Embed Size (px)
Citation preview

Retinoblastoma 67
ACTUALIZACIONES
RETINOBLASTOMA. ACTUALIZACION DE SU TRATAMIENTO
Dres. G. Chantada, A. Fandiño, M. T. G de Dávila, E. Raslawski, J. Manzitti, M. Scopinaro,C. Sánchez y E. Schvartzman
El retinoblastoma es el tumor ocular más fre-cuente en Pediatría. Sin embargo, es lo suficien-temente infrecuente para que la mayoría de los pe-diatras vea pocos casos de esta neoplasia durantesu práctica clínica. En general, los oftalmólogosestablecen el diagnóstico, deciden las modalida-des del tratamiento local y monitorean la respues-ta, mientras que el rol del oncólogo ha crecido enlos últimos años como consecuencia del mayornúmero de indicaciones de quimioterapia. El reti-noblastoma se presenta con una frecuencia deaproximadamente 1 en 15000 – 18000 nacidosvivos en los países desarrollados. Sin embargo,aparentemente es más frecuente en regiones delmundo menos desarrolladas tales como Latino-américa, África y Asia. En tales áreas, el retino-blastoma es diagnosticado tardíamente, cuando ladiseminación extraocular ya ha ocurrido, llevandoa una alta morbilidad (ceguera) y mortalidad. Ennuestro país se evidencia una situación interme-dia, donde el número de pacientes con enferme-dad metastática no supera el 10% del total y lasobreviva a 5 años es mayor del 90% para el to-tal de casos, a costa de un tratamiento intenso delos pacientes con enfermedad regional1,2. Nuestrapoblación de pacientes se caracteriza por presen-tar una mayor proporción de casos con invasiónregional, incluyendo el nervio óptico y la coroidesque en países desarrollados. Nuestro grupo de-mostró que la presencia de enfermedad extraocu-lar al diagnóstico se correlaciona con factoressociales y geográficos 2.
El retinoblastoma puede presentarse de dosformas: hereditaria o no hereditaria y los tumorespueden ser unilaterales o bilaterales. Todos los pa-cientes con retinoblastoma bilateral tienen enfer-medad hereditaria, y por otro lado solo un 10 %de los casos unilaterales tienen la forma hereda-ble. Estos pacientes tienden a ser menores de unaño de edad y frecuentemente presentan tumoresmultifocales. El retinoblastoma no hereditario essiempre unilateral y unifocal. El retinoblastomapuede presentarse de forma familiar o esporádi-ca, pero sólo el 6 al 10% son familiares. La edadmedia de presentación de los pacientes al diag-nóstico es de 24 meses en los casos unilateralesy de 13 meses en los bilaterales. Nuestro Hospi-tal ha investigado activamente esta patología y losdatos más importantes reportados son motivo deesta presentación1,2,3. En nuestro centro existe ungrupo multidisciplinario para el manejo de estaenfermedad que incluye a oftalmólogos, oncólo-gos, radioterapeutas, patólogos, genetistas, clíni-cos, asistentes sociales y psicólogos. Por otraparte, nuestro equipo coordina un grupo de padresde niños que padecen retinoblastoma que funcio-na en forma ininterrumpida desde 1999 en la Fun-dación Natalie D Flexer (www.fundacionflexer.org).
Signos y síntomasLos hallazgos clínicos en todos los estadios del
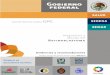
retinoblastoma son numerosos y variados. La granmayoría presenta leucocoria (reflejo pupilar blancoo reflejo de ojo de gato) y/o estrabismo (Figura 1).Por tal razón debe examinarse el fondo de ojo entodos los casos de estrabismo en la niñez. La pre-sencia de reflejo rojo (en especial si se hace sindilatación), no debe ser un reaseguro para supo-
Servicios de Hemato-oncología, Oftalmología , Anatomía Patológicay Terapia Radiante.Hospital de Pediatría Juan P. Garrahan
http://www.medicinainfantil.org.ar
68 Medicina Infantil Vol. XI N° 1 Marzo 2004
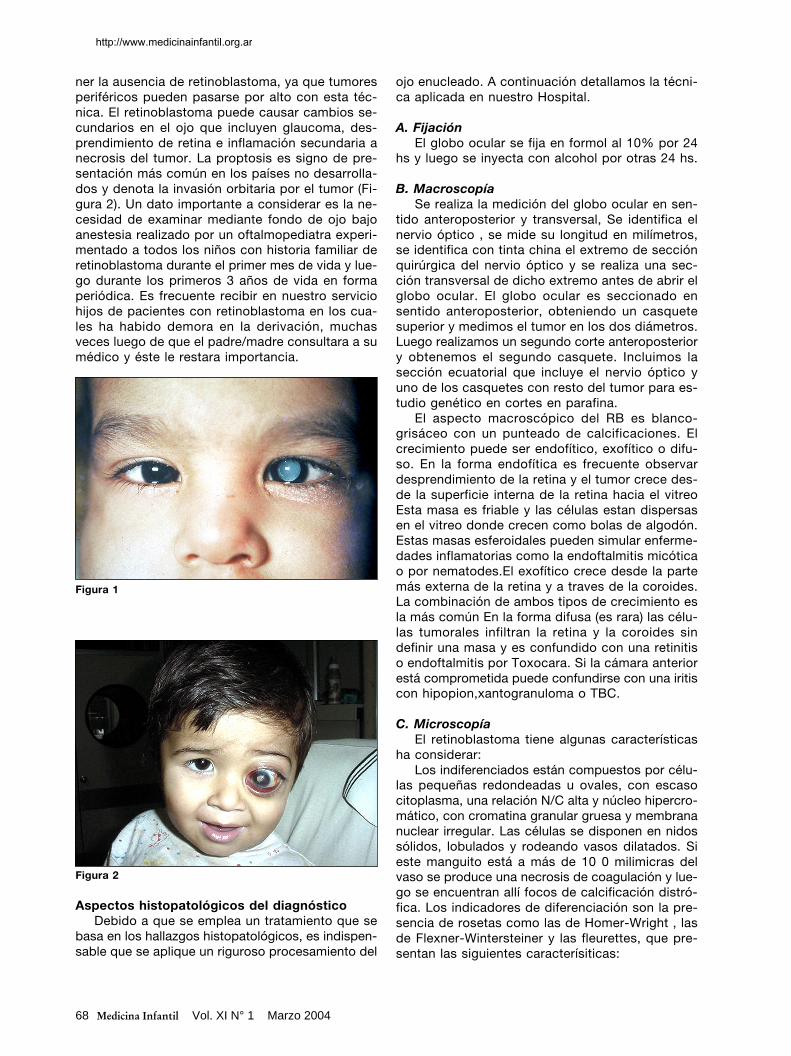
ner la ausencia de retinoblastoma, ya que tumoresperiféricos pueden pasarse por alto con esta téc-nica. El retinoblastoma puede causar cambios se-cundarios en el ojo que incluyen glaucoma, des-prendimiento de retina e inflamación secundaria anecrosis del tumor. La proptosis es signo de pre-sentación más común en los países no desarrolla-dos y denota la invasión orbitaria por el tumor (Fi-gura 2). Un dato importante a considerar es la ne-cesidad de examinar mediante fondo de ojo bajoanestesia realizado por un oftalmopediatra experi-mentado a todos los niños con historia familiar deretinoblastoma durante el primer mes de vida y lue-go durante los primeros 3 años de vida en formaperiódica. Es frecuente recibir en nuestro serviciohijos de pacientes con retinoblastoma en los cua-les ha habido demora en la derivación, muchasveces luego de que el padre/madre consultara a sumédico y éste le restara importancia.
ojo enucleado. A continuación detallamos la técni-ca aplicada en nuestro Hospital.
A. FijaciónEl globo ocular se fija en formol al 10% por 24
hs y luego se inyecta con alcohol por otras 24 hs.
B. MacroscopíaSe realiza la medición del globo ocular en sen-
tido anteroposterior y transversal, Se identifica elnervio óptico , se mide su longitud en milímetros,se identifica con tinta china el extremo de secciónquirúrgica del nervio óptico y se realiza una sec-ción transversal de dicho extremo antes de abrir elglobo ocular. El globo ocular es seccionado ensentido anteroposterior, obteniendo un casquetesuperior y medimos el tumor en los dos diámetros.Luego realizamos un segundo corte anteroposteriory obtenemos el segundo casquete. Incluimos lasección ecuatorial que incluye el nervio óptico yuno de los casquetes con resto del tumor para es-tudio genético en cortes en parafina.
El aspecto macroscópico del RB es blanco-grisáceo con un punteado de calcificaciones. Elcrecimiento puede ser endofítico, exofítico o difu-so. En la forma endofítica es frecuente observardesprendimiento de la retina y el tumor crece des-de la superficie interna de la retina hacia el vitreoEsta masa es friable y las células estan dispersasen el vitreo donde crecen como bolas de algodón.Estas masas esferoidales pueden simular enferme-dades inflamatorias como la endoftalmitis micóticao por nematodes.El exofítico crece desde la partemás externa de la retina y a traves de la coroides.La combinación de ambos tipos de crecimiento esla más común En la forma difusa (es rara) las célu-las tumorales infiltran la retina y la coroides sindefinir una masa y es confundido con una retinitiso endoftalmitis por Toxocara. Si la cámara anteriorestá comprometida puede confundirse con una iritiscon hipopion,xantogranuloma o TBC.
C. MicroscopíaEl retinoblastoma tiene algunas características
ha considerar:Los indiferenciados están compuestos por célu-
las pequeñas redondeadas u ovales, con escasocitoplasma, una relación N/C alta y núcleo hipercro-mático, con cromatina granular gruesa y membrananuclear irregular. Las células se disponen en nidossólidos, lobulados y rodeando vasos dilatados. Sieste manguito está a más de 10 0 milimicras delvaso se produce una necrosis de coagulación y lue-go se encuentran allí focos de calcificación distró-fica. Los indicadores de diferenciación son la pre-sencia de rosetas como las de Homer-Wright , lasde Flexner-Wintersteiner y las fleurettes, que pre-sentan las siguientes caracterísiticas:
Aspectos histopatológicos del diagnósticoDebido a que se emplea un tratamiento que se
basa en los hallazgos histopatológicos, es indispen-sable que se aplique un riguroso procesamiento del
Figura 1
Figura 2
http://www.medicinainfantil.org.ar

Retinoblastoma 69
Homer-Wright (H-W): células tumorales dispues-tas en círculo con una variable maraña de proce-sos celulares fibrilares ligeramente eosinófilos sinuna luz
Flexner-Wintersteiner (F-W): están compuestaspor una hilera de células tumorales rodeando unaluz central con una bien demarcada membranaeosinófila análoga a la capa limitante externa de laretina normal.
Fleurette (F): Es infrecuente, está compuesta porcélulas de mayor tamaño, con abundante citoplas-ma acidófilo y dispuestas en una especial flor delis. En los retinocitomas predominan estas estruc-turas.
Aplicamos un protocolo donde evaluamos:- Grado de diferenciación: indiferenciado, pobre-
mente diferenciado y diferenciado.- Tipos de rosetas: H-W, F-W y F.- Siembras: vítreo y subretinales- Invasión: coroides (parcial o total), iris, cuerpo
ciliar, cámara anterior.- Invasión a la esclera: con o sin extensión
transescleral.- Invasión del nervio óptico: prelaminar, retrolami-
nar y el límite de sección quirúrgica ( libre ocomprometido).
TratamientoDos aspectos del tratamiento del retinoblasto-
ma deben ser considerados; primeramente las op-ciones de la terapéutica local para tratar la enfer-medad intraocular y en segundo lugar el tratamientosistémico para los pacientes con enfermedad ex-traocular, regional o metastásica.
En los países desarrollados, la mayoría de lospacientes presentan enfermedad intraocular conuna sobrevivida de alrededor del 95%. En esoscasos el tratamiento contempla la preservaciónpotencial de la visión tratando de minimizar lassecuelas a largo plazo. El tamaño, número y loca-lización del tumor y el estado del ojo remanentedeben ser tenidos en cuenta en la elección del tra-tamiento. Muchos pacientes con retinoblastomabilateral presentan al ingreso enfermedadintraocular necesitando frecuentemente la enuclea-ción de un ojo. En general, presentan enfermedadmenos avanzada en el ojo contralateral el cualusualmente puede ser preservado. En los países nodesarrollados, el retinoblastoma es usualmentediagnosticado cuando la diseminación extraoculares evidente. En esos casos el tratamiento del reti-noblastoma tiene como objetivo salvar la vida delos pacientes ya que la muerte por enfermedadmetastásica es una amenaza probable.
CirugíaLa enucleación es el tratamiento más simple y
seguro del retinoblastoma. Es imprescindible que
la realice un oftalmólogo pediatra experimentado,ya que es importante obtener un cabo de nervioóptico lo más largo posible. Luego de varias sema-nas, se coloca una prótesis para minimizar los efec-tos cosméticos. Cuando la enucleación es realiza-da en los primeros dos años de vida, en general sedesarrolla una asimetría facial por inhibición delcrecimiento de la órbita.
La enucleación también está indicada cuando nopuede obtenerse una visión adecuada con el tra-tamiento conservador, incluso si los tumores soncontrolados. La mayoría de los ojos estadio V dela clasificación de Reese-Ellsworth (los más avan-zados) (Tabla 1) requiere enucleación especialmen-te cuando no se obtiene una buena visión luego deun tratamiento exitoso del tumor. Sin embargo siel ojo contralateral tiene enfermedad avanzada,puede tomarse una conducta mas conservadora yalgunos ojos podrían ser conservados. La enuclea-ción es mandatoria cuando hay glaucoma, invasiónde la cámara anterior, rubeosis iridis y cuando laterapia local no puede ser evaluada debido a lapresencia de cataratas o a una dificultad en el se-guimiento cercano del paciente. Este hecho es degran importancia en nuestro medio, ya que no debeponerse en juego la vida del paciente con el fin deintentar vanamente la preservación ocular a todacosta en pacientes en quienes no se puede garan-tizar un adecuado seguimiento. La enucleación, porlo tanto no debe ser demorada en casos como losdescriptos, en especial cuando por razones socia-les o geográficas el paciente no puede ser contro-lado adecuadamente.
La enucleación podría ser demorada cuandoexiste evidente extensión extraocular al diagnósti-
TABLA 1: CLASIFICACION DE REESE ELLSWORTH.
Grupo I (Muy Favorable)A: Tumor solitario, menor de 4 diámetros discales a la altura o
posterior al ecuadorB: Tumores múltiples, ninguno mayor de 4 diámetros discales
a la altura o posteriores al ecuadorGrupo II (Favorable)A Tumor solitario de entre 4 a 10 diámetros discales a la altu-
ra o posterior al ecuadorB: Tumores múltiples de entre 4 a 10 diámetros discales poste-
riores al ecuadorGrupo III (Dudoso)A: Cualquier lesión anterior al ecuadorB: Tumores únicos mayores de 10 diámetros discales posterio-
res al ecuadorGrupo IV (Desfavorable)A: Tumores múltiples, algunos mayores de 10 diámetros discalesB: Cualquier lesión extendiéndose anteriormente a la ora serrataGrupo V (Muy desfavorable)A: Tumores involucrando más de la mitad de la retinaB: Siembra vitrea
http://www.medicinainfantil.org.ar

70 Medicina Infantil Vol. XI N° 1 Marzo 2004
co. Las masas orbitarias usualmente disminuyen detamaño después de la quimioterapia neoadyuvante,permitiendo la enucleación, y de este modo evitaruna exenteración orbitaria, la cual conlleva unasecuela cosmética mayor. (4). Hay pocas, si es queaún existen, indicaciones de exenteración orbitariaen el retinoblastoma hoy en día. La cirugíaintraocular, como por ejemplo la vitrectomía estacontraindicada en pacientes en quienes se sospe-che retinoblastoma, ya que esto puede incremen-tar el riesgo de recaída orbitaria5.
Radioterapia externaEl Retinoblastoma es un tumor radiosensible y
la radioterapia externa es la terapéutica local másefectiva para su tratamiento. Sin embargo los efec-tos adversos a largo plazo limitan su uso. La radio-terapia externa se administra con acelerador linealcon dosis de 40 a 45 Gy abarcando la totalidad dela retina. Los lactantes deben ser anestesiados einmovilizados durante el procedimiento. Se requiereuna estrecha cooperación entre el oftalmólogo y elradioterapeuta para planificar los campos a irradiar.La mayoría de los radioterapeutas utilizan un cam-po lateral en forma de D, el cual minimiza el riesgode inducir cataratas con la radiación. No obstantela retina anterior no recibe una dosis plena con estatécnica y pueden aparecer tumores recurrentescercanos a la ora serrata .
El éxito de la terapia de radiación externa conesta técnica depende no sólo del tamaño, sino tam-bién de la localización del tumor. Las secuelas alargo plazo de la radioterapia son preocupantes, locual ha hecho que se busquen nuevas modalida-des de tratamiento. Al igual que la enucleación, laradioterapia externa causa inhibición del crecimien-to del hueso de la órbita ocasionando alteracionescosméticas y más importante, ha sido asociada conel incremento del riesgo de segundas neoplasias.El uso de nuevas técnicas de radioterapiatridimensional no ha sido evaluado completamen-te y debe utilizarse solamente en protocolos deexperimentación.
La radioterapia externa está también indicadapara la enfermedad orbitaria evidente y tambiénpara la invasión del margen de resección final delnervio óptico. En ésta instancia el quiasma debe serincluido en el campo.
Placas de radioterapiaPlacas radioactivas epiesclerales usando 60Co,
106Ru y 125I son usadas en el tratamiento del retino-blastoma6. Las placas son insertadas en el quiró-fano con sutura por fuera de la esclera. Luego sonremovidas en una segunda operación unos díasdespués cuando la dosis en el tumor fue alcanza-da. La placa de radioterapia administra una dosisefectiva en el tumor y de este modo se preservan
los tejidos circundantes de los efectos adversos dela radioterapia. Las placas son usualmente indica-das para tumores de mediano tamaño, no pasiblesde ser tratados con crio o fotocoagulación. El 125Ies el isótopo preferido porque la radiación queemite es segura tanto para el paciente como parael personal médico.
Crio y fotocoagulaciónEstas modalidades de tratamiento son utilizadas
para tratar tumores pequeños (usualmente meno-res de 5 mm) y tumores accesibles. Pueden repe-tirse varias veces hasta que el control local es al-canzado. La crioterapia es usualmente prescriptapara tratar tumores anteriores; es aplicada con unasonda localizada sobre la conjuntiva .Por otro lado,la fotocoagulación es generalmente utilizada paratumores posteriores usando láser de argon oxenon. No obstante, la fotocoagulación debe evi-tarse en tumores cercanos a la mácula porque pue-de causar severas cicatrices ocasionando ambliopíagrave a pesar de lograr el control del tumor. Am-bas modalidades causan poca o ninguna morbili-dad, sin secuelas a largo plazo. El uso determoquimioterapia para tumores del polo posteriorha sido recientemente descripto. Esta técnica sebasa en el efecto sinérgico del carboplatino endo-venoso y la hipertermia utilizada con el diodo lá-ser7.
QuimioterapiaEn el pasado, la quimioterapia, sólo era utiliza-
da para el tratamiento de la enfermedad metastá-sica. Sin embargo, recientemente, la mayoría delos grupos ha usado la quimioterapia como trata-miento primario para la enfermedad intraocular,cuando el tratamiento local no era posible por elgran tamaño tumoral, con el objetivo de disminuirel volumen y hacerlos candidatos para el trata-miento local8,9. Esto se ha dado en llamar “quimio-reducción”, y ha logrado en algunos casos evitarla enucleación o la radioterapia externa en casosselectos. El carboplatino es el agente utilizado másfrecuentemente ya que tiene buena penetración enel ojo. La vincristina y el etopósido son tambiénutilizados, sin embargo su penetración ocular esdesconocida. La mayoría de los tumoresintraoculares usualmente muestran una marcadadisminución en su tamaño luego de la quimiote-rapia sistémica, sin embargo es necesario conso-lidar localmente con otros tratamientos en la ma-yoría de los casos para prevenir una recaída de laenfermedad. La localización del tumor, su tama-ño y la edad del paciente se correlacionan con elgrado de respuesta a la quimioterapia. La mayo-ría de los pacientes con tumores menos avanza-dos (grupo I a IV de la clasificación de Reese-Ellsworth) responden favorablemente a la quimio-
http://www.medicinainfantil.org.ar

Retinoblastoma 71
reducción, obviando de este modo la radioterapiaexterna, que era el tratamiento de elección en elpasado. El grado de preservación ocular tiene unrango de 74% a 100%8,9. Sin embargo, los pacien-tes del grupo V de Reese-Ellsworth, que presen-tan enfermedad avanzada y especialmente aque-llos con siembras vítreas se han obtenido los peo-res resultados con esta modalidad. Luego de ex-periencias alentadoras, fue reportado que la ma-yoría de ellos requiere radioterapia externa para uncontrol efectivo del tumor y muchos son última-mente enucleados luego de haber realizado qui-mioterapia y terapia de radiación externa8. Trata-mientos alternativos para estos pacientes de altoriesgo incluyen la administración de carboplatinoperiocular y la asociación de ciclosporina endove-nosa con carboplatino para evitar la quimioresis-tencia mediada por la glicoproteina P , la cual hasido postulada por algunos grupos como la poten-cial causa de fallo al tratamiento. Debe recordarseque en estos pacientes la quimioterapia se utilizacon el fin de evitar un tratamiento aunque efecti-vo, tóxico a largo plazo, como es la radioterapiaexterna y el uso de drogas con toxicidad tardíacomo los antraciclínicos (Doxorrubicina, Idarubici-na) o los alquilantes (Ciclofosfamida o la Ifosfami-da) están contraindicados.
Aunque la quimioreduccion seguida de trata-miento local es considerada un tratamiento están-dar en la actualidad, hay varias cuestiones no re-sueltas. Los resultados a largo plazo y la seguri-dad son desconocidos, especialmente su poten-cial riesgo de inducir segundas neoplasias. Losagentes quimioterápicos utilizados pueden indu-cir leucemias secundarias, especialmente las epi-podofilotoxinas (como el Etopósido), usadas pormuchos grupos. El régimen óptimo todavía no hasido identificado, así como existe controversiaacerca de la duración del tratamiento y la necesi-dad y tipo de tratamiento local.
Los pacientes con enfermedad unilateral sonfrecuentemente manejados con enucleación. Sinembargo, algunos casos seleccionados podríanbeneficiarse con quimioreducción y terapia localcomo por ejemplo aquellos pacientes en quienes sesospecha enfermedad hereditaria. Finalmente el tra-tamiento con quimioreducción y tratamiento localrequiere un manejo meticuloso el cual solo estadisponible en centros especializados, siendo des-aconsejable para centros con escasa experienciaen esta enfermedad, en especial en la esferaoftalmológica.
El rol de la quimioterapia adyuvante para pa-cientes con factores de riesgo histopatológicospara recaída extraocular es un tema de controver-sia. Es esencial una correcta estatificación histo-patológica para definir los diferentes riesgos derecaída luego de la enucleación 1,3. El oncólogo
pediatra se enfrenta al dilema de prescribir quimio-terapia adyuvante para todos los pacientes conprobables factores de riesgo o abstenerse de in-dicarla en los casos controvertidos y tratar agre-sivamente a aquellos que recaen10. La quimiote-rapia adyuvante no elimina completamente la po-sibilidad de recaída extraocular, y en grupos conbajo riesgo de recaída su beneficio no está pro-bado. Debido a que la tasa de recaída en estapoblación es baja; estudios randomizados compa-rando tratamiento adyuvante versus observaciónrequerirían un número importante de pacientespara su realización. Muchos autores consideran larecaída extraocular como un evento catastrófico,y por lo tanto recomiendan quimioterapia adyuvan-te para todos los pacientes con factores de ries-go histopatológicos11. Sin embargo en los añosrecientes se ha visto que muchos pacientesrecaídos pueden ser salvados con una terapia in-tensiva10-12. Por lo tanto, nuestro grupo y otroscentros en el exterior consideran que evitar laquimioterapia adyuvante para los pacientes conbaja probabilidad de recaída y tratar agresivamen-te a aquellos que recaen, lo cual es una alternati-va razonable10. Hay acuerdo universal en que laquimioterapia adyuvante no es necesaria para lospacientes con enfermedad solo limitada a la reti-na y en aquellos con invasión del nervio ópticoprelaminar10. El rol de la quimioterápia en la inva-sión coroidea aislada es controvertido. Se ha su-gerido que una vez que el tumor esta dentro dela coroides podría ganar acceso a la circulaciónsistémica ocasionando metástasis hematógenas,justificando de este modo el uso de terapia adyu-vante11. Sin embargo en nuestra serie de 40 pa-cientes con invasión coroidea aislada tratados conenucleación como única estrategia, uno sólo ex-perimentó recaída extraocular1. De este modo, deacuerdo a nuestra experiencia, la quimioterapiaadyuvante no es necesaria en ésta población. Lainvasión coroidea tal vez sea relevante en términospronósticos cuando se combina con invasión post-laminar del nervio óptico. La invasión del nervioóptico más allá de la lámina cribosa es un factorde riesgo para recaída extraocular, especialmentecuando el margen de sección está involucrado.Cuando el margen de sección esta libre de tumor,el manejo es controvertido. En nuestro centro, lospacientes con esta condición no reciben otra tera-pia que la enucleación, siempre y cuando no hayainvasión coroidea importante o compromiso de laesclera 1. En nuestra serie publicada de pacientestratados con enucleación y sin terapia adyuvante,ninguno recayó1. Los pacientes con compromisomasivo de la coroides concomitante a la invasiónretrolaminar del nervio óptico con o sin invasiónde la esclera tienen un mayor riesgo de recaída yla quimioterapia adyuvante está indicada. Los pa-
http://www.medicinainfantil.org.ar

72 Medicina Infantil Vol. XI N° 1 Marzo 2004
cientes con invasión del margen de sección delnervio óptico tienen una alto riesgo de recaída. Latasa de sobrevivida en el pasado fue reportada desolamente un 40%13. Sin embargo, con el trata-miento multimodal con quimioterapia adyuvante yradioterapia orbitaria abarcando el quiasma ópti-co, 11de 14 pacientes sobrevivieron en nuestrainstitución9 en el primer estudio y 7 de 10 en elsegundo1,3. El rol de la quimioterapia intratecal enestas situaciones permanece todavía sin ser es-tablecido, existiendo tal vez un espacio para nue-vas drogas como el Topotecan o la Tiotepa. Deacuerdo a nuestra experiencia, los pacientes coninvasión microscópica de la esclera tienen altoriesgo de recaída extraocular y deberían recibirterapia adyuvante1,10.
Cuando existe enfermedad extraocular se debeindicar quimioterapia pre-enucleación4. Doz y co-laboradores fueron los pioneros en el uso delcarboplatino y el etoposido, el cual se convirtió enel tratamiento estándar14. Un estudio de nuestrogrupo demostró una tasa de respuesta del 60% enel retinoblastoma extraocular con el uso de laidarrubicina, la cual sería el antraciclinico de elec-ción 15. La respuesta a otros agentes ha sido estu-diada en series pequeñas o en pacientes únicos.Los agentes más frecuentemente usados son elCisplatino, Ciclofosfamida, Ifosfamida, Vincristina,Doxorubicina, Topotecan y Tiotepa son agentes fre-cuentemente usados. Con una terapia multimodalque combine quimioterapia, cirugía limitada y radio-terapia de las áreas involucradas, nuestro grupoobtuvo una probabilidad de sobrevivida libre deeventos a los 5 años de 84% para los pacientescon enfermedad sólo orbitaria con o sin invasión deganglios preauriculares4. Por otra parte, los pacien-tes con enfermedad extraocular con metástasis adistancia , a pesar de que también alcanzan la re-misión completa con quimioterapia convencional,tienen usualmente una sobrevivida corta y fallecendentro de los primeros 2 años desde diagnósti-co1,3,4. El tratamiento con altas dosis de quimiote-rapia seguidas de rescate con transplante autólogode médula ósea ha mostrado ser eficaz para el tra-tamiento de pacientes con recaída sistémica, peroha dado resultados decepcionantes en el caso dediseminación en SNC12. Los regímenes de condi-cionamiento empleados usualmente incluyen lasdrogas mencionadas a dosis más altas. No hay unaterapia efectiva para estos pacientes y en la ma-yoría de los centros son tratados acorde a proto-colos de investigación o de manera paliativa. Es-
tos pacientes pueden necesitar de radioterapiacraneoespinal para alcanzar un control de la enfer-medad, pero en general la alta frecuencia de se-cuelas neuropsicológicas en los niños pequeñoscontraindica su uso.
Como conclusión cabe resaltar los avances ocu-rridos en los últimos años en el tratamiento del re-tinoblastoma, los que permitieron una mayor tasade sobrevida de pacientes con enfermedad avan-zada, una mayor posibilidad de preservación ocu-lar en aquellos niños con enfermedad intraocular yel descubrimiento de nuevos agentes útiles para eltratamiento de esta enfermedad. Sin embargo, elmayor desafío consiste en lograr diagnosticar estaenfermedad en estadíos tempranos, con lo cual seevitaría tener que utilizar estas terapias costosas ysofisticadas.
REFERENCIAS1. Chantada G, Fandiño A, Dávila MTG, et al. Results of a prospec-
tive protocol for the treatment of retinoblastoma. Cancer 2004;100:834-842
2. Chantada G, Fandiño A, Manzitti J, Urrutia L, Schvartzman E.Latediagnosis of retinoblastoma in a developing country. Arch Dis Child80:171-174,1999.
3. Schvartzman E, Chantada G, Fandiño A, et al (1996). Results of astage-based protocol for the treatment of retinoblastoma. J ClinOncol 1996; 14:1532-1536.
4. Chantada G, Fandiño A, Casak S et al (2003). Treatment of overtextraocular retinoblastoma. Med Pediatr Oncol 40, 158-161.
5. Shields CL, Honavar S, Shields JA, Demirci H, Meadows AT.Vitrectomy in eyes with unsuspected retinoblastoma. Ophthalmol-ogy. 2000;107(12):2250-5.
6. Shields CL, Shields JA, Cater J, Othmane I, Singh AD, Micaily B.Plaque radiotherapy for retinoblastoma: long-term tumor controland treatment complications in 208 tumors. Ophthalmology. 2001;108:2116-21
7. Lumbroso L, Doz F, Urbieta M, et al.(2002). Chemothermo-therapy in the management of retinoblastoma. Ophthalmology109, 1130-6.
8. Shields C, Honavar S, Meadows A et al (2002). Chemoreductionplus focal therapy for retinoblastoma: Factors predictive of needfor treatment with external beam radiotherapy or enucleation. AmJ Ophthalmol 133, 657-664.
9. Kingston J, Hungerford J, Madreperla S, Plowman P (1996). Re-sults of combined chemotherapy and radiotherapy for advancedintraocular retinoblastoma. Arch Ophthalmol 114, 1339-1343.
10. Chantada G, Dunkel I, Dávila MTG, Abramson DH. Retinoblastomawith high risk ocular pathological features: Who needs adjuvanttherapy? Br J Opthalmol 2004 (in Press)
11. Honavar G, Singh A, Shields C, et al (2002). Post enucleation ad-juvant therapy in high risk retinoblastoma. Arch Ophthalmol 120,923-931.
12. Dunkel IJ, Aledo A, Kernan NA et al (2000). Successful treatmentof metastatic retinoblastoma. Cancer 89, 2117-2121.
13. Magramm I, Abramson D, Ellsworth R: Optic nerve involvementin retinoblastoma: Ophthalmology 1989; 96:217-22
14. Doz F, Neuenshwander S, Plantaz D et al (1995). Etoposide andcarboplatin in extraocular retinoblastoma: A study by the SocietéFrançaise d'Oncologie Pédiatrique: J Clin Oncol 13, 902-909.
15. Chantada GL, Fandiño A, Mato G et al (1999). A phase II windowof idarubicin in children with extraocular retinoblastoma. J ClinOncol 17, 1847-1850.
http://www.medicinainfantil.org.ar