Embed Size (px)
Citation preview

GLIOMAS

INTRODUCCIÓN
Los tumores del cerebro siempre han sido una de las enfermedades más devastadoras, debido a que son muy difíciles de tratar y son poco curables.
Los tumores del cerebro puede ser primarios o metastásicos. Los gliomas son tumores cerebrales primarios que se originan en las células gliales, que brindan nutrientes, oxígeno y otro tipo de soporte a las neuronas.
Los gliomas malignos (que se caracterizan por un crecimiento progresivo y descontrolado) son los tumores cerebrales primarios más comunes, responsables de aproximadamente 10,000 tumores cerebrales primarios malignos diagnosticados por año en los Estados Unidos.
American Brain Tumor Association. Gliomas. JAMA 2010; 303 (10): 101 – 104

INTRODUCCIÓN
Los gliomas son tumores de origen glial o neuroepitelial. Con este término se agrupan los astrocitomas, los oligodendrogliomas y los ependimomas.
Los gliomas, que representan el 71% de los tumores intracraneales de los pacientes menores de 9 años, suponen el grupo de tumores sólidos con más prevalencia en este grupo de edad.
Idoate M, Echevaste J. Actualización sobre la biología molecular de los gliomas: hacia
una clasificación patomolecular de los gliomas. Rev Neurol 2007; 44 (4): 217 – 224

INTRODUCCIÓN
Los gliomas son el tipo de tumores más frecuentes entre las neoplasias Cerebrales. Pueden diferenciarse según su grado de malignidad, siguiendo la clasificación en tres grados de la Organización Mundial de la Salud, entre gliomas de bajo grado (grado II) y de alto grado de malignidad (grados III y IV). Ésta distinción es importante, porque el pronóstico de los gliomas de alto grado es mucho peor, como es lógico, y además, el tratamiento indicado es diferente. Para los gliomas de bajo grado basta con la cirugía, mientras que para los gliomas de alto grado es necesario además considerar la quimioterapia y la radioterapia, tratamientos que afectan en gran medida a la calidad de vida de los pacientes.
Lee S, Kim J, Park I, Sung J. Perfusion MR. Imaging in Gliomas: Comparison with
Histologic Tumor Grade. Korean Journal of Radiology 2001; 2 (1): 1 - 7

EPIDEMIOLOGÍA
La incidencia 7 -10/100,0000
habitantes.
2% De todas las neoplasias.
6º Tumores mas frecuentes en adultos.
2º Muerte entre los menores de 15 años.
Tasa de mortalidad 3.8-5.4/100,000
Más comunes Astrocitoma anaplásico y
Gioblastoma multiforme: De 100 000 Hab.
0-4 AÑOS
15-45 AÑOS
65-79 AÑOS
‘Classification of brain tumors’, David Schiff, MD; Uptodate; 2010.

Incidencia:
– Benignos: 6,8 por 100.000 hab./año
– Malignos: 7,3 por 100.000 hab./año
Sobrevida a 5 años global: 33%
– Niños <14 años: 62%
– Adultos >65 años: 4,9%
– Sobrevida 5 años astrocitoma: 30%
– Sobrevida 5 años glioblastoma: 3,3%
‘Classification of brain tumors’, David Schiff, MD; Uptodate; 2010.

EPIDEMIOLOGÍA

EPIDEMIOLOGÍA
No hay alguna predilección por
presentarse estos tumores en alguna
área geográfica determinada.
Kleihues P, Oligaki L. Primary and Secondary Glioblastoma: from concept to clinical
diagnosis. Neuro – Oncology 1999; 1 (1): 44 – 51

Etiología
Se desconocen las causas de los gliomas, si bien se han encontrado en ellos varias mutaciones genéticas adquiridas (no heredadas). Entre ellas están los genes que afectan la copia, la regulación y el crecimiento del ADN. Estas mutaciones pueden hacer que las células se dividan de manera descontrolada.
American Brain Tumor Association. Gliomas. JAMA 2010; 303 (10): 101 – 104

Etiología
No se ha encontrado algún indicio
sobre su etiología aunque se ha
especulado la acción de alguna
energía como las ondas
electromagnéticas o de radio.
Kleihues P, Oligaki L. Primary and Secondary Glioblastoma: from concept to clinical
diagnosis. Neuro – Oncology 1999; 1 (1): 44 – 51

ETIOLOGÍA
ARCH NEUROL/VOL 67 (NO. 3), MAR 2010

Etiopatogenia
Muchos de los tumores cerebrales se desarrollan a partir de células gliales cancerosas y por ello son llamados gliomas. A diferencia de otros cánceres, los gliomas crecen en un espacio reducido en el interior de la cabeza. Para crecer, muchos tipos de cáncer empujan a las células saludables hacia los lados, pero debido a las limitantes de espacio, los gliomas deben destruir a las células normales del cerebro. Para matar a las células saludables, los gliomas liberan grandes cantidades del neurotransmisor glutamato. El exceso de glutamato es tóxico para las neuronas y causa convulsiones hasta al 80% de las personas con gliomas. Dependiendo del tamaño y la localización del tumor, se pueden producir otros síntomas como parálisis, cambios de conducta y mareos.
American Brain Tumor Association. Gliomas. JAMA 2010; 303 (10): 101 – 104

Histología Se estima en relación a cinco variables
1. Heterotipia celular
2. Mitosis
3. Celularidad Grado Pronóstico
4. Necrosis
5. Proliferación vascular
‘Classification of brain tumors’, David Schiff, MD; Uptodate; 2010.
Grado I. Sobrevida de 5 o mas añosGrado II. Sobrevida de 3 a 5 años.Grado III. Sobrevida de 1 a 3 años Grado IV. Sobrevida de menos de un año

Crecimiento Tumoral
Por Expansión
Por Degeneración Quística
Por Infiltración
ARCH NEUROL/VOL 67 (NO. 3), MAR 2010

CLASIFICACIÓN
Los tumores primarios se clasifican según el tipo de células dominantes y se gradúan si presentan o no características patológicas estandarizadas.
Multiplicidad de clasificaciones:
– Virchow (1860).
– Bailey y Cushing (1926).
– Kernohan (1949).
– OMS (1979 – 2007).‘Classification of brain tumors’, David Schiff, MD; Uptodate; 2010.
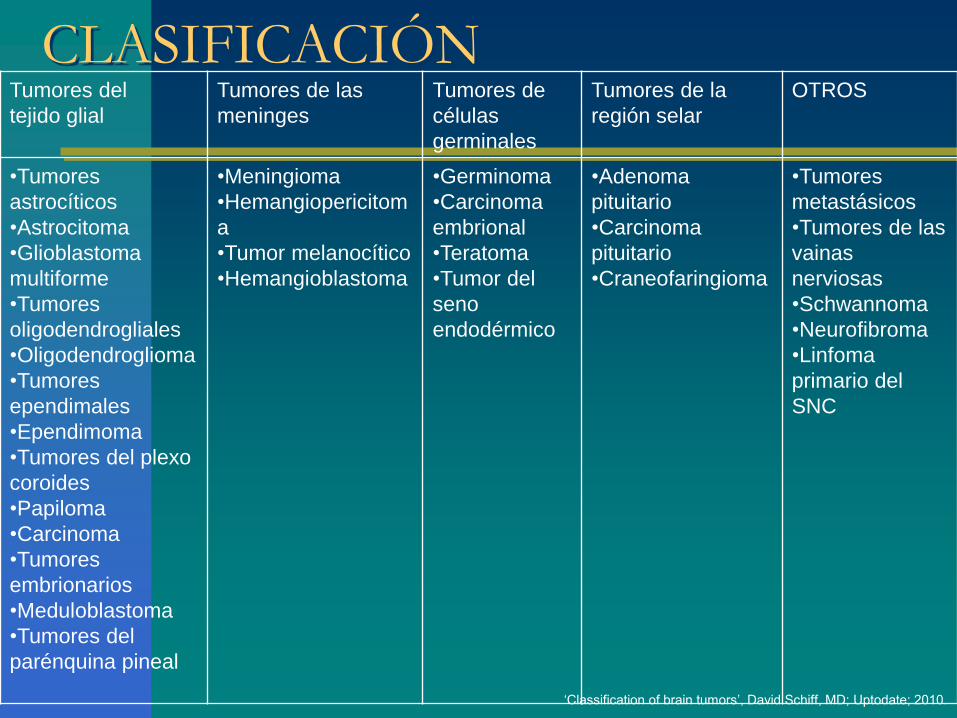
CLASIFICACIÓNTumores del
tejido glial
Tumores de las
meninges
Tumores de
células
germinales
Tumores de la
región selar
OTROS
•Tumores
astrocíticos
•Astrocitoma
•Glioblastoma
multiforme
•Tumores
oligodendrogliales
•Oligodendroglioma
•Tumores
ependimales
•Ependimoma
•Tumores del plexo
coroides
•Papiloma
•Carcinoma
•Tumores
embrionarios
•Meduloblastoma
•Tumores del
parénquina pineal
•Meningioma
•Hemangiopericitom
a
•Tumor melanocítico
•Hemangioblastoma
•Germinoma
•Carcinoma
embrional
•Teratoma
•Tumor del
seno
endodérmico
•Adenoma
pituitario
•Carcinoma
pituitario
•Craneofaringioma
•Tumores
metastásicos
•Tumores de las
vainas
nerviosas
•Schwannoma
•Neurofibroma
•Linfoma
primario del
SNC
‘Classification of brain tumors’, David Schiff, MD; Uptodate; 2010.

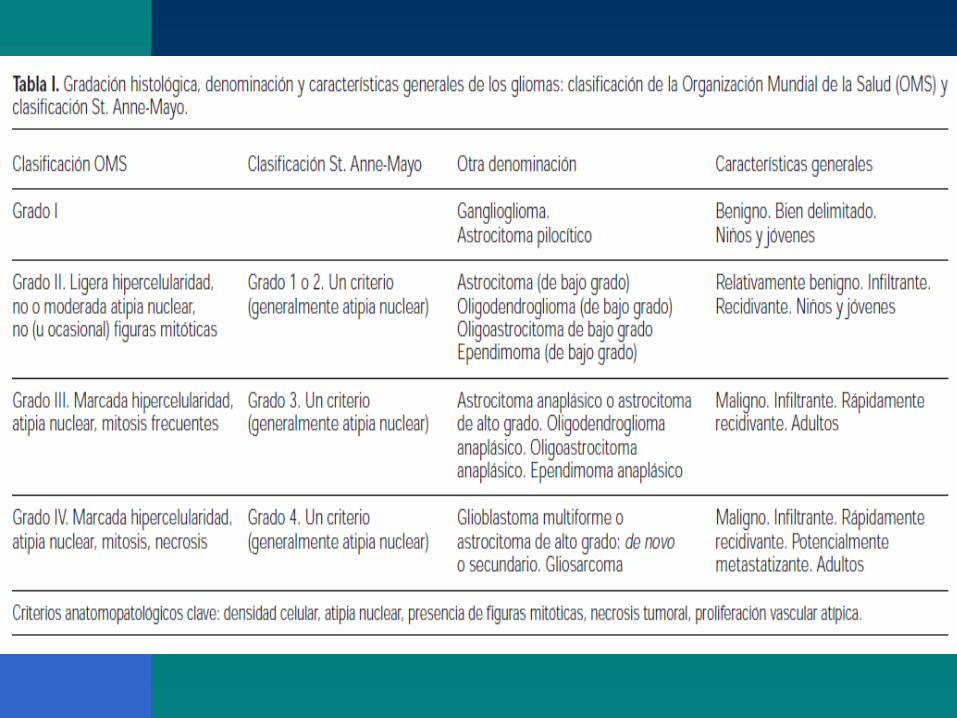
CLASIFICACIÓN OMS Grado I bajo potencial proliferativo, de naturaleza frecuentemente discreta y posibilidad de curación al cabo de la resección quirúrgica sola.
Grado II infiltrantes y de baja actividad mitótica pero que recidivan. Algunos tipos de tumores tienden a avanzar a grados más altos de degeneración.
Grado III lesiones de neoplasia histológica probada, en forma de actividad mitótica, capacidad de infiltración claramente expresada y anaplasia.
Grado IV actvidad mitótica que las hace propensas a la necrosis y, en general, se relacionan con mala evolución prequirúrgica y posquirúrgica.
‘Classification of brain tumors’, David Schiff, MD; Uptodate; 2010.


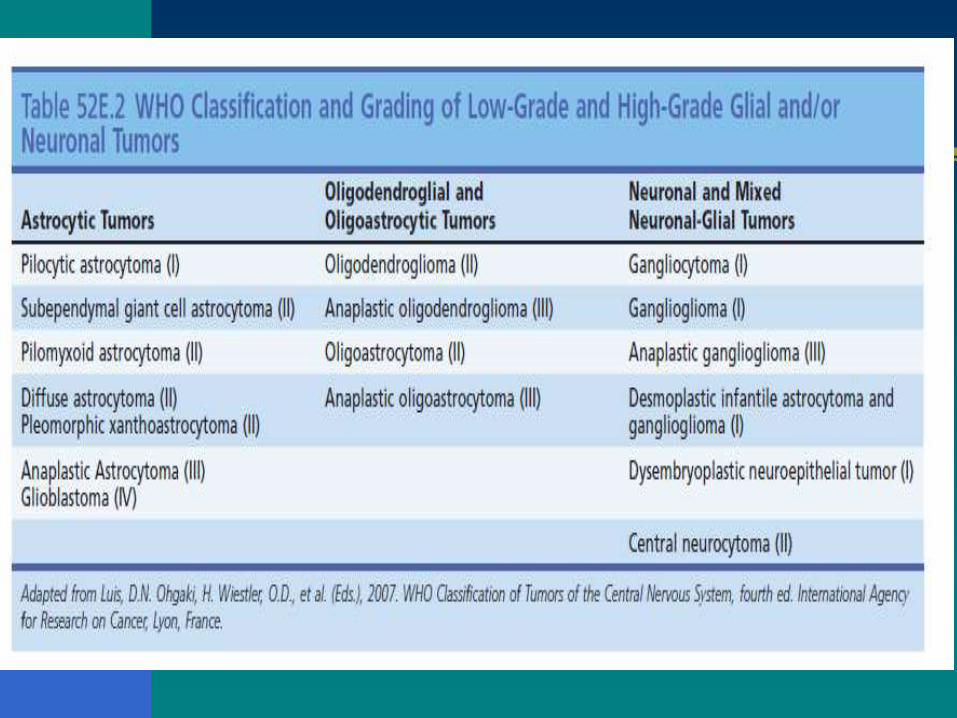
CLASIFICACIÓN ACTUAL DE LOS ASTROCITOMAS
Tipo histopatológico
Desde la clasificación de Bailey y Cushing de los
tumores cerebrales hasta la actual de la OMS ;rasgos
histológicos e inmunohistoquímicos. Un
comportamiento más agresivo.
Grado histopatológico
Agresividad biológica. OMS (grados I a IV).
Esta clasificación ha sido especialmente útil en el caso
de los gliomas.
La gradación de los gliomas se basa en unos
parámetros histológicos.

ASPECTOS GENERALES DE LOS ASTROCITOMAS
La supervivencia : 10-12 meses el glioblastoma
multiforme, 24 meses del astrocitoma anaplásico, y 6-8
años en los astrocitomas de bajo grado.
Glioblastomas secundarios o primarios.
La supervivencia en los astrocitomas de bajo grado
es variable, índice de Karnofsky
Gradación:
Clasificaciones OMS: grados I a IV. El seguimiento
de lo pctes.
La valoración de algunos de estos parámetros
histológicos se presta a la subjetividad y al azar.
Errores de gradación del muestreo(biopsia
estereotáxica) y de la propia heterogeneidad.

CLASIFICACIÓN MOLECULARDE LOS ASTROCITOMAS
Un 5% son hereditarios, asociados la
neurofibromatosis
La radiación.
Genoma de los astrocitomas: alteraciones
genéticas, como se deduce de los estudios de
hibridación genómica comparada.
Tipo de amplificación y deleción: astrocitomas se ha
visto que afectan a prácticamente la totalidad de los
cromosomas.
Mutaciones puntuales y a cambios epigenéticos como
la hipermetilación de genes promotores

La amplificación del gen PDGF/R y la mutación
del gen TP53 como acontecimientos
moleculares iniciales, y la alteración de los
genes EGFR y PTEN como alteraciones
tardías.
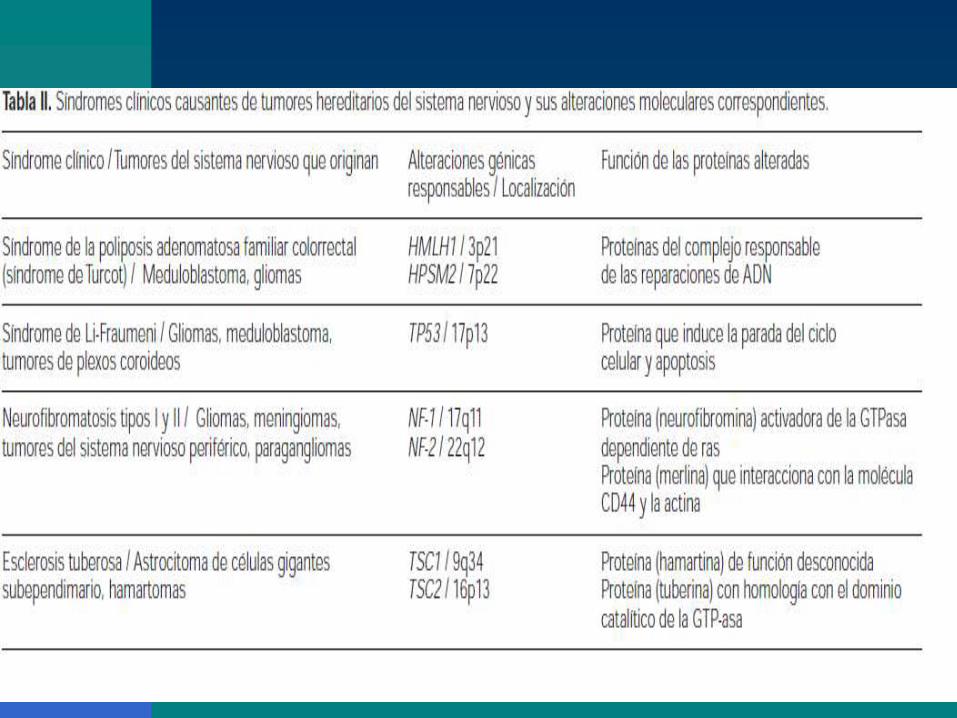
Astrocitomas hereditarios. Se trata de unas
proteínas activadoras de la GTPasa en la
neurofibromatosis (17q11 y 22q12), del
complejo responsable de las reparaciones de
ADN en el síndrome de Turcot (3p21 y 7p22) y
de la proteína p53 en el síndrome de Li-
Fraumeni (17p13).

El gran problema de la heterogeneidad molecular
Heterogeneidad fenotípica y molecular:
marcada cuánto más maligno .
Propiedades biológicas en diferentes áreas:
de los clones; focos eran indicativos de un
cambio en el grado tumoral.
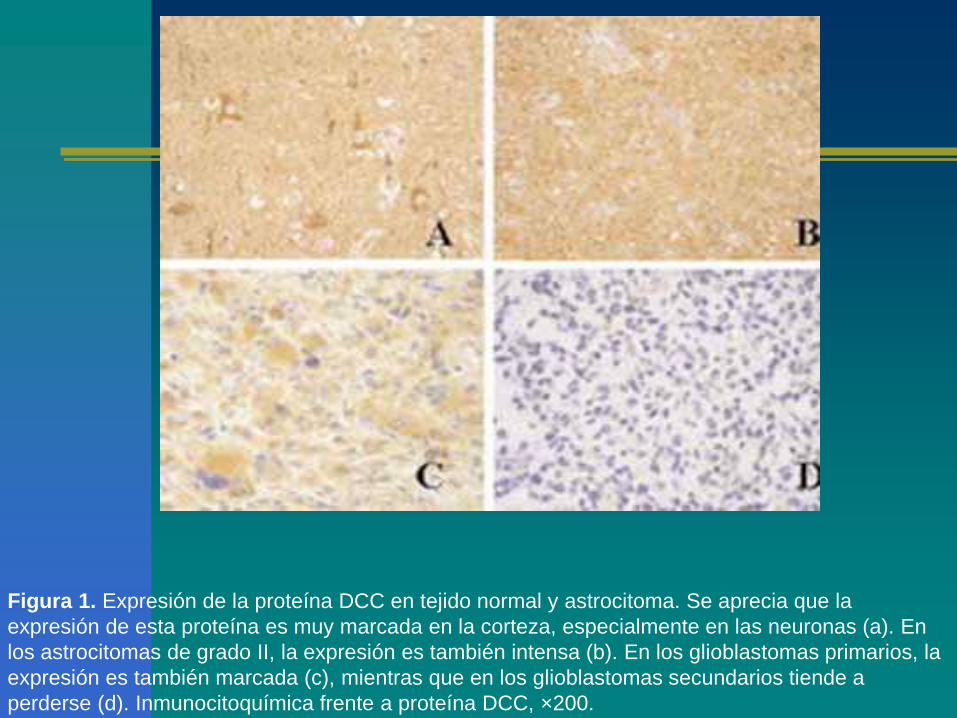
Figura 1. Expresión de la proteína DCC en tejido normal y astrocitoma. Se aprecia que la
expresión de esta proteína es muy marcada en la corteza, especialmente en las neuronas (a). En
los astrocitomas de grado II, la expresión es también intensa (b). En los glioblastomas primarios, la
expresión es también marcada (c), mientras que en los glioblastomas secundarios tiende a
perderse (d). Inmunocitoquímica frente a proteína DCC, ×200.

Los aspectos más relevantes en la valoración de los astrocitomasde bajo grado inestables
Marcadores de proliferación celular: inmunohistoquímica:
antígeno Ki-67, y citofluorometría. Un índice de proliferación
mayor del 5% (los grados II y III) .
La determinación de la ploidía: astrocitomas de bajo grado
tiene valor pronóstico. La aneuplodía en de bajo grado va
ligada a inestabilidad genómica, a la mutación del gen TP53.
Detección de la inactivación de genes supresores y de la
activación de oncogenes en las células tumorales: No
valor pronóstico. Mutación del gen TP53, la deleción del
cromosoma 17p, la amplificación del cromosoma 7, la
deleción del cromosoma 10, la mutación del gen PTEN
(phosphatase and tensin homologue deleted from
chromosome TEN), la deleción del gen DCC, del gen EGFR
y del gen VEGF como el retinoblastoma, el P16, el MDM2,
etc

En resumen: los astrocitomas de bajo
grado aneuploides, no proliferativos y con
el gen TP53 mutado, malignizar.
Astrocitomas grado IV (glioblastomas)
primarios y secundarios.
Según Ohgaki, en una serie de 715
glioblastomas, los glioblastomas
secundarios no superaron el 5%.
La diferenciación clínica o histológica entre
ambos subtipos
La heterogeneidad de grado

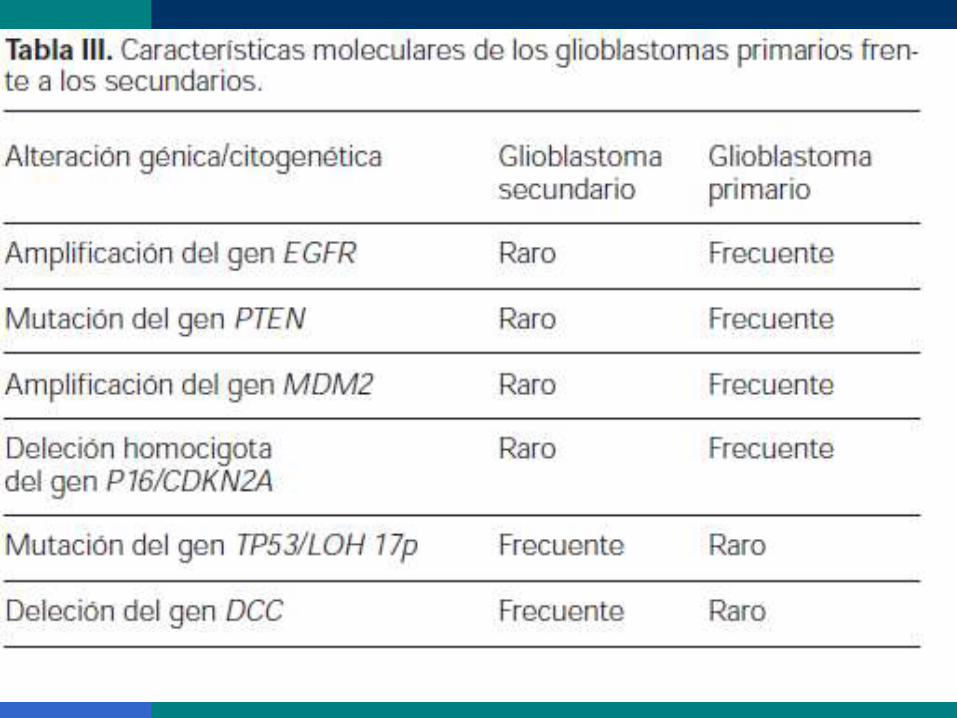
Vías oncogénicas distintas. Así, en el glioblastoma primario:
la amplificación de los genes EGFR y MDM2, la mutación
del gen PTEN y la deleción homocigota del gen
P16/CDKN2A. Por el contrario, la mutación del gen
TP53/LOH 17p y la deleción del gen DCC son propias del
glioblastoma secundario.( En un estudio propio de una serie
de 37 glioblastomas)
27 primarios y 10 secundarios, observamos que el 70% de los
secundarios eran inmunonegativos para la proteína DCC
frente a sólo un 19% en los primarios ( pérdida del gen DCC
)
Los glioblastomas pediátricos y los inducidos por radiación y
glioblastomas secundarios, aunque se manifiestan clínica e
histológicamente como glioblastomas primarios.
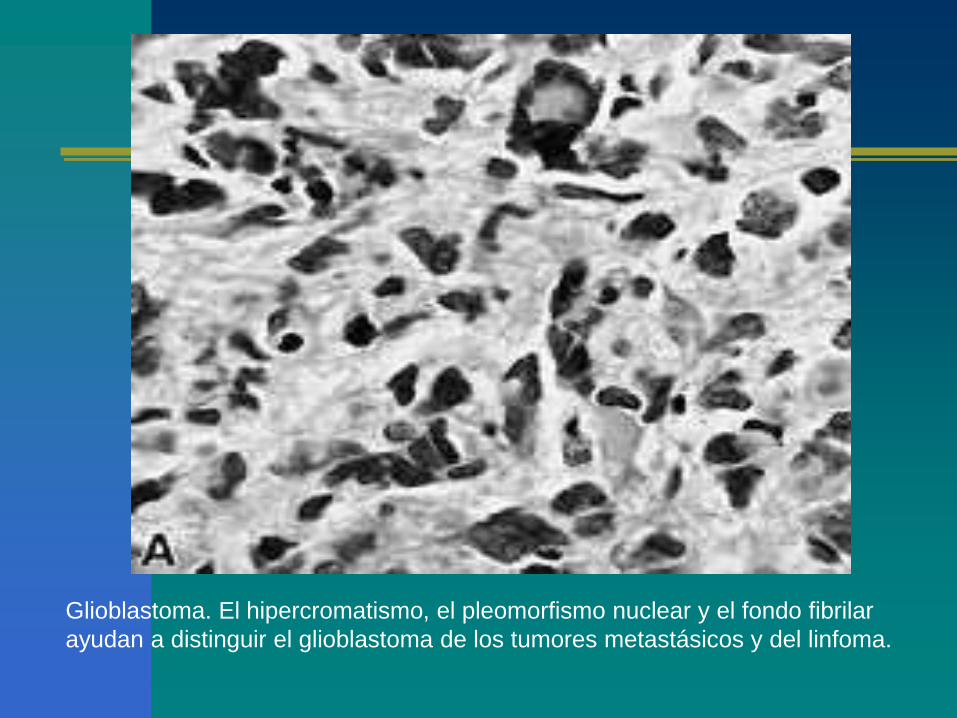
Glioblastoma. El hipercromatismo, el pleomorfismo nuclear y el fondo fibrilar
ayudan a distinguir el glioblastoma de los tumores metastásicos y del linfoma.
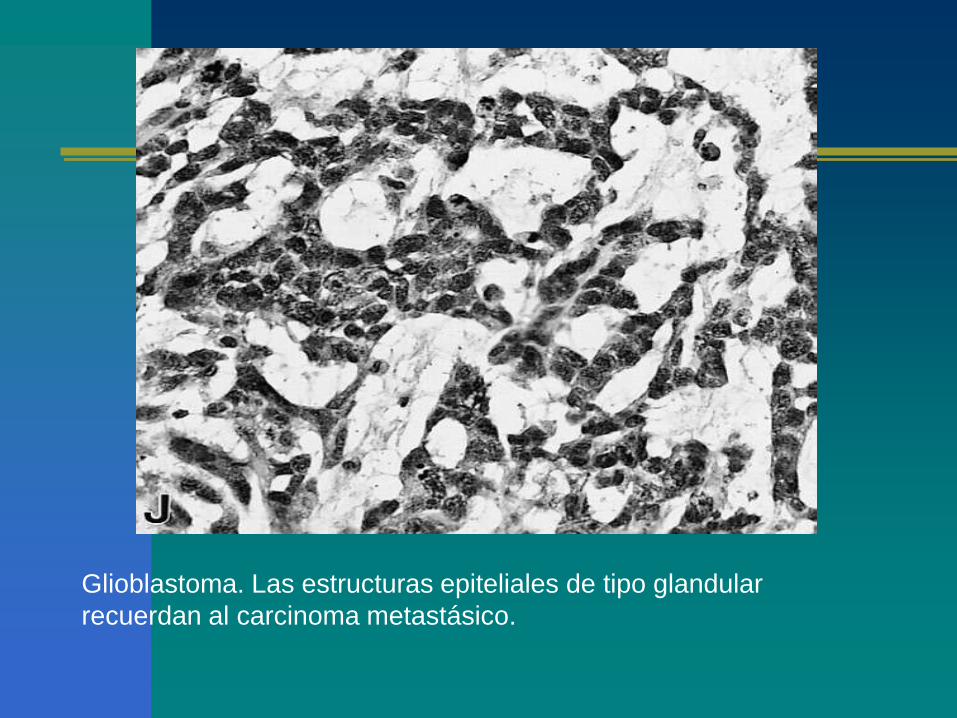
Glioblastoma. Las estructuras epiteliales de tipo glandular
recuerdan al carcinoma metastásico.
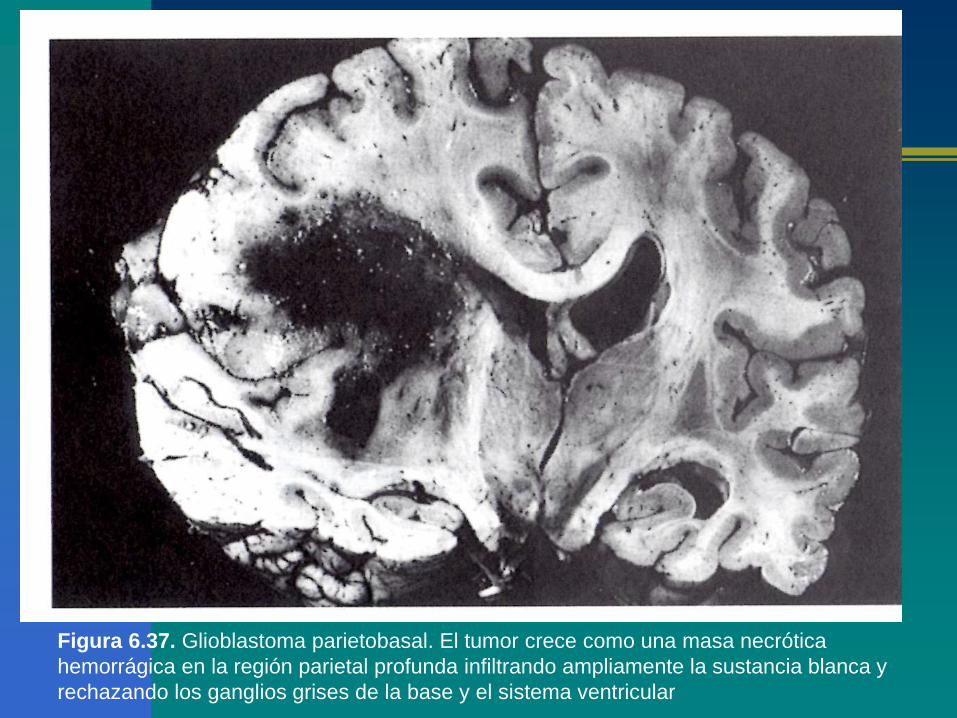
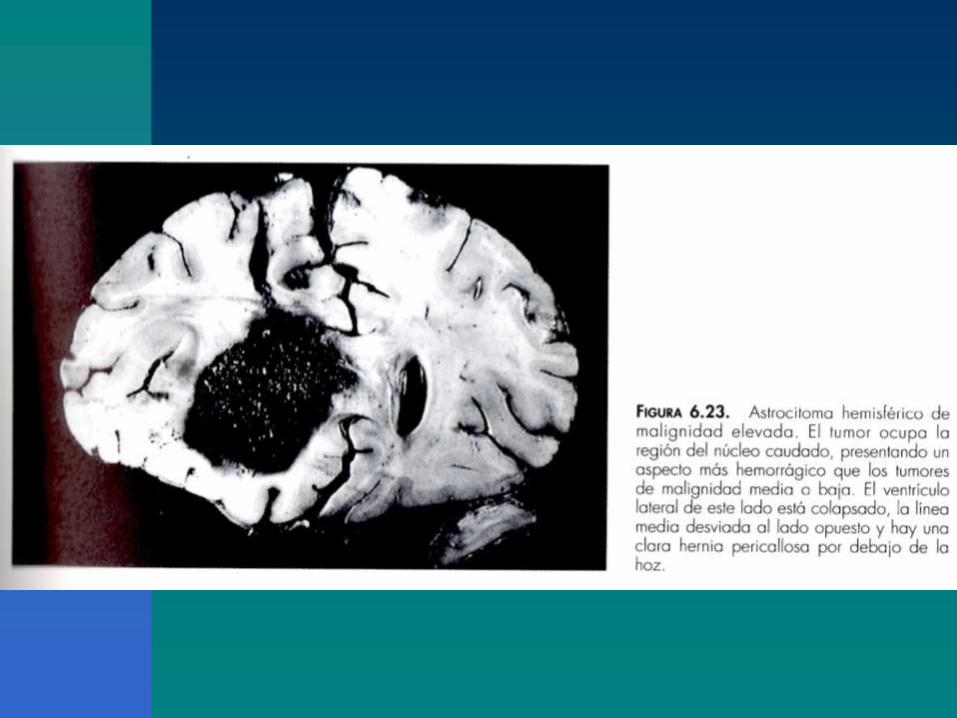
Figura 6.37. Glioblastoma parietobasal. El tumor crece como una masa necrótica
hemorrágica en la región parietal profunda infiltrando ampliamente la sustancia blanca y
rechazando los ganglios grises de la base y el sistema ventricular
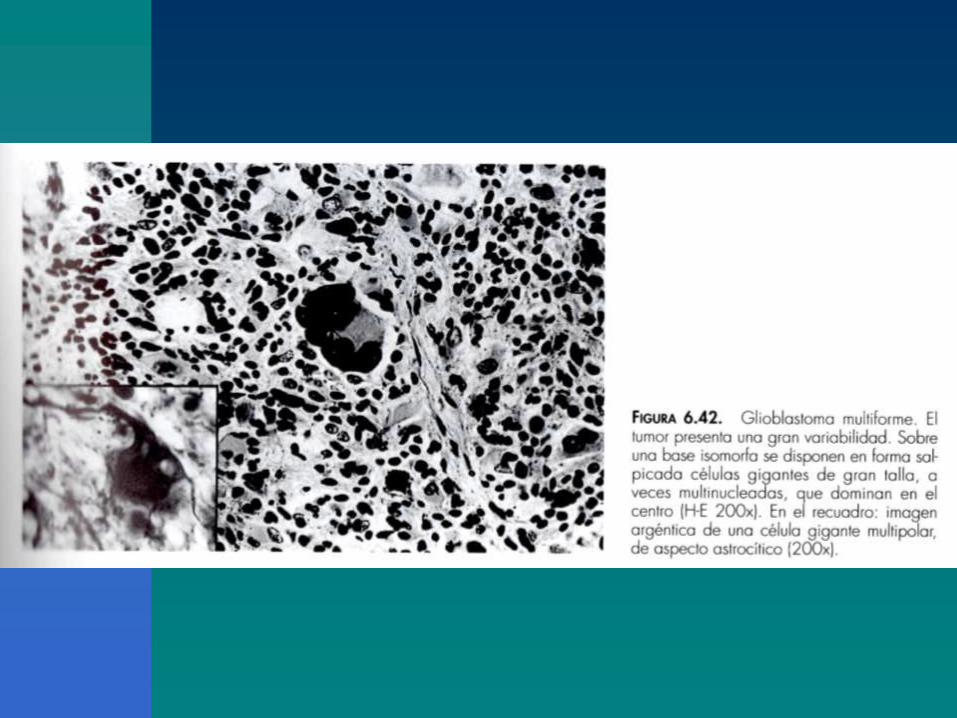
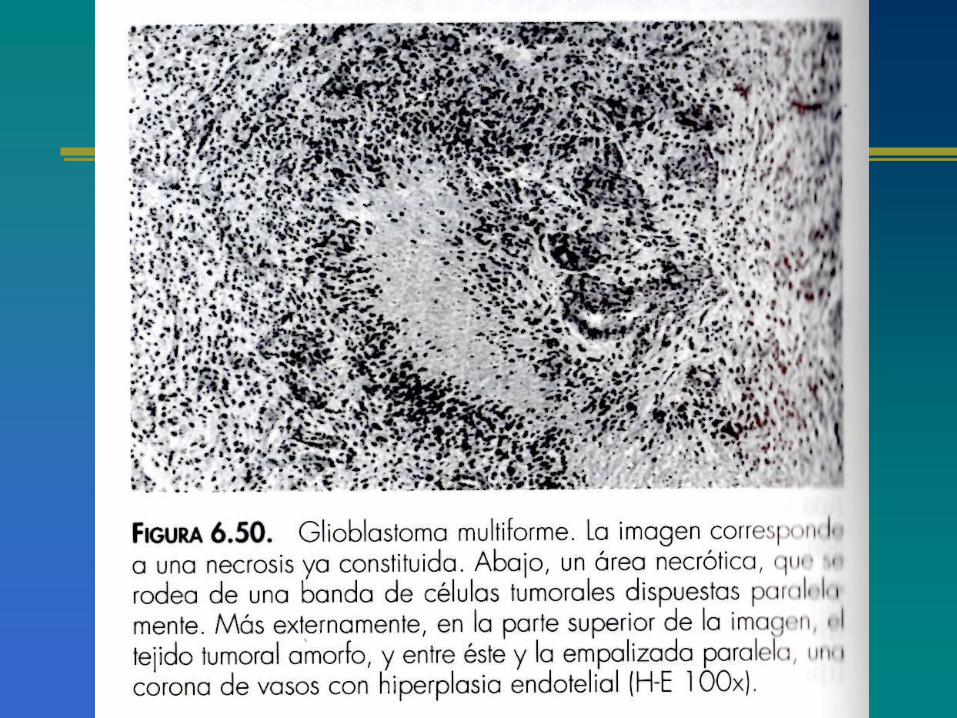
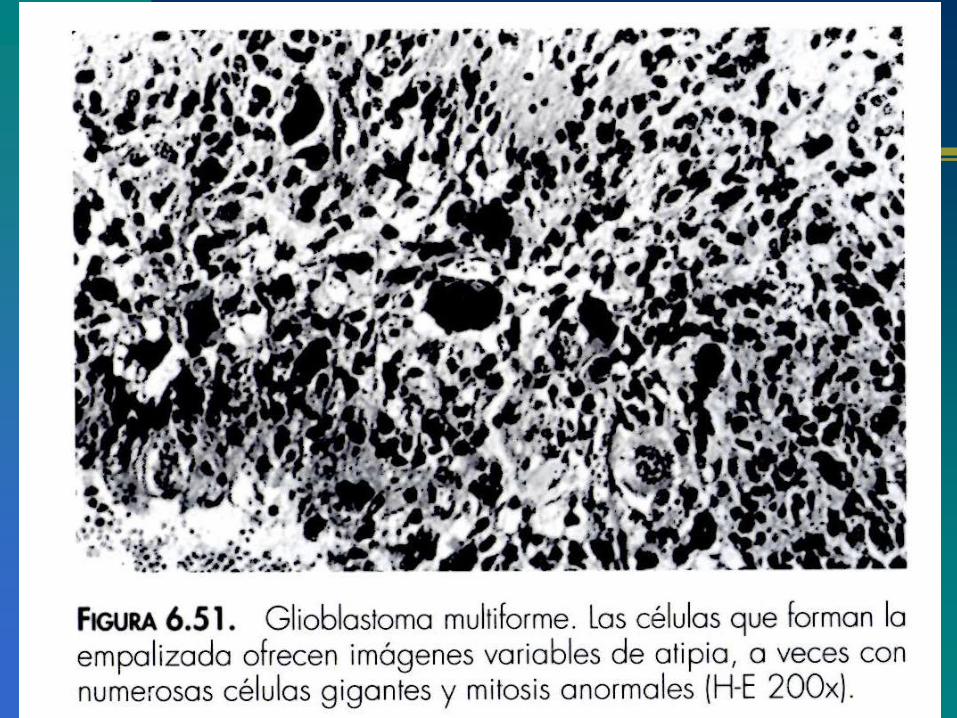

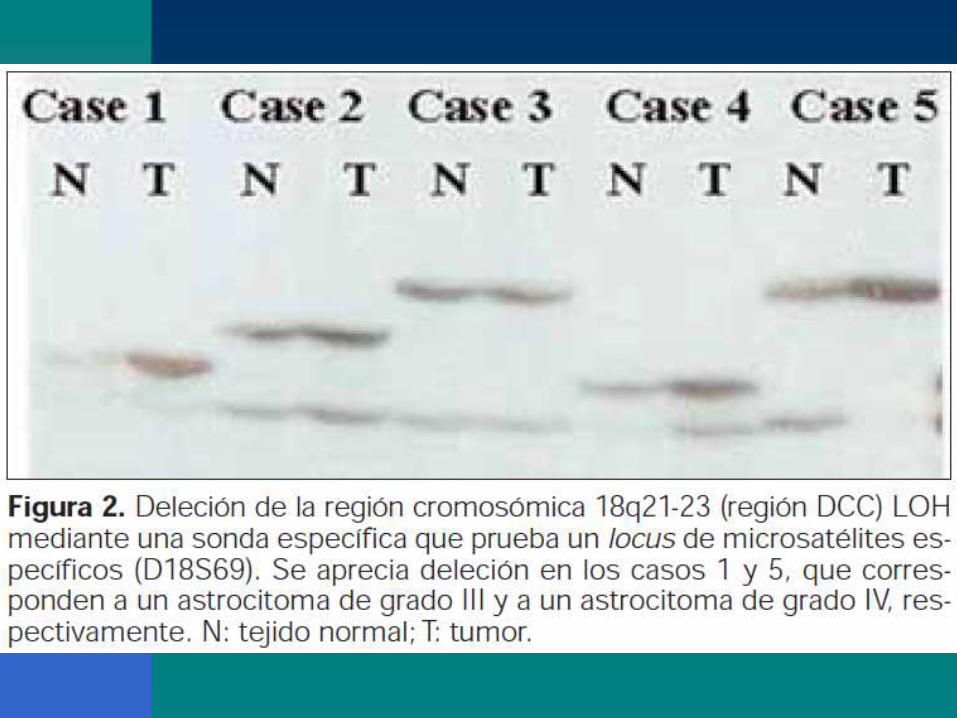
Importancia de la deleción de la región cromosómica10q en la nueva clasificación patomolecular
La región 10q, la región 10p o el cromosoma entero. La
pérdida de heterocigosidad de la región.
Los loci :10q23-24 y el 10q25-pter. La región 10q23 .
En el locus 10q23-24 se localiza el gen PTEN
angiogénesis y proliferativa tumoral.
La proteína PTEN se expresa en la corteza y la
sustancia blanca normales, así como en el grado II,
pero se pierde en los grados III y IV.
La expresión de ARNm del gen PTEN se
correlacionaba con el grado tumoral.
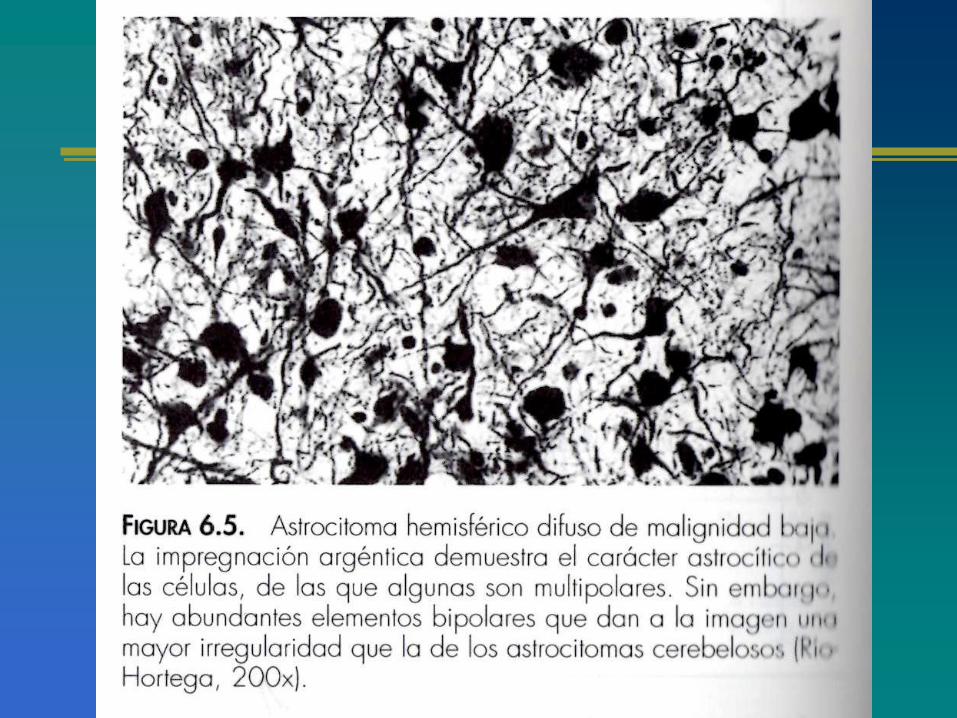

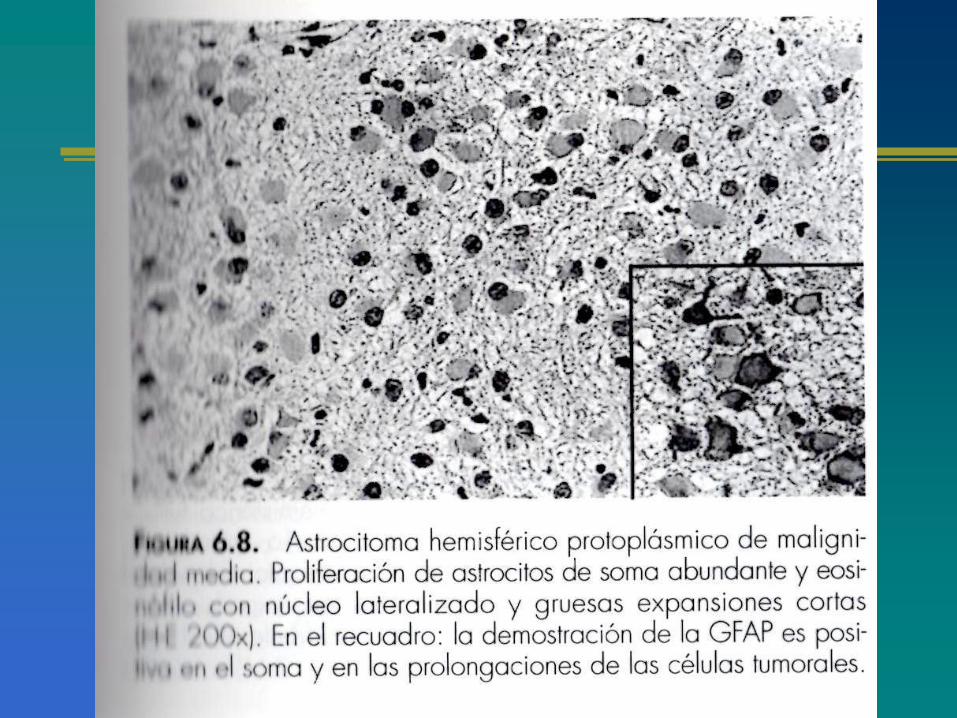
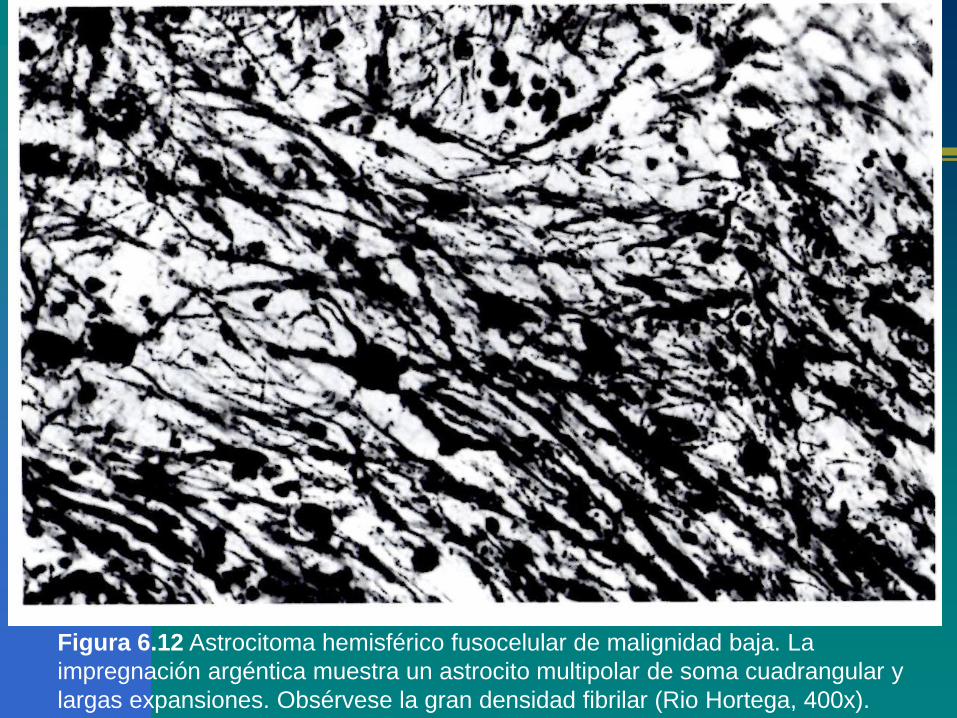
Figura 6.12 Astrocitoma hemisférico fusocelular de malignidad baja. La
impregnación argéntica muestra un astrocito multipolar de soma cuadrangular y
largas expansiones. Obsérvese la gran densidad fibrilar (Rio Hortega, 400x).


OLIGODENDROGLIOMASCaracterísticas generales
5-18% de los gliomas: oligodendrogliomas de bajo grado yanaplásicos.
La incidencia: 0,3 casos por 100.000 habitantes/año.
Dinámica de la malignización de los oligodendrogliomas:
Aspectos moleculares de interés en los oligodendrogliomas
Gradación histopatológica: oligodendrogliomas de bajo grado (gradoII) y anaplásicos (grado III). Supervivencia de los de bajo grado alos 5 años fue del 58%, y del 36% para los anaplásicos.
La presencia de gemistocitos no marca una mayor proclividad a lamalignización
Marcadores de proliferación: Ki-67 mayores del 3% bajo grado.
Un peor pronóstico: La ploidía.
Mutación del gen TP53.
La deleción 1p o combinada 1p/19q es una alteración citogenéticapropia de (lóbulo Temporal).
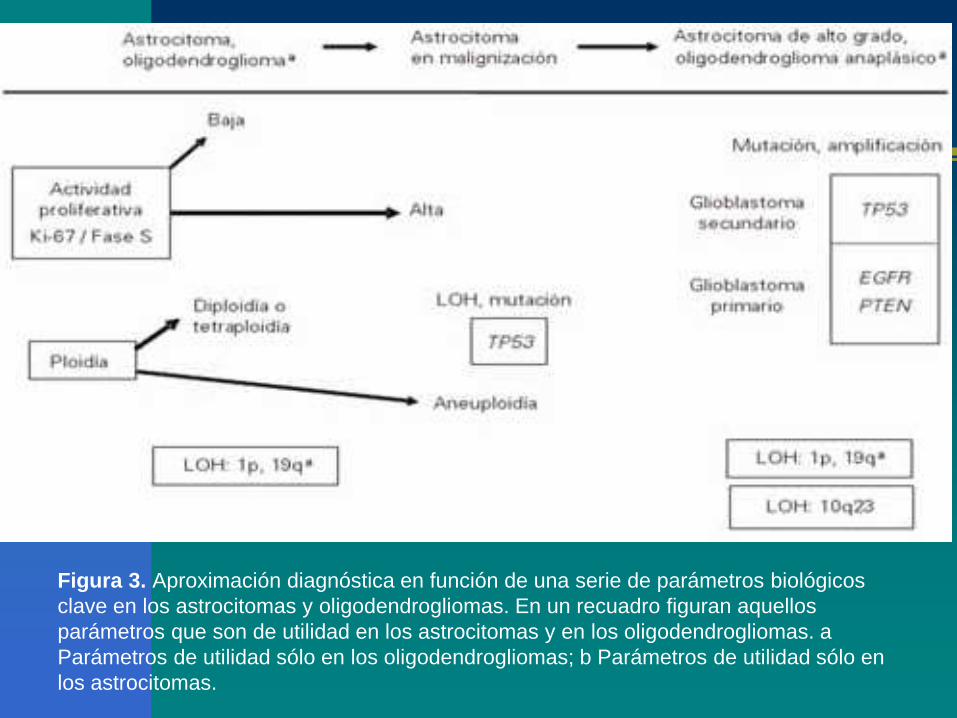
Figura 3. Aproximación diagnóstica en función de una serie de parámetros biológicos
clave en los astrocitomas y oligodendrogliomas. En un recuadro figuran aquellos
parámetros que son de utilidad en los astrocitomas y en los oligodendrogliomas. a
Parámetros de utilidad sólo en los oligodendrogliomas; b Parámetros de utilidad sólo en
los astrocitomas.
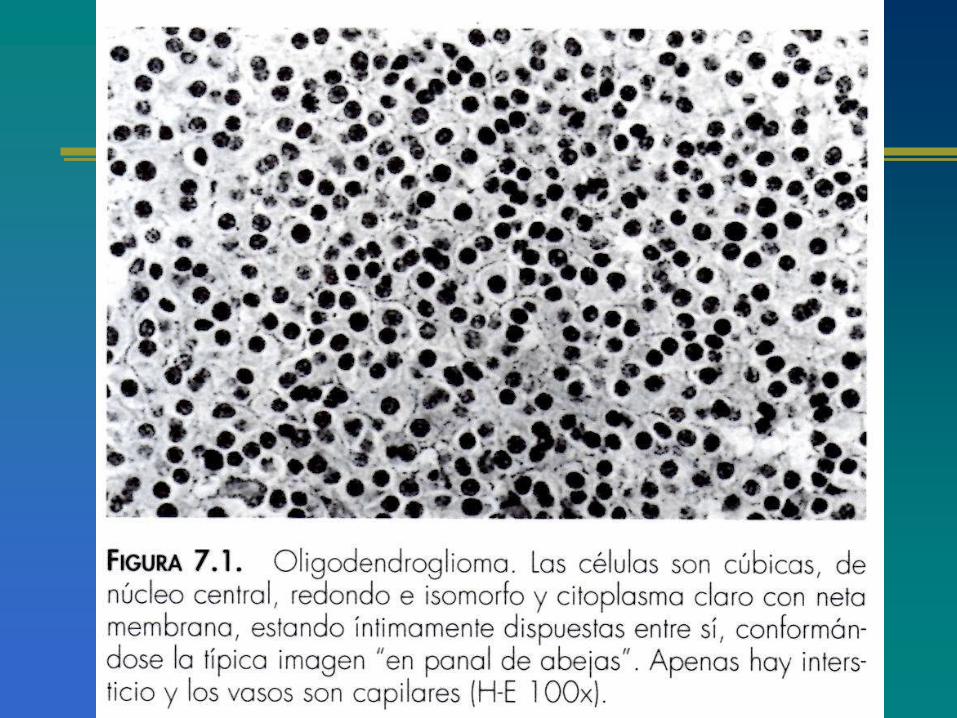
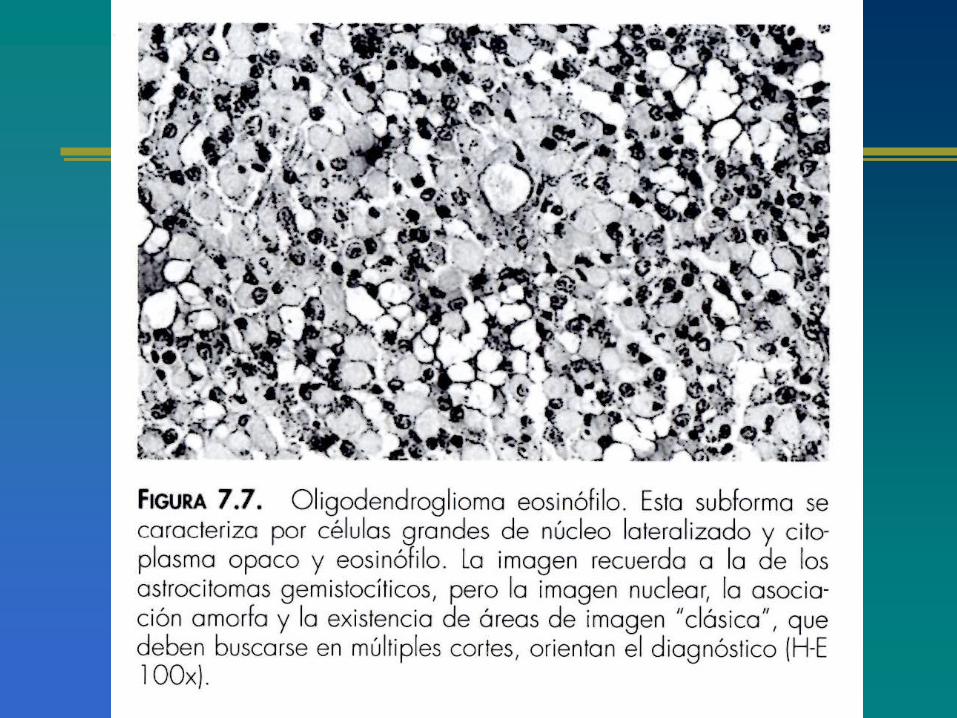
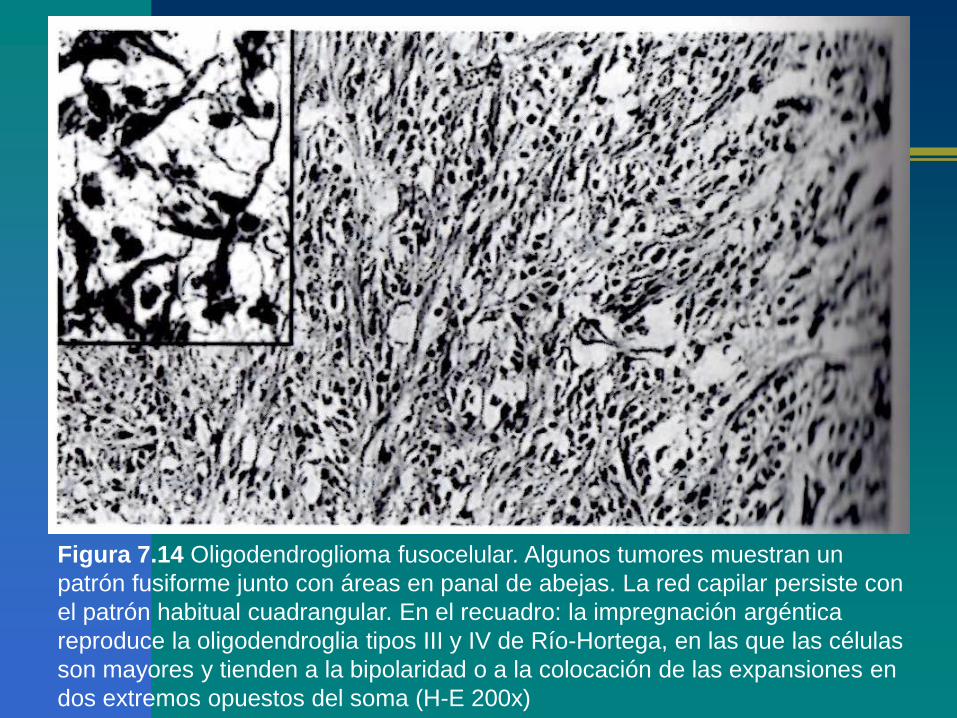
Figura 7.14 Oligodendroglioma fusocelular. Algunos tumores muestran un
patrón fusiforme junto con áreas en panal de abejas. La red capilar persiste con
el patrón habitual cuadrangular. En el recuadro: la impregnación argéntica
reproduce la oligodendroglia tipos III y IV de Río-Hortega, en las que las células
son mayores y tienden a la bipolaridad o a la colocación de las expansiones en
dos extremos opuestos del soma (H-E 200x)
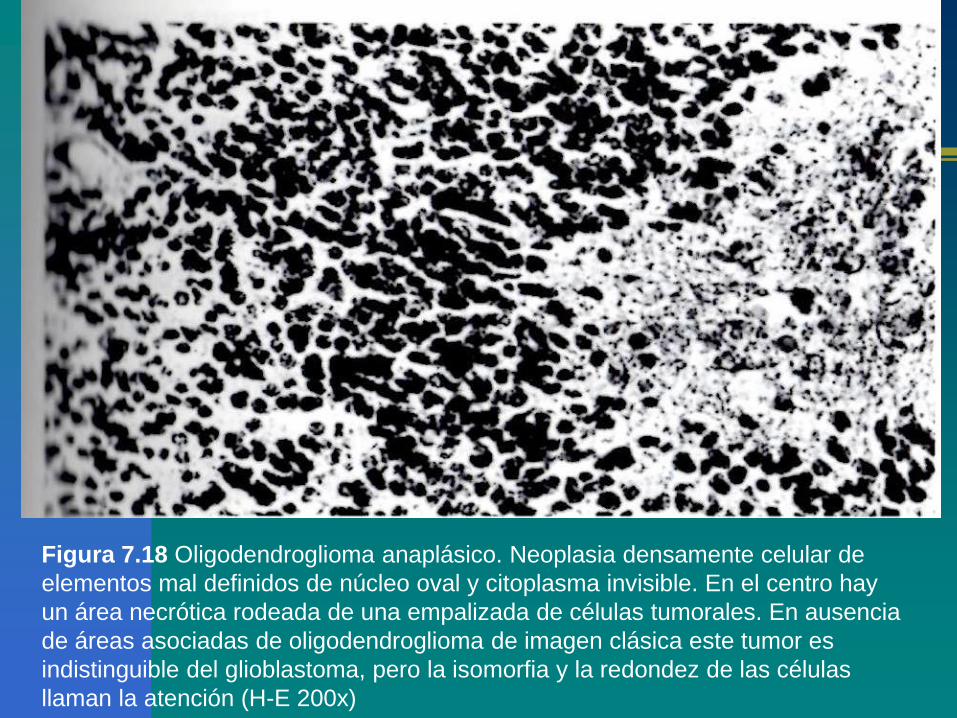
Figura 7.18 Oligodendroglioma anaplásico. Neoplasia densamente celular de
elementos mal definidos de núcleo oval y citoplasma invisible. En el centro hay
un área necrótica rodeada de una empalizada de células tumorales. En ausencia
de áreas asociadas de oligodendroglioma de imagen clásica este tumor es
indistinguible del glioblastoma, pero la isomorfia y la redondez de las células
llaman la atención (H-E 200x)

OLIGOASTROCITOMASCaracterísticas generales
Subtipos histológicos: oligoastrocitomas de bajo
grado (grado II) y oligoastrocitomas
anaplásicos(grado III).
Dinámica de malignización y factores pronósticos
Aspectos moleculares: deleción combinada 1p/19q,
de tal manera que los oligoastrocitomas cerebrales
no temporales muestran esta deleción, como los
oligodendrogliomas convencionales pero los
oligoastrocitomas temporales no presentan
deleción combinada y, además, muestran mutación
del gen TP53.
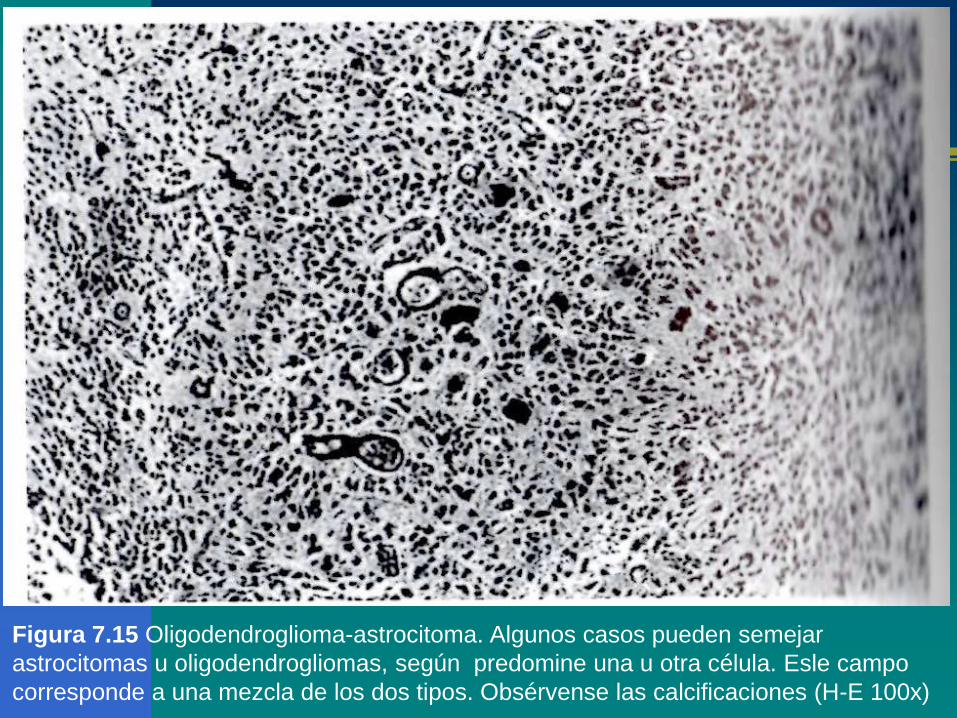
Figura 7.15 Oligodendroglioma-astrocitoma. Algunos casos pueden semejar
astrocitomas u oligodendrogliomas, según predomine una u otra célula. Esle campo
corresponde a una mezcla de los dos tipos. Obsérvense las calcificaciones (H-E 100x)

EPENDIMOMASCaracterísticas generales
El 3-9% neuroepiteliales.
Subtipos histológicos: clásico, anaplásico , mixopapilar ysubependimoma
Dinámica de la malignización del ependimoma
OMS: grado I el subependimoma y el ependimoma mixopapilar; degrado II, el ependimoma de bajo grado, y de grado III, elependimoma anaplásico. La supervivencia: los 10 años de losependimomas clásicos es del 48%, y para los anaplásicos, del25%.
Aspectos moleculares de interés en los ependimomas
– Marcadores de proliferación celular. antígeno Ki-67 puede ser unmarcador
– Hipermetilación de múltiples genes . cromosoma 22
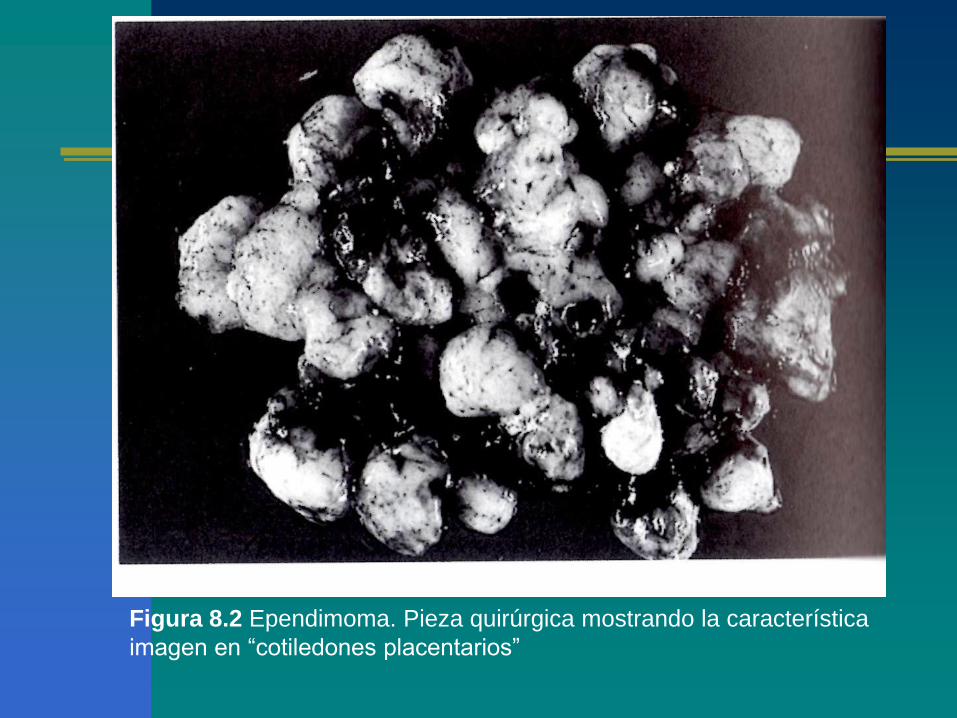
Figura 8.2 Ependimoma. Pieza quirúrgica mostrando la característica
imagen en “cotiledones placentarios”
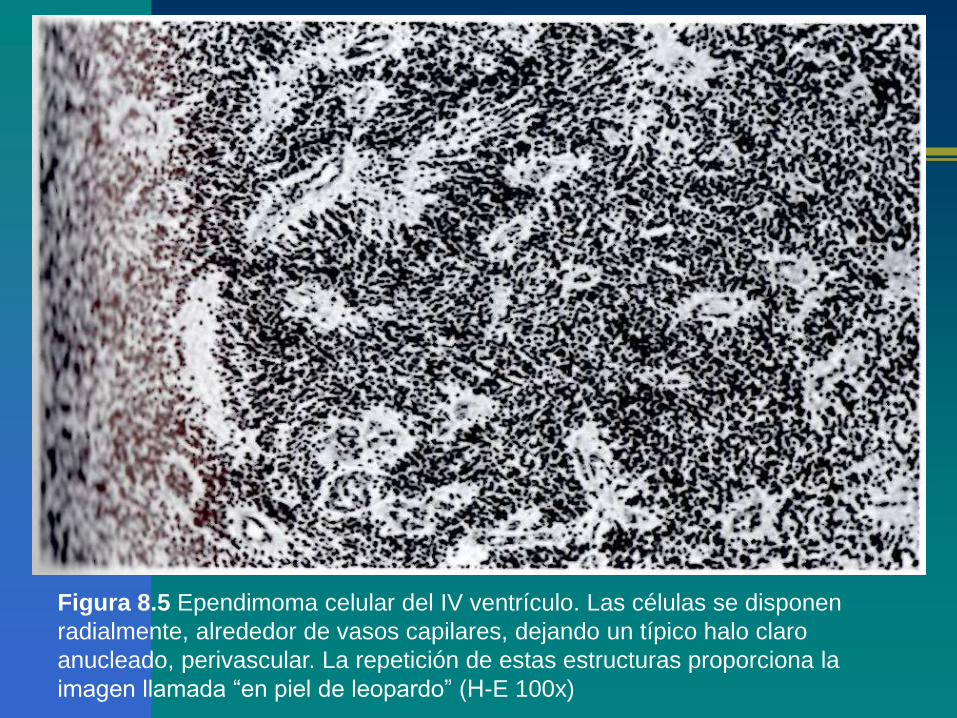
Figura 8.5 Ependimoma celular del IV ventrículo. Las células se disponen
radialmente, alrededor de vasos capilares, dejando un típico halo claro
anucleado, perivascular. La repetición de estas estructuras proporciona la
imagen llamada “en piel de leopardo” (H-E 100x)

NOVEDADES SOBRE LA PREDICCIÓN DERESPUESTA AL TRATAMIENTO DE LOS GLIOMAS
Utilidad de la biología molecular en la valoración
de la respuesta antitumoral: hallazgos
iniciales
Las células de los astrocitomas de alto grado son en
general quimiorradiorresistentes.
La inactivación del gen reparador del ADN,
O6-MGMT, por hipermetilación 10q26.
La radiorresistencia y la resistencia a
citostáticos: la inactivación del gen TP53.
La deleción combinada 1p/19q y la
respuesta al tratamiento con procarbacina,
ciclofosfamida (Citoxan ®) y vincristina.
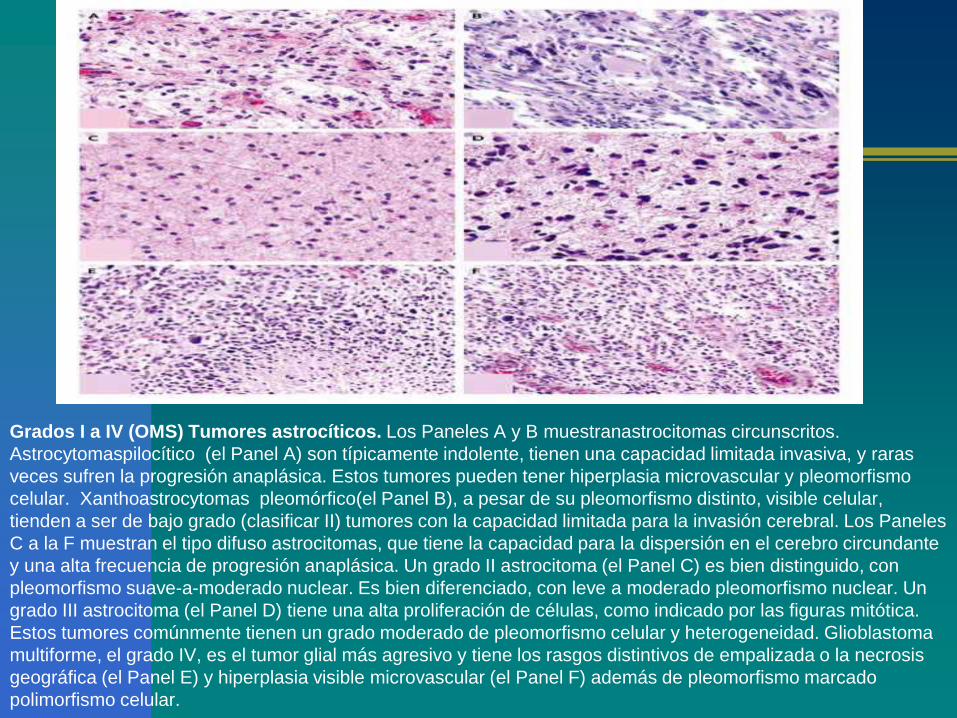
Grados I a IV (OMS) Tumores astrocíticos. Los Paneles A y B muestranastrocitomas circunscritos.
Astrocytomaspilocítico (el Panel A) son típicamente indolente, tienen una capacidad limitada invasiva, y raras
veces sufren la progresión anaplásica. Estos tumores pueden tener hiperplasia microvascular y pleomorfismo
celular. Xanthoastrocytomas pleomórfico(el Panel B), a pesar de su pleomorfismo distinto, visible celular,
tienden a ser de bajo grado (clasificar II) tumores con la capacidad limitada para la invasión cerebral. Los Paneles
C a la F muestran el tipo difuso astrocitomas, que tiene la capacidad para la dispersión en el cerebro circundante
y una alta frecuencia de progresión anaplásica. Un grado II astrocitoma (el Panel C) es bien distinguido, con
pleomorfismo suave-a-moderado nuclear. Es bien diferenciado, con leve a moderado pleomorfismo nuclear. Un
grado III astrocitoma (el Panel D) tiene una alta proliferación de células, como indicado por las figuras mitótica.
Estos tumores comúnmente tienen un grado moderado de pleomorfismo celular y heterogeneidad. Glioblastoma
multiforme, el grado IV, es el tumor glial más agresivo y tiene los rasgos distintivos de empalizada o la necrosis
geográfica (el Panel E) y hiperplasia visible microvascular (el Panel F) además de pleomorfismo marcado
polimorfismo celular.

CUADRO CLÍNICO GENERAL
Objetivos de la evaluación:– Historia minuciosa.
– Examen neurológico detallado.
– Neuroimagenes apropiadas.
Mecanismo de síntomas y signos:– Invasión regional.
– Compresión de estructuras adyacentes.
– Aumento de la presión intracraneal.
Tanto los tumores 1º como las metástasis pueden presentar clínica generalizada como focal.
‘Clinical presentation and diagnosis of brain tumors’, Eric T Wong, MD; Uptodate, 2010.

Cuadro Clínico General
Cefalea 54% : Por tracción directa
tumoral de meninges de la base craneal
(vascular).
Déficit Neurológico 68%: Por
compresión o infiltración.
Crisis convulsivas 26%: Varía según el
tipo de tumor y localización.
‘Clinical presentation and diagnosis of brain tumors’, Eric T Wong, MD; Uptodate, 2010.

Cambios conductuales: Lesiones frontales
Naúseas y vómito
Ataxia y Dismetría: Tumores Cerebelosos
Compromiso de Pares Craneales: Proceso expansivo .
Se dice que los tumores de SNC
presentan manifestaciones cuando
alcanzan un diámetro de 2.5cm.
‘Clinical presentation and diagnosis of brain tumors’, Eric T Wong, MD; Uptodate, 2010.

Criterios de diagnóstico
Se debe considerar en los casos en que se presenten convulsiones tardías o déficit neurológico focal asociado a un síndrome de Hipertensión Endocraneana.
La tomografía axial computarizada del cerebro, la Resonancia magnética y la angiografía son exámenes que permiten un alto grado de diagnóstico.
El diagnóstico final se efectúa con los hallazgos anatomo-patológicos de la biopsia del Tumor
Kleihues P, Oligaki L. Primary and Secondary Glioblastoma: from concept to
clinical diagnosis. Neuro – Oncology 1999; 1 (1): 44 – 51

DIAGNOSTICO NEUROIMAGENOLÓGICO
Resonancia Magnética:
– Usualmente el único examen necesario.
– Superior al TAC en visualizar el tumor y su
relación con el parénquima sano, las
meninges, espacio subaracnoideo, fosa
posterior y trama vascular.
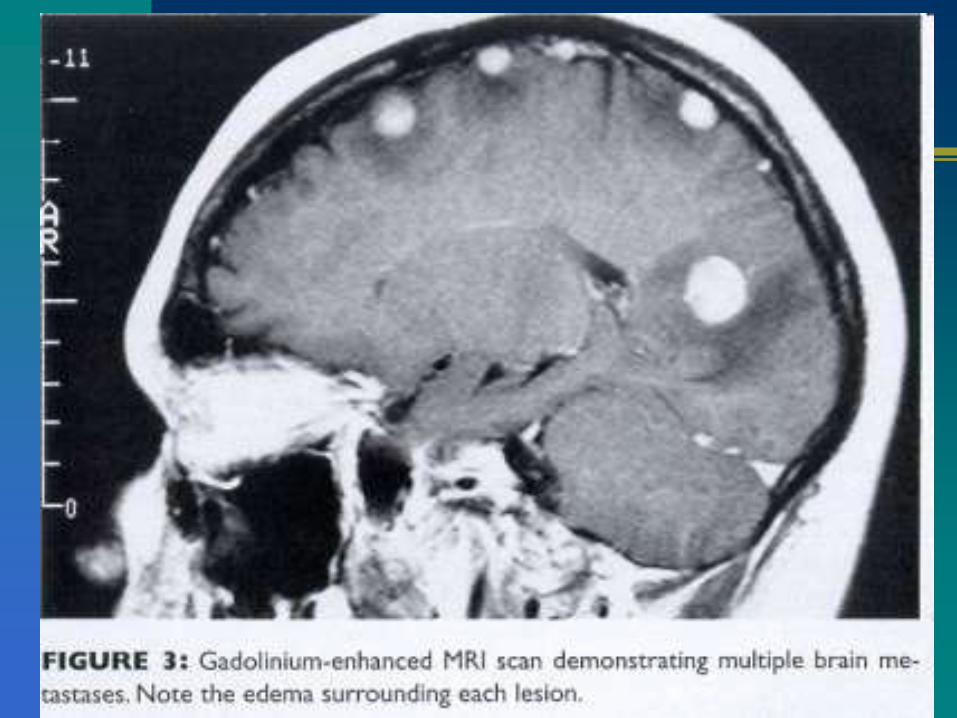
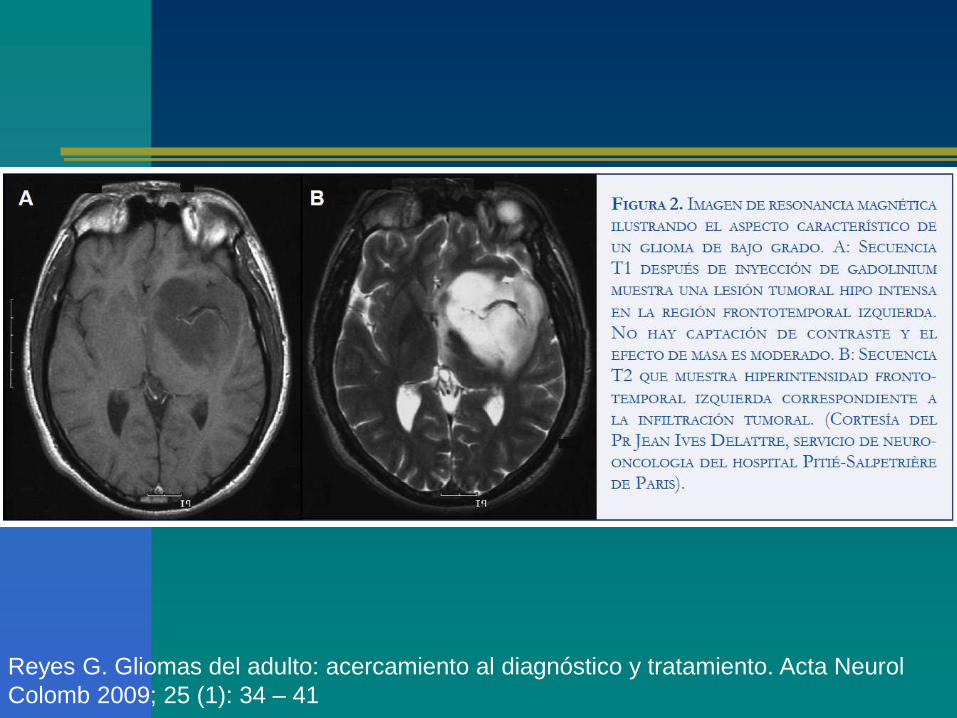
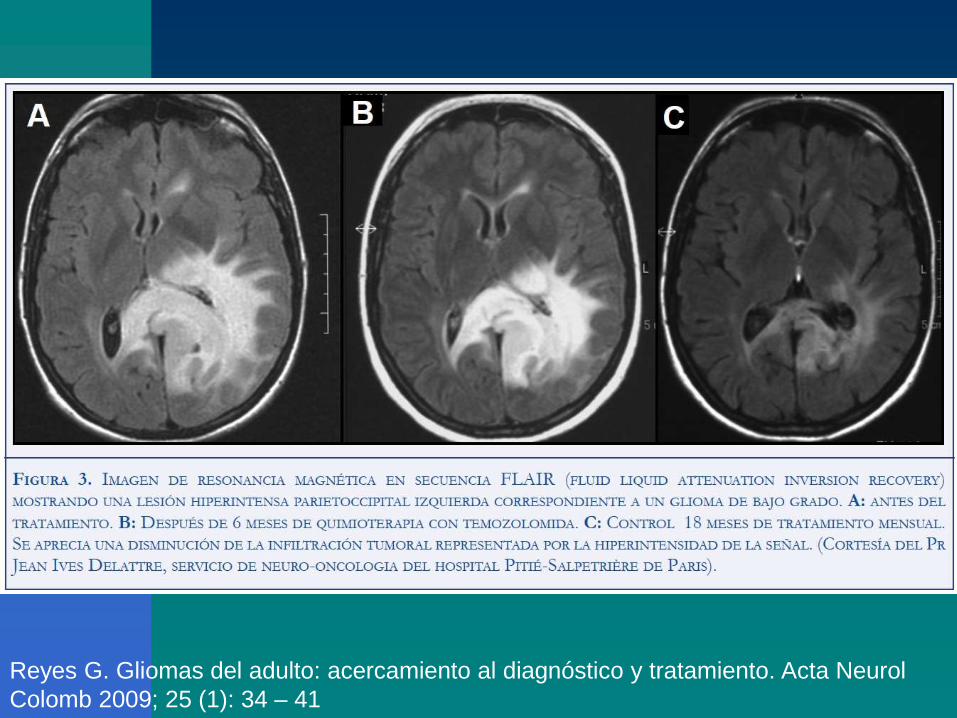
Glioma multiforme: hipointensos en T1.
Astrocitoma: buena visualización en T2
y FLAIR.
‘Clinical presentation and diagnosis of brain tumors’, Eric T Wong, MD; Uptodate, 2010.

Reyes G. Gliomas del adulto: acercamiento al diagnóstico y tratamiento. Acta Neurol
Colomb 2009; 25 (1): 34 – 41
Reyes G. Gliomas del adulto: acercamiento al diagnóstico y tratamiento. Acta Neurol
Colomb 2009; 25 (1): 34 – 41
Reyes G. Gliomas del adulto: acercamiento al diagnóstico y tratamiento. Acta Neurol
Colomb 2009; 25 (1): 34 – 41

DIAGNÓSTICO NEUROQUIRÚRGICO
Un diagnóstico acertivo del tumor
cerebral requiere una muestra de tejido:
– Biopsia estereotáxica.
– Cirugía.
‘Clinical presentation and diagnosis of brain tumors’, Eric T Wong, MD; Uptodate, 2010.

DIAGNÓSTICO NEUROQUIRÚRGICO
Valoracion preoperatoria:
– Las metástasis cerebrales son más comunes que los tumores 1º y pueden ser la manifestación inicial de la enfermedad.
– Si sospecha metástasis se requiere evaluación sistémica, especialmente torácica.
– Al intervenir una metástasis se sopesan los riesgos vs beneficios. Diagnostico histológico.
Resección de una lesión solitaria.
Alivio de síntomas neurológicos.
‘Clinical presentation and diagnosis of brain tumors’, Eric T Wong, MD; Uptodate, 2010.

TRATAMIENTO
Quirúrgico
-Limitada por la localización
-La supervivencia mediana de los pacientes tratados con cirugía exclusiva es de
tan solo 4 a 6 meses.
-Exéresis macroscópicamente completa (cirugía radical)
-Exéresis macroscópicamente incompleta .
-Exéresis parcial - biopsia.

Radioterapia
-Se considera el tratamiento de primera elección en el
caso de los tumores inoperables.

Radiología
La radioterapia es el tratamiento de referencia desde hace más de 20 años. Actualmente se ha determinado por medio de estudios retrospectivos que la dosis de irradiación para una lesión intracraneal no debe sobrepasar los 60 Gy con un fraccionamiento de máximo 2 Gy por sesión. Este esquema de radiación permite limitar la neurotoxicidad retardada que puede presentarse meses después del tratamiento con severas secuelas neurocognitivas.
Reyes G. Gliomas del adulto: acercamiento al diagnóstico y tratamiento. Acta Neurol
Colomb 2009; 25 (1): 34 – 41

Quimioterapia
La quimioterapia adyuvante tras tratamiento local
(cirugía + RT, o RT exclusiva en tumores
irresecables)

Quimioterapia
El papel de la quimioterapia como parte del tratamiento inicial de los gliomas del adulto ha sido el objeto de múltiples estudios multicéntricos en los últimos años. El uso de este tipo de medicamentos ha demostrado su efectividad y actualmente varios estudios de fase III están en curso para tratar de establecer los esquemas terapéuticos mas adaptados según el tipo de tumor.
Reyes G. Gliomas del adulto: acercamiento al diagnóstico y tratamiento. Acta Neurol
Colomb 2009; 25 (1): 34 – 41

Tratamiento de los Gliomas
Desgraciadamente en muchos casos el
tumor se encuentra en localizaciones
que lo hacen inoperable y, en ausencia
de signos clínicos o de progresión
radiológica la recomendación es hacer
un simple seguimiento con un intervalo
entre 3-6 meses.
Reyes G. Gliomas del adulto: acercamiento al diagnóstico y tratamiento. Acta Neurol
Colomb 2009; 25 (1): 34 – 41

Tratamiento de los Gliomas
El tratamiento de los gliomas ha
evolucionado en los últimos años.
Diversos estudios han permitido
establecer la efectividad de la
quimioterapia como una nueva
herramienta terapéutica asociándola a
los tratamientos de referencia que siguen
siendo la cirugía y la radioterapia.
Reyes G. Gliomas del adulto: acercamiento al diagnóstico y tratamiento. Acta Neurol
Colomb 2009; 25 (1): 34 – 41

Tratamiento de los Gliomas
Un aspecto innovador y de mucho
interés actual es la presencia
intratumoral de marcadores moleculares
de quimiosensibilidad.
El MGMT en glioblastomas y la deleción
1p19q en los oligodendrogliomas y
gliomas mixtos se relacionan con una
mejor respuesta a la quimioterapia.
Reyes G. Gliomas del adulto: acercamiento al diagnóstico y tratamiento. Acta Neurol
Colomb 2009; 25 (1): 34 – 41

Los resultados preliminares de las nuevas terapias con anticuerpos monoclonales inhibidores de la proliferación vascular, son prometedores y podrían integrarse en los próximos años dentro de las alternativas terapéuticas de primera línea para el tratamiento de los gliomas de alto grado.
Reyes G. Gliomas del adulto: acercamiento al diagnóstico y tratamiento. Acta Neurol
Colomb 2009; 25 (1): 34 – 41








![ESTUDIO DE LA ACTIVACIÓN DE PI3K/AKT/MTOR, RAS ......de gliomas de alto y bajo grado respectivamente [25], así como en un 12% de rabdomiosarcomas y casos puntuales de neuroblastoma](https://img.pdfslide.es/doc/110x75/6138c1860ad5d2067649741e/estudio-de-la-activacin-de-pi3kaktmtor-ras-de-gliomas-de-alto-y-bajo.jpg)





















