Embed Size (px)
Citation preview

CENTRO DE INVESTIGACIÓN EN MATERIALES
AVANZADOS, S.C.
SIMULACIÓN TEÓRICA DE LA SÍNTESIS Y
ESTUDIO DE LAS PROPIEDADES
MICROESTRUCTURALES DE NANOPARTÍCULAS
DE MAGNETITA OBTENIDAS POR AACVD PARA
SU APLICACIÓN EN LA REMOCIÓN DE
ARSÉNICO.
TESIS QUE COMO REQUISITO PARA OBTENER EL GRADO DE
DOCTORADO
EN
CIENCIA DE MATERIALES
Presenta:
M.C. Blanca Elizabeth Monárrez Cordero
Directores de Tesis:
Dra. Patricia Amézaga Madrid
Dr. Mario Miki Yoshida
Chihuahua, Chih., Mayo 2016.

ÍNDICE GENERAL
CAPITULO I
ESTADO DEL ARTE……………………………………………………………………... 1
1. Introducción……………………………………………………………………………...1
2. Antecedentes……………………………………………………………………………..3
2.1 Problemática ambiental………………………………………………………………..3
2.2 Origen del arsénico en el agua…………………………………………………………3
2.3 Métodos de remoción de arsénico……………………………………………………..5
2.3.1 Métodos convencionales…………………………………………………………...5
2.3.2 Métodos innovadores: Nanotecnología.…………………………......…………….7
2.4 Óxidos de fierro como adsorbentes…………………………………………………...8
2.4.1 Generalidades……………………………………………………………………...8
2.4.2 Nanopartículas de Óxidos de fierro……………………………………………….10
2.4.3 Generalidades de la magnetita……...……………………………………………11
2.4.4 Nanopartículas de magnetita (MNPs)……………………………………………12
2.5 Síntesis de nanopartículas…………………………………………………………….13
2.5.1 Técnicas de síntesis……………………………………………………………..13
2.5.1.1 Microemulsión…………………………………………………………………13
2.5.1.2 Sistemas coloidales…………………………………………………………….14
2.5.1.3 Fases acuosas………………………...……………………………………….. 14
2.5.2 Técnicas de síntesis de partículas nanométricas: químicas y físicas…………….14
2.5.2.1 Técnica AA-CVD (Depósito Químico de Vapor asistido por Aerosol)……….15
2.6 Modelos teóricos de síntesis de nanopartículas…………………...………………...16
2.7 Técnicas de Caracterización Microestructural de Nanopartículas……………..…19
2.7.1 Microscopía electrónica de barrido………………………………………………19
2.7.2 Microscopía electrónica de transmisión………………………………………….20
2.7.3 Difracción de rayos X…………………………………………………………….20
2.7.4 Adsorción y desorción física de gases por el método Brunauer-Emmet-Teller….22
2.7.5 Espectrofotometría de absorción atómica………………………………………...25
2.8 Procesos de adsorción…………………………………………………………………26
2.8.1 Isotermas de adsorción……………………………………………………………27

2.8.2 Energía de adsorción……………………………………………………………..29
3. Justificación…………………………………………………………………………….30
4. Hipótesis………………………………………………………………………………...31
5. Objetivo General……………………………………………………………………….32
5.1 Objetivos particulares……………………………………………………………….32
CAPÍTULO II
METODOLOGÍA EXPERIMENTAL……………………………………………….….33
6. Simulación teórica …………………………………………………………………......33
6.1 Software Solid Works – Fluid Works………………………………….…………….33
6.1.1 Simulaciones del transporte de masa, cantidad de movimiento y energía en el
interior del reactor tubular……………………………………………………………….33
6.2 Software COMSOL MULTIPHYSICS 4.4………………………………………….34
6.2.1 Simulaciones de la evaporación de la gota en su recorrido dentro del reactor
tubular …………………………………………………………………………………..34
7. Síntesis de MNPs………………………………...……………………………………..37
7.1 Caracterización microestructural de las MNPS……………………………………..38
7.1.1 Microscopía electrónica de barrido………………………………………………...38
7.1.2 Microscopia electrónica de transmisión……………………………………………39
7.1.3 Difracción de rayos x.……………………………………………………………...39
7.1.4 Adsorción y desorción física de gases por el método Brunauer-Emmet-Teller …..40
7.2 Correlación de resultados teóricos y experimentales……………………………….41
8. Ruta Química (termoquímica de la formación de magnetita)……………………....42
8.1 Análisis termodinámico………………………………………………………….…...42
8.2 Cinética de descomposición…………………………………………………………..43
8.2.1 Método no isotérmico…………………………………………………………….43
8.2.2 Método isotérmico………………………………………………………………..44
9. Pruebas de remoción de arsénico………………………………………………………45
9.1 Pruebas batch o de contacto………………………………………………………….45
9.1.1 Determinación de la adsorción de arsénico por absorción atómica………………45
9.1.2 Determinación de la adsorción de arsénico por microscopia electrónica…………46

9.2 Desarrollo del mecanismo de adsorción……………………………………………..46
CAPITULO III
RESULTADOS Y DISCUSIÓN…………………………………………………………47
10. Simulación teórica…………………………………………………………………….47
10.1 Solid Works - Fluid Works………………………………………………………….47
10.1.1 Distribución de temperatura del reactor…………………………………………47
10.1.2 Influencia de la temperatura del reactor y del flujo del gas acarreador en la
velocidad del fluido (Ar + aire + vapor de metanol)…………………………………….48
10.1.2.1 Temperatura del reactor………………………………………………………..48
10.1.2.2 Flujo del gas……………………………………………………………………49
10.1.3 Influencia de la temperatura del reactor y del flujo del gas portador en la
velocidad de calentamiento del fluido…………………………………………………..50
10.1.4 Tiempo de residencia en función de la temperatura del horno y flujo de gas…...52
10.2 Simulación teórica por COMSOL Multiphysics 4.4………………………………54
10.2.1 Influencia del disolvente ……………………………………………………… 54
10.2.1.1 Comportamiento y distribución de la concentración de soluto en la gota
utilizando metanol como solvente………………………………………………………54
10.2.1.2 Comportamiento y distribución de la concentración de soluto en la gota
utilizando agua como solvente…………………………………………………………..57
10.2.2 Influencia de la temperatura de la gota…………………………………………..60
10.2.3 Influencia de la concentración del precursor…………………………………….61
10.2.4 Influencia del flujo del gas portador……………………………………………..62
10.2.5 Influencia del diámetro del reactor………………………………………………65
11. Microestructura y caracterización de las MNPs…………………………………... 66
11.1 Microscopia electrónica de barrido…………………………………………………...66
11.2 Microscopia electrónica de transmisión…………………………………………….. 68
11.3 Difracción de rayos x……………………………………………………………….. 70
11.4 Área específica………………………………………………………………………..71
12. Correlación de resultados experimentales con los simulados en Software
COMSOL Multiphysics 4.4………………………………………………………………72

12.1 Microestructura……………………………………………………………………...72
12.1.1 Metanol como disolvente………………………………………………………..73
12.1.2 Agua como disolvente.…………………………………………………………..76
13. Cinética de la reacción del precursor………………………………………………..78
13.1 Mecanismo cinético por método dinámico………………………………………....80
13.1.1 Cálculo de la energía de activación……………………………………………82
13.2 Mecanismo cinético por método isotérmico………………………………………..83
13.2.1 Cálculo de la energía de activación……………………………………………85
14. Mecanismo de adsorción de arsénico………………………………………………..87
14.1 Eficiencia de Adsorción de arsénico en las MNPs………………………………...87
14.1.1 Efecto del tiempo de contacto y la concentración de la solución……………..89
14.2 Isotermas de adsorción……………………………………………………………..89
14.3 Termodinámica del proceso de adsorción …………………………………….......92
14.3.1 Energía de adsorción y energía libre de Gibbs de las MNPs…………………92
14.4 Comprobación de la adsorción de arsénico por microscopía electrónica de
transmisión………………………………………………………………………………..93
CONCLUSIONES………………………………….……………………………………..95
PERSPECTIVAS…………………………………………………………………………97
APORTACIONES………………………………………………………………………...98
REFERENCIAS…………………………………………………………………………..99
ANEXOS..………………………………………………………………………………..109
ARTÍCULOS

ÍNDICE DE FIGURAS
Figura 1. Diagrama Eh – Ph de especies acuosas de arsénico en el sistema As-O2-H2O…..4
Figura 2. Diagrama de pourbaix con la distribución de las especies de hierro en función
de pH- Eh ………………………………………………………………………...9
Figura 3. Movilidad de arsénico en presencia de hierro en función de pH- Eh…………...10
Figura 4. Esquema teórico del proceso de la evaporación de la gota y precipitación de la
sal en la técnica de aspersión pirolítica…………………………………………17
Figura 5. Cambio de la distribución de la concentración en el interior de la gota………...18
Figura 6. Tipos de isotermas……………………………………………………………….23
Figura 7. Tipos de histéresis……………………………………………………………….23
Figura 8. Esquema de la sección transversal de un sólido poroso…………………………25
Figura 9. Modelo General del reactor utilizado para la simulación mediante software
Solid Works……………………………………………………………………...33
Figura 10. Esquema de la técnica AACVD……………………………………………….37
Figura 11. Equipo usado en la síntesis de nanopartículas por la técnica AACVD…………38
Figura 12. Proceso de adsorción de las moléculas de un gas en una superficie porosa……41
Figura 13. Comparación de la distribución de temperatura simulada y experimental
del gas en el interior del reactor………………………………………………..47
Figura 14. Velocidad del fluido en el interior del reactor tubular de 9 mm a diferentes
temperaturas por Solid Works – Fluid Works…………………………………48
Figura 15. Simulación en Solid Works – Fluid Works de la velocidad del fluido en la
coordenada axial z……………………………………………………………..49
Figura 16. Velocidad de calentamiento del fluido en el interior del reactor tubular de
9 mm de diámetro simulados por Solid Works – Fluid Works………………..51
Figura 17. Velocidades de calentamiento en la coordenada axial z, obtenidas por
simulaciones en Solid Works – Fluid Works………………………………….51
Figura 18. Tiempo de residencia para diferentes temperaturas simuladas calculados a
partir de Solid Works - Fluid Works…………………………………………..53
Figura 19. Tiempo de residencia para flujos de 0.808, 1.062 y 1.57 L min-1 simulados por
Solid Works – Fluid Works……………………………………………………53

Figura 20. Distribución de la concentración radial de la sal dentro de la gota, en el
momento de la precipitación para diferentes temperaturas del reactor………..55
Figura 21.- Evolución temporal del radio de la gota para diferentes temperaturas del
reactor…………………………………………………………………………57
Figura 22. Distribución de la concentración de sal precursora dentro de la gota,
cuando la concentración de soluto en la superficie alcanza la CSS utilizando
agua como disolvente a temperaturas del reactor de 423 a 723 K………..……58
Figura 23. Evolución temporal del radio de la gota a temperaturas del reactor de 423
a 723 K, usando agua como disolvente………………………………………...59
Figura 24 Comportamiento de la temperatura de la gota a diferentes temperaturas
del reactor utilizando como precursor: a) agua, b) metanol…………………….60
Figura 25. Distribución de la sal precursora a diferentes concentraciones…………………61
Figura 26. Distribución de la sal dentro de la gota a una temperatura de 723 K,
utilizando diferentes flujos en un reactor tubular de 9 mm…………………….63
Figura 27. Distribución de la sal dentro de la gota a una temperatura de 523 K, utilizando
diferentes flujos en un reactor tubular de 9 mm de diámetro………………….64
Figura 28. Radio de la gota en función del tiempo a temperaturas de 723 K, en los reactores
de 9 y 15 mm de diámetro……………………………………………………..65
Figura 29 . Micrografías por electrones secundarios en microscopio electrónico de
barrido………………………………………………………………………...67
Figura 30. Análisis elemental por EDS……………………………………………………68
Figura 31. Micrografías obtenidas por el microscopio electrónico de transmisión……….69
Figura 32. Difractograma de las nanopartículas sintetizadas a 723 K………………….....70
Figura 33. Isoterma de adsorción obtenida por el método BET ………………………….71
Figura 34. Distribución de tamaño de poro obtenida por el método BET…………………72
Figura 35. Micrografías de los materiales obtenidos a temperaturas de 373 a 523 K,
por electrones secundarios en SEM y contraste-Z en MET, con su histograma de
distribución de la sal incluido…………………………………………………. 73

Figura 36. Micrografías de los materiales obtenidos a temperaturas de 573 a 723 K
por electrones secundarios en SEM y contraste-Z en MET con su histograma
de distribución de la sal incluido……………………………………………….75
Figura 37. Micrografías por electrones secundarios en SEM, de los materiales obtenidos
a temperaturas de 423 a 673 K…………………………………………………77
Figura 38. Micrografías por electrones secundarios en SEM, a una temperatura de
723 K ………………………………………………………………………….78
Figura 39. Temperatura termodinámica posible de la formación de magnetita de acuerdo
a la reacción obtenida por el software HSC…………………………………79
Figura 40. Análisis termogravimétrico del cloruro ferroso tetrahidratado (FeCl2 . 4H2O)..80
Figura 41. Pérdida de peso del precursor de la sal a diferentes velocidades de calentamiento
por medio de análisis termogravimétrico……………………………………….81
Figura 42. Difractograma del análisis termogravimétrico a diferentes velocidades
de calentamiento (5, 10, 15 y 20 K min-1)……………………………………..82
Figura 43. Ajuste lineal de los datos obtenidos por análisis termogravimetrico por el
método de Flynn-Wall-Ozawa …………………………………………………83
Figura 44 . Análisis termogravimétrico del precursor a diferentes temperaturas
(723. 773 y 823 K)……………………………………………………………..84
Figura 45. Difractogramas en rayos X del análisis termogravimetrico (TGA), por el método
isotérmico utilizando temperaturas de 723, 773 y 823 K…………….………..84
Figura 46. Muestra la pérdida de peso de la cinética de formación de la magnetita en el
análisis termogravimétrico partiendo del precursor deshidratado……….…….85
Figura 47. Pendientes de logaritmo de velocidad con logaritmo de peso, para la obtención
de la constante de velocidad a las temperaturas de 723, 773 Y 823 K……….85
Figura 48. Muestra la pendiente obtenida y el factor pre-exponencial para la obtención de
la energía de activación resultante, utilizando temperaturas de 723, 773 y
823 K……………………………………………………………………….....86
Figura 49. Efecto del tiempo de contacto en la eficiencia de remoción de As+3 Y As+5
utilizando 10 mg de MNPs…………………………………………………….87
Figura 50. Mapeo en línea por EDS mediante Microscopio electrónico de transmisión
(MET), de las MNPs después de la remoción de arsénico……………………..94

ÍNDICE DE TABLAS
Tabla 1. Porcentajes de remoción para As+3 y As+5 por tecnologías convencionales………6
Tabla 2. Porcentajes de remoción para As+3 y As+5 por tecnologías innovadoras………….8
Tabla 3. Ecuaciones y parámetros empleados para el montaje de las isotermas de
adsorción en el equilibrio………………………………………………………….28
Tabla 4. Parámetros utilizados en el software Solid Works - Fluid Works usando
diferentes temperaturas y flujos del gas acarreador incluyendo el metanol
en fase gas. ………………………………………………………………………..34
Tabla 5. Condiciones utilizadas para la síntesis de materiales…………………………….42
Tabla 6. Simulación del diámetro de partícula y el espesor de la corteza obtenidos
a diferentes temperaturas por el software COMSOL multiphysics 4.4………….56
Tabla 7. Diámetro de la partícula en el momento de la precipitación usando metanol y
agua como disolventes……………………………………………………………58
Tabla 8. Tiempo de secado de las gotas en el momento de la precipitación usando
metanol y agua como disolventes………………………………………………...60
Tabla 9. Diámetro y espesor de la partícula después de la precipitación, a diversas
concentraciones de la sal precursora.......................................................................62
Tabla 10. Radio y espesor de las partículas simuladas después de la precipitación y su
concentración final a temperaturas de 523 y 723 K con diferentes flujos………..64
Tabla 11. Tiempo de precipitación de la sal a 723 K, utilizando reactores tubulares de 9
y 15 mm de diámetro a diferentes flujos………………………………………...66
Tabla 12. Diámetro promedio de las MNPs, espesor de la corteza y tamaño promedio
del cristal a temperaturas de 723 y 773 K………………………………………..67
Tabla 13. Porosidad de partícula calculada y numero de cristales obtenidos de una gota
a diferentes temperaturas (723 – 773 K). ………………………………………69
Tabla 14. Composición elemental (%) de los materiales obtenidos a diferentes
temperaturas………………………………………………………………...…..76
Tabla 15. Entalpía de reacción y energía de Gibbs, para diferentes rangos de
temperatura, obtenidos por el software HSC. …………………………………..79
Tabla 16. Parámetros de Arrhenius obtenidos por la cinética de reacción a diferentes
pérdidas de peso…………………………………………………………………83

Tabla 17. Resultados de la eliminación de iones de As + 3 y As + 5 del agua tratada……..88
Tabla 18. Condiciones experimentales empleadas en los ensayos de adsorción de
contacto para el cálculo de la capacidad de adsorción de una monocapa……...89
Tabla 19. Resultados del modelo de Langmuir para la adsorción de iones de As + 3 y
As + 5 en la MNPs……………………………………………………………….90
Table 20. Resultados del modelo de Freundlich para la adsorción de iones de As + 3 y
As + 5 en la MNPs……………………………………………………………….91
Tabla 21. Resultados del modelo de Dubinin para la adsorción de iones de As + 3 y As + 5
en la MNPs….…………………………………………………………………..92
Tabla 22. Energía de adsorción en diferentes condiciones experimentales, calculada a partir
del modelo de Dubinin…………………………………………………………..93
Table 23. Energía libre de Gibbs para los iones As + 5 y As + 3 en diferentes condiciones
experimentales, calculada a partir del modelo de Langmuir……………………93

Resumen
La contaminación de arsénico en los cuerpos de agua que se destinan para el consumo
humano, es una problemática a nivel mundial que varios países enfrentan, en América han
reportado la existencia de población expuesta crónicamente a concentraciones de arsénico en
agua, superiores a las previstas por la normatividad de los países. La presencia de arsénico
está relacionada a enfermedades cardiovasculares y diferentes tipos de cáncer. Existen
tecnologías o métodos convencionales para remover arsénico del agua con eficiencias de 70
a 99%, sin embargo los productos secundarios a la remoción son grandes áreas de lodos y se
necesita de alto consumo de energía e infraestructura, para removerlos y llevarlos a su
destino final, esto se traduce en altos costos derivados de los tratamientos secundarios. Por
lo que es necesario optar por alternativas o rutas de remoción de este material de menor costo
y una de ellas es el uso de la nanotecnología, propiamente utilizar nanomateriales en forma
de nanopartículas que sirvan como adsorbentes de este elemento en particular.
Hoy en día, las nanopartículas de magnetita mesoporosas (MNPs), son una clase importante
de nuevos nanomateriales, por lo que ocupan una posición valiosa en la ciencia de los
materiales. Debido a las ventajas que presentan los óxidos de fierro y en especial con
respecto a otros adsorbentes presentan una mayor capacidad de adsorción, por lo tanto existe
un interés creciente hacia la utilización de estos materiales para la eliminación de adsorción
de una variedad de contaminantes, incluyendo colorantes orgánicos de aguas residuales. A
través de la técnica AACVD (Deposito Químico de Vapor Asistido por Aerosol) es posible
sintetizar MNPs huecas, semihuecas y sólidas, las cuales están formadas por la unión de
nanocristales de aproximadamente 20 nm formando una esfera nanoestructura la cual tiene
un espesor de un nanocristal, es por ello que reciben su nombre (nanopartículas). En esta
técnica, la morfología microestructural de las nanopartículas resultantes depende fuertemente
de los parámetros de síntesis. Por lo tanto, con el fin de entender la formación de MNPs por
AACVD, se llevó a cabo un estudio de la ruta química del precursor partiendo del software
HCS, a partir de la cual se determinaron los parámetros de Arrhenius, como la constante de
velocidad, factor de frecuencia y energía de activación, para describir la rapidez térmica de
la reacción a partir del análisis térmico de manera experimental. La influencia de los
parámetros de síntesis fue simulada por 2 softwares: Solid Works - Fluid Works y COMSOL
Multiphysics 4.4, permitiendo comprender la cinética de evaporación de la gota y el

mecanismo de formación de las MNPs. Los resultados de la simulación fueron
correlacionados con los obtenidos de manera experimental.
Las MNPs sintetizadas fueron empleadas para determinar la eficiencia de adsorción de
arsénico por medio de contacto (batch) utilizando el arsénico en sus especies As+3 y As+5
como modelo de estudio para observar la eficiencia de remoción en muestras de agua
preparada en el laboratorio , tomando en cuenta el tiempo de contacto y la concentración
inicial. La cuantificación de As se efectuó por espectroscopia de absorción atómica. Para
obtener un análisis más completo se realizó un estudio al equilibrio, donde se determinó la
capacidad de adsorción de las MNPs, la termodinámica del proceso (Energía de adsorción y
Energía libre de Gibbs) y el tipo de proceso de adsorción (fisisorción o quimisorción), que
se lleva a cabo. Por lo que fue necesario calcular la capacidad de adsorción de la MNPs y
realizar un estudio de las isotermas de Freundlich, Langmuir y Dubinin. Los resultados
mostraron una eficacia global de eliminación de 87% de As + 3 y 98% para As + 5, en un
tiempo de contacto de 15 minutos, resultados que sugieren el uso de MNPs como una
alternativa prometedora en la eliminación de iones de As en el agua.

CAPÍTULO I
ESTADO DEL ARTE
1. Introducción
La presente investigación se ubica en la línea de la Nanotecnología que es de gran interés
actual, en particular en el estudio de materiales nanoestructurados para su aplicación en la
remediación ambiental; lo cual, además del aspecto científico, tiene un gran impacto a nivel
social, ecológico y económico. A través de esta investigación se pretende contribuir con
elementos e información que trate de resolver problemas tecnológicos de la actualidad
relacionados con la remoción de los mismos. Considerando la problemática de que metales
pesados, como el arsénico, se encuentran en los pozos de agua para consumo humano en
diversas partes del mundo [1], en Latinoamérica [2-3] y hablando específicamente de la
situación en nuestro país México, donde en estados principalmente de la zona norte presenta
niveles de arsénico por encima de la norma (0.025 ppm); [4] y aunado a esto que su presencia
está relacionada con enfermedades agudas y crónicas muy peligrosas [5-7], es una
oportunidad tener la alternativa de utilizar la Nanotecnología para poder contribuir a resolver
este tipo de problemáticas y así mejorar la calidad de vida de los seres humanos, por lo que
es necesario el entendimiento de la influencia de los parámetros que determinan la adsorción
de metales pesados, metaloides y su optimización para una mejor remoción y tratamiento.
Se sabe que los óxidos de hierro son excelentes materiales para remover diversos
contaminantes [8-10]. Cuando los compuestos de fierro entran en contacto con el agua se
comportan como coloides positivos, y como tales tienen la capacidad de adsorber los iones
con carga negativa, por ejemplo: el cromo y arsénico (se comportan como oxianiones cuando
están en presencia de agua). Es así que los compuestos de fierro, en particular los óxidos,
poseen la gran capacidad natural de depurar aguas contaminadas con elementos tóxicos [11-
12]. Es por esto que se desea desarrollar un nanomaterial en base de óxido de fierro que sirva
como ruta o alternativa de remoción, ya que, considerando el desarrollo de nanopartículas de
alta superficie específica es posible mejorar considerablemente la capacidad de remoción de
metales pesados en agua para consumo humano. Aunado a esto, analizando la gran desventaja
de las tecnologías convencionales de que producen grandes cantidades de residuos (lodos
con arsénico) [6, 12-14], los cuales están categorizados como peligrosos y requieren de

costosos procesos de tratamientos residuales y disposición final, es necesario desarrollar
estrategias o alternativas diferentes de remoción. Una de ellas, el uso de la nanotecnología,
propiamente el desarrollo de nanopartículas con características microestructurales especiales
que sirvan para estos tratamientos y que además sean reutilizables en los procesos [15-17].
El desarrollo de nanomateriales como las MNPs por la técnica AACVD puede ser una
alternativa de desarrollo en el sector industrial tecnológico y tiene un atractivo interés en el
tratamiento de agua y remediación ambiental [18-19]. La síntesis y caracterización de
nanopartículas es un área de investigación que ha sido ampliamente explorada en los últimos
años, los primeros estudios se enfocaban en producirlas y caracterizarlas, en la actualidad
ahora se quiere producir, caracterizar y aplicar, solo que la demanda en el mercado es para
las nanopartículas que presentan mejores propiedades microestructurales y que se sinteticen
por metodologías de bajo costo y fáciles de escalar industrialmente [20-22]. La investigación
de estos nanomateriales se ha enfocado en buscar aplicaciones para las nanopartículas, las
cuales han sido muy bien aceptadas en el campo de la electrónica, polímeros, textiles, etc.
[23-25]. Uno de los campos que ha recibido mayor atención en los últimos años es la
nanobiotecnología, en él se exploran varios materiales nanoestructurados para diversas
aplicaciones biológicas y ambientales [26-28]. Estructuras en forma de nanopartícula pueden
tener propiedades tan especiales e importantes como alta velocidad de difusión, gran área
superficial, reactividad, tamaño de partícula, que las hacen idóneas para aplicaciones en
diferentes áreas científicas. Para darles propiedades más específicas a este tipo de materiales
y tener un conocimiento más amplio sobre ellos, es importante conocer su mecanismo de
formación, lo cual se facilita utilizando software de simulación, por medio de los cuales se
pueden variar diferentes parámetros de síntesis, obteniendo resultados confiables a lo que
pasaría de manera real como lo es la morfología, sin necesidad de hacerlo experimentalmente,
lo que permite un ahorro en costos y tiempo.
Es por ello que el presente trabajo se enfoca en la simulación teórica de los parámetros que
afectan las propiedades de la MNPs durante las síntesis por AACVD, así como la correlación
con pruebas experimentales y su caracterización microestructural. Además se prueba la
eficiencia de remoción de los materiales obtenidos sobre As+3 y As+5 por contacto o Batch,
realizando un estudio a equilibrio para determinar el tipo de adsorción que se lleva a cabo en
las MNPs.

2. Antecedentes
2.1 Problemática ambiental.
El arsénico (As) es un elemento presente tanto en agua superficial como en agua subterránea
en pequeñas cantidades [29-30], pero lo suficiente para causar efectos adversos en la salud
en animales y seres humanos, ha sido temática de gran interés a nivel mundial debido a la
aparición de enfermedades relacionadas con este elemento, identificándose la magnitud del
problema a partir de que se diagnosticaron enfermedades y envenenamientos en diferentes
poblaciones [6-7,31]. Existen casos de millones de personas afectadas en diversas partes del
mundo, como en Bangladesh en Asia, India, China, Taiwán y Vietnam; en América, la
población está siendo afectada en países como Chile, Argentina, México, El Salvador, y Perú
[32-34].
En México se ha detectado la presencia de As en cuerpos subterráneos de agua que se utilizan
para abastecimiento de la población, como en la Comarca Lagunera comprendida en el estado
de Coahuila y Durango, así como Hidalgo, Guanajuato, Zacatecas Morelos y el estado de
Chihuahua. Localmente los municipios más afectados son Delicias, Meoqui, Camargo y
Jiménez donde se alcanzan concentraciones de As hasta 1 mg L-1 [33].
Esta problemática del As es crítica debido a que los pozos de abastecimiento del agua
subterránea de los municipios del estado presentan concentraciones que superan el límite
máximo permisible de la NOM-127 SSA1 [34] que es de 0.01 mg L-1. La presencia de As en
varios acuíferos de la zona centro y sur del estado de Chihuahua es de origen natural debido
principalmente a la situación geológica del estado [35].
2.2 Origen del arsénico en el agua.
El As es un elemento muy común en el aire, en rocas y suelos, en la hidrosfera y la biosfera.
Es movilizado al medio ambiente a través de una combinación de procesos que incluyen tanto
procesos naturales (meteorización, actividad biológica, emisiones volcánicas), así como
procesos antropogénicos (actividad minera, uso de combustibles fósiles, uso de pesticidas,
herbicidas, desecantes y conservadores de la madera). Se puede encontrar en aguas
subterráneas y en aguas superficiales [36]. El As se encuentra en las aguas naturales como
especie disuelta, la cual se presenta comúnmente como oxianiones con As en dos estados
de oxidación, As+3 y As+5, y con menos frecuencia como As0, As-1 y As-2. As+5 aparece como
H3AsO4 y sus correspondientes productos de disociación (H2AsO4
-, HAsO42- y AsO4
3-). El
As (III) aparece como H3AsO3 y sus correspondientes productos de disociación (H4AsO3+,
H2AsO3-, HAsO3
2- y AsO33-). Aunque tanto el As+5 como el As+3 son móviles en el medio,
es precisamente el As+3 el estado más biotóxico [37]. El estado de oxidación del As, y por
tanto su movilidad, están controlados fundamentalmente por las condiciones redox (potencial
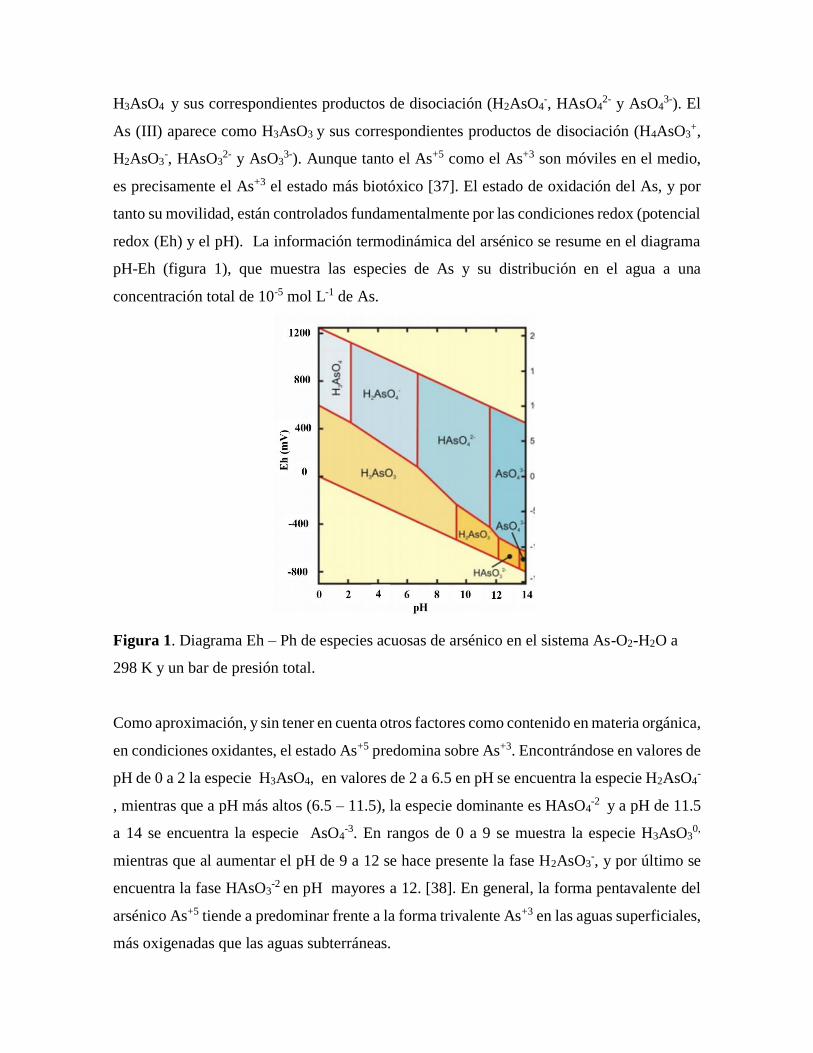
redox (Eh) y el pH). La información termodinámica del arsénico se resume en el diagrama
pH-Eh (figura 1), que muestra las especies de As y su distribución en el agua a una
concentración total de 10-5 mol L-1 de As.
Figura 1. Diagrama Eh – Ph de especies acuosas de arsénico en el sistema As-O2-H2O a
298 K y un bar de presión total.
Como aproximación, y sin tener en cuenta otros factores como contenido en materia orgánica,
en condiciones oxidantes, el estado As+5 predomina sobre As+3. Encontrándose en valores de
pH de 0 a 2 la especie H3AsO4, en valores de 2 a 6.5 en pH se encuentra la especie H2AsO4-
, mientras que a pH más altos (6.5 – 11.5), la especie dominante es HAsO4-2 y a pH de 11.5
a 14 se encuentra la especie AsO4-3. En rangos de 0 a 9 se muestra la especie H3AsO3
0,
mientras que al aumentar el pH de 9 a 12 se hace presente la fase H2AsO3-, y por último se
encuentra la fase HAsO3-2 en pH mayores a 12. [38]. En general, la forma pentavalente del
arsénico As+5 tiende a predominar frente a la forma trivalente As+3 en las aguas superficiales,
más oxigenadas que las aguas subterráneas.

2.3 Métodos de remoción de arsénico
2.3.1 Métodos convencionales
Existen diferentes metodologías convencionales de remoción de As+3 como As+5, sin
embargo la inmensa mayoría suponen un alto costo, complicado uso y mantenimiento,
además algunos procesos presentan el problema de tener un tratamiento previo de oxidación
mediante el uso de algunos agentes químicos oxidantes como el cloro, para que el As+3 se
convierta a As+5, ya que éste último es menos soluble en el agua y por lo tanto más fácil de
remover. Seguido de su separación mediante la explotación de cualquiera de las propiedades
físico-químicas como coagulación-precipitación, ósmosis inversa, intercambio iónico,
nanofiltración, adsorción, etcétera. Las cuales se describen a continuación y se presentan
porcentajes de remoción en la tabla 1.
Coagulación-filtración.-Es un proceso de tratamiento por el cual las cargas eléctricas de las
sustancias coloidales disueltas o suspendidas son neutralizadas con la adición de sustancias
insolubles en el agua, lo que permite la formación de partículas mayores o aglomerados que
pueden ser eliminadas por sedimentación o filtración. Se pueden alcanzar remociones de As+5
de más del 90 % [39].
Osmosis inversa.-Es un proceso para eliminar las sustancias disueltas presentes en el agua,
forzando la circulación del agua por una membrana semipermeable bajo una presión superior
a la osmótica. Tiene una eficiencia de más de 95 % de remoción de As disuelto. Este método
es efectivo para remover arsénico de aguas subterráneas. El rendimiento del proceso con
ósmosis inversa es afectado principalmente por la turbiedad, hierro, manganeso y sílice [14].
Intercambio iónico.-Es un proceso físico y químico, en el cual los iones de una especie dada
son desplazados de un material insoluble de intercambio (resina) por otros iones que se
encuentran en solución, remueve efectivamente el As en el rango de pH entre 8 y 9. Las
consideraciones que se tiene en este proceso comprenden el pH, iones competitivos, tipo de
resina, concentración de As en el afluente, disposición de la resina y los regenerantes usados,
efectos secundarios de la calidad del agua y los parámetros de diseño [6].
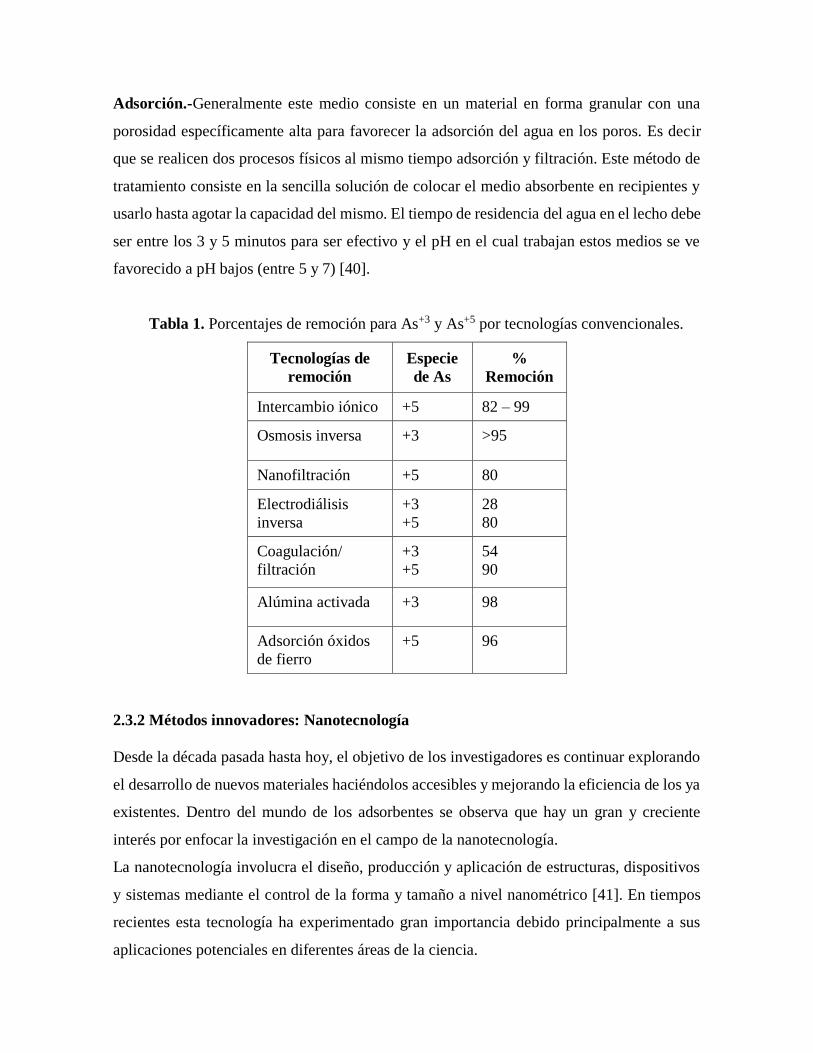
Adsorción.-Generalmente este medio consiste en un material en forma granular con una
porosidad específicamente alta para favorecer la adsorción del agua en los poros. Es decir
que se realicen dos procesos físicos al mismo tiempo adsorción y filtración. Este método de
tratamiento consiste en la sencilla solución de colocar el medio absorbente en recipientes y
usarlo hasta agotar la capacidad del mismo. El tiempo de residencia del agua en el lecho debe
ser entre los 3 y 5 minutos para ser efectivo y el pH en el cual trabajan estos medios se ve
favorecido a pH bajos (entre 5 y 7) [40].
Tabla 1. Porcentajes de remoción para As+3 y As+5 por tecnologías convencionales.
Tecnologías de
remoción
Especie
de As
%
Remoción
Intercambio iónico +5 82 – 99
Osmosis inversa +3 >95
Nanofiltración +5 80
Electrodiálisis
inversa
+3
+5
28
80
Coagulación/
filtración
+3
+5
54
90
Alúmina activada +3 98
Adsorción óxidos
de fierro
+5 96
2.3.2 Métodos innovadores: Nanotecnología
Desde la década pasada hasta hoy, el objetivo de los investigadores es continuar explorando
el desarrollo de nuevos materiales haciéndolos accesibles y mejorando la eficiencia de los ya
existentes. Dentro del mundo de los adsorbentes se observa que hay un gran y creciente
interés por enfocar la investigación en el campo de la nanotecnología.
La nanotecnología involucra el diseño, producción y aplicación de estructuras, dispositivos
y sistemas mediante el control de la forma y tamaño a nivel nanométrico [41]. En tiempos
recientes esta tecnología ha experimentado gran importancia debido principalmente a sus
aplicaciones potenciales en diferentes áreas de la ciencia.

Los nanomateriales pueden ser producidos por diferentes técnicas y de morfologías diversas
(esferas, tubos, hilos, películas delgadas, varillas, grafenos, etc.). A su vez, pueden ser
clasificados en base al tipo de material por ejemplo: metálicos, semiconductores y
poliméricos [42]. Los metales a nivel nanométrico poseen propiedades ópticas, electrónicas,
catalíticas, magnéticas, etc. Los cuales difieren significativamente de aquellas presentes a
mayor escala [43]. Dichas propiedades están fuertemente relacionadas con el tamaño, forma,
composición, cristalinidad y estructura de la partícula.
Algunos investigadores sostienen que la nanotecnología podría purificar el agua contaminada
en agua apta para consumo humano, saneamiento y riego; pues ofrece alternativas más
económicas, eficientes y duraderas, porque podrían mitigar los problemas del agua si se
resuelven los retos técnicos que presenta la remoción de contaminantes como bacterias, virus,
arsénico, mercurio, flúor, pesticidas, etc.
Hoy en día ya se encuentran en el mercado una gran variedad de dispositivos que incorporan
adelantos nanotecnológicos para el tratamiento de aguas, por ejemplo las membranas de
nanofiltración para eliminar sales disueltas y microcontaminantes, pero aún siguen siendo
muy costosas [44]. Un equipo de científicos de la India y EE. UU. desarrollaron filtros hechos
con nanotubos de carbón que tienen el poder de eliminar bacterias y virus con más eficacia
que las membranas de filtración convencionales [45]. Las arcillas (attapulgita) y las zeolitas
naturales que tienen tamaño nanométrico también son usadas en los nanofiltros [46]. Los
nanocatalizadores gracias a su diminuto tamaño pueden degradar los contaminantes
químicamente [47]. Entre los nanomateriales de mayor impacto en el tratamiento de aguas
están las nanopartículas de óxidos de fierro, pues tienen grandes superficies en proporción a
su volumen y se unen con facilidad a contaminantes como el arsénico [48] algunos datos se
muestran en la tabla 2.
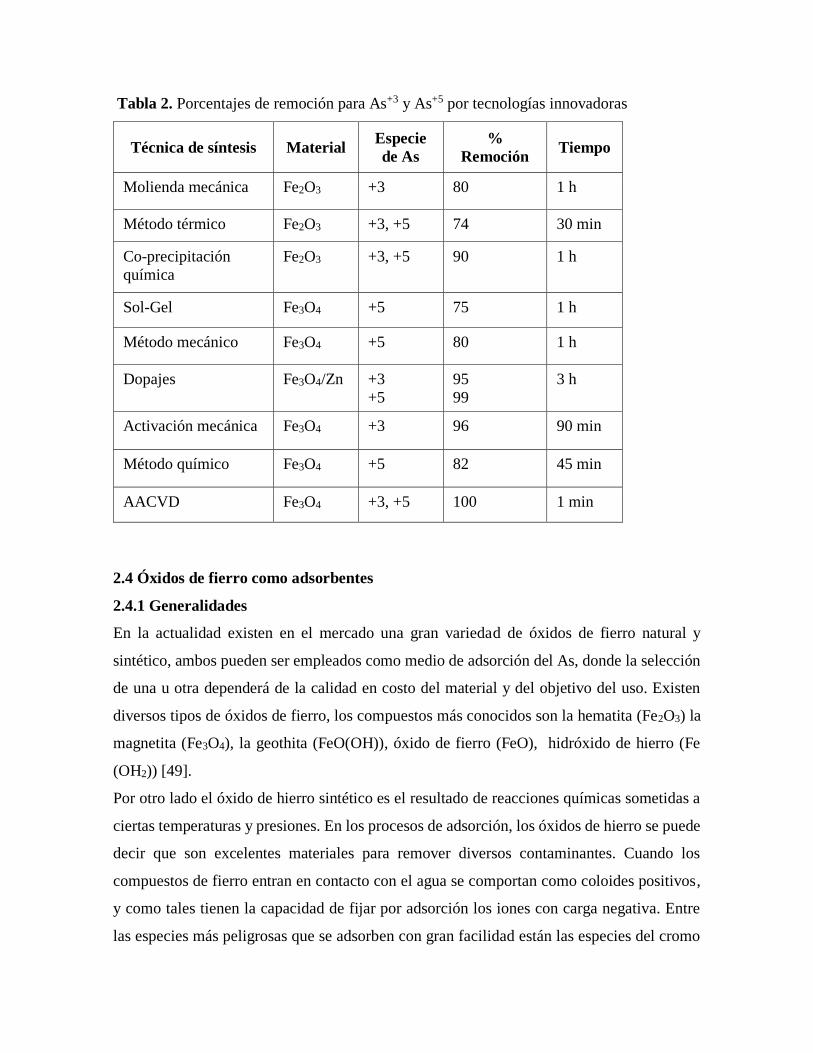
Tabla 2. Porcentajes de remoción para As+3 y As+5 por tecnologías innovadoras
Técnica de síntesis Material Especie
de As
%
Remoción Tiempo
Molienda mecánica Fe2O3 +3 80 1 h
Método térmico Fe2O3 +3, +5 74 30 min
Co-precipitación
química
Fe2O3 +3, +5 90 1 h
Sol-Gel Fe3O4 +5 75 1 h
Método mecánico Fe3O4 +5 80 1 h
Dopajes Fe3O4/Zn +3
+5
95
99
3 h
Activación mecánica Fe3O4 +3 96 90 min
Método químico Fe3O4 +5 82 45 min
AACVD Fe3O4 +3, +5 100 1 min
2.4 Óxidos de fierro como adsorbentes
2.4.1 Generalidades
En la actualidad existen en el mercado una gran variedad de óxidos de fierro natural y
sintético, ambos pueden ser empleados como medio de adsorción del As, donde la selección
de una u otra dependerá de la calidad en costo del material y del objetivo del uso. Existen
diversos tipos de óxidos de fierro, los compuestos más conocidos son la hematita (Fe2O3) la
magnetita (Fe3O4), la geothita (FeO(OH)), óxido de fierro (FeO), hidróxido de hierro (Fe
(OH2)) [49].
Por otro lado el óxido de hierro sintético es el resultado de reacciones químicas sometidas a
ciertas temperaturas y presiones. En los procesos de adsorción, los óxidos de hierro se puede
decir que son excelentes materiales para remover diversos contaminantes. Cuando los
compuestos de fierro entran en contacto con el agua se comportan como coloides positivos,
y como tales tienen la capacidad de fijar por adsorción los iones con carga negativa. Entre
las especies más peligrosas que se adsorben con gran facilidad están las especies del cromo
y del arsénico. Es así que los compuestos de fierro poseen la gran capacidad natural de
depurar aguas contaminadas con elementos tóxicos como el As [11]. La información
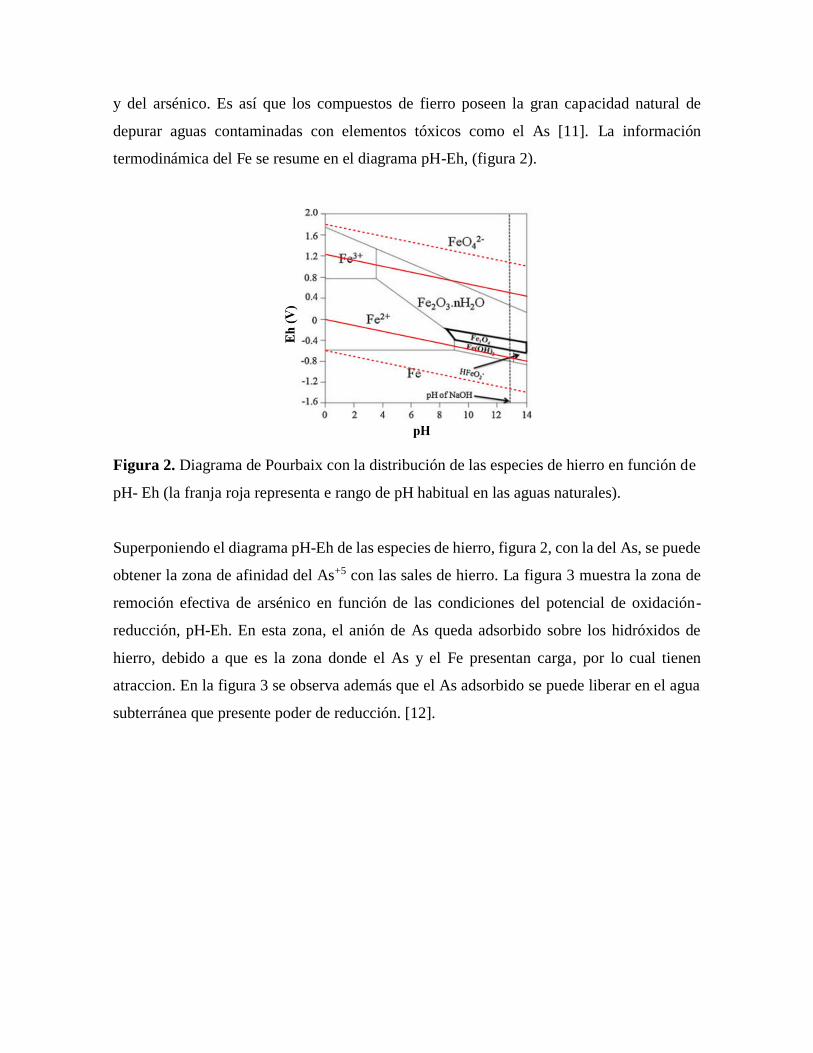
termodinámica del Fe se resume en el diagrama pH-Eh, (figura 2).
Figura 2. Diagrama de Pourbaix con la distribución de las especies de hierro en función de
pH- Eh (la franja roja representa e rango de pH habitual en las aguas naturales).
Superponiendo el diagrama pH-Eh de las especies de hierro, figura 2, con la del As, se puede
obtener la zona de afinidad del As+5 con las sales de hierro. La figura 3 muestra la zona de
remoción efectiva de arsénico en función de las condiciones del potencial de oxidación-
reducción, pH-Eh. En esta zona, el anión de As queda adsorbido sobre los hidróxidos de
hierro, debido a que es la zona donde el As y el Fe presentan carga, por lo cual tienen
atraccion. En la figura 3 se observa además que el As adsorbido se puede liberar en el agua
subterránea que presente poder de reducción. [12].
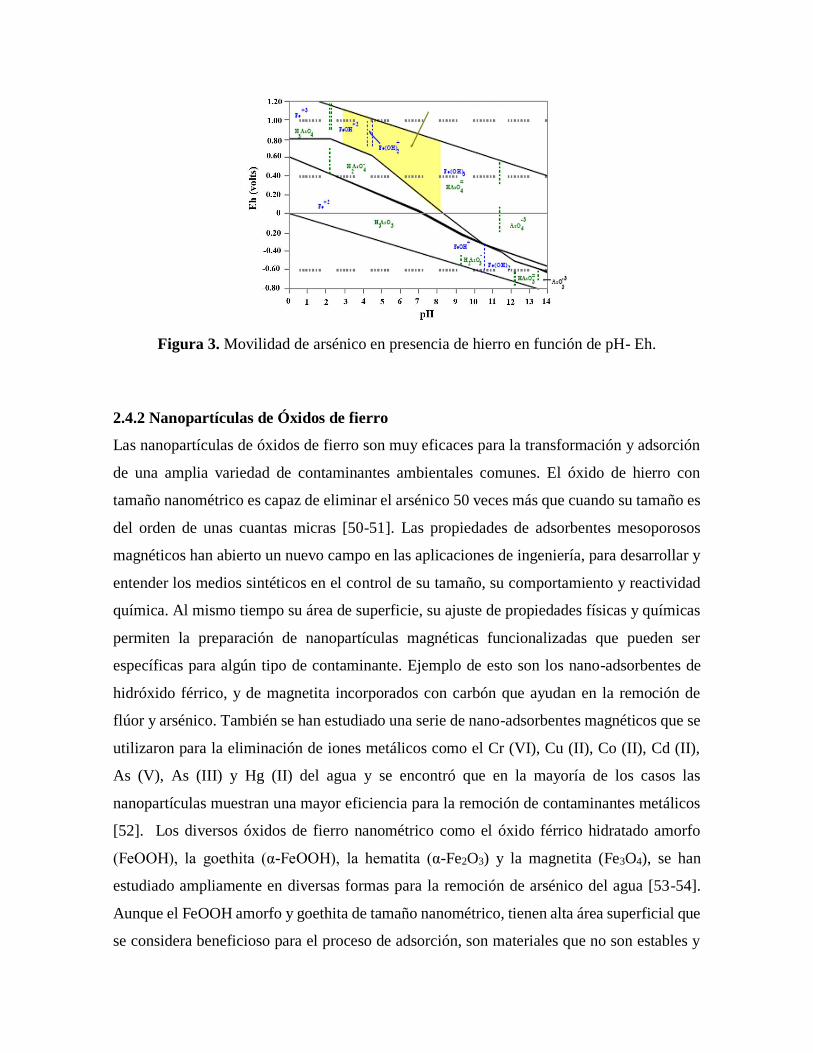
Figura 3. Movilidad de arsénico en presencia de hierro en función de pH- Eh.
2.4.2 Nanopartículas de Óxidos de fierro
Las nanopartículas de óxidos de fierro son muy eficaces para la transformación y adsorción
de una amplia variedad de contaminantes ambientales comunes. El óxido de hierro con
tamaño nanométrico es capaz de eliminar el arsénico 50 veces más que cuando su tamaño es
del orden de unas cuantas micras [50-51]. Las propiedades de adsorbentes mesoporosos
magnéticos han abierto un nuevo campo en las aplicaciones de ingeniería, para desarrollar y
entender los medios sintéticos en el control de su tamaño, su comportamiento y reactividad
química. Al mismo tiempo su área de superficie, su ajuste de propiedades físicas y químicas
permiten la preparación de nanopartículas magnéticas funcionalizadas que pueden ser
específicas para algún tipo de contaminante. Ejemplo de esto son los nano-adsorbentes de
hidróxido férrico, y de magnetita incorporados con carbón que ayudan en la remoción de
flúor y arsénico. También se han estudiado una serie de nano-adsorbentes magnéticos que se
utilizaron para la eliminación de iones metálicos como el Cr (VI), Cu (II), Co (II), Cd (II),
As (V), As (III) y Hg (II) del agua y se encontró que en la mayoría de los casos las
nanopartículas muestran una mayor eficiencia para la remoción de contaminantes metálicos
[52]. Los diversos óxidos de fierro nanométrico como el óxido férrico hidratado amorfo
(FeOOH), la goethita (α-FeOOH), la hematita (α-Fe2O3) y la magnetita (Fe3O4), se han
estudiado ampliamente en diversas formas para la remoción de arsénico del agua [53-54].
Aunque el FeOOH amorfo y goethita de tamaño nanométrico, tienen alta área superficial que
se considera beneficioso para el proceso de adsorción, son materiales que no son estables y

pueden descomponerse o formar fácilmente óxidos de fierro cristalinos durante su síntesis y
uso, lo que reducirá en gran medida su capacidad de remoción de arsénico [55]. La hematita,
tiene una alta estabilidad, alta área superficial (de tamaño nanométrico) y además puede tener
un gran potencial para la eliminación de arsénico, sin embargo tiene alta afinidad al agua y
por lo tanto se dificulta su capacidad de colección una vez terminado el proceso de adsorción
[56], por lo que es preferible trabajar con magnetita ya que se sabe que al aplicar un campo
magnético las partículas tienden a agregarse lo que facilita la separación magnética.
2.4.3 Generalidades de la magnetita
La magnetita pertenece al grupo de la espinela y es clasificada dentro del grupo de los óxidos.
En la naturaleza la encontramos en forma masiva o cristalizada, generalmente, en forma de
octaedros en formaciones sedimentarias de hierro. Está constituida por óxido ferroso-
diférrico (Fe3O4) que debe su nombre de la ciudad griega de Magnesia. Es un mineral muy
denso, frágil, medianamente duro y con propiedades ferromagnéticas, es capaz de atraer al
hierro y otros metales. Su color es negro parduzco, con brillo metálico y se caracteriza,
principalmente por su fuerte magnetismo.
La magnetita puede aparecer en numerosos ambientes: ígneos, sedimentarios o metamórficos
(por metamorfismo de contacto). Se halla diseminada como mineral en muchas rocas ígneas
(sobre todo básicas). En ciertos tipos de segregación magmática es uno de los principales
constituyentes de la roca y forma así grandes masas de mineral. Los mayores depósitos del
mundo se hallan al norte de Suecia. Otros depósitos importantes se encuentran en Chile,
Noruega, Rumania, Siberia, en el complejo ígneo de Bushveld, y en la región del Lago
Superior (EEUU). La magnetita junto a la hematita es la principal mena de hierro. Es
empleada por numerosas especies animales (tortugas, palomas, abejas, moluscos) para su
orientación en el campo magnético terrestre. Se emplea también como material de
contrucción, es un añadido en hormigones, especialmente para protección radiológica. Al ser
un mineral muy estable a alta temperatura, se emplea como protector del interior de los tubos
de las calderas, de igual manera como adsorbente por su afinidad con algunos elementos
como el arsénico.

2.4.4 Nanopartículas de magnetita (MNPs).
Las MNPs son uno de los materiales de óxido de fierro nanoestructurados más importantes,
debido a sus propiedades ópticas, eléctricas, magnéticas y de adsorción [21, 57-59]. Dentro
de su comportamiento magnético presentan la propiedad ferromagnética o
superparamagnética [60-61]. El superparamagnetismo es comúnmente observado a
temperatura ambiente, para nanopartículas de óxido de fierro con tamaños menores o iguales
a 10 nm. Los materiales paramagnéticos se caracterizan por tener sus dipolos magnéticos
orientados al azar, y que pueden ser alineados sólo en la presencia de un campo magnético
externo y a lo largo de su dirección. Bajo el efecto de un campo magnético externo, las
nanopartículas superparamagnéticas responden rápidamente al campo magnético y se
observan altos momentos magnéticos en donde el magnetismo residual (Mr) y la
cohercitividad (HC) están cerca de cero o son nulos [18,60].
El hecho de que las nanopartículas de magnetita presenten la propiedad magnética, ha
permitido que se utilicen para la remoción de materiales contaminantes en medios acuosos.
Debido a que pueden dispersarse homogéneamente en el agua y ofrecen la posibilidad de ser
fácilmente separadas magnéticamente después de su uso [18].
Las nanopartículas de magnetita son un adsorbente prometedor para la eliminación efectiva
de arsénico del agua. La capacidad de adsorción es una función del área superficial lo que
depende del tamaño de partícula del material. Estudios que se centran en el efecto del tamaño
de las MNPs demuestran que el tamaño de las nanopartículas tiene un efecto importante en
la adsorción y desorción de As+3 y As+5 [20, 62]. Los resultados indican que al reducir el
tamaño de las partículas desde 300 nm hasta 12 nm, las capacidades de adsorción de As+3 y
As+5 aumentan casi 200 veces, logrando con esto una mejora en la eficiencia de remoción
[20]. Debido a que tienen una alta afinidad hacia el arsénico y se pueden separar fácilmente
después del tratamiento mediante el uso de imanes, lo que le da una enorme ventaja con
respecto a los métodos convencionales y otros óxidos de fierro.

2.5 Síntesis de nanopartículas
2.5.1 Técnicas de síntesis
Existen diferentes tipos de sistemas o metodologías, que sirven como base para realizar
síntesis de nanopartículas por diferentes técnicas, según los requerimientos de cada una de
ellas, estas metodologías permiten tener nanopartículas con las características deseadas como
forma, tamaño y composición. Pueden ser químicas, físicas, o físico-químicas.
2.5.1.1 Microemulsión
El uso de microemulsiones agua-aceite para la síntesis de nanopartículas es uno de los
métodos más prometedores. La aplicación de esta tecnología permite sintetizar
nanopartículas de muy diferente composición. La metodología de microemulsión ha sido
aplicada para sintetizar nanopartículas de metal puro (Pt, Pd, Ir, Rh, Au, Ag, Cu), así como
para sintetizar nanopartículas binarias (Pt/Pd, Pt/Ru, Pt/Ir, Pt/Rh). La tecnología también
puede ser usada para sintetizar nanopartículas multimetálicas. En el caso de nanopartículas
binarias y multimetálicas, la composición atómica se puede modificar fácilmente en función
de las necesidades. Además, esta metodología se puede usar para preparar distintos tipos de
nanopartículas tales como SiO2, CdS, ZnS, ZrO2, CaCO3, BaCO3, CdSe, TiO2, etc.
El rango de tamaño de las nanopartículas puede ser controlado y variado entre 1-50 nm,
dependiendo en gran medida del tipo de surfactante utilizado. La principal ventaja de este
método radica en la posibilidad de obtener diferentes composiciones y tamaños. Las
propiedades catalíticas y electrocatalíticas de las nanopartículas depende del estado y la
limpieza de su superficie. Por esta razón, es muy importante desarrollar algunos
procedimientos de descontaminación que permitan limpiar la superficie de las nanopartículas
sin modificar ni su estructura inicial ni su composición superficial. Esta descontaminación
permite aplicar las nanopartículas con sus propiedades catalíticas y electrocatalíticas
completas [63-64].

2.5.1.2 Sistemas coloidales
La síntesis de nanopartículas en sistemas coloidales es uno de los métodos mejor conocidos
para sintetizar nanomateriales. Además, esta metodología permite, en algunos casos,
sintetizar nanopartículas con algunas orientaciones preferenciales y se sabe que la forma de
las nanopartículas influye en sus propiedades ópticas, electrónicas, catalíticas y
electrocatalíticas. Este hecho es especialmente importante cuando las nanopartículas se van
a utilizar en reacciones catalíticas y electrocatalíticas que son especialmente sensibles a la
estructura del catalizador. La aplicación de este método de electrocatálisis es un concepto
realmente innovador. Este método tiene como principal ventaja que permite controlar la
forma de las partículas. El tamaño de las nanopartículas varía entre 5-50 nm, y tiene una
fuerte dependencia respecto al agente protector utilizado. Además, se pueden preparar de
diferentes formas (cúbicas, tetraédricas, esféricas, octaédricas truncadas), lo que dará lugar a
diferentes propiedades [65-66].
2.5.1.3 Fases acuosas
Mediante este método, el tamaño de partícula es muy superior al obtenido con otros sistemas
(situado en torno a los 50-500 nm en función del tipo de agente reductor utilizado). Se han
preparado nanopartículas en ausencia de agentes protectores, donde únicamente es necesario
partir de la disolución que contiene a los precursores de la nanopartículas y un agente reductor
(N2H4, H2(g), NaBH4 [67].
2.5.2 Técnicas de síntesis de partículas nanométricas: químicas y físicas.
Las partículas de tamaño nanométrico con propiedades únicas tales como alta reactividad,
baja temperatura de sinterización, y amplia área superficial, han sido obtenidas mediante
diferentes métodos como son aspersión pirolítica [52], descarga plasmática reactiva [21],
depósito químico de vapor (CVD) [68], depósito de vapor electroquímico [50], depósito
químico de vapor asistido por flama (FACVD) [69], CVD asistido por aerosol (AACVD)
[23], sol-gel [70], co-precipitación química, condensación de gas inerte, inmersión química,
descarga plasmática, plasma por microondas, etc. Dentro de todos estos métodos la aspersión
pirolítica es un método atractivo para la preparación de partículas de tamaño nanométrico
por la velocidad de producción, operación continua y uso de equipo relativamente simples.

2.5.2.1 Técnica AA-CVD (Depósito Químico de Vapor asistido por Aerosol).
El proceso de AACVD es una técnica empleada para la elaboración de polvos ultrafinos
como óxidos o compositos metálicos, con propiedades superconductoras, dieléctricas o
piezoeléctricas. Por su sencillez y por ser una técnica económica resalta la AACVD, conocida
comúnmente en nuestro medio como rocío pirolítico, no necesita de una infraestructura
sofisticada para poder implementarse, además de que los materiales obtenidos por este
método tienen propiedades comparables a los obtenidos por otras técnicas, en las cuales se
necesitan costosos sistemas de alto vacío, fuentes de radio frecuencia o de control de gases
para obtener de manera eficiente materiales nanoestructurados en polvo. En la síntesis de
nanopartículas se usan sales precursoras y alcoholes porque a altas temperaturas se
descomponen fácilmente formando óxidos que se depositan en diferentes substratos como se
indica en la siguiente reacción.
– R-O-H + SAL → R-O-- SAL (óxido metálico)
(Metal –O-H) +SAL → Oxido metálico
La técnica consiste básicamente en producir una nube de gotas muy finas de una solución,
formada por el material precursor y un solvente apropiado. Esta nube es transportada, por un
gas vector (aire o argón), hasta una cámara con temperatura controlada, en la que se puede
encontrar el substrato para la obtención de nanopartículas o simplemente un sistema de
recolección del polvo obtenido.
Este proceso se lleva a cabo cuando una solución química, que contiene sales solubles de los
átomos del compuesto deseado es atomizada y rociada mediante una cabeza atomizadora o
pulverizadora sobre un substrato o colector de polvos. Cada gota de rocío llega al colector
efectuándose una reacción química pirolítica formando un material o cúmulos cristalinos del
producto, cuando se alcanza la temperatura de cristalización. El substrato o colector,
proporciona la energía térmica necesaria para realizar la descomposición del precursor
(siempre y cuando se encuentre a la temperatura necesaria para llevarla a cabo) y la
subsecuente recombinación de las especies constituyentes seguida por una sinterización y
recristalización del grupo de cristales, los productos de desecho y los solventes usados para
formar la solución escapan en forma de vapor y no se incorporan en el material formado,

siempre y cuando exista una adecuada remoción de los productos de reacción. Cuando una
solución es atomizada, la solución es transformada en gotas finas usando un dispositivo de
rocío y un gas portador, el cual puede o no jugar un papel activo en la reacción pirolítica. Los
solventes líquidos usados para formar la solución sirven para llevar a los agentes reactivos y
distribuirlos uniformemente sobre el área del substrato o recipiente colector durante el
proceso.
La técnica AACVD, tiene gran importancia porque presenta ventajas como: utilizar metales
nobles, tiene gran homogeneidad, alta calidad y pureza, trabaja con temperaturas
relativamente bajas (200 – 1000 °C), dependiendo del precursor. La composición puede ser
variable a voluntad, por lo que se pueden sintetizar: películas delgadas sobre vidrio o
cerámicos, nanovarillas, nanotubos, grafenos y nanopartículas, de las que se puede tener
control sobre el tamaño de la partícula, su distribución y control sobre la cristalinidad, además
de que se puede trabajar con cualquier óxido nano-estructurado simple o compuesto, puro o
dopado [71].
2.6 Modelos teóricos de síntesis de nanopartículas
La ventaja de contar con modelos teóricos, radica en que se logra tener una idea clara de lo
que puede ocurrir al momento de hacer el diseño experimental además evita el estar haciendo
experimentaciones de acuerdo al ensayo y error. Con lo cual se puede variar una gran
cantidad de parámetros hasta tener las condiciones deseadas y sin desperdiciar material,
gases, reactivos y equipo.
El amplio rango de morfologías que se obtienen en las nanopartículas sintetizadas por las
técnicas de aspersión pirolítica como spray pirolisis y AACVD, entre otros, puede ser
explicado mediante la evolución de la saturación de la gota de la solución precursora. Lo cual
está en función del tamaño de la gota, concentración del soluto, sobresaturación crítica del
precursor (CSS), y la velocidad de evaporación [72]. Dicha evolución de la gota concluye
con la obtención de partículas de tamaño micro o nanométrico, dependiendo del tamaño de
la gota del precursor, y huecas o sólidas, dependiendo de si se excede o no la saturación de
equilibrio del soluto.
Existen reportes donde se explica la formación ahuecada o sólida de partículas esféricas
mediante el uso de modelos teóricos y/o trabajos experimentales [72-74]. En los trabajos

donde realizan ambos, los resultados de la simulación son comprables con los resultados
experimentales. El esquema teórico de la evaporación de la gota y el proceso de precipitación
que se lleva a cabo en el proceso de AACVD se muestra en la Figura 4.
Figura 4. Esquema teórico del proceso de la evaporación de la gota y precipitación de la
sal en la técnica de aspersión pirolítica. [72].
La formación de nanopartículas huecas o sólidas inicia con la atomización de una solución
precursora, posteriormente la nube formada es transportada a través de un reactor que se
encuentra a una temperatura controlada. La temperatura debe ser la adecuada para que la
solución reaccione químicamente y se obtenga el compuesto deseado. Debido al
calentamiento durante dicho proceso ocurre una evaporación del solvente, el tamaño de la
gota disminuye y la concentración de la solución dentro de cada gota aumenta a medida que
el aerosol se desplaza a través del reactor, siendo entonces que la gota original se contrae a
un diámetro más pequeño [73-74]. Con el tiempo, la concentración de soluto en la superficie
de la gota alcanza la concentración de super saturación (CSS) en cuyo punto la nucleación
del soluto y la precipitación puede tener lugar dentro de la gota- partícula dependiendo del
tipo de precursor utilizado [74]. Se propone que la nucleación tiene lugar cuando la
concentración de soluto en la superficie de la gota llega a la CSS [72]. Esto es porque la
evaporación del solvente ocurre en la superficie causando el aumento de la concentración del
soluto y por lo tanto, la concentración del soluto en el centro de la gota resulta menor que en
su superficie. Por otro lado, se asume que ocurre una nucleación homogénea de la sal en la
superficie de la gota cuando alcanza su CSS. Después de la nucleación, la precipitación se
produce sólo en la parte de la gota donde la concentración de soluto es mayor que la

saturación de equilibrio (ES). Luego, las partículas huecas se forman si la concentración de
soluto en el centro de la gota es menor que la saturación de equilibrio del soluto [74].
La Figura 5 muestra un esquema de cómo cambia la concentración del soluto en el interior
de la gota durante la evaporación del disolvente, basado en el formalismo propuesto por [72,
74]. Las gráficas de la Figura 5 muestran cómo varía la concentración de soluto dentro de la
gota. El eje horizontal representa la posición dentro de la gota (adimensional), va de 0 a 1
que corresponde al centro y la superficie de la gota, respectivamente.
Figura 5. Cambio de la distribución de la concentración en el interior de la gota. Adaptado
de la referencia [72].
Inicialmente, la concentración de soluto dentro de la gota es uniforme (C0) (Figura 5a). Con
el tiempo y continuo calentamiento, la evaporación del disolvente hace que la concentración
de soluto incremente en la superficie de las gotas, formando un gradiente de concentración
que resulta en la difusión del soluto hacia el núcleo. Seguido a esto se propone que la
concentración del soluto en la superficie de la gota llegue a la CSS. En este punto, si la
concentración de soluto en el centro de la gota (C(r=0)) es mayor que la concentración de
equilibrio (ES), la fase sólida es propensa a nuclear y los cristalitos crecerán en todo el

volumen de la gota-partícula, lo que resulta en una esfera sólida (Figura 5c). Por otro lado, si
la concentración de soluto en el centro es menor que la ES, se propone que se forme una
partícula hueca (Figura 5d) [74].
Cabe señalar, que la llamada nanopartícula “sólida” es en realidad microporosa, debido a la
porosidad entre las cristalitas de la sal precipitada [72]. Para las nanopartículas huecas,
después de la formación de la costra, la solución atrapada al interior de la gota continua
evaporándose, resultando en un incremento de la presión interna, la cual depende de la
permeabilidad de la sal precipitada. Si la permeabilidad es baja, es previsible la consecuente
explosión de la gota y costra dando como resultado una segunda generación de gotas más
pequeñas y restos fragmentados de la costra original [73]. Por el contrario si la permeabilidad
es alta, los vapores del solvente pueden escapar sin modificar la costra inicial; esto lleva a la
formación de nanopartículas ahuecadas y porosas [72].
2.7 Técnicas de Caracterización Microestructural de las Nanopartículas
2.7.1 Microscopía electrónica de barrido
El microscopio electrónico de barrido (MEB) es uno de los equipos más versátiles en el
estudio y análisis de materiales sólidos. Se usa para caracterizar morfológicamente una
amplia gama de compuestos, entre ellos, la nanopartículas. Permitiendo además conocer su
composición elemental. Algunas de las ventajas que se encuentran al utilizar éste dispositivo
como herramienta y no un microscopio óptico, se deben a la utilización de un mayor número
de señales que provienen de la interacción de los electrones con los sólidos, lo que permite
obtener mayor información sobre: la orientación cristalina, la composición química, la
estructura magnética o el potencial eléctrico del material observado. El principio de
funcionamiento del MEB se basa en el proceso de barrer la muestra con un haz electrónico
de sección transversal pequeña y de alta energía. Para con ayuda de un detector generar una
imagen punto a punto de la muestra [75]. El sistema de epectroscopia de discriminación de
energía de rayos x o mejor conocido como EDS por sus siglas en inglés, es un sistema que
se acopla a los microscopios electrónicos. Los cuales permiten realizar análisis elemental o
caracterización química de una muestra. Las capacidades de caracterización de la técnica de
EDS se deben, en gran parte, al principio fundamental de que cada elemento tiene una
estructura atómica única que genera rayos X característicos con energías diferentes respecto

a otros elementos [75]. Los parámetros que nos permiten conocer la calidad de un MEB son:
la profundidad de foco (depende completamente del instrumento y de las condiciones de
operación), el ruido de la imagen (influye la muestra) y la resolución (el equipo y la muestra
tienen gran influencia). Con éste aparato se puede formar la imagen de los detalles más
profundos de la superficie de la muestra, por ello es útil para estudiar sus características
morfológicas y topográficas (relieve), la ya mencionada composición química, el espesor
(viendo la muestra transversalmente), entre otros.
2.7.2 Microscopía electrónica de transmisión
El microscopio electrónico de transmisión (MET) es un equipo que ha sido usado en todas
las áreas de investigación por su habilidad para observar estructuras muy finas y detectar
ínfimos elementos o compuestos. El principio de operación consta de irradiar una muestra
delgada con un haz de electrones el cual viaja a través de un sistema de vacío para no ser
desviado por el aire y las lentes magnéticas le ayudan a enfocarla y dirigirla hacia la muestra.
La caracterización estructural y química de la muestra es dada al interpretar las interacciones
del haz de electrones con la muestra.
2.7.3 Difracción de rayos X.
La difracción de rayos X (DRX) es una técnica de análisis versátil y no destructiva para la
identificación y determinación cuantitativa de las fases cristalinas presentes en muestras
sólidas o en forma de polvos. Un cristal está formado por un conjunto de átomos que se
repiten en una red tridimensional periódica. Las propiedades de los materiales cristalinos
depende de su composición química (qué tipo de átomos formanel conjunto de átomos que
se repite y cuántos son) y de su estructura (cómo están colocados esos átomos y cómo se
repiten para formar el cristal). Los materiales cristalinos se diferencian unos de otros ya sea
por su composición, o bien, por su estructura.
Un compuesto con una composición y estructura definidas se conoce como fase cristalina.
Así pues, la técnica de DRX nos permite distinguir una fase cristalina de otras y conocer su
estructura. Tal identificación se alcanza comparando patrones de difracción obtenidos a partir
de una muestra desconocida con relación a una base de datos internacional que contiene
patrones de referencia de un gran número de materiales. El fenómeno de la difracción de

rayos x es el resultado de la interacción de un tipo específico de radiación (rayos X) con los
átomos ordenados de los cristales. Se utilizan rayos x porque son del orden de las distancias
interatómicas en los cristales 1Å (Å=1x10-10 m), lo que posibilita la aparición de fenómenos
de interferencia y de direcciones de difracción. La interacción de los rayos x con los cristales
se explica mediante la Ley de Bragg, que se resume a continuación. Si hacemos incidir un
haz de rayos X sobre un cristal, observaremos la aparición de haces difractados según
direcciones discretas y características de dicho cristal de acuerdo con la siguiente ecuación:
𝑛 𝜆 = 2𝑑 𝑠𝑒𝑛𝜃 (1)
Donde n es un número entero de veces de la longitud de onda, 𝜆 es la longitud de onda de
los rayos X, d es la distancia entre los planos de la red cristalina y 𝜃 es el ángulo entre los
rayos incidentes y los planos de dispersión.
En una interpretación sencilla de la Ley de Bragg, las familias de planos cristalográficos
paralelos de un cristal se comportarían como “espejos” que “reflejan” los rayos X para
algunos ángulos de incidencia determinados. Los ángulos (θ) de los rayos X difractados
dependen de la distancia que separa los planos cristalográficos (d) y de la longitud de onda
de los rayos X utilizados (λ). El equipo empleado para hacer éstas mediciones se conoce
como difractómetro y sus resultados son expresados en términos gráficos por medio de un
difractograma, el cual muestra el resultado de realizar un experimento de difracción en el que
se miden las intensidades de los rayos X difractados por un cristal y los ángulos θ donde
aparecen. La aparición de un pico (o reflexión) en el difractograma indica que los cristales
de la muestra tienen una familia de planos con una distancia interplanar tal que se cumple la
ley de Bragg para la longitud de onda utilizada en el ángulo medido. Como en un cristal están
definidos numerosos planos cristalográficos, en los difractogramas de una fase cristalina se
observan diferentes picos para diferentes ángulos. Además, los picos tienen una intensidad
variable, dependiendo de los átomos que componen el cristal y de su posición [77].

2.7.4 Adsorción y desorción física de gases por el método Brunauer-Emmet-Teller
(BET).
El método Brunauer-Emmet-Teller (BET), se basa en la adsorción de un gas inerte sobre una
superficie. La teoría de BET dice que para poder determinar el área superficial es necesario
determinar el número de moléculas del gas requeridas para cubrir la superficie del sólido con
una capa muy fina denominada monocapa. Esta condición se logra cuando la cantidad de gas
adsorbido alcanzo el equilibrio lo cual usualmente se da en un intervalo de presiones menores
a una atmosfera. Los resultados se presentan como isotermas de adsorción, las que se
obtienen graficando los volúmenes de gas adsorbidos a una temperatura constante y para
distintas presiones relativas de gas. Las isotermas de adsorción son una medición útil para la
caracterización de las propiedades texturales de las nanopartículas y de los materiales en
general. Las isotermas están formadas por 2 procesos, el de adsorción y el de desorción. En
el proceso de adsorción las moléculas son transferidas a la superficie interna y externa del
absorbente y en la desorción ocurre una transferencia en sentido contrario, es decir, hay un
descenso en la presión acompañado del desprendimiento de las moléculas del gas de la
superficie.
Ambos procesos forman las isotermas (adsorción y desorción), en donde son formadas las
histéresis debido a que estos procesos no coinciden. Las gráficas de las isotermas están
representadas con una curva con el eje de las ordenadas indicando la cantidad de gas
adsorbido y en el eje de las abscisas la presión relativa de equilibrio (p/p0), la cual va de 0 a
1; la presión de saturación 𝑝0 es la transformación de gas a líquido, p es la presión de
equilibrio del adsorbato. La forma de la isoterma depende del tamaño de poro y del tipo de
poro que presenta la muestra. Existen 6 tipos de isoterma como se observa en la Figura 6.
Aunque los 5 primeros tipos de isotermas fueron propuestos por Brunauer, Deming, Deming
&Teller, como la clasificación BDDT [79], después en 1985 se estableció una sexta
clasificación.

Figura 6. Tipos de isotermas [79].
La isoterma tipo I es característica para sólidos microporosos y la adsorción se produce a
presiones relativas bajas, las isotermas del tipo II son atribuidas a sólidos macroporosos o no
porosos, la pendiente es debida a la adsorción de monocapa-multicapa sobre la superficie
estable. La isoterma tipo III ocurre cuando la interacción adsorbente-adsorbato es baja, es
para sólidos no porosos, y es muy poco frecuente. El isoterma IV es característico de
materiales mesoporosos y presentan un ciclo de histéresis debido a las características de la
porosidad. La isoterma tipo V es igual que la isoterma tipo III, el sólido en este caso es
mesoporoso. El isoterma VI es para sólidos con una superficie no porosa muy uniforme.
De la misma manera como se clasifican los diferentes tipos de isotermas, existe una
clasificación de los diferentes tipos de histéresis de la isoterma, la cual nos proporciona la
forma de los poros, como se muestra en la Figura 7.
Figura 7. Tipos de histéresis [79].
La histéresis tipo H1 (anteriormente tipo A, para poros cilíndricos) se observa en materiales
mesoporosos con distribución de tamaños de poro muy estrecha y en partículas esferoidales

de tamaño uniforme. La histéresis H2 (anteriormente como tipo E, poros en forma de cuello
de botella) ocurre en materiales con distribución de tamaños de poro y morfología no muy
bien definida. La histéresis tipo H3 (anteriormente tipo B) es característico de materiales
compuestos por partículas laminares, con morfología tipo de rendija. Y por último la
histéresis de tipo H4 es característica en sólidos que contienen en forma de rejilla muy
estrechos.
Esta técnica también es utilizada para la determinación de volumen de poro, diámetro de
poro, distribución de diámetro de poro. Cuando se habla de volumen total de poro se refiere
a la cantidad de adsorbato que se requiere para llenar toda la porosidad del material. Esta
cantidad corresponde al volumen del adsorbato cuando p/p0 =1, en ese punto el adsorbato
(nitrógeno) está en un estado condensado. Para determinar el tamaño de poro, es necesario
obtener datos de p/p0 a lo largo de toda la isoterma de adsorción, así, el análisis de
microporos es a p/p0 < 0.1, la adsorción para los mesoporos ocurre entre 0.1< p /p0 < 0.95, y
para la adsorción de macroporos ocurre a p/p0 > 0.95.
La distribución de tamaño de poro es llamado así por la relación que hay entre el tamaño de
poro con respecto a la cantidad de poros que existe de cada intervalo de diámetro de poros,
es decir, la abundancia de poros de un tamaño determinado, estimado por el método de
Barret-Joyner-Halenda (BJH). La IUPAC (International Unión of Pure and Applied
Chemistry) a partir de 1985 ha clasificado los poros de dos maneras por su tamaño y su forma.
Por su tamaño, los poros se clasifican en microporos (anchura de poro media menor a 20 Å),
mesoporos (anchura de poro media 20-500 Å) y macroporos (anchura de poro media mayor
a 500 Å) [80]; en el caso de las nanopartículas de magnetita, los poros se encuentran en el
orden de los mesoporos. En función de la conectividad los poros se clasifican como: poros
cerrados (sin conexiones con otros poros ni con el exterior), abiertos o discontinuos (presenta
una única conexión con el exterior), continuos (presenta más de una conexión), mientras que
en función de la forma como poro cilíndricos (en donde el tamaño de la entrada es igual que
el interior), cuello de botella (el tamaño de la entrada es menor que el interior), forma de
embudo (el tamaño de la entrada es mayor al del interior) y de rendijas.

Figura 8. Esquema de la sección transversal de un sólido poroso. Poros cerrados (a). Poros
abiertos (b, c, d, e, f). Poros discontinuos (b, f). Poros continuos (e). Poros cilíndricos (c, f).
Poros de entrada angosta o cuello de botella (b). Poros en forma de embudo (d). Poros en
forma de rendija (g).
2.7.5 Espectrofotometría de absorción atómica.
Hay algunos elementos que son difíciles de volatilizar con una llama o un horno. Para estos
elementos se utiliza la técnica de generación de vapor, ya sea formando el hidruro metálico
del elemento (As, Bi, Sb, Sn, Se y Te) o directamente vapores como en el caso del Hg. La
generación de vapor aumenta la sensibilidad de la técnica de absorción atómica,
especialmente en estos elementos que tienen importancia como contaminantes ambientales
o en toxicología. Hay dos métodos a través de los cuales se puede formar un hidruro: El
método del borohidruro de sodio, NaBH4, que implica la reacción del elemento analito, en
una solución ácida, con el NaBH4 para formar hidruros gaseosos del elemento en estudio. El
otro método es del cloruro de estaño II (SnCl2), en el cual se agrega K2Cr2O7 a la muestra.
La solución obtenida reacciona con el SnCl2 que está en medio ácido para formar el hidruro
gaseoso del elemento de interés. Una vez formado el hidruro gaseoso, éste es separado de la
solución y transportado por un gas portador hasta una celda de cuarzo, donde es calentado
produciéndose la descomposición térmica (atomización). Como la celda está en el paso
óptico de la radiación emitida por la lámpara de cátodo hueco, se produce una absorción de
la luz por parte de los átomos del analito que será proporcional a su concentración. El éxito
de la técnica se basa en dos características básicas: separa efectivamente el elemento analito
de su matriz química, eliminando así el efecto de interferencias de matriz en el proceso de
atomización y disminuyendo la absorción de fondo; la segunda es que proporciona un medio

más eficiente de atomización para estos elementos. El generador de hidruros HG3000 es un
sistema que genera un flujo continuo de vapor, consta de una bomba peristáltica que
continuamente bombea muestra y reactivos a un tubo múltiple donde se produce la mezcla.
La solución mezcla fluye a través de un tubo serpentín donde se forma el hidruro del elemento
junto con hidrógeno. Con la ayuda del gas portador (Ar o N2) el hidruro (junto con el
hidrógeno) entra a un recipiente separador gas-líquido donde el hidruro gaseoso es removido
de la solución. Posteriormente el hidruro, al estado de vapor es transportado hasta la celda de
absorción de cuarzo, que está montada sobre el mechero y calentada con llama aire-acetileno
o eléctricamente si se dispone del EHG3000. El vapor es atomizado en la llama
produciéndose la absorción de luz. El gas inerte que se usa debe ser de una alta pureza y a
una presión regulada de 30-60 psi (225-455 kPa). El flujo de gas durante la medición debe
ser para el método del borohidruro de sodio, desde el separador gas-líquido de 30 mL min-1.
En el laboratorio la generación de hidruro gaseoso se consigue tratando la muestra que
contiene arsénico con una solución de ácido clorhídrico y borohidruro de sodio. Los hidruros
gaseosos formados son impulsados por un gas inerte de arrastre (argón) hasta el tubo de
cuarzo caliente donde se lleva a cabo la descomposición térmica o atomización del hidruro.
El tubo de cuarzo calentado por la llama, aumenta el tiempo de residencia de los átomos y
evita la dispersión de los mismos y el ruido de fondo. Finalmente las señales detectadas por
el amplificador se reportan por un computador [81].
2.8 Procesos de adsorción
La adsorción es el fenómeno de acumulación de partículas sobre la superficie de un sólido,
la sustancia que se acumula es el adsorbato y el material sobre el cual lo hace es el adsorbente.
Es un proceso selectivo donde al aumentar la superficie de adsorbente y la concentración de
adsorbato, aumenta la cantidad adsorbida. Su velocidad aumenta cuando aumenta la
temperatura, pero desciende cuando aumenta la cantidad adsorbida, esto sucede de forma
espontánea, es decir, que la variación de la energía libre de Gibbs es negativa, es un proceso
exotérmico. De acuerdo a las fuerzas de interacción entre las moléculas del adsorbente y del
adsorbato, existen dos tipos fundamentales de adsorción. Cuando predominan las fuerzas de
atracción electrostáticas o atracciones dipolares entre un átomo o una molécula y la

superficie, se usa el término de adsorción física o fisisorción. Cuando las fuerzas de atracción
son enlaces covalentes, se aplica el término de adsorción química o quimisorción [81].
Fisisorción.-Este proceso de adsorción se caracteriza por ser reversible, debido a las fuerzas
de Van der Waals y no requiere energía de activación. La capa adsorbida puede variar en
espesor pudiéndose extender desde una capa de moléculas a otras. Los calores de adsorción
son numéricamente inferiores a 40 kJ mol-1.
Quimisorción.- Es un proceso que solo se manifiesta cuando el adsorbente y el adsorbato
tienden a formar un compuesto, lo que lo hace no reversible y más lento que la fisisorción.
La quimisorción no puede, por sí misma, dar lugar a una capa de más de una molécula de
espesor, debido a la especificidad del enlace entre el adsorbente y el adsorbato. Sin embargo,
es posible que existan capas subsiguientes adsorbidas físicamente sobre la primera capa. Sus
calores de adsorción química son numéricamente mayores a 40 kJ mol-1.
2.8.1 Isotermas de adsorción
La adsorción en un sistema sólido-líquido, consiste en el incremento de concentración, en la
superficie de un sólido hasta que se establece un equilibrio dinámico entre la concentración
del soluto que permanece en solución y la concentración superficial del soluto. Alcanzado el
equilibrio, se dice que existe una distribución definida del soluto entre las fases sólida y
líquida. La forma más común de manifestar esta distribución superficial consiste en expresar
la cantidad de soluto adsorbido por unidad de peso de adsorbente sólido (qe) como una
función de la concentración del soluto que permanece en solución en el equilibrio (Ce) y a
una temperatura constante. La relación entre qe y Ce, se conoce como isoterma de adsorción
[82]. Existen diversos modelos de adsorción, cada una con diferentes premisas y supuestos,
entre los modelos principales se tienen las isotermas de Langmuir, Freundlich, Brunauer
Emmett Teller (BET) y Dubinin-Radushkevich (DR).
Langmuir .-El modelo de Langmuir, supone que las capas adsorbidas no deben tener un
espesor mayor al de una molécula. Actualmente este supuesto se admite generalmente para
procesos de quimisorción y en determinadas condiciones para la adsorción física (presiones

bajas y temperaturas moderadamente altas) [82] . Solo tiene lugar una adsorción
monomolecular, es localizada, es decir, no existe interacción entre moléculas vecinas.
Freundlich.-Este modelo es una expresión que establece que la energía de adsorción
disminuye logarítmicamente al aumentar la fracción de la superficie ocupada, es muy útil
para describir procesos de adsorción sólido-líquido, en un amplio rango de concentraciones
y que la adsorción es siempre de naturaleza física [83-85]. La hipótesis del modelo de
Freundlich coincide con la de Langmuir cuando se refieren a la adsorción en monocapa; sin
embargo, este modelo si admite la posibilidad de interacción entre las moléculas adsorbidas
lo cual significa que no todos los sitios activos son equivalentes (energías de adsorción
heterogéneas).
Dubinin Rasdushkevich .-Este modelo es aplicado por diferentes autores [86-87], para
entender el mecanismo de adsorción que se está llevando a cabo en cada proceso. Este tipo
de isoterma señala que si se conoce el valor de la energía o calor de adsorción (E), se puede
estimar el tipo de adsorción que se está llevando a cabo. Si E es ≤ 20 kJ mol-1 el tipo de
adsorción podría ser explicado por una fisisorción y si E es ≥ a 40 kJ mol-1 se estaría tratando
de una quimisorción.
Tabla 3. Ecuaciones y parámetros empleados para el montaje de las isotermas de adsorción
en el equilibrio.
Isoterma Ecuación
General Ecuación linealizada Parametros Description
Langmuir
𝑞𝑒 =𝑄0𝐾𝐿𝐶𝑒
𝐶𝑒𝐾𝐿 + 1
𝐶𝑒
𝑞𝑒=
𝐶𝑒
𝑄0+
1
𝐾𝐿𝑄0
Q0 Capacidad máxima
de adsorción
KL
Constante de
Langmuir (Energía
de Adsorción)
Ce Concentración de As
en el equilibrio

2.8.2 Energía de adsorción
La energía libre de adsorción (E) se determinó según la ecuación (2) y representa el cambio
de energía que se requiere para la transferencia de un mol de iones a partir de la solución
sobre la superficie del sólido. La energía libre de Gibbs (∆G) indica la energía necesaria para
desplazar el adsorbato sobre la superficie del adsorbente y se calculó según la ecuación (3).
E = 1
√2𝐾𝐷 (2)
∆G = RT Ln 1
𝐾𝐿 (3)
donde KD es la constante de Dubinin, constante KL Langmuir, R la constante universal de
gases (8,314 J mol-1 K-1) y T la temperatura absoluta.
qe
Capacidad de
adsorción en el
equilibrio
Freundlich
𝑞𝑒 = 𝑄0𝐶𝑒
1𝐾𝐹
ln(𝑞𝑒) = ln (𝑄0) +1
𝐾𝐹ln(𝐶𝑒)
KF
Constante de
Freundlich (Energía
de Adsorción)
Dubinin 𝑞𝑒 = 𝑄0𝑒𝑘Ɛ2
𝑙𝑛 (𝑞𝑒) = ln (𝑄0) − 𝐾𝐷Ɛ2
Ɛ = 𝑅𝑇 ln(1 + 1
𝐶𝑒)
KD
Energía libre de
adsorción por
molécula
R Constante universal
de los gases
Ɛ Potencial de Polany
T Temperatura

3. JUSTIFICACIÓN
Buscar alternativas para disminuir la cantidad de lodos producidos en el tratamiento de aguas
contaminadas con arsénico, por medio de materiales nanoestructurados, para disminuir el
costo en la disposición final del residuo. Donde se tiene tanto aporte científico como
tecnológico.
APORTE CIENTÍFICO
1.- Generación de conocimiento sobre los procesos y mecanismos que determinan la síntesis
de nanopartículas de óxido de fierro de alta área superficial por la técnica AACVD.
2.- Al conocer el proceso de formación de nanopartículas se pueden manipular los parámetros
de síntesis para obtener una morfología deseada para algún tipo de aplicación.
APORTE TECNOLÓGICO
1.- La disminución en la generación de lodos contaminados con residuos de arsénico.
2.- Minimizar el tiempo de remoción.
3.- No utilizar agentes oxidativos de As+3 a As+5 .
4.- Disminución de costos.

4. Hipótesis
La utilización de softwares de simulación permitirá un mejor entendimiento del proceso de
síntesis, modificando parámetros que son importantes en la técnica AACVD y así generar
nanopartículas de magnetita con características microestructurales específicas, para la
adsorción de As.

5. Objetivo General
Realizar el estudio experimental y la simulación teórica de la síntesis por AACVD de
nanopartículas de magnetita para su aplicación en la remoción de arsénico.
5.1 Objetivos particulares
1.-Identificar los parámetros principales involucrados en la síntesis de nanopartículas de
magnetita por la técnica AACVD, para modelar el comportamiento de dichos factores
mediante el uso de softwares especializados como Solid Works – Fluid Works y COMSOL
Multiphysics 4.4 y correlacionar sus efectos con las propiedades microestructurales
obtenidas experimentalmente.
2.- Sintetizar nanopartículas de magnetita (MNPs), con diferentes parámetros de síntesis
(temperatura, flujo del gas de arrastre, concentración del precursor y diámetro de reactor
tubular). Las cuales fuerón caracterizadas por los siguientes métodos:
-Difracción de Rayos X, -MEB (Microscopía Electrónica de Barrido) -MET (Microscopía
electrónica de Transmisión) -BET (método Brunauer-Emmet-Teller).
3.-Desarrollar la ruta química para interpretar el proceso de reacción química hasta la
formación final de las nanopartículas de magnetita, con base en el software HSC
CHEMISTRY (Chemical Reaction and Equilibrium Thermodynamics).
5.-Realizar pruebas de contacto o Batch de las MNPs sintetizadas para las especies de As+3
y As+5 en soluciones con agua tridestilada para determinar :
a) Eficiencia de adsorción de arsénico en las MNPs, variando parámetros como:
- Tiempo de contacto y concentración inicial de As
b) Capacidad de adsorción a una concentración conocida de MNPs As+3 y As+5 en las
MNPs.
6.- Determinar el mecanismo de adsorción que se lleva a cabo por medio de:
a) Determinación de isotermas de Langmuir, Freundlich y Dubinin
b) Calculo de la energía de adsorción y energía libre de Gibbs.

CAPÍTULO II
METODOLOGÍA EXPERIMENTAL
6. Simulación teórica
6.1 Software Solid Works – Fluid Works
6.1.1 Simulaciones del transporte de masa, cantidad de movimiento y energía en el
interior del reactor tubular
Teniendo en cuenta los parámetros que influyen en las características microestructurales de
las MNPs, tales como la temperatura del horno, el flujo de gas portador, y el diámetro del
reactor tubular; fue desarrollado un modelo teórico del transporte de la masa, cantidad de
movimiento y energía. La Figura 9 muestra el modelo utilizado para realizar la simulación,
teniendo en cuenta la siguiente geometría: el tamaño del horno (0,85 m de largo, 0,40 m de
altura y 0,43 m de ancho). Las condiciones utilizadas son: Diámetros de los reactores (de 9
y 15 mm), la longitud del reactor de 0,9 m, distribución de la temperatura del horno de 373
K a 723 K y el flujo de gas portador involucrando al metanol en fase gaseosa; los flujos
fueron de 0.808, 1.062 y 1.569 L min-1. Las condiciones de estas simulaciones se muestran
en la Tabla 4.
Figura 9.- Modelo General del reactor utilizado para la simulación mediante software Solid
Works.
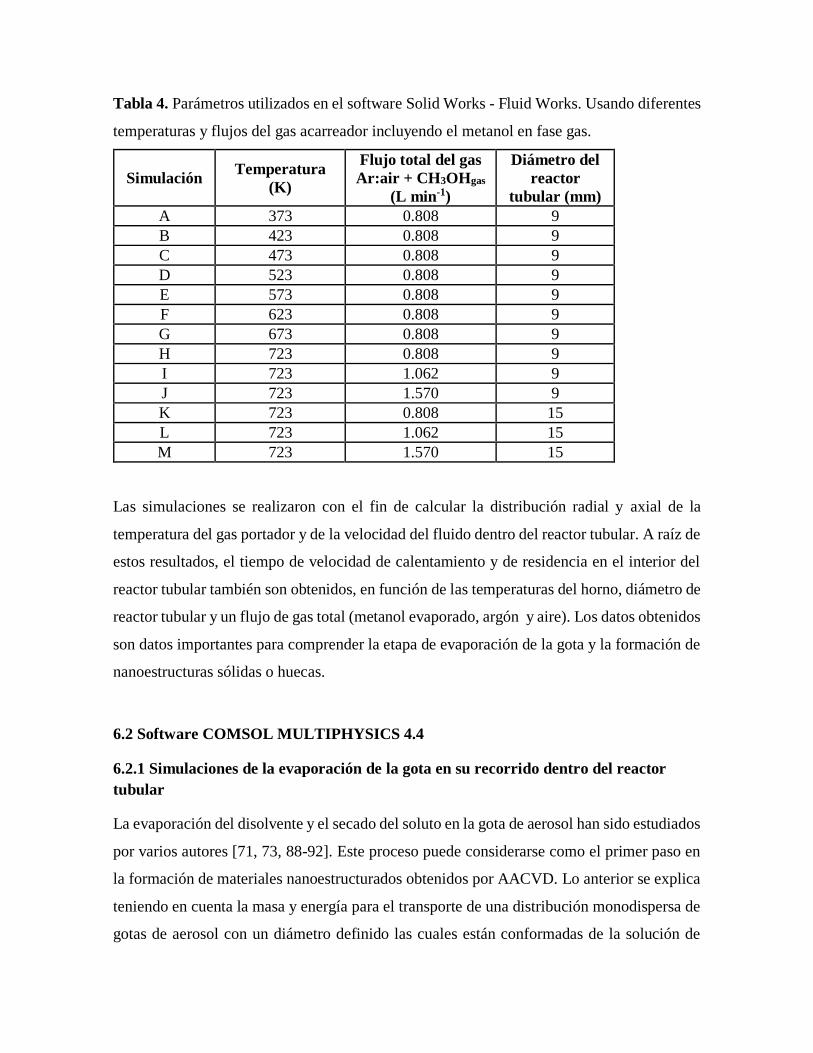
Tabla 4. Parámetros utilizados en el software Solid Works - Fluid Works. Usando diferentes
temperaturas y flujos del gas acarreador incluyendo el metanol en fase gas.
Simulación Temperatura
(K)
Flujo total del gas
Ar:air + CH3OHgas
(L min-1)
Diámetro del
reactor
tubular (mm)
A 373 0.808 9
B 423 0.808 9
C 473 0.808 9
D 523 0.808 9
E 573 0.808 9
F 623 0.808 9
G 673 0.808 9
H 723 0.808 9
I 723 1.062 9
J 723 1.570 9
K 723 0.808 15
L 723 1.062 15
M 723 1.570 15
Las simulaciones se realizaron con el fin de calcular la distribución radial y axial de la
temperatura del gas portador y de la velocidad del fluido dentro del reactor tubular. A raíz de
estos resultados, el tiempo de velocidad de calentamiento y de residencia en el interior del
reactor tubular también son obtenidos, en función de las temperaturas del horno, diámetro de
reactor tubular y un flujo de gas total (metanol evaporado, argón y aire). Los datos obtenidos
son datos importantes para comprender la etapa de evaporación de la gota y la formación de
nanoestructuras sólidas o huecas.
6.2 Software COMSOL MULTIPHYSICS 4.4
6.2.1 Simulaciones de la evaporación de la gota en su recorrido dentro del reactor
tubular
La evaporación del disolvente y el secado del soluto en la gota de aerosol han sido estudiados
por varios autores [71, 73, 88-92]. Este proceso puede considerarse como el primer paso en
la formación de materiales nanoestructurados obtenidos por AACVD. Lo anterior se explica
teniendo en cuenta la masa y energía para el transporte de una distribución monodispersa de
gotas de aerosol con un diámetro definido las cuales están conformadas de la solución de

precursor, con el disolvente y el soluto a determinada concentración. El aerosol es
transportado por un gas portador con un flujo laminar en un reactor de horno tubular, que se
mantiene a temperatura uniforme y constante. En este modelo, las ecuaciones involucradas
incluyen la difusión de solutos en el interior de la gota:
𝜕𝐶(𝑟,𝑡)
𝜕𝑡=
1
𝑟2
𝜕
𝜕𝑟(𝐷𝑠(𝑇𝑑) ∙ 𝑟2 𝜕𝐶(𝑟,𝑡)
𝜕𝑟) (4)
donde C (r, t) es la concentración de soluto dentro de la gota en un radio r en un tiempo t;
Ds(Td) es el coeficiente de difusión de soluto en el disolvente, Td la temperatura de la gota.
Debido al pequeño tamaño de la gota (algunos m) su temperatura Td y el coeficiente de
difusión se consideran uniformes. Otra ecuación que se considera es la de transferencia de
calor en la gota [73]:
𝑑𝑇𝑑
𝑑𝑡=
1
𝑚𝑐𝑝[4𝜋𝑅𝑑(𝑡)𝐾(𝑇𝑔 − 𝑇𝑑)] + 𝜗
𝑑𝑚
𝑑𝑡 (5)
donde m es la masa, cp capacidad calorífica, Rd (t) radio de la gota en el momento t, K es el
coeficiente de transferencia de calor entre la gota y el gas circundante, Tg es la temperatura
del gas circundante, y 𝜗 es el calor latente de evaporación del disolvente. La siguiente
ecuación propone la tasa de evaporación del disolvente a partir de una sola gota [73]:
𝑑𝑚
𝑑𝑡= −
4𝜋𝑅𝑑(𝑡)𝐷𝑣𝑀
𝑁𝐴(𝑛𝑠 − 𝑛𝑔) (6)
donde Dv es el coeficiente de difusión de vapor de disolvente, M es el peso molecular del
disolvente, NA es el número de Avogadro, ns la concentración de vapor disolvente en la
superficie de las gotas, y ng la concentración de vapor de disolvente en el gas circundante.
Para facilitar la solución numérica de la ecuación de difusión, se simplifica el uso de variables
adimensionales utilizando la siguiente ecuación [92]:
𝑦 =𝑠𝑜𝑙𝑢𝑡𝑒 𝑚𝑎𝑠𝑠 𝑡𝑜 𝑟
𝑡𝑜𝑡𝑎𝑙 𝑠𝑜𝑙𝑢𝑡𝑒 𝑚𝑎𝑠𝑠=
𝑚𝑠(𝑟)
𝑚𝑠0 (7)
=𝑠𝑜𝑙𝑣𝑒𝑛𝑡 𝑓𝑟𝑎𝑐𝑡𝑖𝑜𝑛 𝑚𝑎𝑠𝑠 𝑎𝑡 𝑟
𝑠𝑜𝑙𝑢𝑡𝑒 𝑓𝑟𝑎𝑐𝑡𝑖𝑜𝑛 𝑚𝑎𝑠𝑠 𝑎𝑡 𝑟 (8)
que finalmente dan como resultado la ecuación de difusión modificada, considerando ω(r,t)
aproximadamente independiente de y:
𝜕𝜔
𝜕𝑡=
16𝜋2
(𝑚𝑠0)2 ∙𝜕
𝜕𝑦(𝐷𝑠(𝑇𝑑) ∙ 𝑟4 ∙ (
𝜌𝑙
1+𝜔)
2 𝜕𝜔
𝜕𝑦) (9)

donde Ɩ es la densidad de las gotas. El radio de gotas Rd (t) se puede obtener a partir de los
cálculos del volumen de la gota usando las variables 𝑦 y ω [73].
4𝜋
3(𝑅𝑑(𝑡))3 = ∫
𝑚𝑠0
𝜌𝑙
1
0(1 + 𝜔)𝑑𝑦 (10)
Otra ecuación necesaria es la que toma en cuenta el cambio de la concentración de vapor
del disolvente en el reactor debido a la evaporación de las gotas y el transporte por el gas
portador [73]:
𝑑𝑛𝑔
𝑑𝑡= 4𝜋𝑅𝑑(𝑡)𝐷𝑣𝑁0(𝑛𝑠 − 𝑛𝑔) −
2𝐾𝑉
𝑅(𝑛𝑔 − 𝑛𝑤) (11)
donde N0 es la densidad del número de gotas, KV el coeficiente de transferencia de masa de
vapor, R el radio del reactor, y nw es la concentración de vapor en la pared del reactor. Por
último, el cambio de temperatura del gas que rodea la gota, el cual es consecuencia de la
transferencia de calor a la gotitas y de la pared del reactor [73] se pueden calcular:
Φ𝑐𝑝𝑔𝑑𝑇𝑔
𝑑𝑡= 4(𝜋𝑅𝑅𝑑)2𝑁0ℎ𝑑(𝑇𝑑 − 𝑇𝑔) + 2𝜋𝑅ℎ𝑤(𝑇𝑤 − 𝑇𝑔) (12)
donde Φ es la tasa de flujo de gas portador, Cpg es la capacidad de calor del gas portador, hd
y hw representan los coeficientes de la transferencia de calor en la superficie de las gotas y
en la pared del reactor, respectivamente. Tw es la temperatura de la pared del reactor.
Este conjunto de ecuaciones (4-12), con determinadas condiciones iniciales y de contorno,
se puede resolver para determinar la evolución en el tiempo de las características de las gotas,
como: distribución radial de la concentración del soluto C(r,t), radio, masa y temperatura de
la gota. Las condiciones de síntesis más importantes utilizadas en la simulación fueron las
mismas que se utilizaron en la simulación de Solid Works – Fluid Works: Temperaturas (373-
723K; condiciones A-H), reactor de diámetro de 9 mm, sal precursora FeCl2, metanol como
disolvente, concentración inicial del soluto 0,05 mol L-1, diámetro inicial de la gota 2,2
micras y un flujo de gas portador involucrando al fluído (Ar: aire+metanol en fase gas) de
0.808 mL min-1. La lista completa de parámetros están incluidos en el anexo 1.

7. Síntesis de MNPs
El método de síntesis utilizado fue la técnica AACVD; La cual consta de varias partes:
Nebulizador con sus accesorios, regulador de flujo, cámara de reacción, reactor tubular al
interior de un horno cilíndrico, y un sistema de recolección, el cual consta de un serpentín de
acero inoxidable y una trampa de agua; los cuales se muestran en la Figura 10.
Figura 10. Esquema de la técnica AACVD.
Las condiciones de síntesis fueron: Concentración de la solución precursora FeCl2.4H2O
(cloruro ferroso tetrahidrato SIGMA Aldrich) 0,05 mol L-1 y como disolvente metanol
(99,9% JT Baker). el flujo de gas portador consistió en una mezcla de argón y aire a 0.250 y
0.004 L min-1 respectivamente, el reactor tubular utilizado fue de 9 mm de diámetro y de
largo 90 cm. Los materiales sintetizados se recolectaron por medio de un serpentín de acero
inoxidable y una trampa de agua aplicando un campo magnético como se muestra en la figura
11.

Figura 11. Equipo usado en la síntesis de nanopartículas por la técnica AACVD. a) Vista
frontal, b) vista posterior donde se muestra el sistema de recolección.
7.1. Caracterización microestructural de las MNPS
7.1.1 Microscopía electrónica de barrido
Esta técnica fue utilizada para obtener información sobre la morfología y la composición
elemental de la superficie de las nanopartículas. Fue llevada a cabo en el microscopio Jeol,
modelo JSM-7401F, que cuenta con una resolución máxima de 1nm, idónea para visualizar
las fronteras de los cristales. La muestra de nanopartículas fue preparada por dispersión con
CH3OH usando un ultrasonido, con una pipeta Pasteur se depositó una gota de esta solución
en un substrato de silicio y se dejó secar a temperatura ambiente. Para capturar la morfología
y determinar la composición elemental de las nanopartículas se sometió la muestra a ultra
alto vacío. El haz de electrones primarios que genera el cañón de electrones se enfocó sobre
la superficie de la muestra, con un voltaje de 5.0 kV para la obtención de las micrografías y
con 15.0 kV para obtener la composición atómica. La composición elemental de las muestras
de nanopartículas se obtuvo con un detector de energía dispersa EDS OXFORD INCA que
permite colectar los rayos X generados por la muestra en respuesta a ser golpeada por
electrones y realizar diversos análisis semicuantitativos y de distribución de elementos en
superficies.

7.1.2 Microscopia electrónica de transmisión
El uso de esta técnica proporcionó información más detallada de la morfología y la
composición elemental, de la superficie de las nanopartículas y además del interior de las
mismas. Para ello se empleó un microscopio Jeol, modelo JEM-2200FS, que cuenta con una
resolución punto a punto de 0.14 nm en modo TEM y 0.10 nm en modo STEM. Para la
preparación de la muestra fue necesario hacer diluciones con CH3OH de las MNPs, hasta
obtener una solución homogénea y transparente. Seguido a esto con ayuda de un tubo capilar
se depositó una gota sobre una rejilla de cobre con membrana de carbono y se dejó secar bajo
luz incandescente durante 15 minutos. Los análisis detallados de la muestra se llevaron a
cabo en ultra alto vacío en modo TEM y STEM, que incluyeron obtención de micrografías,
patrones de difracción, análisis elemental puntual, lineal, general y mapeo. Para tal propósito,
el haz de electrones se hizo pasar a través de la muestra con una aceleración de 200 kV.
Simultáneamente se recolectaron los electrones que lograron atravesar la muestra, para
generar las imágenes y los patrones de difracción. La composición elemental y mapeo se
obtuvo con la recolección de rayos X que generaron los átomos de la muestra con un detector
de energía dispersa EDS INCA.
7.1.3 Difracción de rayos x
Mediante difracción de rayos x (DRX) se determinaron las fases, planos y estructura
cristalina presentes en las MNPs sintetizadas. Para tal propósito, se utilizó un difractómetro
de rayos x Panalytical, modelo X’Pert PRO. Para preparar la muestra fue necesario dispersar
las nanopartículas en CH3OH, con una pipeta Pasteur se fueron depositando gotas sobre un
substrato de silicio de tamaño 1X1 cm, de tal manera que se formó una capa uniforme de
nanopartículas en toda el área del sustrato. Las muestras se corrieron mediante haz rasante.
Con esta geometría, se fijó el ángulo de incidencia de los rayos x en 0.5 y se hicieron pasar
a través de un sistema de colimación con una máscara de 10 mm, para luego hacerse incidir
sobre la muestra. Siendo así que los haces difractados fueron nuevamente colimados y
monocromados para finalmente ser capturados con el detector. Aunado a esto, se estimaron
los tamaños de cristalita promedio haciendo uso de los difractogramas, el cálculo se hizo por
medio de la ecuación de Scherrer:

(13)
Donde:
= Tamaño promedio de cristalita. Unidades de nm.
K= Constante de Scherrer, su valor es 0.9 para magnetita y maghemita [78, 79].
Adimensional.
= Longitud de onda de los rayos X. Para el difractómetro utilizado es 0.15406
nm.
= Ancho del pico máximo de difracción a la mitad de su altura. Unidades de
radianes. Corregido por el ancho instrumental.
= Posición del pico máximo de difracción.
7.1.4 Adsorción y desorción física de gases por el método Brunauer-Emmet-Teller
(BET).
El uso de este método permitió determinar el área superficial específica, el tamaño y
distribución de los poros de las nanopartículas de magnetita. En función del equipo utilizado
para la caracterización, se puede determinar el rango en el que se encuentran los poros
(microporos, mesoporos o macroporos). En el presente trabajo las propiedades texturales de
las nanopartículas fueron caracterizadas mediante el método BET (Brunauer-Emmett-Teller)
que implicó la fisisorción de nitrógeno líquido a -196°C en el equipo Quantachrome, modelo
Autosorb-1. Antes de la adsorción de nitrógeno las muestras dispuestas en polvo se
desgasificaron en una celda de vidrio, la cual fue calentada bajo vacío a 200°C durante 2
horas para eliminar las impurezas de la superficie. Una vez que la muestra se desgasificó, se
mantuvo a temperatura constante y se incrementó lentamente la presión, promoviendo que
las moléculas se fijaran en la superficie de los poros accesibles del adsorbente, mediante
fuerzas de atracción de Van der Waals entre las moléculas del adsorbente y del adsorbato.
Teniendo como función principal, determinar la cantidad de moléculas del gas que se
depositan en la superficie del sólido o en el interior. El proceso ocurre primeramente con la
formación de una capa fina llamada monocapa en la superficie del material cuando la presión
t =K l
b cosq
t
l
b
q
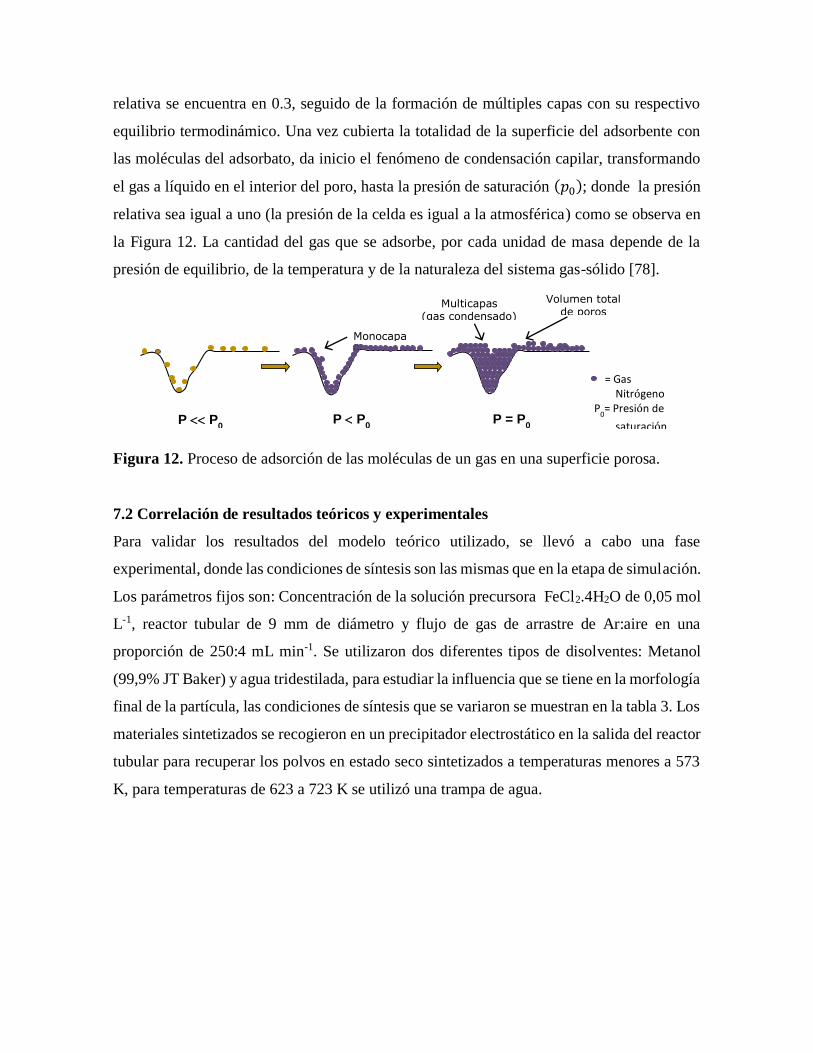
relativa se encuentra en 0.3, seguido de la formación de múltiples capas con su respectivo
equilibrio termodinámico. Una vez cubierta la totalidad de la superficie del adsorbente con
las moléculas del adsorbato, da inicio el fenómeno de condensación capilar, transformando
el gas a líquido en el interior del poro, hasta la presión de saturación (𝑝0); donde la presión
relativa sea igual a uno (la presión de la celda es igual a la atmosférica) como se observa en
la Figura 12. La cantidad del gas que se adsorbe, por cada unidad de masa depende de la
presión de equilibrio, de la temperatura y de la naturaleza del sistema gas-sólido [78].
Figura 12. Proceso de adsorción de las moléculas de un gas en una superficie porosa.
7.2 Correlación de resultados teóricos y experimentales
Para validar los resultados del modelo teórico utilizado, se llevó a cabo una fase
experimental, donde las condiciones de síntesis son las mismas que en la etapa de simulación.
Los parámetros fijos son: Concentración de la solución precursora FeCl2.4H2O de 0,05 mol
L-1, reactor tubular de 9 mm de diámetro y flujo de gas de arrastre de Ar:aire en una
proporción de 250:4 mL min-1. Se utilizaron dos diferentes tipos de disolventes: Metanol
(99,9% JT Baker) y agua tridestilada, para estudiar la influencia que se tiene en la morfología
final de la partícula, las condiciones de síntesis que se variaron se muestran en la tabla 3. Los
materiales sintetizados se recogieron en un precipitador electrostático en la salida del reactor
tubular para recuperar los polvos en estado seco sintetizados a temperaturas menores a 573
K, para temperaturas de 623 a 723 K se utilizó una trampa de agua.
P < P0 P << P
0 P = P
0
Monocapa
Multicapas (gas condensado)
Volumen total de poros
= Gas Nitrógeno
P0= Presión de
saturación
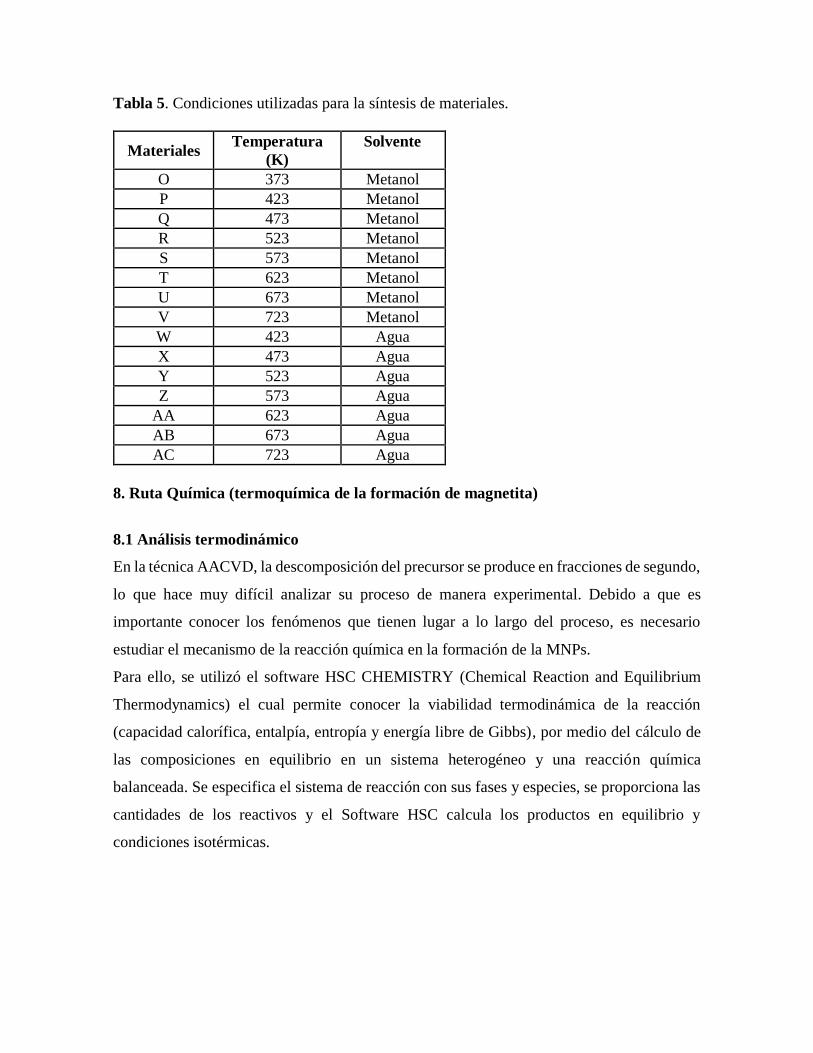
Tabla 5. Condiciones utilizadas para la síntesis de materiales.
Materiales Temperatura
(K)
Solvente
O 373 Metanol
P 423 Metanol
Q 473 Metanol
R 523 Metanol
S 573 Metanol
T 623 Metanol
U 673 Metanol
V 723 Metanol
W 423 Agua
X 473 Agua
Y 523 Agua
Z 573 Agua
AA 623 Agua
AB 673 Agua
AC 723 Agua
8. Ruta Química (termoquímica de la formación de magnetita)
8.1 Análisis termodinámico
En la técnica AACVD, la descomposición del precursor se produce en fracciones de segundo,
lo que hace muy difícil analizar su proceso de manera experimental. Debido a que es
importante conocer los fenómenos que tienen lugar a lo largo del proceso, es necesario
estudiar el mecanismo de la reacción química en la formación de la MNPs.
Para ello, se utilizó el software HSC CHEMISTRY (Chemical Reaction and Equilibrium
Thermodynamics) el cual permite conocer la viabilidad termodinámica de la reacción
(capacidad calorífica, entalpía, entropía y energía libre de Gibbs), por medio del cálculo de
las composiciones en equilibrio en un sistema heterogéneo y una reacción química
balanceada. Se especifica el sistema de reacción con sus fases y especies, se proporciona las
cantidades de los reactivos y el Software HSC calcula los productos en equilibrio y
condiciones isotérmicas.

8.2 Cinética de descomposición
La cinética de la descomposición de la sal ( FeCl2.4H2O) se llevó a cabo en un analizador
termo gravimétrico (TGA) Q500 SDT de TA Instruments, por medio de dos diferentes
métodos (isotérmico y no isotérmico). El análisis se realizó en una atmósfera de agua,
utilizando un saturador de humedad para aportar el oxígeno necesario y controlar la reacción,
ayudando a alcanzar la fase de magnetita. La composición del producto final fue analizada
por difracción de rayos X.
8.2.1 Método no isotérmico
Para determinar los parámetros cinéticos o parámetros de Arrhenius, se llevaron a cabo
análisis termogravimétricos con diferentes velocidades de calentamiento: 5, 7.5, 10, 12.5,
15 y 20 K min-1 desde temperatura ambiente (298 K), hasta 1073 K, utilizando un relación
de flujo argón:aire húmedo de 250:4 ml min-1. La constante cinética (k) se puede determinar
a partir de la ley de Arrhenius [93]
𝑘 = 𝑍 𝑒𝑥𝑝 (−𝐸
𝑅𝑇) (14)
donde k es la constante de velocidad, Z es el factor de frecuencia, E es la energía de
activación, R es la constante universal de los gases y T es la temperatura. Se ajusta
adecuadamente a los datos experimentales en un amplio intervalo de temperaturas y se
considera como la primera estimación adecuada para estudiar la influencia de la temperatura
en la cinética de la ecuación. La descomposición térmica por lo general sigue el modelo
cinético de Arrhenius, propuesto por H. Tanaka [94]:
dα
𝑑𝑡= 𝑓(α) [Z 𝑒𝑥𝑝 (−
𝐸
𝑅𝑇)] (15)
En esta ecuación, α es la fracción de pérdida de peso, T es la temperatura, f (α) es una función
independiente de la temperatura y el tiempo. Sin embargo estudios reportados [95] deducen
que la energía de activación tiene una ligera desviación con la cinética estándar de Arrhenius,
expresada:
𝐸 = −𝑅
𝑏∙
𝑑(ln 𝛽)
𝑑(1
𝑇)
(16)

donde b es un parámetro que depende del valor de E / RT y se puede derivar mediante un
método iterativo de las tablas proporcionadas por Flynn et al. [96]. En este método se elige
un valor preliminar de E, para una temperatura dada, y esto proporciona la primer estimación
de b . Entonces este valor de b provee una nueva estimación de E . Inicia un proceso iterativo
que termina hasta que b y E convergen. El factor de frecuencia (Z) se puede calcular como
(ecuación (4).): Donde n es el orden de reaccion.
ln 𝑧 = ln𝑑α
𝑑𝑡+
𝐸
𝑅𝑇− ln(1 − α)𝑛 (17)
8.2.2 Método isotérmico
En este método se toman en cuenta diferentes temperaturas para determinar los parámetros
de Arrhenius en la cinética de descomposición. Se utilizaron 3 temperaturas diferentes, de
723, 773 y 823 K. Donde se eleva la temperatura a una rampa de calentamiento de 293 K
min-1 y una vez que está en la temperatura deseada se mantiene constante hasta que el soluto
deje de reaccionar y no presente pérdida de peso. La energía de activación de la reacción del
cloruro ferroso para la formación de magnetita, se calcula a partir de las mediciones de TGA
utilizando el método isotérmico con diferentes temperaturas y empleando la ecuación de
Arrhenius [93].
Donde se grafican las pendientes del logaritmo natural de velocidad de calentamiento (ln vel)
& logaritmo natural del peso (ln w), de las diferentes isotermas, a partir del cloruro de fierro
deshidratado. El valor del intercepto con el eje vertical, representa el logaritmo natural de la
constante de velocidad “k”, a las 3 diferentes temperaturas empleadas. Después se realiza
una gráfica del logaritmo de la constante de velocidad (ln k), contra el inverso de la
temperatura (1/T) para emplearlo en el cálculo de la energía de activación. El valor de la
pendiente corresponde a Ea/RT, y el valor del intercepto con el eje vertical al logaritmo
natural del factor pre-exponencial. Para determinar la energía de activación se multiplica el
valor de la pendiente por la constante de los gases ideales R=8.314 J/mol K.

9. Pruebas de remoción de arsénico
Las pruebas se llevaron a cabo por medio de contacto o batch. Para las cuales se emplearon
soluciones de 100 mL de As+3 y As+5 en agua tridestilada. Los precursores de iones de
arsénico utilizados en la preparación de soluciones fueron arsenito de sodio (NaAsO2) y
arsenato dibásico de sodio heptahidratado (Na2HAsO4.7H2O), (99% de pureza, JT Baker)
para el As + 3 y As + 5, respectivamente.
9.1 Pruebas batch o de contacto
Este método consistió en preparar dos soluciones diluidas para cada especie: para As + 3 fue
de 0.152 y 0.164 mg L-1, para el As + 5 0.033 y 0.066 mg L-1. En un matraz Erlen-meyer se
añadieron 10 mg de las MNPs con 100 mL de cada solución de arsénico. Los tiempos de
contacto utilizados fueron 1, 5, 10, 15 y 30 minutos, mediante agitación ultrasónica (todos
por separado). Después de transcurrido este tiempo, con el fin de separar las MNPs del
líquido, las soluciones se centrifugaron a 5000 RPM por 10 minutos. Finalmente, con ayuda
de una micropipeta se retiró la solución y se almacenó en frascos de plástico con tapa de 150
mL. Las pruebas de remoción se hicieron por duplicado y junto con un blanco se mandaron
a analizar por la técnica de absorción atómica para determinar el arsénico y el hierro
remanentes.
9.1.1 Determinación de la adsorción de arsénico por absorción atómica
Las muestras de agua obtenidas en las pruebas de remoción, se analizaron por la técnica de
absorción atómica. Su uso permitió calcular los porcentajes de eficiencia de remoción de
arsénico en las soluciones acuosas, por efecto de las MNPs. Para dicho propósito, se empleó
el espectrómetro de absorción atómica GBC, modelo Avanta Sigma. El análisis se llevó a
cabo a temperatura ambiente. La solución acuosa es succionada y hecha pasar a través de un
capilar para luego ser nebulizada, desolvatada y expuesta a una energía de determinada
longitud de onda emitida por la lámpara de cátodo hueco de hierro y arsénico. Con la cantidad
de luz absorbida el software determina la cantidad de analito existente en la muestra.

9.1.2 Determinación de la adsorción de arsénico por microscopia electrónica
La retención de arsénico en las nanopartículas se determinó mediante análisis elemental y
mapeos en los equipos de microscopía electrónica de barrido y de transmisión mencionados
anteriormente. Para ello se llevaron a cabo pruebas de remoción batch de soluciones de
arsénico con concentraciones fijas de 100 ppm, de la misma forma como se describió en el
apartado 9.1. La muestra se tomó con una micropipeta, depositando unas gotas de la solución
sobre un substrato de silicio para posteriormente ser analizada en el MEB. Con ayuda de un
capilar se tomó una gota de la solución y se depositó sobre una rejilla de cobre con membrana
de carbono para analizarla en el MET, de la misma forma como se describe en la sección
7.1.1 y 7.1.2.
9.2 Desarrollo del mecanismo de adsorción
En función a las características cualitativas y cuantitativas de las isotermas de Langmuir,
Freundlich y Dubinin, que se desarrollan en el presente trabajo (tabla 3), se puede realizar
una descripción del tipo de mecanismo de adsorción de arsénico que se lleva a cabo por las
MNPs. La capacidad de adsorción de las nanopartículas fue calculada a partir de las
concentraciones de arsénico en el equilibrio usando un balance de masa como se muestra en
la ecuacion:
𝑞𝑒 =𝑉 (𝐶0−𝐶𝑒)
𝑀 (18)
Donde qe es la capacidad de adsorción (mg g-1), Co concentración inicial de arsénico en la
solución acuosa (μg L-1), Ce concentración de arsénico al equilibrio (μg L-1), V volumen de
la solución (L), M masa del adsorbente (g).
Los parámetros termodinámicos como la energía libre de adsorción (E) y la energía libre de
Gibbs (ΔGº) fueron calculados a partir de la constante de Dubinin y la constante de Langmuir
respectivamente.
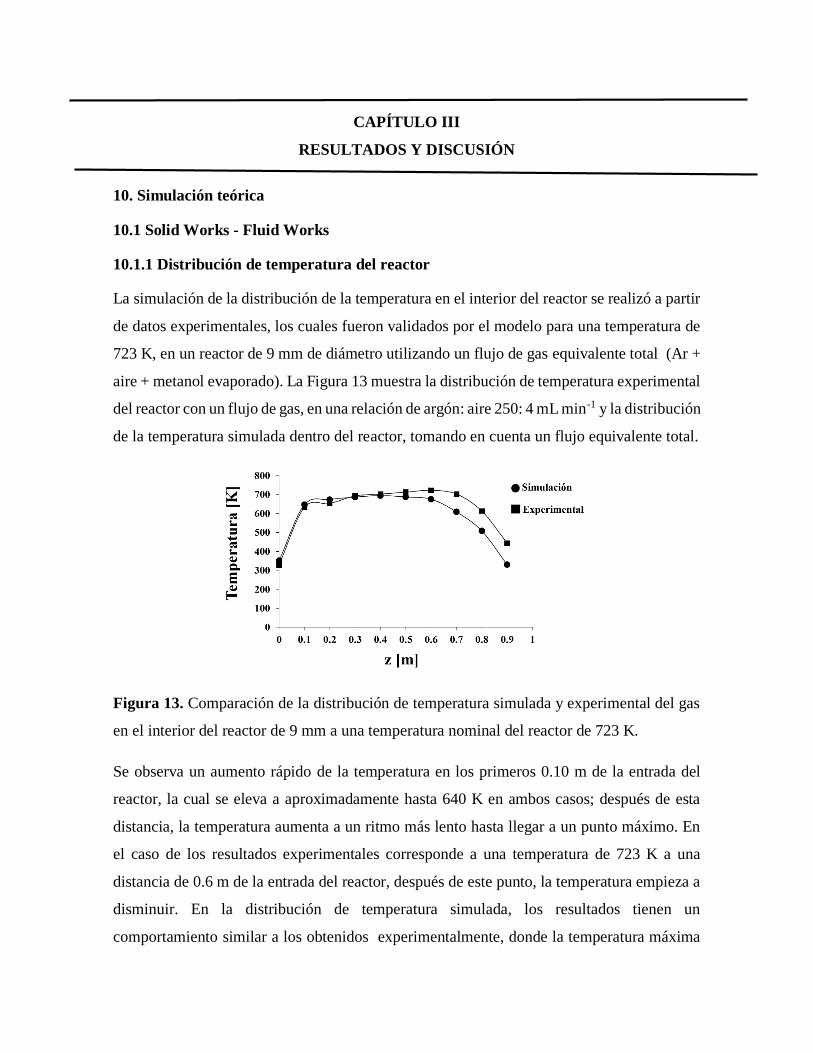
CAPÍTULO III
RESULTADOS Y DISCUSIÓN
10. Simulación teórica
10.1 Solid Works - Fluid Works
10.1.1 Distribución de temperatura del reactor
La simulación de la distribución de la temperatura en el interior del reactor se realizó a partir
de datos experimentales, los cuales fueron validados por el modelo para una temperatura de
723 K, en un reactor de 9 mm de diámetro utilizando un flujo de gas equivalente total (Ar +
aire + metanol evaporado). La Figura 13 muestra la distribución de temperatura experimental
del reactor con un flujo de gas, en una relación de argón: aire 250: 4 mL min-1 y la distribución
de la temperatura simulada dentro del reactor, tomando en cuenta un flujo equivalente total.
Figura 13. Comparación de la distribución de temperatura simulada y experimental del gas
en el interior del reactor de 9 mm a una temperatura nominal del reactor de 723 K.
Se observa un aumento rápido de la temperatura en los primeros 0.10 m de la entrada del
reactor, la cual se eleva a aproximadamente hasta 640 K en ambos casos; después de esta
distancia, la temperatura aumenta a un ritmo más lento hasta llegar a un punto máximo. En
el caso de los resultados experimentales corresponde a una temperatura de 723 K a una
distancia de 0.6 m de la entrada del reactor, después de este punto, la temperatura empieza a
disminuir. En la distribución de temperatura simulada, los resultados tienen un
comportamiento similar a los obtenidos experimentalmente, donde la temperatura máxima
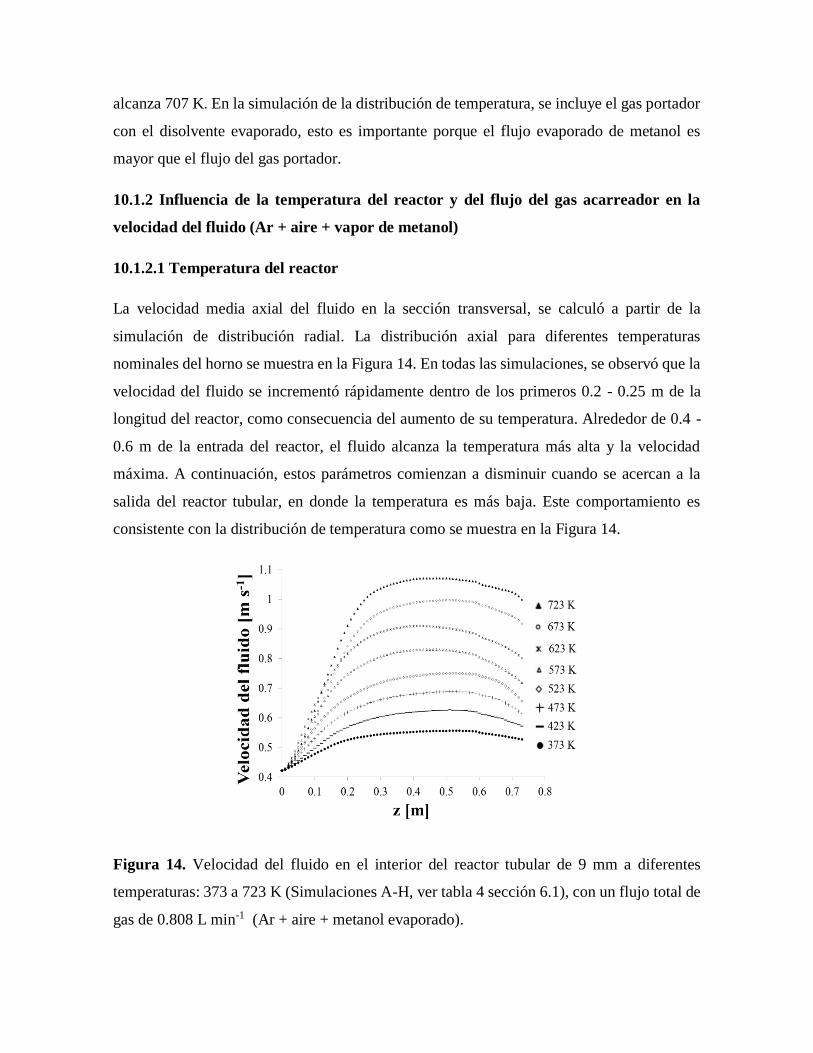
alcanza 707 K. En la simulación de la distribución de temperatura, se incluye el gas portador
con el disolvente evaporado, esto es importante porque el flujo evaporado de metanol es
mayor que el flujo del gas portador.
10.1.2 Influencia de la temperatura del reactor y del flujo del gas acarreador en la
velocidad del fluido (Ar + aire + vapor de metanol)
10.1.2.1 Temperatura del reactor
La velocidad media axial del fluido en la sección transversal, se calculó a partir de la
simulación de distribución radial. La distribución axial para diferentes temperaturas
nominales del horno se muestra en la Figura 14. En todas las simulaciones, se observó que la
velocidad del fluido se incrementó rápidamente dentro de los primeros 0.2 - 0.25 m de la
longitud del reactor, como consecuencia del aumento de su temperatura. Alrededor de 0.4 -
0.6 m de la entrada del reactor, el fluido alcanza la temperatura más alta y la velocidad
máxima. A continuación, estos parámetros comienzan a disminuir cuando se acercan a la
salida del reactor tubular, en donde la temperatura es más baja. Este comportamiento es
consistente con la distribución de temperatura como se muestra en la Figura 14.
Figura 14. Velocidad del fluido en el interior del reactor tubular de 9 mm a diferentes
temperaturas: 373 a 723 K (Simulaciones A-H, ver tabla 4 sección 6.1), con un flujo total de
gas de 0.808 L min-1 (Ar + aire + metanol evaporado).
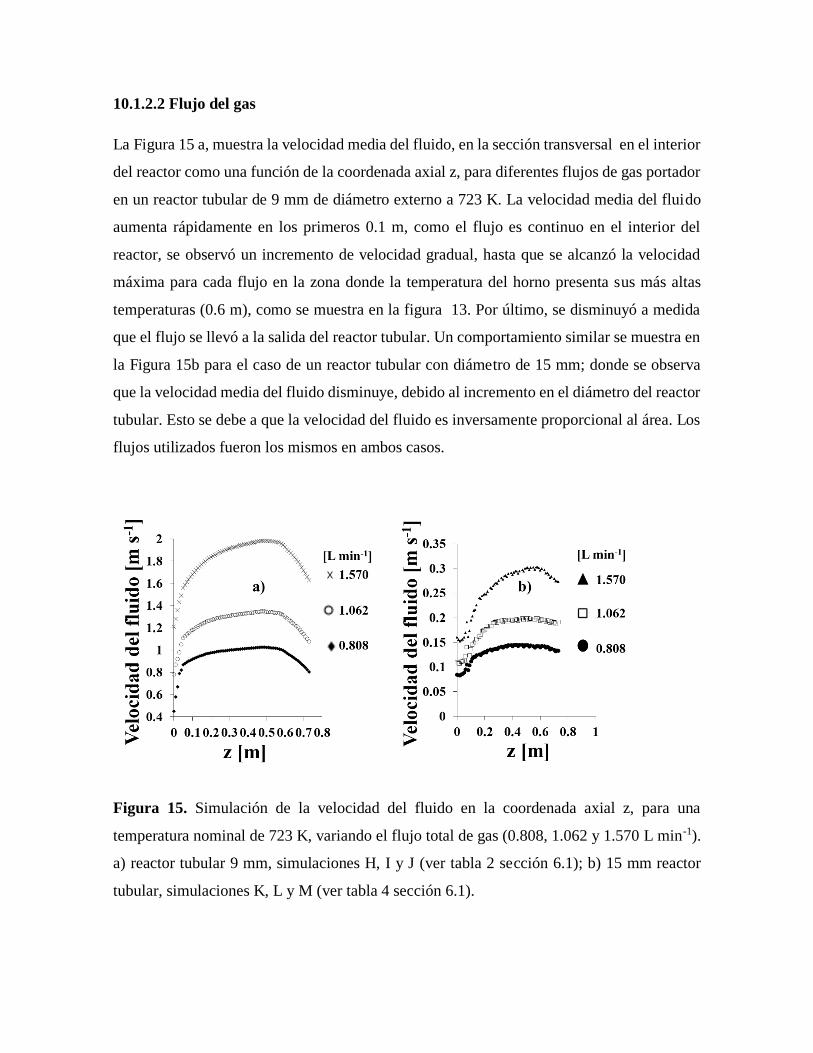
10.1.2.2 Flujo del gas
La Figura 15 a, muestra la velocidad media del fluido, en la sección transversal en el interior
del reactor como una función de la coordenada axial z, para diferentes flujos de gas portador
en un reactor tubular de 9 mm de diámetro externo a 723 K. La velocidad media del fluido
aumenta rápidamente en los primeros 0.1 m, como el flujo es continuo en el interior del
reactor, se observó un incremento de velocidad gradual, hasta que se alcanzó la velocidad
máxima para cada flujo en la zona donde la temperatura del horno presenta sus más altas
temperaturas (0.6 m), como se muestra en la figura 13. Por último, se disminuyó a medida
que el flujo se llevó a la salida del reactor tubular. Un comportamiento similar se muestra en
la Figura 15b para el caso de un reactor tubular con diámetro de 15 mm; donde se observa
que la velocidad media del fluido disminuye, debido al incremento en el diámetro del reactor
tubular. Esto se debe a que la velocidad del fluido es inversamente proporcional al área. Los
flujos utilizados fueron los mismos en ambos casos.
Figura 15. Simulación de la velocidad del fluido en la coordenada axial z, para una
temperatura nominal de 723 K, variando el flujo total de gas (0.808, 1.062 y 1.570 L min-1).
a) reactor tubular 9 mm, simulaciones H, I y J (ver tabla 2 sección 6.1); b) 15 mm reactor
tubular, simulaciones K, L y M (ver tabla 4 sección 6.1).

10.1.3 Influencia de la temperatura del reactor y del flujo del gas portador en la
velocidad de calentamiento del fluido.
El calentamiento del fluido depende de la temperatura del horno y el diámetro del reactor
tubular. La velocidad de calentamiento (HR) se obtuvo por:
𝐻𝑅(𝑧) =∆⟨𝑇𝐶𝑆(𝑧)⟩
∆𝑧⟨𝑣𝑧(𝑧)⟩ (19)
Donde ⟨𝑇𝐶𝑆(𝑧)⟩ es la temperatura media de la sección transversal en z, ∆⟨𝑇𝐶𝑆(𝑧)⟩
∆𝑧 es el
gradiente axial de la temperatura media de la sección transversal en z, y ⟨𝑣𝑧(𝑧)⟩ representa
la velocidad media del fluido en la componente z de la sección transversal. La Figura 16
presenta la influencia de la temperatura del reactor nominal en la velocidad de calentamiento,
como una función de la coordenada axial z del reactor. Para todas las temperaturas la
velocidad de calentamiento aumenta bruscamente en la entrada del reactor, alcanzando su
máximo alrededor de 0.05 m, seguido por una disminución continua hasta la salida del
reactor. A 373 K se tiene una velocidad de calentamiento de 244 K s-1 y en la máxima
temperatura (723 K) se alcanzan más de 1600 K-1 s-1. Sin embargo, a temperaturas ≥ 523 K,
se observa un cambio más pronunciado donde se obtuvo la velocidad de calentamiento cerca
de la entrada del reactor (z ≤ 0.15 m), es decir, que aumenta la velocidad de calentamiento y
disminuye rápidamente en esta zona reducida del reactor.
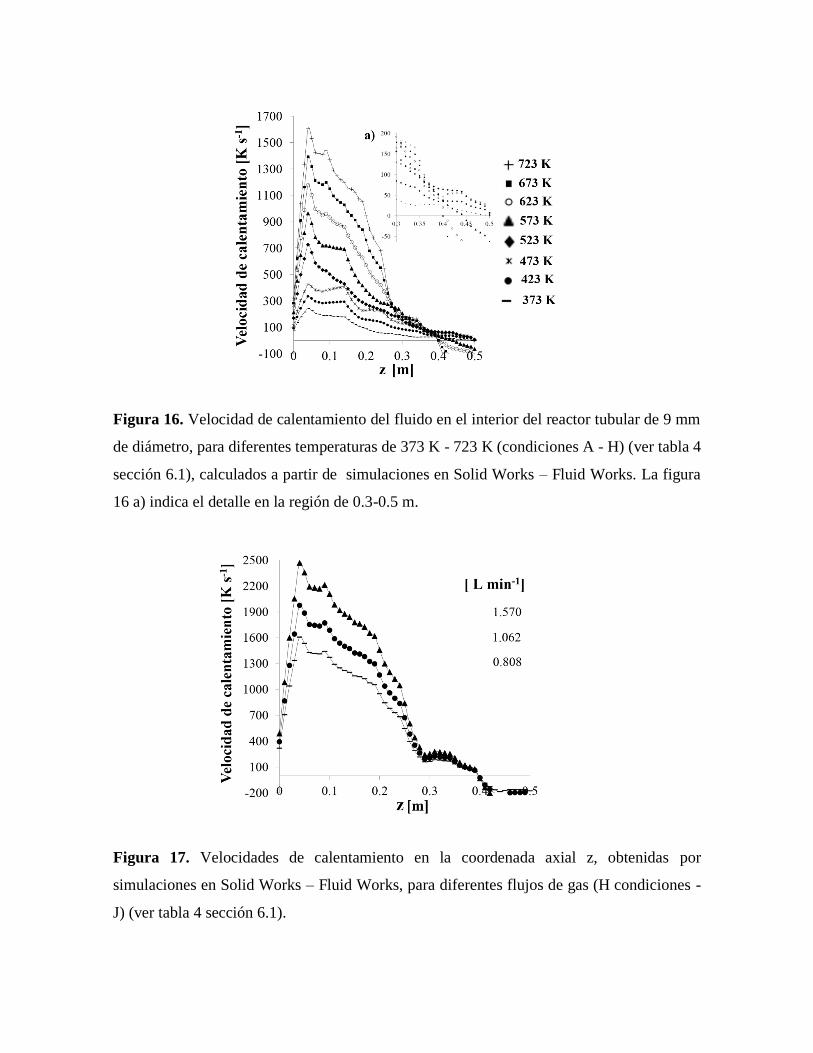
Figura 16. Velocidad de calentamiento del fluido en el interior del reactor tubular de 9 mm
de diámetro, para diferentes temperaturas de 373 K - 723 K (condiciones A - H) (ver tabla 4
sección 6.1), calculados a partir de simulaciones en Solid Works – Fluid Works. La figura
16 a) indica el detalle en la región de 0.3-0.5 m.
Figura 17. Velocidades de calentamiento en la coordenada axial z, obtenidas por
simulaciones en Solid Works – Fluid Works, para diferentes flujos de gas (H condiciones -
J) (ver tabla 4 sección 6.1).

La figura 17 muestra la velocidad de calentamiento calculado en la coordenada axial z, para
una temperatura nominal del horno de 723 K, como una función del flujo de gas. El
comportamiento fue similar en los diferentes flujos utilizados en el reactor de 9 mm, de modo
que hay una relación directa entre la cantidad de flujo inicial y la velocidad de calentamiento,
es decir, un incremento en uno produce un aumento en el otro. Se puede observar que las
velocidades máximas alcanzadas (2469, 1975 y 1606 K s-1) están dentro de los primeros 0,1
m del reactor. Esto es debido a que el mayor aumento de temperatura se produce en este
intervalo, como se muestra en la Figura 13. Enseguida se observa una disminución de la
velocidad de calentamiento del fluido hasta la salida del reactor.
10.1.4 Tiempo de residencia en función de la temperatura del horno y del flujo de gas
portador
El tiempo de residencia (TR) se calculó mediante:
𝑇𝑅 = ∑∆𝑧
⟨𝑣𝑧(𝑧)⟩∆𝑧 (20)
El cual esta dado por la posición en z y la velocidad media del fluido en la componente z de
la sección transversal (⟨𝑣𝑧(𝑧)⟩). En la Figura 18 se puede observar el tiempo de residencia
calculado como una función de la temperatura del horno dentro del reactor tubular de 9 mm.
La disminución del tiempo de residencia que se presenta mediante el aumento de la
temperatura del reactor es una consecuencia directa de la expansión del gas portador con la
temperatura.
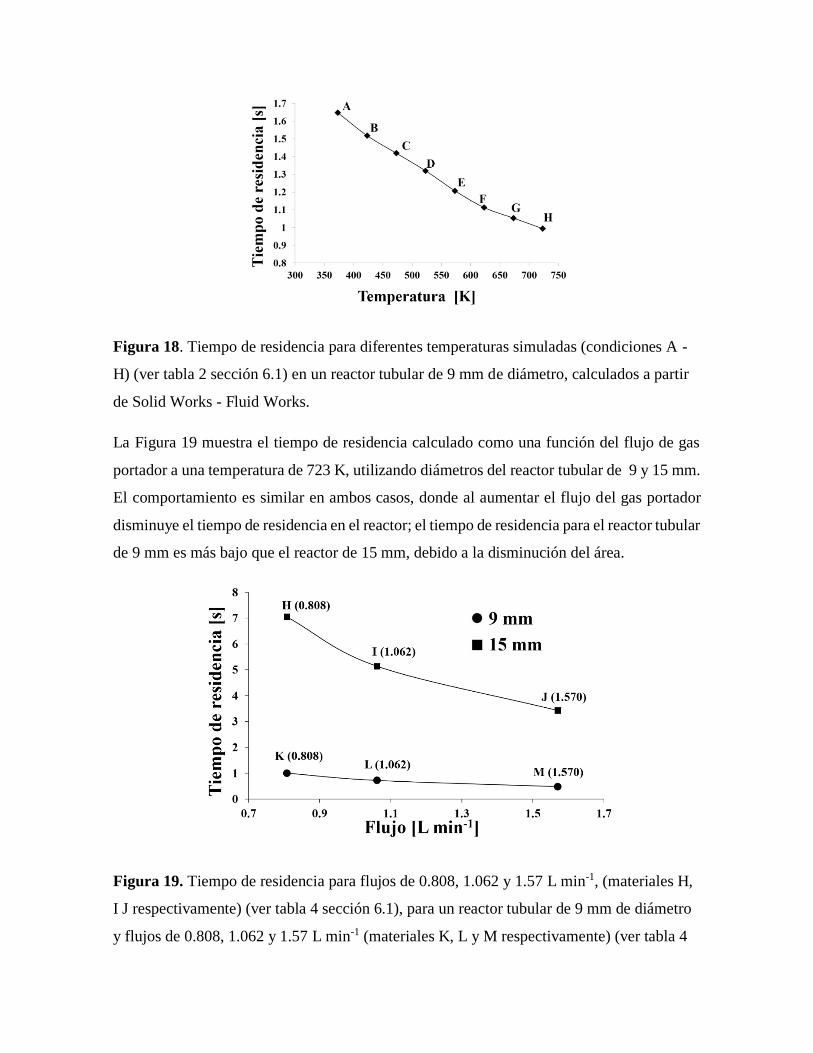
Figura 18. Tiempo de residencia para diferentes temperaturas simuladas (condiciones A -
H) (ver tabla 2 sección 6.1) en un reactor tubular de 9 mm de diámetro, calculados a partir
de Solid Works - Fluid Works.
La Figura 19 muestra el tiempo de residencia calculado como una función del flujo de gas
portador a una temperatura de 723 K, utilizando diámetros del reactor tubular de 9 y 15 mm.
El comportamiento es similar en ambos casos, donde al aumentar el flujo del gas portador
disminuye el tiempo de residencia en el reactor; el tiempo de residencia para el reactor tubular
de 9 mm es más bajo que el reactor de 15 mm, debido a la disminución del área.
Figura 19. Tiempo de residencia para flujos de 0.808, 1.062 y 1.57 L min-1, (materiales H,
I J respectivamente) (ver tabla 4 sección 6.1), para un reactor tubular de 9 mm de diámetro
y flujos de 0.808, 1.062 y 1.57 L min-1 (materiales K, L y M respectivamente) (ver tabla 4

sección 6.1), para un reactor tubular de 15 mm de diámetro, ambos a una temperatura
constante de 723 K, simulados por Solid Works – Fluid Works.
El proceso de formación de nanopartículas es un proceso muy rápido, el tiempo en que pasa
la gota por el sistema hasta la salida varia aproximadamente de 1 a 7 s, dependiendo el
diámetro del reactor tubular (Dato obtenido por Solid Works – Fluid Works), por lo tanto es
necesario simular por medio de software el proceso. Estos resultados que se obtuvieron de
la simulación con Solid Works-Fluid Works, nos sirvieron para comprobar la distribución de
temperatura del gas dentro del reactor tubular, y conocer la velocidad del gas de arrastre, la
velocidad del calentamiento de la gota y el tiempo de residencia dentro del reactor,
parámetros importantes para el estudio del mecanismo de formación de las nanopartículas,
además algunos datos que se obtuvieron fueron necesarios para continuar la simulación por
COMSOL Multhiphysics Software 4.4.
10.2 Simulación teórica por COMSOL Multiphysics 4.4
10.2.1 Influencia del disolvente
10.2.1.1 Comportamiento y distribución de la concentración de soluto en la gota
utilizando metanol como solvente
La Figura 20 muestra la distribución de la concentración de soluto dentro de la gota para
diferentes temperaturas del horno. En el caso de bajas temperaturas (373-473 K) la
distribución de la concentración de soluto es homogénea en toda la gota, se encuentra por
encima de la concentración de saturación (ES) y cerca de la concentración de sobresaturación
(CSS), lo que indica la formación partículas sólidas, estos resultados coinciden con los
reportados por [71]. Con el aumento de la temperatura (≥ 523 K), la concentración de soluto
aumenta en la superficie de la gota y es menor que la ES en el interior de la gota,
consecuencia de una difusión limitada de soluto hacia el centro de la gota y la rápida
evaporación del disolvente en la superficie, por lo que se obtienen partículas huecas. Estos
resultados concuerdan con lo obtenido por [18,19,71,73].
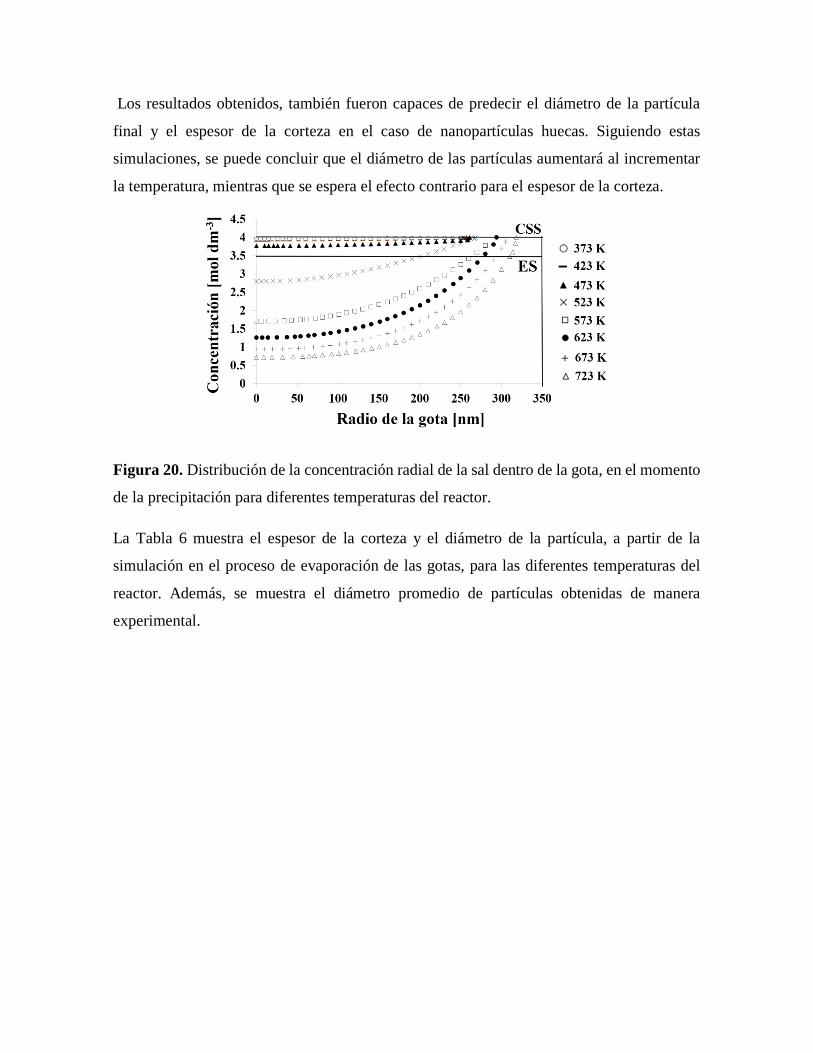
Los resultados obtenidos, también fueron capaces de predecir el diámetro de la partícula
final y el espesor de la corteza en el caso de nanopartículas huecas. Siguiendo estas
simulaciones, se puede concluir que el diámetro de las partículas aumentará al incrementar
la temperatura, mientras que se espera el efecto contrario para el espesor de la corteza.
Figura 20. Distribución de la concentración radial de la sal dentro de la gota, en el momento
de la precipitación para diferentes temperaturas del reactor.
La Tabla 6 muestra el espesor de la corteza y el diámetro de la partícula, a partir de la
simulación en el proceso de evaporación de las gotas, para las diferentes temperaturas del
reactor. Además, se muestra el diámetro promedio de partículas obtenidas de manera
experimental.
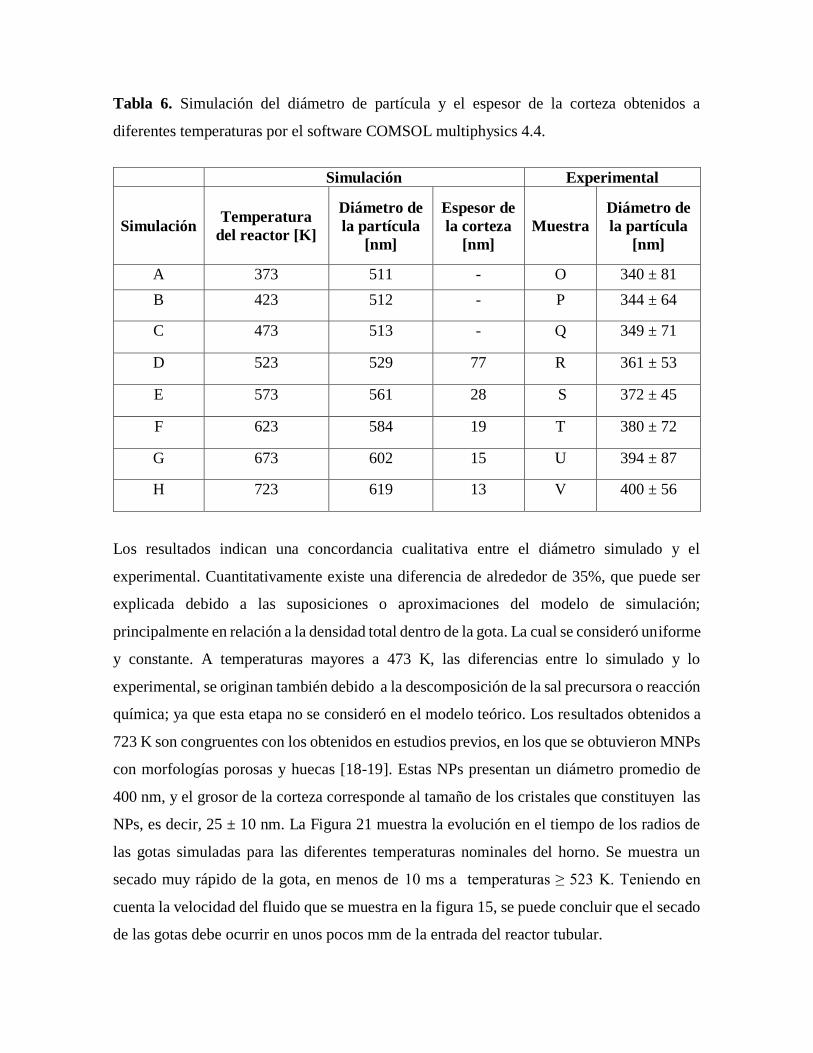
Tabla 6. Simulación del diámetro de partícula y el espesor de la corteza obtenidos a
diferentes temperaturas por el software COMSOL multiphysics 4.4.
Los resultados indican una concordancia cualitativa entre el diámetro simulado y el
experimental. Cuantitativamente existe una diferencia de alrededor de 35%, que puede ser
explicada debido a las suposiciones o aproximaciones del modelo de simulación;
principalmente en relación a la densidad total dentro de la gota. La cual se consideró uniforme
y constante. A temperaturas mayores a 473 K, las diferencias entre lo simulado y lo
experimental, se originan también debido a la descomposición de la sal precursora o reacción
química; ya que esta etapa no se consideró en el modelo teórico. Los resultados obtenidos a
723 K son congruentes con los obtenidos en estudios previos, en los que se obtuvieron MNPs
con morfologías porosas y huecas [18-19]. Estas NPs presentan un diámetro promedio de
400 nm, y el grosor de la corteza corresponde al tamaño de los cristales que constituyen las
NPs, es decir, 25 ± 10 nm. La Figura 21 muestra la evolución en el tiempo de los radios de
las gotas simuladas para las diferentes temperaturas nominales del horno. Se muestra un
secado muy rápido de la gota, en menos de 10 ms a temperaturas ≥ 523 K. Teniendo en
cuenta la velocidad del fluido que se muestra en la figura 15, se puede concluir que el secado
de las gotas debe ocurrir en unos pocos mm de la entrada del reactor tubular.
Simulación Experimental
Simulación Temperatura
del reactor [K]
Diámetro de
la partícula
[nm]
Espesor de
la corteza
[nm]
Muestra
Diámetro de
la partícula
[nm]
A 373 511 - O 340 ± 81
B 423 512 - P 344 ± 64
C 473 513 - Q 349 ± 71
D 523 529 77 R 361 ± 53
E 573 561 28 S 372 ± 45
F 623 584 19 T 380 ± 72
G 673 602 15 U 394 ± 87
H 723 619 13 V 400 ± 56
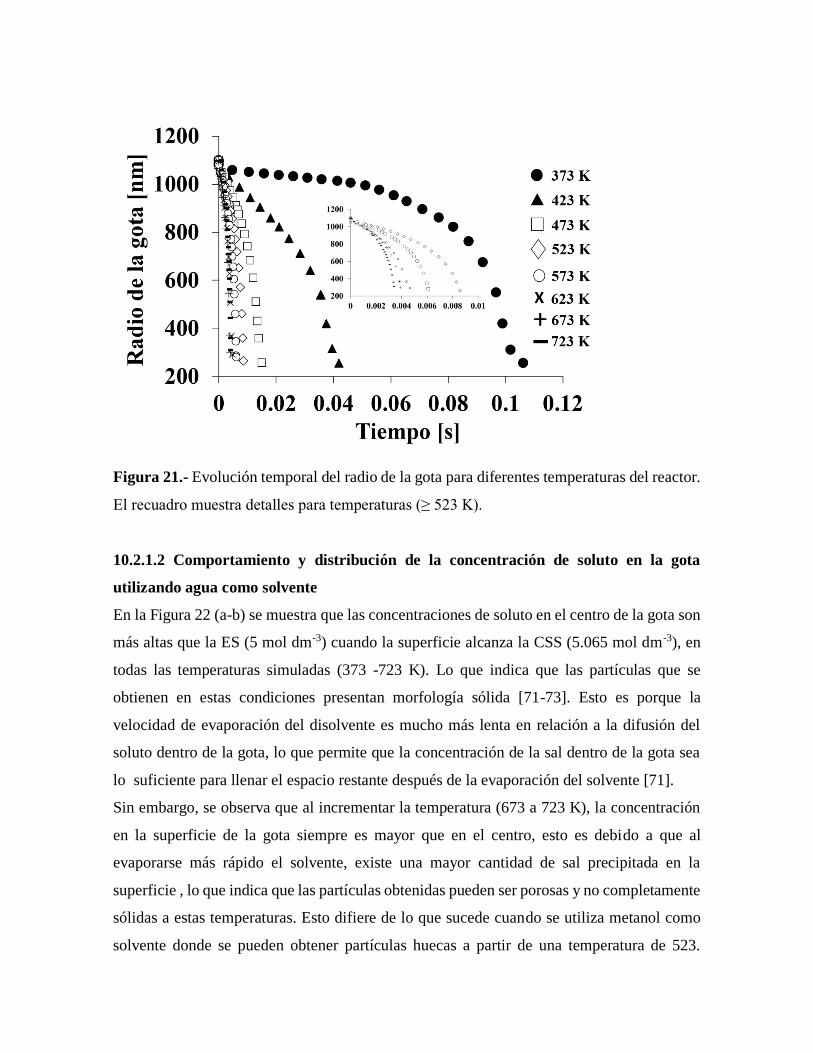
Figura 21.- Evolución temporal del radio de la gota para diferentes temperaturas del reactor.
El recuadro muestra detalles para temperaturas (≥ 523 K).
10.2.1.2 Comportamiento y distribución de la concentración de soluto en la gota
utilizando agua como solvente
En la Figura 22 (a-b) se muestra que las concentraciones de soluto en el centro de la gota son
más altas que la ES (5 mol dm-3) cuando la superficie alcanza la CSS (5.065 mol dm-3), en
todas las temperaturas simuladas (373 -723 K). Lo que indica que las partículas que se
obtienen en estas condiciones presentan morfología sólida [71-73]. Esto es porque la
velocidad de evaporación del disolvente es mucho más lenta en relación a la difusión del
soluto dentro de la gota, lo que permite que la concentración de la sal dentro de la gota sea
lo suficiente para llenar el espacio restante después de la evaporación del solvente [71].
Sin embargo, se observa que al incrementar la temperatura (673 a 723 K), la concentración
en la superficie de la gota siempre es mayor que en el centro, esto es debido a que al
evaporarse más rápido el solvente, existe una mayor cantidad de sal precipitada en la
superficie , lo que indica que las partículas obtenidas pueden ser porosas y no completamente
sólidas a estas temperaturas. Esto difiere de lo que sucede cuando se utiliza metanol como
solvente donde se pueden obtener partículas huecas a partir de una temperatura de 523.
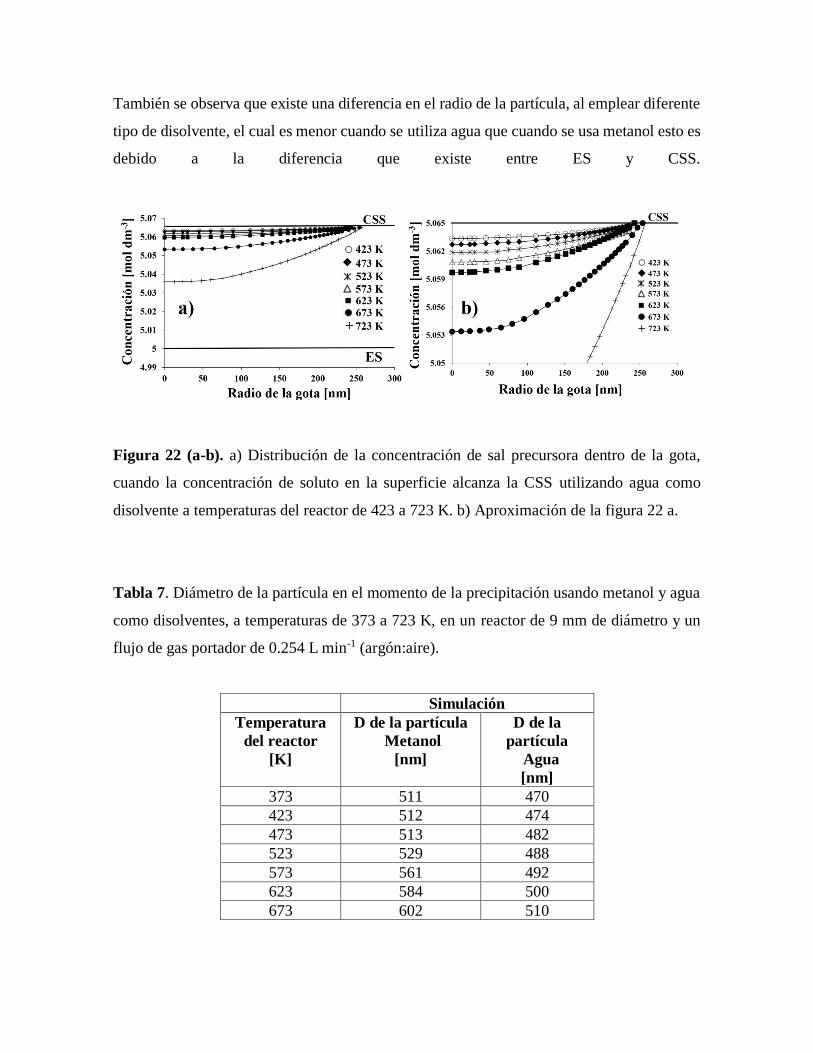
También se observa que existe una diferencia en el radio de la partícula, al emplear diferente
tipo de disolvente, el cual es menor cuando se utiliza agua que cuando se usa metanol esto es
debido a la diferencia que existe entre ES y CSS.
Figura 22 (a-b). a) Distribución de la concentración de sal precursora dentro de la gota,
cuando la concentración de soluto en la superficie alcanza la CSS utilizando agua como
disolvente a temperaturas del reactor de 423 a 723 K. b) Aproximación de la figura 22 a.
Tabla 7. Diámetro de la partícula en el momento de la precipitación usando metanol y agua
como disolventes, a temperaturas de 373 a 723 K, en un reactor de 9 mm de diámetro y un
flujo de gas portador de 0.254 L min-1 (argón:aire).
Simulación
Temperatura
del reactor
[K]
D de la partícula
Metanol
[nm]
D de la
partícula
Agua
[nm]
373 511 470
423 512 474
473 513 482
523 529 488
573 561 492
623 584 500
673 602 510
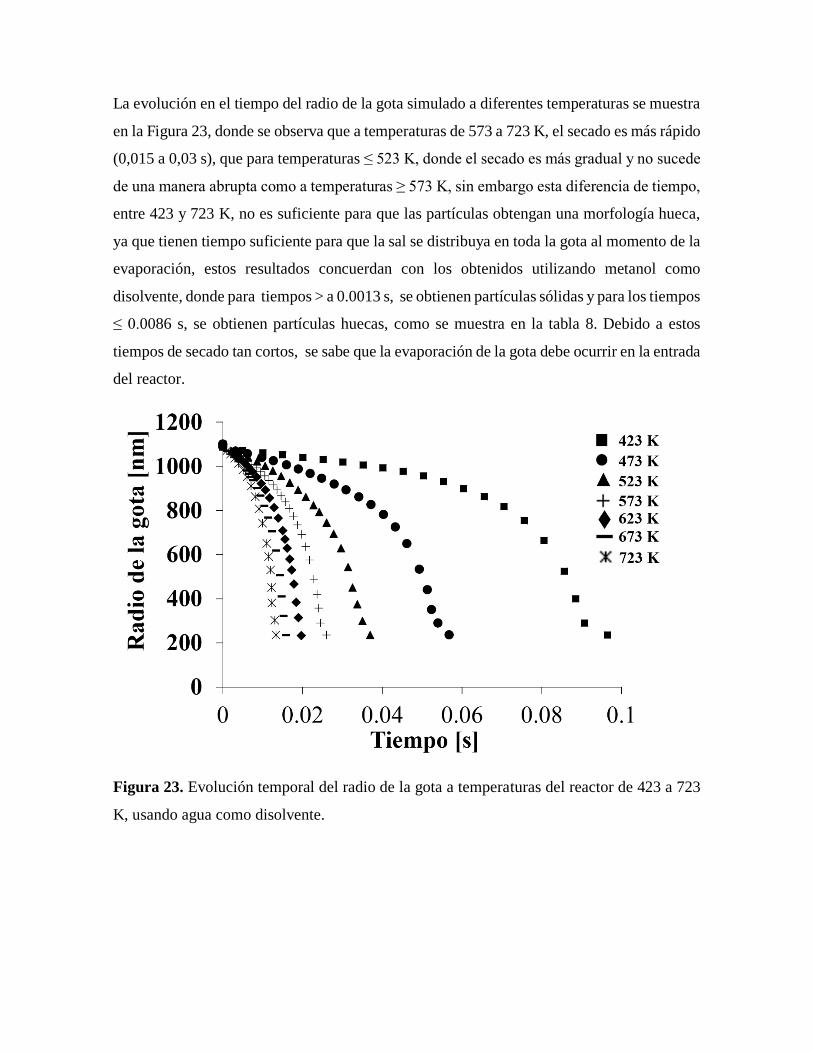
La evolución en el tiempo del radio de la gota simulado a diferentes temperaturas se muestra
en la Figura 23, donde se observa que a temperaturas de 573 a 723 K, el secado es más rápido
(0,015 a 0,03 s), que para temperaturas ≤ 523 K, donde el secado es más gradual y no sucede
de una manera abrupta como a temperaturas ≥ 573 K, sin embargo esta diferencia de tiempo,
entre 423 y 723 K, no es suficiente para que las partículas obtengan una morfología hueca,
ya que tienen tiempo suficiente para que la sal se distribuya en toda la gota al momento de la
evaporación, estos resultados concuerdan con los obtenidos utilizando metanol como
disolvente, donde para tiempos > a 0.0013 s, se obtienen partículas sólidas y para los tiempos
≤ 0.0086 s, se obtienen partículas huecas, como se muestra en la tabla 8. Debido a estos
tiempos de secado tan cortos, se sabe que la evaporación de la gota debe ocurrir en la entrada
del reactor.
Figura 23. Evolución temporal del radio de la gota a temperaturas del reactor de 423 a 723
K, usando agua como disolvente.
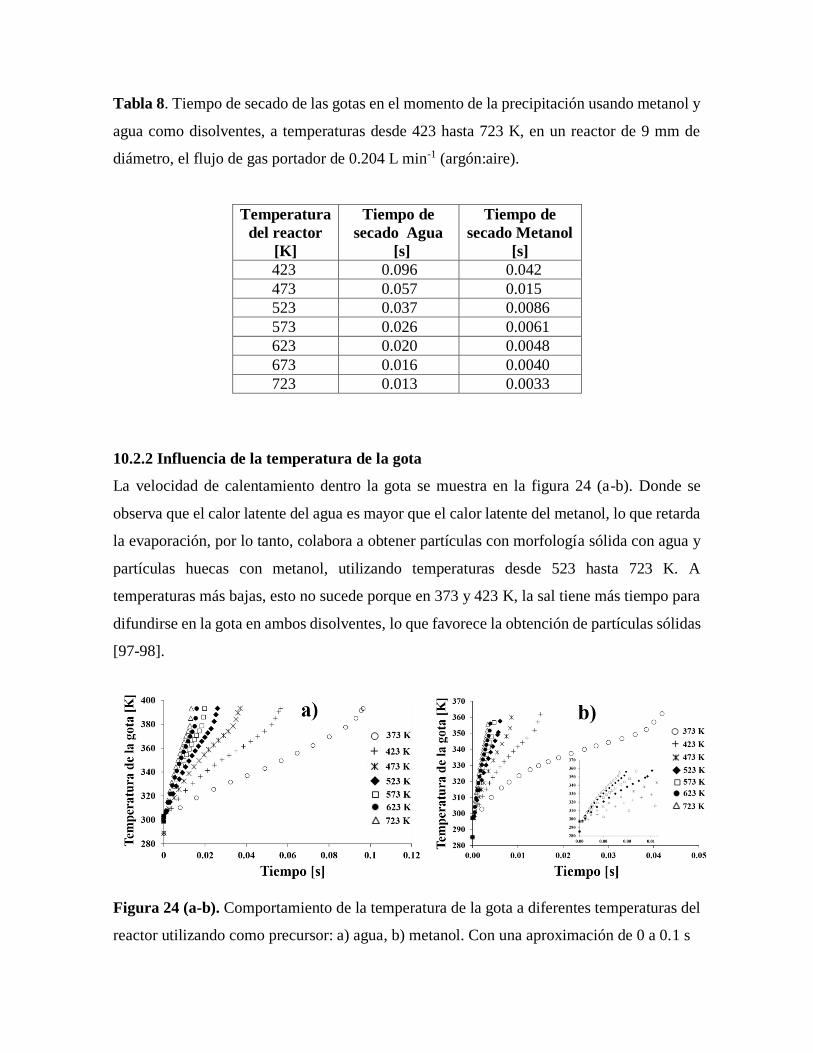
Tabla 8. Tiempo de secado de las gotas en el momento de la precipitación usando metanol y
agua como disolventes, a temperaturas desde 423 hasta 723 K, en un reactor de 9 mm de
diámetro, el flujo de gas portador de 0.204 L min-1 (argón:aire).
Temperatura
del reactor
[K]
Tiempo de
secado Agua
[s]
Tiempo de
secado Metanol
[s]
423 0.096 0.042
473 0.057 0.015
523 0.037 0.0086
573 0.026 0.0061
623 0.020 0.0048
673 0.016 0.0040
723 0.013 0.0033
10.2.2 Influencia de la temperatura de la gota
La velocidad de calentamiento dentro la gota se muestra en la figura 24 (a-b). Donde se
observa que el calor latente del agua es mayor que el calor latente del metanol, lo que retarda
la evaporación, por lo tanto, colabora a obtener partículas con morfología sólida con agua y
partículas huecas con metanol, utilizando temperaturas desde 523 hasta 723 K. A
temperaturas más bajas, esto no sucede porque en 373 y 423 K, la sal tiene más tiempo para
difundirse en la gota en ambos disolventes, lo que favorece la obtención de partículas sólidas
[97-98].
Figura 24 (a-b). Comportamiento de la temperatura de la gota a diferentes temperaturas del
reactor utilizando como precursor: a) agua, b) metanol. Con una aproximación de 0 a 0.1 s
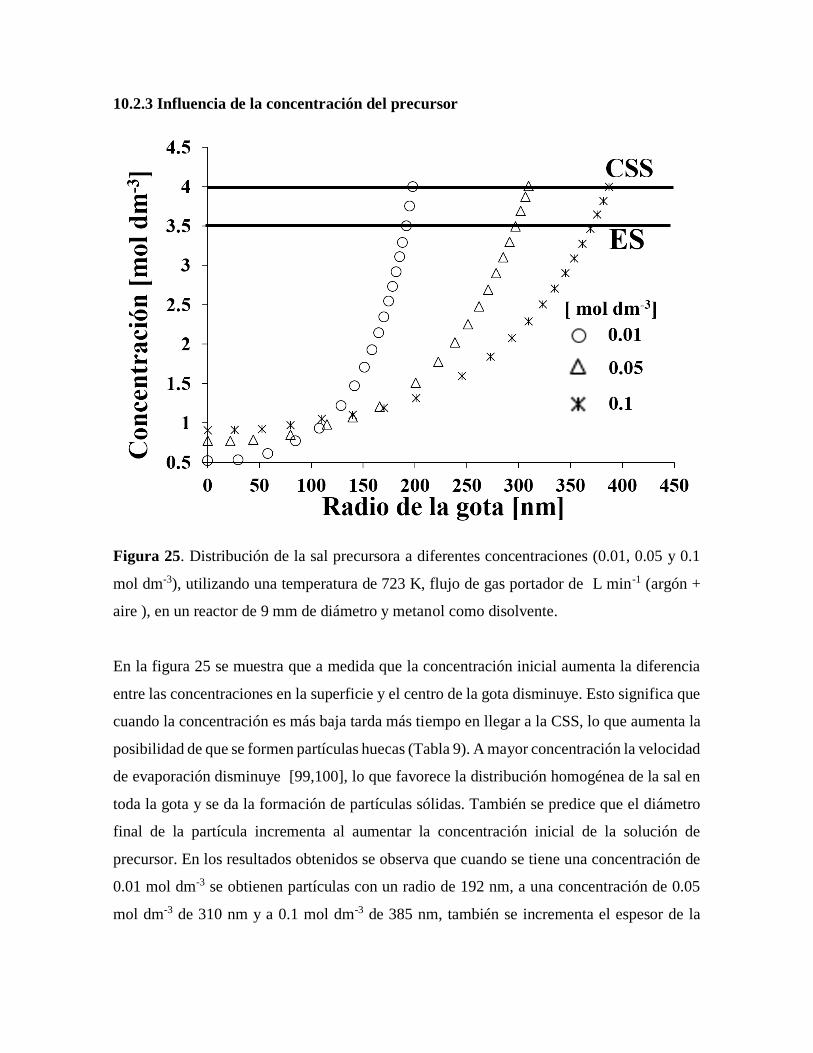
10.2.3 Influencia de la concentración del precursor
Figura 25. Distribución de la sal precursora a diferentes concentraciones (0.01, 0.05 y 0.1
mol dm-3), utilizando una temperatura de 723 K, flujo de gas portador de L min-1 (argón +
aire ), en un reactor de 9 mm de diámetro y metanol como disolvente.
En la figura 25 se muestra que a medida que la concentración inicial aumenta la diferencia
entre las concentraciones en la superficie y el centro de la gota disminuye. Esto significa que
cuando la concentración es más baja tarda más tiempo en llegar a la CSS, lo que aumenta la
posibilidad de que se formen partículas huecas (Tabla 9). A mayor concentración la velocidad
de evaporación disminuye [99,100], lo que favorece la distribución homogénea de la sal en
toda la gota y se da la formación de partículas sólidas. También se predice que el diámetro
final de la partícula incrementa al aumentar la concentración inicial de la solución de
precursor. En los resultados obtenidos se observa que cuando se tiene una concentración de
0.01 mol dm-3 se obtienen partículas con un radio de 192 nm, a una concentración de 0.05
mol dm-3 de 310 nm y a 0.1 mol dm-3 de 385 nm, también se incrementa el espesor de la
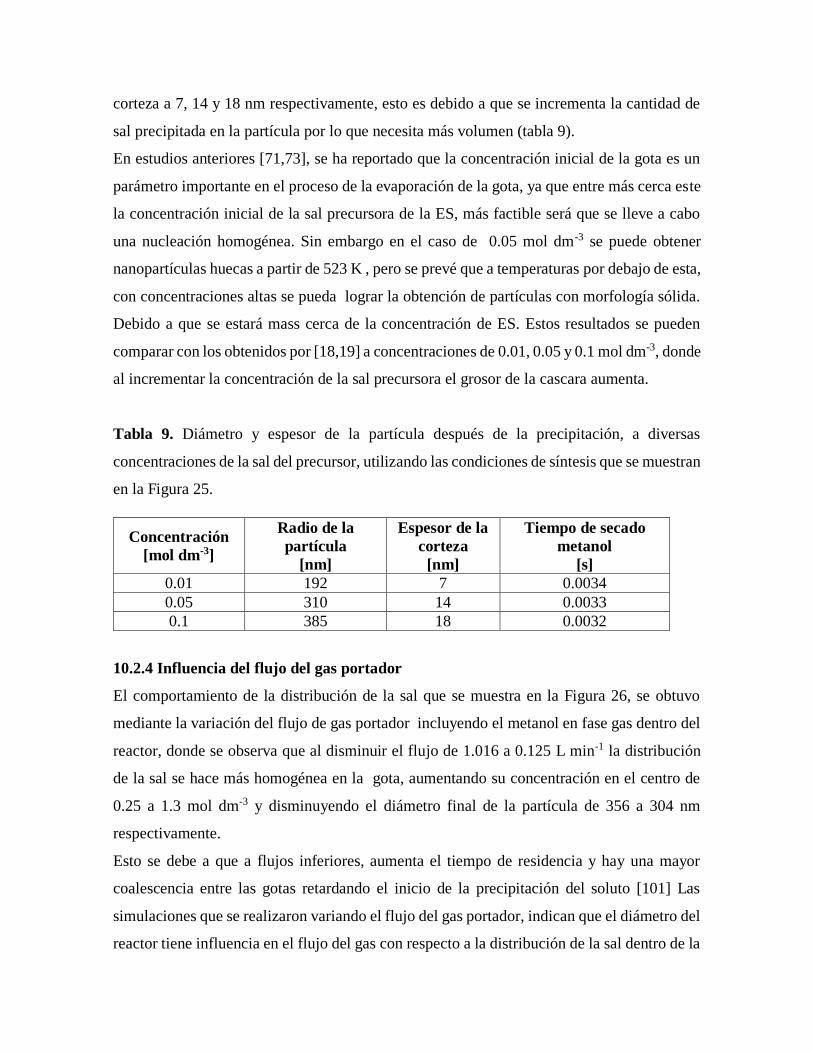
corteza a 7, 14 y 18 nm respectivamente, esto es debido a que se incrementa la cantidad de
sal precipitada en la partícula por lo que necesita más volumen (tabla 9).
En estudios anteriores [71,73], se ha reportado que la concentración inicial de la gota es un
parámetro importante en el proceso de la evaporación de la gota, ya que entre más cerca este
la concentración inicial de la sal precursora de la ES, más factible será que se lleve a cabo
una nucleación homogénea. Sin embargo en el caso de 0.05 mol dm-3 se puede obtener
nanopartículas huecas a partir de 523 K , pero se prevé que a temperaturas por debajo de esta,
con concentraciones altas se pueda lograr la obtención de partículas con morfología sólida.
Debido a que se estará mass cerca de la concentración de ES. Estos resultados se pueden
comparar con los obtenidos por [18,19] a concentraciones de 0.01, 0.05 y 0.1 mol dm-3, donde
al incrementar la concentración de la sal precursora el grosor de la cascara aumenta.
Tabla 9. Diámetro y espesor de la partícula después de la precipitación, a diversas
concentraciones de la sal del precursor, utilizando las condiciones de síntesis que se muestran
en la Figura 25.
10.2.4 Influencia del flujo del gas portador
El comportamiento de la distribución de la sal que se muestra en la Figura 26, se obtuvo
mediante la variación del flujo de gas portador incluyendo el metanol en fase gas dentro del
reactor, donde se observa que al disminuir el flujo de 1.016 a 0.125 L min-1 la distribución
de la sal se hace más homogénea en la gota, aumentando su concentración en el centro de
0.25 a 1.3 mol dm-3 y disminuyendo el diámetro final de la partícula de 356 a 304 nm
respectivamente.
Esto se debe a que a flujos inferiores, aumenta el tiempo de residencia y hay una mayor
coalescencia entre las gotas retardando el inicio de la precipitación del soluto [101] Las
simulaciones que se realizaron variando el flujo del gas portador, indican que el diámetro del
reactor tiene influencia en el flujo del gas con respecto a la distribución de la sal dentro de la
Concentración
[mol dm-3]
Radio de la
partícula
[nm]
Espesor de la
corteza
[nm]
Tiempo de secado
metanol
[s]
0.01 192 7 0.0034
0.05 310 14 0.0033
0.1 385 18 0.0032
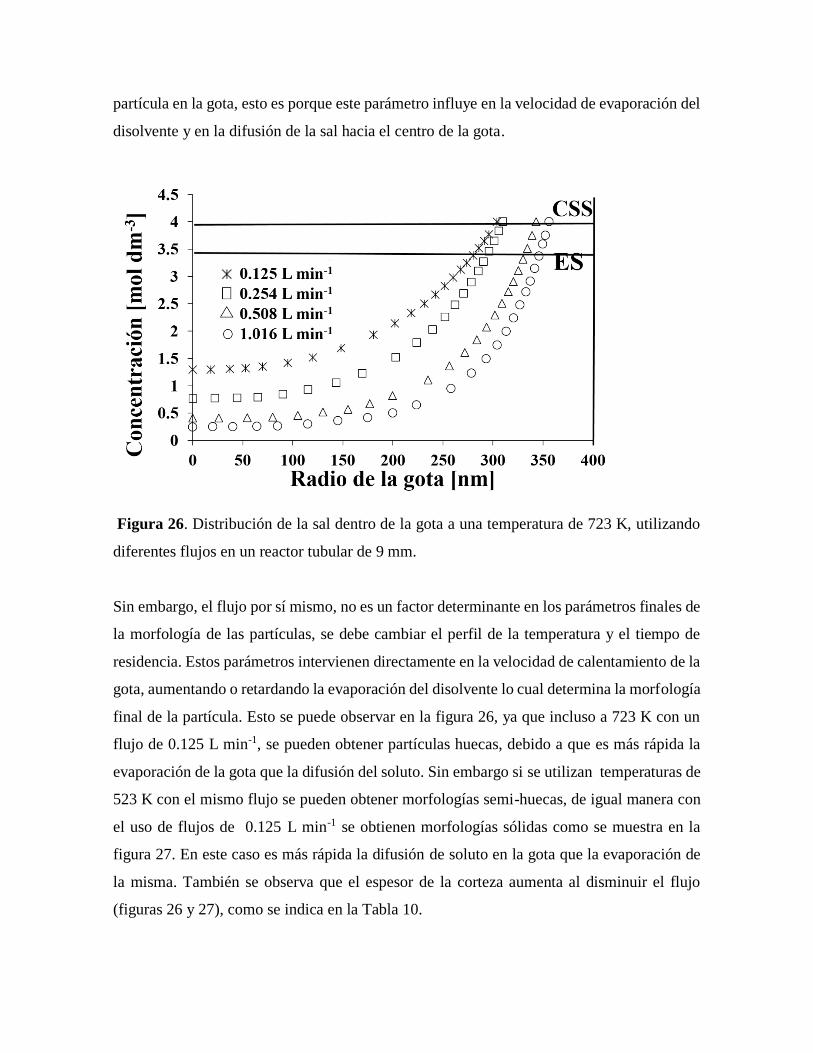
partícula en la gota, esto es porque este parámetro influye en la velocidad de evaporación del
disolvente y en la difusión de la sal hacia el centro de la gota.
Figura 26. Distribución de la sal dentro de la gota a una temperatura de 723 K, utilizando
diferentes flujos en un reactor tubular de 9 mm.
Sin embargo, el flujo por sí mismo, no es un factor determinante en los parámetros finales de
la morfología de las partículas, se debe cambiar el perfil de la temperatura y el tiempo de
residencia. Estos parámetros intervienen directamente en la velocidad de calentamiento de la
gota, aumentando o retardando la evaporación del disolvente lo cual determina la morfología
final de la partícula. Esto se puede observar en la figura 26, ya que incluso a 723 K con un
flujo de 0.125 L min-1, se pueden obtener partículas huecas, debido a que es más rápida la
evaporación de la gota que la difusión del soluto. Sin embargo si se utilizan temperaturas de
523 K con el mismo flujo se pueden obtener morfologías semi-huecas, de igual manera con
el uso de flujos de 0.125 L min-1 se obtienen morfologías sólidas como se muestra en la
figura 27. En este caso es más rápida la difusión de soluto en la gota que la evaporación de
la misma. También se observa que el espesor de la corteza aumenta al disminuir el flujo
(figuras 26 y 27), como se indica en la Tabla 10.
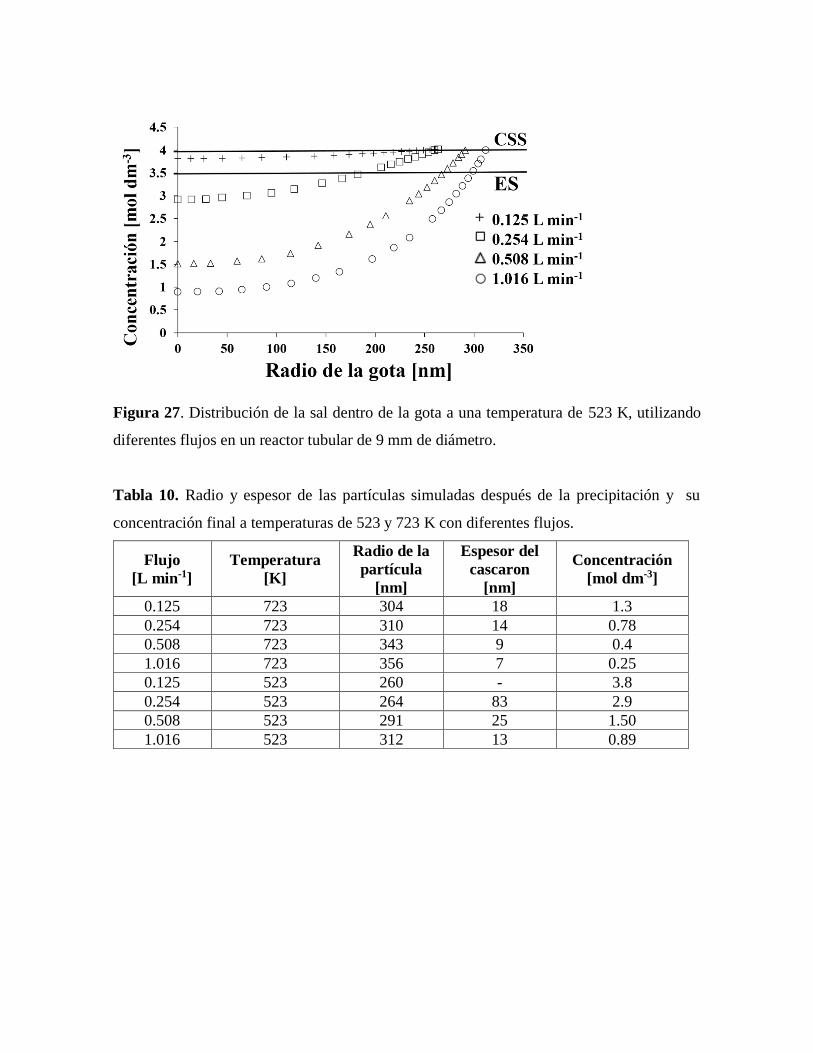
Figura 27. Distribución de la sal dentro de la gota a una temperatura de 523 K, utilizando
diferentes flujos en un reactor tubular de 9 mm de diámetro.
Tabla 10. Radio y espesor de las partículas simuladas después de la precipitación y su
concentración final a temperaturas de 523 y 723 K con diferentes flujos.
Flujo
[L min-1]
Temperatura
[K]
Radio de la
partícula
[nm]
Espesor del
cascaron
[nm]
Concentración
[mol dm-3]
0.125 723 304 18 1.3
0.254 723 310 14 0.78
0.508 723 343 9 0.4
1.016 723 356 7 0.25
0.125 523 260 - 3.8
0.254 523 264 83 2.9
0.508 523 291 25 1.50
1.016 523 312 13 0.89
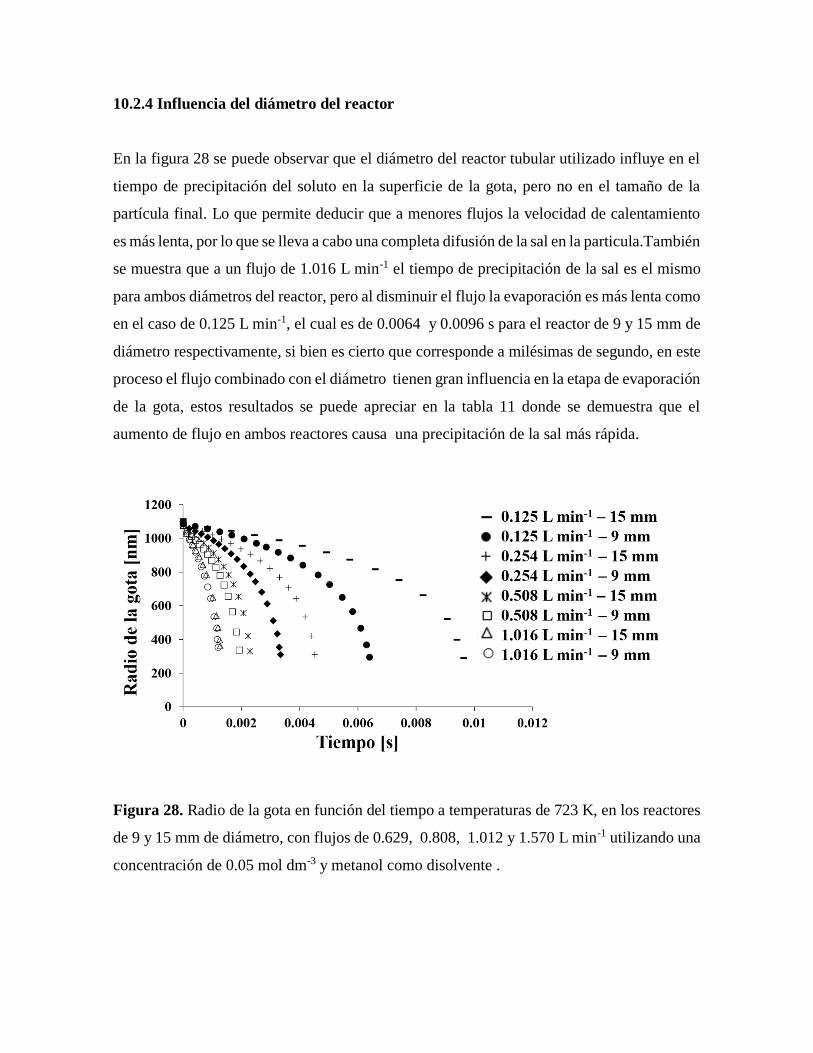
10.2.4 Influencia del diámetro del reactor
En la figura 28 se puede observar que el diámetro del reactor tubular utilizado influye en el
tiempo de precipitación del soluto en la superficie de la gota, pero no en el tamaño de la
partícula final. Lo que permite deducir que a menores flujos la velocidad de calentamiento
es más lenta, por lo que se lleva a cabo una completa difusión de la sal en la particula.También
se muestra que a un flujo de 1.016 L min-1 el tiempo de precipitación de la sal es el mismo
para ambos diámetros del reactor, pero al disminuir el flujo la evaporación es más lenta como
en el caso de 0.125 L min-1, el cual es de 0.0064 y 0.0096 s para el reactor de 9 y 15 mm de
diámetro respectivamente, si bien es cierto que corresponde a milésimas de segundo, en este
proceso el flujo combinado con el diámetro tienen gran influencia en la etapa de evaporación
de la gota, estos resultados se puede apreciar en la tabla 11 donde se demuestra que el
aumento de flujo en ambos reactores causa una precipitación de la sal más rápida.
Figura 28. Radio de la gota en función del tiempo a temperaturas de 723 K, en los reactores
de 9 y 15 mm de diámetro, con flujos de 0.629, 0.808, 1.012 y 1.570 L min-1 utilizando una
concentración de 0.05 mol dm-3 y metanol como disolvente .
Tabla 11. Tiempo de precipitación de la sal a 723 K, utilizando reactores tubulares de 9 y 15
mm de diámetro a diferentes flujos.
Flujo
[L min-1]
Diámetro del
reactor
[mm]
Radio de la
partícula [nm]
Tiempo de
precipitación
[s]
0.125 9 304 0.0064
0.254 9 310 0.0033
0.508 9 343 0.0019
1.016 9 356 0.0012
0.125 15 297 0.0096
0.254 15 308 0.0045
0.508 15 330 0.0023
1.016 15 353 0.0012
Estos resultados obtenidos por el software COMSOL multiphysics, nos permiten comprender
el proceso de evaporación de la gota, parámetro esencial para determinar la morfología final
del nanomaterial, además nos permiten conocer las condiciones en las cuales se pueden
obtener nanomateriales, en este caso Magnetita, con una morfología hueca o sólida, por lo
tanto el obtener estos datos, marca la pauta para manipular las condiciones de síntesis con el
objetivo de obtener el material con una propiedad deseada, en este caso es importante el
obtener nanopartículas huecas y porosas ya que con esto se aumenta el área superficial y la
eficiencia de adsorción.
11 Microestructura y caracterización de las MNPs
11.1.1 Microscopia electrónica de barrido
Con el fin de conocer cómo afecta la temperatura en la morfología de las MNPs, se
sintetizaron nanopartículas a temperaturas de 723 y 773 K. La Figura 29 (a-b) muestra
micrografías obtenidas por electrones secundarios en el microscopio electrónico de barrido,
a partir de las cuales se puede observar la formación de partículas aglomeradas con una
morfología esférica y hueca. La Figura 29 a), muestra las MNPs sintetizadas a 723 K, donde
el tamaño medio de partícula que se obtuvo fue de 300 ± 78 nm, mientras que las que fueron
sintetizadas a 773 K (Fig. 28b) presentaron un diámetro medio de 380 ± 100 nm. Ambas
muestras fueron sintetizadas fijando las condiciones mencionadas en la figura 29.
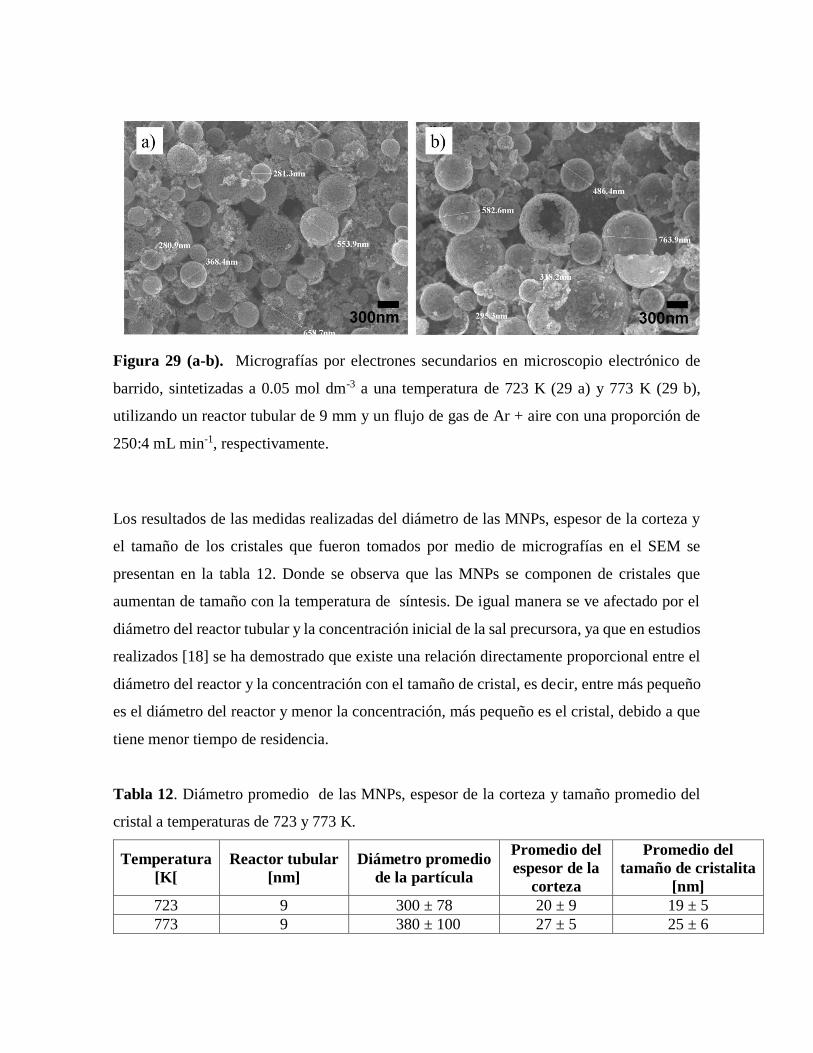
Figura 29 (a-b). Micrografías por electrones secundarios en microscopio electrónico de
barrido, sintetizadas a 0.05 mol dm-3 a una temperatura de 723 K (29 a) y 773 K (29 b),
utilizando un reactor tubular de 9 mm y un flujo de gas de Ar + aire con una proporción de
250:4 mL min-1, respectivamente.
Los resultados de las medidas realizadas del diámetro de las MNPs, espesor de la corteza y
el tamaño de los cristales que fueron tomados por medio de micrografías en el SEM se
presentan en la tabla 12. Donde se observa que las MNPs se componen de cristales que
aumentan de tamaño con la temperatura de síntesis. De igual manera se ve afectado por el
diámetro del reactor tubular y la concentración inicial de la sal precursora, ya que en estudios
realizados [18] se ha demostrado que existe una relación directamente proporcional entre el
diámetro del reactor y la concentración con el tamaño de cristal, es decir, entre más pequeño
es el diámetro del reactor y menor la concentración, más pequeño es el cristal, debido a que
tiene menor tiempo de residencia.
Tabla 12. Diámetro promedio de las MNPs, espesor de la corteza y tamaño promedio del
cristal a temperaturas de 723 y 773 K.
Temperatura
[K[
Reactor tubular
[nm]
Diámetro promedio
de la partícula
Promedio del
espesor de la
corteza
Promedio del
tamaño de cristalita
[nm]
723 9 300 ± 78 20 ± 9 19 ± 5
773 9 380 ± 100 27 ± 5 25 ± 6

En la figura 30 se muestra la composición elemental de las MNPs por EDS. Los resultados
muestran la presencia de carbón (C) el cual se debe al disolvente utilizado (metanol), Fierro
(Fe) y oxígeno (O) que corresponden a las nanopartículas de Fe3O4 obtenidas. En algunas
ocasiones se detecta la presencia de Cloro (Cl) , el cual se puede justificar por la formación
de ácido clorhídrico en la reacción. Finalmente la detección de silicio (Si) se debe al
portamuestras sobre el cual se depositó el material.
Figura 30. Análisis elemental por EDS. Materiales sintetizados a 723 K, concentración de la
sal precursora de 0.05 mol dm-3, con una proporción de flujo de Ar + aire 250:4 L min-1
respectivamente y un reactor tubular de 9 mm.
11.1.2 Microscopia electrónica de transmisión
La figura 31 (a-b) muestra micrografías obtenidas por microscopia electrónica de
transmisión, las cuales fueron sintetizadas a 723 y 773 K, obteniéndose nanoestructuras
esféricas compuestas de cristales. La Figura 31 (a), muestra MNPs obtenidas a una
temperatura de síntesis de 723 K. Donde se aprecia un mayor oscurecimiento en la periferie,
lo que evidencia que son partículas huecas, con cristales que ocupan toda la superficie y con
una porosidad menor que a temperatura de 773 K, debido a que los espacios entre los cristales
son más pequeños. En la Figura 31 (b) se aprecian cristales más definidos y de mayor tamaño
que a temperatura de 723 K. Además se comprueba de manera más detallada la presencia de
cristalitas y su porosidad, sin cambiar su morfología.
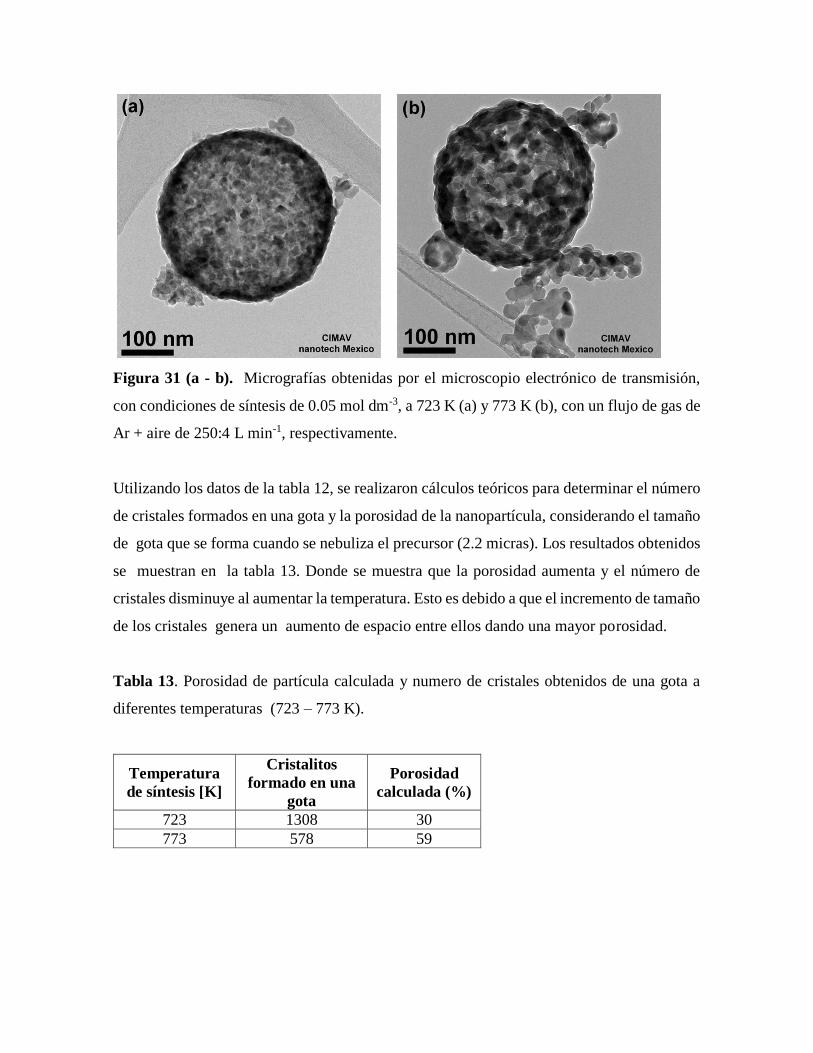
Figura 31 (a - b). Micrografías obtenidas por el microscopio electrónico de transmisión,
con condiciones de síntesis de 0.05 mol dm-3, a 723 K (a) y 773 K (b), con un flujo de gas de
Ar + aire de 250:4 L min-1, respectivamente.
Utilizando los datos de la tabla 12, se realizaron cálculos teóricos para determinar el número
de cristales formados en una gota y la porosidad de la nanopartícula, considerando el tamaño
de gota que se forma cuando se nebuliza el precursor (2.2 micras). Los resultados obtenidos
se muestran en la tabla 13. Donde se muestra que la porosidad aumenta y el número de
cristales disminuye al aumentar la temperatura. Esto es debido a que el incremento de tamaño
de los cristales genera un aumento de espacio entre ellos dando una mayor porosidad.
Tabla 13. Porosidad de partícula calculada y numero de cristales obtenidos de una gota a
diferentes temperaturas (723 – 773 K).
Temperatura
de síntesis [K]
Cristalitos
formado en una
gota
Porosidad
calculada (%)
723 1308 30
773 578 59
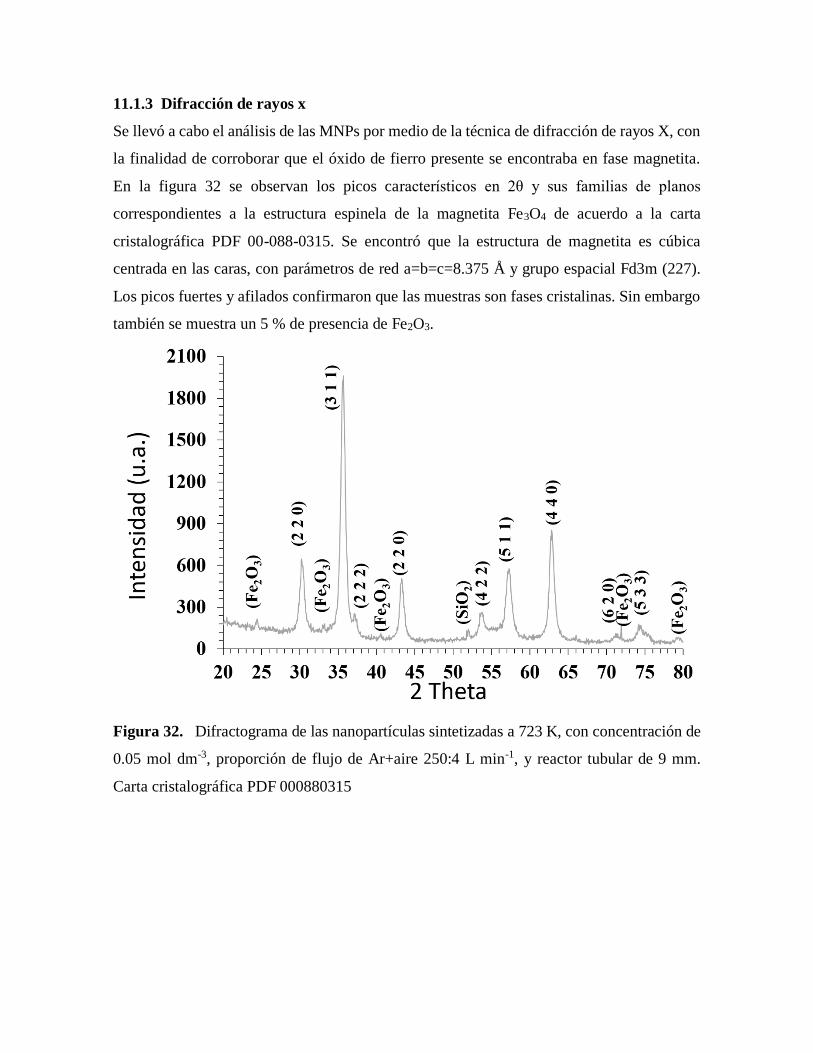
11.1.3 Difracción de rayos x
Se llevó a cabo el análisis de las MNPs por medio de la técnica de difracción de rayos X, con
la finalidad de corroborar que el óxido de fierro presente se encontraba en fase magnetita.
En la figura 32 se observan los picos característicos en 2θ y sus familias de planos
correspondientes a la estructura espinela de la magnetita Fe3O4 de acuerdo a la carta
cristalográfica PDF 00-088-0315. Se encontró que la estructura de magnetita es cúbica
centrada en las caras, con parámetros de red a=b=c=8.375 Å y grupo espacial Fd3m (227).
Los picos fuertes y afilados confirmaron que las muestras son fases cristalinas. Sin embargo
también se muestra un 5 % de presencia de Fe2O3.
Figura 32. Difractograma de las nanopartículas sintetizadas a 723 K, con concentración de
0.05 mol dm-3, proporción de flujo de Ar+aire 250:4 L min-1, y reactor tubular de 9 mm.
Carta cristalográfica PDF 000880315
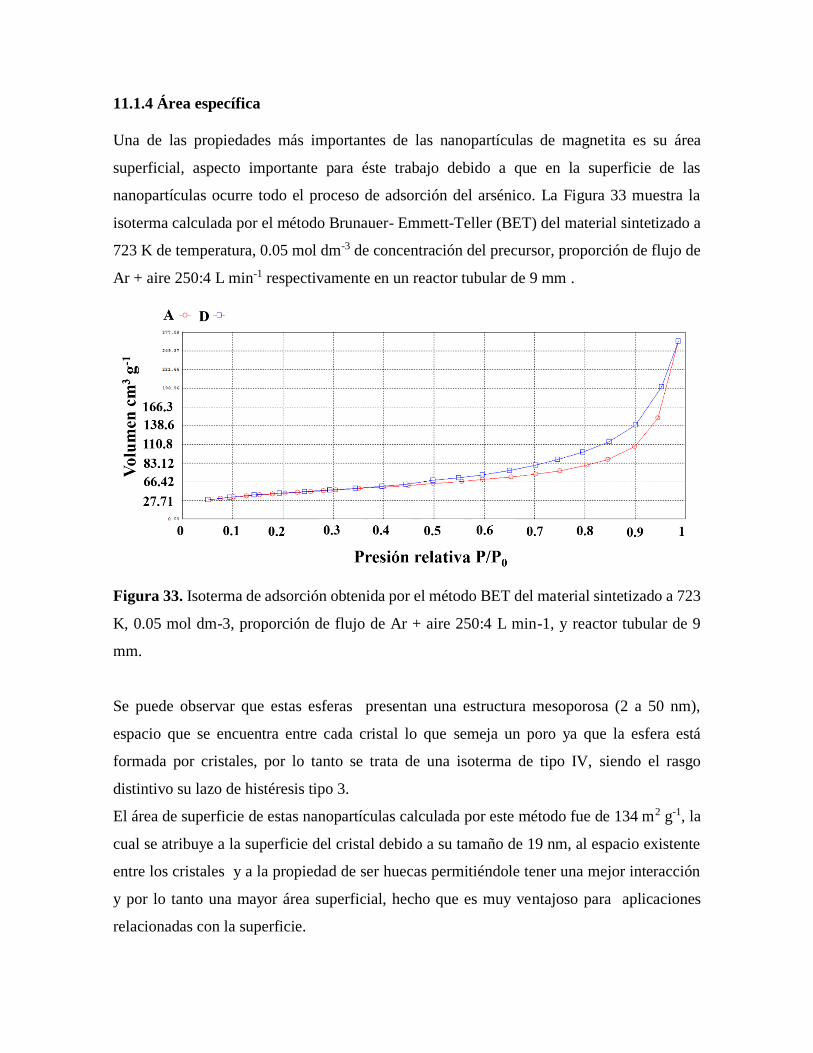
11.1.4 Área específica
Una de las propiedades más importantes de las nanopartículas de magnetita es su área
superficial, aspecto importante para éste trabajo debido a que en la superficie de las
nanopartículas ocurre todo el proceso de adsorción del arsénico. La Figura 33 muestra la
isoterma calculada por el método Brunauer- Emmett-Teller (BET) del material sintetizado a
723 K de temperatura, 0.05 mol dm-3 de concentración del precursor, proporción de flujo de
Ar + aire 250:4 L min-1 respectivamente en un reactor tubular de 9 mm .
Figura 33. Isoterma de adsorción obtenida por el método BET del material sintetizado a 723
K, 0.05 mol dm-3, proporción de flujo de Ar + aire 250:4 L min-1, y reactor tubular de 9
mm.
Se puede observar que estas esferas presentan una estructura mesoporosa (2 a 50 nm),
espacio que se encuentra entre cada cristal lo que semeja un poro ya que la esfera está
formada por cristales, por lo tanto se trata de una isoterma de tipo IV, siendo el rasgo
distintivo su lazo de histéresis tipo 3.
El área de superficie de estas nanopartículas calculada por este método fue de 134 m2 g-1, la
cual se atribuye a la superficie del cristal debido a su tamaño de 19 nm, al espacio existente
entre los cristales y a la propiedad de ser huecas permitiéndole tener una mejor interacción
y por lo tanto una mayor área superficial, hecho que es muy ventajoso para aplicaciones
relacionadas con la superficie.
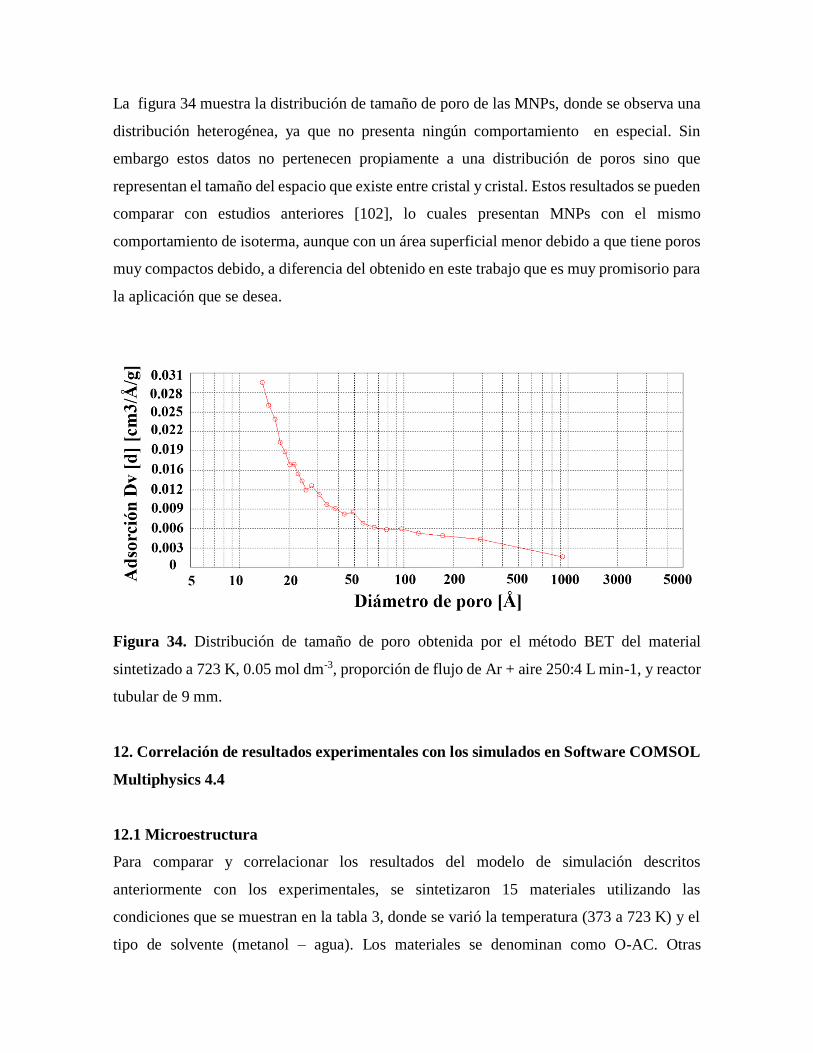
La figura 34 muestra la distribución de tamaño de poro de las MNPs, donde se observa una
distribución heterogénea, ya que no presenta ningún comportamiento en especial. Sin
embargo estos datos no pertenecen propiamente a una distribución de poros sino que
representan el tamaño del espacio que existe entre cristal y cristal. Estos resultados se pueden
comparar con estudios anteriores [102], lo cuales presentan MNPs con el mismo
comportamiento de isoterma, aunque con un área superficial menor debido a que tiene poros
muy compactos debido, a diferencia del obtenido en este trabajo que es muy promisorio para
la aplicación que se desea.
Figura 34. Distribución de tamaño de poro obtenida por el método BET del material
sintetizado a 723 K, 0.05 mol dm-3, proporción de flujo de Ar + aire 250:4 L min-1, y reactor
tubular de 9 mm.
12. Correlación de resultados experimentales con los simulados en Software COMSOL
Multiphysics 4.4
12.1 Microestructura
Para comparar y correlacionar los resultados del modelo de simulación descritos
anteriormente con los experimentales, se sintetizaron 15 materiales utilizando las
condiciones que se muestran en la tabla 3, donde se varió la temperatura (373 a 723 K) y el
tipo de solvente (metanol – agua). Los materiales se denominan como O-AC. Otras
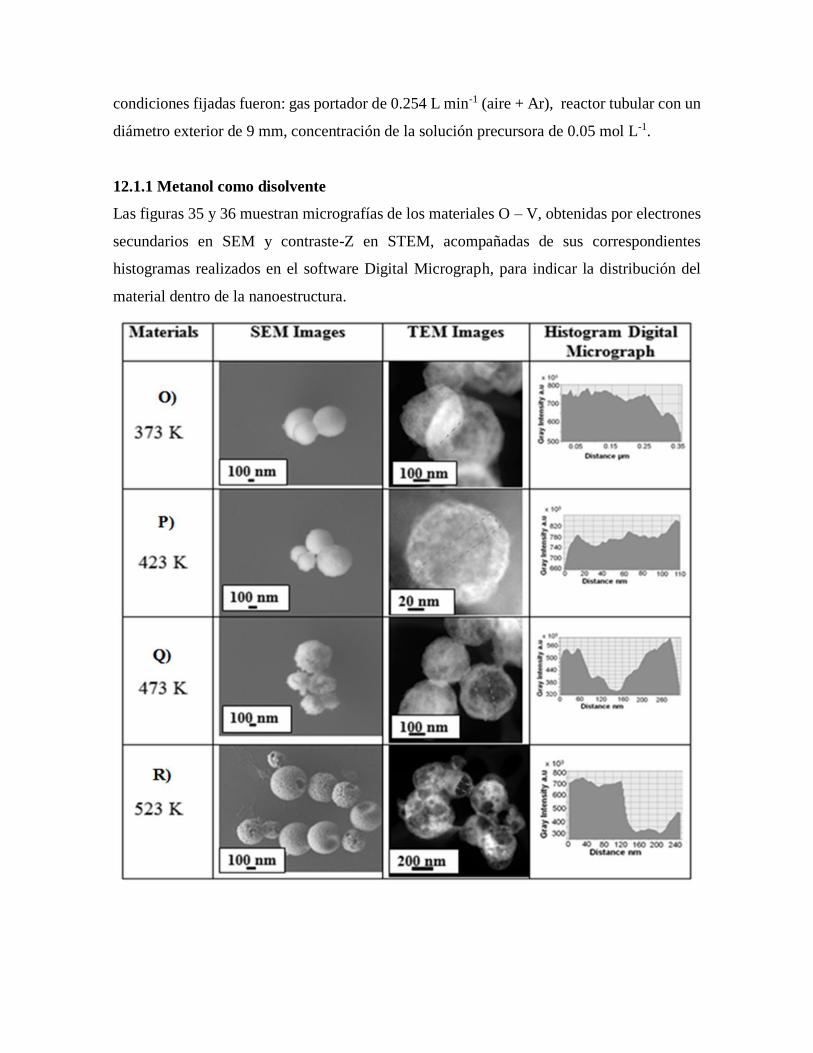
condiciones fijadas fueron: gas portador de 0.254 L min-1 (aire + Ar), reactor tubular con un
diámetro exterior de 9 mm, concentración de la solución precursora de 0.05 mol L-1.
12.1.1 Metanol como disolvente
Las figuras 35 y 36 muestran micrografías de los materiales O – V, obtenidas por electrones
secundarios en SEM y contraste-Z en STEM, acompañadas de sus correspondientes
histogramas realizados en el software Digital Micrograph, para indicar la distribución del
material dentro de la nanoestructura.

Figura 35. Micrografías de los materiales obtenidos a temperaturas de 373 a 523 K
(materiales O-R), por electrones secundarios en SEM y contraste-Z en MET, con su
histograma de distribución de la sal incluido.
La figura 35 muestra los materiales obtenidos de 373 a 523 K, donde se puede observar que
a temperaturas de 373 a 423 K, la morfología de las partículas que se obtiene es esférica y
sólida. Esto sucede porque en el momento de la nucleación, la concentración de soluto dentro
de la gota es mayor que la concentración de ES. Lo anterior se produce porque la velocidad
de evaporación fue lo suficientemente lenta para permitir la difusión del soluto hacia el
interior de la gota, debido a la baja temperatura [71]. A 473 K (material Q), empieza la
descomposición de la sal precursora, disminuyendo la cantidad de cloro e iniciando la
oxidación del Fe. El cloro se desprende de FeCl2 y se une al hidrógeno que se origina a partir
del metanol, para formar HCl en forma de vapor [18].
A esta temperatura (ver figura 35), se observó una morfología sólida y casi esférica; con
aglomerados en su superficie, lo que le da cierta permeabilidad para permitir que se lleve a
cabo una completa evaporación del metanol [72, 103]. Para el material R, la velocidad de
evaporación de metanol aumenta, lo que permite obtener una morfología con partes sólidas
y huecas; estos resultados se pueden correlacionar con los simulados que se muestran en la
Figura 35, donde se indica que a una temperatura de 523 K se pueden obtener partículas con
la misma morfología. De igual manera, se muestra claramente la distribución del material
en la partícula, junto con los histogramas realizadas en cada material presente.
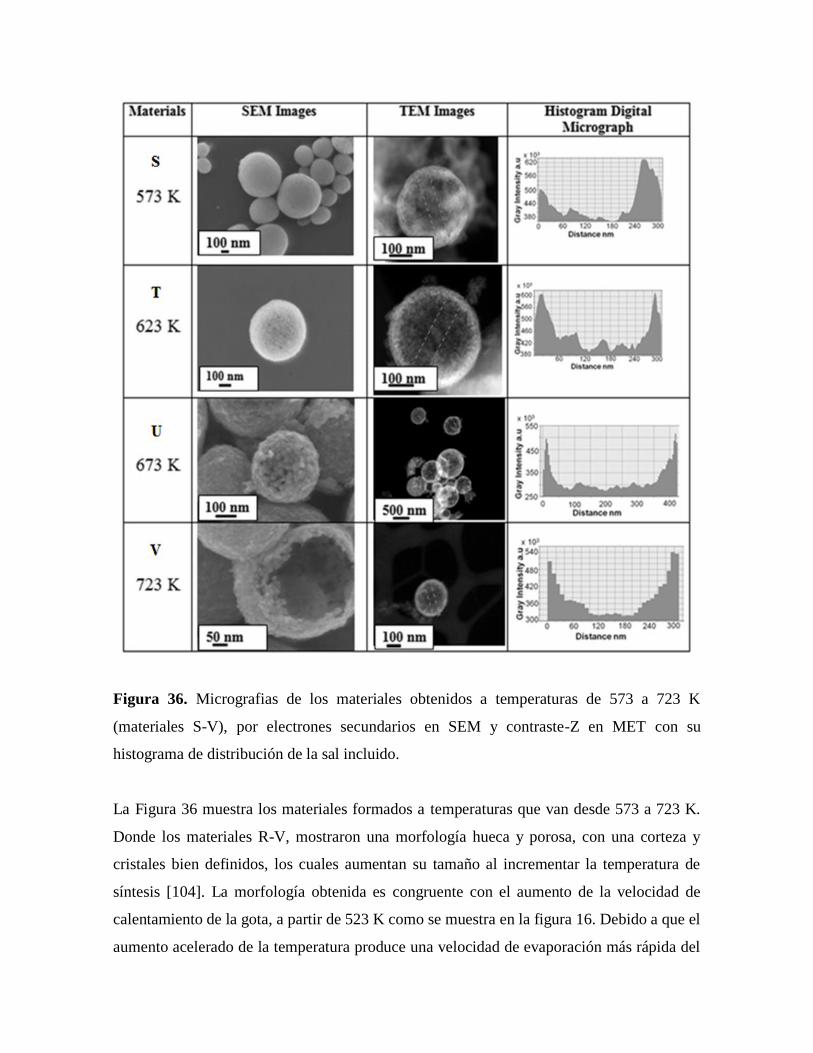
Figura 36. Micrografias de los materiales obtenidos a temperaturas de 573 a 723 K
(materiales S-V), por electrones secundarios en SEM y contraste-Z en MET con su
histograma de distribución de la sal incluido.
La Figura 36 muestra los materiales formados a temperaturas que van desde 573 a 723 K.
Donde los materiales R-V, mostraron una morfología hueca y porosa, con una corteza y
cristales bien definidos, los cuales aumentan su tamaño al incrementar la temperatura de
síntesis [104]. La morfología obtenida es congruente con el aumento de la velocidad de
calentamiento de la gota, a partir de 523 K como se muestra en la figura 16. Debido a que el
aumento acelerado de la temperatura produce una velocidad de evaporación más rápida del

disolvente en la superficie de la gota. que la difusión del soluto hacia el centro de la gota no
es lo suficientemente rápida, la concentración de soluto empieza a ser inferior a la ES, por lo
que se puede generar una morfología hueca y esférica en la partícula esférica [73]. También
se puede observar que el aumento de temperatura provoca la disminución del espesor de la
corteza, como se indica en los histogramas de la Figura 3 y se correlacionan con los resultados
mostrados en la Figura 20. En la tabla 14 se muestran el microanálisis elemental de los
materiales obtenidos a temperaturas ≤ 673 K donde se observa un alto porcentaje de Cl el
cual pertenece al FeCl2, lo que indica que a esas temperaturas, la reacción química no se
lleva a cabo por completo, por lo que se obtienen nanopartículas formadas parcial o
totalmente por la sal precursora.
Tabla 14. Composición elemental (%) de los materiales obtenidos a diferentes temperaturas.
Materiales Temperatura
(K)
% Atómico
Fe Cl O
O 373 34 66 0
Q 473 36 54 10
S 573 31 23 46
U 673 29 16 55
V 723 38 0 62
12.1.2 Agua como disolvente de la solución precursora
En la Figura 37 se muestran las nanoestructuras obtenidas en un intervalo de temperatura de
423 a 673 K (materiales W –AC). Donde se observa que en un rango de 423 a 523 K,
presentan una morfología globular, completamente sólida y sin la presencia de cristales en
la superficie, debido a que la sal comienza su descomposición a estas temperaturas, de
manera que sólo el agua se evapora dejando aglomerados de la sal precursora. Cuando se
tienen temperaturas de 573 a 623 K, la morfología cambia a esferas sólidas porosas, esto es
porque la reacción de descomposición se inicia [104], sin embargo se conserva una
morfologia sólida. Al incrementar la temperatura a 673 y 723 K (Figura 35 y 36 ), se observa
la presencia de cristales de 10 ± 2 nm (Figura 38), los cuales son más pequeños que los que
forman las MNPs sintetizada con metanol, como se muestra en la figura 24 (a-b). Al utilizar
agua como disolvente se obtienen morfologías sólidas, ya que la sal tiene más tiempo para
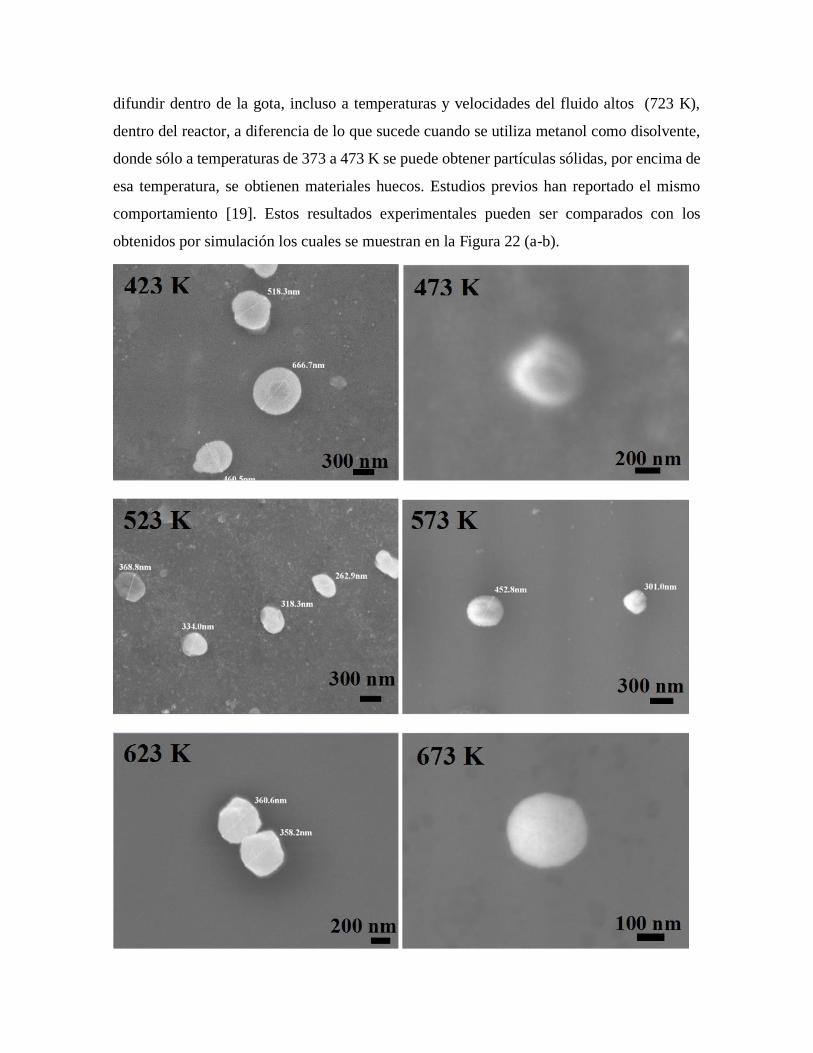
difundir dentro de la gota, incluso a temperaturas y velocidades del fluido altos (723 K),
dentro del reactor, a diferencia de lo que sucede cuando se utiliza metanol como disolvente,
donde sólo a temperaturas de 373 a 473 K se puede obtener partículas sólidas, por encima de
esa temperatura, se obtienen materiales huecos. Estudios previos han reportado el mismo
comportamiento [19]. Estos resultados experimentales pueden ser comparados con los
obtenidos por simulación los cuales se muestran en la Figura 22 (a-b).
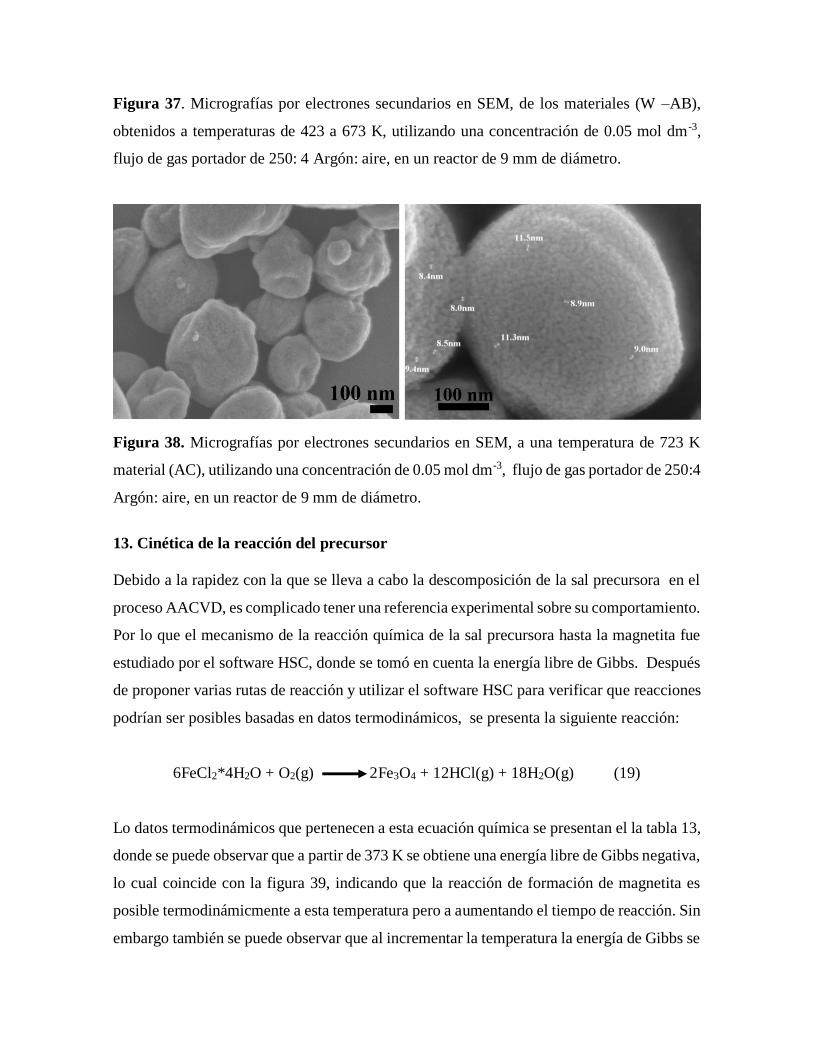
Figura 37. Micrografías por electrones secundarios en SEM, de los materiales (W –AB),
obtenidos a temperaturas de 423 a 673 K, utilizando una concentración de 0.05 mol dm-3,
flujo de gas portador de 250: 4 Argón: aire, en un reactor de 9 mm de diámetro.
Figura 38. Micrografías por electrones secundarios en SEM, a una temperatura de 723 K
material (AC), utilizando una concentración de 0.05 mol dm-3, flujo de gas portador de 250:4
Argón: aire, en un reactor de 9 mm de diámetro.
13. Cinética de la reacción del precursor
Debido a la rapidez con la que se lleva a cabo la descomposición de la sal precursora en el
proceso AACVD, es complicado tener una referencia experimental sobre su comportamiento.
Por lo que el mecanismo de la reacción química de la sal precursora hasta la magnetita fue
estudiado por el software HSC, donde se tomó en cuenta la energía libre de Gibbs. Después
de proponer varias rutas de reacción y utilizar el software HSC para verificar que reacciones
podrían ser posibles basadas en datos termodinámicos, se presenta la siguiente reacción:
6FeCl2*4H2O + O2(g) 2Fe3O4 + 12HCl(g) + 18H2O(g) (19)
Lo datos termodinámicos que pertenecen a esta ecuación química se presentan el la tabla 13,
donde se puede observar que a partir de 373 K se obtiene una energía libre de Gibbs negativa,
lo cual coincide con la figura 39, indicando que la reacción de formación de magnetita es
posible termodinámicmente a esta temperatura pero a aumentando el tiempo de reacción. Sin
embargo también se puede observar que al incrementar la temperatura la energía de Gibbs se
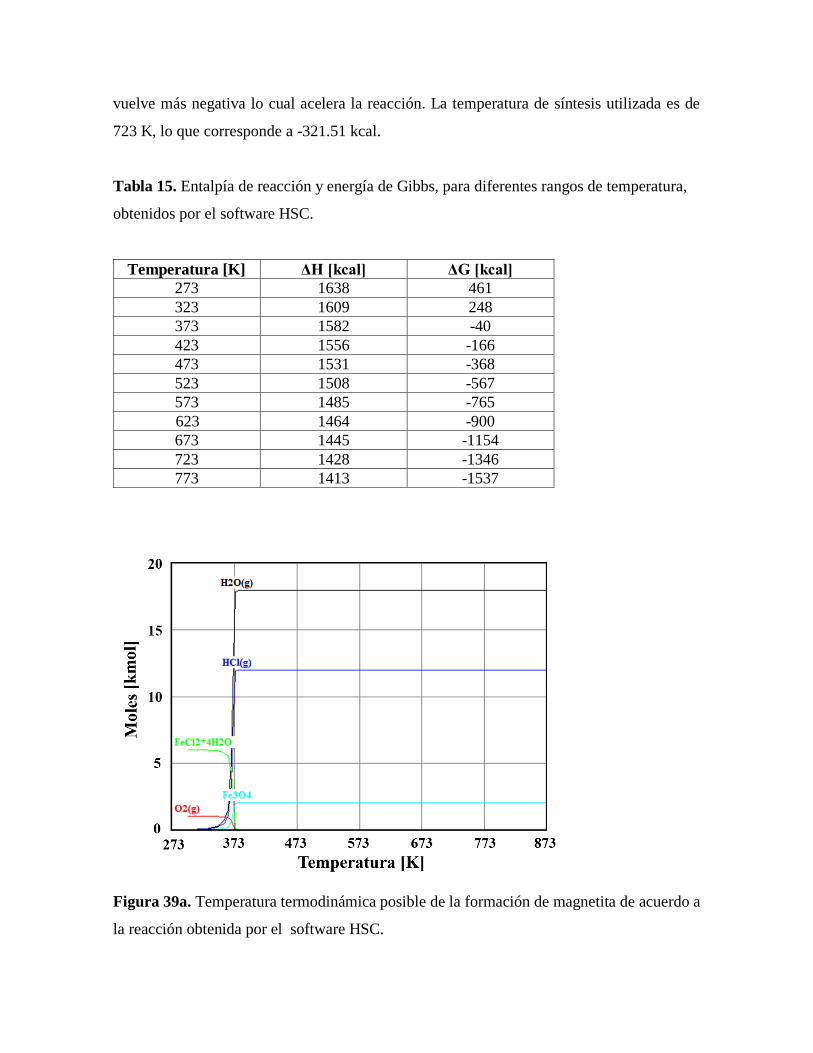
vuelve más negativa lo cual acelera la reacción. La temperatura de síntesis utilizada es de
723 K, lo que corresponde a -321.51 kcal.
Tabla 15. Entalpía de reacción y energía de Gibbs, para diferentes rangos de temperatura,
obtenidos por el software HSC.
Temperatura [K] ΔH [kcal] ΔG [kcal]
273 1638 461
323 1609 248
373 1582 -40
423 1556 -166
473 1531 -368
523 1508 -567
573 1485 -765
623 1464 -900
673 1445 -1154
723 1428 -1346
773 1413 -1537
Figura 39a. Temperatura termodinámica posible de la formación de magnetita de acuerdo a
la reacción obtenida por el software HSC.
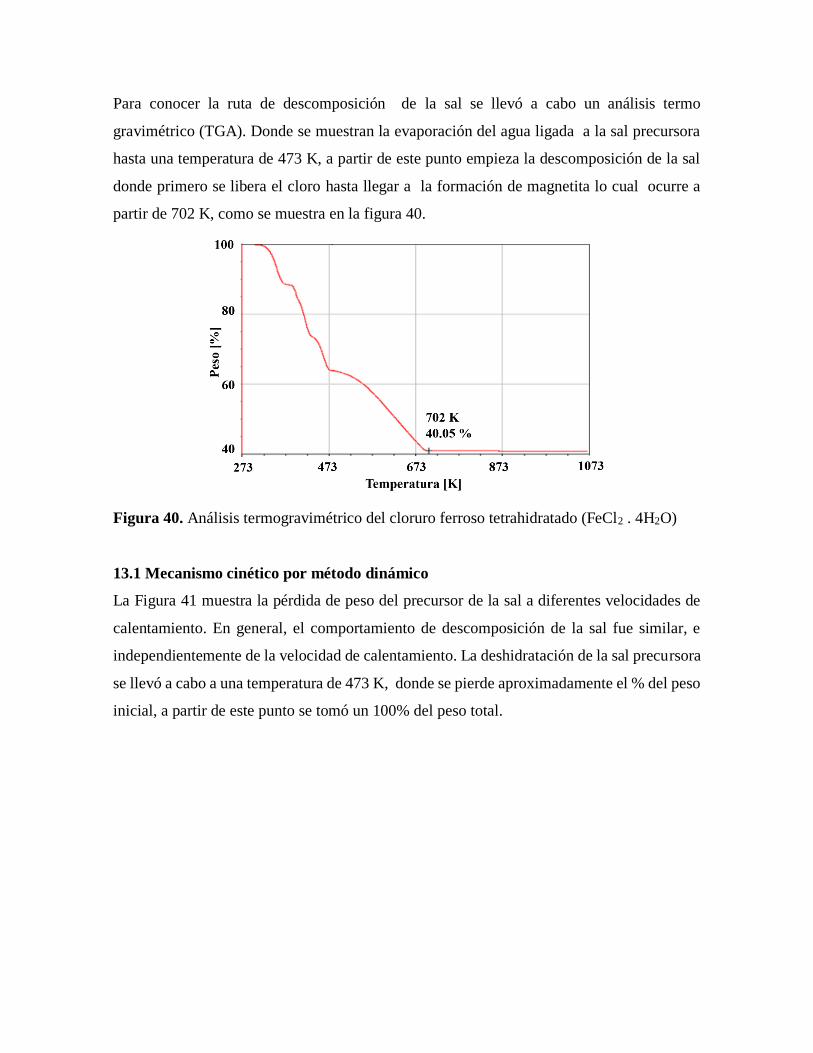
Para conocer la ruta de descomposición de la sal se llevó a cabo un análisis termo
gravimétrico (TGA). Donde se muestran la evaporación del agua ligada a la sal precursora
hasta una temperatura de 473 K, a partir de este punto empieza la descomposición de la sal
donde primero se libera el cloro hasta llegar a la formación de magnetita lo cual ocurre a
partir de 702 K, como se muestra en la figura 40.
Figura 40. Análisis termogravimétrico del cloruro ferroso tetrahidratado (FeCl2 . 4H2O)
13.1 Mecanismo cinético por método dinámico
La Figura 41 muestra la pérdida de peso del precursor de la sal a diferentes velocidades de
calentamiento. En general, el comportamiento de descomposición de la sal fue similar, e
independientemente de la velocidad de calentamiento. La deshidratación de la sal precursora
se llevó a cabo a una temperatura de 473 K, donde se pierde aproximadamente el % del peso
inicial, a partir de este punto se tomó un 100% del peso total.
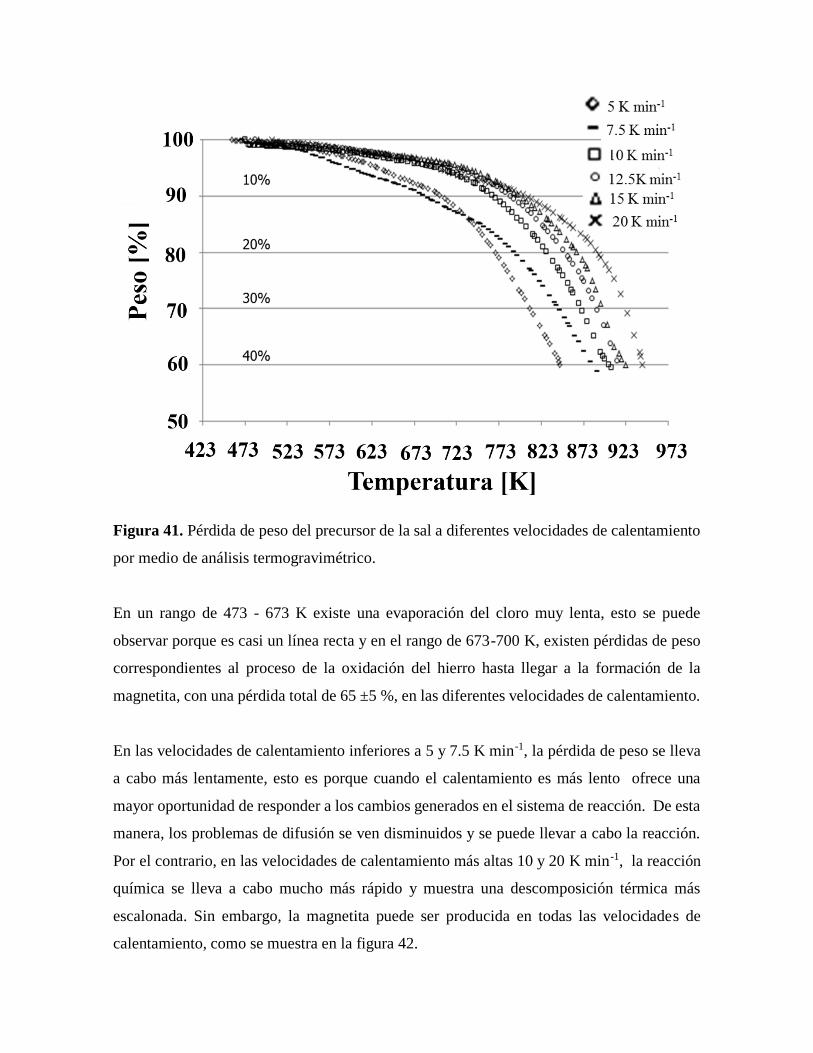
Figura 41. Pérdida de peso del precursor de la sal a diferentes velocidades de calentamiento
por medio de análisis termogravimétrico.
En un rango de 473 - 673 K existe una evaporación del cloro muy lenta, esto se puede
observar porque es casi un línea recta y en el rango de 673-700 K, existen pérdidas de peso
correspondientes al proceso de la oxidación del hierro hasta llegar a la formación de la
magnetita, con una pérdida total de 65 ±5 %, en las diferentes velocidades de calentamiento.
En las velocidades de calentamiento inferiores a 5 y 7.5 K min-1, la pérdida de peso se lleva
a cabo más lentamente, esto es porque cuando el calentamiento es más lento ofrece una
mayor oportunidad de responder a los cambios generados en el sistema de reacción. De esta
manera, los problemas de difusión se ven disminuidos y se puede llevar a cabo la reacción.
Por el contrario, en las velocidades de calentamiento más altas 10 y 20 K min-1, la reacción
química se lleva a cabo mucho más rápido y muestra una descomposición térmica más
escalonada. Sin embargo, la magnetita puede ser producida en todas las velocidades de
calentamiento, como se muestra en la figura 42.
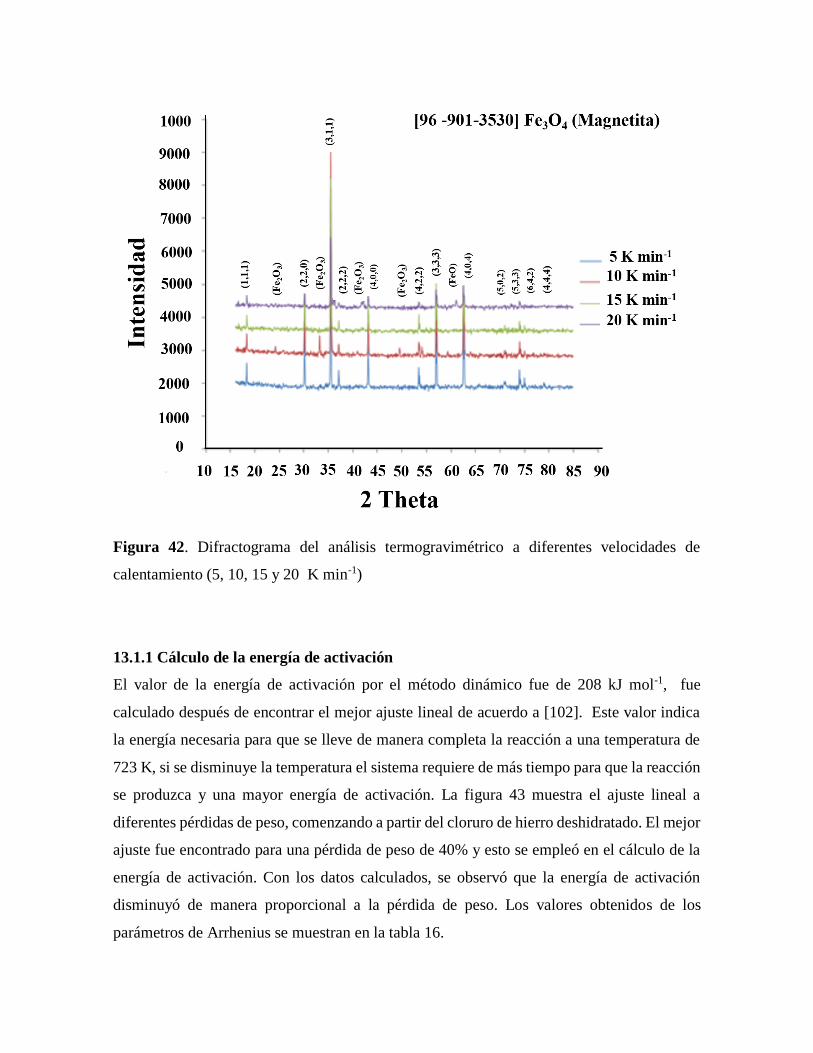
Figura 42. Difractograma del análisis termogravimétrico a diferentes velocidades de
calentamiento (5, 10, 15 y 20 K min-1)
13.1.1 Cálculo de la energía de activación
El valor de la energía de activación por el método dinámico fue de 208 kJ mol-1, fue
calculado después de encontrar el mejor ajuste lineal de acuerdo a [102]. Este valor indica
la energía necesaria para que se lleve de manera completa la reacción a una temperatura de
723 K, si se disminuye la temperatura el sistema requiere de más tiempo para que la reacción
se produzca y una mayor energía de activación. La figura 43 muestra el ajuste lineal a
diferentes pérdidas de peso, comenzando a partir del cloruro de hierro deshidratado. El mejor
ajuste fue encontrado para una pérdida de peso de 40% y esto se empleó en el cálculo de la
energía de activación. Con los datos calculados, se observó que la energía de activación
disminuyó de manera proporcional a la pérdida de peso. Los valores obtenidos de los
parámetros de Arrhenius se muestran en la tabla 16.

Figura 43. Ajuste lineal de los datos obtenidos por análisis termogravimetrico por el método
de Flynn-Wall-Ozawa [104].
Tabla 16. Parámetros de Arrhenius obtenidos por la cinética de reacción a diferentes pérdidas
de peso.
Pérdida de peso [%]
Constante de
velocidad
K [-1]
Factor pre-
exponencial Z
[-1]
Energía de
activación
E [kJ mol -1]
7.3 3.8 E+04 1.1 E+05 64.2
15.1 2.6 E+07 1.6 E+08 108.7
23.2 1.9 E+09 1.9 E+10 38
31.8 1.9 E+11 3.1 E+12 169.3
39.7 3.59 E+13 1 E+15 208
13.2 Mecanismo cinético por método isotérmico
La cinética de la descomposición también fue estudiada por el método isotérmico utilizando
3 diferentes temperaturas (723, 773 y 823 K). Donde se eleva la temperatura a una rampa de
calentamiento de 293 K min-1 y una vez que está en la temperatura deseada se mantiene
constante hasta que el soluto deje de reaccionar y no presente pérdida de peso.
En la figura 44 se observa que en los primeros 10 minutos de la reacción aproximadamente
existen 3 caídas de peso donde se pierde alrededor del 40% del peso inicial, este peso
corresponde a las 4 moléculas de agua ligada que tienen el cloruro ferroso el cual marca su
deshidratación a las diferentes isotermas, en los siguientes 10 minutos hay muy poca
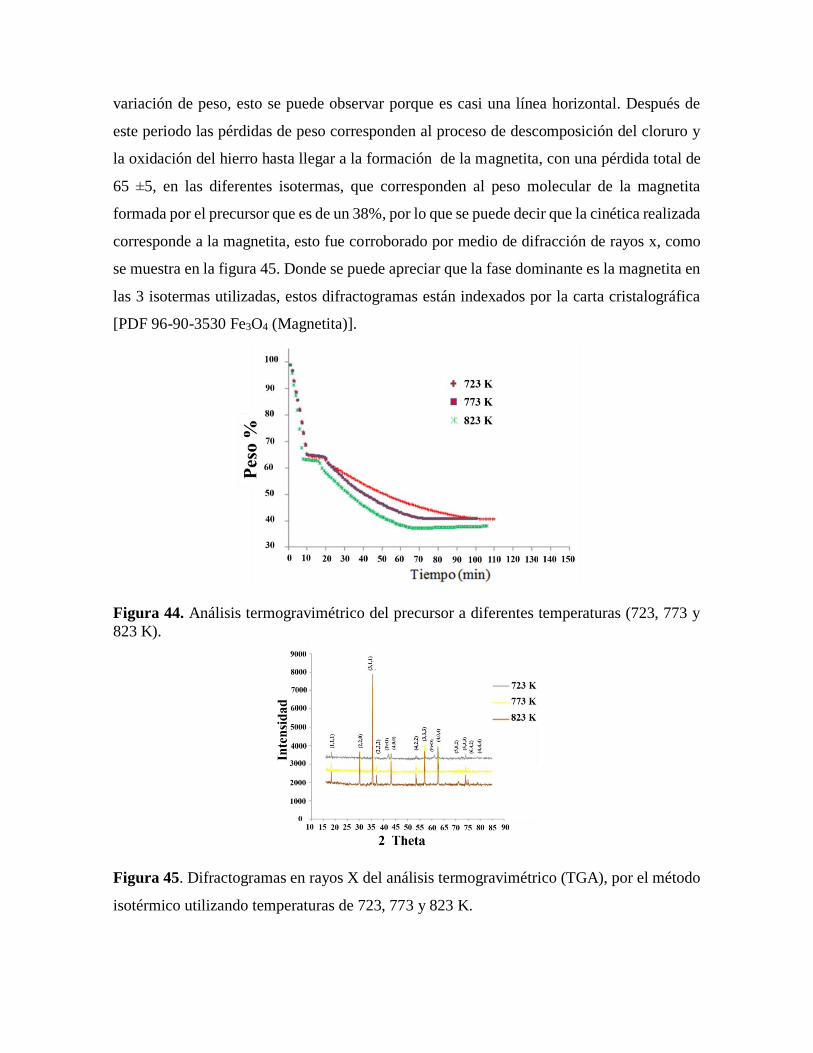
variación de peso, esto se puede observar porque es casi una línea horizontal. Después de
este periodo las pérdidas de peso corresponden al proceso de descomposición del cloruro y
la oxidación del hierro hasta llegar a la formación de la magnetita, con una pérdida total de
65 ±5, en las diferentes isotermas, que corresponden al peso molecular de la magnetita
formada por el precursor que es de un 38%, por lo que se puede decir que la cinética realizada
corresponde a la magnetita, esto fue corroborado por medio de difracción de rayos x, como
se muestra en la figura 45. Donde se puede apreciar que la fase dominante es la magnetita en
las 3 isotermas utilizadas, estos difractogramas están indexados por la carta cristalográfica
[PDF 96-90-3530 Fe3O4 (Magnetita)].
Figura 44. Análisis termogravimétrico del precursor a diferentes temperaturas (723, 773 y
823 K).
Figura 45. Difractogramas en rayos X del análisis termogravimétrico (TGA), por el método
isotérmico utilizando temperaturas de 723, 773 y 823 K.
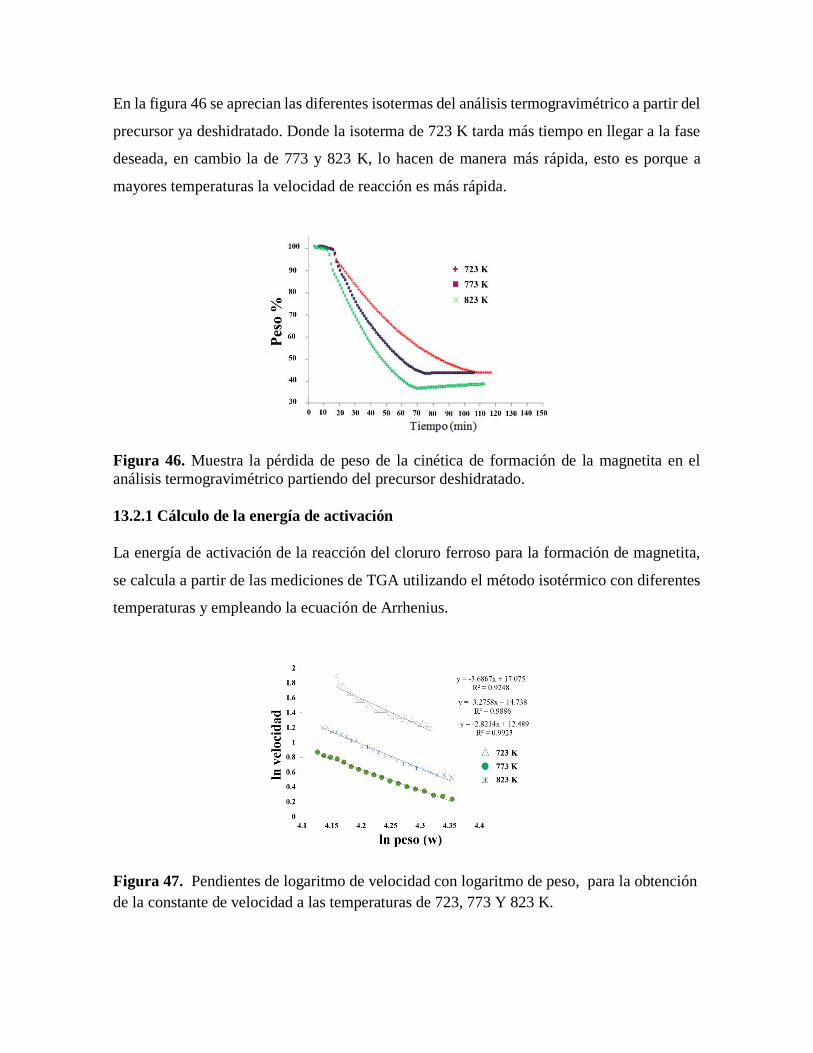
En la figura 46 se aprecian las diferentes isotermas del análisis termogravimétrico a partir del
precursor ya deshidratado. Donde la isoterma de 723 K tarda más tiempo en llegar a la fase
deseada, en cambio la de 773 y 823 K, lo hacen de manera más rápida, esto es porque a
mayores temperaturas la velocidad de reacción es más rápida.
Figura 46. Muestra la pérdida de peso de la cinética de formación de la magnetita en el
análisis termogravimétrico partiendo del precursor deshidratado.
13.2.1 Cálculo de la energía de activación
La energía de activación de la reacción del cloruro ferroso para la formación de magnetita,
se calcula a partir de las mediciones de TGA utilizando el método isotérmico con diferentes
temperaturas y empleando la ecuación de Arrhenius.
Figura 47. Pendientes de logaritmo de velocidad con logaritmo de peso, para la obtención
de la constante de velocidad a las temperaturas de 723, 773 Y 823 K.
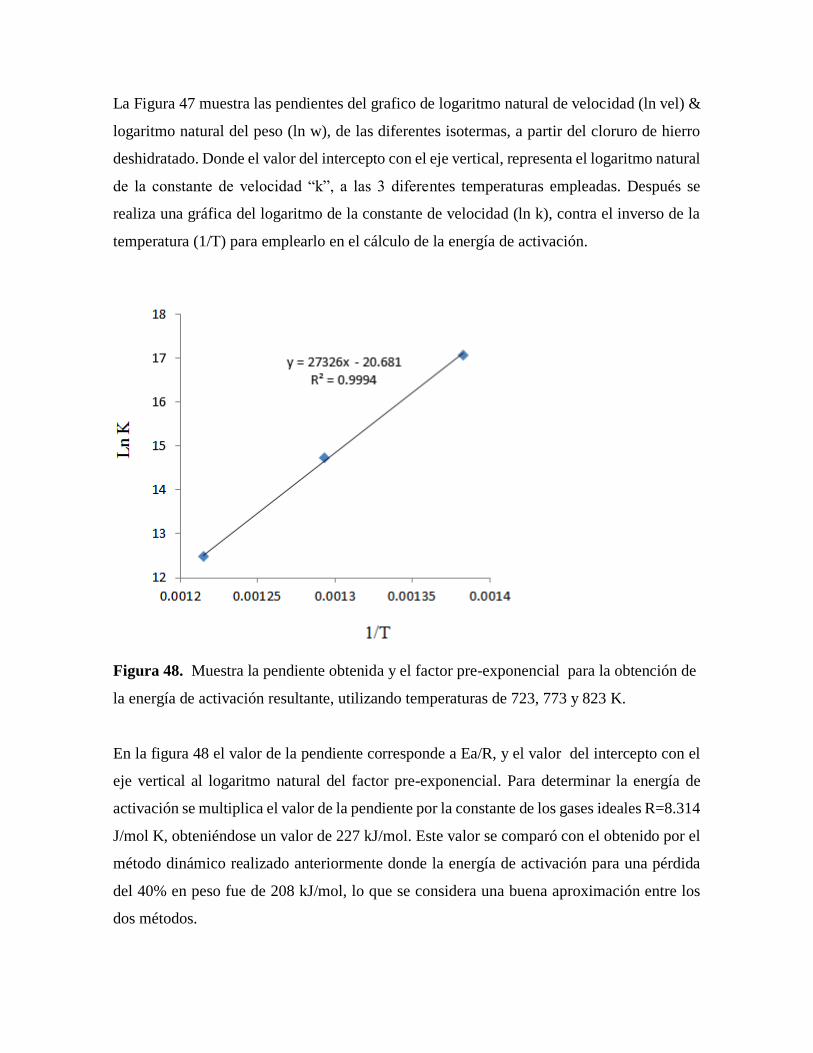
La Figura 47 muestra las pendientes del grafico de logaritmo natural de velocidad (ln vel) &
logaritmo natural del peso (ln w), de las diferentes isotermas, a partir del cloruro de hierro
deshidratado. Donde el valor del intercepto con el eje vertical, representa el logaritmo natural
de la constante de velocidad “k”, a las 3 diferentes temperaturas empleadas. Después se
realiza una gráfica del logaritmo de la constante de velocidad (ln k), contra el inverso de la
temperatura (1/T) para emplearlo en el cálculo de la energía de activación.
Figura 48. Muestra la pendiente obtenida y el factor pre-exponencial para la obtención de
la energía de activación resultante, utilizando temperaturas de 723, 773 y 823 K.
En la figura 48 el valor de la pendiente corresponde a Ea/R, y el valor del intercepto con el
eje vertical al logaritmo natural del factor pre-exponencial. Para determinar la energía de
activación se multiplica el valor de la pendiente por la constante de los gases ideales R=8.314
J/mol K, obteniéndose un valor de 227 kJ/mol. Este valor se comparó con el obtenido por el
método dinámico realizado anteriormente donde la energía de activación para una pérdida
del 40% en peso fue de 208 kJ/mol, lo que se considera una buena aproximación entre los
dos métodos.
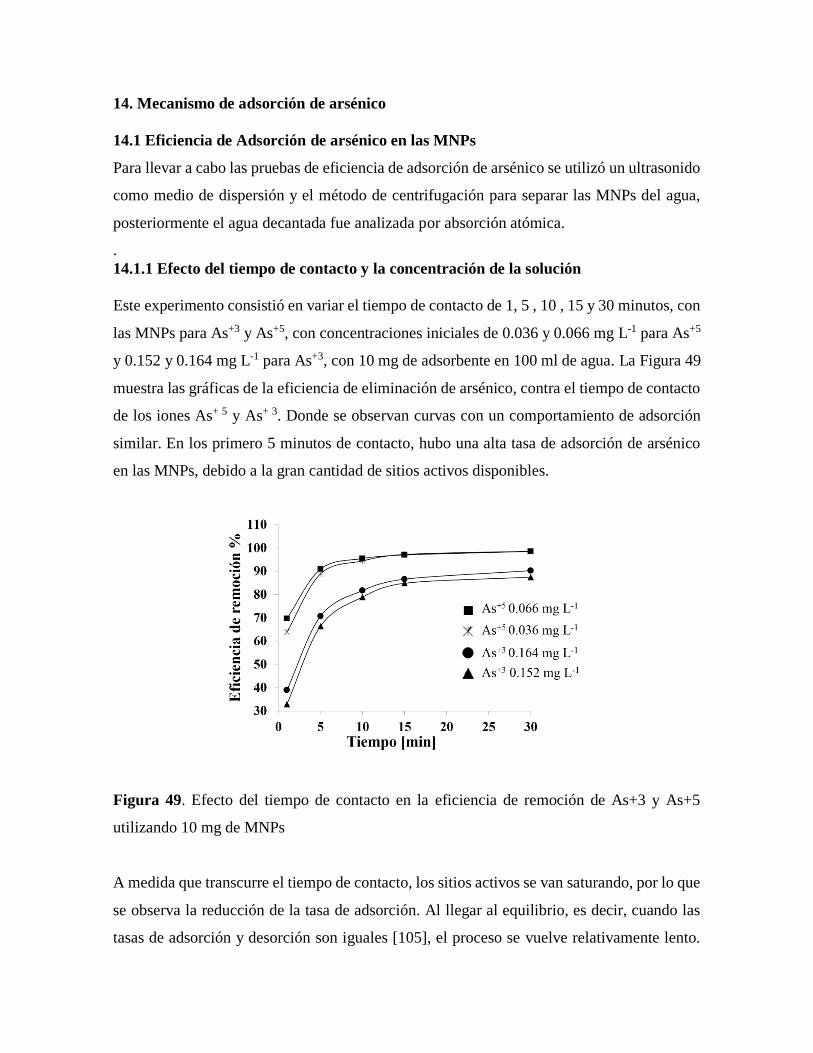
14. Mecanismo de adsorción de arsénico
14.1 Eficiencia de Adsorción de arsénico en las MNPs
Para llevar a cabo las pruebas de eficiencia de adsorción de arsénico se utilizó un ultrasonido
como medio de dispersión y el método de centrifugación para separar las MNPs del agua,
posteriormente el agua decantada fue analizada por absorción atómica.
.
14.1.1 Efecto del tiempo de contacto y la concentración de la solución
Este experimento consistió en variar el tiempo de contacto de 1, 5 , 10 , 15 y 30 minutos, con
las MNPs para As+3 y As+5, con concentraciones iniciales de 0.036 y 0.066 mg L-1 para As+5
y 0.152 y 0.164 mg L-1 para As+3, con 10 mg de adsorbente en 100 ml de agua. La Figura 49
muestra las gráficas de la eficiencia de eliminación de arsénico, contra el tiempo de contacto
de los iones As+ 5 y As+ 3. Donde se observan curvas con un comportamiento de adsorción
similar. En los primero 5 minutos de contacto, hubo una alta tasa de adsorción de arsénico
en las MNPs, debido a la gran cantidad de sitios activos disponibles.
Figura 49. Efecto del tiempo de contacto en la eficiencia de remoción de As+3 y As+5
utilizando 10 mg de MNPs
A medida que transcurre el tiempo de contacto, los sitios activos se van saturando, por lo que
se observa la reducción de la tasa de adsorción. Al llegar al equilibrio, es decir, cuando las
tasas de adsorción y desorción son iguales [105], el proceso se vuelve relativamente lento.
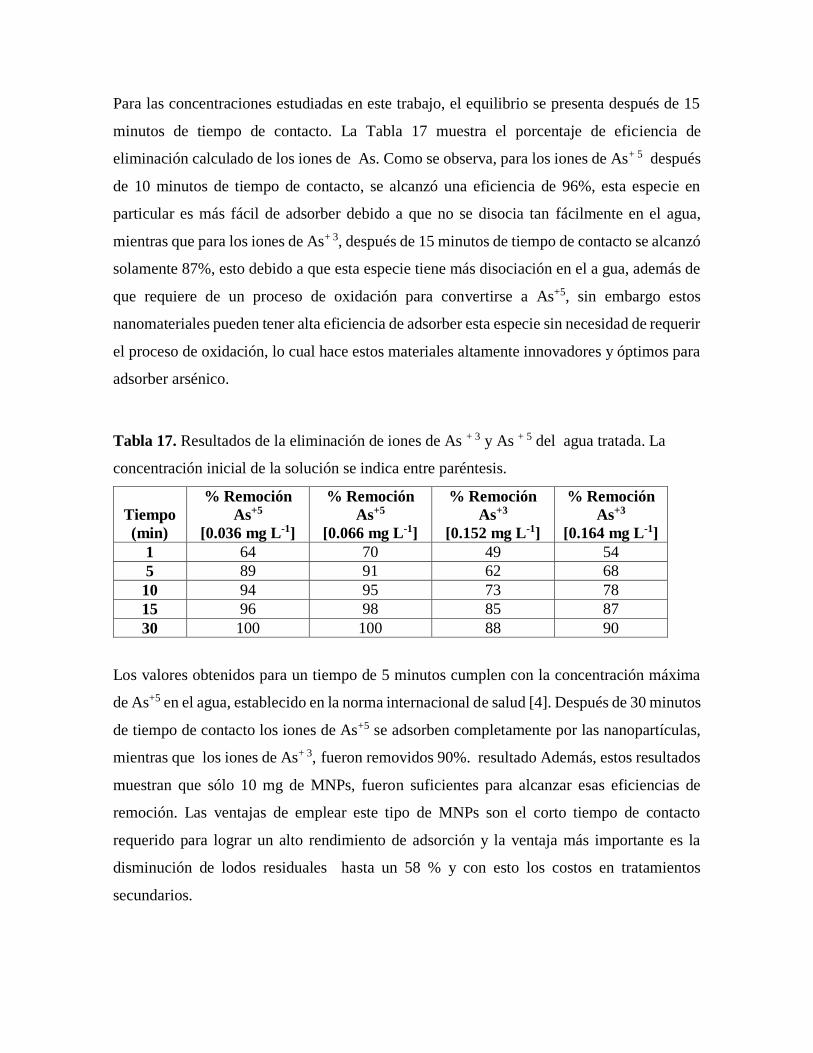
Para las concentraciones estudiadas en este trabajo, el equilibrio se presenta después de 15
minutos de tiempo de contacto. La Tabla 17 muestra el porcentaje de eficiencia de
eliminación calculado de los iones de As. Como se observa, para los iones de As+ 5 después
de 10 minutos de tiempo de contacto, se alcanzó una eficiencia de 96%, esta especie en
particular es más fácil de adsorber debido a que no se disocia tan fácilmente en el agua,
mientras que para los iones de As+ 3, después de 15 minutos de tiempo de contacto se alcanzó
solamente 87%, esto debido a que esta especie tiene más disociación en el a gua, además de
que requiere de un proceso de oxidación para convertirse a As+5, sin embargo estos
nanomateriales pueden tener alta eficiencia de adsorber esta especie sin necesidad de requerir
el proceso de oxidación, lo cual hace estos materiales altamente innovadores y óptimos para
adsorber arsénico.
Tabla 17. Resultados de la eliminación de iones de As + 3 y As + 5 del agua tratada. La
concentración inicial de la solución se indica entre paréntesis.
Tiempo
(min)
% Remoción
As+5
[0.036 mg L-1]
% Remoción
As+5
[0.066 mg L-1]
% Remoción
As+3
[0.152 mg L-1]
% Remoción
As+3
[0.164 mg L-1]
1 64 70 49 54
5 89 91 62 68
10 94 95 73 78
15 96 98 85 87
30 100 100 88 90
Los valores obtenidos para un tiempo de 5 minutos cumplen con la concentración máxima
de As+5 en el agua, establecido en la norma internacional de salud [4]. Después de 30 minutos
de tiempo de contacto los iones de As+5 se adsorben completamente por las nanopartículas,
mientras que los iones de As+ 3, fueron removidos 90%. resultado Además, estos resultados
muestran que sólo 10 mg de MNPs, fueron suficientes para alcanzar esas eficiencias de
remoción. Las ventajas de emplear este tipo de MNPs son el corto tiempo de contacto
requerido para lograr un alto rendimiento de adsorción y la ventaja más importante es la
disminución de lodos residuales hasta un 58 % y con esto los costos en tratamientos
secundarios.

14.2 Isotermas de adsorción
Para demostrar la capacidad de adsorción de las MNPs en el equilibrio se calcularon
isotermas de adsorción (ver tabla 3). La tabla 18 muestra que al aumentar la concentración
inicial tanto en As+5 como en As+3, la capacidad de adsorción aumenta, hecho que puede ser
atribuido a un aumento en la velocidad de transferencia de masa debido al incremento de la
concentración del adsorbato [106]. La interacción soluto-adsorbato fueron calculados por
regresión lineal.
Tabla 18. Condiciones experimentales empleadas en los ensayos de adsorción de contacto
para el cálculo de la capacidad de adsorción de una monocapa.
C0 (mg L-1) V (L) Solución MNPs (g) qe (mg g-1)
As+5 0.036 0.1 0.01 0.355
As+5 0.066 0.1 0.01 0.63
As+3 0.152 0.1 0.01 1.33
As+3 0.164 0.1 0.01 1.38
De acuerdo a los resultados obtenidos por Langmuir en la tabla 19, la capacidad límite de
adsorción para As+5, en concentraciones de 0.036 y 0.066 mg L-1, fue de 0.338 y 0.623 mg g-
1 (Qo), en As+3 se obtuvo 1.052 Y 1.22 mg L-1. El valor de Qo, representa la capacidad de
adsorción de una monocapa, por lo tanto representaría la capacidad máxima del adsorbente,
debido a que Qo es menor que qe, se interpreta que existe una cantidad de adsorbente que
esta adsorbida sobre la monocapa formándose de esta manera, multicapas, lo cual es
representativo de un proceso gobernado por fisisorcion [107]. Los valores obtenidos de la
entalpía de adsorción (KL) son negativos debido a que la superficie del material se encuentra
energéticamente favorable para reaccionar con lo que se encuentre disponible en este caso es
el As, es por ello que la adsorción se da de forma espontánea por lo tanto es un proceso
exotérmico.
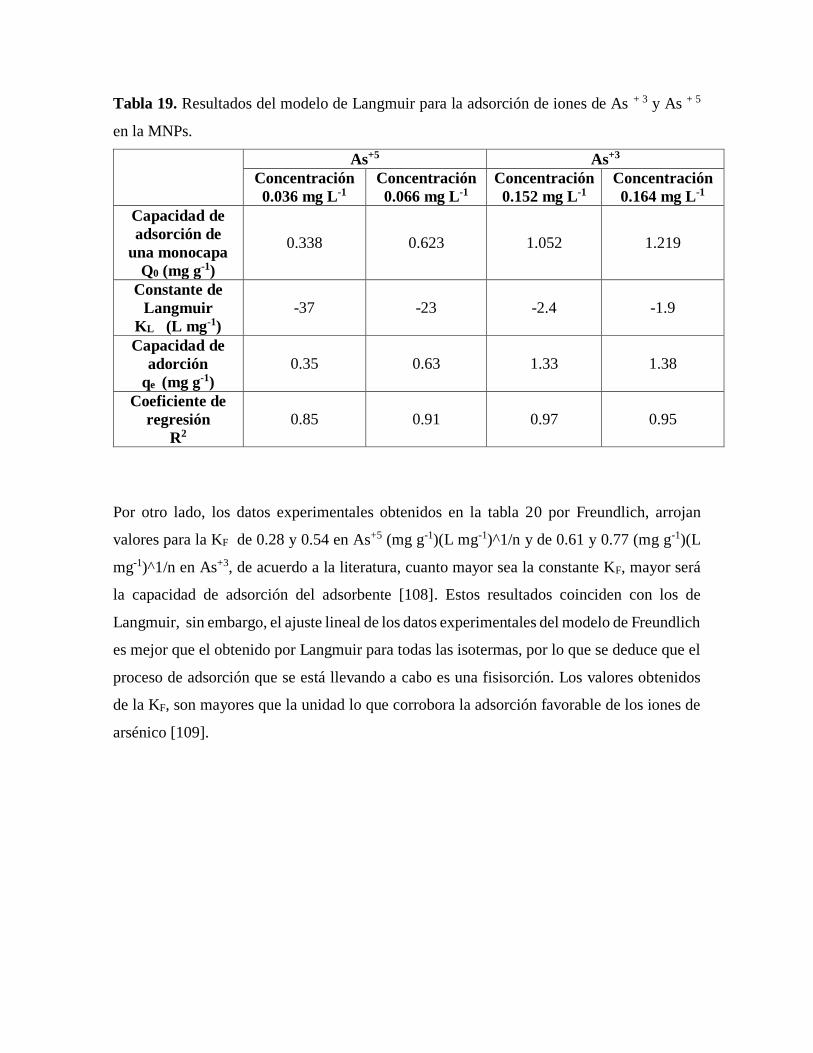
Tabla 19. Resultados del modelo de Langmuir para la adsorción de iones de As + 3 y As + 5
en la MNPs.
As+5 As+3
Concentración
0.036 mg L-1
Concentración
0.066 mg L-1
Concentración
0.152 mg L-1
Concentración
0.164 mg L-1
Capacidad de
adsorción de
una monocapa
Q0 (mg g-1)
0.338 0.623 1.052 1.219
Constante de
Langmuir
KL (L mg-1)
-37 -23 -2.4 -1.9
Capacidad de
adorción
qe (mg g-1)
0.35 0.63 1.33 1.38
Coeficiente de
regresión
R2
0.85 0.91 0.97 0.95
Por otro lado, los datos experimentales obtenidos en la tabla 20 por Freundlich, arrojan
valores para la KF de 0.28 y 0.54 en As+5 (mg g-1)(L mg-1)^1/n y de 0.61 y 0.77 (mg g-1)(L
mg-1)^1/n en As+3, de acuerdo a la literatura, cuanto mayor sea la constante KF, mayor será
la capacidad de adsorción del adsorbente [108]. Estos resultados coinciden con los de
Langmuir, sin embargo, el ajuste lineal de los datos experimentales del modelo de Freundlich
es mejor que el obtenido por Langmuir para todas las isotermas, por lo que se deduce que el
proceso de adsorción que se está llevando a cabo es una fisisorción. Los valores obtenidos
de la KF, son mayores que la unidad lo que corrobora la adsorción favorable de los iones de
arsénico [109].
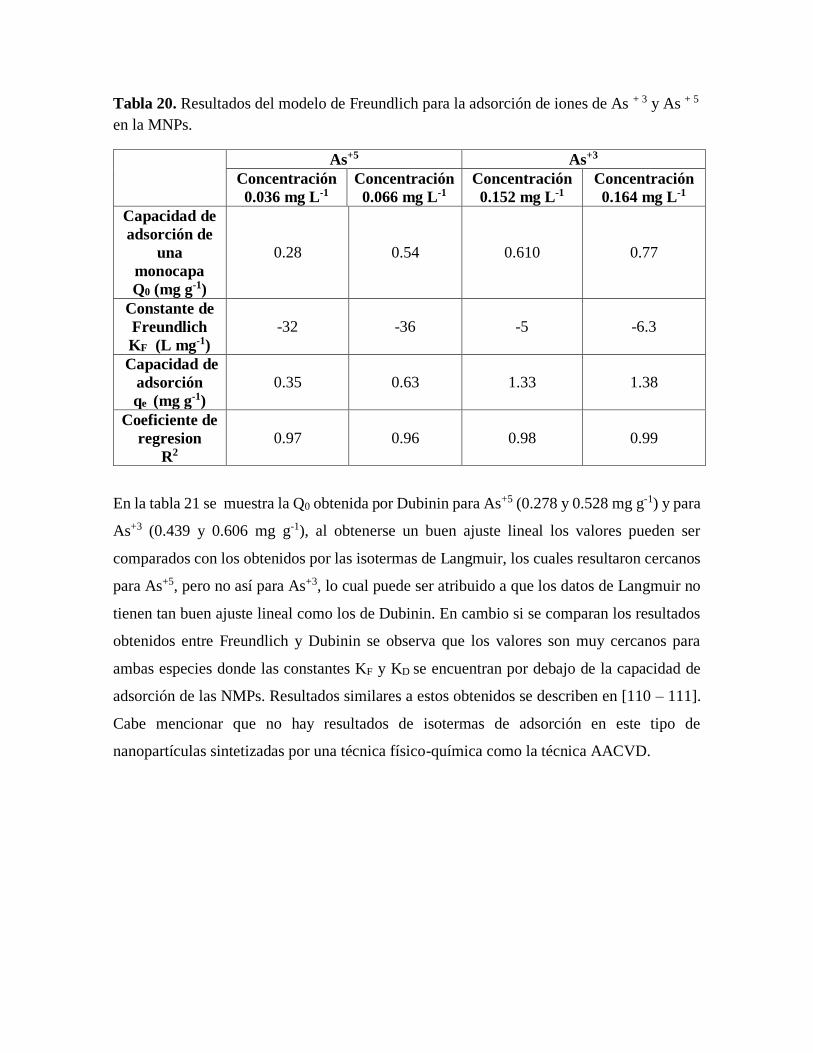
Tabla 20. Resultados del modelo de Freundlich para la adsorción de iones de As + 3 y As + 5
en la MNPs.
As+5 As+3
Concentración
0.036 mg L-1
Concentración
0.066 mg L-1
Concentración
0.152 mg L-1
Concentración
0.164 mg L-1
Capacidad de
adsorción de
una
monocapa
Q0 (mg g-1)
0.28 0.54 0.610 0.77
Constante de
Freundlich
KF (L mg-1)
-32 -36 -5 -6.3
Capacidad de
adsorción
qe (mg g-1)
0.35 0.63 1.33 1.38
Coeficiente de
regresion
R2
0.97 0.96 0.98 0.99
En la tabla 21 se muestra la Q0 obtenida por Dubinin para As+5 (0.278 y 0.528 mg g-1) y para
As+3 (0.439 y 0.606 mg g-1), al obtenerse un buen ajuste lineal los valores pueden ser
comparados con los obtenidos por las isotermas de Langmuir, los cuales resultaron cercanos
para As+5, pero no así para As+3, lo cual puede ser atribuido a que los datos de Langmuir no
tienen tan buen ajuste lineal como los de Dubinin. En cambio si se comparan los resultados
obtenidos entre Freundlich y Dubinin se observa que los valores son muy cercanos para
ambas especies donde las constantes KF y KD se encuentran por debajo de la capacidad de
adsorción de las NMPs. Resultados similares a estos obtenidos se describen en [110 – 111].
Cabe mencionar que no hay resultados de isotermas de adsorción en este tipo de
nanopartículas sintetizadas por una técnica físico-química como la técnica AACVD.
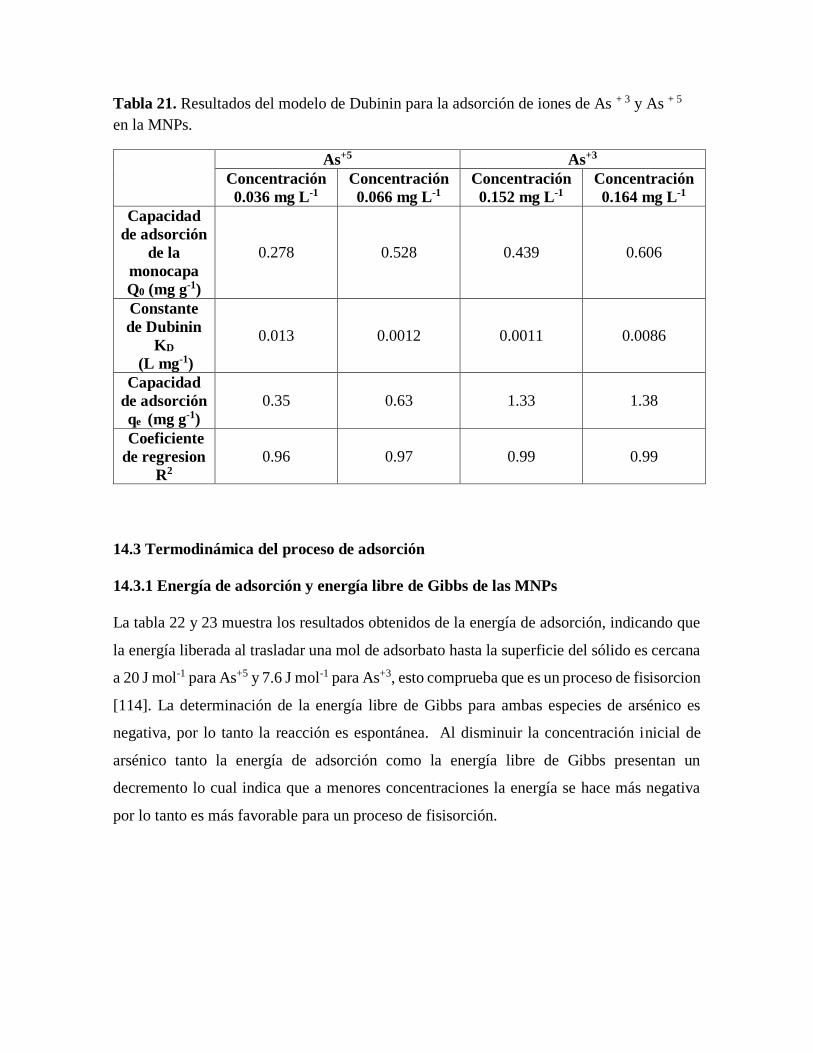
Tabla 21. Resultados del modelo de Dubinin para la adsorción de iones de As + 3 y As + 5
en la MNPs.
As+5 As+3
Concentración
0.036 mg L-1
Concentración
0.066 mg L-1
Concentración
0.152 mg L-1
Concentración
0.164 mg L-1
Capacidad
de adsorción
de la
monocapa
Q0 (mg g-1)
0.278 0.528 0.439 0.606
Constante
de Dubinin
KD
(L mg-1)
0.013 0.0012 0.0011 0.0086
Capacidad
de adsorción
qe (mg g-1)
0.35 0.63 1.33 1.38
Coeficiente
de regresion
R2
0.96 0.97 0.99 0.99
14.3 Termodinámica del proceso de adsorción
14.3.1 Energía de adsorción y energía libre de Gibbs de las MNPs
La tabla 22 y 23 muestra los resultados obtenidos de la energía de adsorción, indicando que
la energía liberada al trasladar una mol de adsorbato hasta la superficie del sólido es cercana
a 20 J mol-1 para As+5 y 7.6 J mol-1 para As+3, esto comprueba que es un proceso de fisisorcion
[114]. La determinación de la energía libre de Gibbs para ambas especies de arsénico es
negativa, por lo tanto la reacción es espontánea. Al disminuir la concentración inicial de
arsénico tanto la energía de adsorción como la energía libre de Gibbs presentan un
decremento lo cual indica que a menores concentraciones la energía se hace más negativa
por lo tanto es más favorable para un proceso de fisisorción.
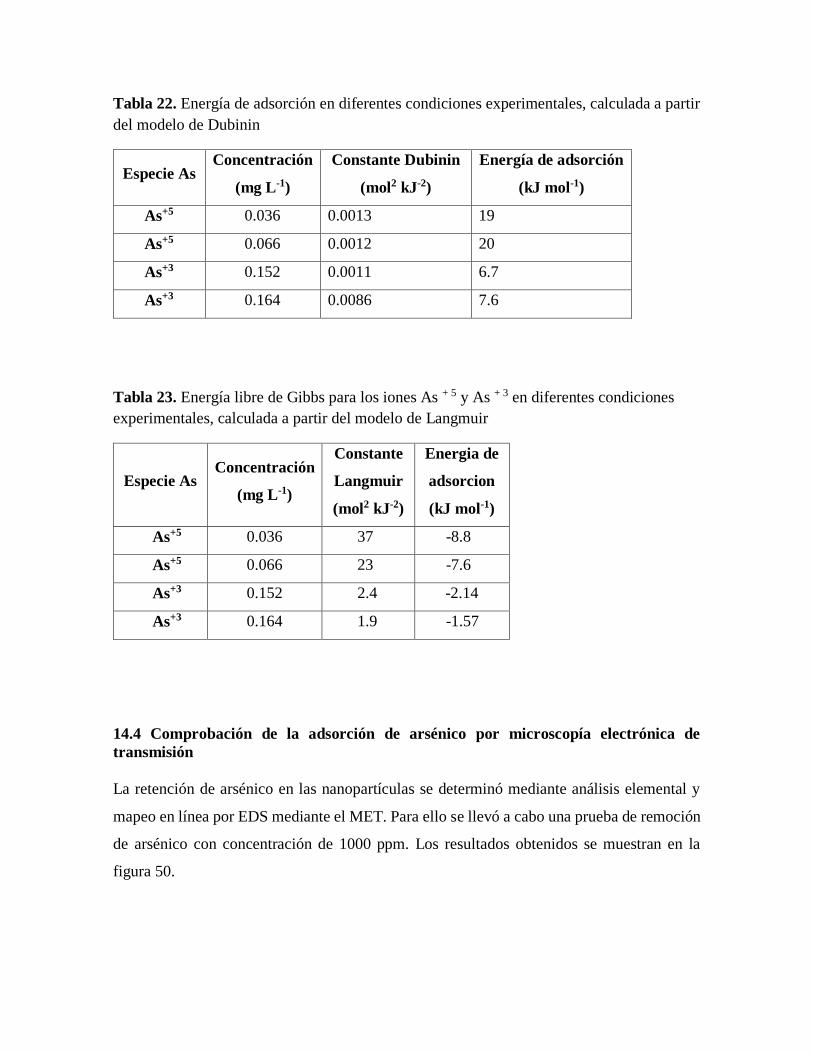
Tabla 22. Energía de adsorción en diferentes condiciones experimentales, calculada a partir
del modelo de Dubinin
Especie As Concentración
(mg L-1)
Constante Dubinin
(mol2 kJ-2)
Energía de adsorción
(kJ mol-1)
As+5 0.036 0.0013 19
As+5 0.066 0.0012 20
As+3 0.152 0.0011 6.7
As+3 0.164 0.0086 7.6
Tabla 23. Energía libre de Gibbs para los iones As + 5 y As + 3 en diferentes condiciones
experimentales, calculada a partir del modelo de Langmuir
Especie As Concentración
(mg L-1)
Constante
Langmuir
(mol2 kJ-2)
Energia de
adsorcion
(kJ mol-1)
As+5 0.036 37 -8.8
As+5 0.066 23 -7.6
As+3 0.152 2.4 -2.14
As+3 0.164 1.9 -1.57
14.4 Comprobación de la adsorción de arsénico por microscopía electrónica de
transmisión
La retención de arsénico en las nanopartículas se determinó mediante análisis elemental y
mapeo en línea por EDS mediante el MET. Para ello se llevó a cabo una prueba de remoción
de arsénico con concentración de 1000 ppm. Los resultados obtenidos se muestran en la
figura 50.
Figura 50. Mapeo en línea por EDS mediante Microscopio electrónico de transmisión
(MET), de las MNPs después de la remoción de arsénico.
En esta figura se demuestra la presencia de arsénico en toda la nanopartícula, donde la mayor
concentración se encuentra en los cristales que forman la costra, más sin embargo, el arsénico
se adsorbe en la superficie de cada cristal, es por ello que en las partes donde existe una
mayor cantidad de cristales se observa más cantidad de As. De igual manera se aprecia la
distribución homogénea, en toda la nanopartícula de los elementos analizados (fierro y
oxígeno), los cuales corresponde a la magnetita.

CONCLUSIONES
1. Etapa de simulación
a) Solid Works- Fluid Works
1.- La zona efectiva de temperatura y velocidad de fluido máxima dentro del reactor tubular,
se obtiene entre los 30 y 60 cm del reactor.
2.-El comportamiento del tiempo de residencia para el reactor tubular de 9 mm es menor que
el reactor de 15 mm, comportamiento inverso a la velocidad de calentamiento del fluido.
b) COMSOL Multiphysics 4.4
1.- Los parámetros de mayor influencia en la morfología final de los materiales sintetizados,
son la temperatura del reactor tubular y el tipo de disolvente utilizado
2.- La morfología general de las nanopartículas obtenidas fueron: Morfología sólidas de 512
nm a temperaturas (<473 K), y huecas porosas de 560 ± 50 nm de diámetro a temperaturas
(≥ 523 K ), con un espesor de corteza de 30 ± 15 nm utilizando metanol como solvente.
3.- Las nanopartículas simuladas con agua como disolvente presentaron morfología solida
con un tamaño de 480 ± 20, utilizando una distribución de temperatura de 373 a 723 K.
2. Etapa experimental
1.-La morfología de las MNPs obtenidas fueron: esferas sólidas de 345 ± 20 nm a
temperaturas (<473 K), huecas porosas de 300 ± 78 nm de diámetro y 19 ± 5 nm de espesor
de la corteza a temperaturas (≥ 523 K ), utilizando metanol como disolvente, presentando
un área superficial de 134 m2 g-1.
2.-Al utilizar agua como disolvente la morfología obtenida fue sólida en una distribución de
temperaturas de 373 723 K, presentando un diámetro de alrededor de 400 ± 56 nm con
presencia de cristales de 10 ± 2 nm, para temperaturas 673 y 723 K.
La energía de activación obtenida para llevar a cabo la reacción fue de 227 kJ mol-1

3.-Etapa de aplicación
1.- El tiempo de eficiencia máxima de remoción es de 10 minutos para ambas especies porque
es donde se llega al equilibrio teniendo un porcentaje de eficiencia de remoción de 95% As+5
y de 80 % de eficiencia para As +3.
2. El modelo que mejor ajuste lineal presentó fue el de Freundlich, cuyos resultados son muy
similares a los obtenidos por Dubinin, por lo tanto se sabe que el tipo de adsorción que se
lleva a cabo es una fisisorción.

PERSPECTIVAS
1. Continuar mejorando el sistema de recolección
2. Iniciar con el estudio de columnas de adsorción de MNPs.
3. Iniciar con el desarrollo de nanoestructuras de óxido de fierro con elementos afines a otros
4. Contaminantes como el aluminio, magnesio y/o titanio para adsorber flúor y arsénico del
agua.

APORTACIONES
1.- No hay reportes sobre el estudio de formación de magnetita por la técnica AACVD, como
resultado de esto se tiene la solicitud de una patente.
2.-Al obtener diferentes morfologías de MNPs se abre su campo de aplicaciones los cuales
pueden ser en electrónica, biomateriales y magnetismo.
3.-El uso de sofwares en los modelos teóricos ayuda a predecir las condiciones de síntesis
para la obtención de materiales con una microestructura deseada.
4.- Los parámetros de Arrhenius para la formación de magnetita son reportados por primera
vez, ya que no se encuentran en la literatura y se aporta la ruta química de la formación de
magnetita por la técnica AACVD.
5.- La eficiencia de adsorción se da sin la necesidad de agregar agentes oxidantes, se obtiene
un alto porcentaje de remoción en poco tiempo para As+3 y As+5 minimizando costos en el
tratamiento de agua.

REFERENCIAS
[1] Peter Ravenscroft, H. Brammer, K. S. Richards, Arsenic pollution: a global synthesis.
Synthesis, Conclusions and Recomendations, Wiley Blackwell, (2009): 500.
[2] Marta I. Litter, María Teresa Alarcón-Herrera, María J. Arenas, María A. Armienta,
Marta Avilés, Roberto E. Cáceres, Henrique Nery Cipriani, Lorena Cornejo, Luiz E. Dias,
Alicia Fernández Cirelli, Elsa M. Farfán, Sofía Garrido, Liliana Lorenzo, María. E. Morgada,
Mario A. Olmos-Márquez, Alejo Pérez-Carrera, Small-scale and household methods to
remove arsenic from water for drinking purposes in Latin America, Science of the Total
Environment, (2012): 429: 107–122.
[3] Jochen Bundschuh, P. Birkle, P. Bhattacharya, J. Matschullat, A. B. Mukherjee, M. A.
Armienta, Occurrence health effects and remediation of arsenic in groundwaters of America
Latina, Natural Arsenic in Groundwater of Latin America, (2006): 20-24.
[4] Norma Oficial Mexicana NOM-127-SSA1-1994, Salud ambiental. Agua para uso y
consumo humano. Límites permisibles de calidad y tratamientos a que debe someterse el
agua para su potabilización.
[5] Morgada Maria E., Levy Ivana K., Salomone Vanesa, Silvia S. Farias, Litter Marta I.,
Lopez Gerardo, Arsenic (V) removal with nanoparticulate zerovalent iron: Effect of UV light
and humic acids, Catalysis Today, (2009): 143: 261-268.
[6] Marta I. Litter, Maria E. Morgada, Jochen Bundschuh, Possible treatments for arsenic
removal in Latin American waters for human consumption, Environmental Pollution, (2010):
158: 1105–1118.
[7] Mustafa Tuzen, Ahmet Sarı, Durali Mendil, Ozgur Dogan Uluozlu, Mustafa Soylak,
Mehmet Dogan, Characterization of biosorption process of As(III) on green algae Ulothrix
cylindricum, Journal of Hazardous Materials, (2009): 165: 566–572.
[8] C. Salazar, M. Villalobos, Ma. Rivas, J. Arenas, J. Alcaraz, M. Gutiérrez,
Characterization and surface reactivity of natural and synthetic magnetites, Chemical
Geology, (2013): 347, 233–245.
[9] Y. Mamindy, C. Hurel, N. Marmier, M. Roméo, Arsenic (V) adsorption from aqueous
solution onto goethite, hematite, magnetite and zero-valent iron: Effects of pH, Concentration
and reversibility, Desalination, (2011): 281, 93–99.
[10] P. Sarkar, P. Pal, D. Bhattacharyay, S. Banerjee, Removal of arsenicfrom drinking water
by ferric hydroxide microcapsule-loaded alginate beads in packed adsorption column,
Journal of Environmental Science and Health Part A, (2010): 45, 1750-1757.

[11] Panigrahi, S: Praharaj, S: Basu, S: Ghost, S. K.: Jana, S.: Pande, S: Vo Dinh, T: Jiang,
H and pal, T. Self, Assembly of silver nanoparticles synthesis, stabilization, optical properties
and application in surface enhanced raman scattering, Journal of Physical chemistry, (2006):
110, 13436-13444.
[12] Liccioni José Rafael, Consideraciones sobre el fierro, Ed: Ministerio del Poder Popular
para las industrias Básicas y Minería, (1997): 41.
[13] D´Ambrosio María Cristina, “Evaluación y selección de tecnologías disponibles para
remoción de arsénico”, IV Congreso Hidrogeológico Argentino, (2005).
[14] M. Sir, M. Podhola, T. Patocka, Z. Honzajkova, P. Kocurek, M. Kubal, M. Kuras, The
effect of humic acids on the reverse osmosis treatment of hazardous landfill leachate, Journal
of Hazardous Materials, (2012): 207–208: 86–90.
[15] David J. Grimshaw, Nanotecnología para obtener agua limpia: hechos y cifras. Red de
Ciencia y Desarrollo Sci Dev.Net. Gales, Inglaterra, (2009).
[16] Olesya Myakonkaya, Clement Guibert, Julian Eastoe and Isabelle Grillo. Recovery of
Nanoparticles Made Easy, Langmuir, (2010): 26 (6): 3794–3797.
[17] Myakonkaya, Olesya, Eastoe Julian, Lowenergy methods of phase separation in
colloidal dispersions and microemulsions, Advances in Colloid and Interface Science,
(2009): 149: 39-46.
[18] Monárrez-Cordero BE, Amézaga-Madrid P, Antúnez-Flores W, Leyva-Porras C, Pizá-
Ruiz P, Miki-Yoshida M, Highly efficient removal of arsenic metal ions with high superficial
area hollow magnetite nanoparticles synthetized by AACVD method, Journal of Alloys and
Compounds, (2014): 586, 520-525.
[19] P.G. Hernández-Salcedo, P. Amézaga-Madrid, B.E. Monárrez-Cordero, W. Antúnez-
Flores, P. Pizá-Ruiz, C. Leyva-Porras, C. Ornelas-Gutiérrez, M. Miki-Yoshida, Theoretical
and experimental influence of aerosol assisted CVD parameters on the microstructural
properties of magnetite nanoparticles and their response on the removal efficiency of arsenic,
J. Alloy Compd., (2015): 643, 287–296.
[20] J. Mayo, C. Yavuz, S. Yean, L. Cong, H. Shipley, W. Yu, J. Falkner, A. Kan, M.
Tomson, V.L. Colvin, The effect of nanocrystalline magnetite size on arsenic removal,
Science and Technology of Advanced Materials, (2007): 8, 71-75.
[21] Z. Huang, F. Tang, Preparation, structure, and magnetic properties of mesoporous
magnetite hollow spheres, Journal of Colloid and Interface Science,(2005): 281, 432-436.
[22] Y.F. Shen, J. Tang, Z.H. Nie, Y.D. Wang, Y. Ren, L. Zuo, Tailoring size and structural
distortion of Fe3O4 nanoparticles for the purification of contaminated water, Bioresource
Technology, (2009): 100, 4139-4146.

[23] Z.M. Cui, L.Y. Jiang, W.G. Song, Y. G.Guo, High-yield gas−liquid interfacial synthesis
of highly dispersed Fe3O4 nanocrystals and their application in lithium-ion batteries, Chem.
Mater., (2009): 21, 1162–1166.
[24] A. Figuerola, R. Di Corato, L. Manna, T. Pellegrino, Review From iron oxide
nanoparticles towards advanced iron based inorganic materials designed for medical
applications, Pharmacological Research, (2010): 62, 126-143.
[25] C. Fan, W.Gao, Z. Chen, H. Fan, M. Li, F. Deng, Z. Chen, Tumor selectivity of stealth
multi-functionalized superparamagnetic iron oxide nanoparticles, International Journal of
Pharmaceutics, (2011): 404, 180-190.
[26] J. Murbe, A. Rechtenbach, J. Topfer, Synthesis and physical characterization of
magnetite nanoparticles for biomedical applications, Materials Chemistry and Physics,
(2008): 110 , 426-433.
[27] S. Guo, D. Li, L. Zhang, J. Li, E. Wang, Monodisperse mesoporous superparamagnetic
single-crystal magnetite nanoparticles for drug delivery, Biomaterials, (2009): 30, 1881-
1889.
[28] B. An, Q. Liang, D. Zhao, Removal of arsenic (V) from spent ion exchange brine using
a new class of starch-bridged magnetite nanoparticles, Water Research, (2011): 45, 1961-
1972.
[29] Fernández Turiel J. L., Galindo Griselda, Parada M. A., D. Gimeno, M. García Valles,
J. Saavedra, Estado actual del conocimiento sobre el arsénico en el agua de Argentina y Chile:
Origen, Movilidad y Tratamiento, IV Congreso Hidrogeológico Argentino y II Seminario
Hispano Latinoamericano sobre temas actuales en hidrológica. Río Cuarto, Argentina,
(2005).
[30] Martínez Luís Dante y Gasquez José Antonio, Determinación del Arsénico en Aguas:
Diferentes Técnicas y Metodologías, IV Congreso Hidrogeológico Argentino, (2005).
[31] Ioannis A. Katsoyiannis, Anastasios I. Zouboulis, Applications of biological procces for
the removal of arsenic from groundwaters, Water research, (2004): 38, 17-26.
[32]Bocanegra Olga C., Bocanegra Martínez Emilia, Álvarez Amilicar, Arsénico en aguas
subterráneas su impacto en la salud, Groundwater and Hurman Development, (2002): 987-
544.
[33] Petkova Simeonova Verguinia, Rivera Huerta Lourdes, Pina Soberanis Maritn, Aviles
Flores Martha y Pérez Castrejon Sara, Evaluación de diversos minerales para la remoción de
arsénico de agua para consumo humano, Instituto Mexicano de tecnología del agua, (1998).
[34] María Socorro Espino-Valdés, Yaravi Barrera-Prieto, Eduardo Herrera-Peraza, Arsenic
presence in North section of Meoqui-Delicias, Aquifero del estado de Chihuahua, Mexico,
Tecnociencia Chihuahua. (2009): Vol. III, No. 1.

[35] Paraguay F., Morales J., Estrada W., Andrade E., Miki Y.M, Influence of Al, In, Cu, Fe
and Sn dopants in the microestructure of Zn oxide obtained by spray pyrolysis, Thin solid
Films, (2000): 366 (1-2), 16-27.
[36] Rodríguez Pineda J., Alfredo Reyes Cortes., Ignacio Hernández Ramírez, Oscar Cruz
medina, Rodybeth y Muñoz, Robles Carlos, Caracterización hidrogeoquimica de la
contaminación de agua subterránea por arsénico en Aldama. Chihuahua Instituto de ecología
y universidad Autónoma de Chihuahua (2004).
[37] Klaassen C, Casarett and Doull's toxicology: the basic science of poisons, New York:
MacMillan Publishing Company, 5th edition, (1996).
[38] Koren H, Illustrated dictionary of environmental health and occupational safety, New
York: CRC Lewis Publishers, (1996)
[39] Maiti Ahijit, Das Gupta sunando, Kumar Basu Jayant, Sirshendu De Adsorption of
arsenite using natural laterite as adsorbent, Separation and purification,(2007): 55, 350-359.
[40] L. Feenstra, L. Vasak, J. Griffioen, Fluoride in groundwater: Overview and evaluation
of removal methods, International Groundwater Resources Assessment Centre INGRAC
Report nr. SP, (2007).
[41] F. Leccabe, R. Panizzieri, Garcia, N. Suarez, J.L. Sánchez, O. Ares y X.R. Hua.
“Magnetic and Mossbauer Study ofRare-Earth-Substituted M-, W- and X- Type Hexagonal
Ferrites”, Journal of Materials Science, (1990): 25, 2765-2770.
[42] J.S. Bradley, B. Tesche, W. Busser, M. Masse, R.T. Reetz, Surface Spectroscopic Study
of the Stabilization Mechanism for Shape-Selectively Synthesized Nanostructured
Transition, Metal Colloids, J Ame Chem Soc , (2000), 122: 4631-4636.
[43] T. Sugimoto, Y.S. Wang, Mechanism of the Shape and Structure Control of
Monodispersed α-Fe2O3Particles by Sulfate Ions, J Colloid Interf Sci, (1998): 207, 137-149.
[44] Rocío Mellado, Nanotecnología de Agua y Saneamiento, Perú: Memorias del seminario
y taller, Consejo Nacional de Ciencia, Tecnología e Innovación Tecnológica, CONCYTEC
(2010).
[45] Robert Pini, Rensselaer Polytechnic Institute (N.Y. USA) y Banaras Hindu University.
Efficient filters produced from carbon nanotubes for remove nano-scale germs from water,
heavy metals and hydrocarbons from petroleum. (2004).
[46] K. Khider, D.E. Akretche, A. Larbot, Purification of water effluent from a milk factory
by ultrafiltration using Algerian clay support, Desalination, (2004): 167, 147-151.
[47] Zhonghou Xu, Xiaoguang Meng, Size effects of nanocrystalline TiO2 on As(V) and
As(III) adsorption and As(III) photooxidation, Journal of Hazardous Materials, (2009): 168,
2-3: 747-752.

[48] Cafer T. Yavuz, J. T. Mayo, William W. Yu, Arjun Prakash, Joshua C. Falkner, Sujin
Yean, Lili Cong, Heather J. Shipley, Amy Kan, Mason Tomson, Douglas Natelson, Vicki L.
Colvin, Low-Field Magnetic Separation of Monodisperse Fe3O4 Nanocrystals, Science,
(2016): 314, Issue 5801: 964-967.
[49] Zheng, Gang; et al. Porphysome nanovesicles generated by porphyrin bilayers for use
as multimodal biophotonic contrast agents, Nature materials, Nature Materials, (2011): 10,
324-332. [50] T. Tuutijärvia, J.Lu, M. Sillanpää, G. Chen. As(V) adsorption on maghemite
nanoparticles. Journal of Hazardous Materials, (2009): 166 Issues 2-3, 1415-1420.
[51]Saidur Rahman Chowdhury, Ernest K. Yanful, Arsenic and chromium removal by mixed
magnetite-maghemite nanoparticles and the effect of phosphate on removal, Journal of
Environmental Management, (2010): 91 Issue 11, 2238-2247.
[52] Y.C. Sharma, V. Srivastava, V.K. Singh, S.N. Kaul, C.H.Weng, Nano-adsorbents for
the removal of metallic pollutants from water and wastewater, Environ. Technology,
(2009): 30, Issue 6, 583–609.
[53] Y. Li, J. Wang, Y. Zhao, Z. Luan, Research on magnetic seeding flocculation for arsenic
removal by superconducting magnetic separation, Separation and Purification Technology,
(2010), 73, Issue 2, 264-270.
[54] I. Ko, A. David, J. Kim, K. Kim, Arsenic Removal by a Colloidal Iron Oxide Coated
Sand. Journal of Environmental Engineering, (2007), 133, Issue 9, 891-898.
[55] J. Giménez, M. Martínez, J. de Pablo, M. Rovira, L. Duro, Arsenic adsorption onto
natural hematite, magnetite, and goethite, Journal of Hazardous Materials, (2007): 141, Issue
3, 575-580.
[56] M.Hua, S. Zhang, B. Pan, W. Zhang, L. Lv, Q. Zhang, Review, Heavy metal removal
from water/wastewater by nanosized metal oxides: Areview. Journal of Hazardous Materials,
(2012): 211-212, Pp 317-331.
[57] M.E. Compean-Jasso, F. Ruiz, J.R. Martinez, A. Herrera-Gomez, Magnetic properties
of magnetite nanoparticles synthesized by forced hydrolysis, Materials Letters, (2008) 62,
Issue 27, 4248-4250.
[58] X. Li, B. Zhang, C. Ju, X. Han, Y. Du, P. Xu, Morphology-controlled synthesis and
electromagnetic properties of porous Fe3O4 nanostructures from iron alkoxide precursors, J.
Phys. Chem. C., (2011): 115, 12350–12357.
[59] A. Andrade, M. Valente, J. Ferreira, J. Fabris, Preparation of sizecontrolled
nanoparticles of magnetite, Journal of Magnetism and Magnetic Materials, (2012): 324 Issue
10, 1753-1757.

[60] A. Reza, M. Al-Sadat, Efficient separation of heavy metal cations by anchoring
polyacrylic acid on superparamagnetic magnetite nanoparticles through surface
modification, Chemical Engineering Journal, (2010): 159 Issues 1-3, 264–271.
[61] G. Goya, T. Berquó, F. Fonseca, Static and dynamic magnetic properties of spherical
magnetite nanoparticles. (2003). Journal of Applied Physics, Volumen 94, Numero 5,
Pp3520-3528.
[62] S. Yean, L. Cong, Effect of magnetite particle size on adsorption and desorption of
arsenite and arsenate, Journal Mater. Res., (2005):20, Issue 12, 3255-3264.
[63] M. Furlan, J. Kluge, M. Mazzotti, M. Lattuada, Preparation of biocompatible magnetite-
PLGA composite nanoparticles using supercritical fluid extraction of emulsions, J. of
Supercritical Fluids, (2010): 54, Issue 3, 348-356.
[64] A.B. Chin, I.I. Yaacob, Synthesis and characterization of magnetic iron oxide
nanoparticles via w/o microemulsion and Massart’s procedure, J. Mater. Process. Technol,
(2007): 191, Isues 1-3, 235-237.
[65] M. Kimata, D. Nakagawa, M. Hasegawa, Preparation of monodisperse magnetic
particles by hidrolysis of iron alkoxide, Powder Technol, (2003) 132, Issues 2-3, 132, 112-
118.
[66] D. Maity, D.C. Agrawal, Synthesis of iron nanoparticles under oxidizing environment
and their stabilization in aqueous and non-aqueous media, J. of Magnetism and Magnetic
Materials, (2007): 308 Issue 1, 46-55.
[67] C. Albornoz, S.E. Jacobo, Preparation of a biocompatible magnetic film from an
aqueous ferrofluid, J. Magn. Mater, (2006):305, Issue 305 12-15.
[68] H.E. Esparza-Ponce, A. Reyes-Rojas, W. Antúnez-Flores, M. Miki Yoshida, Synthesis
and characterization of spherical calcia stabilized zirconia nano-powders obtained by spray
pyrolysis, Mater Sci Eng, (2003): 343, Issues 1-2, 82-88.
[69] P. Tartaj, M.P. Morales, T. Gonzalez, S. Veintenillas, C.J. Serna, Advances in magnetic
nanoparticles for biotechnology applications, J Magn Magn Mater, (2004): 290-291 Part 1,
28-34.
[70] D.K. Kim, M. Mikhaylova, F.H. Wang, J.Kehr, B. Bjelke, Y. Zhang, T. Tsakalakos,M.
Muhammed, Starch-coated superparamagnetic nanoparticles as MR contrast agents, Chem
Mater, (2003): 15, 4343-4351.
[71] G. Jayanthi, S. Zhang, G, Messing, Modeling of solid particle formation during
solution aerosol thermolysis: the evaporation stage, Aerosol Science and Technology,
(1993) 19, Issue 4 , 478-490.

[72] S.C. Zhang, G.L. Messing, M. Borden, Synthesis of solid, spherical zirconia particles
by spray pyrolysis, Journal of the American Ceramic Society, (1990):73, Issue 1, 61–67.
[73] I. Wuled, T. Hata, F. Iskandar, M. Lunden, K. Okuyama, An experimental and modeling
investigation of particle production by spray pyrolysis using a laminar flow aerosol,
ReactorJournal of Material Research, (1993): 15, Issue 03, 733-743.
[74] M.J. Yacamán, J. Reyes, Consejo Nacional de Ciencia y Tecnología. Fondo de Cultura
Económica, (1995): 2-7.
[75] David B. Williams, C. Barry Carter, Transmission Electron Microscopy A Textbook for
Materials Science, 3th Edition. (2009).
[76] Matesanz, E. DRXP en la práctica. Sobre la difracción de rayos x por el método de
polvos (DRXP) y sus aplicaciones. (2010) http://drxp.info/brevisima-introduccion-a-la-
drxp/.
[77] P. G. Hernández-Salcedo, P. Amézaga-Madrid, B. E. Monárrez-Cordero, W. Antúnez-
Flores, P. Pizá-Ruiz, C. Leyva-Porras, C. Ornelas-Gutiérrez, M. Miki-Yoshida, Theoretical
and experimental influence of aerosol assisted CVD parameters on the microstructural
properties of magnetite nanoparticles and their response on the removal efficiency of arsenic.
Journal of Alloys and Compounds, (2015): 643, S287–S296.
[78] S. Brunauer, P.H. Emmett, E. Teller, Adsorption of gases in multimolecular layers, J.
Am. Chem. (1938): 309.
[79] K. S. W. Sing, D.H. Everett, R.A.W. Haul. Reporting physisorption data for gas/solid
systems, Pure Appl. Chem., (1985), 57, Issue 4, 603-619.
[80] Miriam López Paraguay, Adsorción de arsénico y fluoruros en nanopartículas y su
posterior separación del agua tratada (2013).
[81] Walter J. Weber Jr. y Jack A. Borchardt, Control de la calidad del Agua, Procesos
fisicoquímicos, Editorial Reverté S.A. Michigan USA, (2003): 210-262.
[82] López Acevedo Marta y Porta Casanellas Jaime, Estudio experimental de la adsorción,
Edafología para la agricultura y el medio ambiente, Mundi-Prensa Libros, 3era edición.
Madrid, España, (2004): 243-245.
[83] Rodríguez Vidal Francisco Javier, Proceso de filtración - adsorción y efecto del ozono,
Procesos de potabilización del agua e influencia del tratamiento de ozonización, Ediciones
Díaz de Santos. Burgos España, (2003): 196.
[84] A. Blanco Flores, G. Autie Castro, D. Rodríguez Montes de Oca, S. Ricardo Paez, R.
López Cordero, M. Autie Pérez. Características superficiales de un vidrio volcánico cubano
y remoción de Cu+2 desde disoluciones acuosas, Revista Latinoamericana de Recursos
Naturales, (2009): 5(3): 238-252.

[85] Gabriela Huamán Pino, Mauricio Leonardo Torem. Biosorción de metales pesados
contenidos en efluentes utilizando biomasa orgánica, Perumin, (2009): 29.
[86] Sanjoy Kumar Maji, Anjali Pal, Tarasankar Pal. Arsenic removal from real-life
groundwater by adsorption on laterite soil, Journal of Hazardous Materials, (2008): 151: 811–
820.
[87] Indra Deo Mall, Vimal Chandra Srivastava, Nitin Kumar Agarwal, Indra Mani Mishra,
Removal of congo red from aqueous solution by bagasse fly ash and activated carbon:
Kinetics study and equilibrium isotherm analyses, Chemosphere, (2005): 61: 492-501.
[88] Y. Hsuan-Fu, L. Wen-Haur. Evaporation of solution droplets in spray pyrolysis. J. Heat
Mass Transf, (1998): 41: 993-1001.
[89] J. Vicente, J. Pinto, J. Menezes, F. Gaspar, Fundamental analysis of particle formation
in spray drying, Powder Technol, (2013): 247 1-7.
[90] H.S. Kang, Y.C. Kang, H.D. Park, Y.G. Shul, Morphology of particles prepared by spray
pyrolysis from organic precursor solution, Mater. Lett. (2003): 57 1288-1294.
[91] Y. Xiong, T. Kodas, Droplet Evaporation and Solute Precipitation during Spray
Pyrolysis, J. Aerosol Sci, (1993): 24 893-908.
[92] J. van der Lijn, Simulation of heat and mass transfer in spray drying, Agricultural Res.
(1976). Rep. 845 72-85.
[93] Levenspiel, Chemical Reaction Engineering, eighth ed., Reverte, Barcelona. (1986). pp.
3–44.
[94] H. Tanaka, Thermal analysis and kinetics of solid state reactions, Thermochim. Acta,
(1995), 267, 29–44.
[95] J. Opfermann, E. Kaisersberger, An advantageous variant of the Ozawa–Flynn–Wall.
Anal. Thermchim Acta , (1992): 203, 167–175.
[96] J.H. Flynn, L.A. Wall, A. Quick, Direct method for the determination of activation
energy from thermogravimetric data, Polym. Lett., (1966): 4 323–328.
[97] C. Yin, Modelling of heating and evaporation of n-Heptane droplets, Towards a generic
model for fuel droplet/particle conversion, Fuel, (2015): 141, 64–73.
[98] Oberman, G.J. Farell, W. Troy, Modelling of the evaporation of a droplet suspended in
a binary atmosphere, J. Heat Mass Transfer, (2016): 92, 381-393.

[99] Gábor Járvás, János Kontos, Jeno Hancsók, András Dallos, Modeling ethanol–blended
gasoline droplet evaporation using COSMO-RS theory and computation fluid dynamics,
International Journal of Heat and Mass Transfer, (2015): 84, 1019-1029.
[100] Volkov, G.V. Kuznetsov, P.A. Strizhak, Influence of droplet concentration on
evaporation in a high-temperature gas R.S. International Journal of Heat and Mass Transfer,
(2016): 96, 20–28.
[101] N. Reuge, J. Dexpert-Ghys, M. Verelst, B. Caussat, Y2O3:Eu micronic particles
synthesised by spray pyrolysis: Global modelling and optimisation of the evaporation stage.
Chem. Eng. Process, (2008): 47, 731–743.
[102] M. Shusser, T. Vindspoll, D. Weihs, Kinetic theory analysis of explosive boiling of to
liquid droplet, Fluid Dyn, (2000): 27, 353-365.
[103] B. Monárrez-Cordero, P. Amézaga-Madrid, P.G. Hernández-salcedo, W. Antúnez-
flores, C. Leyva-Porras, M. Miki-Yoshida, Theoretical and experimental analysis of
the aerosol assisted CVD synthesis of magnetite hollow nanoparticles, J. Alloy. Compd
(2014); 615, S328-S334.
[104] J.H. Flynn, L.A. Wall, A quick, Direct method for the determination of activation
energy from thermogravimetric data, Polymer let, (1966): 4, 323–328.
[105] Vadivelan V, Kumar KV, Equilibrium, kinetics, mechanism, and process design for
the sorption of methylene blue onto rice husk, Journal of colloid and interface science,
(2015): 286, 90-100.
[106] Hassan K, Heavy metal removal from water by magnetite nanorods, Chemical
Engineering Journal, (2013): 219, 209–216.
[107]. Rodríguez VF, Proceso de filtración - adsorción y efecto del ozono, Procesos de
potabilización del agua e influencia del tratamiento de ozonización. 1er ed. Díaz de Santos.
(2006):196.
[108] Mandal S, Sahu MK, Giri AK, Patel RK, Adsorption studies of chromium (VI) removal
from water by lanthanum diethanolamine hybrid material, Environmental Technology,
(2013): 35:817–832.
[109] Mandal S, Mahapatra S, Patel RK. Neuro fuzzy approach for arsenic (III) and
chromium (VI) removal from water, Journal of Water Process Engineering, (2015): 5:58–75.
[110] Bhaumik M, Noubactep C, Kumar GV , McCrindle R, Maity A. Polyaniline/Fe0
composite nanofibers: An excellent adsorbent for the removal of arsenic from aqueous
solutions, Chemical Engineering Journal, (2015): 271:135–146.

[111] Rahman CS, Yanful EK, Arsenic and chromium removal by mixed magnetite-
maghemite nanoparticles and the effect of phosphate on removal, Journal of Environmental
Management, (2010): 21:2238-2247.
[112] Hokkanen S, Repo E, Lou S, Sillanpa M, Removal of arsenic (V) by magnetic
nanoparticle activated microfibrillated cellulose, Chemical Engineering Journal, (2015):
260:886–894.
ANEXO 1
Parámetros utilizados en la simulación en COMSOL Multiphysics
Parámetro Expresión Descripción
long 0.85[m] Longitud del reactor
R 0.0035[m] Radio del tubo del reactor
vsol 0.1[L] Volumen de la solución
Q 0.254[l/min] Flujo del gas de arrastre
Qm 0.011[mol/min] Flujo molar del gas de arrastre
C0 0.05[mol/l] Concentración inicial de soluto
Fneb 0.0006[L/min] Flujo solución nebulizada
Rp0 0.0000011[m] Radio inicial de gota
volg (4/3)*pi*Rp0^3 Volumen de la gota
N0 Fneb/volg/Q Numero de gotas por metro
cubico
T0 303[K] Temperatura inicial de la gota
Tnom 450[degC] Temperatura nominal horno °C
Maire 28.97[g/mol] Peso molecular del aire
Mg 32.04[g/mol] Peso molecular del solvente
rhoCH3OH 0.791[kg/L] Densidad del metanol
mCH3OH rhoCH3OH*vsol Masa del metanol 100 ml
rhosoluto 3.16[g/cm^3] Densidad soluto FeCl2
Ms 198.81[g/mol] Peso molecular del soluto
msol 0.994[g] Masa soluto en 100 ml de
metanol
rhogota (vsol*rhoCH3OH + msol)/vsol Densidad total de gota
(solvente +soluto)
Parámetro Expresión Descripción
m0 rhogota*volg Masa inicial de gota
z0 C0*m0/rhogota*Ms Masa del soluto en la gota
molFeCl msol/Ms Moles de FeCl en 100 ml
molCH3OH mCH3OH/Mg Moles de CH3OH en 100 ml
moltot molFeCl + molCH3OH Moles totales en 100 ml
fraCH3OH molCH3OH/moltot Fracción molar del metanol
fraFeCl molFeCl/moltot Fracción molar del FeCl2
Cpa 0.53[kJ/K/kg] Calor especifico del aire
húmedo
CpCH3OH 2.6[kJ/K/kg] Calor especifico del metanol
CpFeCl 0.156[kJ/K/kg] Calor especifico de la sal
Cp fraCH3OH*CpCH3OH +
fraFeCl*CpFeCl
Capacidad calorífica de la gota
K 0.019[W/m/K] Conductividad térmica del gas
que rodea la gota
lambda 1100[kJ/kg] Calor latente de la evaporación
del solvente
Km 0[m/s] Coeficiente de transferencia de
masa del vapor al tubo
Ds 1E-10[m^2/s] Coeficiente de difusión del
soluto en la gota
Dv 2.00E-5[m^2/s] Coeficiente de difusión de
solvente-vapor
hw 27.85[W/m^2/K] Coeficiente de transferencia de
calor del gas a reactor
hs 87.73[W/m^2/K] Coeficiente de transferencia de
calor del gas a la gota
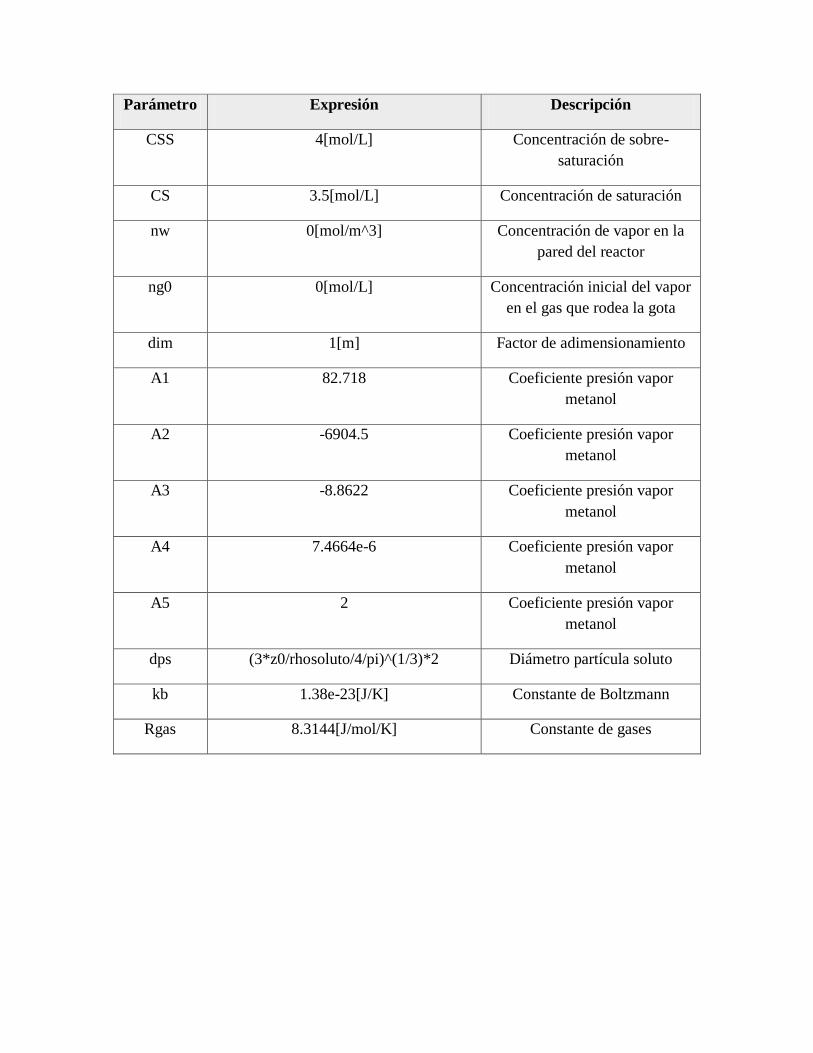
Parámetro Expresión Descripción
CSS 4[mol/L] Concentración de sobre-
saturación
CS 3.5[mol/L] Concentración de saturación
nw 0[mol/m^3] Concentración de vapor en la
pared del reactor
ng0 0[mol/L] Concentración inicial del vapor
en el gas que rodea la gota
dim 1[m] Factor de adimensionamiento
A1 82.718 Coeficiente presión vapor
metanol
A2 -6904.5 Coeficiente presión vapor
metanol
A3 -8.8622 Coeficiente presión vapor
metanol
A4 7.4664e-6 Coeficiente presión vapor
metanol
A5 2 Coeficiente presión vapor
metanol
dps (3*z0/rhosoluto/4/pi)^(1/3)*2 Diámetro partícula soluto
kb 1.38e-23[J/K] Constante de Boltzmann
Rgas 8.3144[J/mol/K] Constante de gases