Embed Size (px)
Citation preview

Servicio de Anatomía Patológica
Hospital Central de Asturias
Dr N. Fuentes

CONSULTA DE NEUROLOGÍA FEBRERO 2007
� Debilidad en miembros inferiores desde Julio 2006
� Dificultad para articular el lenguaje
ANTECENDENTES PERSONALES
� NAMC, No diabetes, No HTA, No hábitos tóxicos….
� Antecedentes paternos
� Sin interes
ANTECENDENTES MATERNOS
� Madre fallecida con diagnóstico de DFT
� 7 tios maternos: Dos sanos
1 Fallecido por enfermedad psiquiátrica
1 Fallecido por ELA
1 Fallecida por DFT-ELA
2 Fallecidas por DFT
� 2 Hermanas menores sanas en la actualidad
�

EXAMEN FÍSICO
•Disartria
•Miembros superiores: Tono y fuerza normal con temblor fino distal
•ROT: Bicipitales y estilo-radiales ++++, tricipitales+++
•Miembros inferiores: Tono muscular aumentado, mas el derecho, fuerza
normal y simétrica, clonus rotuliano y aquileo bilateral. RCP extensor bilateral
•Sensibilidad y Cerebelo: normales
•Signos meningeos: Negativos
•Marcha espástica bilateral.
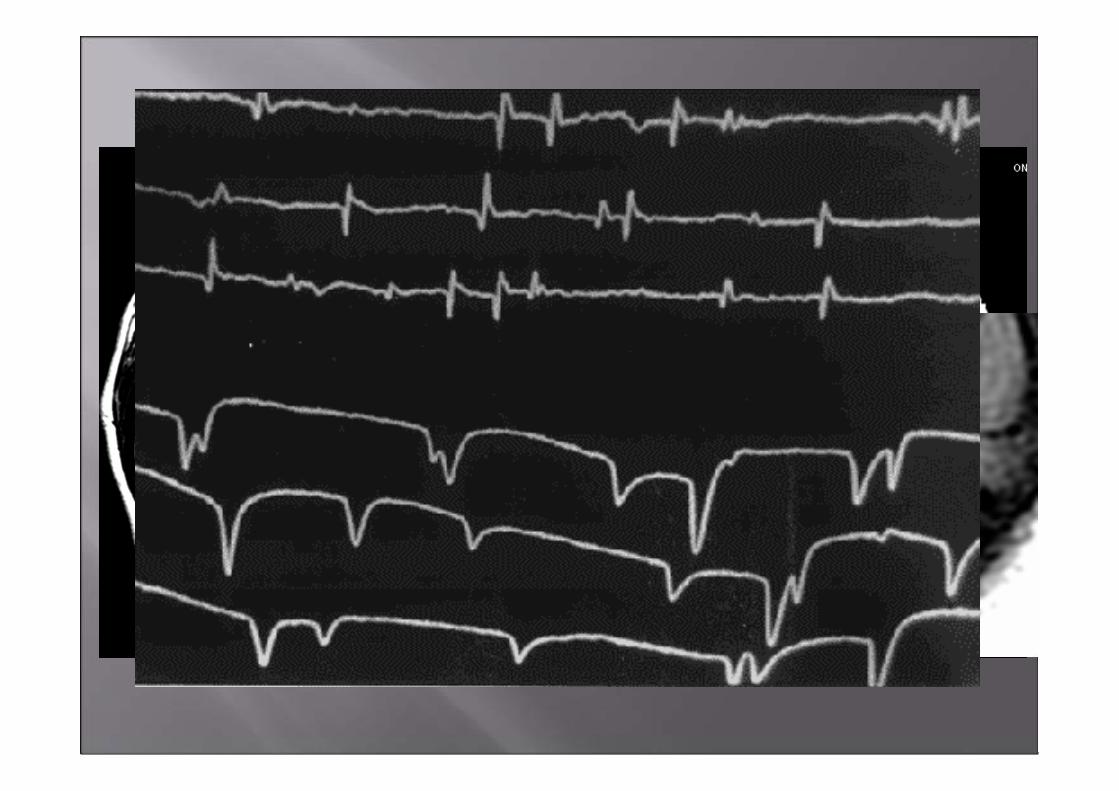
Estudios complementarios
•Hemograma: Normal,
•Bioquímica: Sin alteraciones relevantes
•Serología: Negativas.
• Estudios de autoimunidad: Negativos
•ECG: Normal
•TAC Craneal: Normal
•Estudio de LCR: Normal
•RMN: Prominencia llamativa de los surcos de ambas regiones silvianas
y área parietal. Hiperintensidad bilateral afectando a tractos córtico-
espinales de cáracter inespecífico.
•EMG: Signos de denervación activa en ambos músculos tibiales
anteriores
•ENG: Sin alteraciones específicas.

CLÍNICA
ANTECEDENTES FAMILIARES
PRUEBAS COMPLEMENTARIAS
DEMENCIA FRONTOTEMPORAL CON ELA FAMILIAR
SE PIDIÓ LA MUTACIÓN SOD-1(BARCELONA)

•Ausencia de disnea en reposo•Reflejos regresivos positivos•Reflejo mentoniano policinético•Hiperreflexia en miembros superiores•Trömer, Knips y Hoffman positivos•Espasticidad de los 4 miembros
•Anartria bulbar•Disfagia•Imposibilidad para protuir la lengua•Debilidad labial•Tetraparesia severa•Amiotrofia generalizada.
•Alimentación por Sonda nasogástrica•Infección respiratoria severa•Exitus 23/2/2008
INGRESO EL 18/01/2008

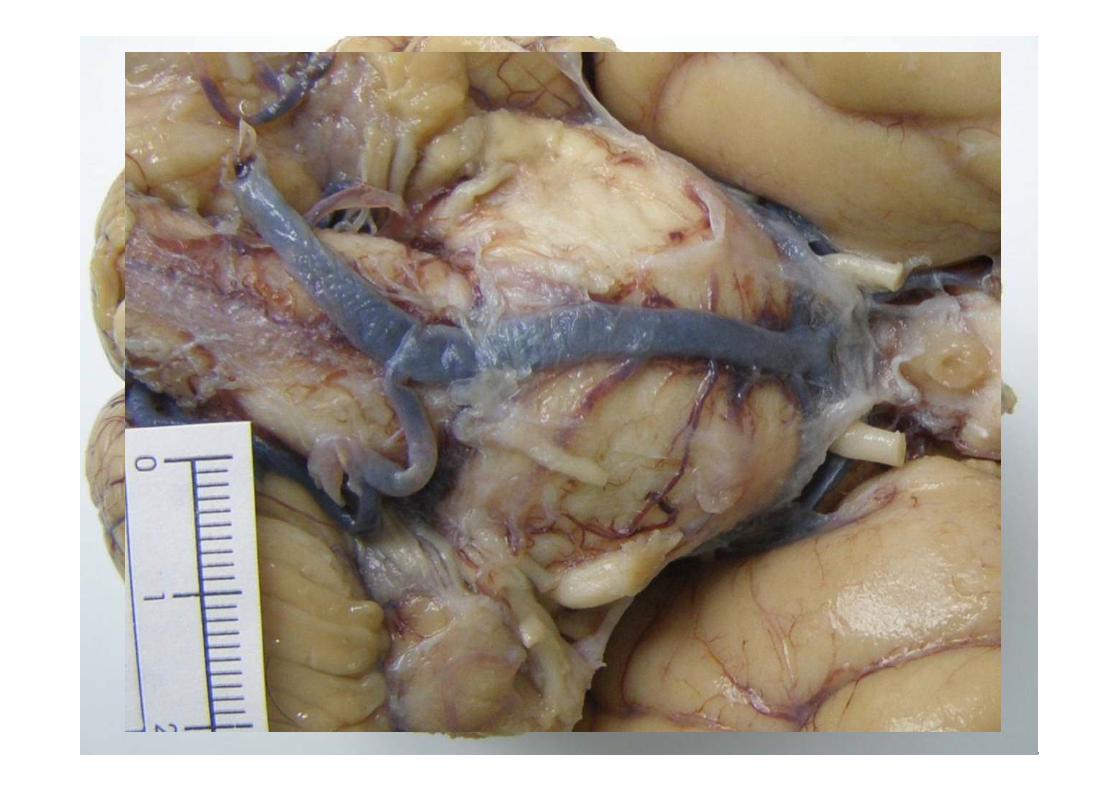
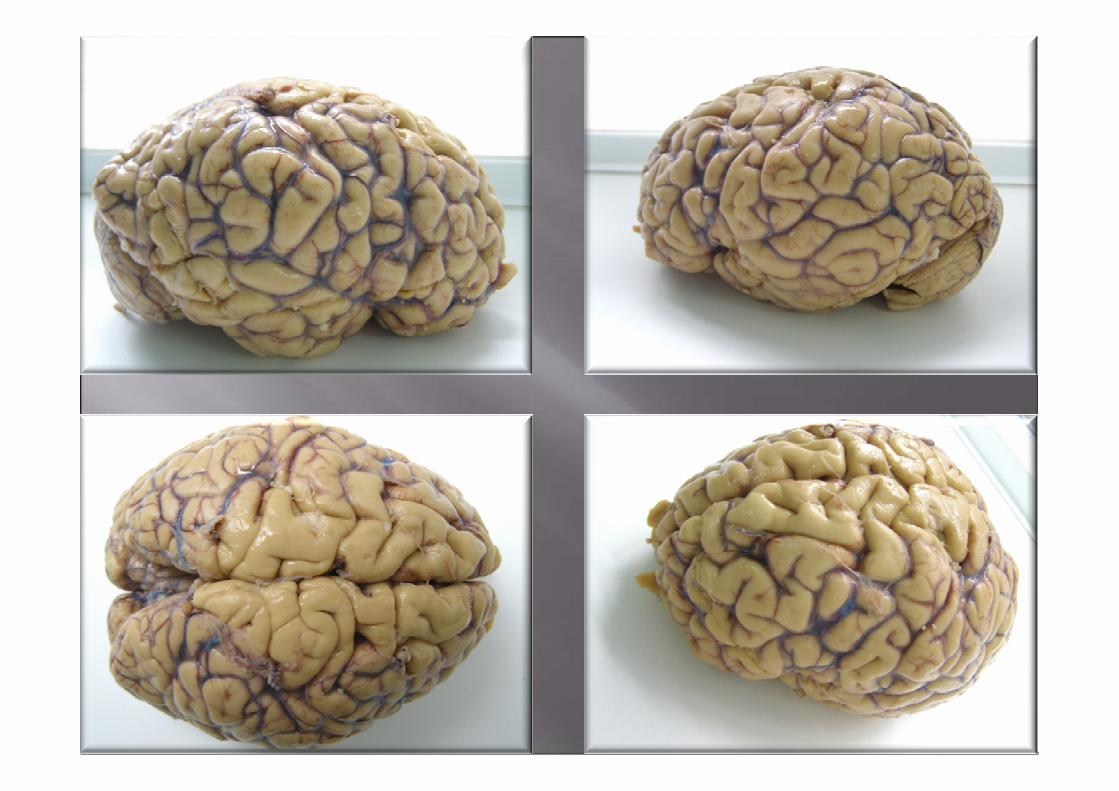
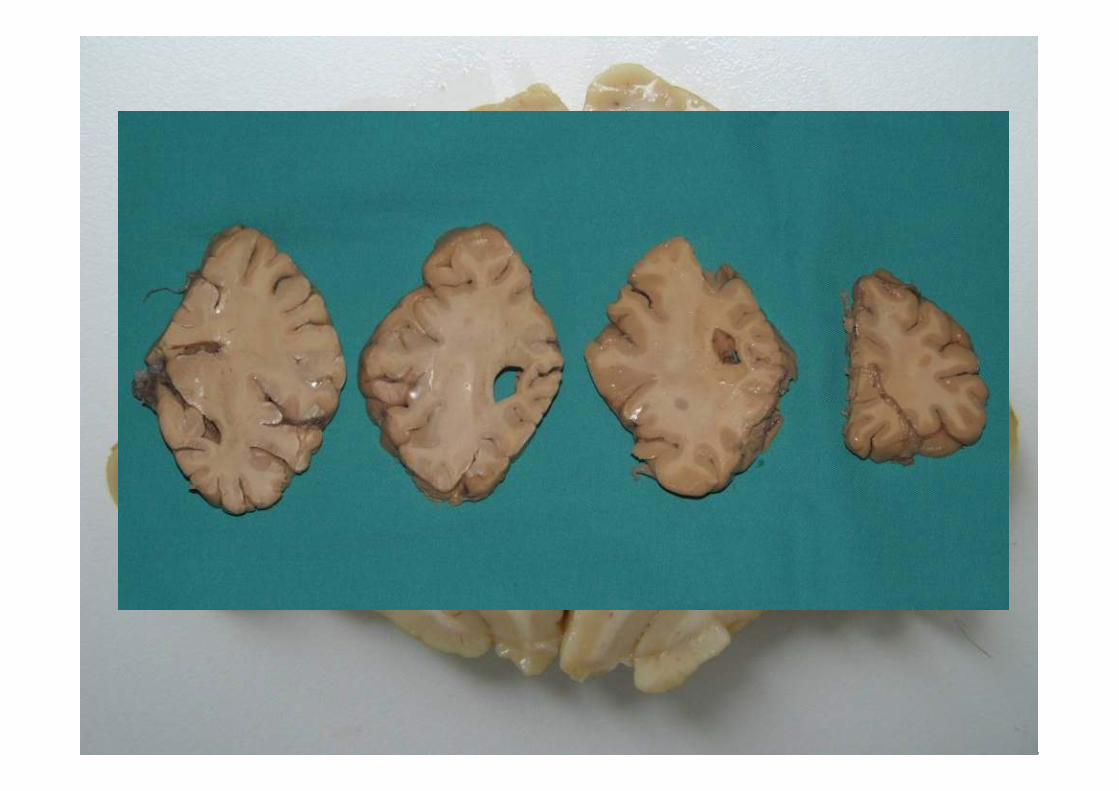
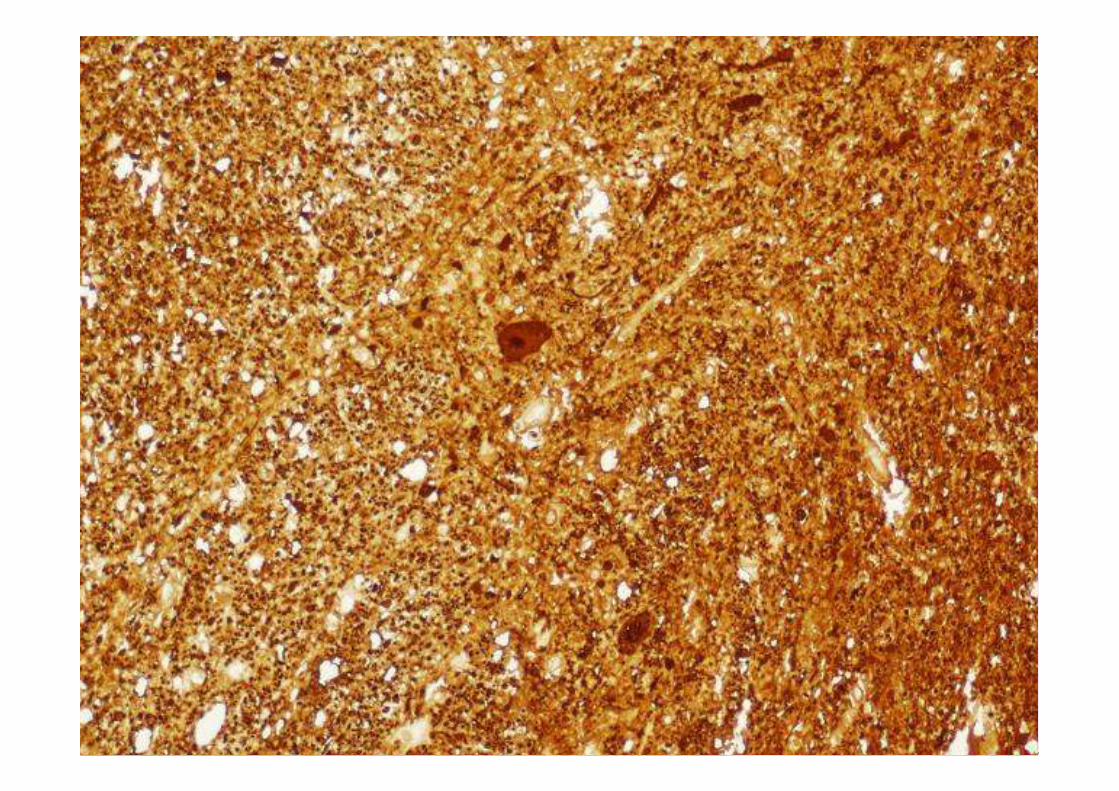
ESTUDIO MACROSCÓPICO


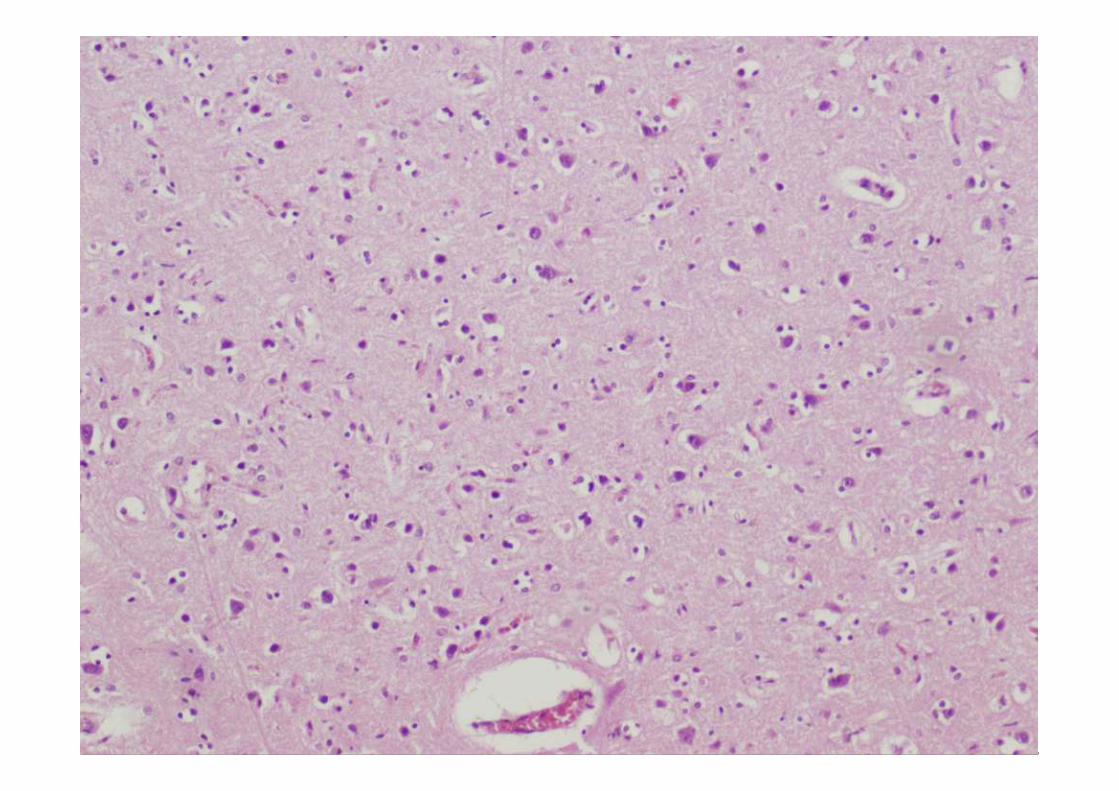
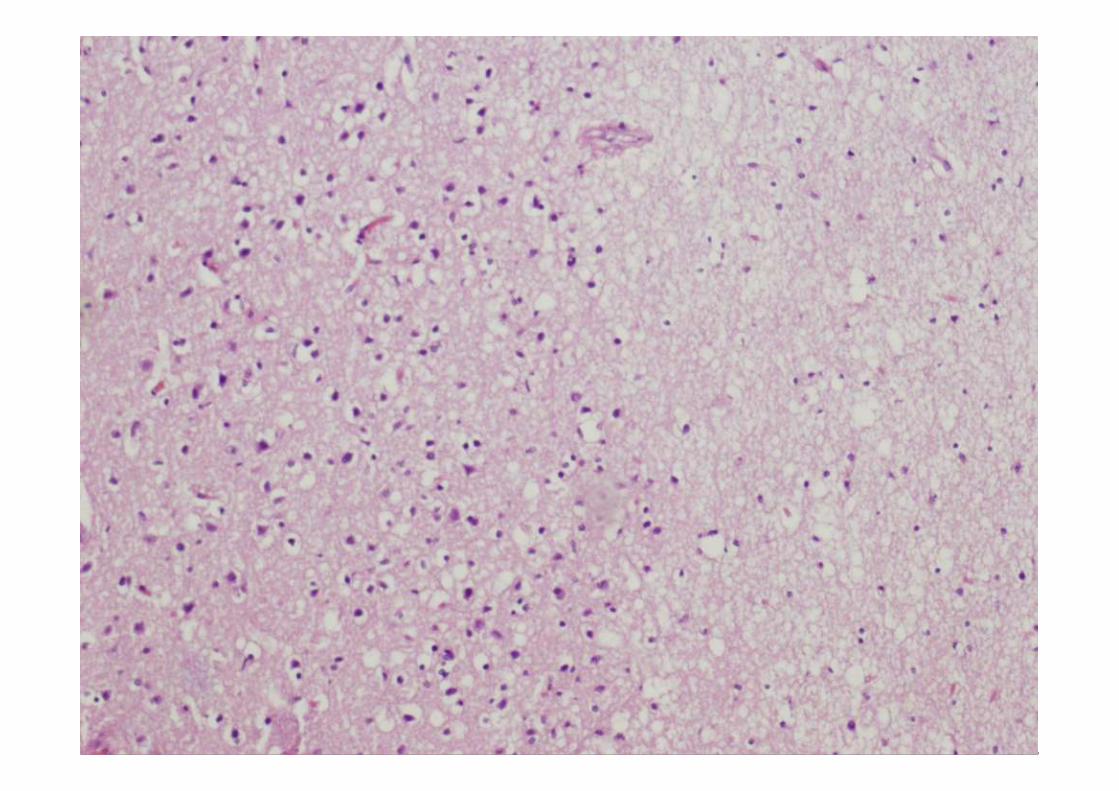
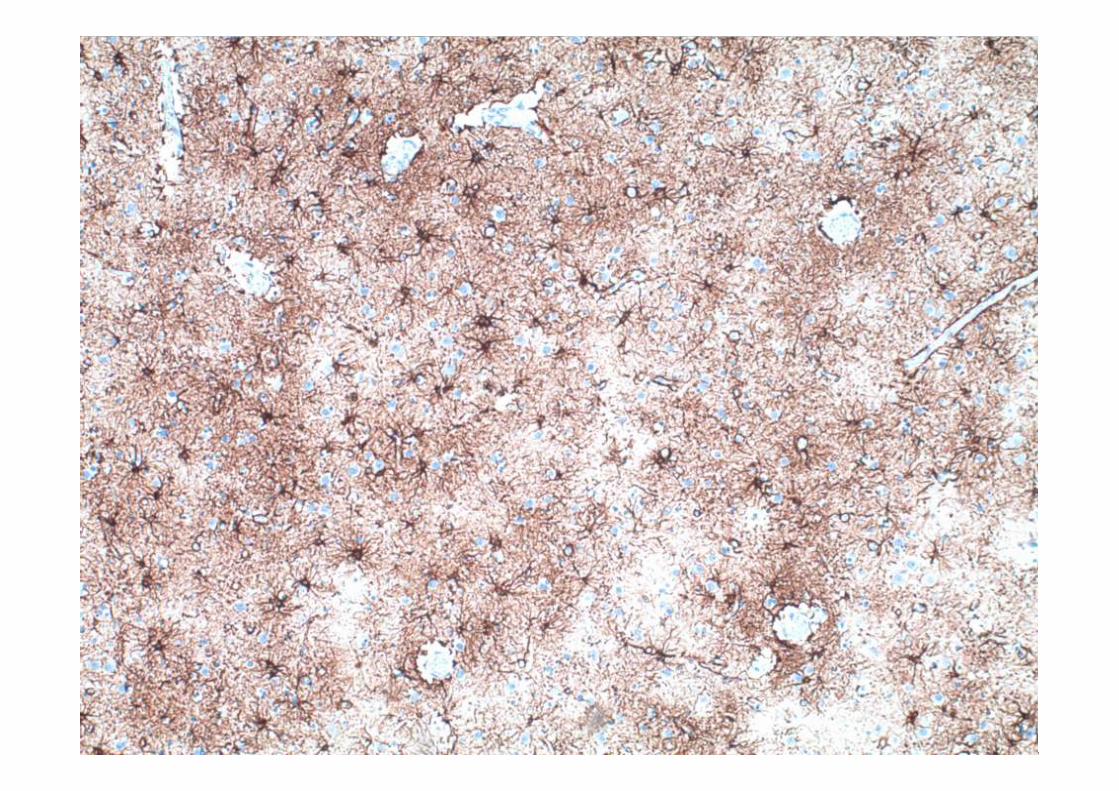
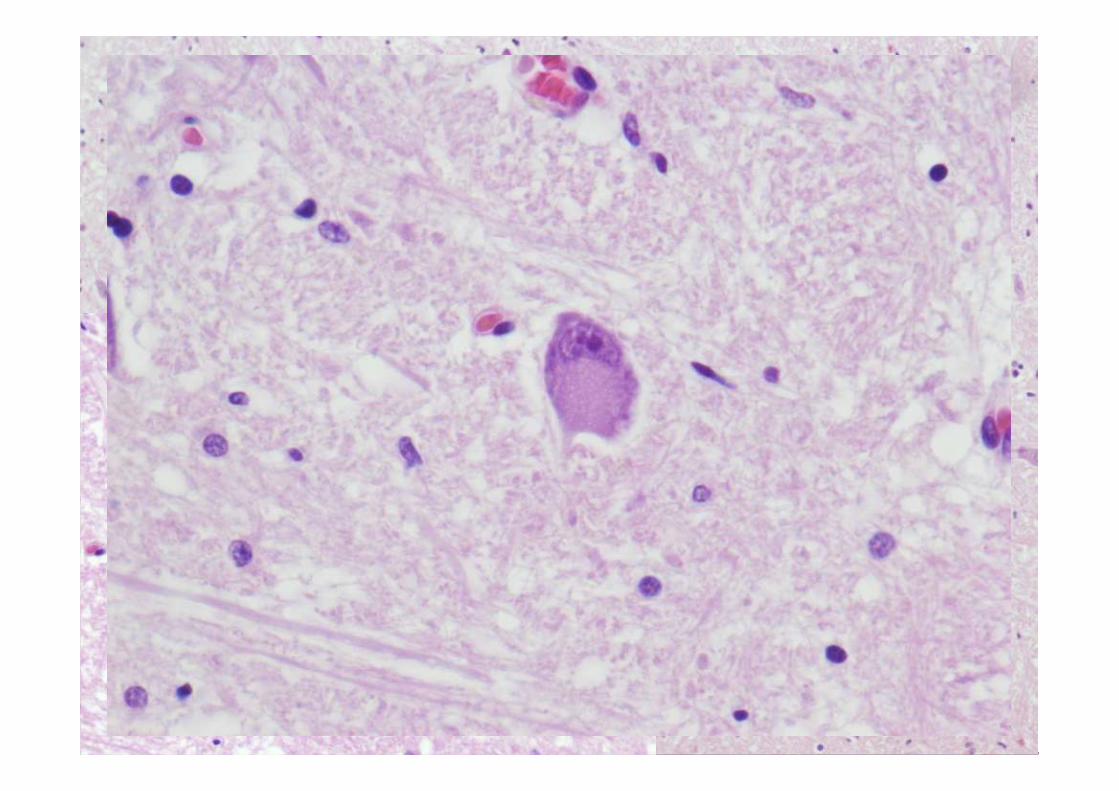
ESTUDIO MICROSCÓPICO

La esclerosis lateral amiotrófica (abreviadamente, ELA) es una enfermedad
degenerativa de tipo neuromuscular por la cual las motoneuronas( primera
y segunda) disminuyen gradualmente su funcionamiento y mueren,
provocando una parálisis muscular progresiva (de pronóstico mortal, pues
en sus etapas avanzadas los pacientes sufren parálisis total) que se
acompaña de una exaltación de los reflejos tendinosos (resultado de la
pérdida de los controles musculares inhibitorios).
ESCLEROSIS LATERAL AMIOTRÓFICA. CONCEPTO

�Descrita en 1869 por primera vez por Jean Martin-Charcot
�No afecta las funciones que no sean motoras, es decir sensitivas e inteligencia
�Las funciones motoras se afectan prácticamente en totalidad.
�La musculatura extrínseca del ojo se afecta en menor grado.
�El núcleo de Onuf no se encuentra afectado.
�Afecta a personas entre 40-70 años
�Varones mas que mujeres
�1-2 casos por cada 100000 habitantes

VARIANTES
ELA esporádica: Aparición al azar, sin poder indentificar
algún factor desencadenante.
ELA familiar: Autosómica dominante, se trata de una
variante familiar, que comprende entre el 5-10% de todos
los casos de ELA.

ETIOLOGIA
•Agentes infecciosos•Alteraciones en el sistema inmunitario
•Herencia•Sustancias tóxicas
•Desnutrición
?PATOGENIA
• Excitotoxicidad mediada por glutamato
•Estrés oxidativo
•Daño mitocondrial
•Alteraciones en el citoesqueleto
•Alteraciones en el transporte axoplasmático
•Neuroinflamación y autoinmunidad

SIGNOS CLÍNICOS
ASOCIADOS A MOTONEURONA SUPERIOR
• DISARTRIA
• DISFAGIA
• SIGNO DE HOFFMANN, BABINSKY Y CLONUS.
ASOCIADOS A MOTONEURONA INFERIOR
• DEBILIDAD MUSCULAR
• FASCICULACIONES
• HIPERREFLEXIA +SIGNO DE HOFFMANN EN MANOS + DEBILIDAD Y
FASCICULACIONES
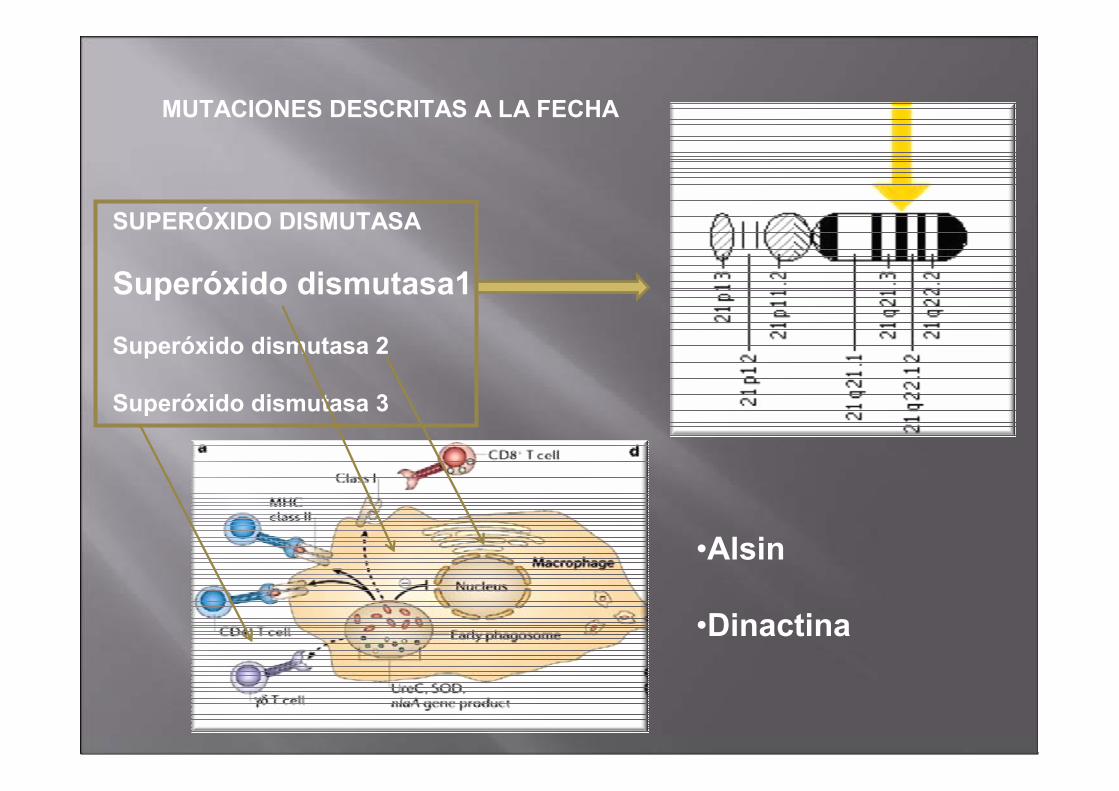
MUTACIONES DESCRITAS A LA FECHA
SUPERÓXIDO DISMUTASA
Superóxido dismutasa1
Superóxido dismutasa 2
Superóxido dismutasa 3
•Alsin
•Dinactina
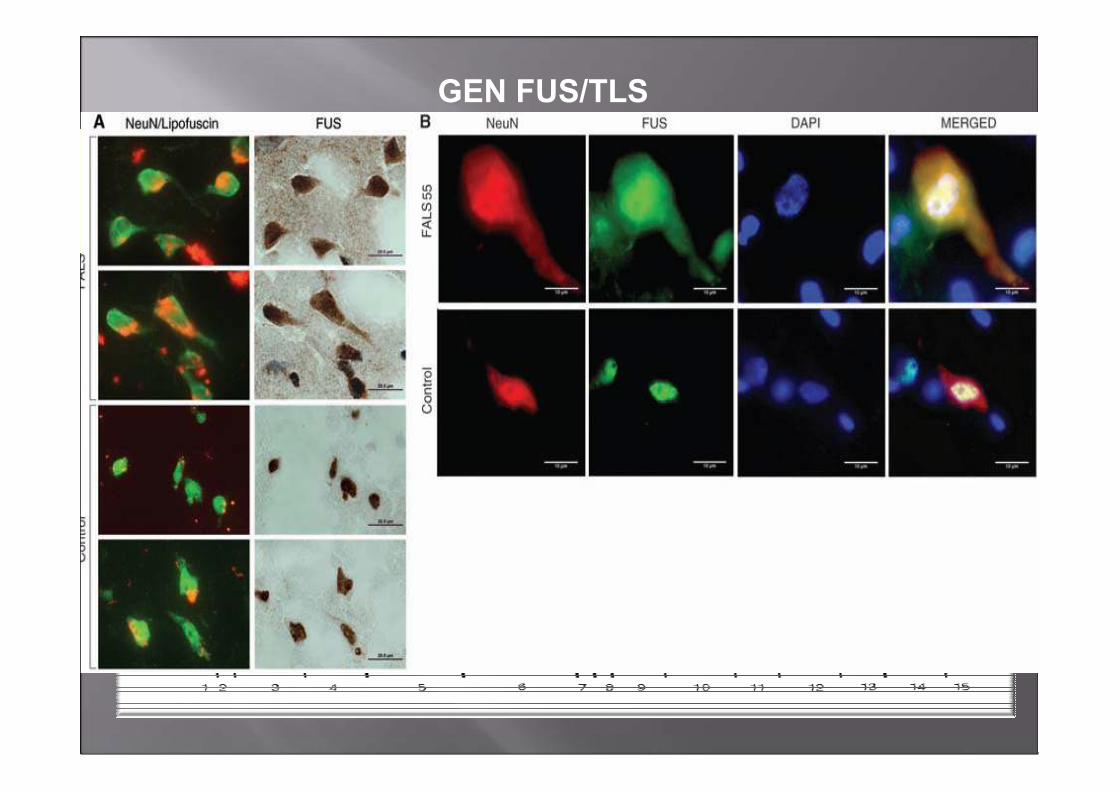
GEN FUS/TLS
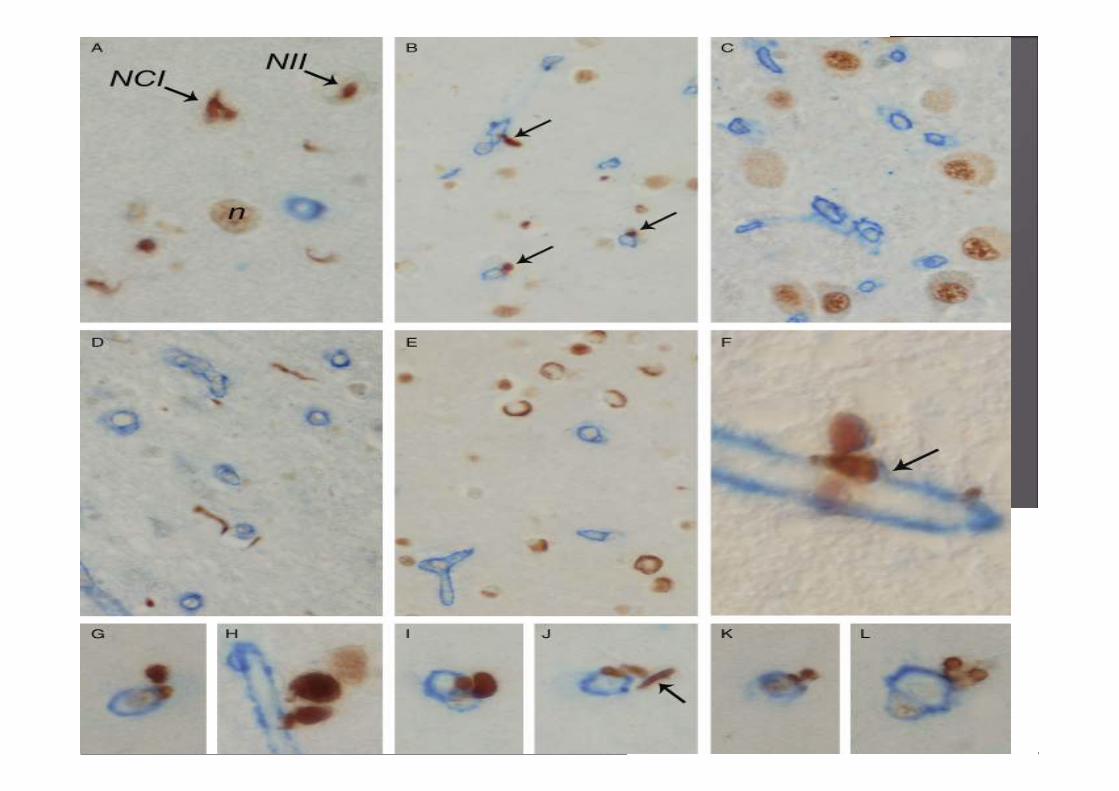
TARDBP
EXON-6
DFT+ELA

Macroscopía•Atrofia frontal y temporal anterior de leve a moderada, puede existir ausencia.
•Perdida de pigmento de la sustancia nigra
•Atrofia del tracto anterior de la médula espinal
Microscopía
• Perdida neuronal de leve a moderada, aunque puede estar ausente
• Gliosis reactiva de las capas II y III con microvacuolización.
• Degeneración que comprende la zona medial del lóbulo temporal, la región para
hipocampal y otras zonas del sistema límbico
• Perdida neuronal de la sustancia nigra y gliosis reactiva que puede ser leve o severa
• Degeneración de motoneuronas superior e inferior con perdida neuronal en el núcleo
hipogloso y cervical y a su vez del asta anterior de la médula espinal.
• Presencia de cuerpos de Bunina
• La degeneración del giro precentral y el tracto piramidal es moderada en comparación
con los casos clásicos de ELA.
• Inclusiones ubicuitina positivas y Tau negativas

DIAGNÓSTICOS DIFERENCIALES
•Neuropatía motora multifocal
•Miastenia gravis
•Esclerosis múltiple
•Páralisis pseudobulbar
•Miopatías
•Síndrome post-polio
•Enfermedad de la motoneurona reversible.

•PACIENTE CON ANTECEDENTES FAMILIARES DE ELA
•SIGNOS CLÍNICOS Y PATOLÓGICOS DE DEMENCIA FRONTOTEMPORAL Y ESCLEROSIS LATERAL AMIOTRÓFICA.
DEMENCIA FRONTOTEMPORAL Y ESCLEROSIS LATERAL AMIOTRÓFICA VARIANTE FAMILIAR

GRACIAS





























