos a
estudiar la capacidad de estos plásmidos de expresión para inducir una
respuesta celular T específica frente a SIINFEKL in vivo. Para ello, tal y como se
ha descrito en materiales y métodos, inyectamos en ratones 50 L de
cardiotoxina (10 nM) en el músculo tibial anterior, a los cuatro días los
inmunizamos con 100 g de plásmido en el mismo lugar, dos semanas después
recibieron una segunda dosis de 100 g de plásmido y a los 10 días se midió la
respuesta celular in vivo mediante el ensayo de in vivo killing, utilizando células
pulsadas con SIINFEKL (10 g/mL). También se cuantificó la producción de
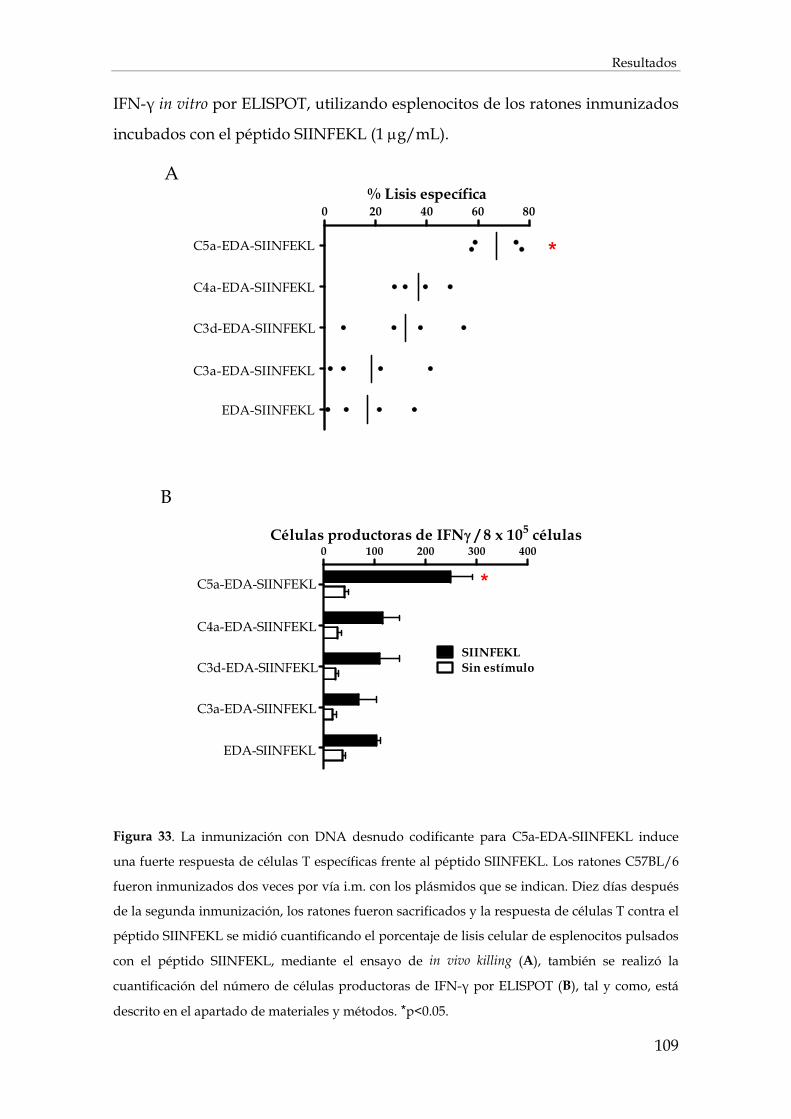
Resultados
109
IFN-γ in vitro por ELISPOT, utilizando esplenocitos de los ratones inmunizados
incubados con el péptido SIINFEKL (1 g/mL).
A
B
Figura 33. La inmunización con DNA desnudo codificante para C5a-EDA-SIINFEKL induce
una fuerte respuesta de células T específicas frente al péptido SIINFEKL. Los ratones C57BL/6
fueron inmunizados dos veces por vía i.m. con los plásmidos que se indican. Diez días después
de la segunda inmunización, los ratones fueron sacrificados y la respuesta de células T contra el
péptido SIINFEKL se midió cuantificando el porcentaje de lisis celular de esplenocitos pulsados
con el péptido SIINFEKL, mediante el ensayo de in vivo killing (A), también se realizó la
cuantificación del número de células productoras de IFN-γ por ELISPOT (B), tal y como, está
descrito en el apartado de materiales y métodos. *p<0.05.
0 100 200 300 400
Sin estímuloSIINFEKL
*C5a-EDA-SIINFEKL
C4a-EDA-SIINFEKL
C3d-EDA-SIINFEKL
C3a-EDA-SIINFEKL
EDA-SIINFEKL
Células productoras de IFN / 8 x 105 células
0 20 40 60 80
*C5a-EDA-SIINFEKL
C4a-EDA-SIINFEKL
C3d-EDA-SIINFEKL
C3a-EDA-SIINFEKL
EDA-SIINFEKL
% Lisis específica

Resultados
110
Observamos que los ratones que recibieron el plásmido pShuttle C5a-
EDA-SIINFEKL desarrollaron una respuesta célular T específica frente a
SIINFEKL significativamente más alta que el resto de grupos. Esta respuesta
celular se midió determinando el porcentaje de muerte celular in vivo de células
pulsadas con el antígeno SIINFEKL mediante la técnica del in vivo killing (Panel
A, Figura 33) o cuantificando el número de células productoras de IFN-γ por
ELISPOT (Panel B, Figura 33). Estos datos sugieren que la incorporación de C5a
a la molécula de fusión EDA-SIINFEKL aumenta su inmunogenicidad.
3.2. Utilización del fragmento C5a como adyuvante de vacunación
Puesto que observamos una inmunogenicidad superior con el plásmido
que expresa la forma secretable C5a-EDA-SIINFEKL con respecto al plásmido
que expresa EDA-SIINFEKL, decidimos producir la proteína recombinante C5a-
EDA-SIINFEKL, con el objetivo de poder confirmar que el fragmento C5a
combinado con la proteína EDA puede aumentar la respuesta celular T
específica inducida por la plataforma EDA unida a antígeno. Estos
experimentos indicaron que la combinación C5a-EDA es una buena estrategia
para el desarrollo de vacunas.
3.2.1. Purificación de las proteínas recombinantes de fusión
Tal y como se detalla en el punto 3.3.1 del apartado de material y
métodos, se procedió a la clonación de los siguientes constructos en el plásmido
pET20b:
EDA-SIINFEKL
C5a-EDA-SIINFEKL
EDA-SIINFEKL-C5a
SIINFEKL-C5a
El plásmido pET20b permite la expresión en bacterias de proteínas de
fusión con una cola de 6 histidinas en el extremo C terminal. Todas las
construcciones fueron confirmadas por secuenciación de DNA.

Resultados
111
Las secuencias de EDA y del fragmento C5a presentaron un 100% de
homología con las ya descritas en las bases de datos públicas.
Secuencia de EDA:
ATGAACATTGATCGCCCTAAAGGACTGGCATTCACTGATGTGGATGTCGATTCC
ATCAAAATTGCTTGGGAAAGCCCACAGGGGCAAGTTTCCAGGTACAGGGTGAC
CTACTCGAGCCCTGAGGATGGAATCCGGGAGCTTTTCCCTGCACCTGATGGTGA
AGACGACACTGCAGAGCTGCAGGGCCTCAGGCCGGGGTCTGAGTACACAGTC
AGTGTGGTTGCCTTGCACGATGATATGGAGAGCCAGCCCCTGATTGGAATCCA
GTCCACAGCGGCCGCACTCGAGCACCACCACCACCACCACTGA
Secuencia de C5a:
AACCTGCATCTCCTAAGGCAGAAAATAGAAGAACAAGCTGCTAAGTACAAAC
ATAGTGTGCCAAAGAAATGCTGCTATGACGGAGCCCGAGTGAACTTCTACGAA
ACCTGTGAGGAGCGAGTGGCCCGGGTTACCATAGGCCCTCTCTGCATCAGGGC
CTTCAACGAGTGCTGTACTATTGCGAACAAGATCCGAAAAGAAAGCCCCCATA
AACCTGTCCAACTGGGAAGGATCCACATTAAGACCCTGTTACCAGTGATGAAG
GCAGATATC
Tanto la proteína EDA-SIINFEKL como la proteína SIINFEKL-C5a tienen
un tamaño teórico de 14 KDa. C5a-EDA-SIINFEKL presenta un tamaño de 25.8
KDa mientras que EDA-SIINFEKL-C5a tiene un tamaño de 27 KDa debido a la
presencia de una secuencia espaciadora (GS(G4S)2GS ) entre SIINFEKL y la cola
de 6 histidinas que permite mejorar la flexibilidad del plegado de la proteína
EDA-SIINFEKL-C5a.
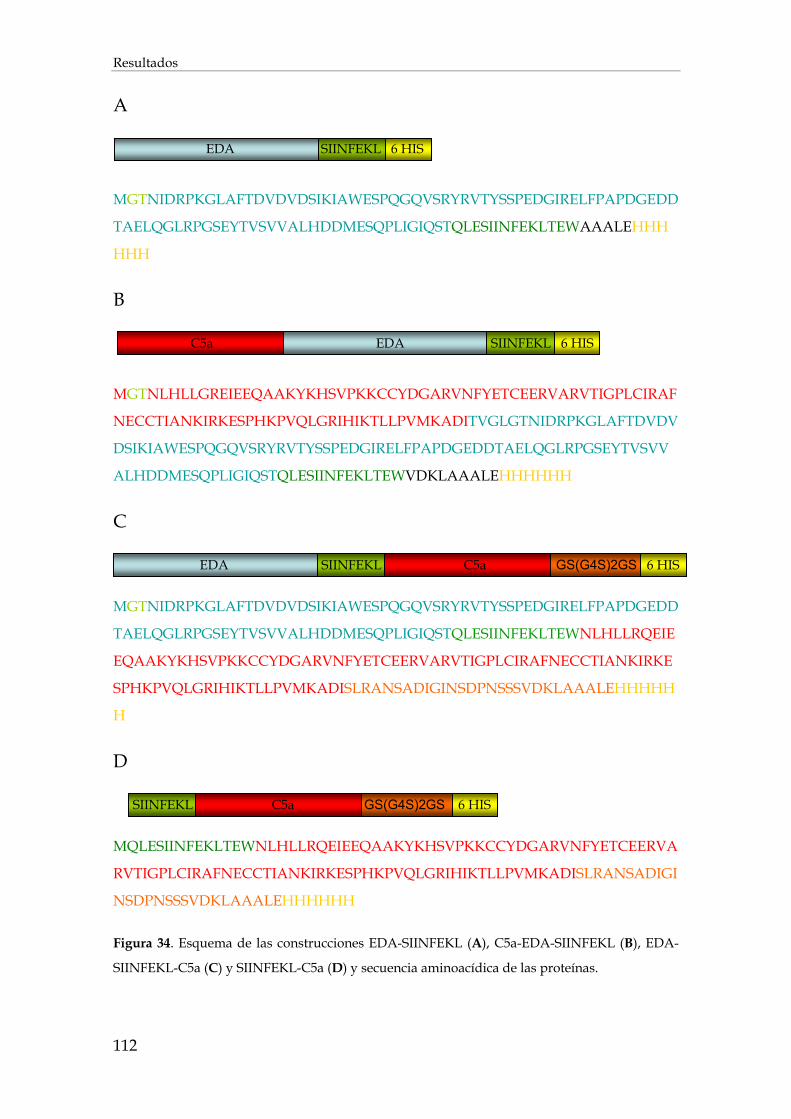
Resultados
112
A
EDA SIINFEKL 6 HISEDA SIINFEKL 6 HIS
MGTNIDRPKGLAFTDVDVDSIKIAWESPQGQVSRYRVTYSSPEDGIRELFPAPDGEDD
TAELQGLRPGSEYTVSVVALHDDMESQPLIGIQSTQLESIINFEKLTEWAAALEHHH
HHH
B
C5a EDA SIINFEKL 6 HISC5a EDA SIINFEKL 6 HIS
MGTNLHLLGREIEEQAAKYKHSVPKKCCYDGARVNFYETCEERVARVTIGPLCIRAF
NECCTIANKIRKESPHKPVQLGRIHIKTLLPVMKADITVGLGTNIDRPKGLAFTDVDV
DSIKIAWESPQGQVSRYRVTYSSPEDGIRELFPAPDGEDDTAELQGLRPGSEYTVSVV
ALHDDMESQPLIGIQSTQLESIINFEKLTEWVDKLAAALEHHHHHH
C
EDA SIINFEKL C5a GS(G4S)2GS 6 HISEDA SIINFEKL C5a GS(G4S)2GS 6 HIS
MGTNIDRPKGLAFTDVDVDSIKIAWESPQGQVSRYRVTYSSPEDGIRELFPAPDGEDD
TAELQGLRPGSEYTVSVVALHDDMESQPLIGIQSTQLESIINFEKLTEWNLHLLRQEIE
EQAAKYKHSVPKKCCYDGARVNFYETCEERVARVTIGPLCIRAFNECCTIANKIRKE
SPHKPVQLGRIHIKTLLPVMKADISLRANSADIGINSDPNSSSVDKLAAALEHHHHH
H
D
SIINFEKL C5a GS(G4S)2GS 6 HISSIINFEKL C5a GS(G4S)2GS 6 HIS
MQLESIINFEKLTEWNLHLLRQEIEEQAAKYKHSVPKKCCYDGARVNFYETCEERVA
RVTIGPLCIRAFNECCTIANKIRKESPHKPVQLGRIHIKTLLPVMKADISLRANSADIGI
NSDPNSSSVDKLAAALEHHHHHH
Figura 34. Esquema de las construcciones EDA-SIINFEKL (A), C5a-EDA-SIINFEKL (B), EDA-
SIINFEKL-C5a (C) y SIINFEKL-C5a (D) y secuencia aminoacídica de las proteínas.
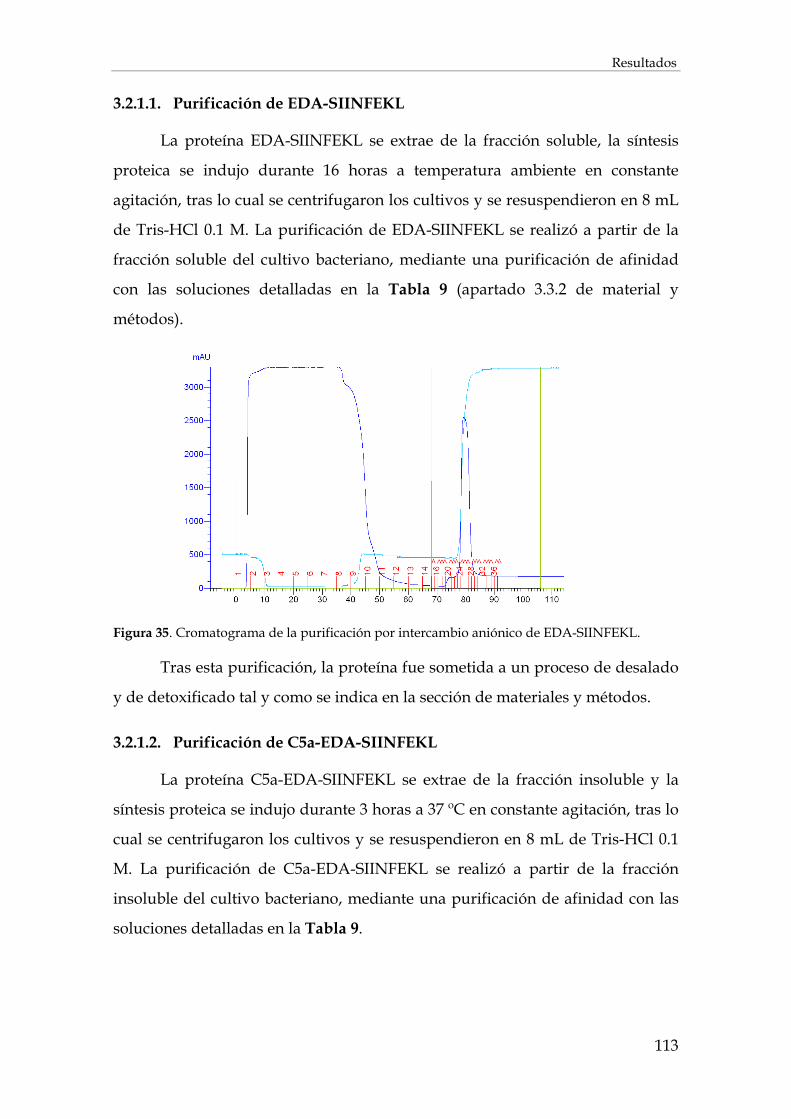
Resultados
113
3.2.1.1. Purificación de EDA-SIINFEKL
La proteína EDA-SIINFEKL se extrae de la fracción soluble, la síntesis
proteica se indujo durante 16 horas a temperatura ambiente en constante
agitación, tras lo cual se centrifugaron los cultivos y se resuspendieron en 8 mL
de Tris-HCl 0.1 M. La purificación de EDA-SIINFEKL se realizó a partir de la
fracción soluble del cultivo bacteriano, mediante una purificación de afinidad
con las soluciones detalladas en la Tabla 9 (apartado 3.3.2 de material y
métodos).
Figura 35. Cromatograma de la purificación por intercambio aniónico de EDA-SIINFEKL.
Tras esta purificación, la proteína fue sometida a un proceso de desalado
y de detoxificado tal y como se indica en la sección de materiales y métodos.
3.2.1.2. Purificación de C5a-EDA-SIINFEKL
La proteína C5a-EDA-SIINFEKL se extrae de la fracción insoluble y la
síntesis proteica se indujo durante 3 horas a 37 ºC en constante agitación, tras lo
cual se centrifugaron los cultivos y se resuspendieron en 8 mL de Tris-HCl 0.1
M. La purificación de C5a-EDA-SIINFEKL se realizó a partir de la fracción
insoluble del cultivo bacteriano, mediante una purificación de afinidad con las
soluciones detalladas en la Tabla 9.
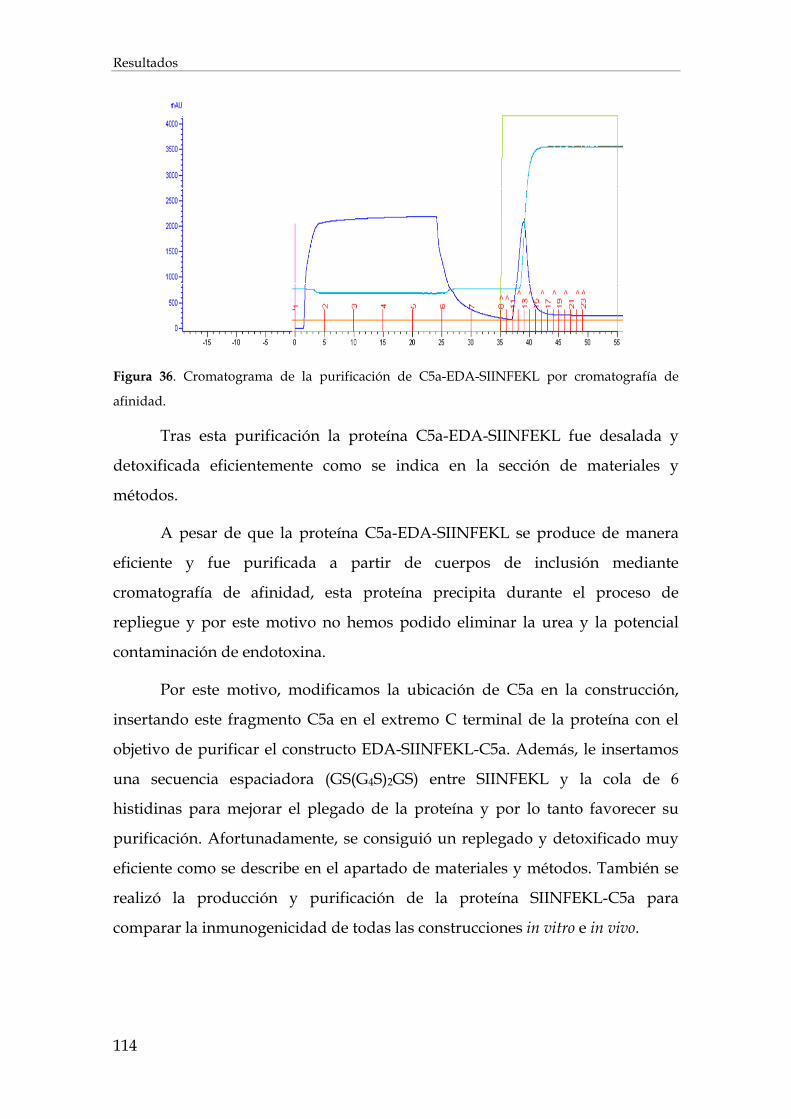
Resultados
114
Figura 36. Cromatograma de la purificación de C5a-EDA-SIINFEKL por cromatografía de
afinidad.
Tras esta purificación la proteína C5a-EDA-SIINFEKL fue desalada y
detoxificada eficientemente como se indica en la sección de materiales y
métodos.
A pesar de que la proteína C5a-EDA-SIINFEKL se produce de manera
eficiente y fue purificada a partir de cuerpos de inclusión mediante
cromatografía de afinidad, esta proteína precipita durante el proceso de
repliegue y por este motivo no hemos podido eliminar la urea y la potencial
contaminación de endotoxina.
Por este motivo, modificamos la ubicación de C5a en la construcción,
insertando este fragmento C5a en el extremo C terminal de la proteína con el
objetivo de purificar el constructo EDA-SIINFEKL-C5a. Además, le insertamos
una secuencia espaciadora (GS(G4S)2GS) entre SIINFEKL y la cola de 6
histidinas para mejorar el plegado de la proteína y por lo tanto favorecer su
purificación. Afortunadamente, se consiguió un replegado y detoxificado muy
eficiente como se describe en el apartado de materiales y métodos. También se
realizó la producción y purificación de la proteína SIINFEKL-C5a para
comparar la inmunogenicidad de todas las construcciones in vitro e in vivo.
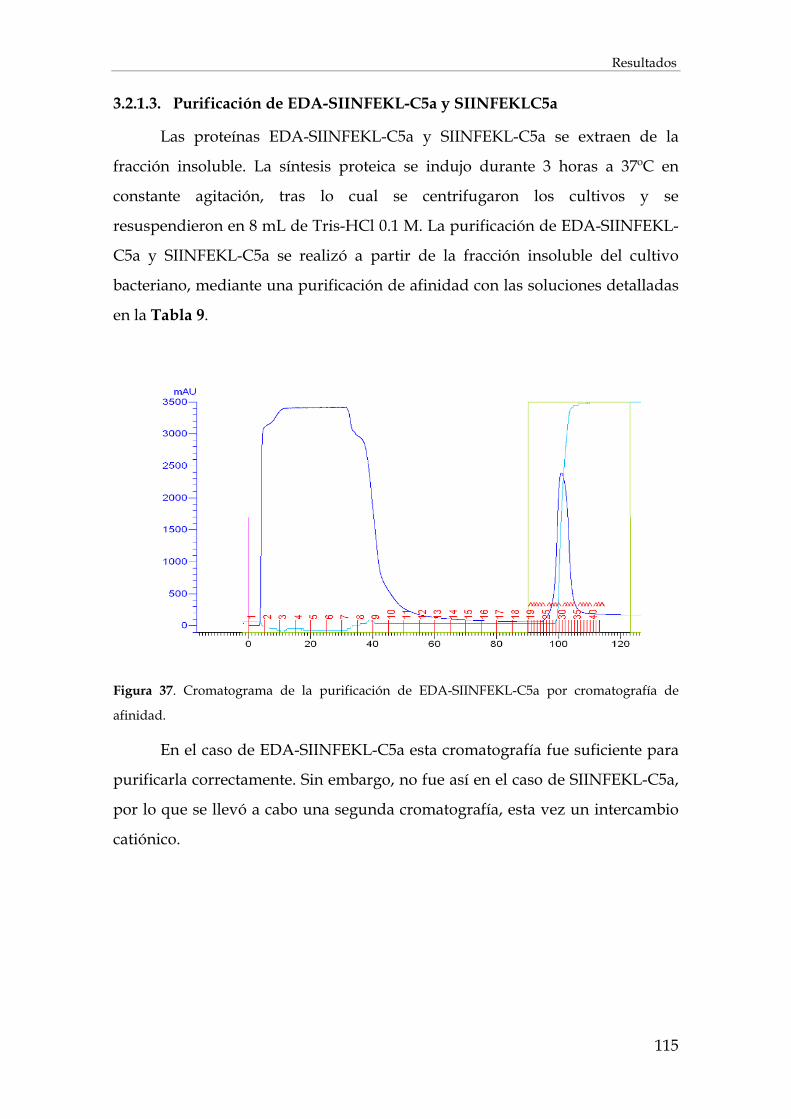
Resultados
115
3.2.1.3. Purificación de EDA-SIINFEKL-C5a y SIINFEKLC5a
Las proteínas EDA-SIINFEKL-C5a y SIINFEKL-C5a se extraen de la
fracción insoluble. La síntesis proteica se indujo durante 3 horas a 37ºC en
constante agitación, tras lo cual se centrifugaron los cultivos y se
resuspendieron en 8 mL de Tris-HCl 0.1 M. La purificación de EDA-SIINFEKL-
C5a y SIINFEKL-C5a se realizó a partir de la fracción insoluble del cultivo
bacteriano, mediante una purificación de afinidad con las soluciones detalladas
en la Tabla 9.
Figura 37. Cromatograma de la purificación de EDA-SIINFEKL-C5a por cromatografía de
afinidad.
En el caso de EDA-SIINFEKL-C5a esta cromatografía fue suficiente para
purificarla correctamente. Sin embargo, no fue así en el caso de SIINFEKL-C5a,
por lo que se llevó a cabo una segunda cromatografía, esta vez un intercambio
catiónico.
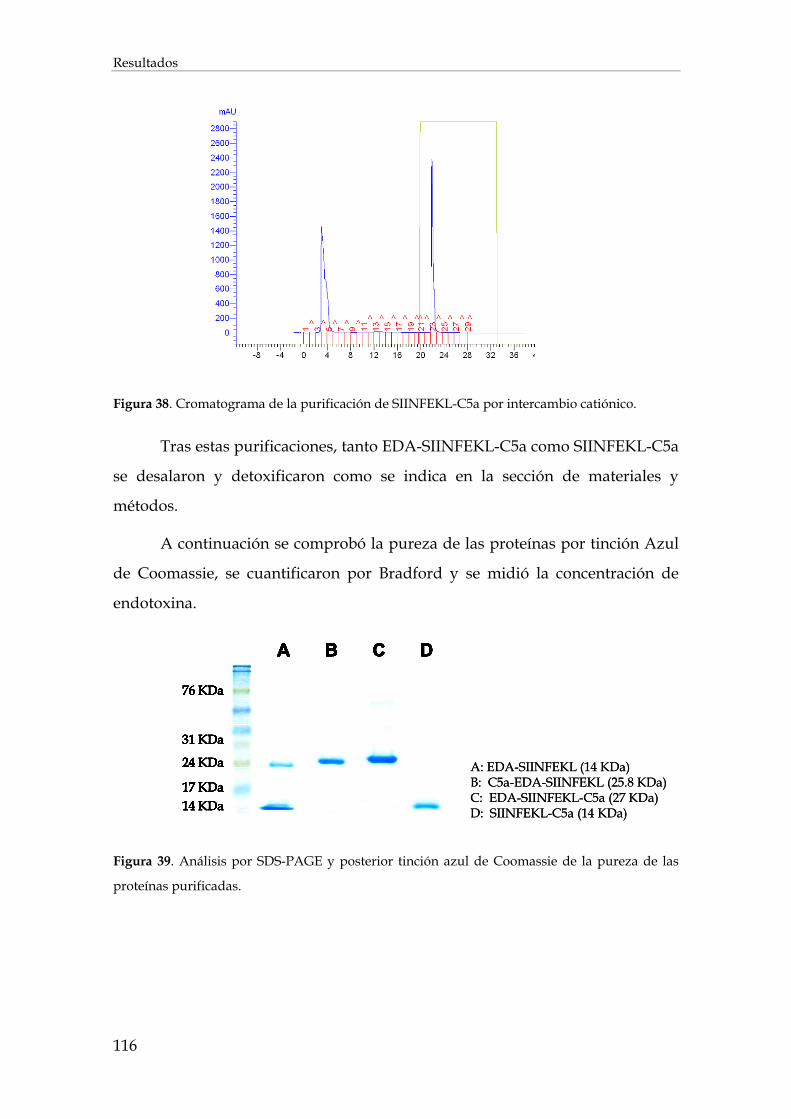
Resultados
116
Figura 38. Cromatograma de la purificación de SIINFEKL-C5a por intercambio catiónico.
Tras estas purificaciones, tanto EDA-SIINFEKL-C5a como SIINFEKL-C5a
se desalaron y detoxificaron como se indica en la sección de materiales y
métodos.
A continuación se comprobó la pureza de las proteínas por tinción Azul
de Coomassie, se cuantificaron por Bradford y se midió la concentración de
endotoxina.
A B C D
14 KDa17 KDa
24 KDa
31 KDa
76 KDa
A B C D
14 KDa17 KDa
24 KDa
31 KDa
76 KDa
A B C D
14 KDa17 KDa
24 KDa
31 KDa
76 KDa
A B C D
14 KDa17 KDa
24 KDa
31 KDa
76 KDa
A: EDA-SIINFEKL (14 KDa)B: C5a-EDA-SIINFEKL (25.8 KDa)C: EDA-SIINFEKL-C5a (27 KDa)D: SIINFEKL-C5a (14 KDa)
A B C D
14 KDa17 KDa
24 KDa
31 KDa
76 KDa
A B C D
14 KDa17 KDa
24 KDa
31 KDa
76 KDa
A B C D
14 KDa17 KDa
24 KDa
31 KDa
76 KDa
A B C D
14 KDa17 KDa
24 KDa
31 KDa
76 KDa
A B C D
14 KDa17 KDa
24 KDa
31 KDa
76 KDa
A B C D
14 KDa17 KDa
24 KDa
31 KDa
76 KDa
A: EDA-SIINFEKL (14 KDa)B: C5a-EDA-SIINFEKL (25.8 KDa)C: EDA-SIINFEKL-C5a (27 KDa)D: SIINFEKL-C5a (14 KDa)
Figura 39. Análisis por SDS-PAGE y posterior tinción azul de Coomassie de la pureza de las
proteínas purificadas.
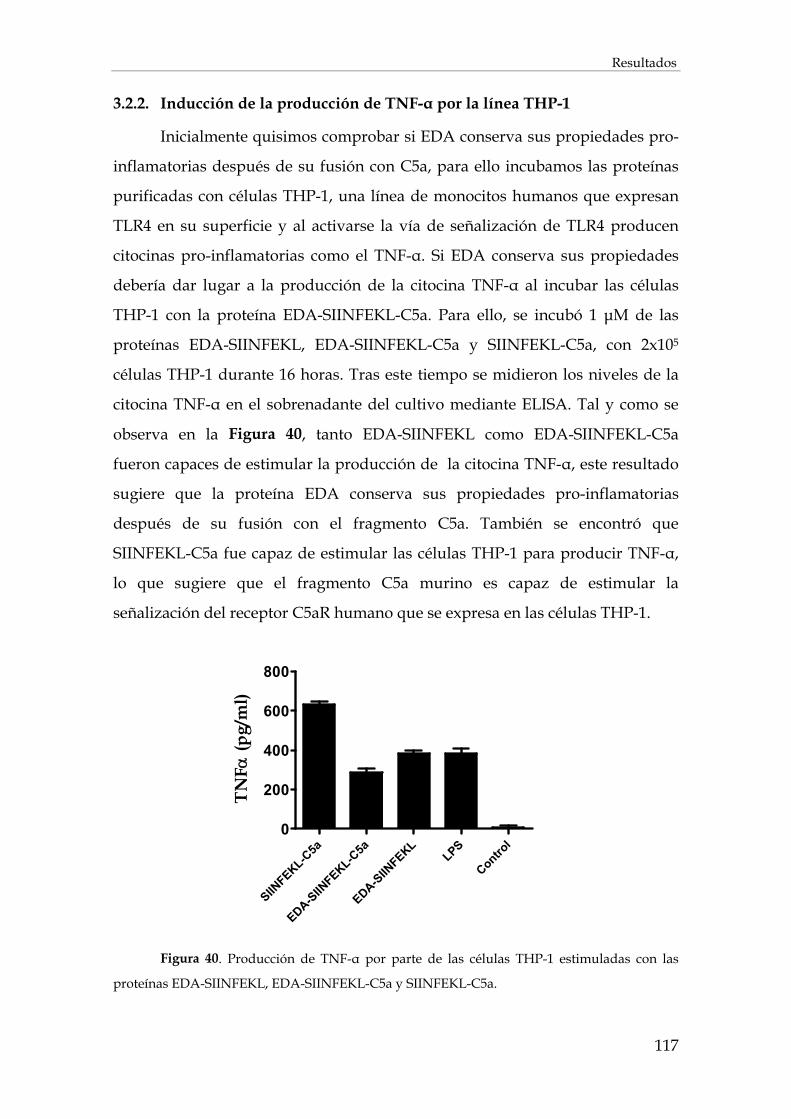
Resultados
117
3.2.2. Inducción de la producción de TNF-α por la línea THP-1
Inicialmente quisimos comprobar si EDA conserva sus propiedades pro-
inflamatorias después de su fusión con C5a, para ello incubamos las proteínas
purificadas con células THP-1, una línea de monocitos humanos que expresan
TLR4 en su superficie y al activarse la vía de señalización de TLR4 producen
citocinas pro-inflamatorias como el TNF-α. Si EDA conserva sus propiedades
debería dar lugar a la producción de la citocina TNF-α al incubar las células
THP-1 con la proteína EDA-SIINFEKL-C5a. Para ello, se incubó 1 μM de las
proteínas EDA-SIINFEKL, EDA-SIINFEKL-C5a y SIINFEKL-C5a, con 2x105
células THP-1 durante 16 horas. Tras este tiempo se midieron los niveles de la
citocina TNF-α en el sobrenadante del cultivo mediante ELISA. Tal y como se
observa en la Figura 40, tanto EDA-SIINFEKL como EDA-SIINFEKL-C5a
fueron capaces de estimular la producción de la citocina TNF-α, este resultado
sugiere que la proteína EDA conserva sus propiedades pro-inflamatorias
después de su fusión con el fragmento C5a. También se encontró que
SIINFEKL-C5a fue capaz de estimular las células THP-1 para producir TNF-α,
lo que sugiere que el fragmento C5a murino es capaz de estimular la
señalización del receptor C5aR humano que se expresa en las células THP-1.
Figura 40. Producción de TNF-α por parte de las células THP-1 estimuladas con las
proteínas EDA-SIINFEKL, EDA-SIINFEKL-C5a y SIINFEKL-C5a.
SIINFEKL-C
5a
EDA-SIIN
FEKL-C5a
EDA-SIIN
FEKLLPS
Control
0
200
400
600
800
TN
F (p
g/m
l)

Resultados
118
3.2.3. Maduración de células dendríticas.
La proteína EDA activa la vía de señalización TLR4 e induce la
maduración de la DC y la producción de citocinas pro-inflamatorias (Lasarte et
al. 2007; Okamura et al. 2001), como la interleucina IL-12 o el TNF-α. Por lo
tanto, queríamos comprobar si las proteínas de fusión SIINFEKL-C5a y EDA-
SIINFEKL-C5a también eran capaces de inducir la activación de las células
dendríticas derivadas de médula ósea tanto de ratones C57BL/6 C5aR+/+
como de ratones C57BL/6 C5aR -/-.
Para ello se analizaron los marcadores de maduración H2Kb, IAb, CD54,
CD80 y CD86 en la superfície de las células dendríticas por citometría de flujo,
así como la inducción de citocinas y quimiocinas por array.
3.2.3.1. Estudio de la expresión de marcadores de superficie en las células
dendríticas
Nuestros resultados muestran que tanto SIINFEKL-C5a como EDA-
SIINFEKL-C5a, inducen la expresión de marcadores de maduración en la
superficie de las DC y que apenas hay diferencias de expresión en la incubación
con la proteína EDA-SIINFEKL. La capacidad de EDA-SIINFEKL-C5a de
inducir la maduración de las DC, es independiente de la presencia de la
molécula C5aR, ya que al incubar esta proteína con las DC de ratones C5aR-KO,
no observamos un descenso en la expresión de los marcadores de superficie. Sin
embargo, las células DC de ratones C5aR-KO incubadas con la proteína
SIINFEKL-C5a, presentan un descenso en la expresión de las moléculas IAb,
CD54 y CD86. Por otra parte, se observa un aumento de la expresión de la
molécula H2Kb en las DC de ratones C5aR-KO cuando son incubadas con
cualquiera de los estímulos indicados (Figura 41).
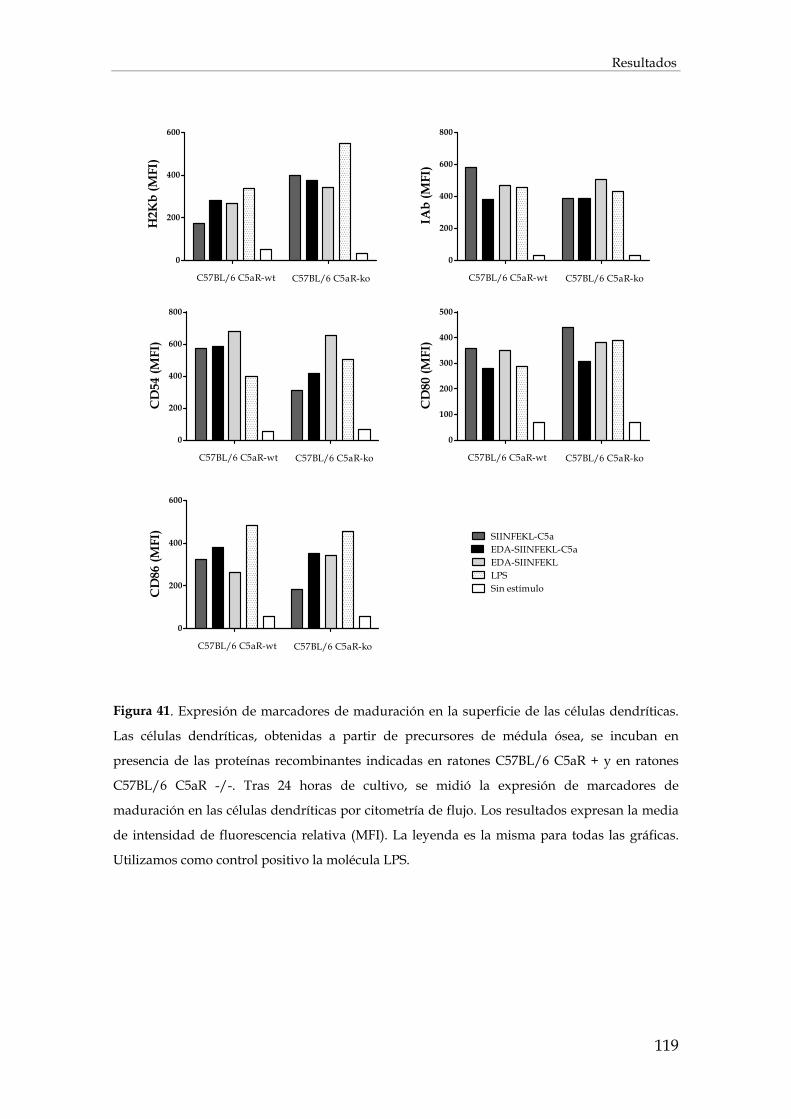
Resultados
119
Figura 41. Expresión de marcadores de maduración en la superficie de las células dendríticas.
Las células dendríticas, obtenidas a partir de precursores de médula ósea, se incuban en
presencia de las proteínas recombinantes indicadas en ratones C57BL/6 C5aR + y en ratones
C57BL/6 C5aR -/-. Tras 24 horas de cultivo, se midió la expresión de marcadores de
maduración en las células dendríticas por citometría de flujo. Los resultados expresan la media
de intensidad de fluorescencia relativa (MFI). La leyenda es la misma para todas las gráficas.
Utilizamos como control positivo la molécula LPS.
0
200
400
600
800
C57BL/6 C5aR-wt C57BL/6 C5aR-ko
CD
54 (M
FI)
0
100
200
300
400
500
C57BL/6 C5aR-wt C57BL/6 C5aR-ko
CD
80 (M
FI)
0
200
400
600
C57BL/6 C5aR-wt C57BL/6 C5aR-ko
SIINFEKL-C5aEDA-SIINFEKL-C5aEDA-SIINFEKLLPSSin estímuloC
D86
(MFI
)
0
200
400
600
800
C57BL/6 C5aR-wt C57BL/6 C5aR-ko
IAb
(MFI
)
0
200
400
600
C57BL/6 C5aR-wt C57BL/6 C5aR-ko
H2K
b (M
FI)

Resultados
120
3.2.3.2. Inducción de citocinas y quimiocinas por las células dendríticas
A continuación pusimos a prueba la capacidad de la proteína EDA-
SIINFEKL-C5a, para activar las células dendríticas y para producir citocinas
pro-inflamatorias, comparando su actividad a la inducida por la proteína EDA-
SIINFEKL. Para ello, las DC de médula ósea derivadas de ratones C57BL/6 se
cultivaron en presencia de EDA-SIINFEKL-C5a (100 nM), EDA-SIINFEKL (100
nM) o medio de cultivo como control. Después de 24 horas de cultivo, se
recogieron los sobrenadantes, y se cuantificó la presencia de citocinas mediante
el uso de un array de citocinas (Raybio). Se encontró que la proteína EDA-
SIINFEKL, así como EDA-SIINFEKL-C5a inducían la producción de varias
citocinas pro-inflamatorias y quimiocinas como Rantes, Mip2, Mip-1beta, Mip-
1alfa, IL-12, IL-6, IL-4 o eotaxina 2 (Panel A y B, Figura 42). Curiosamente,
aunque en la mayoría de los casos la capacidad de inducir moléculas fue
ligeramente inferior para EDA-SIINFEKL-C5a, se comprobó que esta proteína
es capaz de inducir la regulación al alza de las quimiocinas CXCL16, eotaxina,
CTACK y BLC más eficiente que EDA-SIINFEKL (Panel B, Figura 42). Entre
estas quimiocinas destacamos la quimiocina CXCL16 la cual es producida por
las células dendríticas que se encuentran en las zonas de células T de los
órganos linfoides y en la pulpa roja del bazo. Las células que se unen y migran
en respuesta a CXCL16 incluyen varios subconjuntos de células T
(principalmente CD8+ activados) y células NK.
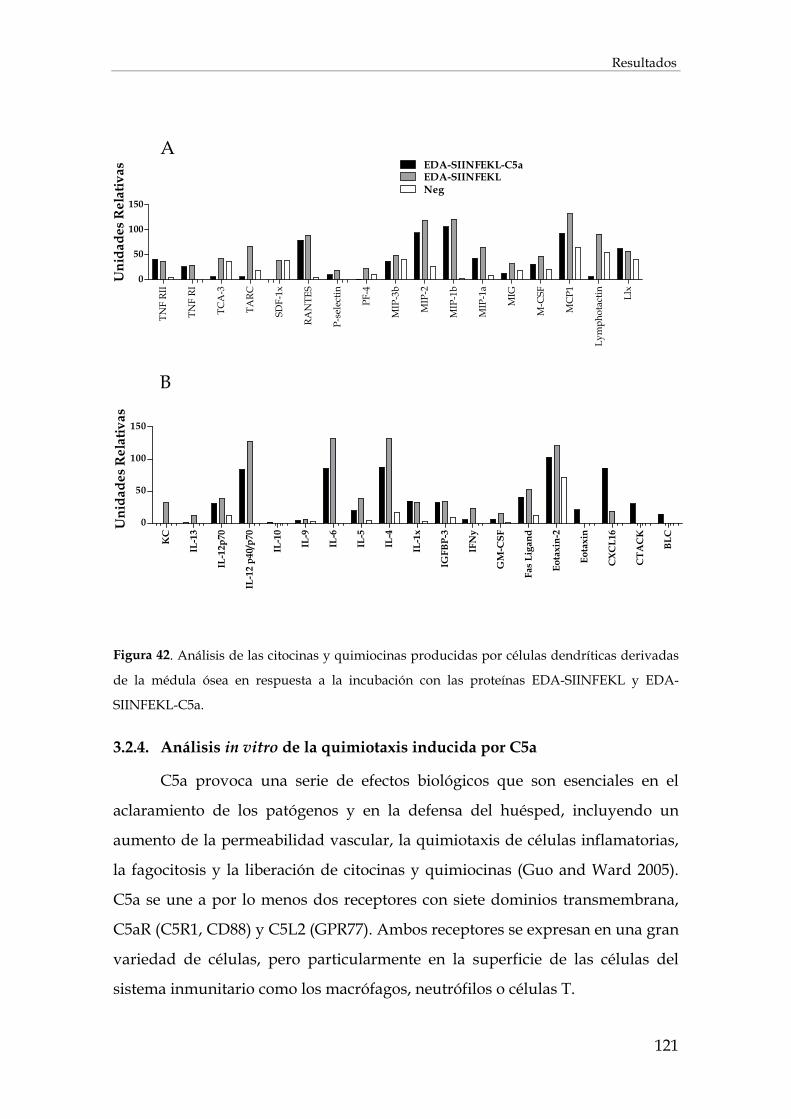
Resultados
121
A
B
Figura 42. Análisis de las citocinas y quimiocinas producidas por células dendríticas derivadas
de la médula ósea en respuesta a la incubación con las proteínas EDA-SIINFEKL y EDA-
SIINFEKL-C5a.
3.2.4. Análisis in vitro de la quimiotaxis inducida por C5a
C5a provoca una serie de efectos biológicos que son esenciales en el
aclaramiento de los patógenos y en la defensa del huésped, incluyendo un
aumento de la permeabilidad vascular, la quimiotaxis de células inflamatorias,
la fagocitosis y la liberación de citocinas y quimiocinas (Guo and Ward 2005).
C5a se une a por lo menos dos receptores con siete dominios transmembrana,
C5aR (C5R1, CD88) y C5L2 (GPR77). Ambos receptores se expresan en una gran
variedad de células, pero particularmente en la superficie de las células del
sistema inmunitario como los macrófagos, neutrófilos o células T.
KC
IL-1
3
IL-1
2p70
IL-1
2 p
40/p
70
IL-1
0
IL-9
IL-6
IL-5
IL-4
IL-1
x
IGFB
P-3
IFN
y
GM
-CS
F
Fas
Lig
and
Eot
axin
-2
Eot
axin
CX
CL
16
CT
AC
K
BL
C
0
50
100
150
U
nid
ades
Rel
ativ
as
TN
F R
II
TN
F R
I
TC
A-3
TA
RC
SDF-
1x
RA
NT
ES
P-s
elec
tin
PF-
4
MIP
-3b
MIP
-2
MIP
-1b
MIP
-1a
MIG
M-C
SF
MC
P1
Lym
phot
acti
n
Llx
0
50
100
150
EDA-SIINFEKLEDA-SIINFEKL-C5a
Neg
U
nid
ades
Rel
ativ
as

Resultados
122
Con el fin de estudiar si C5a conserva su capacidad de inducir la
producción de quimiocinas y el reclutamiento de células mieloides después de
su fusión con EDA-SIINFEKL se llevaron a cabo ensayos de migración celular in
vitro. Por una parte se comprobó el efecto quimiotáctico directo de las proteínas
EDA-SIINFEKL, EDA-SIINFEKL-C5a y SIINFEKL-C5a, utilizando placas
transwell, añadiendo las proteínas (5 μM) en la cámara inferior del pocillo en
presencia de medio RPMI y en la cámara superior esplenocitos de ratones
C57BL/6. Pasadas tres horas de incubación se cuantificó el número de células
presentes en la cámara inferior por citometría de flujo. Por otra parte, se estudio
la capacidad de las proteínas EDA-SIINFEKL, EDA-SIINFEKL-C5a y
SIINFEKL-C5a en inducir la producción de quimiocinas y a su vez inducir la
quimiotaxis celular debida a dicha producción de quimiocinas. Para ello,
utilizamos el sobrenadante de esplenocitos de ratones C57BL/6 cultivados
durante 3 horas con 5 μM de EDA-SIINFEKL, EDA-SIINFEKL-C5a y SIINFEKL-
C5a o con medio RPMI. Los sobrenadantes condicionados se añadieron en las
placas de 24 pocillos y sembramos esplenocitos frescos en la cámara superior
del transwell como se detalla en el apartado 3.3.4.4 de material y métodos.
Después de 3 horas se cuantificó por citometría de flujo el número de
macrófagos (células Ly6G- CD11b+) presentes en la cámara inferior.
Pudimos confirmar que las proteínas que tienen el fragmento C5a
inducen una migración directa de la serie granulocítica, además el efecto
quimiotáctico es mayor con la unión de C5a a la proteína EDA. Sin embargo la
proteína EDA unida a un antígeno no es capaz de inducir la migración de la
serie granulocítica (Panel A, Figura 43). Además, pudimos confirmar que el
sobrenadante de esplenocitos incubados con SIINFEKL-C5a y EDA-SIINFEKL
C5a inducía una fuerte migración de los macrófagos (Panel B, Figura 43), lo que
sugiere que el fragmento C5a conserva su capacidad para inducir la producción
de quimiocinas después de la fusión con la proteína EDA-SIINFEKL. La
proteína EDA-SIINFEKL inducía una migración leve y significativa de los
macrófagos.
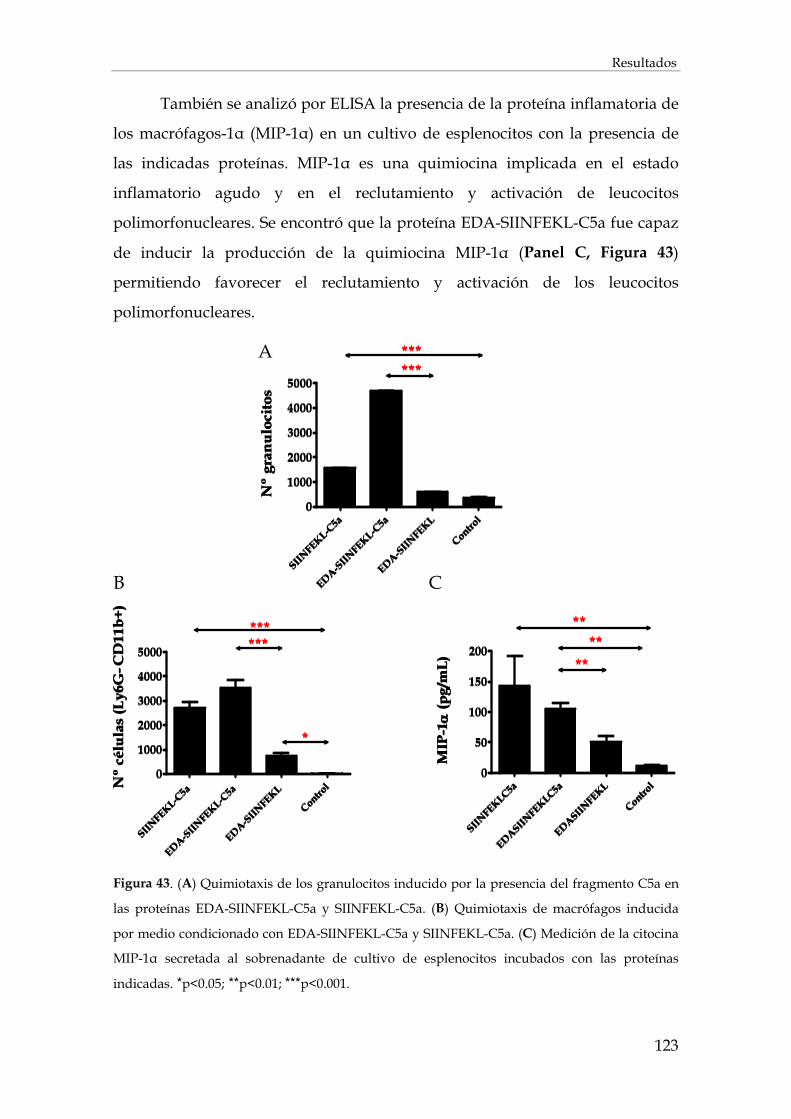
Resultados
123
También se analizó por ELISA la presencia de la proteína inflamatoria de
los macrófagos-1α (MIP-1α) en un cultivo de esplenocitos con la presencia de
las indicadas proteínas. MIP-1α es una quimiocina implicada en el estado
inflamatorio agudo y en el reclutamiento y activación de leucocitos
polimorfonucleares. Se encontró que la proteína EDA-SIINFEKL-C5a fue capaz
de inducir la producción de la quimiocina MIP-1α (Panel C, Figura 43)
permitiendo favorecer el reclutamiento y activación de los leucocitos
polimorfonucleares.
A
B C
Figura 43. (A) Quimiotaxis de los granulocitos inducido por la presencia del fragmento C5a en
las proteínas EDA-SIINFEKL-C5a y SIINFEKL-C5a. (B) Quimiotaxis de macrófagos inducida
por medio condicionado con EDA-SIINFEKL-C5a y SIINFEKL-C5a. (C) Medición de la citocina
MIP-1α secretada al sobrenadante de cultivo de esplenocitos incubados con las proteínas
indicadas. *p<0.05; **p<0.01; ***p<0.001.
SIINFEKL-C
5a
EDA-SIIN
FEKL-C5a
EDA-SIIN
FEKL
Control
0
1000
2000
3000
4000
5000
******
Nº
gran
ulo
cito
s
SIINFEKLC5a
EDASIINFEKLC5a
EDASIINFEKL
Control
0
50
100
150
200 ****
MIP
-1
(p
g/m
L)
**
SIINFEKL-C
5a
EDA-SIIN
FEKL-C5a
EDA-SIIN
FEKL
Control
0
1000
2000
3000
4000
5000
******
*
Nº
célu
las
(Ly6
G-C
D11
b+
)
SIINFEKL-C
5a
EDA-SIIN
FEKL-C5a
EDA-SIIN
FEKL
Control
0
1000
2000
3000
4000
5000
******
Nº
gran
ulo
cito
s
SIINFEKLC5a
EDASIINFEKLC5a
EDASIINFEKL
Control
0
50
100
150
200 ****
MIP
-1
(p
g/m
L)
**
SIINFEKL-C
5a
EDA-SIIN
FEKL-C5a
EDA-SIIN
FEKL
Control
0
1000
2000
3000
4000
5000
******
*
Nº
célu
las
(Ly6
G-C
D11
b+
)

Resultados
124
3.2.5. Presentación antigénica de EDA-SIINFEKL, EDA-SIINFEKL-C5a y
SIINFEKL-C5a
Una respuesta celular T CD8+ (CTL) específica y eficiente requiere que
los antígenos sean presentados en la superfície de las APC, para ello es crucial
que los antígenos sean procesados y posteriormente expuestos en la superficie
celular a través de moléculas de MHC-I.
Por este motivo, potenciar la presentación de un antígeno en la ruta de
presentación antigénica de clase I, es un objetivo en el desarrollo de vacunas
para mejorar la respuesta inmunitaria específica de antígeno. Nuestro grupo ha
descrito que EDA es capaz de favorecer la presentación antigénica (Lasarte et al.
2007), de modo que quisimos estudiar si la unión del fragmento C5a a la
proteína EDA unida covalentemente a un antígeno, podría mejorar su
presentación del antígeno. Para ello, incubamos células B3Z cuya característica
es que reconocen específicamente a SIINFEKL, junto a diferentes
concentraciones de las proteínas recombinates EDA-SIINFEKL, EDA-
SIINFEKL-C5a y SIINFEKL-C5a en presencia de células CD11c+ extraídas de
ratones C57BL/6 WT. La activación de las células B3Z (productoras de
interleucina IL-2) se midió mediante un bioensayo basado en la incubación de
células CTLL2 con los sobrenadantes extraídos de los cultivos de las células B3Z
con las proteínas recombinantes. Al estudiar esta presentación antigénica
observamos que C5a unido covalentemente a un antígeno no es capaz de
favorecer la presentación antigénica. Como esperábamos, encontramos que la
proteína EDA-SIINFEKL permite la presentación antigénica. La inclusión de
C5a no dificultó la capacidad de presentación del antígeno por EDA, si bien se
observó una sensible mejoría en la presentación entre el rango 30 y 250 nM,
resultando ser significativa en la concentración de 125 nM (Figura 44).
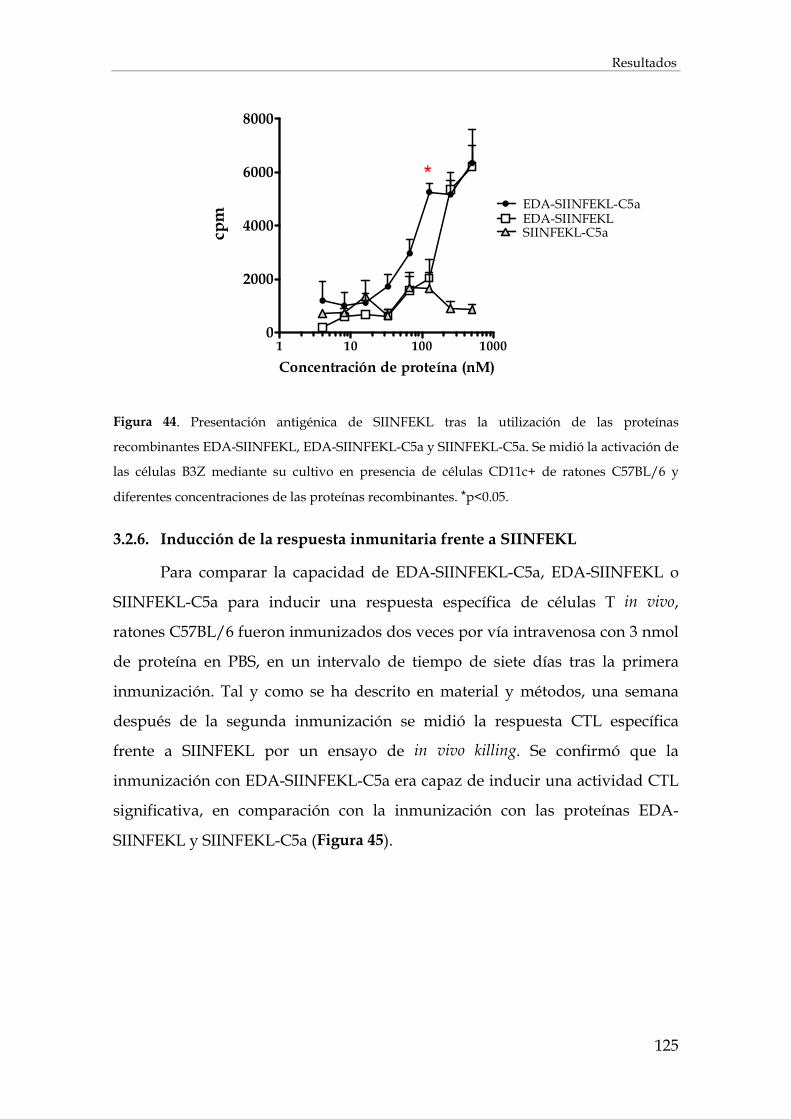
Resultados
125
Figura 44. Presentación antigénica de SIINFEKL tras la utilización de las proteínas
recombinantes EDA-SIINFEKL, EDA-SIINFEKL-C5a y SIINFEKL-C5a. Se midió la activación de
las células B3Z mediante su cultivo en presencia de células CD11c+ de ratones C57BL/6 y
diferentes concentraciones de las proteínas recombinantes. *p<0.05.
3.2.6. Inducción de la respuesta inmunitaria frente a SIINFEKL
Para comparar la capacidad de EDA-SIINFEKL-C5a, EDA-SIINFEKL o
SIINFEKL-C5a para inducir una respuesta específica de células T in vivo,
ratones C57BL/6 fueron inmunizados dos veces por vía intravenosa con 3 nmol
de proteína en PBS, en un intervalo de tiempo de siete días tras la primera
inmunización. Tal y como se ha descrito en material y métodos, una semana
después de la segunda inmunización se midió la respuesta CTL específica
frente a SIINFEKL por un ensayo de in vivo killing. Se confirmó que la
inmunización con EDA-SIINFEKL-C5a era capaz de inducir una actividad CTL
significativa, en comparación con la inmunización con las proteínas EDA-
SIINFEKL y SIINFEKL-C5a (Figura 45).
1 10 100 10000
2000
4000
6000
8000
EDA-SIINFEKLSIINFEKL-C5a
EDA-SIINFEKL-C5a
*
Concentración de proteína (nM)
cpm

Resultados
126
Figura 45. Inducción de una respuesta celular específica in vivo en ratones C57BL/6
inmunizados con las proteínas recombinantes EDA-SIINFEKL, EDA-SIINFEKL-C5a y
SIINFEKL-C5a, utilizando como control el péptido sintético SIINFEKL. ***p<0.001.
A continuación, se estudió si la inmunización con estas proteínas, en
presencia del adyuvante poly(I:C) (ligando de TLR3) era capaz de inducir una
respuesta CTL más eficiente. De modo que, ratones C57BL/6 fueron
inmunizados por vía intravenosa con 3 nmol de EDA-SIINFEKL, EDA-
SIINFEKL-C5a, SIINFEKL-C5a y SIINFEKL en presencia de 50 g del
adyuvante poly(I:C). Como se muestra en la Figura 46 (Panel A), la
inmunización de los ratones con la proteína SIINFEKL-C5a, induce en
combinación con el adyuvante poly(I:C), una fuerte respuesta celular T
específica frente al péptido SIINFEKL. Además, se puede apreciar que la
proteína EDA-SIINFEKL-C5a induce una fuerte actividad CTL in vivo frente a
los esplenocitos pulsados con el péptido SIINFEKL. Esta respuesta fue
significativamente mayor que la alcanzada con las proteínas EDA-SIINFEKL y
SIINFEKL-C5a. El péptido sintético SIINFEKL más poly(I:C) no fue capaz de
inducir respuestas CTL específicas. Se encontraron respuestas similares cuando
se midió por ELISPOT el número de células productoras de IFN- específicas
para SIINFEKL (Panel B, Figura 46).
0 5 10 15 20
***
***
SIINFEKL-C5a
EDA-SIINFEKL-C5a
EDA-SIINFEKL
SIINFEKL
% Lisis específica
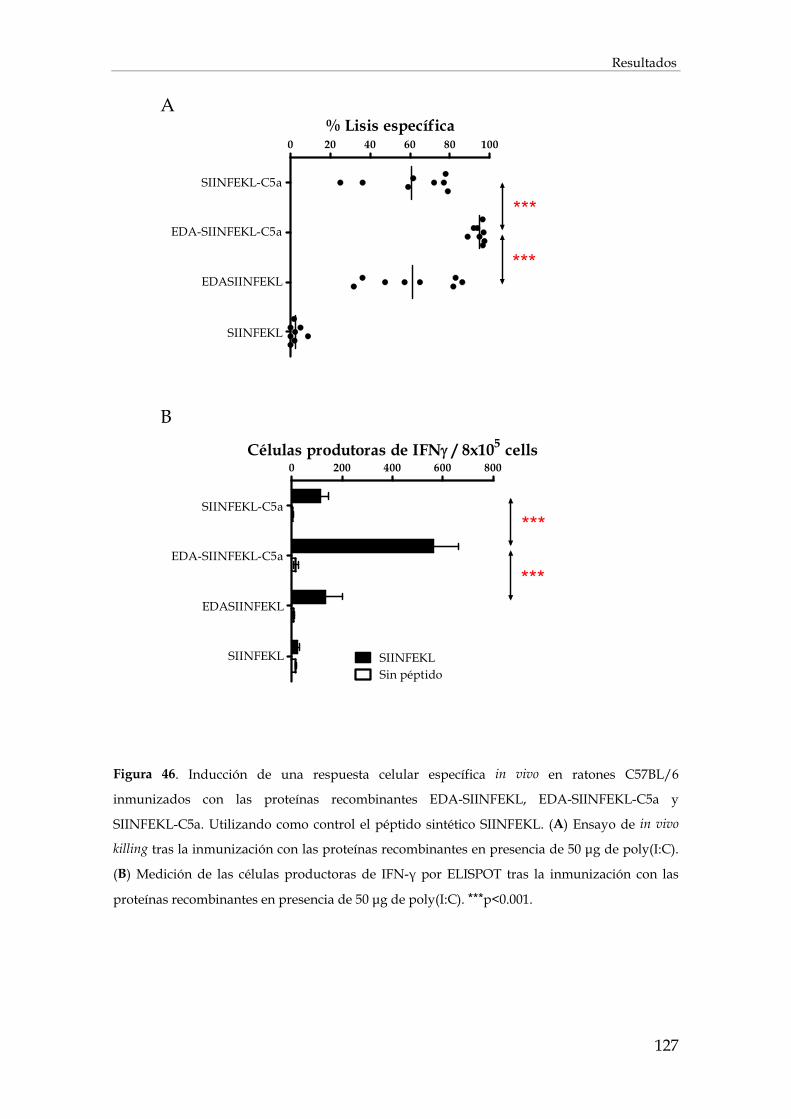
Resultados
127
A
B
Figura 46. Inducción de una respuesta celular específica in vivo en ratones C57BL/6
inmunizados con las proteínas recombinantes EDA-SIINFEKL, EDA-SIINFEKL-C5a y
SIINFEKL-C5a. Utilizando como control el péptido sintético SIINFEKL. (A) Ensayo de in vivo
killing tras la inmunización con las proteínas recombinantes en presencia de 50 μg de poly(I:C).
(B) Medición de las células productoras de IFN-γ por ELISPOT tras la inmunización con las
proteínas recombinantes en presencia de 50 μg de poly(I:C). ***p<0.001.
0 20 40 60 80 100
SIINFEKL-C5a
EDA-SIINFEKL-C5a
EDASIINFEKL
SIINFEKL
***
***
% Lisis específica
0 200 400 600 800
SIINFEKLSin péptido
SIINFEKL-C5a
EDA-SIINFEKL-C5a
EDASIINFEKL
SIINFEKL
***
***
Células produtoras de IFN / 8x105 cells

Resultados
128
3.2.7. Efecto de las proteínas recombinantes sobre la población de las células
T reguladoras en la inducción de la respuesta inmunitaria
Tras observar que C5a unido a un antígeno puede inducir un respuesta
celular específica y que además su presencia como proteína de fusión con EDA
unida a antígeno, mejora la respuesta celular específica, nos preguntamos si la
población de células Treg podría verse afectada por la inmunización con estas
proteínas. Por este motivo, realizamos un marcaje con el anticuerpo anti-FOXP3
PE (eBioscience) de los esplenocitos de ratones obtenidos tras 7 días de
inmunización con las proteínas indicadas y en combinación con 50 μg de
poly(I:C). Estos resultados los analizamos por citometría de flujo. Los datos
indican que la inmunización con la proteína EDA-SIINFEKL-C5a apenas induce
un aumento del porcentaje de células FOXP3+ mientras que sí aumenta tras la
inmunización con las proteínas EDA-SIINFEKL y SIINFEKL-C5a (Figura 47).
Estos resultados se correlacionan con el aumento de respuesta celular T
CD8+ observada con EDA-SIINFEKL-C5a, pudiéndose explicar por una menor
activación y proliferación de las celulas Treg (FOXP3+) que ejercen un efecto
inmunosupresor en la respuesta inmunitaria específica de antígeno.
Figura 47. Marcaje FOXP3 de los esplenocitos de ratones C57BL/6, tras 7 días de la
inmunización con 3 nmol de las proteínas indicadas en combinación con 50 μg de poly(I:C).
*p<0.05.
0
1
2
3
4
Sin estímulopoly(I:C)SIINFEKL-C5aEDA-SIINFEKL-C5aEDA-SIINFEKL
-++--
-+-+-
-+--+
+----
**
ns
% F
OX
P3
/ Esp
len
ocit
os

Resultados
129
Tras estos resultados, comprobamos si la población de células Treg ve
influida su función supresora directa o indirectamente por las proteínas
recombinantes con el fragmento C5a. Para ello, primero quisimos descartar que
tuvieran un efecto sobre la proliferación de las células Tefect. De modo que
realizamos ensayos de proliferación celular in vitro con linfocitos T efectores
CD4+CD25- tanto de ratones C57BL/6 como de ratones BALB/c en presencia
del estímulo anti-CD3 (0.5 μg/mL). Tal y como se muestra en la Figura 48
(Panel A y B) las proteínas recombinantes EDA-SIINFEKL, EDA-SIINFEKL-C5a
y SIINFEKL-C5a no inducen cambios en la proliferación de las células T
efectoras CD4+CD25- ante un estímulo mitogénico. Paralelamente,
comprobamos si la función supresora de las Treg podría verse afectada, para
ello realizamos un ensayo de proliferación celular cocultivando Tefect con Treg
(Panel C y D, Figura 48) en presencia del estímulo anti-CD3 y en presencia de
las proteínas recombinantes indicadas. Nuestros resultados muestran que la
proteína SIINFEKL-C5a es capaz de potenciar el efecto supresor de las Treg,
mientras que no observamos cambios en la función supresora cuando
incubamos con EDA-SIINFEKL-C5a o EDA-SIINFEKL.
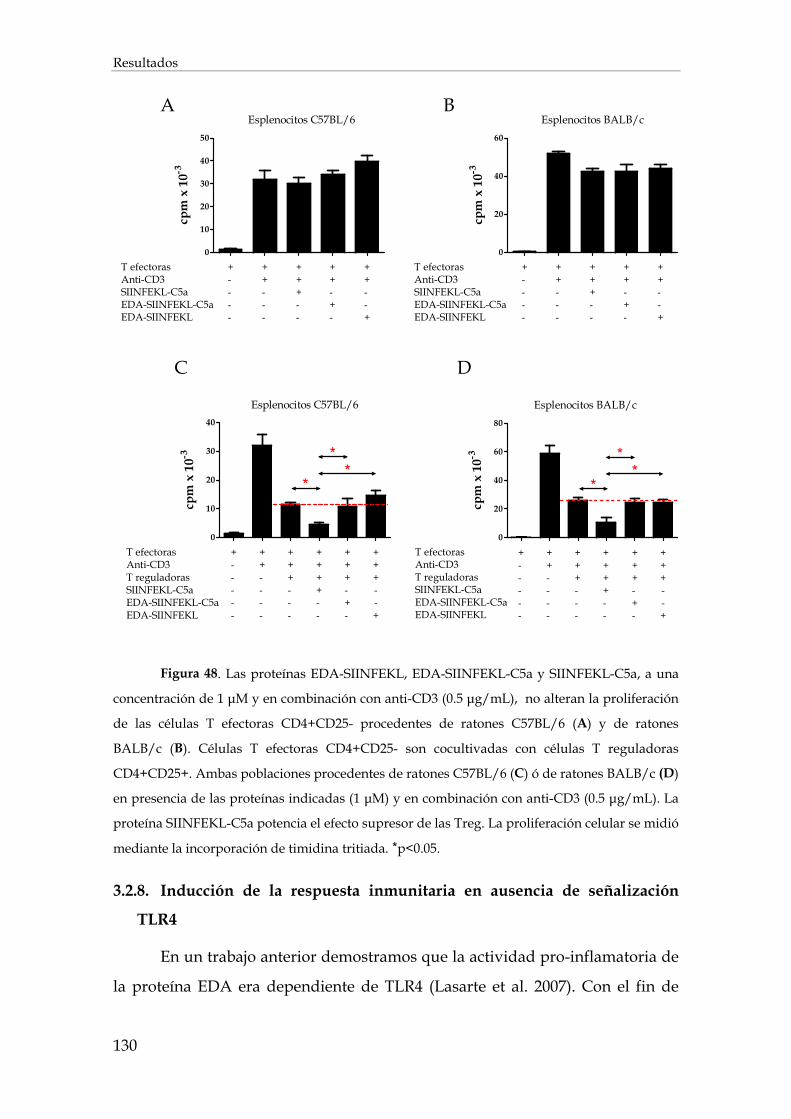
Resultados
130
A B
C D
Figura 48. Las proteínas EDA-SIINFEKL, EDA-SIINFEKL-C5a y SIINFEKL-C5a, a una
concentración de 1 μM y en combinación con anti-CD3 (0.5 μg/mL), no alteran la proliferación
de las células T efectoras CD4+CD25- procedentes de ratones C57BL/6 (A) y de ratones
BALB/c (B). Células T efectoras CD4+CD25- son cocultivadas con células T reguladoras
CD4+CD25+. Ambas poblaciones procedentes de ratones C57BL/6 (C) ó de ratones BALB/c (D)
en presencia de las proteínas indicadas (1 μM) y en combinación con anti-CD3 (0.5 μg/mL). La
proteína SIINFEKL-C5a potencia el efecto supresor de las Treg. La proliferación celular se midió
mediante la incorporación de timidina tritiada. *p<0.05.
3.2.8. Inducción de la respuesta inmunitaria en ausencia de señalización
TLR4
En un trabajo anterior demostramos que la actividad pro-inflamatoria de
la proteína EDA era dependiente de TLR4 (Lasarte et al. 2007). Con el fin de
Esplenocitos C57BL/6
0
10
20
30
40
50
T efectorasAnti-CD3SIINFEKL-C5aEDA-SIINFEKL-C5aEDA-SIINFEKL
+----
++---
+++--
++-+-
++--+
cpm
x 1
0-3
Esplenocitos BALB/c
0
20
40
60
T efectorasAnti-CD3SIINFEKL-C5aEDA-SIINFEKL-C5aEDA-SIINFEKL
+----
++---
+++--
++-+-
++--+
cpm
x 1
0-3
Esplenocitos C57BL/6
0
10
20
30
40
T efectorasAnti-CD3T reguladorasSIINFEKL-C5aEDA-SIINFEKL-C5aEDA-SIINFEKL
+-----
++----
++++--
+++-+-
+++--+
+++---
**
*
cpm
x 1
0-3
Esplenocitos BALB/c
0
20
40
60
80
T efectorasAnti-CD3T reguladorasSIINFEKL-C5aEDA-SIINFEKL-C5aEDA-SIINFEKL
+-----
++----
++++--
+++-+-
+++--+
+++---
**
*cp
m x
10-3

Resultados
131
determinar si la inmunogenicidad de las proteínas de fusión que contienen
EDA o el fragmento C5a dependen de esta vía, se inmunizaron ratones
C57BL/6 y ratones C57BL/6 knockout para TLR4 con EDA-SIINFEKL, EDA-
SIINFEKL-C5a o SIINFEKL-C5a. Estos ensayos se realizaron en colaboración
con las Dras Leclerc y Fayolle del Institute Pasteur (París). Como se muestra en
la Figura 49, la inmunización de ratones C57BL/6 con las citadas proteínas,
induce una fuerte respuesta T citotóxica frente a células diana incubadas con el
péptido SIINFEKL. Como era de esperar, la inmunogenicidad de la proteína
EDA-SIINFEKL se redujo significativamente en los ratones TLR4 KO, aunque es
interesante observar que su actividad no desaparece totalmente. La actividad de
EDA-SIINFEKL-C5a se redujo ligeramente pero se observa que C5a suple con la
ausencia de la activación del receptor TLR4, mientras que la actividad de
SIINFEKL-C5a no se vio afectada por la ausencia de la señalización de TLR4.
Por lo tanto, la capacidad de las proteínas de fusión entre EDA-SIINFEKL y la
proteína C5a para favorecer la inducción de respuestas de células T no requiere
de la señalización TLR4.
Figura 49. Inducción de las respuestas CTL in vivo por las proteínas de fusión recombinantes en
ratones TLR4 KO. Ratones C57BL/6 TLR4+ y ratones TLR4 KO fueron inmunizados con 3 nmol
de las proteínas que se indican con 50 g de poly(I:C). Siete días después de la inmunización,
los ratones fueron sacrificados y la respuesta de células T específica para el péptido SIINFEKL
se midió mediante in vivo killing. **p<0.01.
0 20 40 60 80 100 Ratón wt TLR4Ratón ko TLR4
SIINFEKL-C5a
EDA-SIINFEKL-C5a
EDA-SIINFEKL **
ns
% Lisis específica

Resultados
132
3.2.9. Inducción de la respuesta inmunitaria en ausencia de señalización
C5aR
Como previamente ya se ha comentado, todas las vías de activación del
complemento conducen a la hidrólisis de la molécula C5 del complemento
generándose los fragmentos activos C5a (anafilotoxina) y C5b. C5a ejerce una
actividad predominantemente pro-inflamatoria a través de la interacción con
dos receptores transmembrana de gran afinidad, el receptor C5aR (CD88) que
está acoplado a proteína G, así como el receptor C5L2 (GPR77) que no está
acoplado a proteína G (Manthey et al. 2009; Okinaga et al. 2003). Ambos
receptores se expresan en una gran variedad de células, entre ellas células del
sistema inmunitario como las células dendríticas, macrófagos, monocitos y
linfocitos activados. Sin embargo, el receptor C5L2 se expresa a unos niveles
más bajos que C5aR y además hay controversia con sus funciones, algunos
grupos describen el receptor C5L2 como un scavenger receptor (Gerard et al.
2005) o como modulador negativo de la señal mediada por C5aR (Bamberg et
al. 2010). Sin embargo, otros grupos sugieren acciones pro-inflamatorias para
C5L2 (Rittirsch et al. 2008; Zhang et al. ; Lajoie et al. ; Hashimoto et al.). Por lo
tanto, la función del receptor C5L2 es desconocida o como mínimo está en
controversia.
Por este motivo nos centramos en el receptor C5aR para estudiar el
mecanismo de acción de la proteína EDA-SIINFEKL-C5a. Lo hicimos
estudiando la inmunogenicidad de las proteínas EDA-SIINFEKL, EDA-
SIINFEKL-C5a y SIINFEKL-C5a en ratones knockout para el receptor C5aR y en
ratones tratados con anticuerpo anti-C5aR bloqueante de la función C5aR. Tras
siete días de la inmunización, se midió la respuesta celular específica in vivo
mediante el ensayo de in vivo killling.
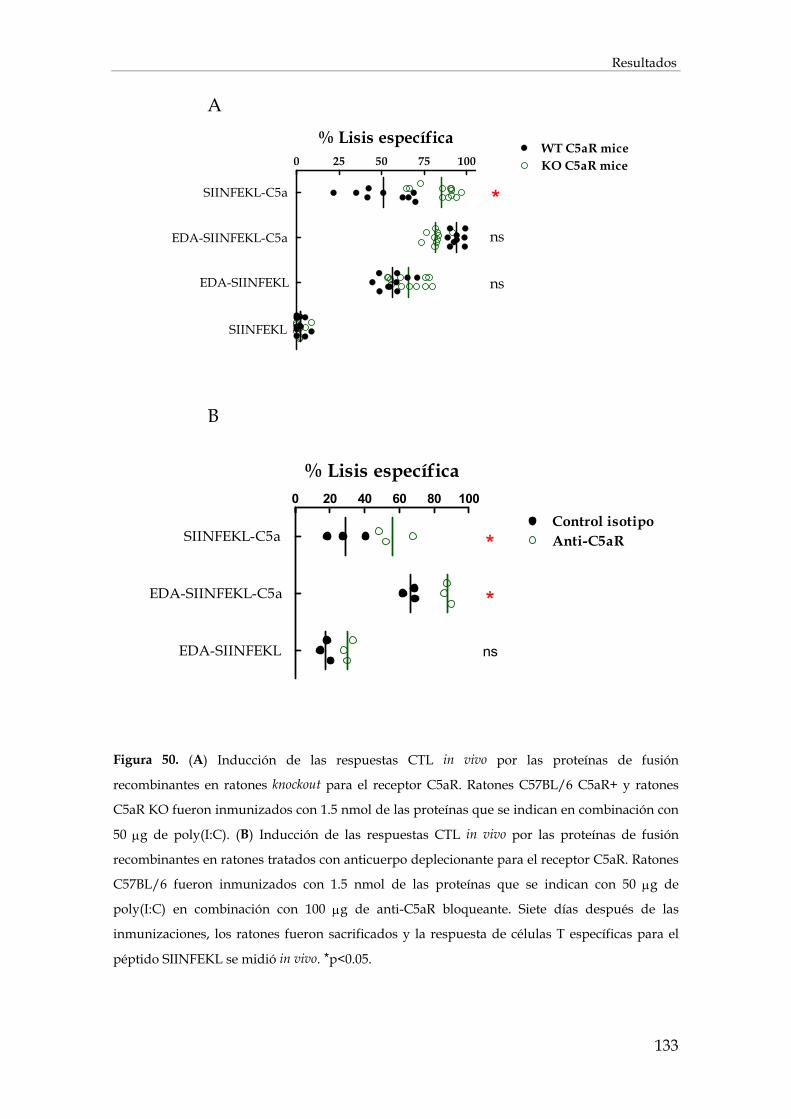
Resultados
133
A
B
0 20 40 60 80 100
Control isotipoAnti-C5aRSIINFEKL-C5a
EDA-SIINFEKL-C5a
EDA-SIINFEKL
*
*
ns
% Lisis específica
Figura 50. (A) Inducción de las respuestas CTL in vivo por las proteínas de fusión
recombinantes en ratones knockout para el receptor C5aR. Ratones C57BL/6 C5aR+ y ratones
C5aR KO fueron inmunizados con 1.5 nmol de las proteínas que se indican en combinación con
50 g de poly(I:C). (B) Inducción de las respuestas CTL in vivo por las proteínas de fusión
recombinantes en ratones tratados con anticuerpo deplecionante para el receptor C5aR. Ratones
C57BL/6 fueron inmunizados con 1.5 nmol de las proteínas que se indican con 50 g de
poly(I:C) en combinación con 100 g de anti-C5aR bloqueante. Siete días después de las
inmunizaciones, los ratones fueron sacrificados y la respuesta de células T específicas para el
péptido SIINFEKL se midió in vivo. *p<0.05.
0 25 50 75 100WT C5aR miceKO C5aR mice
SIINFEKL-C5a
EDA-SIINFEKL-C5a
EDA-SIINFEKL
SIINFEKL
*
ns
ns
% Lisis específica

Resultados
134
Tal y como se aprecia en la Figura 50 (Panel A), la inmunogenicidad de
EDA-SIINFEKL no se vio afectada significativamente por la ausencia de
señalización del receptor C5aR. Sorprendentemente y contrariamente a lo
esperado, la falta de señalización C5aR en los ratones knockout para el receptor
C5aR mejoró la inmunogenicidad de la construcción SIINFEKL-C5a, no así la de
la proteína EDA-SIINFEKL-C5a, para la que no observamos diferencias
significativas. Sin embargo en los ratones tratados con un anticuerpo anti-C5aR,
EDA-SIINFEKL-C5a al igual que SIINFEKL-C5a aumentaba la respuesta frente
a SIINFEKL (Panel B, Figura 50 B). Estos datos sugieren que el efecto
adyuvante del fragmento C5a en estas proteínas recombinantes no es
dependiente del receptor C5aR. Hay que tener en cuenta la existencia de otro
receptor para C5a, el receptor C5L2, que podría ser el responsable de la
capacidad inmunoestimulante de C5a en este modelo.
3.2.10. Inducción de la respuesta inmunitaria NK.
Como se ha descrito anteriormente, la incubación de las células
dendríticas con la proteína EDA-SIINFEKL-C5a mejora la producción de la
quimiocina CXCL16 con respecto a EDA-SIINFEKL, lo que sugiere que las
células NK reclutadas al sitio de deposición del antígeno podrían desempeñar
un papel importante en la inmunogenicidad de la proteína EDA-SIINFEKL-
C5a. Por lo tanto, el fragmento C5a podría ser capaz de potenciar la activación
de las células NK y favorecer la inducción de la respuesta inmunitaria
adaptativa frente al péptido SIINFEKL.
Por este motivo se estudió la actividad NK, determinando el número de
células productoras de IFN-γ mediante ELISPOT 48 horas tras la de
inmunización con las proteínas indicadas. A pesar de que la proteína EDA-
SIINFEKL aumentó la actividad NK, las proteínas SIINFEKL-C5a y EDA-
SIINFEKL-C5a inducían respuestas significativamente mayores (Panel A,
Figura 51). A continuación, se estudió el efecto de la depleción de las células
NK in vivo en la inducción de la respuesta inmunitaria específica de SIINFEKL
tras la inmunización con las proteínas EDA-SIINFEKL, EDA-SIINFEKL-C5a y

Resultados
135
SIINFEKL-C5a (Panel B, Figura 51). Así, los ratones fueron tratados con
anticuerpo anti NK 1.1, consiguiendo una depleción de las células NK superior
al 95%. Con la depleción de las células NK se redujo significativamente la
respuesta inmunitaria inducida por SIINFEKL-C5a. Sin embargo, la depleción
no afecta a la respuesta obtenida con la proteína EDA-SIINFEKL-C5a, mientras
que se observa un ligero incremento en el porcentaje de lisis, así como en el
número de células productoras de IFN-γ en respuesta a SIINFEKL tras la
inmunización con EDA-SIINFEKL (Panel C, Figura 51). Estos datos podrían
sugerir que la actividad inmunoestimuladora del fragmento C5a depende, al
menos parcialmente, de su capacidad para activar las células NK y que la
presencia de EDA en el inmunógeno puede superar la falta de las células NK.

Resultados
136
A
B
C
Figura 51. Implicación de las células NK en la inducción de respuestas específicas de células T
frente a SIINFEKL. (A) ratones C57BL/6 fueron inmunizados con 3 nmol de las proteínas que se
indican con 50 μg de poly(I:C). 48 horas más tarde y utilizando esplenocitos, se midió la
actividad NK frente a células YAC-1, sensibles a la acción lítica de las NK, mediante ELISPOT
(células productoras de IFN-γ). (B y C) Efecto de la depleción de las células NK en la inducción
de la activación celular T in vivo. Los ratones fueron tratados con anti-NK1.1 o anticuerpos de
isotipo control como se describe en el apartado de material y métodos. A continuación, los
ratones fueron inmunizados con el inmunógeno indicado y 7 días más tarde, se midió la
actividad CTL mediante un ensayo de in vivo killing o mediante ELISPOT midiendo el número
de células productoras de IFN-γ espcíficas para el péptido SIINFEKL. *p<0.05; **p<0.01.
0 100 200 300 400 500
**
*
Células productoras de IFN/4x105
SIINFEKL-C5a
EDA-SIINFEKL-C5a
EDA-SIINFEKL
Control
% Lisis específica0 20 40 60 80 100
Control isotipoAnti-NK1.1SIINFEKL-C5a
EDA-SIINFEKL-C5a
EDA-SIINFEKL
*
0 200 400 600 800
Células productoras de IFN/4x105
SIINFEKL-C5a
EDA-SIINFEKL-C5a
EDA-SIINFEKL
Anti-NK1.1Control isotipo
*

Resultados
137
3.2.11. Inducción de protección antitumoral y tratamiento de tumores
establecidos
Teniendo en cuenta que el fragmento del complemento C5a unido
covalentemente a un antígeno (SIINFEKL) y en combinación con el agonista de
TLR3 (poly(I:C)) puede inducir una fuerte respuesta celular T específica,
quisimos determinar su capacidad profiláctica y terapéutica en un modelo
murino de tumor basado en las células tumorales E.G7-OVA. Además,
queríamos evaluar si EDA-SIINFEKL-C5a seguía siendo la mejor estrategia
terapéutica en comparación con las proteínas de fusión monoméricas EDA-
SIINFEKL y SIINFEKL-C5a.
Comenzamos analizando la capacidad profiláctica de las proteínas
indicadas. Para ello, inmunizamos ratones con 3 nmol del inmunógeno
indicado en combinación con poly(I:C) por vía intravenosa. Como control del
crecimiento tumoral un grupo de ratones recibió una solución salina y otro
grupo recibió el antígeno SIINFEKL en combinación con poly(I:C). Siete días
después de la inmunización, inoculamos subcutáneamente 5x105 células
tumorales E.G7-OVA y realizamos el seguimiento del tumor dos veces por
semana utilizando un calibre. En la Figura 52, se representa una gráfica de
Kaplan-Meier de supervivencia y observamos como la proteína SIINFEKL-C5a
confiere un 35% de protección frente a la proteína EDA-SIINFEKL cuya
protección es del 28.57%. Por otra parte, la capacidad de EDA-SIINFEKL-C5a en
inducir una mejor respuesta celular T específica que la proteína EDA-
SIINFEKL, también se traduce en unos niveles significativamente más altos de
protección tumoral frente al tumor E.G7-OVA, observándose un 61.5% de
protección tumoral.
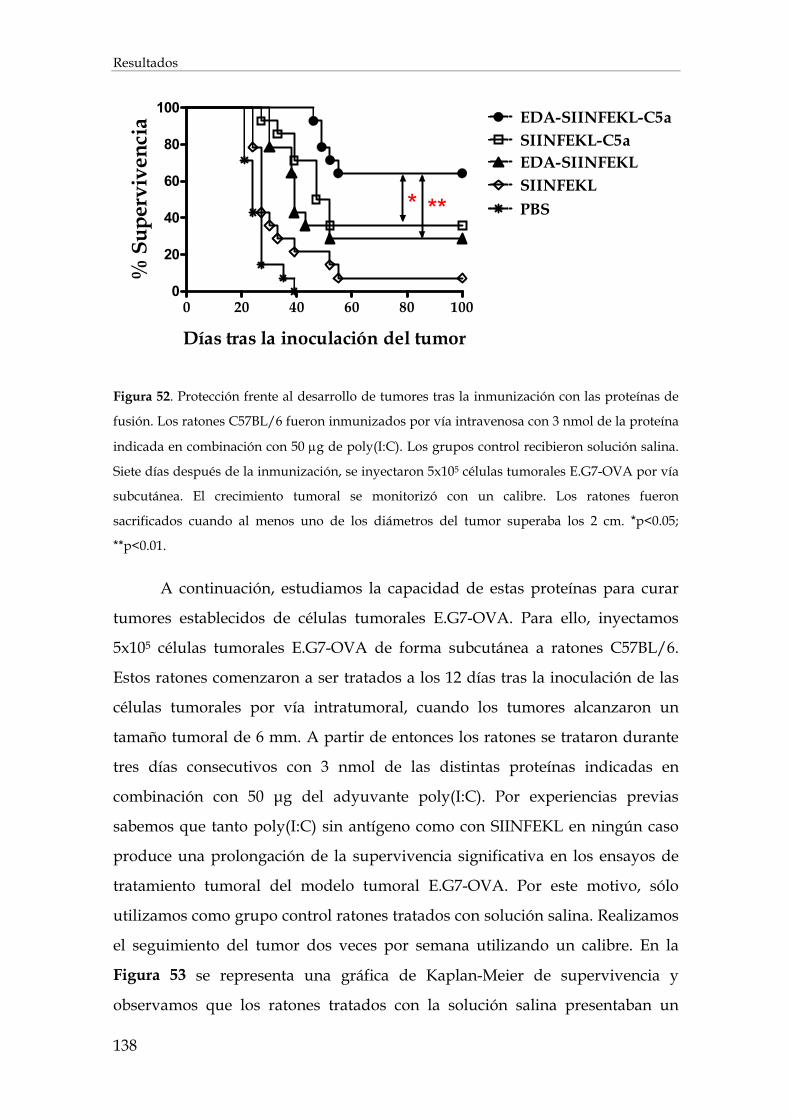
Resultados
138
Figura 52. Protección frente al desarrollo de tumores tras la inmunización con las proteínas de
fusión. Los ratones C57BL/6 fueron inmunizados por vía intravenosa con 3 nmol de la proteína
indicada en combinación con 50 g de poly(I:C). Los grupos control recibieron solución salina.
Siete días después de la inmunización, se inyectaron 5x105 células tumorales E.G7-OVA por vía
subcutánea. El crecimiento tumoral se monitorizó con un calibre. Los ratones fueron
sacrificados cuando al menos uno de los diámetros del tumor superaba los 2 cm. *p<0.05;
**p<0.01.
A continuación, estudiamos la capacidad de estas proteínas para curar
tumores establecidos de células tumorales E.G7-OVA. Para ello, inyectamos
5x105 células tumorales E.G7-OVA de forma subcutánea a ratones C57BL/6.
Estos ratones comenzaron a ser tratados a los 12 días tras la inoculación de las
células tumorales por vía intratumoral, cuando los tumores alcanzaron un
tamaño tumoral de 6 mm. A partir de entonces los ratones se trataron durante
tres días consecutivos con 3 nmol de las distintas proteínas indicadas en
combinación con 50 μg del adyuvante poly(I:C). Por experiencias previas
sabemos que tanto poly(I:C) sin antígeno como con SIINFEKL en ningún caso
produce una prolongación de la supervivencia significativa en los ensayos de
tratamiento tumoral del modelo tumoral E.G7-OVA. Por este motivo, sólo
utilizamos como grupo control ratones tratados con solución salina. Realizamos
el seguimiento del tumor dos veces por semana utilizando un calibre. En la
Figura 53 se representa una gráfica de Kaplan-Meier de supervivencia y
observamos que los ratones tratados con la solución salina presentaban un
0 20 40 60 80 1000
20
40
60
80
100
PBSSIINFEKLEDA-SIINFEKLSIINFEKL-C5aEDA-SIINFEKL-C5a
* **
Días tras la inoculación del tumor
% S
uper
vive
ncia
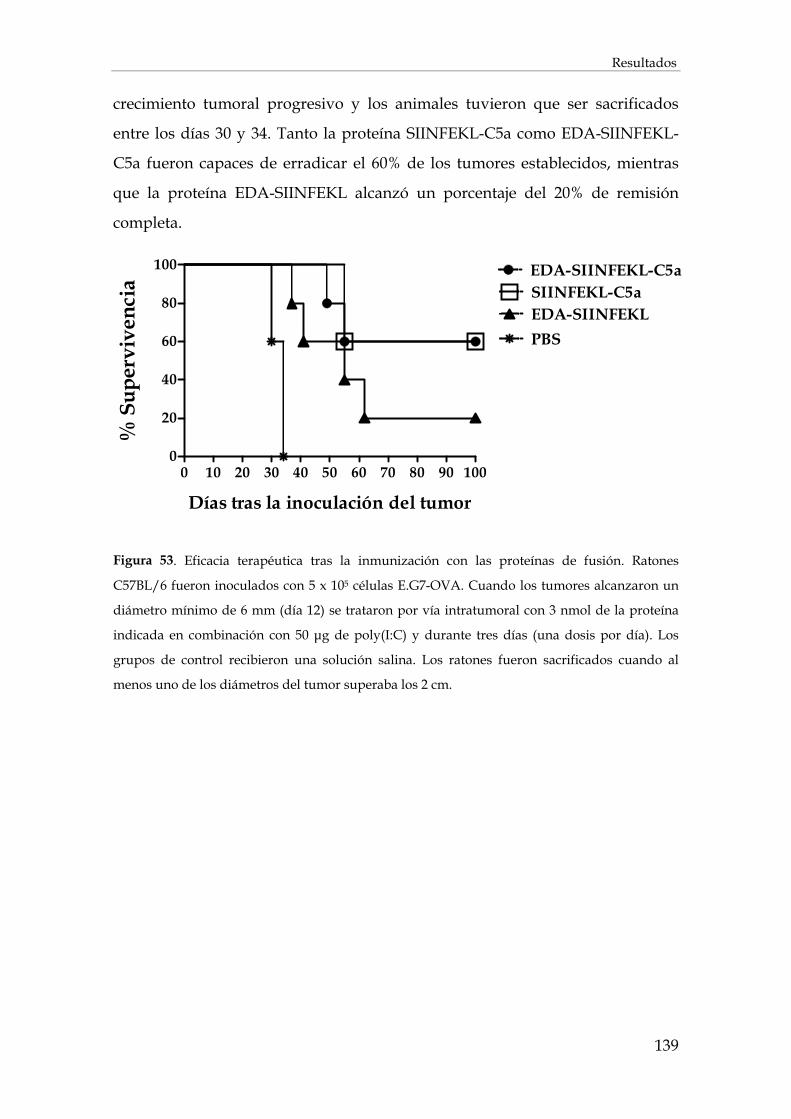
Resultados
139
crecimiento tumoral progresivo y los animales tuvieron que ser sacrificados
entre los días 30 y 34. Tanto la proteína SIINFEKL-C5a como EDA-SIINFEKL-
C5a fueron capaces de erradicar el 60% de los tumores establecidos, mientras
que la proteína EDA-SIINFEKL alcanzó un porcentaje del 20% de remisión
completa.
Figura 53. Eficacia terapéutica tras la inmunización con las proteínas de fusión. Ratones
C57BL/6 fueron inoculados con 5 x 105 células E.G7-OVA. Cuando los tumores alcanzaron un
diámetro mínimo de 6 mm (día 12) se trataron por vía intratumoral con 3 nmol de la proteína
indicada en combinación con 50 μg de poly(I:C) y durante tres días (una dosis por día). Los
grupos de control recibieron una solución salina. Los ratones fueron sacrificados cuando al
menos uno de los diámetros del tumor superaba los 2 cm.
0 10 20 30 40 50 60 70 80 90 1000
20
40
60
80
100
PBS
EDA-SIINFEKL
EDA-SIINFEKL-C5aSIINFEKL-C5a
Días tras la inoculación del tumor
% S
uper
vive
ncia


DISCUSIÓN


Discusión
143
En este trabajo nos hemos planteamos dos estrategias para potenciar el
desarrollo de vacunas. Por un lado, la inhibición de la función supresora de las
células T reguladoras y por otro lado, la potenciación de la capacidad de EDA
para inducir respuestas citotóxicas frente a un antígeno modelo. A
continuación, se discuten los resultados obtenidos con ambas estrategias.
1. Estudio del desarrollo de inhibidores de la acción de las células
T reguladoras
La señal primaria para la activación de los linfocitos T es el aumento
bifásico de la concentración intracelular de Ca2+, inducido por la molécula IP3
tras el reconocimiento de un estímulo mitogénico. Por un lado, se vacían los
reservorios de Ca2+ del retículo endoplasmático a través de los receptores para
IP3 y por otro lado, se produce un influjo de Ca2+ en la membrana plasmática
(canales CRAC) sostenido y activado por el vaciado de los reservorios
intracelulares de Ca2+ a través de la proteína estromal STIM1. La rapidez y el
mantenimiento de influjo de Ca2+ es crucial para la activación de la señalización
celular responsable de la activación celular (Oh-hora 2009; Vig and Kinet 2009).
Durante la activación celular también se producen cambios en el potencial de
membrana plasmática y en el pH intracelular.
Los canales CRAC son independientes del potencial de membrana pero
hay otros transportadores de iones Ca2+ que son dependientes de voltaje. Estos
canales de calcio dependientes de voltaje, son bloqueados por la disminución
del potencial de membrana que se produce como consecuencia del influjo de
Ca2+ llevado a cabo por los canales CRAC. Además, los intercambiadores de
iones Cl- y K+ ayudan a mantener el potencial de membrana negativo
favoreciendo el bloqueo de estos canales de Ca2+ activados por voltaje
(Kerschbaum et al. 1997). Este hecho hace pensar que los canales de Cl- y K+
podrían tener un efecto directo sobre los canales de Ca2+ CRAC. Es lógico
pensar que se pueda producir una interacción entre los sistemas de transporte
de iones Cl- y Ca2+ en la activación y proliferación celular. De hecho ya está
descrito que los canales de iones cloruro podrían estar implicados en la

Discusión
144
proliferación de diversos tipos celulares (Rouzaire-Dubois et al. 2000; Xiao et al.
2002); incluso está descrito que participarían en la inhibición de la proliferación
celular T (Phipps et al. 1996) y que este efecto estaría mediado por la interacción
de los canales de Cl- sobre la activación de los canales CRAC y en consecuencia
en la activación celular T (Wang et al. 2006).
Por lo que al pH intracelular se refiere, cuando un linfocito murino se
activa tras un estímulo mitogénico, tienen lugar dos estados de alcalinización
secuenciales. El primero corresponde a la respuesta primaria de activación
celular en respuesta al estímulo mitogénico mientras que el segundo estado de
alcalinización tiene lugar durante el proceso de proliferación celular (Gerson et
al. 1982). Hay datos descritos en la literatura que indican que el grado de
alcalinización influye en la magnitud e intensidad del aumento de la
concentración de Ca2+ intracelular y por lo tanto, en el grado de activación
celular de los linfocitos T (Rosoff and Terres 1986).
En los años 90 se describió que el intercambiador aniónico AE2 está
implicado en la regulación del pH intracelular, en el control del volumen
celular e incluso en el control de la excitabilidad de la célula, pudiendo, por
tanto, jugar un papel importante en los procesos de activación celular y del
sistema inmunitario. En el año 93 se observó que la expresión de AE2 estaba
disminuida en las biopsias hepáticas de pacientes con cirrosis biliar primaria
(CBP), una enfermedad caracterizada por fenómenos de autoinmunidad (Prieto
et al. 1993; Medina et al. 1997). Y más recientemente se ha descrito que los
ratones knockout para el gen AE2 presentan un fenotipo anormal del sistema
inmunitario caracterizado por un descenso en el número de células T
reguladoras CD4+CD25+, un aumento en la producción de citocinas pro-
inflamatorias, expansión de los linfocitos T CD8+, producción de
autoanticuerpos y un descenso en el pH intracelular de los linfocitos (Salas et al.
2008). Estos datos suguieren que los intercambiadores aniónicos podrían estar
implicados en la homeostasis de las células T reguladoras. Por ello, nos
planteamos el bloqueo de estos intercambiadores para su utilización como
inhibidores de la acción de las células T reguladoras y por lo tanto como

Discusión
145
potenciadores de la respuesta inmunitaria en enfermedades como el cáncer o
infecciones crónicas, donde las células T reguladoras juegan un papel
inmunosupresor.
En estos ensayos hemos demostrado que los bloqueantes de los
intercambiadores de iones Cl-, lejos de inhibir la proliferación celular de los
linfocitos T efectores, pueden revertir ligeramente la función supresora de las
células Treg CD4+CD25+. Tanto el DIDS como el NPPB y el NFA pueden
restaurar la activación y proliferación de las células T efectoras en presencia de
células T reguladoras, así como la expresión de interleucina IL-2. Por lo tanto,
podemos pensar que los intercambiadores de Cl- podrían estar implicados en la
activación celular linfocitaria.
También hemos demostrado que los linfocitos T efectores y las células T
reguladoras presentan una diferente susceptibilidad a la convanavalina A,
presentando un mayor influjo en la entrada de Ca2+ en las células T efectoras
CD4+CD25- que las T reguladoras. Todo esto sugiere que hay diferencias en las
vías de señalización mediadas por Ca2+ de ambos tipos celulares y que
cualquier bloqueo en estos canales podría tener un efecto más pronunciado
sobre las células T reguladoras. Hay que mencionar que el inhibidor DIDS
afecta al flujo de entrada de Ca2+ en los linfocitos T efectores activados con anti-
CD3, mientras que en las reguladoras no vemos diferencias significativas con el
DIDS. Este dato también corrobora las diferencias en la regulación de la entrada
de Ca2+ celular y posterior activación celular de ambos tipos celulares.
Nuestros datos indican que los canales de Cl- parecen estar implicados en
la activación celular y teniendo en cuenta el fenotipo de los ratones knockout
para AE2, hemos diseñado y sintetizado unos péptidos dirigidos al
intercambiador de aniones cloruro AE2 con el fin de bloquearlo y conseguir una
reversión de la supresión mediada por los linfocitos T reguladores. Hemos
demostrado que algunos interaccionan con AE2 y que tienen una acción
mitogénica en presencia del estímulo anti-CD3.

Discusión
146
Nuestros datos indican que la actividad inmunosupresora de los
linfocitos Treg puede ser regulada o bloqueada, de forma transitoria o
temporal, mediante el uso de inhibidores del intercambio aniónico celular, así
como por un péptido sintético capaz de unirse al intercambiador AE2. Hemos
demostrado que el péptido p17AE2, tiene un efecto potenciador sobre la
proliferación de las esplenocitos ante el estímulo de anti-CD3 + IL-2, al mismo
tiempo que inhibe la proliferación de los linfocitos T reguladores ante el
estímulo de anti-CD3 + IL-2. Por lo tanto, la función supresora de las células
Treg se ve afectada por la presencia del péptido p17AE2. Esta inhibición de la
función Treg tiene lugar incluso en presencia de una concentración de
tapsigargina, que potencia el efecto inmunosupresor de las células Treg.
El efecto p17AE2 sobre la actividad Treg podría estar asociado a su
capacidad de inducir la apoptosis en este tipo celular. Por este motivo,
decidimos cuantificar el porcentaje de células apoptóticas tras la estimulación
de células T efectoras o células T reguladoras con anti-CD3 e IL-2. La presencia
de p17AE2 en los cultivos de células T efectoras no tiene un efecto significativo
sobre la apoptosis celular. Sin embargo, cuando el p17AE2 se cultiva con los
linfocitos T reguladores durante 16 horas, un 43% de ellos entran en apoptosis
celular. Estos resultados justifican el efecto observado sobre la proliferación de
las Treg, que era significativamente inhibida por el péptido p17AE2. Por lo
tanto, el péptido p17AE2 in vitro, es capaz de potenciar la proliferación de las
células T efectoras ante un estímulo, al mismo tiempo que induce la apoptosis
de las células T reguladoras ante el mismo estímulo.
Hemos realizado estudios preliminares in vivo con el péptido p17AE2.
Aunque los resultados sugieren que el p17AE2 reduce el número de células
Treg tras su administración por vía intraperitoneal, estos resultados no son
concluyentes y no se han incluído en esta tesis. Estos trabajos requieren de
nuevos ensayos de optimización de dosis, rutas y pautas de administración
para elaborar una conclusión firme. La vida media de los péptidos in vivo es por
lo general extremadamente corta. El diseño de péptidos miméticos más
resistentes a la acción de las proteasas sería de gran utilidad en este proyecto.

Discusión
147
2. Estudio del efecto adyuvante de las proteínas del complemento
El sistema del complemento se compone de más de 30 proteínas que se
activan en respuesta a una lesión tisular, la infección por patógenos u otras
sustancias. Durante la activación del complemento, se generan fragmentos
solubles activos de aproximadamente 9 KDa (74-77 residuos) que son liberados
de los componentes C4, C3 y C5. Estas moléculas presentan potentes
propiedades pro-inflamatorias y se conocen con el nombre de anafilotoxinas.
Las anafilotoxinas señalizan mediante la unión a sus distintos receptores que se
localizan en una gran variedad de células, principalmente en células del sistema
inmunitario. Además, como ya hemos citado, está descrita la interacción entre
el sistema del complemento y la señalización de los receptores TLR, que en
muchos casos es una interacción de sinergia entre ambos sistemas
(Hajishengallis and Lambris 2010). Esta sinergia va dirigida hacia un aumento o
potenciación de la respuesta innata y en consecuencia también de la respuesta
adaptativa, puesto que ambas respuestas están conectadas y una influye en la
otra. Por este motivo, la activación de ambos sistemas de la respuesta innata
constituye una herramienta interesante en las estrategias utilizadas para el
desarrollo de vacunas capaces de inducir una respuesta adaptativa eficaz.
En este trabajo se ha estudiado el posible efecto adyuvante de las
anafilotoxinas C3a, C4a y C5a cuando se combinan con el dominio extra A de la
fibronectina, un ligando del receptor TLR4 que se utiliza para vehiculizar los
antígenos a las células profesionales presentadoras de antígeno que expresan el
receptor TLR4, para inducir respuestas T antígeno específicas in vivo. Además,
quisimos comprobar si las anafilotoxinas como proteínas de fusión
monoméricas, es decir, unidas covalentemente con un antígeno, podrían ser
una plataforma para el desarrollo de vacunas como lo es la proteína EDA unida
a un antígeno. Esta estrategia podría ser considerada para el desarrollo de
nuevas vacunas, ya sea la proteína C5a unida a un antígeno y combinada con
un adyuvante o preferiblemente combinada con la proteína EDA y un
adyuvante.

Discusión
148
Inicialmente con el fin de determinar qué fragmento activo del
complemento podríamos utilizar para mejorar la capacidad
inmunoestimuladora de EDA, realizamos un cribado con plásmidos que
expresan las formas secretables de la proteína EDA-SIINFEKL unida con
distintos fragmentos activos del complemento, concretamente con las
anafilotoxinas C3a, C4a, C5a y el fragmento activo C3d, el cual tras unirlo con
un antígeno, está descrito que puede ser eficaz para inducir respuestas de
anticuerpo específicas de antígeno (Dempsey et al. 1996; Kolla et al. 2007).
Tras la obtención de dichos plásmidos comprobamos la expresión de las
proteínas y fuimos capaces de detectar la expresión de todas las proteínas con la
excepción de EDA-SIINFEKL. Este hecho, puede ser debido a una rápida
degradación de la proteína o bien a una conformación de la proteína tal que
impida la unión de los anticuerpos anti-His y anti-EDA utilizados para detectar
las proteínas, de modo que los puntos de unión con los anticuerpos queden
escondidos.
Una vez confirmada la expresión de las formas secretables por los
plásmidos diseñados, procedimos a medir la respuesta celular in vivo, a pesar
de no detectar la expresión de nuestra proteína control EDA-SIINFEKL.
Nuestros resultados muestran que los ratones C57BL/6 inmunizados con el
plásmido pShuttle C5a-EDA-SIINFEKL desarrollaron una respuesta célula T
específica frente a SIINFEKL significativamente superior a la respuesta
inducida por el plásmido que expresa la forma secretable de EDA-SIINFEKL.
También demostramos que el plásmido pShuttle-EDA-SIINFEKL expresa la
proteína EDA-SIINFEKL puesto que podemos ver respuesta específica en los
ratones inmunizados con este plásmido. Por otra parte, la inmunización con los
plásmidos de expresión tiene la desventaja de que no podemos controlar el
nivel de expresión de cada plásmido y por lo tanto la cantidad de proteína
(inmunógeno) que se expresa.
En resumen, este cribado, aunque no es determinante, sugiere que el
fragmento C5a induce una potenciacion del efecto inmunomodulador de EDA.

Discusión
149
Estos datos constituyen un primer indicio favorable para pensar que C5a puede
ser un buen adyuvante en la vacunación con EDA.
Por este motivo y tras obtener estos resultados nos planteamos purificar
la proteína C5a-EDA-SIINFEKL para analizar su capacidad inmunógena. A
pesar de repetidos intentos y utilizando distintas estrategias de purificación, la
proteína C5a-EDA-SIINFEKL precipitaba durante el proceso de replegado, con
lo cual, solamente recuperábamos proteína precipitada o bien proteína en
solución con urea, que nos impedía poder reducir a límites aceptables los
niveles de endotoxina. Por este motivo, y como se ha indicado anteriormente,
decidimos cambiar la estructura del constructo insertando el fragmento C5a en
el extremo C terminal y además, insertamos una secuencia espaciadora entre el
antígeno y el fragmento C5a, para facilitar el correcto plegamiento de la
proteína. Finalmente purificamos el constructo EDA-SIINFEKL-C5a con éxito y
como controles purificamos las proteínas EDA-SIINFEKL y SIINFEKL-C5a.
Tras haber producido estas proteínas recombinantes realizamos ensayos in vitro
para medir su actividad pro-inflamatoria y facilitadora de la presentación de
antígenos. Para posteriormente, evaluar su potencialidad in vivo en protocolos
de vacunación y protección frente al desarrollo de tumores.
El primer paso tras la purificación de las proteínas recombinantes fue
comprobar que la proteína EDA-SIINFEKL-C5a era activa y que el fragmento
EDA mantenía su actividad. Por este motivo, incubamos las proteínas indicadas
con células THP-1 (línea monocítica humana) para comprobar que inducían la
producción de TNFα como indicador de la activación de estas proteínas.
Nuestros resultados muestran claramente que la proteína recombiante es capaz
de activar la señalización TRL4 y por lo tanto que el fragmento EDA se
mantiene activo, sin embargo la proteína SIINFEKL-C5a también activa las
células THP-1, lo cual nos hace pensar que el receptor C5aR humano reconoce
al fragmento C5a murino. Por este motivo, no podemos asegurar que el
fragmento EDA de la proteína EDA-SIINFEKL-C5a mantenga su actividad
puesto que la actividad observada mediante la producción de TNFα puede
corresponder a la activación de la señalización del receptor C5aR humano

Discusión
150
presente en las células THP-1. Sin embargo, este ensayo nos permite afirmar si
la proteína es o no activa. En este caso, confirmamos que habíamos purificado
unas proteínas activas con capacidad pro-inflamatoria.
Nuestro grupo ha demostrado que EDA-SIINFEKL es capaz de permitir
la incorporación, procesamiento y presentación del determinante citotóxico de
la proteína OVA (SIINFEKL) a los linfocitos T específicos del antígeno, así como
inducir una maduración de las DC que permite potenciar la respuesta celular T
específica (Lasarte et al. 2007). Además, está descrito que el fragmento C5a
actúa directamente sobre el receptor C5aR presente en las células dendríticas
dando lugar a una activación celular y en consecuencia, potenciando la
capacidad de la célula dendrítica para activar la respuesta celular T (Li et al.
2011; Peng et al. 2009).
Con estos datos descritos, el siguiente paso que nos planteamos era
poder confirmar si la proteína EDA-SIINFEKL-C5a también es capaz de inducir
la activación de las células dendríticas. Con esta finalidad estudiamos la
capacidad de la proteína EDA-SIINFEKL-C5a de inducir maduración en las DC,
conocer el patrón de citocinas y quimiocinas que se inducían y analizar la
capacidad de favorecer la presentación del determinante T citotóxico SIINFEKL.
Nuestros resultados confirman tal y como esperábamos, que la proteína
EDA-SIINFEKL-C5a es capaz de inducir la activación y la maduración de las
células dendríticas. Además, EDA-SIINFEKL-C5a presenta la capacidad de
inducir citocinas tipo Th2 (IL-13, IL-6, IL-5, IL-4) de forma ligeramente inferior a
EDA-SIINFEKL y a su vez produce una regulación al alza de ciertas
quimiocinas como CXCL16, cuya expresión es inducida por citocinas pro-
inflamatorias como IFNγ y TNF-α. Como ya hemos comentado, CXCL16 es una
quimiocina que induce el reclutamiento de subpoblaciones de linfocitos T
activados, principalmente linfocitos CD8+ y linfocitos CD4+ Th1, así como
también células NKT y células NK (estas dos últimas expresan
constitutivamente el receptor CXCR6) (Abel et al. 2004). Esta quimiocina está
relacionada con el rechazo de injertos transplantados y con un pronóstico

Discusión
151
favorable en algunos modelos tumorales (Jiang et al. ; Jiang et al. 2005; Hojo et
al. 2007). Por otra parte, también se observa que en presencia de EDA-
SIINFEKL-C5a hay un aumento del ratio IL-12p40/p70, lo cual nos hace pensar
en una mayor bioactividad de la IL-12 en presencia de la proteína EDA-
SIINFEKL-C5a. Por otro lado, la proteína EDA-SIINFEKL-C5a mejora la
presentación antigénica in vitro de manera sutil pero no significativa. Sin
embargo, la proteína SIINFEKL-C5a induce la presentación del antígeno in vitro
de forma muy pobre cuando cocultivamos células CD11c+ con la línea celular
B3Z. Pero ya que induce la maduración de la célula dendrítica, podemos pensar
que C5a unido a un antígeno potencia la inflamación favoreciendo la
maduración de la célula dendrítica pero con independencia de la captación y
del procesamiento antigénico.
Puesto que nuestros datos confirman que EDA-SIINFEKL-C5a es capaz
de inducir la producción de factores quimiotácticos como la quimiocina
CXCL16 y a su vez está descrito que las anafilotoxinas inducen quimiotaxis
celular, nos planteamos realizar ensayos in vitro de quimiotaxis, con el fin de
demostrar que el fragmento C5a presente en la proteína EDA-SIINFEKL-C5a es
activo y capaz de inducir esta función quimiotáctica. Nuestros resultados
muestran que las proteínas EDA-SIINFEKL-C5a y SIINFEKL-C5a son capaces
de inducir quimiotaxis de la serie granulocítica in vitro, además de inducir la
producción de factores quimiotácticos que permiten inducir la migración de los
macrófagos (Ly6G- CD11b+). A su vez, hemos demostrado la inducción de la
quimiocina MIP-1α por parte de ambas proteínas recombinantes. MIP-1α
también conocida como CCL3, está involucrada en estados agudos de
inflamación y en el reclutamiento y activación de leucocitos polimorfonucleares
(Maurer and von Stebut 2004).
Una vez comprobada la capacidad pro-inflamatoria de la proteína EDA-
SIINFEKL-C5a, analizamos la capacidad de inducir y potenciar la respuesta
inmunitaria adaptativa. La inmunización con esta proteína recombinante
inducía una respuesta citotóxica específica de SIINFEKL superior a la obtenida
con la proteína EDA-SIINFEKL, tanto si la inmunización se llevaba a cabo con

Discusión
152
el adyuvante poly(I:C) como en su ausencia. Además, la unión del antígeno
SIINFEKL al fragmento C5a, representado por la proteína SIINFEKL-C5a
inducía unas respuestas similares a las obtenidas por la proteína EDA-
SIINFEKL.
Con estos resultados quisimos comprobar si la capacidad adyuvante del
fragmento C5a era dependiente de la señalización de su receptor C5aR y a su
vez del receptor TLR4. Para ello inmunizamos ratones knockout para estos
receptores.
La respuesta inducida por los ratones knockout para C5aR muestra que no
hay diferencias significativas con EDA-SIINFEKL-C5a pero, sin embargo, sí las
hay con SIINFEKL-C5a, y al contrario de lo esperado, apreciamos un aumento
de la respuesta inmunitaria. Para descartar que estos efectos pudieran ser
debidos a mecanismos compensatorios presentes en el ratón deficiente del
receptor C5aR, utilizamos un anticuerpo bloqueante frente a C5aR. En estos
ensayos, observamos que tanto EDA-SIINFEKL-C5a como SIINFEKL-C5a
aumentaban su capacidad para inducir respuesta inmunitaria. Basándonos en
estos datos parece que el efecto inmunopotenciador observado con las proteínas
que contienen C5a no es dependiente del receptor C5aR y además el bloqueo de
este receptor podría favorecer el efecto potenciador de nuestras proteínas
recombinantes EDA-SIINFEKL-C5a y SIINFEKL-C5a. Una posible explicación
de nuestros datos la podríamos encontrar en el receptor C5L2, cuya función está
actualmente en controversia. C5L2 podría favorecer el efecto de estas proteínas
recombinantes, de modo que la ausencia del receptor C5aR influyera en
potenciar la acción de las proteínas recombinantes C5a a través del receptor
C5L2.
En cuanto a la inmunización de los ratones knockout para el receptor
TLR4, observamos que las proteínas EDA-SIINFEKL-C5a y SIINFEKL-C5a
actúan independientemente de la señalización TLR4, aunque en el caso de la
proteína EDA-SIINFEKL-C5a podemos observar un ligero descenso de la
respuesta en los ratones deficientes de TLR4, que parece estar indicando que el

Discusión
153
fragmento C5a suple la ausencia de actividad de la proteína EDA vía
señalización TLR4. Por otro lado, también hay que destacar que la proteína
EDA-SIINFEKL induce una respuesta celular específica disminuida en los
ratones deficientes en TLR4, lo cual puede indicar que la proteína EDA ejerce
parte de su acción potenciadora de la respuesta inmunitaria de forma
independiente del receptor TRL4. Está descrito que la proteína EDA
interacciona con la integrina α9β1 de membrana y es posible que dicha
interacción produzca efectos celulares (Shinde et al. 2008), por lo tanto, no
podemos descartar que la respuesta detectada en los ratones TLR4-/-, puede
deberse a efectos independientes de TLR4 de la proteína EDA. Otra posible
explicación y, quizás más plausible, es que dicho efecto podría ser debido al
adyuvante poly(I:C) cuando se combina con una proteína. Sabemos por
experiencias anteriores que la proteína OVA combinada con poly(I:C) induce
una respuesta celular específica del antígeno SIINFEKL (determinante T
citotóxico de la proteína OVA) y, sin embargo, cuando se combina el adyuvante
poly(I:C) con un péptido como SIINFEKL no se induce respuesta celular
específica.
Quisimos averiguar si las células NK tenían alguna implicación en la
mejora de la respuesta inmunitaria observada con la utilización del fragmento
C5a como adyuvante. Por este motivo, realizamos un estudio de la respuesta
innata así como un estudio de la respuesta adaptativa pero en este caso en
ausencia de células NK1.1, utilizando anticuerpos deplecionantes anti-NK1.1.
Observamos que las proteínas que contienen el fragmento C5a inducen una
respuesta NK más potente. Además, la depleción de esta población celular
provocó un descenso de la respuesta adaptativa tras la inmunización con la
proteína SIINFEKL-C5a, no así con la proteína EDA-SIINFEKL-C5a, que al
tener el fragmento EDA suple la carencia de las células NK1.1. Por lo tanto,
destacamos que el efecto adyuvante de C5a sea dependiente de la potenciación
en la actividad NK.
La utilización del adyuvante poly(I:C), un ligando de TLR3, en las
inmunizaciones puede tener un impacto en la activación de las células T

Discusión
154
reguladoras. Quisimos estudiar en nuestros experimentos el efecto que podría
tener el inmunógeno en la expansión de ésta subpoblación. Resultó
sorprendente encontrar que la inmunización con la proteína EDA-SIINFEKL-
C5a previno la expansión de las células Treg tras la inmunización, un efecto que
no se observó con las proteínas EDA-SIINFEKL como SIINFEKL-C5a. Además,
cuando analizamos el efecto de las proteínas en la actividad Treg in vitro,
encontramos, que la presencia de SIINFEKL-C5a en el cultivo incrementó la
actividad supresora de las células Treg. Todavía no tenemos una explicación
clara para estos hallazgos, pero podrían justificarse por el mayor efecto
inmunopotenciador de la proteína de fusión EDA-SIINFEKL-C5a in vivo. Es
necesario realizar más estudios para confirmar y entender estos resultados.
Finalmente, se destaca que la inmunización con la proteína EDA-
SIINFEKL-C5a protege a los ratones del desafío con células tumorales E.G7-
OVA, apreciando que además SIINFEKL-C5 parece ser un buen candidato para
el tratamiento tumoral en el modelo E.G7-OVA.
En resumen hemos demostrado que cuando al fragmento C5a se le une
un antígeno es capaz de inducir, en combinación con el agonista del receptor
TLR3 (poly(I:C)) una fuerte respuesta de células T específica frente al antígeno.
Pero lo más interesante es que la combinación de C5a con la proteína EDA
unida a un antígeno, induce una fuerte respuesta celular capaz de inducir
protección tumoral frente a las células tumorales E.G7-OVA, pudiendo ser un
vector proteico muy adecuado para la inducción de respuestas celulares frente a
un antígeno de interés. La construcción de proteínas de fusión basadas en el
fragmento C5a unido a EDA y a un antígeno podría suponer una estrategia
apropiada en los protocolos de vacunación frente a enfermedades tumorales o
enfermedades causadas por agentes infecciosos.

CONCLUSIONES


Conclusiones
157
1. Las células T efectoras y T reguladoras presentan diferencias en la entrada
de Ca2+ tras la estimulación del TCR.
2. Los canales e intercambiadores de Cl- están implicados en la activación
celular linfocitaria. Hemos encotrado que el bloqueo de los
canales/intercambiadores de Cl- tiene un efecto inhibidor en la acción
inmunosupresora de las células T reguladoras.
3. El péptido p17AE2, que es capaz de unirse al bucle 3 del intercambiador
intercambiador de aniones Cl-/HCO3- AE2, favorece la proliferación de los
linfocitos T efectores (CD4+CD25) y la apoptosis de las células T reguladoras
(CD4+CD25+) tras la estimulación del TCR.
4. La inmunización con SIINFEKL-C5a en combinación con poly(I:C) permite
la inducción de respuestas citotóxicas específicas frente al antígeno.
5. El fragmento C5a induce la producción de moléculas quimiotácticas para
macrófagos, la migración de granulocitos y la activación de células NK.
Estas propiedades pueden favorecer la activación de una respuesta
inmunitaria adaptativa frente al antígeno.
6. La anafilotoxina C5a altera la función de las células T reguladoras
CD4+CD25+FOXP3+. Aunque C5a parece favorecer la actividad Treg, la
unión a EDA reduce este efecto.
7. La incorporación de C5a a la proteína de fusión EDA-SIINFEKL aumenta la
capacidad de la proteína para inducir in vivo una respuesta inmunitaria
celular frente al antígeno SIINFEKL. En combinación con el adyuvante
poly(I:C), la administración de la proteína EDA-SIINFEKL-C5a tienen un
efecto antitumoral frente a la inyección subcutánea de la línea E.G7OVA.
Por lo tanto, la combinación de C5a con la proteína EDA y un antígeno viral
o tumoral podría constituir una alternativa en el desarrollo de vacunas
profilácticas o terapéuticas.


BIBLIOGRAFÍA


Bibliografía
Abdollahi-Roodsaz, S., L. A. Joosten, M. I. Koenders, I. Devesa, M. F. Roelofs, T. R. Radstake, M. Heuvelmans-Jacobs, S. Akira, M. J. Nicklin, F. Ribeiro-Dias, and W. B. van den Berg. 2008. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest 118 (1):205-216.
Abel, S., C. Hundhausen, R. Mentlein, A. Schulte, T. A. Berkhout, N. Broadway, D. Hartmann, R. Sedlacek, S. Dietrich, B. Muetze, B. Schuster, K. J. Kallen, P. Saftig, S. Rose-John, and A. Ludwig. 2004. The transmembrane CXC-chemokine ligand 16 is induced by IFN-gamma and TNF-alpha and shed by the activity of the disintegrin-like metalloproteinase ADAM10. J Immunol 172 (10):6362-6372.
Aderem, A., and R. J. Ulevitch. 2000. Toll-like receptors in the induction of the innate immune response. Nature 406 (6797):782-787.
Ahmed, R., and D. Gray. 1996. Immunological memory and protective immunity: understanding their relation. Science 272 (5258):54-60.
Akira, S., and K. Takeda. 2004. Toll-like receptor signalling. Nat Rev Immunol 4 (7):499-511.
Amara, U., D. Rittirsch, M. Flierl, U. Bruckner, A. Klos, F. Gebhard, J. D. Lambris, and M. Huber-Lang. 2008. Interaction between the coagulation and complement system. Adv Exp Med Biol 632:71-79.
Ames, R. S., Y. Li, H. M. Sarau, P. Nuthulaganti, J. J. Foley, C. Ellis, Z. Zeng, K. Su, A. J. Jurewicz, R. P. Hertzberg, D. J. Bergsma, and C. Kumar. 1996. Molecular cloning and characterization of the human anaphylatoxin C3a receptor. J Biol Chem 271 (34):20231-20234.
Aranda, F., D. Llopiz, N. Diaz-Valdes, J. I. Riezu-Boj, J. Bezunartea, M. Ruiz, M. Martinez, M. Durantez, C. Mansilla, J. Prieto, J. J. Lasarte, F. Borras-Cuesta, and P. Sarobe. 2011. Adjuvant Combination and Antigen Targeting as a Strategy to Induce Polyfunctional and High-Avidity T-Cell Responses against Poorly Immunogenic Tumors. Cancer Res 71 (9):3214-3224.
Arase, H., N. Arase, and T. Saito. 1996. Interferon gamma production by natural killer (NK) cells and NK1.1+ T cells upon NKR-P1 cross-linking. J Exp Med 183 (5):2391-2396.
Bachmann, M. F., R. M. Zinkernagel, and A. Oxenius. 1998. Immune responses in the absence of costimulation: viruses know the trick. J Immunol 161 (11):5791-5794.
Bamberg, C. E., C. R. Mackay, H. Lee, D. Zahra, J. Jackson, Y. S. Lim, P. L. Whitfeld, S. Craig, E. Corsini, B. Lu, C. Gerard, and N. P. Gerard. 2010. The C5a receptor (C5aR) C5L2 is a modulator of C5aR-mediated signal transduction. J Biol Chem 285 (10):7633-7644.
Banchereau, J., and R. M. Steinman. 1998. Dendritic cells and the control of immunity. Nature 392 (6673):245-252.

Bibliografía
162
Belmonte, L., C. Parodi, P. Bare, M. Baston, M. M. Bracco, and B. Ruibal-Ares. 2007. [The role of dendritic cells in the infection by HIV and HCV]. Medicina (B Aires) 67 (1):63-70.
Bennett, S. R., F. R. Carbone, F. Karamalis, R. A. Flavell, J. F. Miller, and W. R. Heath. 1998. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 393 (6684):478-480.
Bhakdi, S., F. Hugo, and J. Tranum-Jensen. 1990. Functions and relevance of the terminal complement sequence. Blut 60 (6):309-318.
Billet, S. E., S. A. Grando, and M. R. Pittelkow. 2006. Paraneoplastic autoimmune multiorgan syndrome: review of the literature and support for a cytotoxic role in pathogenesis. Autoimmunity 39 (7):617-630.
Binder, R. J., and P. K. Srivastava. 2005. Peptides chaperoned by heat-shock proteins are a necessary and sufficient source of antigen in the cross-priming of CD8+ T cells. Nat Immunol 6 (6):593-599.
Bohnsack, J. F., J. J. O'Shea, T. Takahashi, and E. J. Brown. 1985. Fibronectin-enhanced phagocytosis of an alternative pathway activator by human culture-derived macrophages is mediated by the C4b/C3b complement receptor (CR1). J Immunol 135 (4):2680-2686.
Bokisch, V. A., and H. J. Muller-Eberhard. 1970. Anaphylatoxin inactivator of human plasma: its isolation and characterization as a carboxypeptidase. J Clin Invest 49 (12):2427-2436.
Bonifaz, L. C., D. P. Bonnyay, A. Charalambous, D. I. Darguste, S. Fujii, H. Soares, M. K. Brimnes, B. Moltedo, T. M. Moran, and R. M. Steinman. 2004. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J Exp Med 199 (6):815-824.
Borras-Cuesta, F., J. Golvano, P. Sarobe, J. J. Lasarte, I. Prieto, A. Szabo, J. L. Guillaume, and J. G. Guillet. 1991. Insights on the amino acid side-chain interactions of a synthetic T-cell determinant. Biologicals 19 (3):187-190.
Bothwell, A. L. 1999. Characterization of the human antiporcine immune response: a prerequisite to xenotransplantation. Immunol Res 19 (2-3):233-243.
Bourgeois, C., H. Veiga-Fernandes, A. M. Joret, B. Rocha, and C. Tanchot. 2002. CD8 lethargy in the absence of CD4 help. Eur J Immunol 32 (8):2199-2207.
Bower, J. F., T. D. Green, and T. M. Ross. 2004. DNA vaccines expressing soluble CD4-envelope proteins fused to C3d elicit cross-reactive neutralizing antibodies to HIV-1. Virology 328 (2):292-300.
Cahalan, M. D., H. Wulff, and K. G. Chandy. 2001. Molecular properties and physiological roles of ion channels in the immune system. J Clin Immunol 21 (4):235-252.
Cain, S. A., and P. N. Monk. 2002. The orphan receptor C5L2 has high affinity binding sites for complement fragments C5a and C5a des-Arg(74). J Biol Chem 277 (9):7165-7169.

Bibliografía
163
Casares, N., L. Arribillaga, P. Sarobe, J. Dotor, A. Lopez-Diaz de Cerio, I. Melero, J. Prieto, F. Borras-Cuesta, and J. J. Lasarte. 2003. CD4+/CD25+ regulatory cells inhibit activation of tumor-primed CD4+ T cells with IFN-gamma-dependent antiangiogenic activity, as well as long-lasting tumor immunity elicited by peptide vaccination. J Immunol 171 (11):5931-5939.
Casares, N., F. Rudilla, L. Arribillaga, D. Llopiz, J. I. Riezu-Boj, T. Lozano, J. Lopez-Sagaseta, L. Guembe, P. Sarobe, J. Prieto, F. Borras-Cuesta, and J. J. Lasarte. 2010. A peptide inhibitor of FOXP3 impairs regulatory T cell activity and improves vaccine efficacy in mice. J Immunol 185 (9):5150-5159.
Cella, M., D. Jarrossay, F. Facchetti, O. Alebardi, H. Nakajima, A. Lanzavecchia, and M. Colonna. 1999. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med 5 (8):919-923.
Colombo, M. P., and S. Piconese. 2007. Regulatory-T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nat Rev Cancer 7 (11):880-887.
Corr, M., H. Tighe, D. Lee, J. Dudler, M. Trieu, D. C. Brinson, and D. A. Carson. 1997. Costimulation provided by DNA immunization enhances antitumor immunity. J Immunol 159 (10):4999-5004.
Cuadros, C., F. J. Lopez-Hernandez, A. L. Dominguez, M. McClelland, and J. Lustgarten. 2004. Flagellin fusion proteins as adjuvants or vaccines induce specific immune responses. Infect Immun 72 (5):2810-2816.
Chandy, K. G., H. Wulff, C. Beeton, M. Pennington, G. A. Gutman, and M. D. Cahalan. 2004. K+ channels as targets for specific immunomodulation. Trends Pharmacol Sci 25 (5):280-289.
Chen, X., O. M. Howard, and J. J. Oppenheim. 2007. Pertussis toxin by inducing IL-6 promotes the generation of IL-17-producing CD4 cells. J Immunol 178 (10):6123-6129.
Cherwinski, H. M., J. H. Schumacher, K. D. Brown, and T. R. Mosmann. 1987. Two types of mouse helper T cell clone. III. Further differences in lymphokine synthesis between Th1 and Th2 clones revealed by RNA hybridization, functionally monospecific bioassays, and monoclonal antibodies. J Exp Med 166 (5):1229-1244.
Chomczynski, P., and N. Sacchi. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162 (1):156-159.
Delneste, Y., G. Magistrelli, J. Gauchat, J. Haeuw, J. Aubry, K. Nakamura, N. Kawakami-Honda, L. Goetsch, T. Sawamura, J. Bonnefoy, and P. Jeannin. 2002. Involvement of LOX-1 in dendritic cell-mediated antigen cross-presentation. Immunity 17 (3):353-362.
Dempsey, P. W., M. E. Allison, S. Akkaraju, C. C. Goodnow, and D. T. Fearon. 1996. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science 271 (5247):348-350.

Bibliografía
164
Depla, E., A. Van der Aa, B. D. Livingston, C. Crimi, K. Allosery, V. De Brabandere, J. Krakover, S. Murthy, M. Huang, S. Power, L. Babe, C. Dahlberg, D. McKinney, A. Sette, S. Southwood, R. Philip, M. J. Newman, and L. Meheus. 2008. Rational design of a multiepitope vaccine encoding T-lymphocyte epitopes for treatment of chronic hepatitis B virus infections. J Virol 82 (1):435-450.
Dieckmann, D., H. Plottner, S. Berchtold, T. Berger, and G. Schuler. 2001. Ex vivo isolation and characterization of CD4(+)CD25(+) T cells with regulatory properties from human blood. J Exp Med 193 (11):1303-1310.
Dunkelberger, J. R., and W. C. Song. 2010. Complement and its role in innate and adaptive immune responses. Cell Res 20 (1):34-50.
Durantez, M., C. Fayolle, N. Casares, V. Belsue, J. I. Riezu-Boj, P. Sarobe, J. Prieto, F. Borras-Cuesta, C. Leclerc, and J. J. Lasarte. 2010. Tumor therapy in mice by using a tumor antigen linked to modulin peptides from Staphylococcus epidermidis. Vaccine 28 (44):7146-7154.
Durantez, M., A. B. Lopez-Vazquez, A. L. de Cerio, E. Huarte, N. Casares, J. Prieto, F. Borras-Cuesta, J. J. Lasarte, and P. Sarobe. 2009. Induction of multiepitopic and long-lasting immune responses against tumour antigens by immunization with peptides, DNA and recombinant adenoviruses expressing minigenes. Scand J Immunol 69 (2):80-89.
Engvall, E., and E. Ruoslahti. 1977. Binding of soluble form of fibroblast surface protein, fibronectin, to collagen. Int J Cancer 20 (1):1-5.
Fang, C., T. Miwa, H. Shen, and W. C. Song. 2007. Complement-dependent enhancement of CD8+ T cell immunity to lymphocytic choriomeningitis virus infection in decay-accelerating factor-deficient mice. J Immunol 179 (5):3178-3186.
Fang, C., X. Zhang, T. Miwa, and W. C. Song. 2009. Complement promotes the development of inflammatory T-helper 17 cells through synergistic interaction with Toll-like receptor signaling and interleukin-6 production. Blood 114 (5):1005-1015.
Fayolle, C., D. Ladant, G. Karimova, A. Ullmann, and C. Leclerc. 1999. Therapy of murine tumors with recombinant Bordetella pertussis adenylate cyclase carrying a cytotoxic T cell epitope. J Immunol 162 (7):4157-4162.
Feske, S. 2007. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol 7 (9):690-702.
Feske, S., J. Giltnane, R. Dolmetsch, L. M. Staudt, and A. Rao. 2001. Gene regulation mediated by calcium signals in T lymphocytes. Nat Immunol 2 (4):316-324.
Freeman, G. J., F. Borriello, R. J. Hodes, H. Reiser, J. G. Gribben, J. W. Ng, J. Kim, J. M. Goldberg, K. Hathcock, G. Laszlo, and et al. 1993. Murine B7-2, an alternative CTLA4 counter-receptor that costimulates T cell proliferation and interleukin 2 production. J Exp Med 178 (6):2185-2192.

Bibliografía
165
Fuenmayor, J., K. Perez-Vazquez, D. Perez-Witzke, M. L. Penichet, and R. F. Montano. 2010. Decreased survival of human breast cancer cells expressing HER2/neu on in vitro incubation with an anti-HER2/neu antibody fused to C5a or C5a desArg. Mol Cancer Ther 9 (8):2175-2185.
Gao, H., T. A. Neff, R. F. Guo, C. L. Speyer, J. V. Sarma, S. Tomlins, Y. Man, N. C. Riedemann, L. M. Hoesel, E. Younkin, F. S. Zetoune, and P. A. Ward. 2005. Evidence for a functional role of the second C5a receptor C5L2. Faseb J 19 (8):1003-1005.
Gelman, A. E., J. Zhang, Y. Choi, and L. A. Turka. 2004. Toll-like receptor ligands directly promote activated CD4+ T cell survival. J Immunol 172 (10):6065-6073.
Gerard, N. P., B. Lu, P. Liu, S. Craig, Y. Fujiwara, S. Okinaga, and C. Gerard. 2005. An anti-inflammatory function for the complement anaphylatoxin C5a-binding protein, C5L2. J Biol Chem 280 (48):39677-39680.
Germain, R. N., A. J. Sant, N. S. Braunstein, and F. Ronchese. 1988. The molecular basis of antigen presentation. Princess Takamatsu Symp 19:179-191.
Gerson, D. F., H. Kiefer, and W. Eufe. 1982. Intracellular pH of mitogen-stimulated lymphocytes. Science 216 (4549):1009-1010.
Gil-Torregrosa, B. C., A. Raul Castano, and M. Del Val. 1998. Major histocompatibility complex class I viral antigen processing in the secretory pathway defined by the trans-Golgi network protease furin. J Exp Med 188 (6):1105-1116.
Governa, M., M. Amati, I. Fenoglio, M. Valentino, S. Coloccini, L. Bolognini, G. Carlo Botta, M. Emanuelli, F. Pierella, A. R. Volpe, P. Astolfi, M. Carmignani, and B. Fubini. 2005. Variability of biological effects of silicas: different degrees of activation of the fifth component of complement by amorphous silicas. Toxicol Appl Pharmacol 208 (1):68-77.
Governa, M., M. Amati, M. Valentino, I. Visona, B. Fubini, G. C. Botta, A. R. Volpe, and M. Carmignani. 2000. In vitro cleavage by asbestos fibers of the fifth component of human complement through free-radical generation and kallikrein activation. J Toxicol Environ Health A 59 (7):539-552.
Guermonprez, P., N. Khelef, E. Blouin, P. Rieu, P. Ricciardi-Castagnoli, N. Guiso, D. Ladant, and C. Leclerc. 2001. The adenylate cyclase toxin of Bordetella pertussis binds to target cells via the alpha(M)beta(2) integrin (CD11b/CD18). J Exp Med 193 (9):1035-1044.
Guermonprez, P., J. Valladeau, L. Zitvogel, C. Thery, and S. Amigorena. 2002. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol 20:621-667.
Guo, R. F., N. C. Riedemann, and P. A. Ward. 2004. Role of C5a-C5aR interaction in sepsis. Shock 21 (1):1-7.
Guo, R. F., and P. A. Ward. 2005. Role of C5a in inflammatory responses. Annu Rev Immunol 23:821-852.

Bibliografía
166
Guy, B. 2007. The perfect mix: recent progress in adjuvant research. Nat Rev Microbiol 5 (7):505-517.
Hackl, D., J. Loschko, T. Sparwasser, W. Reindl, and A. B. Krug. 2011. Activation of dendritic cells via TLR7 reduces Foxp3 expression and suppressive function in induced Tregs. Eur J Immunol 41 (5):1334-1343.
Hackl, N. J., C. Bersch, P. Feick, C. Antoni, A. Franke, M. V. Singer, and I. A. Nakchbandi. 2010. Circulating fibronectin isoforms predict the degree of fibrosis in chronic hepatitis C. Scand J Gastroenterol 45 (3):349-356.
Hajishengallis, G., and J. D. Lambris. 2010. Crosstalk pathways between Toll-like receptors and the complement system. Trends Immunol 31 (4):154-163.
Harty, J. T., A. R. Tvinnereim, and D. W. White. 2000. CD8+ T cell effector mechanisms in resistance to infection. Annu Rev Immunol 18:275-308.
Hashimoto, M., K. Hirota, H. Yoshitomi, S. Maeda, S. Teradaira, S. Akizuki, P. Prieto-Martin, T. Nomura, N. Sakaguchi, J. Kohl, B. Heyman, M. Takahashi, T. Fujita, T. Mimori, and S. Sakaguchi. Complement drives Th17 cell differentiation and triggers autoimmune arthritis. J Exp Med 207 (6):1135-1143.
Hawlisch, H., Y. Belkaid, R. Baelder, D. Hildeman, C. Gerard, and J. Kohl. 2005. C5a negatively regulates toll-like receptor 4-induced immune responses. Immunity 22 (4):415-426.
Heath, W. R., and F. R. Carbone. 2001. Cross-presentation in viral immunity and self-tolerance. Nat Rev Immunol 1 (2):126-134.
Heinzel, F. P., M. D. Sadick, B. J. Holasday, R. L. Coffman, and R. M. Locksley. 1989. Reciprocal expression of interferon gamma or interleukin 4 during the resolution or progression of murine leishmaniasis. Evidence for expansion of distinct helper T cell subsets. J Exp Med 169 (1):59-72.
Hojo, S., K. Koizumi, K. Tsuneyama, Y. Arita, Z. Cui, K. Shinohara, T. Minami, I. Hashimoto, T. Nakayama, H. Sakurai, Y. Takano, O. Yoshie, K. Tsukada, and I. Saiki. 2007. High-level expression of chemokine CXCL16 by tumor cells correlates with a good prognosis and increased tumor-infiltrating lymphocytes in colorectal cancer. Cancer Res 67 (10):4725-4731.
Holme, E. R., and K. Whaley. 1989. Complement and related clinical disorders. Blood Rev 3 (2):120-129.
Hopken, U., M. Mohr, A. Struber, H. Montz, H. Burchardi, O. Gotze, and M. Oppermann. 1996. Inhibition of interleukin-6 synthesis in an animal model of septic shock by anti-C5a monoclonal antibodies. Eur J Immunol 26 (5):1103-1109.
Huber-Lang, M., J. V. Sarma, D. Rittirsch, H. Schreiber, M. Weiss, M. Flierl, E. Younkin, M. Schneider, H. Suger-Wiedeck, F. Gebhard, S. D. McClintock, T. Neff, F. Zetoune, U. Bruckner, R. F. Guo, P. N. Monk, and P. A. Ward. 2005. Changes in the novel orphan, C5a receptor (C5L2), during experimental sepsis and sepsis in humans. J Immunol 174 (2):1104-1110.

Bibliografía
167
Huber-Lang, M., J. V. Sarma, F. S. Zetoune, D. Rittirsch, T. A. Neff, S. R. McGuire, J. D. Lambris, R. L. Warner, M. A. Flierl, L. M. Hoesel, F. Gebhard, J. G. Younger, S. M. Drouin, R. A. Wetsel, and P. A. Ward. 2006. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med 12 (6):682-687.
Hynes, R. 1990. Fibronectins 1st ed. New York: Springer-Verlag
Ishioka, G. Y., J. Fikes, G. Hermanson, B. Livingston, C. Crimi, M. Qin, M. F. del Guercio, C. Oseroff, C. Dahlberg, J. Alexander, R. W. Chesnut, and A. Sette. 1999. Utilization of MHC class I transgenic mice for development of minigene DNA vaccines encoding multiple HLA-restricted CTL epitopes. J Immunol 162 (7):3915-3925.
Jalili, A., N. Shirvaikar, L. Marquez-Curtis, Y. Qiu, C. Korol, H. Lee, A. R. Turner, M. Z. Ratajczak, and A. Janowska-Wieczorek. Fifth complement cascade protein (C5) cleavage fragments disrupt the SDF-1/CXCR4 axis: further evidence that innate immunity orchestrates the mobilization of hematopoietic stem/progenitor cells. Exp Hematol 38 (4):321-332.
Janeway, C. A., Jr., and R. Medzhitov. 2002. Innate immune recognition. Annu Rev Immunol 20:197-216.
Janssen, E. M., E. E. Lemmens, T. Wolfe, U. Christen, M. G. von Herrath, and S. P. Schoenberger. 2003. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 421 (6925):852-856.
Jensen, P. E. 2007. Recent advances in antigen processing and presentation. Nat Immunol 8 (10):1041-1048.
Jiang, X., T. Shimaoka, S. Kojo, M. Harada, H. Watarai, H. Wakao, N. Ohkohchi, S. Yonehara, M. Taniguchi, and K. Seino. 2005. Cutting edge: critical role of CXCL16/CXCR6 in NKT cell trafficking in allograft tolerance. J Immunol 175 (4):2051-2055.
Jiang, X., W. Sun, L. Zhu, D. Guo, H. Jiang, D. Ma, J. Jin, Y. Zhao, and J. Liang. Expression of CXCR6 on CD8(+) T cells was up-regulated in allograft rejection. Transpl Immunol 22 (3-4):179-183.
Jonuleit, H., E. Schmitt, M. Stassen, A. Tuettenberg, J. Knop, and A. H. Enk. 2001. Identification and functional characterization of human CD4(+)CD25(+) T cells with regulatory properties isolated from peripheral blood. J Exp Med 193 (11):1285-1294.
Kaczorowski, D. J., A. Afrazi, M. J. Scott, J. H. Kwak, R. Gill, R. D. Edmonds, Y. Liu, J. Fan, and T. R. Billiar. 2010. Pivotal advance: The pattern recognition receptor ligands lipopolysaccharide and polyinosine-polycytidylic acid stimulate factor B synthesis by the macrophage through distinct but overlapping mechanisms. J Leukoc Biol 88 (4):609-618.
Kaisho, T., and S. Akira. 2002. Toll-like receptors as adjuvant receptors. Biochim Biophys Acta 1589 (1):1-13.

Bibliografía
168
Kalant, D., R. MacLaren, W. Cui, R. Samanta, P. N. Monk, S. A. Laporte, and K. Cianflone. 2005. C5L2 is a functional receptor for acylation-stimulating protein. J Biol Chem 280 (25):23936-23944.
Kaspar, M., L. Zardi, and D. Neri. 2006. Fibronectin as target for tumor therapy. Int J Cancer 118 (6):1331-1339.
Kawai, T., and S. Akira. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11 (5):373-384.
Kemper, C., and J. P. Atkinson. 2007. T-cell regulation: with complements from innate immunity. Nat Rev Immunol 7 (1):9-18.
Kerschbaum, H. H., P. A. Negulescu, and M. D. Cahalan. 1997. Ion channels, Ca2+ signaling, and reporter gene expression in antigen-specific mouse T cells. J Immunol 159 (4):1628-1638.
Kim, A. H., I. D. Dimitriou, M. C. Holland, D. Mastellos, Y. M. Mueller, J. D. Altman, J. D. Lambris, and P. D. Katsikis. 2004. Complement C5a receptor is essential for the optimal generation of antiviral CD8+ T cell responses. J Immunol 173 (4):2524-2529.
Kim, S. K., D. S. Reed, S. Olson, M. J. Schnell, J. K. Rose, P. A. Morton, and L. Lefrancois. 1998. Generation of mucosal cytotoxic T cells against soluble protein by tissue-specific environmental and costimulatory signals. Proc Natl Acad Sci U S A 95 (18):10814-10819.
Klinman, D. M., A. K. Yi, S. L. Beaucage, J. Conover, and A. M. Krieg. 1996. CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12, and interferon gamma. Proc Natl Acad Sci U S A 93 (7):2879-2883.
Koch, F., U. Stanzl, P. Jennewein, K. Janke, C. Heufler, E. Kampgen, N. Romani, and G. Schuler. 1996. High level IL-12 production by murine dendritic cells: upregulation via MHC class II and CD40 molecules and downregulation by IL-4 and IL-10. J Exp Med 184 (2):741-746.
Koleva, M., G. Schlaf, R. Landmann, O. Gotze, K. Jungermann, and H. L. Schieferdecker. 2002. Induction of anaphylatoxin C5a receptors in rat hepatocytes by lipopolysaccharide in vivo: mediation by interleukin-6 from Kupffer cells. Gastroenterology 122 (3):697-708.
Kolla, R. V., S. Chintalapati, M. Sabet, E. Santelli, R. C. Liddington, M. David, J. Fierer, D. Guiney, and R. C. Rickert. 2007. Complement C3d conjugation to anthrax protective antigen promotes a rapid, sustained, and protective antibody response. PLoS One 2 (10):e1044.
Krug, A., R. Veeraswamy, A. Pekosz, O. Kanagawa, E. R. Unanue, M. Colonna, and M. Cella. 2003. Interferon-producing cells fail to induce proliferation of naive T cells but can promote expansion and T helper 1 differentiation of antigen-experienced unpolarized T cells. J Exp Med 197 (7):899-906.
Kumar, H., T. Kawai, and S. Akira. 2009. Toll-like receptors and innate immunity. Biochem Biophys Res Commun 388 (4):621-625.

Bibliografía
169
Kumar, V., S. R. Ali, S. Konrad, J. Zwirner, J. S. Verbeek, R. E. Schmidt, and J. E. Gessner. 2006. Cell-derived anaphylatoxins as key mediators of antibody-dependent type II autoimmunity in mice. J Clin Invest 116 (2):512-520.
Lai, M. Z., D. T. Ross, J. G. Guillet, T. J. Briner, M. L. Gefter, and J. A. Smith. 1987. T lymphocyte response to bacteriophage lambda repressor cI protein. Recognition of the same peptide presented by Ia molecules of different haplotypes. J Immunol 139 (12):3973-3980.
Lajoie, S., I. P. Lewkowich, Y. Suzuki, J. R. Clark, A. A. Sproles, K. Dienger, A. L. Budelsky, and M. Wills-Karp. Complement-mediated regulation of the IL-17A axis is a central genetic determinant of the severity of experimental allergic asthma. Nat Immunol 11 (10):928-935.
Lalli, P. N., M. G. Strainic, M. Yang, F. Lin, M. E. Medof, and P. S. Heeger. 2008. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood 112 (5):1759-1766.
Lambrecht, B. N. 2006. An unexpected role for the anaphylatoxin C5a receptor in allergic sensitization. J Clin Invest 116 (3):628-632.
Lappegard, K. T., D. Christiansen, A. Pharo, E. B. Thorgersen, B. C. Hellerud, J. Lindstad, E. W. Nielsen, G. Bergseth, D. Fadnes, T. G. Abrahamsen, E. A. Hoiby, L. Schejbel, P. Garred, J. D. Lambris, M. Harboe, and T. E. Mollnes. 2009. Human genetic deficiencies reveal the roles of complement in the inflammatory network: lessons from nature. Proc Natl Acad Sci U S A 106 (37):15861-15866.
Lasarte, J. J., N. Casares, M. Gorraiz, S. Hervas-Stubbs, L. Arribillaga, C. Mansilla, M. Durantez, D. Llopiz, P. Sarobe, F. Borras-Cuesta, J. Prieto, and C. Leclerc. 2007. The extra domain A from fibronectin targets antigens to TLR4-expressing cells and induces cytotoxic T cell responses in vivo. J Immunol 178 (2):748-756.
Lasarte, J. J., P. Sarobe, J. Prieto, and F. Borras-Cuesta. 1995. In vivo cytotoxic T-lymphocyte induction may take place via CD8 T helper lymphocytes. Res Immunol 146 (1):35-44.
Latz, E., A. Visintin, E. Lien, K. A. Fitzgerald, T. Espevik, and D. T. Golenbock. 2003. The LPS receptor generates inflammatory signals from the cell surface. J Endotoxin Res 9 (6):375-380.
Laudes, I. J., J. C. Chu, M. Huber-Lang, R. F. Guo, N. C. Riedemann, J. V. Sarma, F. Mahdi, H. S. Murphy, C. Speyer, K. T. Lu, J. D. Lambris, F. S. Zetoune, and P. A. Ward. 2002. Expression and function of C5a receptor in mouse microvascular endothelial cells. J Immunol 169 (10):5962-5970.
Levings, M. K., R. Sangregorio, and M. G. Roncarolo. 2001. Human cd25(+)cd4(+) t regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J Exp Med 193 (11):1295-1302.
Li, K., K. J. Anderson, Q. Peng, A. Noble, B. Lu, A. P. Kelly, N. Wang, S. H. Sacks, and W. Zhou. 2008. Cyclic AMP plays a critical role in C3a-receptor-mediated

Bibliografía
170
regulation of dendritic cells in antigen uptake and T-cell stimulation. Blood 112 (13):5084-5094.
Li, K., H. Fazekasova, N. Wang, P. Sagoo, Q. Peng, W. Khamri, C. Gomes, S. H. Sacks, G. Lombardi, and W. Zhou. 2011. Expression of complement components, receptors and regulators by human dendritic cells. Mol Immunol 48 (9-10):1121-1127.
Li, Q., D. Huang, K. Nacion, H. Bu, and F. Lin. 2009. Augmenting DAF levels in vivo ameliorates experimental autoimmune encephalomyelitis. Mol Immunol 46 (15):2885-2891.
Linsley, P. S., and J. A. Ledbetter. 1993. The role of the CD28 receptor during T cell responses to antigen. Annu Rev Immunol 11:191-212.
Liu, H., M. Komai-Koma, D. Xu, and F. Y. Liew. 2006. Toll-like receptor 2 signaling modulates the functions of CD4+ CD25+ regulatory T cells. Proc Natl Acad Sci U S A 103 (18):7048-7053.
Liu, Y. J. 2005. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol 23:275-306.
MacLaren, R., W. Cui, and K. Cianflone. 2008. Adipokines and the immune system: an adipocentric view. Adv Exp Med Biol 632:1-21.
Mandelboim, O., S. Kent, D. M. Davis, S. B. Wilson, T. Okazaki, R. Jackson, D. Hafler, and J. L. Strominger. 1998. Natural killer activating receptors trigger interferon gamma secretion from T cells and natural killer cells. Proc Natl Acad Sci U S A 95 (7):3798-3803.
Mangsbo, S. M., J. Sanchez, K. Anger, J. D. Lambris, K. N. Ekdahl, A. S. Loskog, B. Nilsson, and T. H. Totterman. 2009. Complement activation by CpG in a human whole blood loop system: mechanisms and immunomodulatory effects. J Immunol 183 (10):6724-6732.
Mansilla, C., M. Gorraiz, M. Martinez, N. Casares, L. Arribillaga, F. Rudilla, I. Echeverria, J. I. Riezu-Boj, P. Sarobe, F. Borras-Cuesta, J. Prieto, and J. J. Lasarte. 2009. Immunization against hepatitis C virus with a fusion protein containing the extra domain A from fibronectin and the hepatitis C virus NS3 protein. J Hepatol 51 (3):520-527.
Manthey, H. D., T. M. Woodruff, S. M. Taylor, and P. N. Monk. 2009. Complement component 5a (C5a). Int J Biochem Cell Biol 41 (11):2114-2117.
Marder, S. R., D. E. Chenoweth, I. M. Goldstein, and H. D. Perez. 1985. Chemotactic responses of human peripheral blood monocytes to the complement-derived peptides C5a and C5a des Arg. J Immunol 134 (5):3325-3331.
Markiewski, M. M., B. Nilsson, K. N. Ekdahl, T. E. Mollnes, and J. D. Lambris. 2007. Complement and coagulation: strangers or partners in crime? Trends Immunol 28 (4):184-192.

Bibliografía
171
Martin, M., K. Rehani, R. S. Jope, and S. M. Michalek. 2005. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol 6 (8):777-784.
Maruo, K., T. Akaike, T. Ono, T. Okamoto, and H. Maeda. 1997. Generation of anaphylatoxins through proteolytic processing of C3 and C5 by house dust mite protease. J Allergy Clin Immunol 100 (2):253-260.
Marzo, A. L., B. F. Kinnear, R. A. Lake, J. J. Frelinger, E. J. Collins, B. W. Robinson, and B. Scott. 2000. Tumor-specific CD4+ T cells have a major "post-licensing" role in CTL mediated anti-tumor immunity. J Immunol 165 (11):6047-6055.
Maurer, M., and E. von Stebut. 2004. Macrophage inflammatory protein-1. Int J Biochem Cell Biol 36 (10):1882-1886.
McMichael, A. J. 1979. Lymphocytes. 1. Function. Genetic restrictions in the immune response. J Clin Pathol Suppl (R Coll Pathol) 13:30-38.
McMullen, M. E., M. L. Hart, M. C. Walsh, J. Buras, K. Takahashi, and G. L. Stahl. 2006. Mannose-binding lectin binds IgM to activate the lectin complement pathway in vitro and in vivo. Immunobiology 211 (10):759-766.
Medina, J. F., A. Martinez, J. J. Vazquez, and J. Prieto. 1997. Decreased anion exchanger 2 immunoreactivity in the liver of patients with primary biliary cirrhosis. Hepatology 25 (1):12-17.
Medzhitov, R. 2001. Toll-like receptors and innate immunity. Nat Rev Immunol 1 (2):135-145.
Mellman, I. 2005. Antigen processing and presentation by dendritic cells: cell biological mechanisms. Adv Exp Med Biol 560:63-67.
Merrifield, R. B. 1964. Solid-Phase Peptide Synthesis. 3. An Improved Synthesis Of Bradykinin. Biochemistry 3:1385-1390.
Monk, P. N., A. M. Scola, P. Madala, and D. P. Fairlie. 2007. Function, structure and therapeutic potential of complement C5a receptors. Br J Pharmacol 152 (4):429-448.
Morgan, B. P. 1989. Mechanisms of tissue damage by the membrane attack complex of complement. Complement Inflamm 6 (2):104-111.
Mosmann, T. R., and R. L. Coffman. 1989. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol 7:145-173.
O'Neill, L. A., and A. G. Bowie. 2007. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 7 (5):353-364.
Oh-hora, M. 2009. Calcium signaling in the development and function of T-lineage cells. Immunol Rev 231 (1):210-224.
Okamura, Y., M. Watari, E. S. Jerud, D. W. Young, S. T. Ishizaka, J. Rose, J. C. Chow, and J. F. Strauss, 3rd. 2001. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem 276 (13):10229-10233.

Bibliografía
172
Okinaga, S., D. Slattery, A. Humbles, Z. Zsengeller, O. Morteau, M. B. Kinrade, R. M. Brodbeck, J. E. Krause, H. R. Choe, N. P. Gerard, and C. Gerard. 2003. C5L2, a nonsignaling C5A binding protein. Biochemistry 42 (31):9406-9415.
Onizuka, S., I. Tawara, J. Shimizu, S. Sakaguchi, T. Fujita, and E. Nakayama. 1999. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res 59 (13):3128-3133.
Onoe, K., N. Arase, H. Arase, T. Takayanagi, H. Nishihori, K. Iwabuchi, K. Ogasawara, and R. A. Good. 1997. Influence of graft versus host reaction on the T cell repertoire differentiating from bone marrow precursors following allogeneic bone marrow transplantation. Transpl Immunol 5 (2):75-82.
Pardoll, D. M. 1993. New strategies for enhancing the immunogenicity of tumors. Curr Opin Immunol 5 (5):719-725.
Partiseti, M., F. Le Deist, C. Hivroz, A. Fischer, H. Korn, and D. Choquet. 1994. The calcium current activated by T cell receptor and store depletion in human lymphocytes is absent in a primary immunodeficiency. J Biol Chem 269 (51):32327-32335.
Pasare, C., and R. Medzhitov. 2004. Toll-like receptors: linking innate and adaptive immunity. Microbes Infect 6 (15):1382-1387.
Peng, Q., K. Li, N. Wang, Q. Li, E. Asgari, B. Lu, T. M. Woodruff, S. H. Sacks, and W. Zhou. 2009. Dendritic cell function in allostimulation is modulated by C5aR signaling. J Immunol 183 (10):6058-6068.
Perianayagam, M. C., V. S. Balakrishnan, A. J. King, B. J. Pereira, and B. L. Jaber. 2002. C5a delays apoptosis of human neutrophils by a phosphatidylinositol 3-kinase-signaling pathway. Kidney Int 61 (2):456-463.
Phipps, D. J., D. R. Branch, and L. C. Schlichter. 1996. Chloride-channel block inhibits T lymphocyte activation and signalling. Cell Signal 8 (2):141-149.
Pierres, M., C. Goridis, and P. Golstein. 1982. Inhibition of murine T cell-mediated cytolysis and T cell proliferation by a rat monoclonal antibody immunoprecipitating two lymphoid cell surface polypeptides of 94 000 and 180 000 molecular weight. Eur J Immunol 12 (1):60-69.
Prieto, J., C. Qian, N. Garcia, J. Diez, and J. F. Medina. 1993. Abnormal expression of anion exchanger genes in primary biliary cirrhosis. Gastroenterology 105 (2):572-578.
Przybysz, M., K. Borysewicz, and I. Katnik-Prastowska. 2009. Differences between the early and advanced stages of rheumatoid arthritis in the expression of EDA-containing fibronectin. Rheumatol Int 29 (12):1397-1401.
Ratajczak, M. Z., R. Reca, M. Wysoczynski, J. Yan, and J. Ratajczak. 2006. Modulation of the SDF-1-CXCR4 axis by the third complement component (C3)--implications for trafficking of CXCR4+ stem cells. Exp Hematol 34 (8):986-995.

Bibliografía
173
Riedemann, N. C., R. F. Guo, V. J. Sarma, I. J. Laudes, M. Huber-Lang, R. L. Warner, E. A. Albrecht, C. L. Speyer, and P. A. Ward. 2002. Expression and function of the C5a receptor in rat alveolar epithelial cells. J Immunol 168 (4):1919-1925.
Rittirsch, D., M. A. Flierl, B. A. Nadeau, D. E. Day, M. Huber-Lang, C. R. Mackay, F. S. Zetoune, N. P. Gerard, K. Cianflone, J. Kohl, C. Gerard, J. V. Sarma, and P. A. Ward. 2008. Functional roles for C5a receptors in sepsis. Nat Med 14 (5):551-557.
Rock, K. L., and L. Shen. 2005. Cross-presentation: underlying mechanisms and role in immune surveillance. Immunol Rev 207:166-183.
Romero, M. F., C. M. Fulton, and W. F. Boron. 2004. The SLC4 family of HCO 3 - transporters. Pflugers Arch 447 (5):495-509.
Rosoff, P. M., and G. Terres. 1986. Cyclosporine A inhibits Ca2+-dependent stimulation of the Na+/H+ antiport in human T cells. J Cell Biol 103 (2):457-463.
Rouzaire-Dubois, B., J. B. Milandri, S. Bostel, and J. M. Dubois. 2000. Control of cell proliferation by cell volume alterations in rat C6 glioma cells. Pflugers Arch 440 (6):881-888.
Rybak, J. N., C. Roesli, M. Kaspar, A. Villa, and D. Neri. 2007. The extra-domain A of fibronectin is a vascular marker of solid tumors and metastases. Cancer Res 67 (22):10948-10957.
Saito, S., N. Yamaji, K. Yasunaga, T. Saito, S. Matsumoto, M. Katoh, S. Kobayashi, and Y. Masuho. 1999. The fibronectin extra domain A activates matrix metalloproteinase gene expression by an interleukin-1-dependent mechanism. J Biol Chem 274 (43):30756-30763.
Sakaguchi, S., N. Sakaguchi, M. Asano, M. Itoh, and M. Toda. 1995. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 155 (3):1151-1164.
Salas, J. T., J. M. Banales, S. Sarvide, S. Recalde, A. Ferrer, I. Uriarte, R. P. Oude Elferink, J. Prieto, and J. F. Medina. 2008. Ae2a,b-deficient mice develop antimitochondrial antibodies and other features resembling primary biliary cirrhosis. Gastroenterology 134 (5):1482-1493.
Salem, M. L., A. N. Kadima, D. J. Cole, and W. E. Gillanders. 2005. Defining the antigen-specific T-cell response to vaccination and poly(I:C)/TLR3 signaling: evidence of enhanced primary and memory CD8 T-cell responses and antitumor immunity. J Immunother 28 (3):220-228.
Sancho, D., D. Mourao-Sa, O. P. Joffre, O. Schulz, N. C. Rogers, D. J. Pennington, J. R. Carlyle, and C. Reis e Sousa. 2008. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J Clin Invest 118 (6):2098-2110.
Scola, A. M., A. Higginbottom, L. J. Partridge, R. C. Reid, T. Woodruff, S. M. Taylor, D. P. Fairlie, and P. N. Monk. 2007. The role of the N-terminal domain of the

Bibliografía
174
complement fragment receptor C5L2 in ligand binding. J Biol Chem 282 (6):3664-3671.
Schnare, M., G. M. Barton, A. C. Holt, K. Takeda, S. Akira, and R. Medzhitov. 2001. Toll-like receptors control activation of adaptive immune responses. Nat Immunol 2 (10):947-950.
Schumacher, W. A., J. C. Fantone, S. E. Kunkel, R. C. Webb, and B. R. Lucchesi. 1991. The anaphylatoxins C3a and C5a are vasodilators in the canine coronary vasculature in vitro and in vivo. Agents Actions 34 (3-4):345-349.
Schwarz, K., T. Storni, V. Manolova, A. Didierlaurent, J. C. Sirard, P. Rothlisberger, and M. F. Bachmann. 2003. Role of Toll-like receptors in costimulating cytotoxic T cell responses. Eur J Immunol 33 (6):1465-1470.
Selander, B., U. Martensson, A. Weintraub, E. Holmstrom, M. Matsushita, S. Thiel, J. C. Jensenius, L. Truedsson, and A. G. Sjoholm. 2006. Mannan-binding lectin activates C3 and the alternative complement pathway without involvement of C2. J Clin Invest 116 (5):1425-1434.
Shedlock, D. J., and H. Shen. 2003. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science 300 (5617):337-339.
Shimizu, J., S. Yamazaki, and S. Sakaguchi. 1999. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol 163 (10):5211-5218.
Shinde, A. V., C. Bystroff, C. Wang, M. G. Vogelezang, P. A. Vincent, R. O. Hynes, and L. Van De Water. 2008. Identification of the peptide sequences within the EIIIA (EDA) segment of fibronectin that mediate integrin alpha9beta1-dependent cellular activities. J Biol Chem 283 (5):2858-2870.
Shushakova, N., J. Skokowa, J. Schulman, U. Baumann, J. Zwirner, R. E. Schmidt, and J. E. Gessner. 2002. C5a anaphylatoxin is a major regulator of activating versus inhibitory FcgammaRs in immune complex-induced lung disease. J Clin Invest 110 (12):1823-1830.
Siegal, F. P., N. Kadowaki, M. Shodell, P. A. Fitzgerald-Bocarsly, K. Shah, S. Ho, S. Antonenko, and Y. J. Liu. 1999. The nature of the principal type 1 interferon-producing cells in human blood. Science 284 (5421):1835-1837.
Steinman, R. M., and J. W. Young. 1991. Signals arising from antigen-presenting cells. Curr Opin Immunol 3 (3):361-372.
Steitz, J., J. Bruck, J. Lenz, J. Knop, and T. Tuting. 2001. Depletion of CD25(+) CD4(+) T cells and treatment with tyrosinase-related protein 2-transduced dendritic cells enhance the interferon alpha-induced, CD8(+) T-cell-dependent immune defense of B16 melanoma. Cancer Res 61 (24):8643-8646.
Stewart, A. K., C. E. Kurschat, and S. L. Alper. 2007. Role of nonconserved charged residues of the AE2 transmembrane domain in regulation of anion exchange by pH. Pflugers Arch 454 (3):373-384.

Bibliografía
175
Strainic, M. G., J. Liu, D. Huang, F. An, P. N. Lalli, N. Muqim, V. S. Shapiro, G. R. Dubyak, P. S. Heeger, and M. E. Medof. 2008. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity 28 (3):425-435.
Strieter, R. M., K. Kasahara, R. M. Allen, T. J. Standiford, M. W. Rolfe, F. S. Becker, S. W. Chensue, and S. L. Kunkel. 1992. Cytokine-induced neutrophil-derived interleukin-8. Am J Pathol 141 (2):397-407.
Sun, S., X. Zhang, D. F. Tough, and J. Sprent. 1998. Type I interferon-mediated stimulation of T cells by CpG DNA. J Exp Med 188 (12):2335-2342.
Surh, C. D., and J. Sprent. 2008. Homeostasis of naive and memory T cells. Immunity 29 (6):848-862.
Sutmuller, R. P., M. H. den Brok, M. Kramer, E. J. Bennink, L. W. Toonen, B. J. Kullberg, L. A. Joosten, S. Akira, M. G. Netea, and G. J. Adema. 2006. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest 116 (2):485-494.
Sutmuller, R. P., L. M. van Duivenvoorde, A. van Elsas, T. N. Schumacher, M. E. Wildenberg, J. P. Allison, R. E. Toes, R. Offringa, and C. J. Melief. 2001. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J Exp Med 194 (6):823-832.
Takahashi, T., Y. Kuniyasu, M. Toda, N. Sakaguchi, M. Itoh, M. Iwata, J. Shimizu, and S. Sakaguchi. 1998. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol 10 (12):1969-1980.
Takeda, K., T. Kaisho, and S. Akira. 2003. Toll-like receptors. Annu Rev Immunol 21:335-376.
Thornton, A. M., and E. M. Shevach. 1998. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med 188 (2):287-296.
Tighe, H., K. Takabayashi, D. Schwartz, R. Marsden, L. Beck, J. Corbeil, D. D. Richman, J. J. Eiden, Jr., H. L. Spiegelberg, and E. Raz. 2000. Conjugation of protein to immunostimulatory DNA results in a rapid, long-lasting and potent induction of cell-mediated and humoral immunity. Eur J Immunol 30 (7):1939-1947.
Townsend, A., and H. Bodmer. 1989. Antigen recognition by class I-restricted T lymphocytes. Annu Rev Immunol 7:601-624.
Ulmer, J. B., J. J. Donnelly, S. E. Parker, G. H. Rhodes, P. L. Felgner, V. J. Dwarki, S. H. Gromkowski, R. R. Deck, C. M. DeWitt, A. Friedman, and et al. 1993. Heterologous protection against influenza by injection of DNA encoding a viral protein. Science 259 (5102):1745-1749.

Bibliografía
176
van Leeuwen, J. E., and L. E. Samelson. 1999. T cell antigen-receptor signal transduction. Curr Opin Immunol 11 (3):242-248.
Vazeux, R., P. A. Hoffman, J. K. Tomita, E. S. Dickinson, R. L. Jasman, T. St John, and W. M. Gallatin. 1992. Cloning and characterization of a new intercellular adhesion molecule ICAM-R. Nature 360 (6403):485-488.
Vergani, D. 1986. Complement. Diabet Med 3 (4):306-311.
Vig, M., and J. P. Kinet. 2009. Calcium signaling in immune cells. Nat Immunol 10 (1):21-27.
Villa, A., E. Trachsel, M. Kaspar, C. Schliemann, R. Sommavilla, J. N. Rybak, C. Rosli, L. Borsi, and D. Neri. 2008. A high-affinity human monoclonal antibody specific to the alternatively spliced EDA domain of fibronectin efficiently targets tumor neo-vasculature in vivo. Int J Cancer 122 (11):2405-2413.
Villadangos, J. A., and P. Schnorrer. 2007. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat Rev Immunol 7 (7):543-555.
Wang, G. L., Y. Qian, Q. Y. Qiu, X. J. Lan, H. He, and Y. Y. Guan. 2006. Interaction between Cl- channels and CRAC-related Ca2+ signaling during T lymphocyte activation and proliferation. Acta Pharmacol Sin 27 (4):437-446.
Wang, L., J. O. Sunyer, and L. J. Bello. 2004. Fusion to C3d enhances the immunogenicity of the E2 glycoprotein of type 2 bovine viral diarrhea virus. J Virol 78 (4):1616-1622.
Ward, P. A. 2004. The dark side of C5a in sepsis. Nat Rev Immunol 4 (2):133-142.
Watanabe, I., T. M. Ross, S. Tamura, T. Ichinohe, S. Ito, H. Takahashi, H. Sawa, J. Chiba, T. Kurata, T. Sata, and H. Hasegawa. 2003. Protection against influenza virus infection by intranasal administration of C3d-fused hemagglutinin. Vaccine 21 (31):4532-4538.
Werling, D., and T. W. Jungi. 2003. TOLL-like receptors linking innate and adaptive immune response. Vet Immunol Immunopathol 91 (1):1-12.
Wetsel, R. A. 1995. Expression of the complement C5a anaphylatoxin receptor (C5aR) on non-myeloid cells. Immunol Lett 44 (2-3):183-187.
Winslow, M. M., J. R. Neilson, and G. R. Crabtree. 2003. Calcium signalling in lymphocytes. Curr Opin Immunol 15 (3):299-307.
Xiao, G. N., Y. Y. Guan, and H. He. 2002. Effects of Cl- channel blockers on endothelin-1-induced proliferation of rat vascular smooth muscle cells. Life Sci 70 (19):2233-2241.
Xu, G. L., K. Q. Zhang, B. Guo, T. T. Zhao, F. Yang, M. Jiang, Q. H. Wang, Y. H. Shang, and Y. Z. Wu. 2010. Induction of protective and therapeutic antitumor immunity by a DNA vaccine with C3d as a molecular adjuvant. Vaccine 28 (44):7221-7227.

Bibliografía
177
Young, H. A., and K. J. Hardy. 1995. Role of interferon-gamma in immune cell regulation. J Leukoc Biol 58 (4):373-381.
Zhang, J., K. V. Salojin, J. X. Gao, M. J. Cameron, I. Bergerot, and T. L. Delovitch. 1999. p38 mitogen-activated protein kinase mediates signal integration of TCR/CD28 costimulation in primary murine T cells. J Immunol 162 (7):3819-3829.
Zhang, X., Y. Kimura, C. Fang, L. Zhou, G. Sfyroera, J. D. Lambris, R. A. Wetsel, T. Miwa, and W. C. Song. 2007. Regulation of Toll-like receptor-mediated inflammatory response by complement in vivo. Blood 110 (1):228-236.
Zhang, X., I. Schmudde, Y. Laumonnier, M. K. Pandey, J. R. Clark, P. Konig, N. P. Gerard, C. Gerard, M. Wills-Karp, and J. Kohl. A critical role for C5L2 in the pathogenesis of experimental allergic asthma. J Immunol 185 (11):6741-6752.
Zhang, X., S. Sun, I. Hwang, D. F. Tough, and J. Sprent. 1998. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity 8 (5):591-599.
Zinkernagel, R. M., and A. Althage. 1977. Antiviral protection by virus-immune cytotoxic T cells: infected target cells are lysed before infectious virus progeny is assembled. J Exp Med 145 (3):644-651.
Zuckerman, L. A., L. Pullen, and J. Miller. 1998. Functional consequences of costimulation by ICAM-1 on IL-2 gene expression and T cell activation. J Immunol 160 (7):3259-3268.


ANEXOS


Anexos
181
Anexo 1: Artículo
Gil-Guerrero L, Dotor J, Huibregtse IL, Casares N, López-Vázquez AB, Rudilla F,
Riezu-Boj JI, López-Sagaseta J, Hermida J, Van Deventer S, Bezunartea J, Llopiz D,
Sarobe P, Prieto J, Borrás-Cuesta F, Lasarte JJ.Journal of Immunology, 2008,
181:126-135 In vitro and in vivo down-regulation of regulatory T cell activity
with a peptide inhibitor of TGF-beta1.
Anexo 2: Artículo
Mansilla C, Gorraiz M, Martinez M, Casares N, Arribillaga L, Rudilla F,
Echeverria I, Riezu-Boj JI, Sarobe P, Borrás-Cuesta F, Prieto J, Lasarte JJ. Journal
of Hepatology, 2009, 51: 520-527. Immunization against hepatitis C virus
with a fusion protein containing the extra domain A from fibronectin and
the hepatitis C virus NS3 protein.
Anexo 3: Artículo
Casares N, Rudilla F, Arribillaga L, Llopiz D, Riezu-Boj JI, Lozano T, López-
Sagaseta J, Guembe L, Sarobe P, Prieto J, Borrás-Cuesta F, Lasarte JJ. Journal of
Immunology, 2010, 185:5150-5159. A peptide inhibitor of FOXP3 impairs
regulatory T cell activity and improves vaccine efficacy in mice.


In Vitro and In Vivo Down-Regulation of Regulatory T CellActivity with a Peptide Inhibitor of TGF-�11
Lucıa Gil-Guerrero,2* Javier Dotor,2* Inge Louise Huibregtse,‡ Noelia Casares,*Ana Belen Lopez-Vazquez,* Francesc Rudilla,* Jose Ignacio Riezu-Boj,* Jacinto Lopez-Sagaseta,†
Jose Hermida,† Sander Van Deventer,‡ Jaione Bezunartea,* Diana Llopiz,* Pablo Sarobe,*Jesus Prieto,* Francisco Borras-Cuesta,3* and Juan Jose Lasarte3,4*
Down-regulation of CD4�CD25� regulatory T (Treg) cell function might be beneficial to enhance the immunogenicity of viral andtumor vaccines or to induce breakdown of immunotolerance. Although the mechanism of suppression used by Treg cells remainscontroversial, it has been postulated that TGF-�1 mediates their immunosuppressive activity. In this study, we show that P17, ashort synthetic peptide that inhibits TGF-�1 and TGF-�2 developed in our laboratory, is able to inhibit Treg activity in vitro andin vivo. In vitro studies demonstrate that P17 inhibits murine and human Treg-induced unresponsiveness of effector T cells toanti-CD3 stimulation, in an MLR or to a specific Ag. Moreover, administration of P17 to mice immunized with peptide vaccinescontaining tumor or viral Ags enhanced anti-vaccine immune responses and improved protective immunogenicity against tumorgrowth or viral infection or replication. When CD4� T cells purified from OT-II transgenic mice were transferred into C57BL/6mice bearing s.c. EG.7-OVA tumors, administration of P17 improved their proliferation, reduced the number of CD4�Foxp3� Tcells, and inhibited tumor growth. Also, P17 prevented development of immunotolerance induced by oral administration of OVAby genetically modified Lactococcus lactis in DO11.10 transgenic mice sensitized by s.c. injection of OVA. These findings dem-onstrate that peptide inhibitors of TGF-� may be a valuable tool to enhance vaccination efficacy and to break tolerance againstpathogens or tumor Ags. The Journal of Immunology, 2008, 181: 126–135.
D uring the last years, CD4�CD25� regulatory T (Treg)5
cells have been the subject of intense study. This interestis because their function appears to be critical in the
maintenance of peripheral tolerance and regulation of immune re-sponses to non-self Ags. Treg cells can inhibit activation of otherT cells (1) and are needed for protection against autoimmune dis-eases and prevention of rejection of allogeneic transplants. How-ever, immunoregulatory function of Treg cells may hinder the in-duction of immune responses against cancer and infectious agents.Thus, the presence of Treg cells within tumors may prevent acti-vation of antitumor immune responses favoring tumor growth.This effect suggests that counteracting Treg activity could evoke
effective antitumor immunity (2–5). Treg cells capable of sup-pressing the in vitro function of tumor-reactive T cells have beenfound in humans in tumors such as melanoma (6, 7), lung (8),ovary (8, 9), pancreas and breast (10) cancers as well as hepato-cellular carcinoma (11, 12). Moreover, recent findings suggest thatTreg cells infiltrating neoplastic tissues might be associated with ahigher death hazard and reduced survival (7, 9, 12). In infectiousdiseases, the control exerted by Treg cells may limit the magnitudeof effector T cell responses and may result in failure to controlinfection. Indeed, it has been shown that some viruses, such ashepatitis B (13), hepatitis C (14–17), and HIV (18–21), may ex-ploit Treg cells to dampen the antiviral response to favor the per-sistence of the infection.
Although Treg cells require Ag exposure to initiate suppressiveactivity, the effector phase seems to be mediated by an Ag non-specific mechanism (22). The mechanism of suppression by Tregcells remains controversial, with differences between in vitro andin vivo experiments in terms of the relative contribution of solublecytokines with respect to cell-to-cell contact. In many experimen-tal systems, multiple subsets of Treg cells seem to function in vivoby secreting immunosuppressive cytokines such as TGF-� andIL-10 (23–26). It has been suggested that TGF-� produced by Tregcells or bound to the cell membrane may mediate suppression of Tcells (24). Moreover, it has been described that CD4�CD25� cell-mediated suppression of autoimmune or antitumor CD8� cells re-quires an intact TGF-� receptor II on the CD8� cells (27, 28).TGF-� is also important in the homeostasis of Treg cells becauseit may contribute to the generation and proliferation of Treg cells(29). In addition, in the context of TCR stimulation, TGF-� is ableto convert peripheral CD4�CD25� naive T cells to CD4�CD25�
Treg cells via induction of transcription factor Foxp3 (30, 31).These data suggest that inhibition of TGF-�, in particular by smallmolecules that might penetrate the interface between contacting T
*Center for Applied Medical Research, Division of Hepatology and Gene Therapy,and †Division of Cardiovascular Sciences, University of Navarra, Pamplona, Spain;and ‡Academic Medical Center, Center for Experimental and Molecular Medicine,University of Amsterdam, Amsterdam, The Netherlands
Received for publication September 27, 2007. Accepted for publication April24, 2008.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by grants from Fundacion Mutua Madrilena and Gobiernode Navarra (to J.J.L.), by Grants SAF2007-61432 (to J.J.L.), SAF2004-01680 (toP.S.), and SAF2003-04751 (to F.B.-C.) from Ministerio de Educacion y Ciencia, andby Union Temporal de Empresas-proyecto Centro de Investigacion Medica Aplicada.2 L.G.-G. and J.D. contributed equally to this work.3 F.B.-C. and J.J.L. are senior authors on this work.4 Address correspondence and reprint requests to Dr. Juan Jose Lasarte, Area deHepatologıa y Terapia Genica, Centro de Investigacion Medica Aplicada, AvenidaPıo XII no. 55, 31008 Pamplona, Spain. E-mail address: [email protected] Abbreviations used in this paper: Treg, regulatory T; TCd, cytotoxic T cell deter-minant; LAP, latency-associated peptide; HCV, hepatitis C virus; DTH, delayed-typehypersensitivity.
Copyright © 2008 by The American Association of Immunologists, Inc. 0022-1767/08/$2.00
The Journal of Immunology
www.jimmunol.org

cells, would be a valuable tool to inhibit Treg activity and con-comitantly foster antiviral or antitumor immunotherapies.
Using a phage-displayed random 15-mer peptide library wehave identified a peptide inhibitor of TGF-�1, named P17. Thispeptide blocks the inhibition activity of TGF-�1 on the growth ofMv-1-Lu cells in vitro and also inhibits TGF-�1-dependent ex-pression of collagen type I mRNA in the liver of mice orally chal-lenged with carbon tetrachloride (32). In the present study weshow that P17 is able to inhibit Treg cells in vitro and improve theefficacy of vaccination in vivo. Also, P17 reduces the number ofCD4�Foxp3� T cells and augments proliferation of OT-II-derivedCD4� T cells after their adoptive transfer in mice bearing EG.7-OVA tumors. Moreover P17 efficiently blocked the induction ofAg-specific oral tolerance induced in vivo after intragastric sup-plementation of the OVA secreting Lactococcus lactis (LL-OVA)in DO11.10 mice (33). Thus, as we discuss in more detail, P17clearly holds promise to boost immune responses by down-regu-lation of CD4�CD25� Treg cells, an effect that can be used toenhance the effectiveness of vaccination.
Materials and MethodsPeptides
KRIWFIPRSSWYERA (P17) is a peptide inhibitor of TGF-� developed inour laboratory (32). Peptide SPSYVYHQF (herein AH1) is a cytotoxic Tcell determinant (TCd) expressed by CT26 cells and presented by H-2Ld
MHC class I molecules (34). Peptide p1073 (CVNGVCWTV) from hep-atitis C virus (HCV) NS3 protein containing a TCd presented by HLA.A2.1class I molecules (35), the H2-Kb-restricted OVA TCd peptide SIINFEKL,and an irrelevant control peptide (AKAVVKTFHETLDCC) from humanCD81 molecule were synthesized manually in a multiple peptide synthe-sizer using F-moc chemistry as previously described (36). The purity ofpeptides was �90% as judged by HPLC.
Mice
Female BALB/c and C57BL/6 mice were purchased from IFFA-Credo. Abreeding pair of HHD transgenic mice expressing human HLA.A2 mole-cule was provided by Dr. F. Lemonnier (Institut Pasteur, Paris, France),and were bred and maintained in pathogen-free conditions. OVA-specificTCR transgenic mice (DO11.10) on a BALB/c background were providedby Dr. J. N. Samson (Vrije Universiteit, Amsterdam, The Netherlands) andbred at the Academic Medical Center (Amsterdam, The Netherlands).Eight- to 10-wk-old DO11.10 mice were used for the experiments. OVA-specific TCR transgenic mice OT-II and OT-I (C57BL/6 background) wereprovided by Dr. I. Melero (Centro de Investigacion Medica Aplicada, Pam-plona, Spain). All mice were housed in a conventional animal facility underroutine laboratory conditions. All experiments performed followed insti-tutional guidelines and were approved by the institutional ethicalcommittee.
Cell culture
BSC-1 cells, provided by Dr. J. A. Berzofsky (National Institutes of Health,Bethesda, MD) were used for titration of vaccinia virus in ovaries. T2 andP815 cells from the American Type Culture Collection (ATCC) were usedas target cells in chromium release assays with CTL from HHD or BALB/cmice, respectively. OVA-transfected E.G7-OVA cells (H-2b) (37) andCT26 tumor cells (H-2d) were purchased from ATCC and used in vivo fortumor protection and treatment experiments. They were cultured in com-plete medium (RPMI 1640 containing 10% FCS, 100 U/ml penicillin, 100�g/ml streptomycin, 2 mM glutamine, and 50 �M 2-ME). Medium forE.G7-OVA cells also contained 400 �g/ml G418.
Purification of Treg cells
Isolation of murine and human CD4�, CD4�CD25�, and CD4�CD25� Tcells was performed from murine spleen cells or from human PBMC byusing murine and human Treg isolation kits (Miltenyi Biotec), respectively,according to the manufacturer’s instructions. The purity of the resulting Tcell populations was confirmed to be �95% by flow cytometry. Expressionof membrane-associated TGF-�1 on the surface of CD4�CD25� T cellswas measured by flow cytometry using anti-latency-associated peptide (anti-LAP, TGF-�1) Abs (R&D Systems).
RNA extraction and real-time RT-PCR
Total RNA extraction from CD4�CD25� or from CD4�CD25� T cellswas performed using the Nucleic Acid Purification Lysis Solution (AppliedBiosystems) and the semiautomated system ABI PRISM 6100 NucleicAcid PrepStation (Applied Biosystems). DNase treatment, reverse tran-scription, and quantitative real-time PCR for CTLA4, glucocorticoid-in-duced TNFR-related gene, Foxp3, and IL-10 were conducted as described(38). mRNA values were represented by the 2�Ct formula, where �Ct in-dicates the difference in threshold cycle between control (� actin) andtarget genes.
In vitro assays for murine or human Treg function
Inhibitory activity of murine or human Treg cells was measured in threedifferent in vitro assays of T cell stimulation. CD25-depleted spleen cells(105 cells/well) from BALB/c mice or human PBMC (105) were stimulatedin vitro with the following: 0.5 �g/ml of anti-mouse or anti-human CD3 Ab(BD Pharmingen), respectively; bone marrow dendritic cells (103) fromC57BL/6 mice (prepared as previously described (39)) or human PBMC(105) from a different donor (to induce a MLR); or a specific Ag. To studythe effect of Treg cells on Ag-specific T cell stimulation, 105 OVA-specificCD8� T cells from TCR transgenic OT-I mice were incubated with 103
bone marrow dendritic cells (from C57BL/6 mice) pulsed with SIINFEKLpeptide (1 �g/ml), whereas CD4�CD25� effector T cells (105/well) iso-lated from human PBMC were cultured in the presence or absence oftetanus toxoid Ag (5 limit of flocculation units/ml). All T cell stimulationwas conducted in the presence or absence of CD4�CD25� Treg cells (104),and the indicated concentrations of peptide P17 or control peptide wereadded to the cultures. To assess whether CD4�CD25� T cells exert theirregulatory function through direct cell contact or through release of solublefactors, we performed a series of Transwell experiments. Once purified,CD4�CD25� T cells were added at a ratio of 1:10 to autologousCD4�CD25� T cells seeded at 5 � 105 cells/well in the lower chamber ofa 24-well plate. CD4�CD25� T cells were either cultured in the lowerchamber directly in contact with the target cells or in the upper chamberseparated from the target cells by a 0.4-�m pore membrane (DiscoveryLabware; BD Biosciences), which allows diffusion of small molecules,such as cytokines, but not of cells. T cell proliferation was tested after 3days of culture by measuring [methyl-3H]thymidine incorporation. Brieflyat the second day of culture, 0.5 �Ci of [methyl-3H]thymidine was addedto each well and incubated overnight. Cells were harvested (Filtermate 96harvester; Packard Instrument), and incorporated radioactivity was mea-sured using a scintillation counter (TopCount; Packard Instrument). IFN-�secretion to the culture supernatant was measured by ELISA (BD Pharm-ingen) according to the manufacturer’s instructions.
In vitro assays of T cell proliferation in the presence ofTGF-�1 or TGF-�2
Recombinant human TGF-�1 or TGF-�2 inhibit proliferation of murine- orhuman-derived effector T cells stimulated with anti-CD3. The IC50 forhuman TGF-�1 was 20 and 1 pg/ml when using murine- and human-derived effector T cells, respectively. For TGF-�2, the IC50 was 0.25 pg/mlwhen using murine splenocytes. Splenocytes from C57BL/6 female mice,or PBLs from human blood donors, were cultured (105 cells/well) in thepresence or absence of 0.5 �g/ml anti-mouse or anti-human CD3 Abs andin the presence or absence of the corresponding IC50 of exogenously addedhuman TGF-�1 or human TGF-�2 (R&D Systems). P17 or control peptidewas added to the cultures at the concentrations indicated in each experi-ment. T cell proliferation was tested after 3 days of culture by measuring[methyl-3H]thymidine incorporation as described.
Biomolecular interaction analysis
Screening of peptide binding to TGF-�1, TGF-�2, and TGF-�3 was per-formed by surface plasmon resonance using a BIAcore X Biosensor.TGF-� isoforms (R&D Systems) as well as an irrelevant protein werecovalently immobilized onto the surface of flow cell 2 of CM5 chips (BIA-core) as described (40). Flow cell 1, which does not contain immobilizedTGF-�, was used as the reference flow cell. Individual peptide solutions (5�M) were injected three times in 10 mM HEPES, 150 mM NaCl, 0.005%(v/v) Tween 20, 0.1 mg/ml BSA (pH 7.4), at a flow of 30 �l/min. Masstransport limitation was excluded. Curves were processed by subtractingthe response in flow cell 1 from that in flow cell 2.
127The Journal of Immunology
Immunization experiments and measurement of T cell activation
BALB/c or HHD mice were immunized s.c. at day 0 with 50 nmol TCdpeptides AH1 or p1073, respectively, which were emulsified in IFA. Pep-tide P17 (50 nmol/mouse) was administered i.p. on days 6–9 after immu-nization. At day 10, mice were sacrificed, and spleens were removed, ho-mogenized, and 8 � 105 cells cultured in 96-well plates in completemedium in the presence or absence of 10 nM of the corresponding TCdpeptide. Peptide-specific CTL responses were measured using a conven-tional cytotoxicity assay as previously described (41). IFN-� produced in
response to the TCd was measured by ELISA (BD Pharmingen) in culturesupernatants (50 �l) harvested after 48 h of culture, according to the man-ufacturer’s instructions.
Effect of P17 in antitumor peptide vaccination
Animals immunized with 50 nmol AH1 peptide emulsified in IFA as previ-ously described (42) were treated with 50 nmol P17 peptide or with saline ondays 6–10. Ten days after immunization, mice were challenged by s.c. injec-tion with 5 � 105 CT26 tumor cells. In an independent experiment, 100 �g per
FIGURE 1. P17 inhibits immunosuppression caused by TGF-� or by murine Treg cells. A, Inhibition of TGF-�1. Spleen cells from BALB/c mice werestimulated with anti-CD3 mAbs in the presence or absence of exogenously added recombinant human TGF-�1 (20 pg/ml) and the indicated concentrationof P17, control peptide (Pcont), or polyclonal anti-TGF-� or the corresponding isotype control Abs (2 �g/ml). B, Characterization of purified CD4�CD25�
T cells. Expression of TGF-�1 bound to LAP on their membrane was measured by flow cytometry, and mRNA expression of Treg-associated genes wasmeasured by real-time PCR. C, Immunosuppression caused by purified CD4�CD25� T cells is contact-dependent. Effector T cells were stimulated withanti-CD3 in the presence of purified CD4�CD25� T cells either in direct contact or separated by a 0.4-�m pore membrane. D–F, Inhibition of murine Tregcells by P17. D, Spleen cells from BALB/c mice were stimulated with anti-CD3 mAbs in the presence or absence of purified CD4�CD25� Treg cells. E,MLR using spleen cells from BALB/c mice (105 cells/well) and bone marrow-derived dendritic cells from C57BL/6 mice (103 cells/well). F, T cells fromOT-I mice were incubated with DC pulsed with SIINFEKL peptide, with or without CD4�CD25� Treg cells. Different concentrations of P17 (12.5–100�M) were tested in D to measure inhibition of Treg activity, whereas a single concentration (100 �M) was tested in E and F. A concentration of 2 �g/mlpolyclonal anti-TGF-� or the corresponding isotype control Abs (2 �g/ml) was used in A and D. Cell proliferation (A and C–F) was analyzed by measuringtritiated thymidine incorporation in the harvested cells using a scintillation counter. IFN-� released to the culture was quantified by ELISA (E). �, p � 0.05by t test comparison between the indicated group and the corresponding control of immunosuppression in the absence of inhibitors. Results are repre-sentative of at least three different experiments per each result.
128 DOWN-REGULATION OF Treg CELLS BY A TGF-� INHIBITOR PEPTIDE
dose of neutralizing anti-TGF-� polyclonal Ab from rabbit, or the correspond-ing isotype control (R&D Systems), were administered i.p. to mice using thesame schedule as for P17. Tumor size was monitored twice a week with acaliper and was expressed according to the formula: volume in cubic milli-meters � (length � (width)2)/2). Mice were sacrificed when tumor sizereached a volume greater than 4 cm3.
Effect of P17 in “in vivo” T cell transfer experiments inE.G7-OVA tumor-bearing mice
Groups of five C57BL/6 mice were challenged with 5 � 105 E.G7-OVAtumor cells. When tumors reached 12.5 mm3 in size, mice were adoptivelytransferred with 3 � 106 CFSE-labeled CD4� T cells isolated from OT-IItransgenic mice. CFSE cell labeling was conducted by incubation with 1 �MCFSE for 10 min at room temperature followed by three washes with PBS.After T cell transfer, a group of mice was treated daily with 50 nmol P17 byi.p. route. Five days after T cell transfer, mice were immunized i.v. with 50 �gof OVA (Sigma-Aldrich) in PBS and sacrificed 3 days after immunization.Splenocytes were isolated, and the analysis of CFSE-labeled CD4� T cellproliferation and CD4�Foxp3� cell staining was conducted by flow cytom-etry. Analysis of CD4�Foxp3� cells was conducted using the mouse Treg cellstaining kit (eBioscience) according to the manufacturer’s instructions. Tumorsize was measured the day of sacrifice as described.
Effect of P17 in in vivo protection against infection with arecombinant vaccinia virus expressing HCV proteins
HHD mice immunized with peptide p1073 as described, were treated i.p.with 50 nmol P17 or with saline at days 6–9 after immunization. They werechallenged i.p. at day 10 with 5 � 106 PFU of the recombinant vacciniavHCV1–3011 expressing HCV polyprotein. Three days after vaccinia chal-lenge, mice were sacrificed, and viral titer was measured as described (43).
Effect of P17 in a model of hypersensitivity
The L. lactis MG1363 strain was genetically modified and used throughoutthis study, as previously described (44, 45). Bacteria were cultured in
GM17E medium, i.e., M17 broth (Difco) supplemented with 0.5% glucoseand 5 �g/ml erythromycin (Abbott B.V.). Bacteria were diluted 200-fold inGM17E medium, incubated at 30°C overnight, harvested by centrifugation,and concentrated in BM9 medium at 2 � 109 bacteria/100 �l. Treatedmice, received 100 �l of this suspension daily by intragastric catheter (46).
DO11.10 mice were sensitized by s.c. injection of 100 �g of OVA in 50�l of a 1:1 CFA (Difco) saline solution in the tail base at day 1 (47). Micewere fed BM9 as a control or LL-OVA (both at days 1–5 and days 8–12)administrations using a stainless 18-gauge animal feeding needle. Everyother day, starting at day 0, mice received 50 nmol P17 peptide each.Eleven days after sensitization, Ag-specific delayed-type hypersensitiv-ity (DTH) responses were assessed. For DTH measurement, mice werechallenged with 10 �g of OVA in 10 �l of saline in the auricle of oneear and 10 �l of saline in the other. The increase in ear thickness wasmeasured in a blinded fashion using an engineer’s micrometer (Mitu-toyo) at 24 h after challenge. DTH responses were expressed as thedifference in increase between the OVA-injected and saline-injected earthickness, following subtraction of ear thickness before the challenge(DTH response � OVA � saline � baseline). Intact, LPS-free OVAgrade V protein was used as Ag (Sigma-Aldrich).
For cytokine measurements, 2 � 105 cells of splenocytes were culturedin 96-well U-bottom plates in a total volume of 200 �l of complete mediumwith 100 �g/ml OVA. Cells were cultured at 37°C in a 5% CO2 humidifiedincubator. After 72 h, culture supernatants were collected and frozen at�20°C until cytokine analysis was performed. Cytokine production wasquantified using the Mouse Inflammation Cytometric Bead Assay (BDBiosciences).
Statistical analysis
Normality was assessed with Shapiro-Wilk W test. Statistical analyses wereperformed using parametric (Student’s t test and one-way ANOVA) andnonparametric (Kruskal-Wallis and Mann-Whitney U test) tests. For alltests, a value for p � 0.05 was considered statistically significant. Descrip-tive data for continuous variables are reported as mean � SD. SPSS 9.0 forWindows was used for statistical analysis.
FIGURE 2. P17 inhibits immunosuppression caused by TGF-� or by human Treg cells. A, Inhibition of TGF-�1. Human PBMC were stimulated withanti-CD3 mAbs in the presence or absence of exogenously added recombinant human TGF-�1 (1 pg/ml) and the indicated concentration of P17 orpolyclonal anti-TGF-� or the corresponding isotype control Abs (2 �g/ml). B–D, Inhibition of human Treg cells. B, Human PBMC were stimulated withanti-CD3 mAbs in the presence or absence of purified CD4�CD25� Treg cells. C, MLR using PBMC isolated from two donors in the presence or absenceof CD4�CD25� Treg cells isolated from one of the human donors. D, Human PBMC were cultured with or without tetanus toxoid in the presence orabsence of human CD4�CD25� Treg cells. Different concentrations of P17 (12–100 �M) were tested in B to measure inhibition of Treg activity, whereasa single concentration (100 �M) was tested in C and D. Cell proliferation was analyzed by measuring tritiated thymidine incorporation in the harvestedcells using a scintillation counter. �, p � 0.05 t test comparison between the indicated group and the corresponding control of immunosuppression in theabsence of inhibitors. Results are representative of at least three different experiments per each result.
129The Journal of Immunology
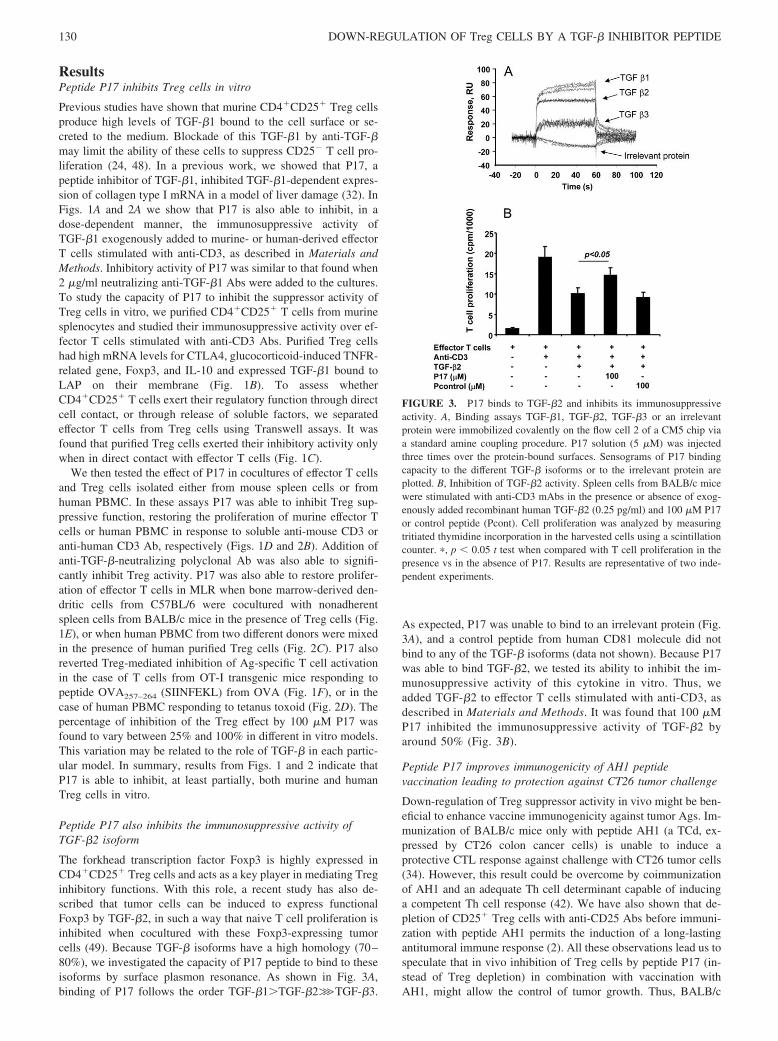
ResultsPeptide P17 inhibits Treg cells in vitro
Previous studies have shown that murine CD4�CD25� Treg cellsproduce high levels of TGF-�1 bound to the cell surface or se-creted to the medium. Blockade of this TGF-�1 by anti-TGF-�may limit the ability of these cells to suppress CD25� T cell pro-liferation (24, 48). In a previous work, we showed that P17, apeptide inhibitor of TGF-�1, inhibited TGF-�1-dependent expres-sion of collagen type I mRNA in a model of liver damage (32). InFigs. 1A and 2A we show that P17 is also able to inhibit, in adose-dependent manner, the immunosuppressive activity ofTGF-�1 exogenously added to murine- or human-derived effectorT cells stimulated with anti-CD3, as described in Materials andMethods. Inhibitory activity of P17 was similar to that found when2 �g/ml neutralizing anti-TGF-�1 Abs were added to the cultures.To study the capacity of P17 to inhibit the suppressor activity ofTreg cells in vitro, we purified CD4�CD25� T cells from murinesplenocytes and studied their immunosuppressive activity over ef-fector T cells stimulated with anti-CD3 Abs. Purified Treg cellshad high mRNA levels for CTLA4, glucocorticoid-induced TNFR-related gene, Foxp3, and IL-10 and expressed TGF-�1 bound toLAP on their membrane (Fig. 1B). To assess whetherCD4�CD25� T cells exert their regulatory function through directcell contact, or through release of soluble factors, we separatedeffector T cells from Treg cells using Transwell assays. It wasfound that purified Treg cells exerted their inhibitory activity onlywhen in direct contact with effector T cells (Fig. 1C).
We then tested the effect of P17 in cocultures of effector T cellsand Treg cells isolated either from mouse spleen cells or fromhuman PBMC. In these assays P17 was able to inhibit Treg sup-pressive function, restoring the proliferation of murine effector Tcells or human PBMC in response to soluble anti-mouse CD3 oranti-human CD3 Ab, respectively (Figs. 1D and 2B). Addition ofanti-TGF-�-neutralizing polyclonal Ab was also able to signifi-cantly inhibit Treg activity. P17 was also able to restore prolifer-ation of effector T cells in MLR when bone marrow-derived den-dritic cells from C57BL/6 were cocultured with nonadherentspleen cells from BALB/c mice in the presence of Treg cells (Fig.1E), or when human PBMC from two different donors were mixedin the presence of human purified Treg cells (Fig. 2C). P17 alsoreverted Treg-mediated inhibition of Ag-specific T cell activationin the case of T cells from OT-I transgenic mice responding topeptide OVA257–264 (SIINFEKL) from OVA (Fig. 1F), or in thecase of human PBMC responding to tetanus toxoid (Fig. 2D). Thepercentage of inhibition of the Treg effect by 100 �M P17 wasfound to vary between 25% and 100% in different in vitro models.This variation may be related to the role of TGF-� in each partic-ular model. In summary, results from Figs. 1 and 2 indicate thatP17 is able to inhibit, at least partially, both murine and humanTreg cells in vitro.
Peptide P17 also inhibits the immunosuppressive activity ofTGF-�2 isoform
The forkhead transcription factor Foxp3 is highly expressed inCD4�CD25� Treg cells and acts as a key player in mediating Treginhibitory functions. With this role, a recent study has also de-scribed that tumor cells can be induced to express functionalFoxp3 by TGF-�2, in such a way that naive T cell proliferation isinhibited when cocultured with these Foxp3-expressing tumorcells (49). Because TGF-� isoforms have a high homology (70–80%), we investigated the capacity of P17 peptide to bind to theseisoforms by surface plasmon resonance. As shown in Fig. 3A,binding of P17 follows the order TGF-�1�TGF-�2 TGF-�3.
As expected, P17 was unable to bind to an irrelevant protein (Fig.3A), and a control peptide from human CD81 molecule did notbind to any of the TGF-� isoforms (data not shown). Because P17was able to bind TGF-�2, we tested its ability to inhibit the im-munosuppressive activity of this cytokine in vitro. Thus, weadded TGF-�2 to effector T cells stimulated with anti-CD3, asdescribed in Materials and Methods. It was found that 100 �MP17 inhibited the immunosuppressive activity of TGF-�2 byaround 50% (Fig. 3B).
Peptide P17 improves immunogenicity of AH1 peptidevaccination leading to protection against CT26 tumor challenge
Down-regulation of Treg suppressor activity in vivo might be ben-eficial to enhance vaccine immunogenicity against tumor Ags. Im-munization of BALB/c mice only with peptide AH1 (a TCd, ex-pressed by CT26 colon cancer cells) is unable to induce aprotective CTL response against challenge with CT26 tumor cells(34). However, this result could be overcome by coimmunizationof AH1 and an adequate Th cell determinant capable of inducinga competent Th cell response (42). We have also shown that de-pletion of CD25� Treg cells with anti-CD25 Abs before immuni-zation with peptide AH1 permits the induction of a long-lastingantitumoral immune response (2). All these observations lead us tospeculate that in vivo inhibition of Treg cells by peptide P17 (in-stead of Treg depletion) in combination with vaccination withAH1, might allow the control of tumor growth. Thus, BALB/c
FIGURE 3. P17 binds to TGF-�2 and inhibits its immunosuppressiveactivity. A, Binding assays TGF-�1, TGF-�2, TGF-�3 or an irrelevantprotein were immobilized covalently on the flow cell 2 of a CM5 chip viaa standard amine coupling procedure. P17 solution (5 �M) was injectedthree times over the protein-bound surfaces. Sensograms of P17 bindingcapacity to the different TGF-� isoforms or to the irrelevant protein areplotted. B, Inhibition of TGF-�2 activity. Spleen cells from BALB/c micewere stimulated with anti-CD3 mAbs in the presence or absence of exog-enously added recombinant human TGF-�2 (0.25 pg/ml) and 100 �M P17or control peptide (Pcont). Cell proliferation was analyzed by measuringtritiated thymidine incorporation in the harvested cells using a scintillationcounter. �, p � 0.05 t test when compared with T cell proliferation in thepresence vs in the absence of P17. Results are representative of two inde-pendent experiments.
130 DOWN-REGULATION OF Treg CELLS BY A TGF-� INHIBITOR PEPTIDE
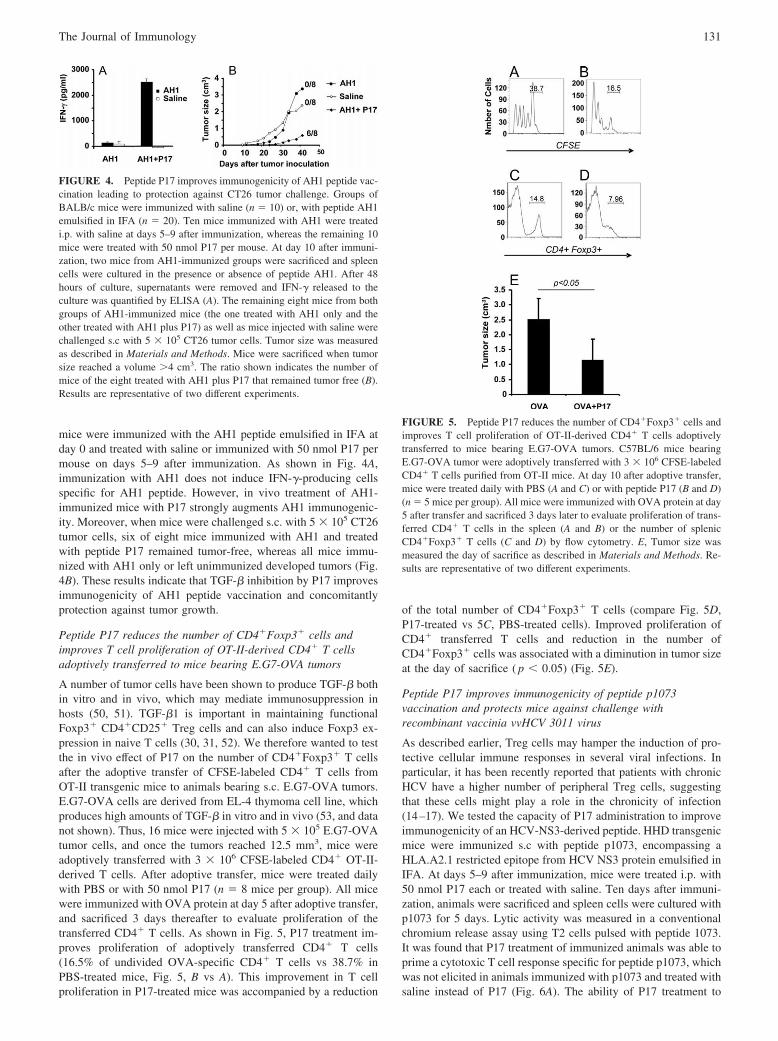
mice were immunized with the AH1 peptide emulsified in IFA atday 0 and treated with saline or immunized with 50 nmol P17 permouse on days 5–9 after immunization. As shown in Fig. 4A,immunization with AH1 does not induce IFN-�-producing cellsspecific for AH1 peptide. However, in vivo treatment of AH1-immunized mice with P17 strongly augments AH1 immunogenic-ity. Moreover, when mice were challenged s.c. with 5 � 105 CT26tumor cells, six of eight mice immunized with AH1 and treatedwith peptide P17 remained tumor-free, whereas all mice immu-nized with AH1 only or left unimmunized developed tumors (Fig.4B). These results indicate that TGF-� inhibition by P17 improvesimmunogenicity of AH1 peptide vaccination and concomitantlyprotection against tumor growth.
Peptide P17 reduces the number of CD4�Foxp3� cells andimproves T cell proliferation of OT-II-derived CD4� T cellsadoptively transferred to mice bearing E.G7-OVA tumors
A number of tumor cells have been shown to produce TGF-� bothin vitro and in vivo, which may mediate immunosuppression inhosts (50, 51). TGF-�1 is important in maintaining functionalFoxp3� CD4�CD25� Treg cells and can also induce Foxp3 ex-pression in naive T cells (30, 31, 52). We therefore wanted to testthe in vivo effect of P17 on the number of CD4�Foxp3� T cellsafter the adoptive transfer of CFSE-labeled CD4� T cells fromOT-II transgenic mice to animals bearing s.c. E.G7-OVA tumors.E.G7-OVA cells are derived from EL-4 thymoma cell line, whichproduces high amounts of TGF-� in vitro and in vivo (53, and datanot shown). Thus, 16 mice were injected with 5 � 105 E.G7-OVAtumor cells, and once the tumors reached 12.5 mm3, mice wereadoptively transferred with 3 � 106 CFSE-labeled CD4� OT-II-derived T cells. After adoptive transfer, mice were treated dailywith PBS or with 50 nmol P17 (n � 8 mice per group). All micewere immunized with OVA protein at day 5 after adoptive transfer,and sacrificed 3 days thereafter to evaluate proliferation of thetransferred CD4� T cells. As shown in Fig. 5, P17 treatment im-proves proliferation of adoptively transferred CD4� T cells(16.5% of undivided OVA-specific CD4� T cells vs 38.7% inPBS-treated mice, Fig. 5, B vs A). This improvement in T cellproliferation in P17-treated mice was accompanied by a reduction
of the total number of CD4�Foxp3� T cells (compare Fig. 5D,P17-treated vs 5C, PBS-treated cells). Improved proliferation ofCD4� transferred T cells and reduction in the number ofCD4�Foxp3� cells was associated with a diminution in tumor sizeat the day of sacrifice ( p � 0.05) (Fig. 5E).
Peptide P17 improves immunogenicity of peptide p1073vaccination and protects mice against challenge withrecombinant vaccinia vvHCV 3011 virus
As described earlier, Treg cells may hamper the induction of pro-tective cellular immune responses in several viral infections. Inparticular, it has been recently reported that patients with chronicHCV have a higher number of peripheral Treg cells, suggestingthat these cells might play a role in the chronicity of infection(14–17). We tested the capacity of P17 administration to improveimmunogenicity of an HCV-NS3-derived peptide. HHD transgenicmice were immunized s.c with peptide p1073, encompassing aHLA.A2.1 restricted epitope from HCV NS3 protein emulsified inIFA. At days 5–9 after immunization, mice were treated i.p. with50 nmol P17 each or treated with saline. Ten days after immuni-zation, animals were sacrificed and spleen cells were cultured withp1073 for 5 days. Lytic activity was measured in a conventionalchromium release assay using T2 cells pulsed with peptide 1073.It was found that P17 treatment of immunized animals was able toprime a cytotoxic T cell response specific for peptide p1073, whichwas not elicited in animals immunized with p1073 and treated withsaline instead of P17 (Fig. 6A). The ability of P17 treatment to
FIGURE 4. Peptide P17 improves immunogenicity of AH1 peptide vac-cination leading to protection against CT26 tumor challenge. Groups ofBALB/c mice were immunized with saline (n � 10) or, with peptide AH1emulsified in IFA (n � 20). Ten mice immunized with AH1 were treatedi.p. with saline at days 5–9 after immunization, whereas the remaining 10mice were treated with 50 nmol P17 per mouse. At day 10 after immuni-zation, two mice from AH1-immunized groups were sacrificed and spleencells were cultured in the presence or absence of peptide AH1. After 48hours of culture, supernatants were removed and IFN-� released to theculture was quantified by ELISA (A). The remaining eight mice from bothgroups of AH1-immunized mice (the one treated with AH1 only and theother treated with AH1 plus P17) as well as mice injected with saline werechallenged s.c with 5 � 105 CT26 tumor cells. Tumor size was measuredas described in Materials and Methods. Mice were sacrificed when tumorsize reached a volume �4 cm3. The ratio shown indicates the number ofmice of the eight treated with AH1 plus P17 that remained tumor free (B).Results are representative of two different experiments.
FIGURE 5. Peptide P17 reduces the number of CD4�Foxp3� cells andimproves T cell proliferation of OT-II-derived CD4� T cells adoptivelytransferred to mice bearing E.G7-OVA tumors. C57BL/6 mice bearingE.G7-OVA tumor were adoptively transferred with 3 � 106 CFSE-labeledCD4� T cells purified from OT-II mice. At day 10 after adoptive transfer,mice were treated daily with PBS (A and C) or with peptide P17 (B and D)(n � 5 mice per group). All mice were immunized with OVA protein at day5 after transfer and sacrificed 3 days later to evaluate proliferation of trans-ferred CD4� T cells in the spleen (A and B) or the number of splenicCD4�Foxp3� T cells (C and D) by flow cytometry. E, Tumor size wasmeasured the day of sacrifice as described in Materials and Methods. Re-sults are representative of two different experiments.
131The Journal of Immunology
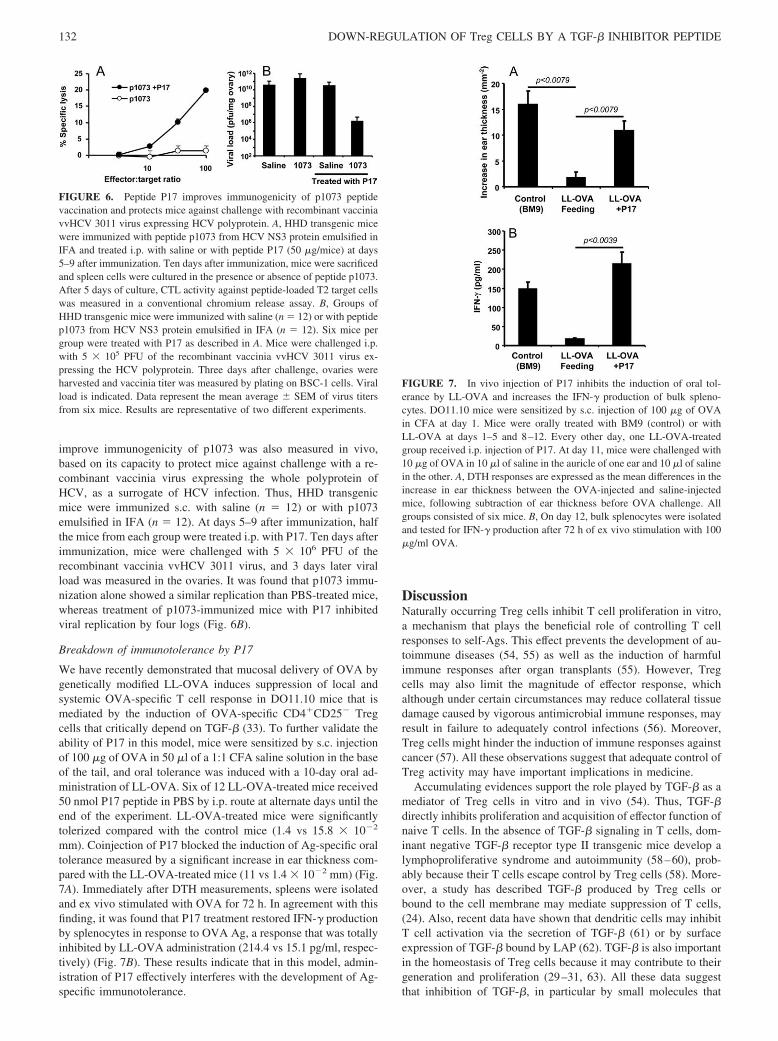
improve immunogenicity of p1073 was also measured in vivo,based on its capacity to protect mice against challenge with a re-combinant vaccinia virus expressing the whole polyprotein ofHCV, as a surrogate of HCV infection. Thus, HHD transgenicmice were immunized s.c. with saline (n � 12) or with p1073emulsified in IFA (n � 12). At days 5–9 after immunization, halfthe mice from each group were treated i.p. with P17. Ten days afterimmunization, mice were challenged with 5 � 106 PFU of therecombinant vaccinia vvHCV 3011 virus, and 3 days later viralload was measured in the ovaries. It was found that p1073 immu-nization alone showed a similar replication than PBS-treated mice,whereas treatment of p1073-immunized mice with P17 inhibitedviral replication by four logs (Fig. 6B).
Breakdown of immunotolerance by P17
We have recently demonstrated that mucosal delivery of OVA bygenetically modified LL-OVA induces suppression of local andsystemic OVA-specific T cell response in DO11.10 mice that ismediated by the induction of OVA-specific CD4�CD25� Tregcells that critically depend on TGF-� (33). To further validate theability of P17 in this model, mice were sensitized by s.c. injectionof 100 �g of OVA in 50 �l of a 1:1 CFA saline solution in the baseof the tail, and oral tolerance was induced with a 10-day oral ad-ministration of LL-OVA. Six of 12 LL-OVA-treated mice received50 nmol P17 peptide in PBS by i.p. route at alternate days until theend of the experiment. LL-OVA-treated mice were significantlytolerized compared with the control mice (1.4 vs 15.8 � 10�2
mm). Coinjection of P17 blocked the induction of Ag-specific oraltolerance measured by a significant increase in ear thickness com-pared with the LL-OVA-treated mice (11 vs 1.4 � 10�2 mm) (Fig.7A). Immediately after DTH measurements, spleens were isolatedand ex vivo stimulated with OVA for 72 h. In agreement with thisfinding, it was found that P17 treatment restored IFN-� productionby splenocytes in response to OVA Ag, a response that was totallyinhibited by LL-OVA administration (214.4 vs 15.1 pg/ml, respec-tively) (Fig. 7B). These results indicate that in this model, admin-istration of P17 effectively interferes with the development of Ag-specific immunotolerance.
DiscussionNaturally occurring Treg cells inhibit T cell proliferation in vitro,a mechanism that plays the beneficial role of controlling T cellresponses to self-Ags. This effect prevents the development of au-toimmune diseases (54, 55) as well as the induction of harmfulimmune responses after organ transplants (55). However, Tregcells may also limit the magnitude of effector response, whichalthough under certain circumstances may reduce collateral tissuedamage caused by vigorous antimicrobial immune responses, mayresult in failure to adequately control infections (56). Moreover,Treg cells might hinder the induction of immune responses againstcancer (57). All these observations suggest that adequate control ofTreg activity may have important implications in medicine.
Accumulating evidences support the role played by TGF-� as amediator of Treg cells in vitro and in vivo (54). Thus, TGF-�directly inhibits proliferation and acquisition of effector function ofnaive T cells. In the absence of TGF-� signaling in T cells, dom-inant negative TGF-� receptor type II transgenic mice develop alymphoproliferative syndrome and autoimmunity (58–60), prob-ably because their T cells escape control by Treg cells (58). More-over, a study has described TGF-� produced by Treg cells orbound to the cell membrane may mediate suppression of T cells,(24). Also, recent data have shown that dendritic cells may inhibitT cell activation via the secretion of TGF-� (61) or by surfaceexpression of TGF-� bound by LAP (62). TGF-� is also importantin the homeostasis of Treg cells because it may contribute to theirgeneration and proliferation (29–31, 63). All these data suggestthat inhibition of TGF-�, in particular by small molecules that
FIGURE 6. Peptide P17 improves immunogenicity of p1073 peptidevaccination and protects mice against challenge with recombinant vacciniavvHCV 3011 virus expressing HCV polyprotein. A, HHD transgenic micewere immunized with peptide p1073 from HCV NS3 protein emulsified inIFA and treated i.p. with saline or with peptide P17 (50 �g/mice) at days5–9 after immunization. Ten days after immunization, mice were sacrificedand spleen cells were cultured in the presence or absence of peptide p1073.After 5 days of culture, CTL activity against peptide-loaded T2 target cellswas measured in a conventional chromium release assay. B, Groups ofHHD transgenic mice were immunized with saline (n � 12) or with peptidep1073 from HCV NS3 protein emulsified in IFA (n � 12). Six mice pergroup were treated with P17 as described in A. Mice were challenged i.p.with 5 � 105 PFU of the recombinant vaccinia vvHCV 3011 virus ex-pressing the HCV polyprotein. Three days after challenge, ovaries wereharvested and vaccinia titer was measured by plating on BSC-1 cells. Viralload is indicated. Data represent the mean average � SEM of virus titersfrom six mice. Results are representative of two different experiments.
FIGURE 7. In vivo injection of P17 inhibits the induction of oral tol-erance by LL-OVA and increases the IFN-� production of bulk spleno-cytes. DO11.10 mice were sensitized by s.c. injection of 100 �g of OVAin CFA at day 1. Mice were orally treated with BM9 (control) or withLL-OVA at days 1–5 and 8–12. Every other day, one LL-OVA-treatedgroup received i.p. injection of P17. At day 11, mice were challenged with10 �g of OVA in 10 �l of saline in the auricle of one ear and 10 �l of salinein the other. A, DTH responses are expressed as the mean differences in theincrease in ear thickness between the OVA-injected and saline-injectedmice, following subtraction of ear thickness before OVA challenge. Allgroups consisted of six mice. B, On day 12, bulk splenocytes were isolatedand tested for IFN-� production after 72 h of ex vivo stimulation with 100�g/ml OVA.
132 DOWN-REGULATION OF Treg CELLS BY A TGF-� INHIBITOR PEPTIDE

might penetrate the interface between contacting T cells, might beuseful to potentiate antiviral or antitumor immunotherapies.
We have shown that P17, a TGF-� inhibitor peptide developedin our laboratory (32), is able to inhibit murine- or human-derivedTreg activity in vitro in three different experimental settings. Thus,P17 was able to restore murine or human T cell proliferation inresponse to anti-CD3 stimulation, which was inhibited by the ad-dition of Treg cells. Similarly, P17 restored, at least partially, Tcell proliferation in an MLR inhibited by Treg cells. P17 alsoinhibited Treg activity over specific T cells stimulated by an Ag.These results prompted us to test P17 in vivo. In a previous workwe showed that in vivo CD25� T cell depletion improved immu-nogenicity of AH1 peptide in vaccination and protected miceagainst tumor challenge (2). We found that in vivo P17 adminis-tration, instead of Treg depletion, was also able to enhance immu-nogenicity of AH1 peptide vaccination, and protected mice fromCT26 tumor challenge. Down-regulation of Treg suppressor activ-ity in vivo may be beneficial to enhance immunogenicity of avaccine (2). Similarly, P17 administration improved the immuno-genicity of a peptide vaccine consisting of the immunization ofpeptide p1073, which encompasses a HLA.A2.1 restricted epitopefrom HCV NS3 protein, with the outcome being a reduction ofrecombinant vaccinia vvHCV 3011 virus replication after vacci-nation with p1073. These results are in agreement with previousreports showing an enhancement of immunogenicity of a vaccineby the depletion of Treg cells (2–5, 64). However, we believe thatin vivo inhibition of Treg activity by P17, instead of Treg deple-tion, might allow a better control of Treg function, reducing therisk of autoimmune diseases that may be favored in the absence ofTreg cells (65). When we compared the effect of anti-TGF-� Abswith P17 in vivo, both molecules were effective. Indeed, AH1-immunized mice remained protected from CT26 tumor challengeif they were treated with anti-TGF-�1 polyclonal Abs (five admin-istrations of 100 �g of anti-TGF-�1 per mouse from day 5 to 9after AH1 immunization). Similarly, in vivo administration of anti-TGF-�1 Abs reverted immunotolerance induced by LL-OVA ad-ministration (data not shown). Although both molecules are effi-cient TGF-� inhibitors, the use of peptides might have advantages.Thus, the relative short life of peptides would allow a finer controlon the inhibition of TGF-� during the required period in vivo. Thisuse would reduce the potential toxic effects of long-term inhibitionof the cytokine.
Tumors produce factors such as prostaglandins, IL-10, vascularendothelial growth factor, and TGF-�, which may create an im-munosuppressive microenvironment and may hamper immuno-therapy. This microenvironment, and in particular TGF-�1, mightfavor Treg development. Indeed, it has been widely described thatduring tumor progression in humans, Treg cells accumulate in tu-mors and secondary lymphoid organs (6–12). This increase in thenumber of Treg cells may be favored by a recruitment of naturallyoccurring Treg cells, as well as by a conversion of CD4� effectorTh cells into Treg cells, in this particular TGF-�-enriched tumormicroenvironment (66, 67). In addition, it has been recently de-scribed that the TGF-�2 isoform may induce Foxp3 expression inpancreatic carcinoma cells, enabling these tumor cells to suppressT cell proliferation (49). Thus, inhibition of TGF-�1 and TGF-�2isoforms would have an impact in this adverse environment byreducing the number of Treg cells and Foxp3 expression and byfavoring effector T cell proliferation and function. We show in thisstudy that P17 is able to inhibit the immunosuppressive activity ofTGF-�1 as well as TGF-�2 in vitro. P17 administration, afteradoptive transfer of CFSE-labeled CD4� T cells from OT-II trans-genic mice to C57BL/6 mice bearing EG.7-OVA tumors, im-proved proliferation of transferred T cells and reduced the number
of CD4�Foxp3� T cells. Moreover, P17 treatment after the adop-tive transfer of CD4� T cells inhibited tumor growth, suggestingthat this TGF-� inhibitor might be very useful for the developmentof anti-cancer therapies.
TGF-� plays a central role in oral tolerance. This takes place viaregulation of mucosal inflammation and mediation of active sup-pression against orally administered Ags (reviewed in Ref. 68).Thus, TGF-� knockout mice develop chronic inflammation inmany tissues, including the gastrointestinal tract (69). In addition,recent findings suggest that TGF-� may be a primary link betweendistinct populations of Treg cells that are induced by feeding. Wehave recently shown that mucosal delivery of OVA by geneticallymodified LL-OVA induces OVA-specific CD4�CD25� Tregcells, which in turn suppress OVA-specific T cell responses inDO11.10 mice, in a process critically dependent on TGF-� (33).We have found that P17 administration is able to inhibit inductionof oral tolerance in this model, suggesting that P17 may have im-portant applications to enhance the immunogenicity of orallyadministered Ags.
In summary, we have shown that inhibition of TGF-� by P17 isable to inhibit the immunosuppressive activity of murine- and hu-man-derived Treg cells in vitro. Also, and most importantly, invivo experiments using P17 show that this peptide fosters the im-munogenicity of peptide vaccination when administrated 5 daysafter vaccination. Moreover, P17 was able to improve proliferationof adoptively transferred T cells, and reduce the number ofCD4�Foxp3� T cells in vivo in mice bearing a TGF-� producingtumor. In addition, P17 was also able to inhibit Treg function in anAg-specific model of oral-induced tolerance. In summary, our re-sults demonstrate that inhibition of TGF-�1 and TGF-�2 with asmall synthetic peptide can be a useful therapeutic strategy to en-hance the immunogenicity of vaccines or to break toleranceagainst pathogens or tumor Ags.
DisclosuresJavier Dotor is currently an employee of Digna-Biotech, the company hold-ing patent rights on P17.
References1. Shevach, E. M. 2001. Certified professionals: CD4�CD25� suppressor T cells.
J. Exp. Med. 193: F41–F46.2. Casares, N., L. Arribillaga, P. Sarobe, J. Dotor, A. Lopez-Diaz de Cerio,
I. Melero, J. Prieto, F. Borras-Cuesta, and J. J. Lasarte. 2003. CD4�/CD25�
regulatory cells inhibit activation of tumor-primed CD4� T cells with IFN-�-dependent antiangiogenic activity, as well as long-lasting tumor immunity elic-ited by peptide vaccination. J. Immunol. 171: 5931–5939.
3. Onizuka, S., I. Tawara, J. Shimizu, S. Sakaguchi, T. Fujita, and E. Nakayama.1999. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2receptor �) monoclonal antibody. Cancer Res. 59: 3128–3133.
4. Shimizu, J., S. Yamazaki, and S. Sakaguchi. 1999. Induction of tumor immunityby removing CD25�CD4� T cells: a common basis between tumor immunityand autoimmunity. J. Immunol. 163: 5211–5218.
5. Takahashi, T., T. Tagami, S. Yamazaki, T. Uede, J. Shimizu, N. Sakaguchi,T. W. Mak, and S. Sakaguchi. 2000. Immunologic self-tolerance maintained byCD25�CD4� regulatory T cells constitutively expressing cytotoxic T lympho-cyte-associated antigen 4. J. Exp. Med. 192: 303–310.
6. Wang, H. Y., G. Peng, Z. Guo, E. M. Shevach, and R. F. Wang. 2005. Recog-nition of a new ARTC1 peptide ligand uniquely expressed in tumor cells byantigen-specific CD4� regulatory T cells. J. Immunol. 174: 2661–2670.
7. Viguier, M., F. Lemaître, O. Verola, M. S. Cho, G. Gorochov, L. Dubertret,H. Bachelez, P. Kourilsky, and L. Ferradini. 2004. Foxp3 expressingCD4�CD25high regulatory T cells are overrepresented in human metastatic mel-anoma lymph nodes and inhibit the function of infiltrating T cells. J. Immunol.173: 1444–1453.
8. Woo, E. Y., C. S. Chu, T. J. Goletz, K. Schlienger, H. Yeh, G. Coukos,S. C. Rubin, L. R. Kaiser, and C. H. June. 2001. Regulatory CD4�CD25� T cellsin tumors from patients with early-stage non-small cell lung cancer and late-stageovarian cancer. Cancer Res. 61: 4766–4772.
9. Curiel, T. J., G. Coukos, L. Zou, X. Alvarez, P. Cheng, P. Mottram,M. Evdemon-Hogan, J. R. Conejo-Garcia, L. Zhang, M. Burow, et al. 2004.Specific recruitment of regulatory T cells in ovarian carcinoma fosters immuneprivilege and predicts reduced survival. Nat. Med. 10: 942–949.
133The Journal of Immunology

10. Liyanage, U. K., T. T. Moore, H. G. Joo, Y. Tanaka, V. Herrmann, G. Doherty,J. A. Drebin, S. M. Strasberg, T. J. Eberlein, P. S. Goedegebuure, andD. C. Linehan. 2002. Prevalence of regulatory T cells is increased in peripheralblood and tumor microenvironment of patients with pancreas or breast adeno-carcinoma. J. Immunol. 169: 2756–2761.
11. Ormandy, L. A., T. Hillemann, H. Wedemeyer, M. P. Manns, T. F. Greten, andF. Korangy. 2005. Increased populations of regulatory T cells in peripheral bloodof patients with hepatocellular carcinoma. Cancer Res. 65: 2457–2464.
12. Kobayashi, N., N. Hiraoka, W. Yamagami, H. Ojima, Y. Kanai, T. Kosuge,A. Nakajima, and S. Hirohashi. 2007. FoxP3� regulatory T cells affect the de-velopment and progression of hepatocarcinogenesis. Clin. Cancer Res. 13:902–911.
13. Xu, D., J. Fu, L. Jin, H. Zhang, C. Zhou, Z. Zou, J. M. Zhao, B. Zhang, M. Shi,X. Ding, et al. 2006. Circulating and liver resident CD4�CD25� regulatory Tcells actively influence the antiviral immune response and disease progression inpatients with hepatitis B. J. Immunol. 177: 739–747.
14. Boettler, T., H. C. Spangenberg, C. Neumann-Haefelin, E. Panther, S. Urbani,C. Ferrari, H. E. Blum, F. von Weizsacker, and R. Thimme. 2005. T cells witha CD4�CD25� regulatory phenotype suppress in vitro proliferation of virus-specific CD8� T cells during chronic hepatitis C virus infection. J. Virol. 79:7860–7867.
15. Cabrera, R., Z. Tu, Y. Xu, R. J. Firpi, H. R. Rosen, C. Liu, and D. R. Nelson.2004. An immunomodulatory role for CD4�CD25� regulatory T lymphocytes inhepatitis C virus infection. Hepatology 40: 1062–1071.
16. Rushbrook, S. M., S. M. Ward, E. Unitt, S. L. Vowler, M. Lucas, P. Klenerman,and G. J. Alexander. 2005. Regulatory T cells suppress in vitro proliferation ofvirus-specific CD8� T cells during persistent hepatitis C virus infection. J. Virol.79: 7852–7859.
17. Sugimoto, K., F. Ikeda, J. Stadanlick, F. A. Nunes, H. J. Alter, and K. M. Chang.2003. Suppression of HCV-specific T cells without differential hierarchy dem-onstrated ex vivo in persistent HCV infection. Hepatology 38: 1437–1448.
18. Aandahl, E. M., J. Michaelsson, W. J. Moretto, F. M. Hecht, and D. F. Nixon.2004. Human CD4�CD25� regulatory T cells control T-cell responses to humanimmunodeficiency virus and cytomegalovirus antigens. J. Virol. 78: 2454–2459.
19. Kinter, A. L., M. Hennessey, A. Bell, S. Kern, Y. Lin, M. Daucher, M. Planta,M. McGlaughlin, R. Jackson, S. F. Ziegler, and A. S. Fauci. 2004. CD25�CD4�
regulatory T cells from the peripheral blood of asymptomatic HIV-infected in-dividuals regulate CD4� and CD8� HIV-specific T cell immune responses invitro and are associated with favorable clinical markers of disease status. J. Exp.Med. 200: 331–343.
20. Oswald-Richter, K., S. M. Grill, N. Shariat, M. Leelawong, M. S. Sundrud,D. W. Haas, and D. Unutmaz. 2004. HIV infection of naturally occurring andgenetically reprogrammed human regulatory T-cells. PLoS Biol. 2: E198.
21. Weiss, L., V. Donkova-Petrini, L. Caccavelli, M. Balbo, C. Carbonneil, andY. Levy. 2004. Human immunodeficiency virus-driven expansion ofCD4�CD25� regulatory T cells, which suppress HIV-specific CD4 T-cell re-sponses in HIV-infected patients. Blood 104: 3249–3256.
22. Thornton, A. M., and E. M. Shevach. 2000. Suppressor effector function ofCD4�CD25� immunoregulatory T cells is antigen nonspecific. J. Immunol. 164:183–190.
23. Huang, X., J. Zhu, and Y. Yang. 2005. Protection against autoimmunity in non-lymphopenic hosts by CD4�CD25� regulatory T cells is antigen-specific andrequires IL-10 and TGF-�. J. Immunol. 175: 4283–4291.
24. Nakamura, K., A. Kitani, and W. Strober. 2001. Cell contact-dependent immu-nosuppression by CD4�CD25� regulatory T cells is mediated by cell surface-bound transforming growth factor �. J. Exp. Med. 194: 629–644.
25. Somasundaram, R., L. Jacob, R. Swoboda, L. Caputo, H. Song, S. Basak,D. Monos, D. Peritt, F. Marincola, D. Cai, et al. 2002. Inhibition of cytolytic Tlymphocyte proliferation by autologous CD4�/CD25� regulatory T cells in acolorectal carcinoma patient is mediated by transforming growth factor-�. Can-cer Res. 62: 5267–5272.
26. Roncarolo, M. G., R. Bacchetta, C. Bordignon, S. Narula, and M. K. Levings.2001. Type 1 T regulatory cells. Immunol. Rev. 182: 68–79.
27. Chen, M. L., M. J. Pittet, L. Gorelik, R. A. Flavell, R. Weissleder,H. von Boehmer, and K. Khazaie. 2005. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-� signals in vivo. Proc. Natl. Acad.Sci. USA 102: 419–424.
28. Green, E. A., L. Gorelik, C. M. McGregor, E. H. Tran, and R. A. Flavell. 2003.CD4�CD25� T regulatory cells control anti-islet CD8� T cells through TGF-�-TGF-� receptor interactions in type 1 diabetes. Proc. Natl. Acad. Sci. USA 100:10878–10883.
29. Huber, S., C. Schramm, H. A. Lehr, A. Mann, S. Schmitt, C. Becker,M. Protschka, P. R. Galle, M. F. Neurath, and M. Blessing. 2004. Cutting edge:TGF-� signaling is required for the in vivo expansion and immunosuppressivecapacity of regulatory CD4�CD25� T cells. J. Immunol. 173: 6526–6531.
30. Chen, W., W. Jin, N. Hardegen, K. J. Lei, L. Li, N. Marinos, G. McGrady, andS. M. Wahl. 2003. Conversion of peripheral CD4�CD25� naive T cells toCD4�CD25� regulatory T cells by TGF-� induction of transcription factorFoxp3. J. Exp. Med. 198: 1875–1886.
31. Fantini, M. C., C. Becker, G. Monteleone, F. Pallone, P. R. Galle, andM. F. Neurath. 2004. Cutting edge: TGF-� induces a regulatory phenotype inCD4�CD25� T cells through Foxp3 induction and down-regulation of Smad7.J. Immunol. 172: 5149–5153.
32. Dotor, J., A. B. Lopez-Vazquez, J. J. Lasarte, P. Sarobe, M. Garcıa-Granero,J. I. Riezu-Boj, A. Martınez, E. Feijoo, J. Lopez-Sagaseta, J. Hermida, et al. 2007.Identification of peptide inhibitors of transforming growth factor �1 using aphage-displayed peptide library. Cytokine 39: 106–115.
33. Huibregtse, I. L., V. Snoeck, A. de Creus, H. Braat, E. C. de Jong,S. J. Van Deventer, and P. Rottiers. 2007. Induction of ovalbumin-specific tol-erance by oral administration of Lactococcus lactis secreting ovalbumin. Gas-troenterology 133: 517–528.
34. Huang, A. Y., P. H. Gulden, A. S. Woods, M. C. Thomas, C. D. Tong, W. Wang,V. H. Engelhard, G. Pasternack, R. Cotter, D. Hunt, et al. 1996. The immuno-dominant major histocompatibility complex class I-restricted antigen of a murinecolon tumor derives from an endogenous retroviral gene product. Proc. Natl.Acad. Sci. USA 93: 9730–9735.
35. Cerny, A., J. G. McHutchison, C. Pasquinelli, M. E. Brown, M. A. Brothers,B. Grabscheid, P. Fowler, M. Houghton, and F. V. Chisari. 1995. Cytotoxic Tlymphocyte response to hepatitis C virus-derived peptides containing the HLAA2.1 binding motif. J. Clin. Invest. 95: 521–530.
36. Borras-Cuesta, F., J. Golvano, P. Sarobe, J. J. Lasarte, I. Prieto, A. Szabo,J. L. Guillaume, and J. G. Guillet. 1991. Insights on the amino acid side-chaininteractions of a synthetic T-cell determinant. Biologicals 19: 187–190.
37. Moore, M. W., F. R. Carbone, and M. J. Bevan. 1988. Introduction of solubleprotein into the class I pathway of antigen processing and presentation. Cell 54:777–785.
38. Zabala, M., J. J. Lasarte, C. Perret, J. Sola, P. Berraondo, M. Alfaro, E. Larrea,J. Prieto, and M. G. Kramer. 2007. Induction of immunosuppressive moleculesand regulatory T cells counteracts the antitumor effect of interleukin-12-basedgene therapy in a transgenic mouse model of liver cancer. J. Hepatol. 47:807–815.
39. Sarobe, P., J. J. Lasarte, A. Zabaleta, L. Arribillaga, A. Arina, I. Melero,F. Borras-Cuesta, and J. Prieto. 2003. Hepatitis C virus structural proteins impairdendritic cell maturation and inhibit in vivo induction of cellular immune re-sponses. J. Virol. 77: 10862–10871.
40. De Crescenzo, G., S. Grothe, J. Zwaagstra, M. Tsang, and M. D. O’Connor-McCourt. 2001. Real-time monitoring of the interactions of transforming growthfactor-� (TGF-�) isoforms with latency-associated protein and the ectodomainsof the TGF-� type II and III receptors reveals different kinetic models and sto-ichiometries of binding. J. Biol. Chem. 276: 29632–29643.
41. Lasarte, J. J., P. Sarobe, A. Gullon, J. Prieto, and F. Borras-Cuesta. 1992. Induc-tion of cytotoxic T lymphocytes in mice against the principal neutralizing domainof HIV-1 by immunization with an engineered T-cytotoxic-T-helper syntheticpeptide construct. Cell Immunol. 141: 211–218.
42. Casares, N., J. J. Lasarte, A. L. de Cerio, P. Sarobe, M. Ruiz, I. Melero, J. Prieto,and F. Borras-Cuesta. 2001. Immunization with a tumor-associated CTL epitopeplus a tumor-related or unrelated Th1 helper peptide elicits protective CTL im-munity. Eur. J. Immunol. 31: 1780–1789.
43. Arribillaga, L., P. Sarobe, A. Arina, M. Gorraiz, F. Borras-Cuesta, J. Ruiz,J. Prieto, L. Chen, I. Melero, and J. J. Lasarte. 2005. Enhancement of CD4 andCD8 immunity by anti-CD137 (4-1BB) monoclonal antibodies during hepatitis Cvaccination with recombinant adenovirus. Vaccine 23: 3493–3499.
44. Gasson, M. J. 1983. Plasmid complements of Streptococcus lactis NCDO 712and other lactic streptococci after protoplast-induced curing. J. Bacteriol.154: 1–9.
45. Steidler, L., W. Hans, L. Schotte, S. Neirynck, F. Obermeier, W. Falk, W. Fiers,and E. Remaut. 2000. Treatment of murine colitis by Lactococcus lactis secretinginterleukin-10. Science 289: 1352–1355.
46. Vandenbroucke, K., W. Hans, J. Van Huysse, S. Neirynck, P. Demetter,E. Remaut, P. Rottiers, and L. Steidler. 2004. Active delivery of trefoil factors bygenetically modified Lactococcus lactis prevents and heals acute colitis in mice.Gastroenterology 127: 502–513.
47. Tobagus, I. T., W. R. Thomas, and P. G. Holt. 2004. Adjuvant costimulationduring secondary antigen challenge directs qualitative aspects of oral toleranceinduction, particularly during the neonatal period. J. Immunol. 172: 2274–2285.
48. Nakamura, K., A. Kitani, I. Fuss, A. Pedersen, N. Harada, H. Nawata, andW. Strober. 2004. TGF-�1 plays an important role in the mechanism ofCD4�CD25� regulatory T cell activity in both humans and mice. J. Immunol.172: 834–842.
49. Hinz, S., L. Pagerols-Raluy, H.-H. Oberg, O. Ammerpohl, S. Grussel, B. Sipos,R. Grutzmann, C. Pilarsky, H. Ungefroren, H. D. Saeger, et al. 2007. Foxp3expression in pancreatic carcinoma cells as a novel mechanism of immune eva-sion in cancer. Cancer Res. 67: 8344–8350.
50. Chang, H. L., N. Gillett, I. Figari, A. R. Lopez, M. A. Palladino, and R. Derynck.1993. Increased transforming growth factor � expression inhibits cell prolifera-tion in vitro, yet increases tumorigenicity and tumor growth of Meth A sarcomacells. Cancer Res. 53: 4391–4398.
51. Young, M. R., M. A. Wright, M. Coogan, M. E. Young, and J. Bagash. 1992.Tumor-derived cytokines induce bone marrow suppressor cells that mediate im-munosuppression through transforming growth factor �. Cancer Immunol. Im-munother. 35: 14–18.
52. Marie, J. C., J. J. Letterio, M. Gavin, and A. Y. Rudensky. 2005. TGF-�1 main-tains suppressor function and Foxp3 expression in CD4�CD25� regulatory Tcells. J. Exp. Med. 201: 1061–1067.
53. Maeda, H., and A. Shiraishi. 1996. TGF-� contributes to the shift toward Th2-type responses through direct and IL-10-mediated pathways in tumor-bearingmice. J. Immunol. 156: 73–78.
54. Maloy, K. J., and F. Powrie. 2001. Regulatory T cells in the control of immunepathology. Nat. Immunol. 2: 816–822.
55. Sakaguchi, S. 2004. Naturally arising CD4� regulatory T cells for immunologicself-tolerance and negative control of immune responses. Annu. Rev. Immunol.22: 531–562.
56. Belkaid, Y., and B. T. Rouse. 2005. Natural regulatory T cells in infectiousdisease. Nat. Immunol. 6: 353–360.
134 DOWN-REGULATION OF Treg CELLS BY A TGF-� INHIBITOR PEPTIDE

57. Beyer, M., and J. L. Schultze. 2006. Regulatory T cells in cancer. Blood 108:804–811.
58. Fahlen, L., S. Read, L. Gorelik, S. D. Hurst, R. L. Coffman, R. A. Flavell, andF. Powrie. 2005. T cells that cannot respond to TGF-� escape control byCD4�CD25� regulatory T cells. J. Exp. Med. 201: 737–746.
59. Gorelik, L., and R. A. Flavell. 2000. Abrogation of TGF� signaling in T cellsleads to spontaneous T cell differentiation and autoimmune disease. Immunity 12:171–181.
60. Lucas, P. J., S. J. Kim, S. J. Melby, and R. E. Gress. 2000. Disruption of T cellhomeostasis in mice expressing a T cell-specific dominant negative transforminggrowth factor � II receptor. J. Exp. Med. 191: 1187–1196.
61. Coombes, J. L., K. R. Siddiqui, C. V. Arancibia-Carcamo, J. Hall, C. M. Sun,Y. Belkaid, and F. Powrie. 2007. A functionally specialized population of mu-cosal CD103� DCs induces Foxp3� regulatory T cells via a TGF-�- and retinoicacid-dependent mechanism. J. Exp. Med. 204: 1757–1764.
62. Gandhi, R., D. E. Anderson, and H. L. Weiner. 2007. Cutting edge: immaturehuman dendritic cells express latency-associated peptide and inhibit T cell acti-vation in a TGF-�-dependent manner. J. Immunol. 178: 4017–4021.
63. Yamagiwa, S., J. D. Gray, S. Hashimoto, and D. A. Horwitz. 2001. A role forTGF-� in the generation and expansion of CD4�CD25� regulatory T cells fromhuman peripheral blood. J. Immunol. 166: 7282–7289.
64. Litzinger, M. T., R. Fernando, T. J. Curiel, D. W. Grosenbach, J. Schlom, andC. Palena. 2007. The IL-2 immunotoxin denileukin diftitox reduces regulatory Tcells and enhances vaccine-mediated T-cell immunity. Blood 110: 3192–3201.
65. Stephens, L. A., D. Gray, and S. M. Anderton. 2005. CD4�CD25� regulatory Tcells limit the risk of autoimmune disease arising from T cell receptor crossre-activity. Proc. Natl. Acad. Sci. USA 102: 17418–17423.
66. Liu, V. C., L. Y. Wong, T. Jang, A. H. Shah, I. Park, X. Yang, Q. Zhang,S. Lonning, B. A. Teicher, and C. Lee. 2007. Tumor evasion of the immunesystem by converting CD4�CD25� T cells into CD4�CD25� T regulatory cells:role of tumor-derived TGF-�. J. Immunol. 178: 2883–2892.
67. Ghiringhelli, F., P. E. Puig, S. Roux, A. Parcellier, E. Schmitt, E. Solary,G. Kroemer, F. Martin, B. Chauffert, and L. Zitvogel. 2005. Tumor cells convertimmature myeloid dendritic cells into TGF-�-secreting cells inducingCD4�CD25� regulatory T cell proliferation. J. Exp. Med. 202: 919–929.
68. Ohtsuka, Y., and I. R. Sanderson. 2000. Transforming growth factor-�: an im-portant cytokine in the mucosal immune response. Curr. Opin. Gastroenterol. 16:541–545.
69. Shull, M. M., I. Ormsby, A. B. Kier, S. Pawlowski, R. J. Diebold, M. Yin,R. Allen, C. Sidman, G. Proetzel, D. Calvin, et al. 1992. Targeted disruption ofthe mouse transforming growth factor-�1 gene results in multifocal inflammatorydisease. Nature 359: 693–699.
135The Journal of Immunology

Author's personal copy
Immunization against hepatitis C virus with a fusionprotein containing the extra domain A from fibronectin and
the hepatitis C virus NS3 proteinq
Cristina Mansilla, Marta Gorraiz, Marta Martinez, Noelia Casares, Laura Arribillaga,Francesc Rudilla, Iciar Echeverria, Jose Ignacio Riezu-Boj, Pablo Sarobe,
Francisco Borras-Cuesta, Jesus Prieto, Juan Jose Lasarte*
University of Navarra, Centre for Applied Medical Research (CIMA), Division of Hepatology and Gene Therapy, Avenida Pıo XII 55,
31008 Pamplona, Spain
Background/Aims: Vaccination strategies able to induce strong T-cell responses might contribute to eradicate hepatitisC virus (HCV) infection. We previously demonstrated that fusion of an antigen to the extra domain A from fibronectin
(EDA) targets the antigen to TLR4-expressing dendritic cells (DC) and improves its immunogenicity. Here, we studied
if fusion of EDA with the non-structural HCV protein NS3 might constitute an effective immunogen against HCV.
Methods: Recombinant NS3 and the fusion protein EDA-NS3 were produced and purified from E. coli, and tested
in vitro for their capacity to activate maturation of DC and to favour antigen presentation. HHD transgenic mice express-
ing the human HLA-A2 molecule were immunized with recombinant proteins in the absence or presence of poly(I:C) and
anti-CD40 agonistic antibodies and responses elicited by vaccination were tested in vitro, and in vivo, by their capacity to
downregulate intrahepatic expression of HCV-NS3 RNA.Results: EDA-NS3, but not NS3 alone, upregulated the expression of maturation markers, as well as Delta-like 1 and
Delta-like 4 Notch ligands in DC and induced the production of IL-12. Mice immunized with EDA-NS3 had strong and
long lasting NS3-specific CD4+ and CD8+ T-cell responses and, in combination with poly(I:C) and anti-CD40, downreg-
ulated intrahepatic expression of HCV-NS3 RNA.
Conclusions: Recombinant EDA-NS3 may be considered for the development of vaccines against HCV infection.
� 2009 European Association for the Study of the Liver. Published by Elsevier B.V. All rights reserved.
Keywords: Hepatitis C; EDA; Adjuvant; Dendritic cell; Vaccine
1. Introduction
There is no effective vaccine against hepatitis C virus(HCV), a pathogen which is thought to infect 170 mil-lion people worldwide. HCV causes a high rate ofchronic infection, which can lead to liver cirrhosisand hepatocellular carcinoma. The outcome of HCVinfection appears to be modulated by a complex inter-play of immunological and virological factors, andboth antiviral humoral and cellular immune responsesseem to be required to control the infection. Thus,rapid induction of neutralizing antibodies after viral
0168-8278/$36.00 � 2009 European Association for the Study of the Liver. Published by Elsevier B.V. All rights reserved.
doi:10.1016/j.jhep.2009.06.005
Received 6 April 2009; received in revised form 15 May 2009; accepted 1
June 2009; available online 23 June 2009
Associate Editor: V. Barnabaq The authors who have taken part in this study declared that they do
not have anything to declare regarding funding from industry orconflict of interest with respect to this manuscript.
* Corresponding author. Tel.: +34 948 194700; fax: +34 948 194717.E-mail address: [email protected] (J.J. Lasarte).Abbreviations: HCV, hepatitis C virus; EDA, extra domain A from
fibronectin; DC, dendritic cell; Poly(I:C), polyinosinic-polycytidylicacid; BMDC, bone marrow derived dendritic cell; DLL1 and DLL4,Delta-like 1 and Delta-like 4 Notch ligands.
www.elsevier.com/locate/jhep
Journal of Hepatology 51 (2009) 520–527

Author's personal copy
infection may have an impact on viral clearance, andalthough HCV may continuously escape from thisresponse, broadly neutralizing antibodies may have animpact on the protection against heterologous viralinfection (reviewed in [1]). On the other hand, HCVchronic infection is associated with a low or absent T-cellresponse against viral antigens, and in contrast, thoseindividuals who clear viral infection display potent andwide antiviral CD4 and CD8 T-cell responses (reviewedin [2]). In particular, immune responses against non-structural protein NS3 from HCV have been associatedwith viral clearance [3]. Thus, induction of a vigorousanti-HCV T-cell response directed against NS3 isconsidered an important feature of a candidate HCVvaccine.
Dendritic cells (DCs) are the professional antigenpresenting cells (APCs) of the immune system. Theyorchestrate a repertoire of immune responses from toler-ance to self-antigens to resistance to infectious patho-gens depending on their maturation status (reviewed in[4]). Thus, it is generally accepted that efficient activa-tion of T-cell immune responses are dependent on DCmaturation triggered by a combination of stimuliderived from microbial products or inflammatory sig-nals. For these reasons, several vaccination strategiesincorporate ligands for pattern recognition receptors(PRRs) [5–8], agonistic antibodies against costimulatorymolecules [9,10] or cytokines [11] able to trigger matura-tion of DC. We have previously reported [6] that fusionof an antigen with the extra domain A from fibronectin(EDA) leads to antigen targeting to TLR4-expressingDC, enhancing cross-presentation and immunogenicity.To test if EDA-NS3 might behave as an immunogencapable of eliciting robust anti-HCV responses, we pre-pared this fusion protein and tested its capacity to acti-vate DC maturation in vitro and its immunogenicityin vivo. We also studied the effect of combining EDA-NS3 and other DC stimulators, specifically polyino-sinic-polycytidylic acid (poly(I:C)), a TLR3 ligand [12]and anti-CD40, an antibody mimicking the effect ofCD40L present on activated T-cells. Our results suggestthat EDA-NS3 combined with these adjuvants may beconsidered for the development of a vaccine againstHCV infection.
2. Materials and methods
2.1. Mice
Female C57BL/6 mice, 6–8 weeks old, from Harlan (Barcelona,Spain) and HHD mice, transgenic for human HLA-A2.1 and beta-2 microglobulin molecules [13], kindly provided by Dr. F. Lemonnier(Institute Pasteur, Paris, France), were housed in appropriate animalcare facilities during the experimental period and handled followingthe international guidelines required for experimentation withanimals.
2.2. Cell lines
T2 cells were used as target cells in chromium release assays withCTL from HHD mice. THP1 cells (American Type Culture Collection,Manassas, VA) were used for in vitro assays of monocyte activation byEDA-containing fusion proteins. They were grown in complete med-ium (CM) (RPMI 1640 containing 10% fetal calf serum, 100 U/mLpenicillin, 100 lg/mL streptomycin, 2 mM glutamine and 50 lM 2-mercaptoethanol (Life Technologies, Paisley, UK).
2.3. Peptides
HLA-A2-restricted peptides p1073 (CVNGVCWTV) and p1038(GLLGCIITSL) [14] from HCV NS3 protein were synthesized manu-ally in a multiple peptide synthesizer using Fmoc-chemistry. Their pur-ity was always above 90% as determined by HPLC.
2.4. Recombinant proteins
Plasmid pET20b-EDA, expressing the extra domain A from fibro-nectin, [6] was used for the construction of the expression plasmidpET20b-EDA-NS3, to express a fusion protein containing the first195 amino acids from HCV-NS3 protein (corresponding to the serineprotease domain of NS3 [15]) linked to the C-terminus of EDA andcarrying six histidines at the carboxy-terminus. Plasmid pET45b-NS3expressing the 195 first amino acids from HCV-NS3 protein linkedto a six histidine tag at the N-terminus was constructed using conven-tional cloning techniques. EDA-NS3 was purified from inclusionbodies by affinity chromatography (Histrap, GE Healthcare, Uppsala,Sweden) followed by ion exchange chromatography (DEAE-Sephar-ose column, GE Healthcare). NS3 was purified also from inclusionbodies by a semi-preparative isoelectrofocussing followed by affinitychromatography. Resulting proteins were refolded in a sepharoseG25 column using a urea gradient size-exclusion chromatographyand purified from endotoxins by using Endotrap columns (ProfosAg, Regensburg, Germany) until endotoxin levels were below0.1 EU/mg protein (tested by Quantitative Chromogenic LimulusAmebocyte Lysate assay (Cambrex, Walkersville, MD, USA)). Puri-fied recombinant proteins were analyzed by SDS–PAGE and stainedwith Coomassie blue (Bio-Safe Coomassie reagent, Bio-Rad, Hercules,CA).
2.5. Plasmids
Plasmids pShuttle-HTLP-EDA-NS3 (pEDA-NS3) and pShuttle-HTLP-NS3 (pNS3) expressing secretable versions of EDA-NS3 andNS3 were constructed by PCR using plasmid pET20b-EDA-NS3 asa template and specific primers to introduce at the N-terminus of theprotein a sequence coding for the human tyrosinase leader peptide(HTLP). Resulting PCR products were cloned in pShuttle plasmid(Clontech, BD Biosciences).
2.6. In vitro analysis of THP-1 monocyte cell line
activation
THP-1 cells were plated at 106 cells/well and cultured with EDA-NS3 (2 lM), NS3 (2 lM), LPS (1 lg/mL) or culture medium for15 h. In some cases EDA-NS3 was previously digested with protein-ase-K using agarose-proteinase K beads according to manufacturer’sinstructions (Sigma, St. Louis). Human TNF-a released to the mediumby THP-1 cells in response to the stimuli was quantified using a com-mercial ELISA assay (BD-Pharmingen).
2.7. In vitro analysis of DC stimulation
Bone marrow DC (BMDC) were generated from C57Bl6 micefemur marrow cell cultures as previously described [6]. Non-adherentDC were harvested at day 6 and cultured in the presence or absence
C. Mansilla et al. / Journal of Hepatology 51 (2009) 520–527 521
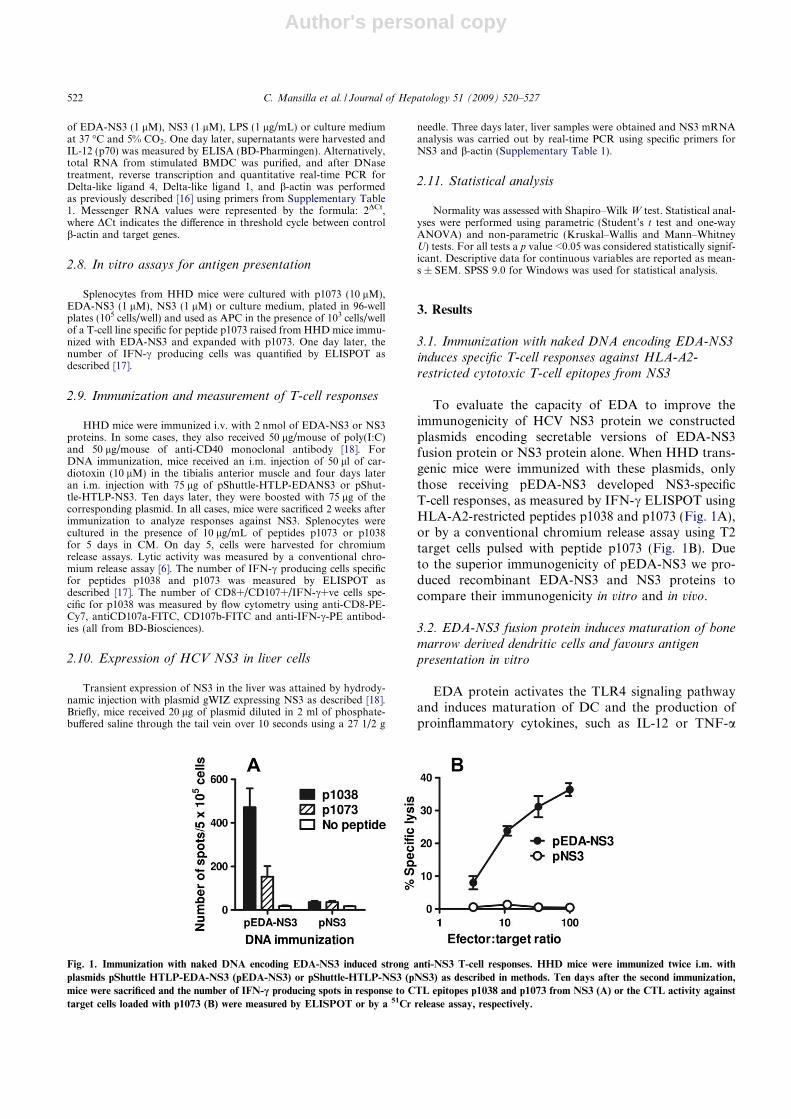
Author's personal copy
of EDA-NS3 (1 lM), NS3 (1 lM), LPS (1 lg/mL) or culture mediumat 37 �C and 5% CO2. One day later, supernatants were harvested andIL-12 (p70) was measured by ELISA (BD-Pharmingen). Alternatively,total RNA from stimulated BMDC was purified, and after DNasetreatment, reverse transcription and quantitative real-time PCR forDelta-like ligand 4, Delta-like ligand 1, and b-actin was performedas previously described [16] using primers from Supplementary Table1. Messenger RNA values were represented by the formula: 2DCt,where DCt indicates the difference in threshold cycle between controlb-actin and target genes.
2.8. In vitro assays for antigen presentation
Splenocytes from HHD mice were cultured with p1073 (10 lM),EDA-NS3 (1 lM), NS3 (1 lM) or culture medium, plated in 96-wellplates (105 cells/well) and used as APC in the presence of 103 cells/wellof a T-cell line specific for peptide p1073 raised from HHD mice immu-nized with EDA-NS3 and expanded with p1073. One day later, thenumber of IFN-c producing cells was quantified by ELISPOT asdescribed [17].
2.9. Immunization and measurement of T-cell responses
HHD mice were immunized i.v. with 2 nmol of EDA-NS3 or NS3proteins. In some cases, they also received 50 lg/mouse of poly(I:C)and 50 lg/mouse of anti-CD40 monoclonal antibody [18]. ForDNA immunization, mice received an i.m. injection of 50 ll of car-diotoxin (10 lM) in the tibialis anterior muscle and four days lateran i.m. injection with 75 lg of pShuttle-HTLP-EDANS3 or pShut-tle-HTLP-NS3. Ten days later, they were boosted with 75 lg of thecorresponding plasmid. In all cases, mice were sacrificed 2 weeks afterimmunization to analyze responses against NS3. Splenocytes werecultured in the presence of 10 lg/mL of peptides p1073 or p1038for 5 days in CM. On day 5, cells were harvested for chromiumrelease assays. Lytic activity was measured by a conventional chro-mium release assay [6]. The number of IFN-c producing cells specificfor peptides p1038 and p1073 was measured by ELISPOT asdescribed [17]. The number of CD8+/CD107+/IFN-c+ve cells spe-cific for p1038 was measured by flow cytometry using anti-CD8-PE-Cy7, antiCD107a-FITC, CD107b-FITC and anti-IFN-c-PE antibod-ies (all from BD-Biosciences).
2.10. Expression of HCV NS3 in liver cells
Transient expression of NS3 in the liver was attained by hydrody-namic injection with plasmid gWIZ expressing NS3 as described [18].Briefly, mice received 20 lg of plasmid diluted in 2 ml of phosphate-buffered saline through the tail vein over 10 seconds using a 27 1/2 g
needle. Three days later, liver samples were obtained and NS3 mRNAanalysis was carried out by real-time PCR using specific primers forNS3 and b-actin (Supplementary Table 1).
2.11. Statistical analysis
Normality was assessed with Shapiro–Wilk W test. Statistical anal-yses were performed using parametric (Student’s t test and one-wayANOVA) and non-parametric (Kruskal–Wallis and Mann–WhitneyU) tests. For all tests a p value <0.05 was considered statistically signif-icant. Descriptive data for continuous variables are reported as mean-s ± SEM. SPSS 9.0 for Windows was used for statistical analysis.
3. Results
3.1. Immunization with naked DNA encoding EDA-NS3
induces specific T-cell responses against HLA-A2-
restricted cytotoxic T-cell epitopes from NS3
To evaluate the capacity of EDA to improve theimmunogenicity of HCV NS3 protein we constructedplasmids encoding secretable versions of EDA-NS3fusion protein or NS3 protein alone. When HHD trans-genic mice were immunized with these plasmids, onlythose receiving pEDA-NS3 developed NS3-specificT-cell responses, as measured by IFN-c ELISPOT usingHLA-A2-restricted peptides p1038 and p1073 (Fig. 1A),or by a conventional chromium release assay using T2target cells pulsed with peptide p1073 (Fig. 1B). Dueto the superior immunogenicity of pEDA-NS3 we pro-duced recombinant EDA-NS3 and NS3 proteins tocompare their immunogenicity in vitro and in vivo.
3.2. EDA-NS3 fusion protein induces maturation of bone
marrow derived dendritic cells and favours antigen
presentation in vitro
EDA protein activates the TLR4 signaling pathwayand induces maturation of DC and the production ofproinflammatory cytokines, such as IL-12 or TNF-a
Fig. 1. Immunization with naked DNA encoding EDA-NS3 induced strong anti-NS3 T-cell responses. HHD mice were immunized twice i.m. with
plasmids pShuttle HTLP-EDA-NS3 (pEDA-NS3) or pShuttle-HTLP-NS3 (pNS3) as described in methods. Ten days after the second immunization,
mice were sacrificed and the number of IFN-c producing spots in response to CTL epitopes p1038 and p1073 from NS3 (A) or the CTL activity against
target cells loaded with p1073 (B) were measured by ELISPOT or by a 51Cr release assay, respectively.
522 C. Mansilla et al. / Journal of Hepatology 51 (2009) 520–527
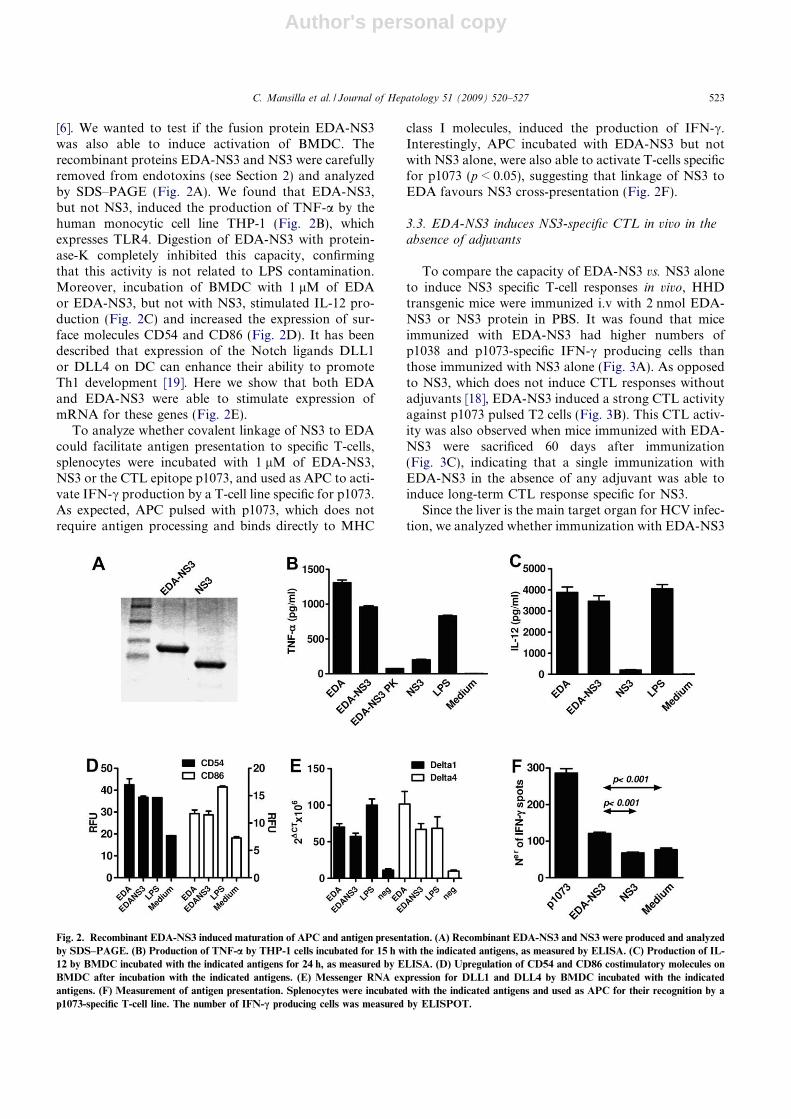
Author's personal copy
[6]. We wanted to test if the fusion protein EDA-NS3was also able to induce activation of BMDC. Therecombinant proteins EDA-NS3 and NS3 were carefullyremoved from endotoxins (see Section 2) and analyzedby SDS–PAGE (Fig. 2A). We found that EDA-NS3,but not NS3, induced the production of TNF-a by thehuman monocytic cell line THP-1 (Fig. 2B), whichexpresses TLR4. Digestion of EDA-NS3 with protein-ase-K completely inhibited this capacity, confirmingthat this activity is not related to LPS contamination.Moreover, incubation of BMDC with 1 lM of EDAor EDA-NS3, but not with NS3, stimulated IL-12 pro-duction (Fig. 2C) and increased the expression of sur-face molecules CD54 and CD86 (Fig. 2D). It has beendescribed that expression of the Notch ligands DLL1or DLL4 on DC can enhance their ability to promoteTh1 development [19]. Here we show that both EDAand EDA-NS3 were able to stimulate expression ofmRNA for these genes (Fig. 2E).
To analyze whether covalent linkage of NS3 to EDAcould facilitate antigen presentation to specific T-cells,splenocytes were incubated with 1 lM of EDA-NS3,NS3 or the CTL epitope p1073, and used as APC to acti-vate IFN-c production by a T-cell line specific for p1073.As expected, APC pulsed with p1073, which does notrequire antigen processing and binds directly to MHC
class I molecules, induced the production of IFN-c.Interestingly, APC incubated with EDA-NS3 but notwith NS3 alone, were also able to activate T-cells specificfor p1073 (p < 0.05), suggesting that linkage of NS3 toEDA favours NS3 cross-presentation (Fig. 2F).
3.3. EDA-NS3 induces NS3-specific CTL in vivo in the
absence of adjuvants
To compare the capacity of EDA-NS3 vs. NS3 aloneto induce NS3 specific T-cell responses in vivo, HHDtransgenic mice were immunized i.v with 2 nmol EDA-NS3 or NS3 protein in PBS. It was found that miceimmunized with EDA-NS3 had higher numbers ofp1038 and p1073-specific IFN-c producing cells thanthose immunized with NS3 alone (Fig. 3A). As opposedto NS3, which does not induce CTL responses withoutadjuvants [18], EDA-NS3 induced a strong CTL activityagainst p1073 pulsed T2 cells (Fig. 3B). This CTL activ-ity was also observed when mice immunized with EDA-NS3 were sacrificed 60 days after immunization(Fig. 3C), indicating that a single immunization withEDA-NS3 in the absence of any adjuvant was able toinduce long-term CTL response specific for NS3.
Since the liver is the main target organ for HCV infec-tion, we analyzed whether immunization with EDA-NS3
Fig. 2. Recombinant EDA-NS3 induced maturation of APC and antigen presentation. (A) Recombinant EDA-NS3 and NS3 were produced and analyzed
by SDS–PAGE. (B) Production of TNF-a by THP-1 cells incubated for 15 h with the indicated antigens, as measured by ELISA. (C) Production of IL-
12 by BMDC incubated with the indicated antigens for 24 h, as measured by ELISA. (D) Upregulation of CD54 and CD86 costimulatory molecules on
BMDC after incubation with the indicated antigens. (E) Messenger RNA expression for DLL1 and DLL4 by BMDC incubated with the indicated
antigens. (F) Measurement of antigen presentation. Splenocytes were incubated with the indicated antigens and used as APC for their recognition by a
p1073-specific T-cell line. The number of IFN-c producing cells was measured by ELISPOT.
C. Mansilla et al. / Journal of Hepatology 51 (2009) 520–527 523
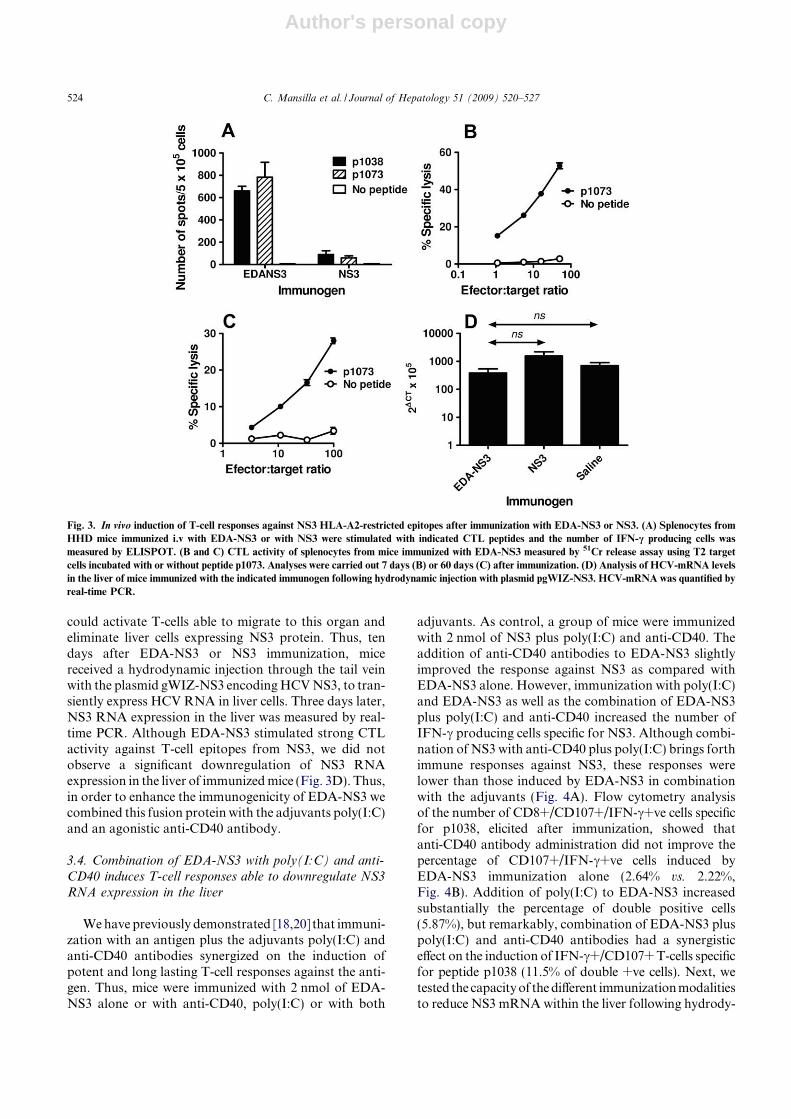
Author's personal copy
could activate T-cells able to migrate to this organ andeliminate liver cells expressing NS3 protein. Thus, tendays after EDA-NS3 or NS3 immunization, micereceived a hydrodynamic injection through the tail veinwith the plasmid gWIZ-NS3 encoding HCV NS3, to tran-siently express HCV RNA in liver cells. Three days later,NS3 RNA expression in the liver was measured by real-time PCR. Although EDA-NS3 stimulated strong CTLactivity against T-cell epitopes from NS3, we did notobserve a significant downregulation of NS3 RNAexpression in the liver of immunized mice (Fig. 3D). Thus,in order to enhance the immunogenicity of EDA-NS3 wecombined this fusion protein with the adjuvants poly(I:C)and an agonistic anti-CD40 antibody.
3.4. Combination of EDA-NS3 with poly(I:C) and anti-
CD40 induces T-cell responses able to downregulate NS3
RNA expression in the liver
We have previously demonstrated [18,20] that immuni-zation with an antigen plus the adjuvants poly(I:C) andanti-CD40 antibodies synergized on the induction ofpotent and long lasting T-cell responses against the anti-gen. Thus, mice were immunized with 2 nmol of EDA-NS3 alone or with anti-CD40, poly(I:C) or with both
adjuvants. As control, a group of mice were immunizedwith 2 nmol of NS3 plus poly(I:C) and anti-CD40. Theaddition of anti-CD40 antibodies to EDA-NS3 slightlyimproved the response against NS3 as compared withEDA-NS3 alone. However, immunization with poly(I:C)and EDA-NS3 as well as the combination of EDA-NS3plus poly(I:C) and anti-CD40 increased the number ofIFN-c producing cells specific for NS3. Although combi-nation of NS3 with anti-CD40 plus poly(I:C) brings forthimmune responses against NS3, these responses werelower than those induced by EDA-NS3 in combinationwith the adjuvants (Fig. 4A). Flow cytometry analysisof the number of CD8+/CD107+/IFN-c+ve cells specificfor p1038, elicited after immunization, showed thatanti-CD40 antibody administration did not improve thepercentage of CD107+/IFN-c+ve cells induced byEDA-NS3 immunization alone (2.64% vs. 2.22%,Fig. 4B). Addition of poly(I:C) to EDA-NS3 increasedsubstantially the percentage of double positive cells(5.87%), but remarkably, combination of EDA-NS3 pluspoly(I:C) and anti-CD40 antibodies had a synergisticeffect on the induction of IFN-c+/CD107+ T-cells specificfor peptide p1038 (11.5% of double +ve cells). Next, wetested the capacity of the different immunization modalitiesto reduce NS3 mRNA within the liver following hydrody-
Fig. 3. In vivo induction of T-cell responses against NS3 HLA-A2-restricted epitopes after immunization with EDA-NS3 or NS3. (A) Splenocytes from
HHD mice immunized i.v with EDA-NS3 or with NS3 were stimulated with indicated CTL peptides and the number of IFN-c producing cells was
measured by ELISPOT. (B and C) CTL activity of splenocytes from mice immunized with EDA-NS3 measured by 51Cr release assay using T2 target
cells incubated with or without peptide p1073. Analyses were carried out 7 days (B) or 60 days (C) after immunization. (D) Analysis of HCV-mRNA levels
in the liver of mice immunized with the indicated immunogen following hydrodynamic injection with plasmid pgWIZ-NS3. HCV-mRNA was quantified by
real-time PCR.
524 C. Mansilla et al. / Journal of Hepatology 51 (2009) 520–527
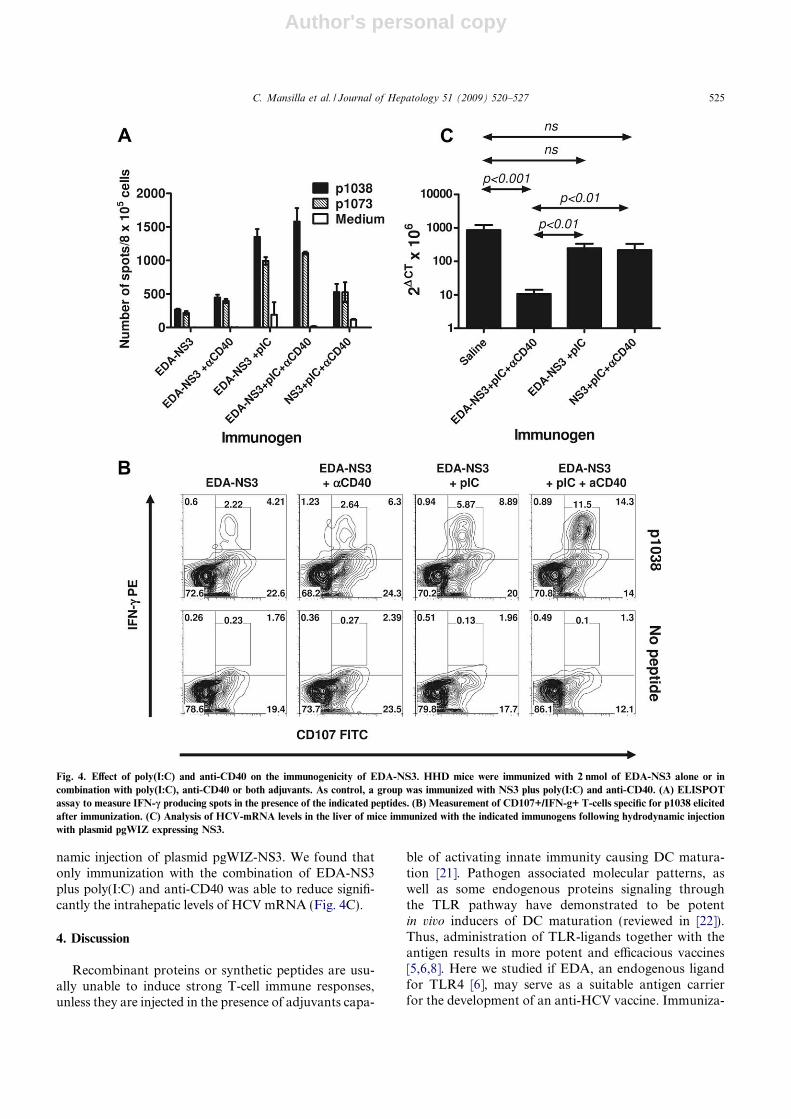
Author's personal copy
namic injection of plasmid pgWIZ-NS3. We found thatonly immunization with the combination of EDA-NS3plus poly(I:C) and anti-CD40 was able to reduce signifi-cantly the intrahepatic levels of HCV mRNA (Fig. 4C).
4. Discussion
Recombinant proteins or synthetic peptides are usu-ally unable to induce strong T-cell immune responses,unless they are injected in the presence of adjuvants capa-
ble of activating innate immunity causing DC matura-tion [21]. Pathogen associated molecular patterns, aswell as some endogenous proteins signaling throughthe TLR pathway have demonstrated to be potentin vivo inducers of DC maturation (reviewed in [22]).Thus, administration of TLR-ligands together with theantigen results in more potent and efficacious vaccines[5,6,8]. Here we studied if EDA, an endogenous ligandfor TLR4 [6], may serve as a suitable antigen carrierfor the development of an anti-HCV vaccine. Immuniza-
Fig. 4. Effect of poly(I:C) and anti-CD40 on the immunogenicity of EDA-NS3. HHD mice were immunized with 2 nmol of EDA-NS3 alone or in
combination with poly(I:C), anti-CD40 or both adjuvants. As control, a group was immunized with NS3 plus poly(I:C) and anti-CD40. (A) ELISPOT
assay to measure IFN-c producing spots in the presence of the indicated peptides. (B) Measurement of CD107+/IFN-g+ T-cells specific for p1038 elicited
after immunization. (C) Analysis of HCV-mRNA levels in the liver of mice immunized with the indicated immunogens following hydrodynamic injection
with plasmid pgWIZ expressing NS3.
C. Mansilla et al. / Journal of Hepatology 51 (2009) 520–527 525

Author's personal copy
tion with a plasmid encoding a secretable version ofEDA-NS3 fusion protein induced more IFN-c produc-ing cells specific for NS3 T-cell epitopes than when ani-mals received the plasmid encoding NS3 alone. Theseinitial promising data prompted us to produce recombi-nant EDA-NS3 and NS3 proteins to compare theirimmunogenicity in vitro and in vivo. EDA-NS3 was ableto stimulate the production of high amounts of IL-12 byBMDC, and to induce in these cells potent upregulationof maturation markers as well as the Notch ligands DlL1and DlL4. Moreover, BMDC incubated with EDA-NS3,but not with NS3 alone, capture and process antigen inan efficient way leading to enhanced presentation of theCTL epitope p1073 to specific T-cells. Thus, covalentlinkage of EDA to NS3 not only facilitates bindingof the immunogen to DC but also, activates theimmunostimulatory functions of these cells resulting inmore potent T-cell responses. These results may haveimportant implications in vaccination since it has beenshown that when DC fail to mature after taking up theantigen the result may be tolerance rather than immunity[23,24].
Immunization of mice with EDA-NS3 in theabsence of additional adjuvants induced a strong cellu-lar immune response against the immunodominantCTL cell epitopes from NS3, a response which wasnot elicited by immunization with NS3 alone. How-ever, this response did not reduce significantly theintrahepatic levels of NS3-mRNA in our model ofhydrodynamic injection, suggesting that additional sig-nals were required to increase the generation of effectorT-cells able to clear efficiently the transducedhepatocytes.
It has been shown that stimulation of CD8 T-cellsby CD40 ligation increases primary and memory cyto-toxic T-cell responses [25,26]. However, immunizationwith EDA-NS3 plus anti-CD40 antibodies did notenhance substantially T-cell immunoreactivity againstNS3. When EDA-NS3 was combined with poly(I:C),which promotes T-cell proliferation and survivalthrough the production of type I IFN [27,28], animportant improvement in the number of IFN-c pro-ducing T-cells specific for NS3 peptides was found.However, this increased anti-NS3 immune responsewas not accompanied by a significant reduction ofintrahepatic NS3 mRNA levels. Importantly, althoughimmunization of mice with NS3 protein together withanti-CD40 and poly(I:C) increased the number ofIFN-c producing cells (Fig. 4 and [18]) only immuniza-tion with EDA-NS3 plus the two adjuvants was able tostrongly increase IFN-c producing cells and induce asignificant reduction in the intrahepatic levels of NS3-mRNA. These data suggest that combined administra-tion of TLR3 (poly(I:C)) and TLR4 (EDA) agonistsplus anti-CD40 may have synergistic effects on DCfunctions in vivo, as it has been demonstrated to occur
in vitro [29,30]. In addition, poly(I:C) and anti-CD40may stimulate other cell populations, such us CD4+T-cells [31] or NK cells [32], and all these effects mightcontribute to the induction multifunctional CD8+ T-cell responses. Notch signaling plays important rolesin Th1 polarization in vitro and in vivo [19,33] and ithas been described that TLR ligation and activationof MyD88-dependent pathway can induce upregulationof the Notch ligands DLL1 and DLL4 [34]. EDA-dependent upregulation of DLL1 and DLL4 expressionon BMDC might be related to its ability to activateTLR-4 dependent MyD88 signaling pathway (datanot shown), as described for LPS [35]. This capacitymight favour the induction of anti-NS3 T-cellresponses and synergize with poly(I:C), which signalsthrough the adaptor molecule TRIF [36], and CD40ligation, which involves the interactions of CD40 withvarious members of the TRAF family [37].
Several prophylactic HCV vaccine formulationshave been evaluated in preclinical studies in chimpan-zees and, although protection against chronic infectionwas limited, most of them succeeded in inducingimmune responses capable of reducing acute-phaseviral load (reviewed in [1]). Some of them have alsobeen evaluated in clinical trials in HCV patients, butvery little impact on the disease has been observed,suggesting that vaccine strategies need to be improvedand should be combined with the available antiviraltherapies.
Here we show that the recombinant fusion proteinEDA-NS3 induces DC maturation, enhances antigenpresentation and activation of long-term antiviralimmune responses in vivo. In combination withpoly(I:C) and anti-CD40 antibodies, EDA-NS3 wasable to downregulate HCV RNA expression in theliver in a murine model using hydrodynamic transduc-tion of hepatocytes with HCV NS3. These results sug-gest that EDA-NS3 protein may be considered as acandidate for the development of vaccines againstHCV infection.
Acknowledgements
We thank Dr. C. Leclerc for helpful discussion. Wealso thank Elena Ciordia and Eneko Elizalde for excellentanimal care. This work was supported by grants fromGobierno de Navarra and from Proyecto Euroinnova(Evidencia) to J.J.L., from Instituto de Salud Carlos IIIto C.M. and by ‘‘UTE project CIMA”.
Appendix A. Supplementary data
Supplementary data associated with this article canbe found, in the online version, at doi:10.1016/j.jhep.2009.06.005.
526 C. Mansilla et al. / Journal of Hepatology 51 (2009) 520–527

Author's personal copy
References
[1] Stoll-Keller F, Barth H, Fafi-Kremer S, Zeisel MB, Baumert TF.Development of hepatitis C virus vaccines: challenges andprogress. Expert Rev Vaccines 2009;8:333–345.
[2] Rehermann B, Nascimbeni M. Immunology of hepatitis B virusand hepatitis C virus infection. Nat Rev Immunol 2005;5:215–229.
[3] Diepolder HM, Zachoval R, Hoffmann RM, Wierenga EA,Santantonio T, Jung MC, et al. Possible mechanism involving T-lymphocyte response to non-structural protein 3 in viral clearancein acute hepatitis C virus infection. Lancet 1995;346:1006–1007.
[4] Reis e Sousa C. Dendritic cells in a mature age. Nat Rev Immunol2006;6:476–483.
[5] Cuadros C, Lopez-Hernandez FJ, Dominguez AL, McClelland M,Lustgarten J. Flagellin fusion proteins as adjuvants or vaccinesinduce specific immune responses. Infect Immun 2004;72:2810–2816.
[6] Lasarte JJ, Casares N, Gorraiz M, Hervas-Stubbs S, ArribillagaL, Mansilla C, et al. The extra domain A from fibronectin targetsantigens to TLR4-expressing cells and induces cytotoxic T cellresponses in vivo. J Immunol 2007;178:748–756.
[7] LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F,Slack EC, Tsoni SV, et al. Syk- and CARD9-dependent couplingof innate immunity to the induction of T helper cells that produceinterleukin 17. Nat Immunol 2007;8:630–638.
[8] Tighe H, Takabayashi K, Schwartz D, Marsden R, Beck L, CorbeilJ, et al. Conjugation of protein to immunostimulatory DNA resultsin a rapid, long-lasting and potent induction of cell-mediated andhumoral immunity. Eur J Immunol 2000;30:1939–1947.
[9] Ahonen CL, Doxsee CL, McGurran SM, Riter TR, Wade WF,Barth RJ, et al. Combined TLR and CD40 triggering inducespotent CD8+ T cell expansion with variable dependence on type IIFN. J Exp Med 2004;199:775–784.
[10] Wen T, Bukczynski J, Watts TH. 4-1BB ligand-mediated costi-mulation of human T cells induces CD4 and CD8 T cellexpansion, cytokine production, and the development of cytolyticeffector function. J Immunol 2002;168:4897–4906.
[11] Ahlers JD, Belyakov IM, Berzofsky JA. Cytokine, chemokine,and costimulatory molecule modulation to enhance efficacy ofHIV vaccines. Curr Mol Med 2003;3:285–301.
[12] Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognitionof double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001;413:732–738.
[13] Pascolo S, Bervas N, Ure JM, Smith AG, Lemonnier FA,Perarnau B. HLA-A2.1-restricted education and cytolytic activityof CD8(+) T lymphocytes from beta2 microglobulin (beta2m)HLA-A2.1 monochain transgenic H-2Db beta2m double knock-out mice. J Exp Med 1997;185:2043–2051.
[14] Cerny A, McHutchison JG, Pasquinelli C, Brown ME, BrothersMA, Grabscheid B, et al. Cytotoxic T lymphocyte response tohepatitis C virus-derived peptides containing the HLA A2.1binding motif. J Clin Invest 1995;95:521–530.
[15] Vishnuvardhan D, Kakiuchi N, Urvil PT, Shimotohno K, KumarPK, Nishikawa S. Expression of highly active recombinant NS3protease domain of hepatitis C virus in E. coli. FEBS Lett1997;402:209–212.
[16] Zabala M, Lasarte JJ, Perret C, Sola J, Berraondo P, Alfaro M,et al. Induction of immunosuppressive molecules and regulatory Tcells counteracts the antitumor effect of interleukin-12-based genetherapy in a transgenic mouse model of liver cancer. J Hepatol2007;47:807–815.
[17] Durantez M, Lopez-Vazquez AB, de Cerio AL, Huarte E, CasaresN, Prieto J, et al. Induction of multiepitopic and long-lastingimmune responses against tumour antigens by immunization withpeptides, DNA and recombinant adenoviruses expressing minig-enes. Scand J Immunol 2009;69:80–89.
[18] Zabaleta A, Arribillaga L, Llopiz D, Dotor J, Lasarte JJ, Prieto J,et al. Induction of potent and long-lasting CD4 and CD8 T-cellresponses against hepatitis C virus by immunization with viralantigens plus poly(I:C) and anti-CD40. Antiviral Res 2007;74:25–35.
[19] Amsen D, Blander JM, Lee GR, Tanigaki K, Honjo T, Flavell RA.Instruction of distinct CD4 T helper cell fates by different Notchligands on antigen-presenting cells. Cell 2004;117:515–526.
[20] Llopiz D, Dotor J, Zabaleta A, Lasarte JJ, Prieto J, Borras-CuestaF, et al. Combined immunization with adjuvant moleculespoly(I:C) and anti-CD40 plus a tumor antigen has potentprophylactic and therapeutic antitumor effects. Cancer ImmunolImmunother 2008;57:19–29.
[21] Audibert FM, Lise LD. Adjuvants: current status, clinical perspec-tives and future prospects. Immunol Today 1993;14:281–284.
[22] Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immuneresponses. Nat Rev Immunol 2004;4:469–478.
[23] Bonifaz L, Bonnyay D, Mahnke K, Rivera M, Nussenzweig MC,Steinman RM. Efficient targeting of protein antigen to thedendritic cell receptor DEC-205 in the steady state leads toantigen presentation on major histocompatibility complex class Iproducts and peripheral CD8+ T cell tolerance. J Exp Med2002;196:1627–1638.
[24] Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic den-dritic cells. Annu Rev Immunol 2003;21:685–711.
[25] Bourgeois C, Rocha B, Tanchot C. A role for CD40 expression onCD8+ T cells in the generation of CD8+ T cell memory. Science2002;297:2060–2063.
[26] Lefrancois L, Altman JD, Williams K, Olson S. Soluble antigenand CD40 triggering are sufficient to induce primary and memorycytotoxic T cells. J Immunol 2000;164:725–732.
[27] Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allowclonal expansion and memory formation in response to viralinfection. J Exp Med 2005;202:637–650.
[28] Tough DF, Borrow P, Sprent J. Induction of bystander T cellproliferation by viruses and type I interferon in vivo. Science1996;272:1947–1950.
[29] Lapointe R, Toso JF, Butts C, Young HA, Hwu P. Humandendritic cells require multiple activation signals for the efficientgeneration of tumor antigen-specific T lymphocytes. Eur JImmunol 2000;30:3291–3298.
[30] Schulz O, Edwards AD, Schito M, Aliberti J, Manickasingham S,Sher A, et al. CD40 triggering of heterodimeric IL-12 p70production by dendritic cells in vivo requires a microbial primingsignal. Immunity 2000;13:453–462.
[31] Gelman AE, Zhang J, Choi Y, Turka LA. Toll-like receptorligands directly promote activated CD4+ T cell survival. JImmunol 2004;172:6065–6073.
[32] Sivori S, Falco M, Della Chiesa M, Carlomagno S, Vitale M,Moretta L, et al. CpG and double-stranded RNA trigger humanNK cells by Toll-like receptors: induction of cytokine release andcytotoxicity against tumors and dendritic cells. Proc Natl Acad SciUSA 2004;101:10116–10121.
[33] Maekawa Y, Tsukumo S, Chiba S, Hirai H, Hayashi Y, Okada H,et al. Delta1-Notch3 interactions bias the functional differentia-tion of activated CD4+ T cells. Immunity 2003;19:549–559.
[34] Sun J, Krawczyk CJ, Pearce EJ. Suppression of Th2 celldevelopment by Notch ligands Delta1 and Delta4. J Immunol2008;180:1655–1661.
[35] Skokos D, Nussenzweig MC. CD8-DCs induce IL-12-indepen-dent Th1 differentiation through Delta 4 Notch-like ligand inresponse to bacterial LPS. J Exp Med 2007;204:1525–1531.
[36] Akira S, Takeda K. Toll-like receptor signalling. Nat RevImmunol 2004;4:499–511.
[37] Bishop GA, Moore CR, Xie P, Stunz LL, Kraus ZJ. TRAFproteins in CD40 signaling. Adv Exp Med Biol 2007;597:131–151.
C. Mansilla et al. / Journal of Hepatology 51 (2009) 520–527 527

The Journal of Immunology
A Peptide Inhibitor of FOXP3 Impairs Regulatory T CellActivity and Improves Vaccine Efficacy in Mice
Noelia Casares,* Francesc Rudilla,* Laura Arribillaga,* Diana Llopiz,*
Jose Ignacio Riezu-Boj,* Teresa Lozano,* Jacinto Lopez-Sagaseta,† Laura Guembe,‡,x
Pablo Sarobe,* Jesus Prieto,*,‡,x Francisco Borras-Cuesta,* and Juan Jose Lasarte*
Immunosuppressive activity of regulatory T cells (Treg) may contribute to the progression of cancer or infectious diseases by pre-
venting the induction of specific immune responses. Using a phage-displayed random peptide library, we identified a 15-mer syn-
thetic peptide, P60, able to bind to forkhead/winged helix transcription factor 3 (FOXP3), a factor required for development and
function of Treg. P60 enters the cells, inhibits FOXP3 nuclear translocation, and reduces its ability to suppress the transcription
factors NF-kB and NFAT. In vitro, P60 inhibited murine and human-derived Treg and improved effector T cell stimulation. P60
administration to newborn mice induced a lymphoproliferative autoimmune syndrome resembling the reported pathology in
scurfy mice lacking functional Foxp3. However, P60 did not cause toxic effects in adult mice and, when given to BALB/c mice
immunized with the cytotoxic T cell epitope AH1 from CT26 tumor cells, it induced protection against tumor implantation.
Similarly, P60 improved the antiviral efficacy of a recombinant adenovirus expressing NS3 protein from hepatitis C virus.
Functional inhibition of Treg by the FOXP3-inhibitory peptide P60 constitutes a strategy to enhance antitumor and antiviral
immunotherapies. The Journal of Immunology, 2010, 185: 5150–5159.
Regulatory T cells (Tregs) are a distinct lymphocyte line-age endowed with inhibitory properties that affect theactivation of the immune system. Tregs are characterized
by the expression of CD25 and the Treg-specific forkhead/wingedhelix transcription factor 3 (FOXP3), which is required for theirdevelopment and function (1, 2). These cells can inhibit activationof other T cells (3) and are needed for protection against autoim-mune diseases and prevent rejection of allogeneic transplants. Inhumans, mutations in FOXP3 result in an autoimmune syndrometermed IPEX (immunodysregulation, polyendocrinopathy entero-pathy, X-linked syndrome), an X-linked immunodeficiency syn-drome characterized by insulin-dependent diabetes, thyroiditis,massive T cell infiltration in multiple organs, and chronic wasting(4–6). However, immunoregulatory function of Treg may hinderthe induction of immune responses against cancer and infectiousagents (7–13). Indeed, Tregs capable of suppressing the in vitrofunction of tumor-reactive T cells have been found in humans inmany types of tumors (9–13) and have been associated with a high
death hazard and reduced survival (9, 11). It has been shown thatcounteracting Treg activity can evoke effective antitumor immu-nity (14–16), and it is acknowledged that inhibition of Treg func-tion in patients with cancer is an essential step to improve theefficacy of antitumoral therapies, especially those based in immu-notherapeutic approaches (reviewed in Ref. 17).Several strategies have been proposed to neutralize Treg activity.
Thus, it has been shown that administration of low doses of cy-clophosphamide is able to deplete Treg and favors antitumortherapies (18–20). Also, targeting the a-subunit of the IL-2R byusing a fusion protein between the IL-2 and the diphtheria toxinhas been tested in clinical trials with different results (21, 22).However, these strategies lack of high specificity and mighteliminate both effector T cells and Tregs. In addition, depletion ofTregs by these strategies also raises the possibility of autoimmunity(23). An alternative way to control Tregs is their functional in-activation rather than their depletion. Because sustained FOXP3expression in mature Tregs is necessary for maintenance of theTreg phenotype and suppressor function, and ablation of FOXP3or reducing its expression results in the loss of their suppressivefunction (1, 2), we decided to search for peptide inhibitors ofFOXP3 using a phage-displayed random peptide library. Using thisstrategy, we identified peptide P60, which is able to inhibit Tregfunctions in vitro and in vivo.
Materials and MethodsPreparation of the recombinant FOXP3 protein
Plasmid pDEST15-FOXP3 (provided by Dr. Casal, Centro Nacional deInvestigaciones Oncologicas, Madrid, Spain) encoding the fusion proteinGST-FOXP3 (GST fused to the isoform A from human FOXP3 gene), wastransformed into BL21 (DE3; Novagen, Schwalbach, Germany) cells forthe expression of the recombinant protein. GST-FOXP3 protein was pu-rified from the soluble fraction of cell extracts, by affinity chromatography(GSTrap; Amersham, Piscataway, NJ) using a fast protein liquid chro-matography platform (AKTA; Amersham). Purified recombinant proteinwas analyzed by SDS-PAGE using Coomassie blue (Bio-Safe Coomassiereagent; Bio-Rad, Hercules, CA) and by Western blot using anti-FOXP3
*Gene Therapy and Hepatology Area, †Cardiovascular Sciences Area, and ‡Department of Morphology, Center for Applied Medical Research, University of Navarra; andxCIBERehd, Pamplona, Spain
Received for publication April 7, 2010. Accepted for publication August 21, 2010.
This work was supported by Fundacion Mutua Madrilena, Ministerio de Educacion yCiencia (Grants SAF2007-61432 and SAF2010-15060 to J.J.L.), and “UTE projectCIMA”. N.C. is a recipient of a “Ramon y Cajal” contract (Ministerio de Ciencia eInnovacion).
Address correspondence and reprint requests to Dr. Juan Jose Lasarte, Gene Therapyand Hepatology Area, Center for Applied Medical Research, Avenida Pıo XII, 55,31008, Pamplona, Spain. E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this paper: CF, carboxyfluorescein diacetate; CPP, cell-pene-trating peptide; ctrl pept, control peptide; EGFP, enhanced GFP; FKH, forkheaddomain; FOXP3, forkhead/winged helix transcription factor 3; HCV, hepatitis Cvirus; ICR, imprinting control region; K, Karpas cells; Luc, luciferase; MFI, maxi-mum fluorescence intensity; NS3, nonstructural protein 3 from hepatitis C virus;RAdNS3, recombinant adenovirus expressing NS3; RLU, relative luciferase units;ROR, retinoic acid-related orphan receptor; Treg, regulatory T cell.
Copyright� 2010 by TheAmericanAssociation of Immunologists, Inc. 0022-1767/10/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1001114
on October 21, 2010
ww
w.jim
munol.org
Dow
nloaded from

Abs (clone 236A/E7, provided by Dr. Roncador, Centro Nacional de In-vestigaciones Oncologicas).
Identification of potential peptide inhibitors of FOXP3 using aphage-displayed random peptide library
Phages from phage library (fUSE5/15-mer library) expressing 15-merrandom peptides near the N terminus of phage surface protein pIII (a gift ofDr. Smith, University of Missouri-Columbia, Columbia, MO) were allowedto interact with GST-FOXP3. Bound phages were then eluted and amplifiedin Escherichia coli K91Kan. After three rounds of elution-amplificationat decreasing concentrations of FOXP3, best binder phages were selectedand their DNA region coding for their corresponding 15-mer peptide weresequenced as previously described (24) with minor modifications. Bindingcapacity of the amplified phages was tested by ELISA using GST-FOXP3–coated plates and an anti-M13 Ab-HRP conjugated (Amersham PharmaciaBiotech, Uppsala, Sweden). Phages were selected when the OD ratio be-tween GSF-FOXP3 and BSA was .2. Fourteen phages were selected andthe corresponding 15-mer peptides were synthesized as described later.
Peptide synthesis
Peptides were synthesized by the solid phase method of Merrifield usingthe fluorenylmethyloxycarbonyl alternative, as previously described (25).To study peptide internalization into the cells, we labeled peptide P60,as well as peptide OVA(235–264), at the N terminus by the addition of 5(6)-carboxyfluorescein N-hydroxysuccinimide ester (Sigma-Aldrich, Steim-heim, Germany) at the end of the synthesis. The purity of peptides was.90% as judged by HPLC. Peptide P60 (RDFQSFRKMWPFFAM) andpeptide AH1 (SPSYVYHQF) containing a cytotoxic T cell determinant(TCd) expressed by CT26 cells and presented by H-2Ld MHC class Imolecule (26) were synthesized by Neomps (Strasbourg, France). PeptideP60Scramble (MKMFFDAFPQRRSWF), which corresponds to a scram-bled sequence of P60, was also synthesized and used as a control peptide.
Biomolecular interaction analysis
Screening of peptide binding to FOXP3 was performed by surfaceplasmon resonance using Biacore T100 (GE Healthcare, Piscataway, NJ)and ProteOn XPR36 (Bio-Rad) optical biosensors. When we used theBiacore T100, GST-FOXP3 fusion protein was covalently immobilizedonto the surface of flow cell 2 of a CM5 chip (Biacore) as described pre-viously (24). Flow cell 1, in which surface there is no immobilized FOXP3,was used as the reference flow cell. When we used the ProteOn XPR36system, GST-FOXP3 and GST proteins were covalently immobilized ontothe surface of two independent channels of a GLC sensor chips (Bio-Rad)using the coupling reagents N-hydroxysulfosuccinimide and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (Bio-Rad). After proteinimmobilization, chip surface was treated with ethanolamine to deactivate theexcess of reactive esters. In both systems, individual peptide solutions (5mM)were injected two times in running buffer (PBS, 0.005% [v/v] Tween 20, pH7.4) at a flow rate of 30 ml/min. In the experiments carried out in the BiacoreT100, curves were processed by subtracting the response in flow cell 1 fromthat in flow cell 2. In the ProteOn XPR36 system, the signal obtained in thechannel immobilized with GST protein was used as reference. Associationand dissociation phases were monitored for 60 and 30 s, respectively.
Cell lines and viruses
Karpas-299 cell line, a human T cell lymphoma that shows characteristicstypical of natural Tregs (27), was purchased from DSMZ (Braunschweig,Germany). CT26 murine tumor cells (H-2d) from American Type CultureCollection were used in vivo for tumor protection and treatment experi-ments. BSC-1 cells (provided by Dr. Berzofsky, National Institutes ofHealth, Bethesda, MD) were used to titrate vaccinia virus PFU in the ovariesfrom infected animals. They were cultured in complete medium (RPMI1640 containing 10% FCS, 100 U/ml penicillin, 100 mg/ml streptomycin,2 mM glutamine, and 50 mM 2-ME). Human embryonic kidney 293 (In-vivogen, Toulouse, France) and Jurkat (American Type Culture Collection)cell lines used for transfection experiments were cultured in DMEM sup-plemented with 10% FCS and antibiotics.
Recombinant adenovirus expressing hepatitis C virus (HCV) NS3 pro-tein (RAdNS3) was generated as described previously (28). Vaccinia virusvHCV1-3011–expressing HCV polyprotein was provided by Dr. C.M. Rice(Washington University School of Medicine, St. Louis, MO).
Confocal microscopy
Jurkat cells (105 cells/well) were plated into eight-chamber glass slides(IBIDI GmbH, Munich, Germany). They were allowed to settle beforeadding carboxyfluorescein diacetate (CF)-P60 or CF-OVA(235–264) pep-
tides. Intracellular distribution of the fluorescence was analyzed at dif-ferent time points after adding the peptide (10 mM) using a Zeiss con-focal microscope (LSM 510 Meta, Munich, Germany) equipped with aNA 1.4363 oil-immersion Plan-Apochromat objective lens. Percentage ofthe maximum fluorescence intensity (MFI; extracellular mean fluorescenceunits after addition of CF-labeled peptides) was calculated using the for-mula: %MFI = 100*(mfiexp 2 mfibasal)/(MFI2mfibasal), where mfiexp is themean fluorescence intensity per cell and mfibasal is the mean fluorescenceintensity of the background. Mean fluorescence intensity at each time pointwas calculated individually in 20 cells per assay using LSM 510 Metasoftware (Carl Zeiss, Munich, Germany).
Plasmids and transfections
Plasmids used in this work were pNFAT-luciferase (provided by Dr. Revilla,Autonomous University of Madrid, Madrid, Spain), pNF-kB–luciferase(BD Clontech, Palo Alto, CA), pcDNA-FOXP3 (provided by Dr. Ziegler,Seattle, WA), pMIG-R1 Foxp3 (provided by Dr. Mailer, Max DelbruckCenter for Molecular Medicine, Berlin, Germany), pRenilla (provided byDr. Fortes, Center for Applied Medical Research, Pamplona, Spain), andpFOXP3-enhanced GFP (EGFP), which was prepared by cloning FOXP3 inpEGFP-N1 (BD Clontech). Transfections were performed with Lipofect-amine 2000 (Invitrogen, Carlsbad, CA) following the manufacturer’sinstructions and using 1 mg of the indicated plasmids. After 24 h of cul-ture, cells were analyzed by means of the dual luciferase assay (Promega,Madison, WI) with relative normalization for Renilla.
Mice
Female BALB/c mice and imprinting control region (ICR; CD1) mice werepurchased by Harlan (Barcelona, Spain) and bred at the animal facility ofCenter for Applied Medical Research. All experiments performed followedinstitutional guidelines and were approved by the institutional ethicalcommittees.
Treg purification
Isolation of murine and human CD4+CD25+ and CD4+CD252 T cells wasperformed from murine spleen cells or from human PBMCs by usingmurine and human Treg isolation kits (Miltenyi Biotec, Bergisch Glad-bach, Germany), respectively, and according to manufacturer’s instruc-tions. The purity of the resulting T cell populations was confirmed to be.95% by flow cytometry.
Immunohistochemistry
Paraffin liver sections (3 mm thick) were cut, dewaxed, and hydrated. Agretrieval was performed for 30 min at 95˚C in 0.01 M Tris, 1 mM EDTAbuffer (pH 9) in a Pascal pressure chamber (S2800; Dako, Carpinteria, CA).Slides were allowed to cool for 20 min; then endogenous peroxidase wasblocked with 3% H2O2 in deionized water for 10 min, and sections werewashed in TBST. Sections were incubated overnight at 4˚C with anti-mouse/rat-Foxp3 (1:100, clon FJK-16s; eBiosciences, San Diego, CA).After rinsing in TBST, sections were incubated with rabbit anti-rat (E0468;Dako) and then with goat anti-rabbit (K4003; Dako)–labeled polymer bothfor 30 min at room temperature. Peroxidase activity was revealed usingdiaminobenzidine (DAB+; K3468; Dako), and sections were lightly coun-terstained with Harris hematoxylin. Finally, slides were dehydrated ingraded series of ethanol, cleared in xylene, and mounted in distyrene,plasticizer, and xylene.
RNA extraction and quantitative real-time RT-PCR
Total RNA extraction from cell cultures was performed as previouslydescribed (29). Murine Foxp3, IL-10, IFN-g, and b-actin expression wasmeasured by quantitative real-time PCR using specific primers for eachgene as described previously (30). The level of expression of b-actin wasused to normalize gene expression.
In vitro assays for murine or human Treg function
Inhibition of murine or human Treg was measured in two different in vitroassays of T cell stimulation. CD25-depleted spleen cells (105 cells/well)from BALB/c mice or 105 human PBMCs/well were stimulated in vitrowith 1) 0.5 mg/ml anti-mouse CD3 Ab (Pharmingen, San Diego, CA)or with anti-human CD3/CD28 beads (Dynal, Oslo, Norway), respectively;or with 2) 103 bone marrow dendritic cells from C57BL/6 mice (preparedas described previously [31]) or with 105 human PBMCs (from a differentdonor) to induce an MLR, in the presence or absence of purified Tregs (104
cells/well). T cell proliferation was measured 3 d later, as previously de-scribed (32). In some experiments, Karpas cell line was used as a sourceof human Tregs in MLR reactions. Thus, 105 PBMCs from two different
The Journal of Immunology 5151
on October 21, 2010
ww
w.jim
munol.org
Dow
nloaded from

donors were mixed with 104 Karpas cells in the presence or absence of 100mM of different peptides. IFN-g secretion to the culture supernatant wasmeasured by ELISA (Pharmingen) according tomanufacturer’s instructions.
Antitumor peptide vaccination
Animals immunized with 50 nmol/mouse of peptide AH1 emulsified in IFA,as previously described (14), were treated i.p. with 50 nmol/mouse of peptideP60, control peptide, or saline daily during 10 d. At day 10, splenocytes fromimmunized animals were cultured in the presence or absence of peptideAH1 for 48 h. IFN-g released to the supernatant was measured by a com-mercial ELISA (Pharmingen), according to manufacturer’s instructions.Alternatively, micewere injected s.c. with 53 105 CT26 tumor cells. Tumorsize was monitored twice a week with a caliper, and it was expressedaccording to the formula: V = (length 3 width2)/2. Mice were killed whentumor size reached a volume .4 cm3. Alternatively, splenocytes from im-munized animals were cultured in the presence or absence of peptide AH1for 48 h. IFN-g released to the supernatant was measured by a commercialELISA (Pharmingen) according to manufacturer’s instructions.
In vivo protection against infection with a recombinantvaccinia virus expressing HCV proteins
Mice immunized i.p. with 5 3 108 PFU of the recombinant adenovirusRAdNS3 were treated i.p. with 50 nmol/mouse of peptide P60 or with salinefrom days 0–10. As control animals, nonimmunized mice were treated withP60 or with an irrelevant peptide from days 0–10. At day 10, mice werechallenged i.p. with 53 106 PFU of the recombinant vaccinia vHCV1-3011expressing HCV polyprotein. Three days after vaccinia challenge, micewere killed and ovaries removed, weighted, homogenized, sonicated, andassayed for vHCV1-3011 titer by plating serial 10-fold dilutions of sam-ples on a plate of BSC-1 indicator cells. After 2 d of culture, cells werestained with crystal violet to detect PFU at each serial dilution.
Statistical analysis
Normality was assessed with Shapiro–Wilk W test. Statistical analyseswere performed using parametric (Student t test and one-way ANOVA)and nonparametric (Kruskal–Wallis and Mann–Whitney U) tests. For alltests, p , 0.05 was considered statistically significant. Descriptive data forcontinuous variables are reported as means6 SEM. SPSS 9.0 for Windows(SPSS, Chicago, IL) was used for statistical analysis.
ResultsIdentification of potential peptide inhibitors of FOXP3
To identify peptide inhibitors of FOXP3 of potential use in therapy,we used a phage-displayed random 15-mer peptide library. Thislibrary was allowed to interact with GST-FOXP3 immobilized onplastic plates. After three cycles of elution-amplification at de-creasing concentrations of GST-FOXP3, a total of 100 bacterialclones (infected with a unique phage) were obtained, and their phageDNA insert coding for their corresponding 15-mer peptides wassequenced. The binding capacity of 14 of the selected phages wasconfirmed by ELISA using plates coated with GST-FOXP3 (notshown). Fifteen-mer peptides codified by these 14 phages weresynthesized and tested in an in vitro assay of inhibition of the im-munosuppressive activity of the natural regulatory T cell line Karpas(27) in an MLR. Thus, PBMCs from two different healthy donors(PBMC1 and PBMC2) were mixed in culture to stimulate the pro-duction of IFN-g in the presence or absence of Karpas cells. After48 h of culture, we measured the IFN-g released to the supernatantby ELISA. As shown in Fig. 1A, the addition of Karpas cells to thesecultures was able to strongly inhibit the production of IFN-g. Wescreened in this assay the capacity of the 14 peptides to restore theIFN-g production. Peptide P60 (RDFQSFRKMWPFFAM)was ableto overcome the immunosuppressive effect of Karpas cells and wasselected as the best inhibitor of Treg activity. Thus, we focused onthe characterization of the inhibitory capacity of this peptide. Thebinding capacity of P60 to GST-FOXP3 was analyzed and con-firmed by surface plasmon resonance using GST-FOXP3 and GSTproteins covalently immobilized onto the chip surface. As control,we also injected the irrelevant peptide VKQFYDQALQQAVVD (aa123–137 from human receptor CD81) or the P60-scrambled peptide
MKMFFDAFPQRRSWF. It was found that P60, but not the controlpeptide, binds specifically to FOXP3 protein (Fig. 1B). We concludethat P60 binds specifically to FOXP3 because no positive signal wasobserved when it was injected through a chip surface coated withGST protein. Moreover, the P60-scrambled peptide was unable tointeract with GST-FOXP3 (data not shown).
Peptide P60 enters into the cells, downregulates FOXP3nuclear translocation, and inhibits human or murine Tregin vitro
Peptides that are able to translocate across cell membranes areknown as cell-penetrating peptides (CPPs). They are characterizedas short (,30 aa), water-soluble, and cationic or amphipathic innature (reviewed in Ref. 33). Membrane translocation of the po-tential FOXP3 inhibitory peptide P60 is necessary to reach in-hibition of Treg activity. To address whether P60 was able to enterinto the cell, we synthesized the CF-P60 peptide containing amolecule of CF linked to the N terminus of the peptide, to visualizedirectly peptide internalization in live Jurkat cells by confocalfluorescence microscopy. As control, we used the irrelevant CF-labeled peptide CF-OVA(235–264). Time-course imaging analysisof the cells revealed a significant increase in their intracellular meanfluorescence intensity after the addition of CF-P60 peptide, but notof CF-OVA(235–264), reaching a maximum after 10 min of in-cubation (Fig. 1C, 1D), suggesting that peptide P60 is able to enterinto the cell.The FOXP3 protein encodes at least three discernible functional
domains, a carboxyl-termination forkhead domain (FKH), a singleC2H2 zinc finger, and a leucine zipper-like motif. The ability ofFOXP3 to act as a transcriptional repressor requires the presenceof the FKH domain, which is critical for the nuclear translocation ofFOXP3 and for its DNA-binding capacity (34). To study whetherbinding of P60 to FOXP3 could prevent its nuclear localization, wegenerated a plasmid expressing FOXP3-EGFP fusion protein. Theconstruct was transfected into 293 cells to evaluate by fluorescencemicroscopy the subcellular localization of FOXP3-EGFP. It wasfound that 24 h after transfection, GFP was mainly localized intothe nucleus of the cells. However, the addition of P60, but not theirrelevant peptide, to the transfected cells reduced significantlyFOXP3-EGFP nuclear translocation (Fig. 1E). The addition ofP60 to the cultures did not affect the level of expression of FOXP3-GFP, which was equivalent for P60 and for the control peptide(mean fluorescence intensity of 321 6 19.1 versus 317 6 15.6, re-spectively). Similar results were obtained when we studied by im-munohistochemistry the cellular localization of Foxp3 in primarysplenocytes incubated for 24 h in the presence or absence of P60or the control peptide. Indeed, whereas Foxp3 staining was locatedinto the nucleus when cells were incubated with culture mediumalone or with control peptide, it was found that incubation of cellswith P60 favored cytoplasm localization of Foxp3 staining (Sup-plemental Fig. 1), suggesting that P60 can inhibit nuclear trans-location of Foxp3 in primary Treg.
P60 overcomes the inhibitory effect of FOXP3 on the activity ofthe transcription factors NF-kB and NFAT
It has been described that FOXP3 interacts with NFATand with NF-kB to repress cytokine gene expression and effector functions of Thelper cells (35). When FOXP3 is overexpressed in 293 cells, itinhibits the basal level of NF-kB activation (35). When we mea-sured activation of NF-kB on 293 cells transfected with the re-porter plasmid NF-kB–luciferase in the presence or absence ofa plasmid coding for FOXP3, we confirmed its inhibitory activity.However, the addition of P60 to the cultures was able to restore, atleast in part, NF-kB activity (Fig. 2A). Similarly, when Jurkat cells
5152 DEVELOPMENT OF A FOXP3 INHIBITORY PEPTIDE FOR VACCINATION
on October 21, 2010
ww
w.jim
munol.org
Dow
nloaded from
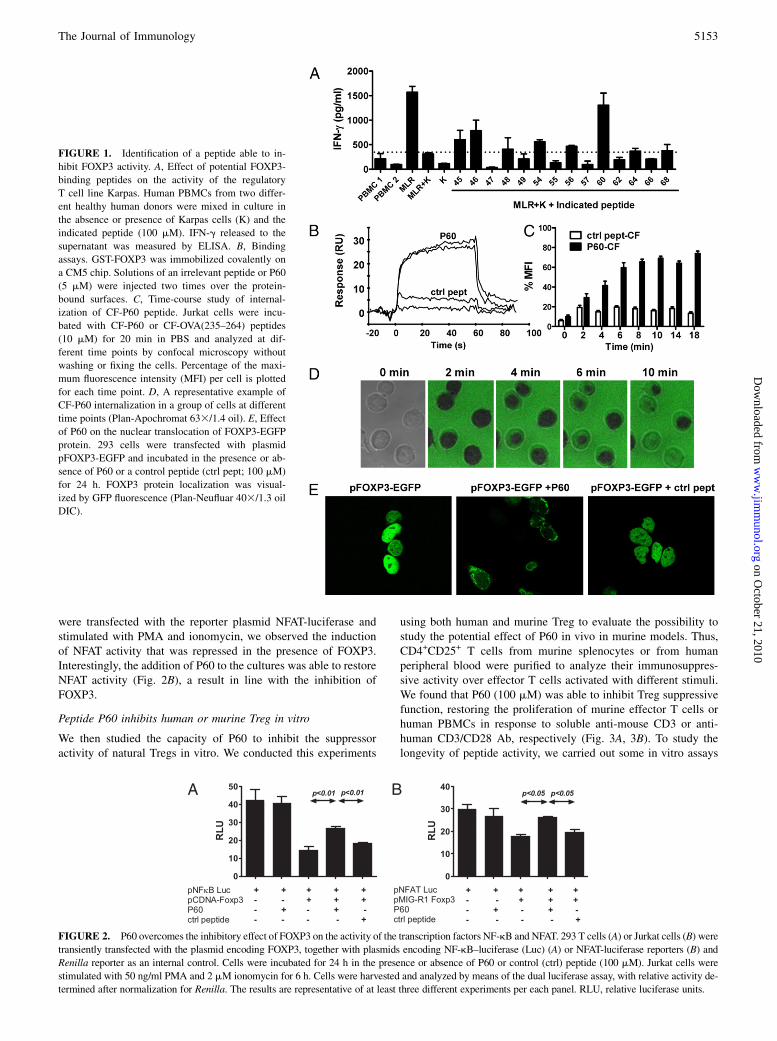
were transfected with the reporter plasmid NFAT-luciferase andstimulated with PMA and ionomycin, we observed the inductionof NFAT activity that was repressed in the presence of FOXP3.Interestingly, the addition of P60 to the cultures was able to restoreNFAT activity (Fig. 2B), a result in line with the inhibition ofFOXP3.
Peptide P60 inhibits human or murine Treg in vitro
We then studied the capacity of P60 to inhibit the suppressoractivity of natural Tregs in vitro. We conducted this experiments
using both human and murine Treg to evaluate the possibility tostudy the potential effect of P60 in vivo in murine models. Thus,CD4+CD25+ T cells from murine splenocytes or from humanperipheral blood were purified to analyze their immunosuppres-sive activity over effector T cells activated with different stimuli.We found that P60 (100 mM) was able to inhibit Treg suppressivefunction, restoring the proliferation of murine effector T cells orhuman PBMCs in response to soluble anti-mouse CD3 or anti-human CD3/CD28 Ab, respectively (Fig. 3A, 3B). To study thelongevity of peptide activity, we carried out some in vitro assays
FIGURE 1. Identification of a peptide able to in-
hibit FOXP3 activity. A, Effect of potential FOXP3-
binding peptides on the activity of the regulatory
T cell line Karpas. Human PBMCs from two differ-
ent healthy human donors were mixed in culture in
the absence or presence of Karpas cells (K) and the
indicated peptide (100 mM). IFN-g released to the
supernatant was measured by ELISA. B, Binding
assays. GST-FOXP3 was immobilized covalently on
a CM5 chip. Solutions of an irrelevant peptide or P60
(5 mM) were injected two times over the protein-
bound surfaces. C, Time-course study of internal-
ization of CF-P60 peptide. Jurkat cells were incu-
bated with CF-P60 or CF-OVA(235–264) peptides
(10 mM) for 20 min in PBS and analyzed at dif-
ferent time points by confocal microscopy without
washing or fixing the cells. Percentage of the maxi-
mum fluorescence intensity (MFI) per cell is plotted
for each time point. D, A representative example of
CF-P60 internalization in a group of cells at different
time points (Plan-Apochromat 633/1.4 oil). E, Effect
of P60 on the nuclear translocation of FOXP3-EGFP
protein. 293 cells were transfected with plasmid
pFOXP3-EGFP and incubated in the presence or ab-
sence of P60 or a control peptide (ctrl pept; 100 mM)
for 24 h. FOXP3 protein localization was visual-
ized by GFP fluorescence (Plan-Neufluar 403/1.3 oil
DIC).
FIGURE 2. P60 overcomes the inhibitory effect of FOXP3 on the activity of the transcription factors NF-kB and NFAT. 293 T cells (A) or Jurkat cells (B) were
transiently transfected with the plasmid encoding FOXP3, together with plasmids encoding NF-kB–luciferase (Luc) (A) or NFAT-luciferase reporters (B) and
Renilla reporter as an internal control. Cells were incubated for 24 h in the presence or absence of P60 or control (ctrl) peptide (100 mM). Jurkat cells were
stimulated with 50 ng/ml PMA and 2 mM ionomycin for 6 h. Cells were harvested and analyzed by means of the dual luciferase assay, with relative activity de-
termined after normalization for Renilla. The results are representative of at least three different experiments per each panel. RLU, relative luciferase units.
The Journal of Immunology 5153
on October 21, 2010
ww
w.jim
munol.org
Dow
nloaded from
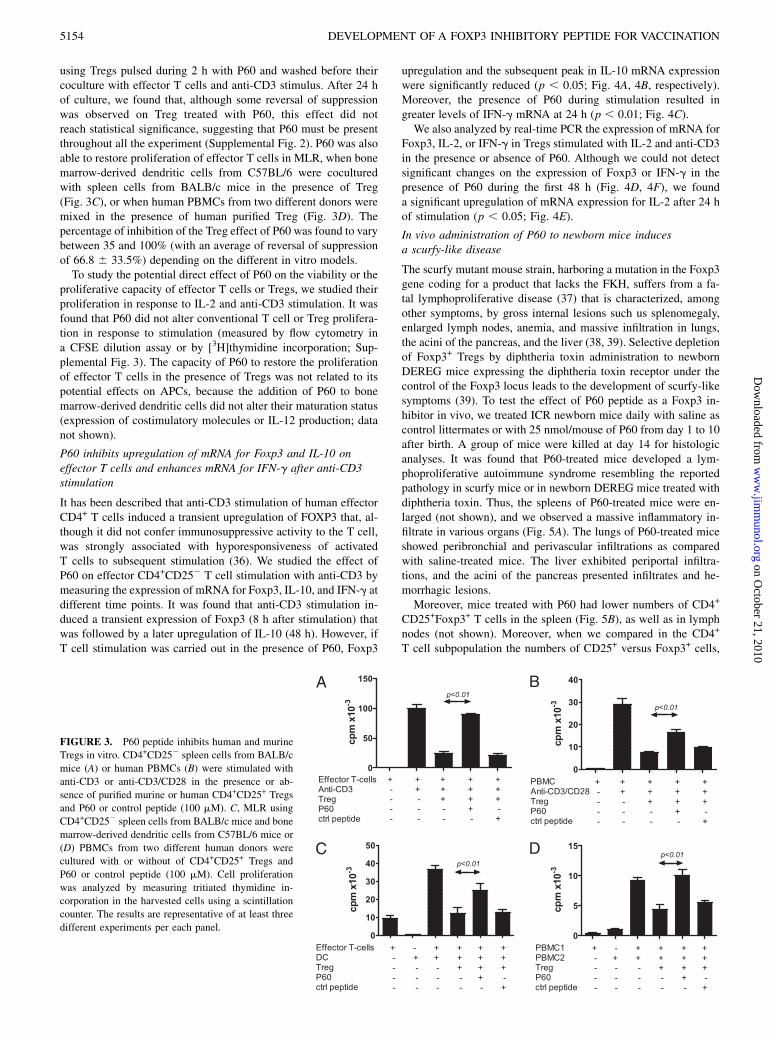
using Tregs pulsed during 2 h with P60 and washed before theircoculture with effector T cells and anti-CD3 stimulus. After 24 hof culture, we found that, although some reversal of suppressionwas observed on Treg treated with P60, this effect did notreach statistical significance, suggesting that P60 must be presentthroughout all the experiment (Supplemental Fig. 2). P60 was alsoable to restore proliferation of effector T cells in MLR, when bonemarrow-derived dendritic cells from C57BL/6 were coculturedwith spleen cells from BALB/c mice in the presence of Treg(Fig. 3C), or when human PBMCs from two different donors weremixed in the presence of human purified Treg (Fig. 3D). Thepercentage of inhibition of the Treg effect of P60 was found to varybetween 35 and 100% (with an average of reversal of suppressionof 66.8 6 33.5%) depending on the different in vitro models.To study the potential direct effect of P60 on the viability or the
proliferative capacity of effector T cells or Tregs, we studied theirproliferation in response to IL-2 and anti-CD3 stimulation. It wasfound that P60 did not alter conventional T cell or Treg prolifera-tion in response to stimulation (measured by flow cytometry ina CFSE dilution assay or by [3H]thymidine incorporation; Sup-plemental Fig. 3). The capacity of P60 to restore the proliferationof effector T cells in the presence of Tregs was not related to itspotential effects on APCs, because the addition of P60 to bonemarrow-derived dendritic cells did not alter their maturation status(expression of costimulatory molecules or IL-12 production; datanot shown).
P60 inhibits upregulation of mRNA for Foxp3 and IL-10 oneffector T cells and enhances mRNA for IFN-g after anti-CD3stimulation
It has been described that anti-CD3 stimulation of human effectorCD4+ T cells induced a transient upregulation of FOXP3 that, al-though it did not confer immunosuppressive activity to the T cell,was strongly associated with hyporesponsiveness of activatedT cells to subsequent stimulation (36). We studied the effect ofP60 on effector CD4+CD252 T cell stimulation with anti-CD3 bymeasuring the expression of mRNA for Foxp3, IL-10, and IFN-g atdifferent time points. It was found that anti-CD3 stimulation in-duced a transient expression of Foxp3 (8 h after stimulation) thatwas followed by a later upregulation of IL-10 (48 h). However, ifT cell stimulation was carried out in the presence of P60, Foxp3
upregulation and the subsequent peak in IL-10 mRNA expressionwere significantly reduced (p , 0.05; Fig. 4A, 4B, respectively).Moreover, the presence of P60 during stimulation resulted ingreater levels of IFN-g mRNA at 24 h (p , 0.01; Fig. 4C).We also analyzed by real-time PCR the expression of mRNA for
Foxp3, IL-2, or IFN-g in Tregs stimulated with IL-2 and anti-CD3in the presence or absence of P60. Although we could not detectsignificant changes on the expression of Foxp3 or IFN-g in thepresence of P60 during the first 48 h (Fig. 4D, 4F), we founda significant upregulation of mRNA expression for IL-2 after 24 hof stimulation (p , 0.05; Fig. 4E).
In vivo administration of P60 to newborn mice inducesa scurfy-like disease
The scurfy mutant mouse strain, harboring a mutation in the Foxp3gene coding for a product that lacks the FKH, suffers from a fa-tal lymphoproliferative disease (37) that is characterized, amongother symptoms, by gross internal lesions such us splenomegaly,enlarged lymph nodes, anemia, and massive infiltration in lungs,the acini of the pancreas, and the liver (38, 39). Selective depletionof Foxp3+ Tregs by diphtheria toxin administration to newbornDEREG mice expressing the diphtheria toxin receptor under thecontrol of the Foxp3 locus leads to the development of scurfy-likesymptoms (39). To test the effect of P60 peptide as a Foxp3 in-hibitor in vivo, we treated ICR newborn mice daily with saline ascontrol littermates or with 25 nmol/mouse of P60 from day 1 to 10after birth. A group of mice were killed at day 14 for histologicanalyses. It was found that P60-treated mice developed a lym-phoproliferative autoimmune syndrome resembling the reportedpathology in scurfy mice or in newborn DEREG mice treated withdiphtheria toxin. Thus, the spleens of P60-treated mice were en-larged (not shown), and we observed a massive inflammatory in-filtrate in various organs (Fig. 5A). The lungs of P60-treated miceshowed peribronchial and perivascular infiltrations as comparedwith saline-treated mice. The liver exhibited periportal infiltra-tions, and the acini of the pancreas presented infiltrates and he-morrhagic lesions.Moreover, mice treated with P60 had lower numbers of CD4+
CD25+Foxp3+ T cells in the spleen (Fig. 5B), as well as in lymphnodes (not shown). Moreover, when we compared in the CD4+
T cell subpopulation the numbers of CD25+ versus Foxp3+ cells,
FIGURE 3. P60 peptide inhibits human and murine
Tregs in vitro. CD4+CD252 spleen cells from BALB/c
mice (A) or human PBMCs (B) were stimulated with
anti-CD3 or anti-CD3/CD28 in the presence or ab-
sence of purified murine or human CD4+CD25+ Tregs
and P60 or control peptide (100 mM). C, MLR using
CD4+CD252 spleen cells from BALB/c mice and bone
marrow-derived dendritic cells from C57BL/6 mice or
(D) PBMCs from two different human donors were
cultured with or without of CD4+CD25+ Tregs and
P60 or control peptide (100 mM). Cell proliferation
was analyzed by measuring tritiated thymidine in-
corporation in the harvested cells using a scintillation
counter. The results are representative of at least three
different experiments per each panel.
5154 DEVELOPMENT OF A FOXP3 INHIBITORY PEPTIDE FOR VACCINATION
on October 21, 2010
ww
w.jim
munol.org
Dow
nloaded from
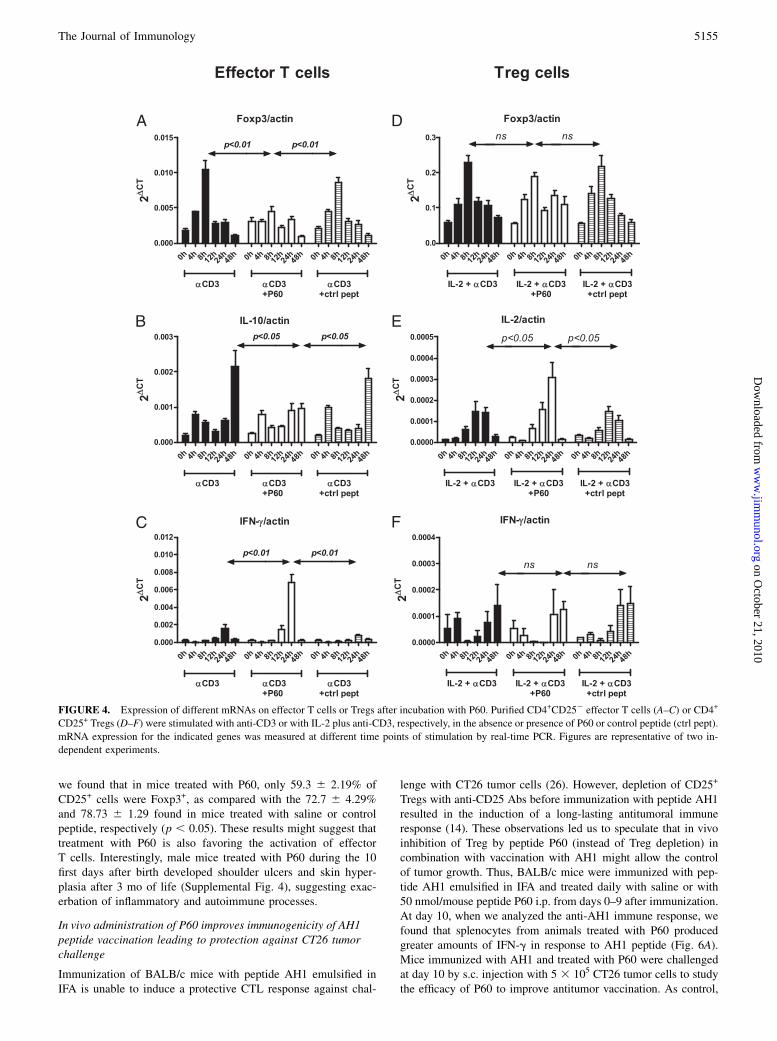
we found that in mice treated with P60, only 59.3 6 2.19% ofCD25+ cells were Foxp3+, as compared with the 72.7 6 4.29%and 78.73 6 1.29 found in mice treated with saline or controlpeptide, respectively (p , 0.05). These results might suggest thattreatment with P60 is also favoring the activation of effectorT cells. Interestingly, male mice treated with P60 during the 10first days after birth developed shoulder ulcers and skin hyper-plasia after 3 mo of life (Supplemental Fig. 4), suggesting exac-erbation of inflammatory and autoimmune processes.
In vivo administration of P60 improves immunogenicity of AH1peptide vaccination leading to protection against CT26 tumorchallenge
Immunization of BALB/c mice with peptide AH1 emulsified inIFA is unable to induce a protective CTL response against chal-
lenge with CT26 tumor cells (26). However, depletion of CD25+
Tregs with anti-CD25 Abs before immunization with peptide AH1resulted in the induction of a long-lasting antitumoral immuneresponse (14). These observations led us to speculate that in vivoinhibition of Treg by peptide P60 (instead of Treg depletion) incombination with vaccination with AH1 might allow the controlof tumor growth. Thus, BALB/c mice were immunized with pep-tide AH1 emulsified in IFA and treated daily with saline or with50 nmol/mouse peptide P60 i.p. from days 0–9 after immunization.At day 10, when we analyzed the anti-AH1 immune response, wefound that splenocytes from animals treated with P60 producedgreater amounts of IFN-g in response to AH1 peptide (Fig. 6A).Mice immunized with AH1 and treated with P60 were challengedat day 10 by s.c. injection with 5 3 105 CT26 tumor cells to studythe efficacy of P60 to improve antitumor vaccination. As control,
FIGURE 4. Expression of different mRNAs on effector T cells or Tregs after incubation with P60. Purified CD4+CD252 effector T cells (A–C) or CD4+
CD25+ Tregs (D–F) were stimulated with anti-CD3 or with IL-2 plus anti-CD3, respectively, in the absence or presence of P60 or control peptide (ctrl pept).
mRNA expression for the indicated genes was measured at different time points of stimulation by real-time PCR. Figures are representative of two in-
dependent experiments.
The Journal of Immunology 5155
on October 21, 2010
ww
w.jim
munol.org
Dow
nloaded from
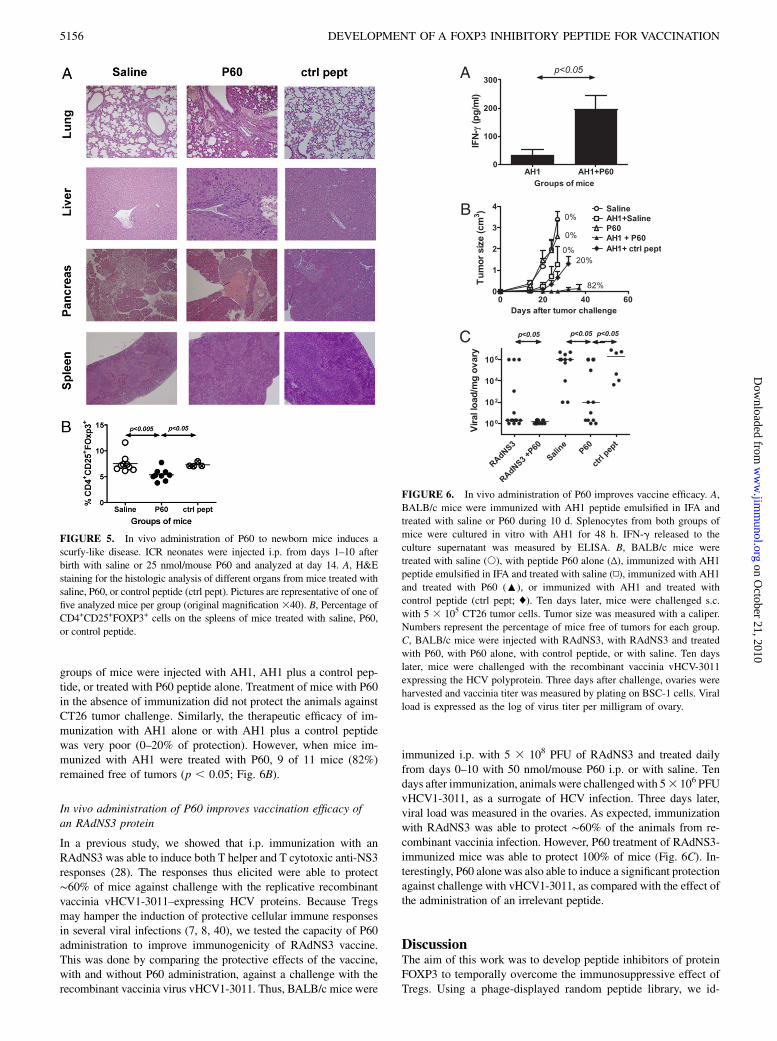
groups of mice were injected with AH1, AH1 plus a control pep-tide, or treated with P60 peptide alone. Treatment of mice with P60in the absence of immunization did not protect the animals againstCT26 tumor challenge. Similarly, the therapeutic efficacy of im-munization with AH1 alone or with AH1 plus a control peptidewas very poor (0–20% of protection). However, when mice im-munized with AH1 were treated with P60, 9 of 11 mice (82%)remained free of tumors (p , 0.05; Fig. 6B).
In vivo administration of P60 improves vaccination efficacy ofan RAdNS3 protein
In a previous study, we showed that i.p. immunization with anRAdNS3 was able to induce both T helper and T cytotoxic anti-NS3responses (28). The responses thus elicited were able to protect∼60% of mice against challenge with the replicative recombinantvaccinia vHCV1-3011–expressing HCV proteins. Because Tregsmay hamper the induction of protective cellular immune responsesin several viral infections (7, 8, 40), we tested the capacity of P60administration to improve immunogenicity of RAdNS3 vaccine.This was done by comparing the protective effects of the vaccine,with and without P60 administration, against a challenge with therecombinant vaccinia virus vHCV1-3011. Thus, BALB/c mice were
immunized i.p. with 5 3 108 PFU of RAdNS3 and treated dailyfrom days 0–10 with 50 nmol/mouse P60 i.p. or with saline. Tendays after immunization, animals were challengedwith 53 106 PFUvHCV1-3011, as a surrogate of HCV infection. Three days later,viral load was measured in the ovaries. As expected, immunizationwith RAdNS3 was able to protect ∼60% of the animals from re-combinant vaccinia infection. However, P60 treatment of RAdNS3-immunized mice was able to protect 100% of mice (Fig. 6C). In-terestingly, P60 alonewas also able to induce a significant protectionagainst challenge with vHCV1-3011, as compared with the effect ofthe administration of an irrelevant peptide.
DiscussionThe aim of this work was to develop peptide inhibitors of proteinFOXP3 to temporally overcome the immunosuppressive effect ofTregs. Using a phage-displayed random peptide library, we id-
FIGURE 5. In vivo administration of P60 to newborn mice induces a
scurfy-like disease. ICR neonates were injected i.p. from days 1–10 after
birth with saline or 25 nmol/mouse P60 and analyzed at day 14. A, H&E
staining for the histologic analysis of different organs from mice treated with
saline, P60, or control peptide (ctrl pept). Pictures are representative of one of
five analyzed mice per group (original magnification 340). B, Percentage of
CD4+CD25+FOXP3+ cells on the spleens of mice treated with saline, P60,
or control peptide.
FIGURE 6. In vivo administration of P60 improves vaccine efficacy. A,
BALB/c mice were immunized with AH1 peptide emulsified in IFA and
treated with saline or P60 during 10 d. Splenocytes from both groups of
mice were cultured in vitro with AH1 for 48 h. IFN-g released to the
culture supernatant was measured by ELISA. B, BALB/c mice were
treated with saline (s), with peptide P60 alone (Δ), immunized with AH1
peptide emulsified in IFA and treated with saline (N), immunized with AH1
and treated with P60 (:), or immunized with AH1 and treated with
control peptide (ctrl pept; ♦). Ten days later, mice were challenged s.c.
with 5 3 105 CT26 tumor cells. Tumor size was measured with a caliper.
Numbers represent the percentage of mice free of tumors for each group.
C, BALB/c mice were injected with RAdNS3, with RAdNS3 and treated
with P60, with P60 alone, with control peptide, or with saline. Ten days
later, mice were challenged with the recombinant vaccinia vHCV-3011
expressing the HCV polyprotein. Three days after challenge, ovaries were
harvested and vaccinia titer was measured by plating on BSC-1 cells. Viral
load is expressed as the log of virus titer per milligram of ovary.
5156 DEVELOPMENT OF A FOXP3 INHIBITORY PEPTIDE FOR VACCINATION
on October 21, 2010
ww
w.jim
munol.org
Dow
nloaded from

entified a number of potential binders to FOXP3. Among them, P60was found to be able to bind to FOXP3 and inhibit Treg activityin vitro and in vivo.Because the activity of FOXP3 takes place inside the cell, we
speculated that P60 was capable of entering into the cell. Indeed,experiments conducted with CF-labeled peptide suggest that P60 isable to enter the cell as a CPP. Although the exact mechanismof cellular entry of these peptides is still elusive, P60 fulfils thesequence requirements described for CPPs (reviewed in Ref. 33).Indeed, P60 has cationic character and contains arginines, as wellas tryptophan, and it is also predicted as an amphipathic helix (41).Most importantly, P60 is able to reduce FOXP3 nuclear trans-location and consequently to inhibit Treg activity. This is shown byits capacity to restore proliferation of human or murine effectorT cells, in response to anti-CD3 in the presence of inhibitory Tregs,and also by its ability to enhance human or murine T cell pro-liferation in an MLR inhibited by Treg.FOXP3 exists as an homo-oligomeric component of a large
supramolecular complex that may contain different proteins andtranscription factors, such as BRG-1, HDAC7, TIP60, NFATc2,NF-kB, FOXP1, retinoic acid-related orphan receptor (ROR)-a,ROR-gt, MEF2D, or AML1 (35, 42–46). It is clear that interactionsbetween these proteins and FOXP3 are crucial for the final functionof FOXP3, and thus disruption of one or more of these interactionsmight impair the potential effects of this supramolecular complex.We found that P60 reduced the inhibitory effect of FOXP3 onNF-kB and NFAT activity, suggesting that the binding of P60 toFOXP3 might take place in a region affecting the interaction withthese transcription factors. These data are consistent with the ca-pacity of P60 to upregulate the expression of mRNA for IL-2 onTregs, which has been shown to be repressed by the interactionbetween NFAT and FOXP3 (47). We could not detect any effect ofP60 on the capacity of FOXP3 to inhibit the transcriptional ac-tivation of the ROR-a (44) (data not shown). It has been describedthat the region encoded by exon 2 from FOXP3 is involved in theinteraction with ROR-a (44). Thus, it is likely that differentregions of FOXP3 may be involved in the interaction with thedifferent transcription factors, and consequently, P60 could onlybe able to inhibit some of them. The capacity of P60 to inhibitFOXP3 nuclear translocation might suggest that the peptide bindsto FKH domain or to a region affecting its ability to target FOXP3to the nucleus. However, we do not have direct evidence to sup-port this hypothesis. We are currently attempting to determine theregion of interaction between P60 and FOXP3.It has been described that activation of human naive T cells
through TCR cross-linking can activate FOXP3 expression (re-viewed in Ref. 48). In most cases, this expression is transient, andalthough it does not confer immunosuppressive activity to theT cell, it is strongly associated with hyporesponsiveness of acti-vated T cells to subsequent stimulation (36). We identified a tran-sient expression of Foxp3 soon after stimulation with anti-CD3,which was accompanied by a peak on the expression of mRNA forIL-10. Thus, P60 could have a direct effect on conventional T cellsafter their activation. Indeed, the addition of P60 reduced thisFoxp3 expression and the subsequent peak of IL-10. Moreover,P60 increased IFN-g induced by anti-CD3, suggesting that P60could favor T cell activation after TCR stimulation by inhibitingFoxp3 expression. The regulation of FOXP3 transcription is acomplex process, and there is considerable uncertainty on themultiple signal pathways implicated (reviewed in Ref. 49). Thecharacterization of the core promoter of human FOXP3 revealedthe presence of six NFAT and AP-1 binding sites, which are posi-tively regulating the transactivation of the FOXP3 promoter aftertriggering of the TCR (50). Because FOXP3 can cooperate in a
DNA-binding complex with NFAT to regulate the transcription of
target genes (47), it is tempting to postulate that FOXP3 might also
be implicated in its own transcription, and thus explain the effect of
P60 at this level.The scurfy mutant mouse strain, harboring a mutation in the
Foxp3 gene coding for a product that lacks the FKH, suffers from
a fatal lymphoproliferative disease (37). Lack of functional Tregs
has been suggested to cause the disease in scurfy mice, which is
characterized, among other symptoms, by gross internal lesions
such us splenomegaly, enlarged lymph nodes, anemia, and mas-
sive infiltration in lungs, the acini of the pancreas and the liver (38,
39). It has recently been shown that selective depletion of Foxp3+
Tregs by diphtheria toxin administration to newborn DEREG mice
(expressing the diphtheria toxin receptor under the control of the
Foxp3 locus) leads to the development of scurfy-like symptoms
(39). We have found that newborn ICR mice treated daily with
P60 during 10 d after birth develop a lymphoproliferative auto-
immune syndrome, resembling the reported pathology in scurfy
mice or in newborn DEREG mice treated with diphtheria toxin.
Treatment with P60 was also able to reduce the number of CD4+
CD25+Foxp3+ T cells in the spleen, suggesting that inhibition of
FOXP3 might have an effect in the homeostasis of Tregs. P60
administration was limited to the first 10 d after birth. This treat-
ment did not lead to an early death (15–25 d) as occurred in the
scurfy mutant mice (38). However, survival of male mice was
highly compromised, with ∼50% of deaths 3 mo after birth. It is
interesting to note that all male mice treated with P60 developed
shoulder ulcers after 3 mo of life, with skin hyperplasia (Supple-
mental Fig. 4) and persisting peribronchial and perivascular infil-
trations in the lungs, indicating the presence of an unremitting and
uncontrolled inflammatory syndrome. But interestingly, in adult
mice, neither Treg depletion with anti-CD25 Abs (15), depletion of
total Foxp3+ Tregs by diphtheria toxin treatment in DEREG mice
(39), nor treatment of mice with P60 peptide resulted in the de-
velopment of inflammatory autoimmune disorders (Supplemental
Fig. 5). As it has been suggested by others, this finding could be
explained by the lack of lymphopenia-induced proliferation,
a physiologic process that regularly occurs during early postnatal
life (51), preventing the activation of autoreactive T cells (52).Downregulation of Treg suppressor activity in vivo in adults may
be beneficial to enhance the immunogenicity of a vaccine (14). In a
previous work, we showed that in vivo CD25+ T cell depletion
improved immunogenicity of AH1 peptide vaccination and pro-
tected mice against CT26 tumor challenge (14). In this work, we
have found that in vivo administration of P60, instead of Treg de-
pletion, was also able to protect mice from tumor challenge. Simi-
larly, in vivo administration of P60 improved the efficacy of an
RAdNS3-based vaccine against infection with a recombinant rep-
licative vaccinia vHCV1-3011 expressing the HCV polyprotein.
These results are in agreement with previous reports showing an
enhancement of immunogenicity of a vaccine by Treg depletion
(14, 16). However, P60 administration in vivo might allow a finer
control on the intensity and duration of Treg suppression, in par-
ticular after vaccination, avoiding the risk for autoimmunity asso-
ciated with depletion of Treg.In summary, we have identified peptide P60, which is able to enter
into the cells, bind to FOXP3, reduce its nuclear translocation, and
inhibit its ability to downregulate the activity of the transcription
factors NF-kB and NFAT. P60 is able to inhibit the immunosup-
pressive activity of murine and human-derived Tregs and enhances
the effector T cell stimulation in vitro. Also, and most importantly,
we show that P60 can improve the immunogenicity of cancer and
viral vaccines.
The Journal of Immunology 5157
on October 21, 2010
ww
w.jim
munol.org
Dow
nloaded from

AcknowledgmentsWe thank Elena Ciordia and Eneko Elizalde for excellent animal care, Vir-
ginia Belsue for peptide synthesis, Marta Martınez and Dr. L. Manterola
for technical assistance, Dr. Jose L. Romero for assistance in the histologic
analyses, and the Servicio de Morfologıa e Imagen (CIMA) for assistance
in confocal microscopy.
DisclosuresThe authors have no financial conflicts of interest.
References1. Wan, Y. Y., and R. A. Flavell. 2007. Regulatory T-cell functions are subverted
and converted owing to attenuated Foxp3 expression. Nature 445: 766–770.2. Williams, L. M., and A. Y. Rudensky. 2007. Maintenance of the Foxp3-
dependent developmental program in mature regulatory T cells requires con-tinued expression of Foxp3. Nat. Immunol. 8: 277–284.
3. Shevach, E. M. 2001. Certified professionals: CD4(+)CD25(+) suppressorT cells. J. Exp. Med. 193: F41–F46.
4. Bennett, C. L., J. Christie, F. Ramsdell, M. E. Brunkow, P. J. Ferguson,L. Whitesell, T. E. Kelly, F. T. Saulsbury, P. F. Chance, and H. D. Ochs. 2001.The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syn-drome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 27: 20–21.
5. Chatila, T. A., F. Blaeser, N. Ho, H. M. Lederman, C. Voulgaropoulos, C. Helms,and A. M. Bowcock. 2000. JM2, encoding a fork head-related protein, is mutatedin X-linked autoimmunity-allergic disregulation syndrome. J. Clin. Invest. 106:R75–R81.
6. Wildin, R. S., F. Ramsdell, J. Peake, F. Faravelli, J. L. Casanova, N. Buist,E. Levy-Lahad, M. Mazzella, O. Goulet, L. Perroni, et al. 2001. X-linked neo-natal diabetes mellitus, enteropathy and endocrinopathy syndrome is the humanequivalent of mouse scurfy. Nat. Genet. 27: 18–20.
7. Aandahl, E. M., J. Michaelsson, W. J. Moretto, F. M. Hecht, and D. F. Nixon.2004. Human CD4+ CD25+ regulatory T cells control T-cell responses to humanimmunodeficiency virus and cytomegalovirus antigens. J. Virol. 78: 2454–2459.
8. Cabrera, R., Z. Tu, Y. Xu, R. J. Firpi, H. R. Rosen, C. Liu, and D. R. Nelson. 2004.An immunomodulatory role for CD4(+)CD25(+) regulatory T lymphocytes inhepatitis C virus infection. Hepatology 40: 1062–1071.
9. Viguier, M., F. Lemaıtre, O. Verola, M. S. Cho, G. Gorochov, L. Dubertret,H. Bachelez, P. Kourilsky, and L. Ferradini. 2004. Foxp3 expressing CD4+CD25(high) regulatory T cells are overrepresented in humanmetastatic melanoma lymphnodes and inhibit the function of infiltrating T cells. J. Immunol. 173: 1444–1453.
10. Woo, E. Y., C. S. Chu, T. J. Goletz, K. Schlienger, H. Yeh, G. Coukos,S. C. Rubin, L. R. Kaiser, and C. H. June. 2001. Regulatory CD4(+)CD25(+)T cells in tumors from patients with early-stage non-small cell lung cancer andlate-stage ovarian cancer. Cancer Res. 61: 4766–4772.
11. Curiel, T. J., G. Coukos, L. Zou, X. Alvarez, P. Cheng, P. Mottram, M. Evdemon-Hogan, J. R. Conejo-Garcia, L. Zhang, M. Burow, et al. 2004. Specific re-cruitment of regulatory T cells in ovarian carcinoma fosters immune privilegeand predicts reduced survival. Nat. Med. 10: 942–949.
12. Liyanage, U. K., T. T. Moore, H. G. Joo, Y. Tanaka, V. Herrmann, G. Doherty,J. A. Drebin, S. M. Strasberg, T. J. Eberlein, P. S. Goedegebuure, andD. C. Linehan. 2002. Prevalence of regulatory T cells is increased in peripheralblood and tumor microenvironment of patients with pancreas or breast adeno-carcinoma. J. Immunol. 169: 2756–2761.
13. Ormandy, L. A., T. Hillemann, H. Wedemeyer, M. P. Manns, T. F. Greten, andF. Korangy. 2005. Increased populations of regulatory T cells in peripheral bloodof patients with hepatocellular carcinoma. Cancer Res. 65: 2457–2464.
14. Casares, N., L. Arribillaga, P. Sarobe, J. Dotor, A. Lopez-Diaz de Cerio,I. Melero, J. Prieto, F. Borras-Cuesta, and J. J. Lasarte. 2003. CD4+/CD25+regulatory cells inhibit activation of tumor-primed CD4+ T cells with IFN-gamma-dependent antiangiogenic activity, as well as long-lasting tumor im-munity elicited by peptide vaccination. J. Immunol. 171: 5931–5939.
15. Onizuka, S., I. Tawara, J. Shimizu, S. Sakaguchi, T. Fujita, and E. Nakayama.1999. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2receptor alpha) monoclonal antibody. Cancer Res. 59: 3128–3133.
16. Shimizu, J., S. Yamazaki, and S. Sakaguchi. 1999. Induction of tumor immunityby removing CD25+CD4+ T cells: a common basis between tumor immunityand autoimmunity. J. Immunol. 163: 5211–5218.
17. Colombo, M. P., and S. Piconese. 2007. Regulatory-T-cell inhibition versus de-pletion: the right choice in cancer immunotherapy. Nat. Rev. Cancer 7: 880–887.
18. Berraondo, P., C. Nouze, X. Preville, D. Ladant, and C. Leclerc. 2007. Eradi-cation of large tumors in mice by a tritherapy targeting the innate, adaptive, andregulatory components of the immune system. Cancer Res. 67: 8847–8855.
19. Ghiringhelli, F., C. Menard, P. E. Puig, S. Ladoire, S. Roux, F. Martin, E. Solary,A. Le Cesne, L. Zitvogel, and B. Chauffert. 2007. Metronomic cyclophospha-mide regimen selectively depletes CD4+CD25+ regulatory T cells and restoresT and NK effector functions in end stage cancer patients. Cancer Immunol.Immunother. 56: 641–648.
20. Hermans, I. F., T. W. Chong, M. J. Palmowski, A. L. Harris, and V. Cerundolo.2003. Synergistic effect of metronomic dosing of cyclophosphamide combinedwith specific antitumor immunotherapy in a murine melanoma model. CancerRes. 63: 8408–8413.
21. Attia, P., A. V. Maker, L. R. Haworth, L. Rogers-Freezer, and S. A. Rosenberg.2005. Inability of a fusion protein of IL-2 and diphtheria toxin (Denileukin
Diftitox, DAB389IL-2, ONTAK) to eliminate regulatory T lymphocytes inpatients with melanoma. J. Immunother. 28: 582–592.
22. Dannull, J., Z. Su, D. Rizzieri, B. K. Yang, D. Coleman, D. Yancey, A. Zhang,P. Dahm, N. Chao, E. Gilboa, and J. Vieweg. 2005. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatoryT cells. J. Clin. Invest. 115: 3623–3633.
23. Ruddle, J. B., C. A. Harper, D. Honemann, J. F. Seymour, and H. M. Prince.2006. A denileukin diftitox (Ontak) associated retinopathy? Br. J. Ophthalmol.90: 1070–1071.
24. Dotor, J., A. B. Lopez-Vazquez, J. J. Lasarte, P. Sarobe, M. Garcıa-Granero,J. I. Riezu-Boj, A. Martınez, E. Feijoo, J. Lopez-Sagaseta, J. Hermida, et al.2007. Identification of peptide inhibitors of transforming growth factor beta 1using a phage-displayed peptide library. Cytokine 39: 106–115.
25. Borras-Cuesta, F., J. Golvano, P. Sarobe, J. J. Lasarte, I. Prieto, A. Szabo,J. L. Guillaume, and J. G. Guillet. 1991. Insights on the amino acid side-chaininteractions of a synthetic T-cell determinant. Biologicals 19: 187–190.
26. Huang, A. Y., P. H. Gulden, A. S. Woods, M. C. Thomas, C. D. Tong, W. Wang,V. H. Engelhard, G. Pasternack, R. Cotter, D. Hunt, et al. 1996. The immuno-dominant major histocompatibility complex class I-restricted antigen of a murinecolon tumor derives from an endogenous retroviral gene product. Proc. Natl.Acad. Sci. USA 93: 9730–9735.
27. Wolke, C., J. Tadje, A. Bukowska, M. Tager, U. Bank, A. Ittenson, S. Ansorge,and U. Lendeckel. 2006. Assigning the phenotype of a natural regulatory T-cellto the human T-cell line, KARPAS-299. Int. J. Mol. Med. 17: 275–278.
28. Arribillaga, L., A. L. de Cerio, P. Sarobe, N. Casares, M. Gorraiz, A. Vales,O. Bruna-Romero, F. Borras-Cuesta, G. Paranhos-Baccala, J. Prieto, et al. 2002.Vaccination with an adenoviral vector encoding hepatitis C virus (HCV) NS3protein protects against infection with HCV-recombinant vaccinia virus. Vaccine21: 202–210.
29. Larrea, E., J. I. Riezu-Boj, L. Gil-Guerrero, N. Casares, R. Aldabe, P. Sarobe,M. P. Civeira, J. L. Heeney, C. Rollier, B. Verstrepen, et al. 2007. Upregulationof indoleamine 2,3-dioxygenase in hepatitis C virus infection. J. Virol. 81: 3662–3666.
30. Zabala, M., J. J. Lasarte, C. Perret, J. Sola, P. Berraondo, M. Alfaro, E. Larrea,J. Prieto, and M. G. Kramer. 2007. Induction of immunosuppressive moleculesand regulatory T cells counteracts the antitumor effect of interleukin-12-basedgene therapy in a transgenic mouse model of liver cancer. J. Hepatol. 47: 807–815.
31. Sarobe, P., J. J. Lasarte, A. Zabaleta, L. Arribillaga, A. Arina, I. Melero,F. Borras-Cuesta, and J. Prieto. 2003. Hepatitis C virus structural proteins impairdendritic cell maturation and inhibit in vivo induction of cellular immuneresponses. J. Virol. 77: 10862–10871.
32. Gil-Guerrero, L., J. Dotor, I. L. Huibregtse, N. Casares, A. B. Lopez-Vazquez,F. Rudilla, J. I. Riezu-Boj, J. Lopez-Sagaseta, J. Hermida, S. Van Deventer, et al.2008. In vitro and in vivo down-regulation of regulatory T cell activity witha peptide inhibitor of TGF-beta1. J. Immunol. 181: 126–135.
33. Patel, L. N., J. L. Zaro, and W. C. Shen. 2007. Cell penetrating peptides: in-tracellular pathways and pharmaceutical perspectives. Pharm. Res. 24: 1977–1992.
34. Lopes, J. E., T. R. Torgerson, L. A. Schubert, S. D. Anover, E. L. Ocheltree,H. D. Ochs, and S. F. Ziegler. 2006. Analysis of FOXP3 reveals multipledomains required for its function as a transcriptional repressor. J. Immunol. 177:3133–3142.
35. Bettelli, E., M. Dastrange, and M. Oukka. 2005. Foxp3 interacts with nuclearfactor of activated T cells and NF-kappa B to repress cytokine gene expressionand effector functions of T helper cells. Proc. Natl. Acad. Sci. USA 102: 5138–5143.
36. Wang, J., A. Ioan-Facsinay, E. I. van der Voort, T. W. Huizinga, and R. E. Toes.2007. Transient expression of FOXP3 in human activated nonregulatory CD4+T cells. Eur. J. Immunol. 37: 129–138.
37. Brunkow, M. E., E. W. Jeffery, K. A. Hjerrild, B. Paeper, L. B. Clark,S. A. Yasayko, J. E. Wilkinson, D. Galas, S. F. Ziegler, and F. Ramsdell. 2001.Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatallymphoproliferative disorder of the scurfy mouse. Nat. Genet. 27: 68–73.
38. Godfrey, V. L., J. E. Wilkinson, and L. B. Russell. 1991. X-linked lymphor-eticular disease in the scurfy (sf) mutant mouse. Am. J. Pathol. 138: 1379–1387.
39. Lahl, K., C. Loddenkemper, C. Drouin, J. Freyer, J. Arnason, G. Eberl,A. Hamann, H. Wagner, J. Huehn, and T. Sparwasser. 2007. Selective depletionof Foxp3+ regulatory T cells induces a scurfy-like disease. J. Exp. Med. 204: 57–63.
40. Boettler, T., H. C. Spangenberg, C. Neumann-Haefelin, E. Panther, S. Urbani,C. Ferrari, H. E. Blum, F. von Weizsacker, and R. Thimme. 2005. T cells witha CD4+CD25+ regulatory phenotype suppress in vitro proliferation of virus-specific CD8+ T cells during chronic hepatitis C virus infection. J. Virol. 79:7860–7867.
41. Scheller, A., J. Oehlke, B. Wiesner, M. Dathe, E. Krause, M. Beyermann,M. Melzig, and M. Bienert. 1999. Structural requirements for cellular uptake ofalpha-helical amphipathic peptides. J. Pept. Sci. 5: 185–194.
42. Li, B., A. Samanta, X. Song, K. T. Iacono, P. Brennan, T. A. Chatila,G. Roncador, A. H. Banham, J. L. Riley, Q. Wang, et al. 2007. FOXP3 is a homo-oligomer and a component of a supramolecular regulatory complex disabled inthe human XLAAD/IPEX autoimmune disease. Int. Immunol. 19: 825–835.
43. Ono, M., H. Yaguchi, N. Ohkura, I. Kitabayashi, Y. Nagamura, T. Nomura,Y. Miyachi, T. Tsukada, and S. Sakaguchi. 2007. Foxp3 controls regulatoryT-cell function by interacting with AML1/Runx1. Nature 446: 685–689.
5158 DEVELOPMENT OF A FOXP3 INHIBITORY PEPTIDE FOR VACCINATION
on October 21, 2010
ww
w.jim
munol.org
Dow
nloaded from

44. Du, J., C. Huang, B. Zhou, and S. F. Ziegler. 2008. Isoform-specific inhibition ofROR alpha-mediated transcriptional activation by human FOXP3. J. Immunol.180: 4785–4792.
45. Zhang, F., G. Meng, and W. Strober. 2008. Interactions among the transcriptionfactors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin17-producing T cells. Nat. Immunol. 9: 1297–1306.
46. Zhou, L., J. E. Lopes, M. M. Chong, I. I. Ivanov, R. Min, G. D. Victora, Y. Shen,J. Du, Y. P. Rubtsov, A. Y. Rudensky, et al. 2008. TGF-beta-induced Foxp3inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Na-ture 453: 236–240.
47. Wu, Y., M. Borde, V. Heissmeyer, M. Feuerer, A. D. Lapan, J. C. Stroud,D. L. Bates, L. Guo, A. Han, S. F. Ziegler, et al. 2006. FOXP3 controls regu-latory T cell function through cooperation with NFAT. Cell 126: 375–387.
48. Ziegler, S. F. 2007. FOXP3: not just for regulatory T cells anymore. Eur. J.Immunol. 37: 21–23.
49. Shen, Z., L. Chen, F. Hao, and J. Wu. 2009. Transcriptional regulation of Foxp3gene: multiple signal pathways on the road. Med. Res. Rev. 29: 742–766.
50. Mantel, P. Y., N. Ouaked, B. Ruckert, C. Karagiannidis, R. Welz, K. Blaser, andC. B. Schmidt-Weber. 2006. Molecular mechanisms underlying FOXP3 in-duction in human T cells. J. Immunol. 176: 3593–3602.
51. Min, B., R. McHugh, G. D. Sempowski, C. Mackall, G. Foucras, and W. E. Paul.2003. Neonates support lymphopenia-induced proliferation. Immunity 18: 131–140.
52. McHugh, R. S., and E. M. Shevach. 2002. Cutting edge: depletion of CD4+CD25+ regulatory T cells is necessary, but not sufficient, for induction of organ-specific autoimmune disease. J. Immunol. 168: 5979–5983.
The Journal of Immunology 5159
on October 21, 2010
ww
w.jim
munol.org
Dow
nloaded from




























