Embed Size (px)
Citation preview

Silvia Pérez Casas Termodinámica de Soluciones
1
TERMODINÁMICA DE SOLUCIONES.
Una disolución es una mezcla homogénea de especies químicas
dispersas a escala molecular. Una disolución puede ser gaseosa, líquida o
sólida. Se dice que las soluciones son binarias cuando están formadas por
dos constituyentes; terciarias, cuando están formadas por tres
constituyentes, etc. El constituyente más abundante se conoce como
disolvente y el menos abundante como soluto, aunque por conveniencia
puede hacerse esta designación de otra manera.
I) Potencial químico
En este capítulo vamos a estudiar la termodinámica de disoluciones de
no electrolitos en las cuales no ocurre ninguna reacción. Recordamos que la
ecuación fundamental para la energía libre de Gibbs para una sustancia pura
está dada por dG = VdP – SdT.
Para una disolución en la cual el número de moles de las sustancias
presentes varía, G = G(T, P, n1, n2, ...), y por lo tanto
...2
,,21
,,1,,
dnn
Gdn
n
GdP
P
GdT
T
GdG
jjii nPTnPTnTnP
donde ni significa que todos los números de moles son constantes en la
diferenciación, mientras que nj significa que todos los números de moles,
excepto el de esa derivada, permanecen constantes en la diferenciación.
En esta expresión identificamos a ST
G
inP
,
y a VP
G
inT
,
y
definimos al potencial químico como:
PTii n
G
,
de esta manera tenemos:

Silvia Pérez Casas Termodinámica de Soluciones
2
...2211 dndnVdPSdTdG
Observamos que para una sustancia pura a temperatura y presión
constantes, el potencial químico es su energía libre molar de Gibbs:
n
G
Podemos encontrar la definición de potencial químico en función de otras
propiedades termodinámicas de la manera siguiente:
TSHG y PVUH , entonces TSPVUG y por lo tanto:
SdTTdSVdPPdVdGdU
SdTTdSVdPPdVdUdG
y sustituyendo la expresión para dG tenemos:
TdSSdTVdPPdVdndnVdPSdTdU ...2211
i
iidnTdSPdVdU
por lo tanto podemos definir al potencial químico también como:
, , j
ii S V n
U
n
Dado que PVUH , tenemos que VdPPdVdUdH y sustituyendo
la diferencial de la energía interna, VdPPdVdnTdSPdVdHi
ii , por
lo tanto:
i
ii dnTdSVdPdH
y entonces podemos expresar al potencial químico también como:
jnSPii n
H
,,
Finalmente, como PVATSPVUTSHG , tenemos que
VdPPdVdAdG . Despejando dA y sustituyendo la dG obtenemos:
VdPPdVdGdA
VdPPdVdnVdPSdTdA ii
i

Silvia Pérez Casas Termodinámica de Soluciones
3
ii dnSdTPdVdA
jnTVii n
A
,,
Dado que su definición es: PTi
i n
G
,
, el potencial químico es el
criterio de espontaneidad y equilibrio entre fases ya que los cambios de
fase se realizan a temperatura y presión constantes. Los cambios de fase
espontáneos son aquéllos en los cuales el cambio de potencial químico es
negativo y para que dos o más fases estén en equilibrio es necesario que
el potencial químico no cambie.
Tomando como ejemplo sencillo el agua, si tenemos cubitos de hielo a
250C y 1 atm, sabemos por experiencia que todo el hielo se fundirá
después de un tiempo, es decir, el proceso es espontáneo, por lo que el
cambio de potencial químico es negativo:
H2O (sólida) H2O (líquida)
(250C, 1 atm) (250C, 1 atm)
0 es decir: (líquido) - (sólido) 0; por lo tanto (líquido) (sólido).
De aquí concluimos que la fase más estable es aquélla que presenta el
menor potencial químico. Si queremos tener en equilibrio hielo y agua
líquida a 1 atm, sabemos que la temperatura deberá ser de 00C:
H2O (sólida) H2O (líquida)
(00C, 1 atm) (00C, 1 atm)
para que el hielo y el agua líquida estén en equilibrio, es necesario que
= 0, es decir, (líquido) - (sólido) = 0, entonces (líquido) = (sólido) y
concluimos que para dos o más fases de una sustancia estén en equilibrio,
es necesario que el potencial químico de la sustancia tenga el mismo valor
en todas las fases en que esté presente.
En una disolución de i constituyentes, i
iinG , entonces

Silvia Pérez Casas Termodinámica de Soluciones
4
i
iii
iiii dnVdPSdTdndndG
VdPSdTdndGi
ii
Esta expresión se conoce con el nombre de ecuación de Gibbs-Duhem
y a temperatura y presión constantes se convierte en:
0i
ii dn
Lo cual significa que si la composición varía, los potenciales químicos
no cambian independientemente sino de una manera relacionada.
Se puede demostrar mediante un procedimiento análogo que las
variaciones con la composición de cualquiera de las cantidades molares
parciales ( iJ ) están relacionadas por la ecuación:
i
ii Jdn
a temperatura y presión constantes.
II) Propiedades termodinámicas de las disoluciones ideales.
a) Cambios de energía libre, volumen, entalpía y entropía.
Consideremos que tenemos una disolución de gases ideales. Para el
gas A a una presión P antes de ser mezclado:
00 ln
P
PRTnGG
P
dPnRTVdPdG
AAiA
donde 0AG es la energía estándar del gas A (a la presión estándar P0).
Después de haberse mezclado los gases manteniendo la presión total
constante P y la temperatura constante T, para el gas A tenemos:
00 ln
P
PRTnGG fA
AAfA

Silvia Pérez Casas Termodinámica de Soluciones
5
es decir, estamos considerando que la presión final de la disolución es
igual a la presión que tenía cada uno de los gases antes de mezclarse,
como se muestra en la figura siguiente:
H2 , 4 atm y 298.15K
Ar, 4 atm y 298.15K H2 + Ar, 4 atm y 298.15K
Para cada uno de los gases que se mezclan, se aplican ecuaciones
equivalentes a las anteriores.
Para obtener el cambio de energía libre de la disolución tenemos:
ifmexcla GGG
00 ln
P
PRTnG fA
AA +0
0 lnP
PRTnG fB
BB +...0
0 lnP
PRTnG AA ...ln
00
P
PRTnG BB
iiimexcla
BB
AAfB
Bfa
Amezcla
xxNRTG
xRTN
NnxRT
N
Nn
P
PRTn
P
PRTnG
ln
...lnln...lnln
donde N es el número total de moles de la disolución.
En esta ecuación se observa que el cambio de energía libre de Gibbs
para una disolución ideal, siempre es negativo (dado que los logaritmos de
las fracciones molares siempre son negativos), por lo tanto sus
componentes se mezclan en todas proporciones de manera espontánea.
A partir de esta ecuación podemos calcular fácilmente los cambio de
entropía, volumen y entalpía de mezclado de manera sencilla:

Silvia Pérez Casas Termodinámica de Soluciones
6
iiT
mezcla xxNRT
GS ln
0
T
mezcla P
GV
0 mezclamezclamezcla STGH
De estas ecuaciones podemos ver que para una disolución ideal el
cambio de entropía es siempre positivo, el volumen no cambia y no se
genera ni se absorbe calor. ¡Obviamente es un comportamiento ideal! La
mayoría de las disoluciones no se comporta así.
b) Ley de Raoult.
Consideremos ahora una disolución formada por un soluto sólido (es
decir, no volátil, cuya presión de vapor se puede considerar cero) y un
disolvente volátil (con alta presión de vapor). Si colocamos el líquido
volátil en un recipiente previamente evacuado y mantenemos la
temperatura constante, el espacio por encima del líquido será ocupado por
el vapor del líquido puro, y la presión será la presión de vapor del líquido
puro. Al añadir un soluto no volátil, la presión de vapor disminuye
proporcionalmente a la cantidad de soluto añadido siguiendo la Ley de
Raoult:
02
002
0 1 xPxPPxPP
donde P es la solución de la disolución, P0 es la presión de vapor del
disolvente puro, x es la fracción molar del disolvente y x2 es la fracción
molar del soluto. Se considera que la disolución se comporta idealmente si
cada uno de sus constituyentes cumple la ley de Raoult en todo el
intervalo de concentraciones. La figura
c) Ley de Henry.

Silvia Pérez Casas Termodinámica de Soluciones
7
Mientras que en una disolución ideal la presión de vapor del
componente presente en pequeñas concentraciones (el soluto) también
cumple la ley de Roult, en soluciones reales existen desviaciones. No
obstante, es conveniente suponer que la presión de vapor del soluto
depende aproximadamente en forma lineal de la cantidad de soluto
presente a bajas concentraciones. Esta suposición es el contenido de la
ley de Henry, que establece que a bajas concentraciones la presión de
vapor del soluto cumple la ley de Henry:
HxkP
Los sistemas que obedecen la ley de Henry son menos ideales que los
que cumplen la ley de Raoult y se denominan soluciones ideales diluidas.
III) Propiedades termodinámicas de disoluciones reales.
El modelo más simple de una disolución es aquél en el cual todos los
componentes obedecen la ley de Raoult en todo el intervalo de
concentraciones. Aunque hay algunas disoluciones que efectivamente se
comportan de esta manera, la mayoría no lo hace así. Veremos que
mientras el comportamiento de los gases no ideales puede ser descrito
mediante la fugacidad, las disoluciones líquidas pueden describirse en
función de la actividad.
Propiedades molares parciales.
Si tenemos un recipiente muy grande de agua, digamos 1000 L, y le
agregamos 1 mol (18 g, o sea 18 mL) de agua, el volumen de nuestro
sistema se verá aumentado en 18 mL, es decir, el volumen del agua
después de la adición será de 1000.018 L. Por otro lado, si tenemos un
recipiente de 1000 L de etanol y a este sistema le agregamos 1 mol de agua
(18 mL), el volumen de nuestro sistema se verá aumentado solamente en
14 mL, es decir, el volumen final del sistema será de 1000.014 L, en este

Silvia Pérez Casas Termodinámica de Soluciones
8
caso 1 mol de agua agregada al alcohol solamente contribuye con 14 mL al
volumen total, decimos que el volumen molar parcial del agua en esta
disolución es de 14 mL mol-1. Definimos el volumen molar parcial de una
sustancia i en una disolución como:
ji
nTPn
VV j
i
i
,,
y el volumen total de la disolución resultante está dado por:
i
iiVnV
VnVnV ...2211
El volumen molar parcial de un componente en una disolución depende
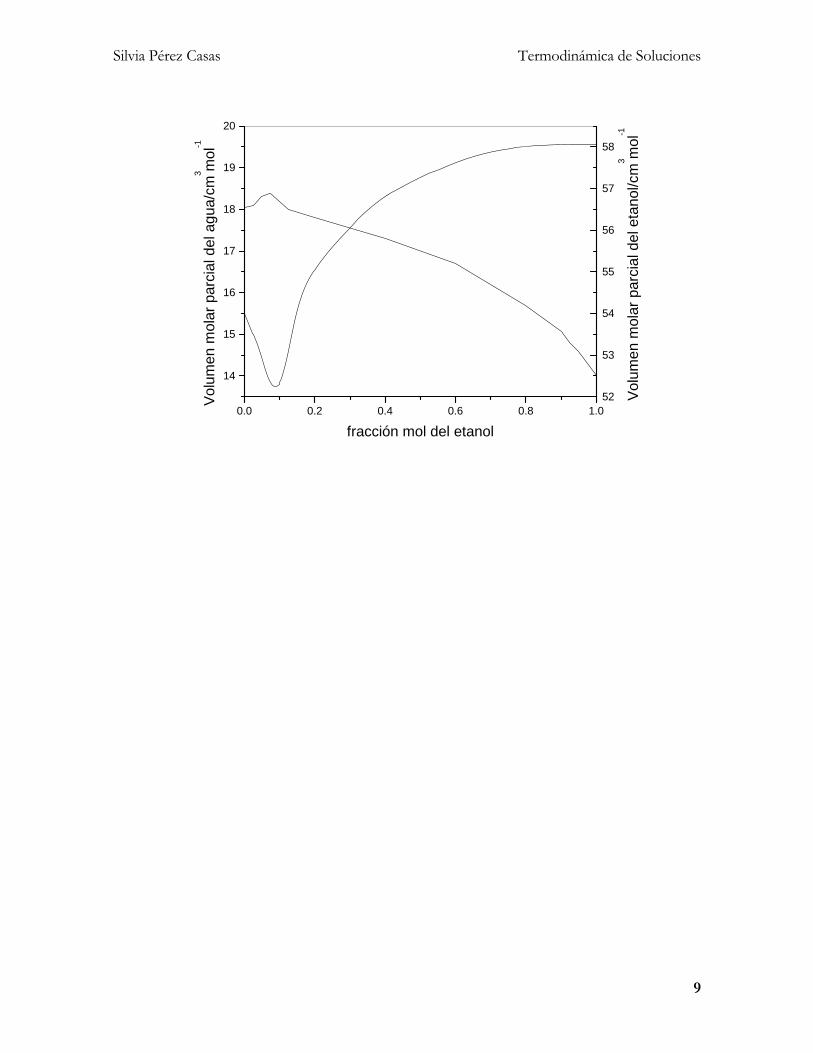
de la naturaleza de la disolución así como de su composición. La figura
siguiente representa el comportamiento de la disolución etanol + agua. Si
mezclamos 4 moles de etanol con 6 moles de agua, xetanol=0.4 y xagua=0.6.
De la gráfica leemos que el volumen molar parcial del agua para esta
disolución es aproximadamente 17 cm3 mol-1, mientras que para el etanol
es de aproximadamente 57 cm3 mol-1, entonces el volumen total de la
disolución obtenida se obtiene así:
)17(6)57(4 1313 molcmmolmolcmmolV
Silvia Pérez Casas Termodinámica de Soluciones
9
0.0 0.2 0.4 0.6 0.8 1.0
14
15
16
17
18
19
20
Vol
umen
mol
ar p
arci
al d
el e
tano
l/cm
3 mol
-1
Vol
umen
mol
ar p
arci
al d
el a
gua
/cm
3 mol
-1
fracción mol del etanol
52
53
54
55
56
57
58

Silvia Pérez Casas Termodinámica de Soluciones
10
De manera similar definimos la entalpía molar parcial del componente i
como:
jnPTi
in
HH
,,
, la energía molar parcial del componente i en la
disolución como:
jnPTi
in
UU
,,
, la entropía molar parcial del
componente i en la disolución como:
jnPTi
in
SS
,,
, la energía libre de
Helmholtz molar parcial del componente i en la disolución
como:
jnPTi
in
AA
,,
, y la energía libre de Gibbs molar parcial del
componente i en la disolución como:
jnPTi
in
GG
,,
. Se observa que en
el caso de la energía libre de Gibbs molar parcial del componente i en la
disolución, su definición coincide con la definición del potencial químico
del componente i en la disolución. Este es el único caso en que ambas
cantidades coinciden, pero no es así con las demás propiedades molares
parciales. En general, para toda propiedad extensiva (X) de una disolución
podemos escribir que:
i
iXXnXnX ...2211
donde iX es la cantidad molar parcial del componente i en la disolución.
Esta expresión se conoce como regla de aditividad.
a) Fugacidad.
La fugacidad es la medida de la no idealidad de un gas.
Para un gas ideal a temperatura constante tenemos que:

Silvia Pérez Casas Termodinámica de Soluciones
11
1ln
ln
0
10
PRT
P
dPRTd
PRTddPP
RTd
dPVd
VdPdG
P
donde la presión inicial (P0) es 1 bar y 0 es el potencial estándar, que en
este caso ha sido tomado como 1 bar.
Para estudiar los gases reales se parte de esta ecuación y se define la
fugacidad f de la manera siguiente:
fRT
fRTddPVd
ln
ln0
de donde se deduce que la relación entre la fugacidad y la presión es:
Pf
donde es el coeficiente de fugacidad.
Dado que para el gas ideal 0P
PRTddPV ideal y para el gas real
0f
fRTddPV real , la diferencia entre ellos es:
00
lnln)(0 P
P
f
fRTdPVV
P
P
idealreal
donde tomamos a P0 como una presión muy baja que tiende a 0, de tal
manera que en estas condiciones la presión y la fugacidad se igualan.
dPVVPf
fPRT
P
idealreal
00
0
ln
dividimos todo entre RT y el miembro de la derecha lo multiplicamos y
dividimos por P:
dPP
VPVP
RTP
f
RT
RT Pidealreal
0
1ln
Silvia Pérez Casas Termodinámica de Soluciones
12
en esta expresión reconocemos al factor de compresibilidad RT
VPZ
real y
también que 1RT
VP ideal, por lo tanto tenemos:
ln1
ln0
dPP
Z
P
f P
a partir de esta ecuación podemos obtener la fugacidad o el coeficiente de
fugacidad de un gas. Esta expresión también la podemos escribir en
función de la presión reducida y así nos queda:
R
P
R
dPP
ZR
0
1ln
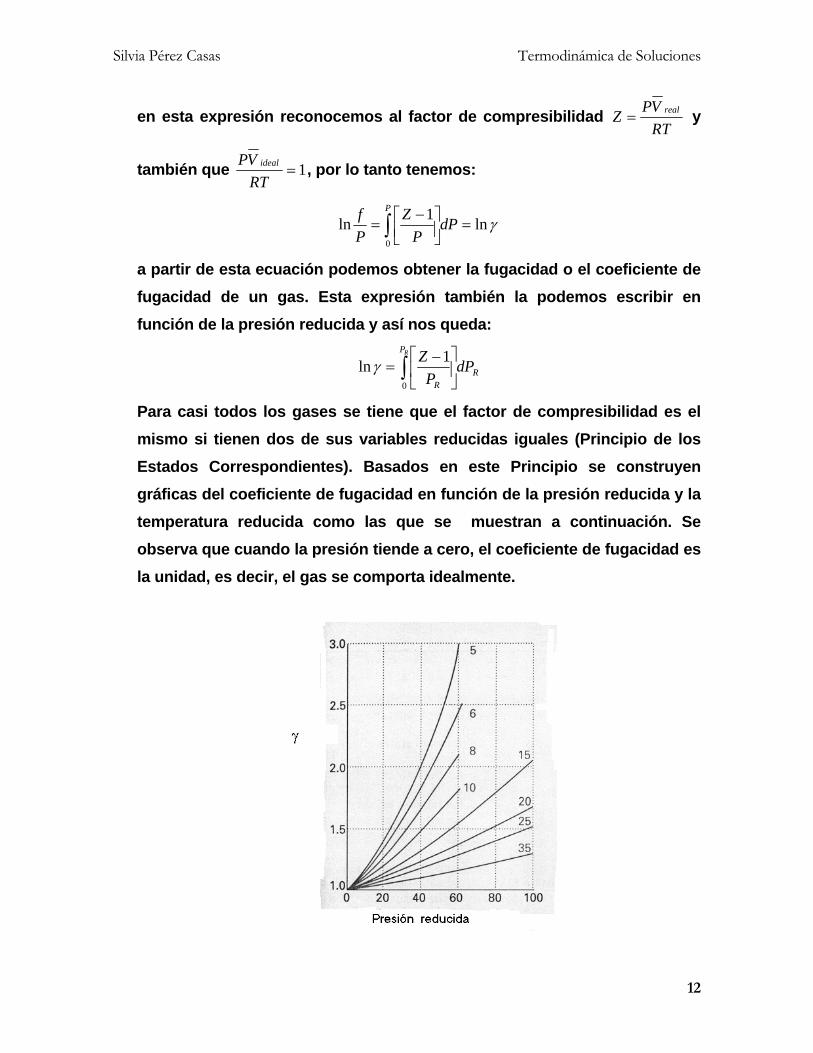
Para casi todos los gases se tiene que el factor de compresibilidad es el
mismo si tienen dos de sus variables reducidas iguales (Principio de los
Estados Correspondientes). Basados en este Principio se construyen
gráficas del coeficiente de fugacidad en función de la presión reducida y la
temperatura reducida como las que se muestran a continuación. Se
observa que cuando la presión tiende a cero, el coeficiente de fugacidad es
la unidad, es decir, el gas se comporta idealmente.
Silvia Pérez Casas Termodinámica de Soluciones
13
b) Funciones de Exceso.
Las funciones de exceso se definen como la diferencia entre el valor
de una propiedad termodinámica para una disolución y el valor de esa
misma propiedad para una disolución ideal en las mismas condiciones de
temperatura, presión y composición. En consecuencia, la energía libre de
Gibbs de exceso, GE, se define como:
idealxPT
realxPT
E GGG ,,,,
Las relaciones entre las funciones de exceso son idénticas a las
existentes entre funciones totales:
EEE
EEE
EEE
TSUATSHGPVUH
y las derivadas parciales de las funciones de exceso extensivas son
análogas a las funciones totales, por ejemplo:
E
xT
E
VP
G
,

Silvia Pérez Casas Termodinámica de Soluciones
14
Las funciones de exceso molares parciales se definen de manera
análoga a las propiedades termodinámicas molares parciales. Entonces, si
M es una propiedad termodinámica extensiva, la propiedad molar parcial
de exceso, se define como: jnPTi
EEi
n
Mm
,,
y de acuerdo al teorema de
Euler:
i
ii mnM
i
Eii
E mnM
c) Actividad.
El potencial químico de un componente i en una solución líquida está
dado por:
00ln ln
i
isoi P
PRT
Si asumimos que las presiones de vapor involucradas son
suficientemente bajas de tal manera que los vapores pueden ser
considerados como gases ideales (de no ser así, deberíamos considerar
las fugacidades), se cumpliría la ley de Raoult, 0xPP y entonces
tendríamos para la solución ideal:
iisoi xRT ln0ln
En el caso de las soluciones no ideales formadas por dos
constituyentes se encuentra empíricamente que para el constituyente 1:
...exp 32
22
0111 xxPxP
y por lo tanto :
...ln 32
221
01
ln RTxRTxxRTsoi
que es una expresión mucho más complicada. Con la finalidad de
simplificar las expresiones que describen el comportamiento de una
disolución real, Lewis introdujo la actividad, a.

Silvia Pérez Casas Termodinámica de Soluciones
15
iisoi aRT ln0ln
que tiene la misma forma simple de la ecuación que describe el
comportamiento de las disoluciones ideales.
La actividad de un sustancia indica qué tan “activa” es una sustancia
en relación con su estado estándar porque es una medida de la diferencia
entre el potencial químico de la sustancia en el estado de interés y el de su
estado estándar a la misma temperatura.
Comparando esta ecuación con aquélla que describe el
comportamiento ideal, vemos que para un vapor ideal:
01P
Pa i
i
aplicamos la ley de Raoult y tenemos que:
101
011 x
P
Pxai
Sin embargo para las disoluciones reales de dos constituyentes queda
representada empíricamente por la ecuación:
...exp 32
2211 xxxa
en donde se ve claramente que 11 a cuando 11 x y por lo tanto 02 x .
Para medir la desviación de la solución del comportamiento ideal,
definimos el coeficiente de actividad, , como:
x
aii
Si =1 para todo el intervalo de soluciones, la disolución es ideal. Si 1,
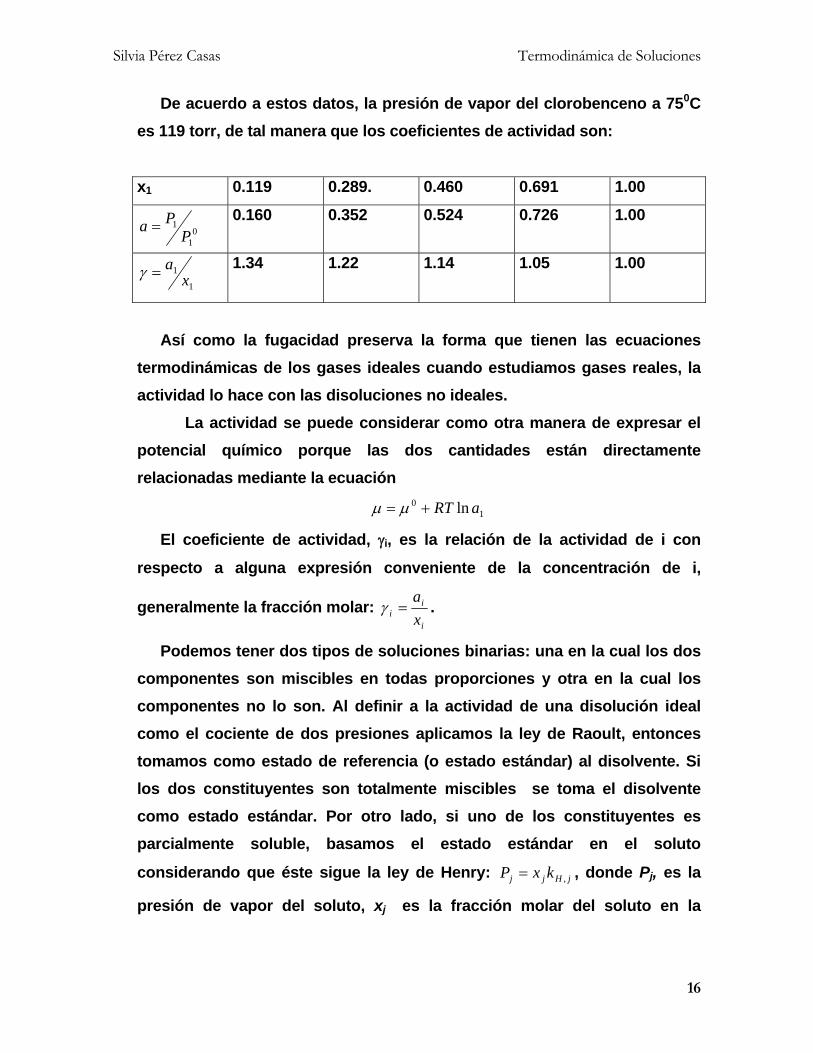
la solución no es ideal. Por ejemplo, las presiones parciales del
clorobenceno en equilibrio con 1-nitropropano a 750C son:
x1 0.119 0.289. 0.460 0.691 1.00
P1/torr 19.0 41.9 62.4 86.4 119
Silvia Pérez Casas Termodinámica de Soluciones
16
De acuerdo a estos datos, la presión de vapor del clorobenceno a 750C
es 119 torr, de tal manera que los coeficientes de actividad son:
x1 0.119 0.289. 0.460 0.691 1.00
01
1
PPa
0.160 0.352 0.524 0.726 1.00
1
1x
a 1.34 1.22 1.14 1.05 1.00
Así como la fugacidad preserva la forma que tienen las ecuaciones
termodinámicas de los gases ideales cuando estudiamos gases reales, la
actividad lo hace con las disoluciones no ideales.
La actividad se puede considerar como otra manera de expresar el
potencial químico porque las dos cantidades están directamente
relacionadas mediante la ecuación
10 ln aRT
El coeficiente de actividad, i, es la relación de la actividad de i con
respecto a alguna expresión conveniente de la concentración de i,
generalmente la fracción molar: i
ii x
a .
Podemos tener dos tipos de soluciones binarias: una en la cual los dos
componentes son miscibles en todas proporciones y otra en la cual los
componentes no lo son. Al definir a la actividad de una disolución ideal
como el cociente de dos presiones aplicamos la ley de Raoult, entonces
tomamos como estado de referencia (o estado estándar) al disolvente. Si
los dos constituyentes son totalmente miscibles se toma el disolvente
como estado estándar. Por otro lado, si uno de los constituyentes es
parcialmente soluble, basamos el estado estándar en el soluto
considerando que éste sigue la ley de Henry: jHjj kxP , , donde Pj, es la
presión de vapor del soluto, xj es la fracción molar del soluto en la

Silvia Pérez Casas Termodinámica de Soluciones
17
disolución y Kj es la constante de Henry, la cual depende de la naturaleza
del soluto así como de la temperatura, entonces tenemos:
0
,00
0ln lnlnj
jHjj
j
jj
soj P
kxRT
P
PRT cuando 0jx
jj
jHj
soj RTlx
P
kRT
0
,0ln ln
y ahora definimos a la actividad del constituyente j como:
jj
jHj
soj aRT
P
kRT lnln
0
.0ln
de tal manera que jj xa cuando 0jx . En el límite cuando esto sucede,
0lnj
soj , entonces la actividad de un vapor ideal queda definida como:
jH
jj k
Pa
,
y escogemos el estado estándar de tal manera que 0, jjH Pk . Este estado
estándar puede no existir en la práctica por lo que es denominado estado
estándar hipotético.
El valor numérico de la actividad o del coeficiente de actividad depende
del estado estándar elegido.
d) Cálculo de la energía libre de Gibbs utilizando los coeficientes de
actividad.
Considerando que para el constituyente 1
jjjjjsoj RTxRTaRT lnlnln 00ln
fácilmente se puede demostrar que
iii
iiimezcla xxxRTG lnln
donde la primera suma representa la energía libre de Gibbs de una
disolución ideal. Además
i
iiE xRTG ln

Silvia Pérez Casas Termodinámica de Soluciones
18
Problemas.
1) La entalpía molar de vaporización del benceno en su punto normal de
ebullición (80.090C) es 30.72 kJ mol-1. Calcule el cambio de energía libre
molar de vaporización del benceno a 750C, 80.090C y 850C asumiendo
que el cambio de entalpía y el cambio de entropía para este proceso
permanecen constantes en este intervalo de temperaturas. Interprete
los resultados físicamente.
2) Resuelva el problema 1) considerando que el cambio de entalpía y el
cambio de entropía para la vaporización cambian con la temperatura. El
Cp para el benceno líquido es 136.3 J K-1 mol –1 y para el benceno
gaseoso es 82.4 J K-1 mol-1. Comparar los resultados con el problema
anterior. ¿Cambia la interpretación física de los resultados? ¿Por qué?
3) En una disolución donde la fracción molar del cloroformo es 0.4693, los
volúmenes molares parciales de la acetona y el cloroformo son 74.166
cm3 mol-1 y 80.235 cm3 mol-1 respectivamente. ¿Cuál es el volumen de
una solución cuya masa total es de 1.000 kg?
4) En una disolución de líquidos A y B la fracción mol de A es 0.3713 y los
volúmenes molares de A y B son 188.2 cm3 mol-1 y 176.14 cm3 mol-1
respectivamente. Las masas molares de A y B son 241.1 g mol-1 y 198.2
g mol-1. ¿Cuál es el volumen de una solución de 2.000 kg de masa?
5) A 250C, la densidad de una solución al 50% en peso de etanol en agua
es 0.914 g cm-3. Dado que el volumen molar parcial del agua en la
solución es 14.4 cm3 mol-1, calcule el volumen molar parcial del etanol.
6) A 200C, la densidad de una solución de etanol al 20% en peso en agua
es de 968.7 kg m-3. dado que el volumen molar parcial del etanol en la
solución es de 52.2 cm3 mol-1, calcule el volumen molar parcial del
agua.
7) El volumen de una solución acuosa de cloruro de sodio a 250C fue
medida a diferentes molalidades b, y se encontró que el volumen puede
expresarse como V/cm3 = 1003 + 16.62b + 1.77b3/2 + 0.12b2 donde V es el

Silvia Pérez Casas Termodinámica de Soluciones
19
volumen de una solución formada por 1.000 kg de agua y b es la
molalidad. Calcule el volumen molar parcial de los componentes en una
solución de molalidad 0.100 mol kg-1.
8) A 180C el volumen total de una solución formada por sulfato de
magnesio y 1.000 kg de agua queda expresado por la ecuación: V =
1001.21 + 34.69(b - 0.070)2. Calcule el volumen molar parcial de la sal y
del disolvente en una solución cuya concentración es 0.05 mol kg-1.
9) ¿En qué proporciones deberían mezclarse el etanol y el agua para
preparar 100 g de una disolución al 50% en masa de etanol?
10) La energía libre de exceso del metilciclohexano y el tetrahidrofurano a
303.15K sigue la siguiente ecuación:
GE = RTx (1-x) ( 0.4857 – 0.1077(2x-1) + 0.0191 (2x – 1)2 )
Donde x es la fracción mol del metilciclohexano. Calcule la energía libre
de Gibbs de la disolución de 1 mol de metilciclohexano y 3 moles de
tetrahidrofurano.
11) A continuación se presentan las densidades de diversas soluciones de
sulfato de cobre (II) a 200C. Determine y grafique los volúmenes molares
parciales del sulfato de cobre (II) en ese intervalo de concentraciones
m (CuSO4) / g 5 10 15 20
/ (g cm-3) 1.051 1.107 1.167 1.230
Donde m(CuSO4) es la masa del sulfato cuproso disuelta en 100 g de
solución.
12) Obtener ln usando la expresión de la expansión virial.
13) Encuentre la expresión para el coeficiente de fugacidad para un gas que
obedece la ecuación de estado: V
qT
RT
VP 1 donde q es una constante.
Haga la gráfica de contra 4Pq/R.
14) La energía libre de Gibbs de exceso de una disolución binaria es igual a
gRTx(1-x) donde g es una constante y x es la fracción molar del soluto
A. Encuentre una expresión para el potencial químico de A en la
disolución y bosqueje su dependencia con la composición.

Silvia Pérez Casas Termodinámica de Soluciones
20
15) Use la ecuación de Gibbs-Duhem para derivar la ecuación de Gibbs-
Duhem-Margules: TPB
B
TPA
A
x
f
x
f
,,ln
ln
ln
ln
donde f es la fugacidad. Use
esta ecuación para demostrar que cuando las fugacidades son
reemplazadas por presiones, la ley de Raoul es cumplida por cada
componente.
16) Use la ecuación de Gibbs-Duhem para demostrar que el volumen molar
parcial (o cualquier propiedad molar parcial) de un componente B
puede ser obtenida si el volumen molar parcial (u otra propiedad) de A
es conocida para toda concentración . Haga esto demostrando que
A
A
V
V AA
ABB dV
x
xVV
0 10
17) Goodwin reporta los siguientes datos para el tolueno (J. Phys. Chem.
Ref. Data 18, 1565 (1989)) en función del facor de compresibilidad Z, a
diferentes presiones y temperaturas. A partir de estos datos determinar
el coeficiente de fugacidad del tolueno a 600K y a) 30 bar, b) 1000 bar
P/bar 0.500 1.013 2.00 3.00 5.00
Z 0.99412 0.98896 0.97942 0.96995 0.95133
P/bar 10.00 20.00 30.00 42.4 50.0
Z 0.90569 0.81227 0.70177 0.47198 0.22376
P/bar 70.0 100.0 200 300 500 1000
Z 0.26520 0.34920 0.62362 0.88288 1.37109 2.48836
18) El factor de compresibilidad para el etano a 600 K se ajusta a la
siguiente expresión:
Z=1.0000-0.000612 (P/bar) +2.661 x 10-6 (P/bar)2 – 1.390 x 10-9 (P/bar)3
– 1.077 x 10-13 (P/bar)4
Para 0 P/bar 600. Use esta expresión para obtener el coeficiente de
fugacidad para el etano como función de la presión a 600 K.

Silvia Pérez Casas Termodinámica de Soluciones
21
19) Si la presión de vapor de los dos constituyentes de una disolución
binaria están dados por: RTxPxP /exp 22
0111 y RTxPxP /exp 2
10
222 ,
demostrar que
21221121 lnln/ xxxxxRT
nnGG mezclamezcla
221121 lnln// xxxxRnnSRS mezcla
2121/ xxnnHH mezclamezcla
Una disolución que satisface estas ecuaciones es llamada solución
regular. Un modelo de termodinámica estadística muestra que para una
solución regular binaria, está dada por:
122211 AN
donde ij es la energía de interacción entre los componentes i y j. Hay que
observar que =0 si
22211
12
, lo cual significa que energéticamente
las interacciones entre las moléculas 1 y 2 son similares a las
interacciones entre dos moléculas del mismo componente.
20) Demostrar que meclaG , mezclaS y mezclaH de una disolución regular son
asimétricas con respecto al punto x1=x2=1/2.
21) La Ley de Henry sirve para determinar diversas cosas y proporciona
una manera simple de encontrar la solubilidad de gases en líquidos. Por
ejemplo, ¿Cuál es la solubilidad del dióxido de carbono en agua a 250C
cuando su presión parcial es: a) 0.1 atm, b) 1.0 atm? Para el CO2 la
constante de Henry es 1.25 x 106 mmHg.
22) Se midieron a 350C la presión de vapor y las presiones parciales de
vapor de una mezcla de acetona y cloroformo en el intervalo que va
desde la acetona pura al cloroformo puro. Los resultados fueron los
siguientes:
x(cloroformo) 0.00 0.20 0.40 0.60 0.80 1.00
P(cloroformo)/mmHg 0 35 82 142 219 293
P(acetona)/mmHg 347 270 185 102 37 0

Silvia Pérez Casas Termodinámica de Soluciones
22
Confirme que la mezcla sigue la ley de Raoult para el componente en
mayor proporción y la ley de Henry para el componente en menor
proporción.
Bibliografía:
1) Atkins, P. Physical Chemistry. Sixth Edition. W.H. Freeman and Company.
New York, 1999. (también disponible en español en varias ediciones).
2) Castellan, G. Fisicoquímica. 2ª edición. Fondo Educativo Interamericano.
España 1987.
3) Levine, I. N. Fisicoquímica. 4ª edición. Vol. 2. McGraw-Hill. España, 1995.
4) McQuarrie, D. A.; Simon, J. D. Physical Chemistry. A Molecular Approach.
University Science Books. Sausalito, California, 1997.
5) Prausnitz, J. M., Lichtenthaler, R. N., Gomes de Azevedo, E. Termodinámica
Molecular de los Equilibrios de fases. Tercera edición. Prentice Hall.
España 2000.





























