Embed Size (px)
Citation preview

UNIVERSIDAD CENTRAL DEL ECUADOR
FACULTAD DE CIENCIAS QUÍMICAS
INSTITUTO DE INVESTIGACIÓN Y POSGRADO
MAESTRIA EN QUIMICA ANALITICA
Desarrollo de un método analítico por espectrofotometría visible para determinar
detergentes aniónicos en aguas limpias y residuales
Trabajo de investigación previo la obtención del título de
Magister en Química Analítica
Autor: Químico. Salomón Chacha Palango
Tutor: Dr. Iván Luis Tapia Calvopiña. Msc.
Quito, julio 2018

ii
DERECHOS DE AUTOR
Yo SALOMON CHACHA PALANGO, en calidad de autor del trabajo de investigación:
“DESARROLLO DE UN MÉTODO ANALÍTICO POR ESPECTROFOTOMETRÍA
VISIBLE PARA DETERMINAR DETERGENTES ANIÓNICOS EN AGUAS
LIMPIAS Y RESIDUALES”, autorizo a la Universidad Central del Ecuador hacer uso de
todos los contenidos que me pertenecen o parte de los que contiene esta obra, con fines
estrictamente académicos o de investigación.
Los derechos que como autor me corresponden, con excepción de la presente
autorización, seguirán vigentes a mi favor, de conformidad con lo establecido en los
artículos 5, 6, 8, 19 y demás pertinentes de la Ley de Propiedad intelectual y su
Reglamento.
Asimismo, autorizo a la Universidad Central del Ecuador para que realice la digitalización
y publicación de este trabajo de investigación en el repositorio virtual, de conformidad a
lo dispuesto en el Art. 144 de la Ley Orgánica de Educación Superior.
En la ciudad de Quito a los 13 días del mes de julio de 2018

iii
APROBACIÓN DEL TUTOR DEL TRABAJO DE TITULACIÓN

iv
APROBACIÓN DEL TRABAJO FINAL POR EL TRIBUNAL
El tribunal constituido por: …………………………………………………….
……………………………………………………………………………………………
……………………………………………………………………………………………
………………. luego de receptar la presentación oral del trabajo de titulación previo a
la obtención del título de Maestría en Química Analítica presentado por el sr Salomón
Chacha Palango. Con el título: “Desarrollo de un Método Analítico por
Espectrofotometría Visible para Determinar Detergentes Aniónicos en Aguas
Limpias y Residuales”
Emite el siguiente veredicto (Aprobado/Reprobado)…………………………………
Fecha: ……………………………………….
Para constancia de lo actuado firman:
Nombres Apellidos Calificación Firma
Presidente: ………………………………… ……………. …………………
Vocal 1: …………………………………… ……………… ………………….
Vocal 2: …………………………………… …………….. ……………………

v
DEDICATORIA
A mi esposa Dolores del Rocío y a mis hijos:
Alexander, Boris, Kevin, Edwin, Isaac y Carolina
Con todo mi amor.

vi
AGRADECIMIENTO
Agradezco a la Universidad Central del Ecuador, al personal del Laboratorio OSP,
de la Facultad de Ciencias Químicas, al personal del Laboratorio de Ingeniería Ambiental
de la Facultad de Ingeniería en Geología, Minas, Petróleo y Ambiental, por permitirme
llevar a cabo el desarrollo de la presente investigación. A mi Tutor de Tesis, Dr. Iván
Tapia, por brindarme sus conocimientos, apoyo e interés en este proyecto de
investigación.

vii
ÍNDICE DE CONTENIDO
DERECHOS DE AUTOR .................................................................................... II
APROBACIÓN DEL TUTOR DEL TRABAJO DE TITULACIÓN ............... III
APROBACIÓN DEL TRABAJO FINAL POR EL TRIBUNAL ..................... IV
DEDICATORIA ................................................................................................... V
AGRADECIMIENTO ........................................................................................ VI
ÍNDICE DE CONTENIDO ............................................................................... VII
ÍNDICE DE TABLAS ....................................................................................... XI
ÍNDICE DE FIGURAS .................................................................................... XIII
RESUMEN ........................................................................................................ XV
ABSTRACT .................................................................................................... XVI
INTRODUCCION ............................................................................................... 1
Planteamiento del Problema. ...................................................................... 1
Formulación del Problema.......................................................................... 2
Objetivos ..................................................................................................... 2
1.3.1 General ................................................................................................... 2
1.3.2 Específicos ............................................................................................. 2
Justificación de la Investigación ................................................................. 2
Limitaciones ............................................................................................... 3
MARCO TEÓRICO ............................................................................................. 4
Antecedentes de la Investigación ............................................................... 4

viii
2.1.1 Método 1: colorimétrico ......................................................................... 4
2.1.2 Método 2: colorimétrico ......................................................................... 4
2.1.3 Método 3: Absorción Atómica ............................................................... 5
2.1.4 Método 4: Cromatografía Liquida de Alta Resolución (HPLC) ............ 5
Detergentes ................................................................................................. 6
2.2.1 Desarrollo Histórico ............................................................................... 6
2.2.2 Clasificación ........................................................................................... 8
2.2.3 Detergentes en aguas naturales y en aguas residuales .......................... 11
2.2.4 Biodegradabilidad de los detergentes ................................................... 11
Química Verde .......................................................................................... 12
2.3.1 Solventes verdes ................................................................................... 13
Serie eluotrópica ....................................................................................... 17
2.4.1 Índice de polaridad de Snyder (P) ........................................................ 17
Método de extracción ............................................................................... 18
2.5.1 Generalidades ....................................................................................... 18
Equilibrio Químico ................................................................................... 20
Pares iónicos ............................................................................................. 21
Fuerza Iónica ............................................................................................ 23
Espectrometría de Absorción Ultravioleta – Visible ................................ 24
2.9.1 Interacción de la luz con la materia ...................................................... 24
2.9.2 Espectroscopía de absorción molecular uv-visible .............................. 25
2.9.3 Ley de Beer .......................................................................................... 27
2.9.4 Aplicaciones de la Ley de Beer a mezclas ........................................... 28
2.9.5 Limitaciones de la Ley de Lambert-Beer ............................................. 29
2.9.6 Efectos del Ruido instrumental ............................................................ 30
2.9.7 Instrumentación .................................................................................... 32
Desarrollo de Métodos Analíticos ............................................................ 35
Selección de un Método Analítico............................................................ 36
Calibración de métodos instrumentales .................................................... 36
2.12.1 Comparación con estándares .............................................................. 37
2.12.2 Calibración de un estándar externo .................................................... 37
2.12.3 Características de desempeño de los instrumentos ............................. 38

ix
Parámetros de calidad ............................................................................... 40
2.13.1 Linealidad ........................................................................................... 40
2.13.2 Intervalo lineal .................................................................................... 43
2.13.3 Selectividad ........................................................................................ 43
2.13.4 Sensibilidad ........................................................................................ 43
2.13.5 Límite de detección ............................................................................ 45
2.13.6 Límite de Cuantificación (LC) ........................................................... 47
2.13.7 Precisión ............................................................................................. 47
2.13.8 Diseño de Factores Anidados: Una variación del tema ...................... 49
2.13.9 Exactitud. ............................................................................................ 54
Z-score ............................................................................................................ 56
Hipótesis De Investigación ....................................................................... 59
Variables ................................................................................................... 60
CAPITULO III ................................................................................................... 61
METODOLOGIA .............................................................................................. 61
Diseño metodológico ................................................................................ 61
3.1.1 Tipo de Investigación ........................................................................... 61
Población y Muestra ................................................................................. 61
3.2.1 Población .............................................................................................. 61
3.2.2 Muestra ................................................................................................. 61
3.2.3 Diseño muestral .................................................................................... 61
Diseño experimental ................................................................................. 62
3.3.1 Puesta a punto del método analítico propuesto .................................... 62
3.3.2 Parámetros de Calidad .......................................................................... 62
Diseño estadístico ..................................................................................... 63
Técnicas analíticas empleadas en el trabajo de investigación .................. 64
Técnicas e Instrumentos de Recolección de Datos ................................... 73
3.6.1 Técnica: ................................................................................................ 73
3.6.2 Instrumento: ......................................................................................... 73
3.6.3 Descripción de instrumentos ................................................................ 74
CAPITULO IV ................................................................................................... 75

x
4 RESULTADOS Y DISCUSIÓN .................................................................. 75
Puesta a punto del método analítico propuesto ........................................ 75
4.1.1 Determinación de la longitud de onda. ................................................. 75
4.1.2 Efecto de la mezcla de solventes .......................................................... 75
4.1.3 Efecto del pH en la formación y extracción del par iónico .................. 79
4.1.4 Efecto de la fuerza iónica en la formación de la especie química ........ 80
4.1.5 Determinación de la concentración óptima del colorante CV .............. 82
4.1.6 Determinación del intervalo dinámico ................................................. 83
Parámetros de calidad ............................................................................... 85
4.2.1 Determinación de límite de detección y límite cuantificación ............. 85
4.2.2 Determinación de la linealidad ............................................................. 86
Sensibilidad .............................................................................................. 91
Precisión ................................................................................................... 91
Exactitud ................................................................................................... 93
CAPITULO V .................................................................................................... 95
CONCLUSIONES Y RECOMENDACIONES ................................................. 95
Conclusiones ............................................................................................. 95
Recomendaciones ..................................................................................... 96
6 BIBLIOGRAFIA .......................................................................................... 97
7 ANEXO A .................................................................................................. 103
ANEXO B ........................................................................................................ 107
ANEXO C ........................................................................................................ 110

xi
ÍNDICE DE TABLAS
Tabla 1.- Propiedades relevantes de n-hexano, tolueno, diclorometano, benceno y d-
limoneno .................................................................................................................. 16
Tabla 2.- Propiedades de los solventes orgánicos Fuerza de elución y polaridad. ......... 18
Tabla 3.- Criterios numéricos para la seleccionar métodos analíticos ........................... 39
Tabla 4.- Otras características a tener en cuenta en la elección del método ................... 39
Tabla 5.- Parámetros de calidad para la precisión de los métodos analíticos. ................ 48
Tabla 6.- Para el Análisis Simple de la Varianza ........................................................... 50
Tabla 7.- Análisis de Varianza ...................................................................................... 51
Tabla 8.- Variables Independiente y dependiente .......................................................... 60
Tabla 9.- Parámetros considerados para el diseño Experimental .................................. 63
Tabla 10.- Tamaño de la muestra en función de la concentración de SAAM. ............... 68
Tabla 11.- Preparación de la curva de calibrado ............................................................ 72
Tabla 12.- Procedimiento para el análisis de muestras ................................................... 72
Tabla 13.- Resultados en Absorbancia de la Extracción del par iónico con mezclas de
disolventes ............................................................................................................... 76
Tabla 14.- Resultados en absorbancia de la Extracción del colorante catiónico sin el
analito con mezclas de disolventes.......................................................................... 77
Tabla 15.- Resultados en absorbancia(A), de la especie extraída con L-X en diferentes
proporciones. ........................................................................................................... 78
Tabla 16.- Resultados en Absorbancia del Efecto a diferentes valores de pH en la
formación y extracción del par iónico. .................................................................... 79
Tabla 17.-Resultado en absorbancia del Efecto de la fuerza Iónica en la formación y
extracción del par iónico ......................................................................................... 81

xii
Tabla 18.- Resultados en Absorbancia de la concentración optima del CV................... 82
Tabla 19.- Resultados en absorbancia para la determinación del intervalo dinámico .... 83
Tabla 20.- Resultados corregidos para el intervalo dinámico ........................................ 84
Tabla 21.- Resultados en Absorbancia (A) y la concentración en ppm obtenidos a partir
de 20 blancos ........................................................................................................... 85
Tabla 22.- Resultados en Absorbancia de la lectura de los estándares de diferente
concentración en cinco días..................................................................................... 86
Tabla 23.- Determinación de los Parámetros estadísticos básicos ................................. 87
Tabla 24.- Función Respuesta Instrumental y Análisis de las curvas de calibración ..... 89
Tabla 25.- Resultados de la CURVA GLOBAL y de los Límites superior e inferior .... 89
Tabla 26.- Función respuesta de la curva global ............................................................ 90
Tabla 27.- Resultados de las pendientes de las curvas de calibración ............................ 91
Tabla 28.- Resultados de la Desviación estándar de la Repetibilidad para los seis niveles
................................................................................................................................. 92
Tabla 29.-Resultados de la Desviación estándar de la Reproducibilidad para los seis
niveles...................................................................................................................... 92
Tabla 30.- Resultados de la F calculada ......................................................................... 92
Tabla 31.- Resultados del porcentaje de recuperación para evaluar la exactitud ........... 93

xiii
ÍNDICE DE FIGURAS
Fig 1.- Porcentaje estimado de volumen de venta anual mundial de agentes activos de
superficie para 2006 por región, basado en un total de 13 millones de toneladas
métricas. .................................................................................................................... 8
Fig 2.- Ejemplos de tensioactivos aniónicos: a) Sulfonato de alquílbenceno lineal, b)
Sulfonato de 1-n-alquilo, c) Sulfonato de alquilbenceno ramificado, d) Sulfonato de
4-(1-n-octil) benceno. ................................................................................................ 9
Fig. 3.- Ejemplos de Tensoactivos catiónicos: a) Bromuro de cetilpiridinio, b) Bromuro
de trimetilhexadecilamonio. .................................................................................... 10
Fig 4.- Ejemplo de tensioactivo no iónico: Alcohol etoxilado. ...................................... 10
Fig. 5.- Ejemplos de tensioactivos anfóteros. a) Alquilbetaína, b)
Alquilamidopropilbetaína........................................................................................ 11
Fig. 6.- Estructura química de d-limoneno .................................................................... 15
Fig. 7.- Espectro electromagnetico. ................................................................................ 25
Fig. 8.- Pérdidas por reflexión y dispersión.................................................................... 26
Fig. 9.- La radiación de inicial Po y la radiación de salida P luego de atravesar la celda
que contiene la solución con el analito.................................................................... 27
Fig. 10.- Espectrofotómetro Jasco V-630. ...................................................................... 67
Fig. 11.- Espectrofotómetro HACH DR/4000. ............................................................... 71
Fig. 12. Espectro visible de la longitud de onda de máxima absorbancia de la especie
detergente aniónico- cristal violeta ......................................................................... 75
Fig. 13.- Medida de la absorbancia del par iónico extraído con diferentes mezclas de
solventes. ................................................................................................................. 77

xiv
Fig. 14.- Extracción del colorante catiónico con diferentes mezclas de solventes y
medida de la absorbancia. ....................................................................................... 78
Fig. 15.- Evaluación de la mezcla óptima para la extracción del par iónico. ................. 79
Fig. 16.- Influencia del pH en la formación y extracción del par iónico ........................ 80
Fig. 17.- Variación de la Absorbancia por efecto de la Fuerza iónica (µm) ................... 81
Fig 18.- Variación de la Absorbancia en función de la concentración del colorante
catiónico. ................................................................................................................. 82
Fig 19.- Gráfica para determinar el Intervalo útil para el método analítico propuesto .. 83
Fig 20.- Intervalo dinámico del método propuesto......................................................... 84
Fig 21.- Curva de calibración día 2. ............................................................................... 87
Fig. 22.- Curva de calibración día 3. .............................................................................. 88
Fig.23.- Curva de calibración día 4 ................................................................................ 88
Fig.24.- Curva de calibración día 5. ............................................................................... 88
Fig. 25.- Gráfica de la Curva Global y los Límites superior e inferior .......................... 90

xv
“DESARROLLO DE UN MÉTODO ANALÍTICO POR
ESPECTROFOTOMETRÍA VISIBLE PARA DETERMINAR DETERGENTES
ANIÓNICOS EN AGUAS LIMPIAS Y RESIDUALES”
RESUMEN
El presente trabajo de investigación tiene como propósito el desarrollo de un método
ecológico; para determinar detergentes aniónicos en aguas limpias y residuales como una
alternativa a los métodos que se encuentran vigentes en los laboratorios de ensayo y de
investigación, los mismo que en la mayoría de los casos no han tomado en cuenta los
riesgos de toxicidad de los disolventes utilizados (benceno y cloroformo) que son
considerados altamente tóxicos.
El desarrollo del método se realizó mediante un Espectrofotómetro DR/4000 a una
longitud de onda de 605 nm, balanza analítica, materiales de vidrio clase A, reactivos
estándar lauril sulfato de sodio, cloruro de sodio, cristal violeta, limoneno y xileno.
El método oficial usado en el laboratorio de Química Ambiental OSP es el Método
estandarizado 5540 C Surfactantes Amónicos como SAAM. “La extracción se realiza en
cloroformo (CHCl3) a partir de medio acuoso ácido que contenga azul de metileno en
exceso, seguidas de lavado por contracorriente con agua, y la determinación del color
azul en el CHCl3 por espectrofotometría a 652 nm.”
Las fases de la investigación: 1) bibliográfica. 2) experimental: a) puesta a punto del
método y b) evaluar los parámetros de calidad. Los valores de desviación estándar relativa
para la repetibilidad y reproducibilidad son menores al 5%. Para la exactitud se obtuvo
una media de % de recuperación de 98% y una prueba de t-student que dio un valor t
experimental de -0,558 y el t crítico de 3,18. Para los límites de detección 0,007 ppm y
de cuantificación 0,025 ppm. Para la linealidad se encontró un coeficiente de correlación
r=0.99. Los parámetros fueron sometidas a pruebas estadísticas; concluyendo que el
método analítico por espectroscopia visible para la determinación de detergentes
aniónicos en aguas cumple con los objetivos de la investigación.
Palabras claves: DETERGENTES ANIÓNICOS, DESARROLLO, FUERZA IÓNICA,
ESPECTROFOTOMETRÍA, PARÁMETROS DE CALIDAD, DISOLVENTES,
MATERIAL DE REFERENCIA.

xvi
TOPIC: DEVELOPMENT OF AN ANALYTICAL METHOD BY
VISIBLE SPECTROPHOTOMETRY TO DETERMINE ANIONIC
DETERGENTS IN CLEAN AND WASTEWATER
ABSTRACT
The purpose of this research is to develop an ecological method to determine
anionic detergents in clean and residual waters as an alternative to the mandatory
techniques used in research laboratories. Most of these methods do not consider the
toxicity risks of the used solvents (benzene and chloroform) which are considered
highly toxic. The development of the method was carried out by a DR / 4000
Spectrophotometer at a wavelength of 605 nm, analytical scale, class A glass materials,
standard agents sodium lauryl sulfate, sodium chloride, violet crystal, limonene, and
xylene. The official method used in the OSP Environmental Chemistry laboratory is
the Standardized Method 5540 C Ammonia Surfactants as SAAM. "The extraction is
carried out in chloroform (CHC13) from aqueous acidic medium containing excess
methylene blue, followed by backwashing with water, and the determination of
the blue color in CHCI3 by spectrophotometry at 652 nm." The phases of
research: 1) literature review, 2) experimental: a) tuning the method and b) evaluating
the quality of the parameters. The values of relative standard deviation for repeatability
and reproducibility are less than 5%. For accuracy, we obtained an average recovery of
98% and a t-student test that gave a t experimental value of -0.558 and a t critical of 3.18.
For detection limits 0.007 ppm and quantification 0.025 ppm. For linearity, a correlation
coefficient r = 0.99 was found. The parameters evaluated were subjected to statistical
tests; concluding that the analytical method by visible spectroscopy for the
determination of anionic detergents in waters fulfills the objectives of the investigation.
KEYWORDS: ANIONIC DETERGENTS, DEVELOPMENT IONIC STRENGTH,
SPECTROPHOTOMETRY, QUALITY PARAMETERS, SOLVENTS, REFERENCE
MATERIAL
xvii

1
CAPITULO I
INTRODUCCION
Planteamiento del Problema.
Para la determinación físico-química de los detergentes aniónicos en aguas limpias y
residuales en el Laboratorio de ensayos de Química Ambiental “OSP” de la Facultad de
Ciencias Químicas de la Universidad Central del Ecuador, se ha utilizado desde el comienzo
de funcionamiento del Laboratorio (1998), un Método Colorimétrico rápido, en cual se
emplea “benceno” como disolvente de extracción de la especie química, en la actualidad se
dispone de otro Método Colorimétrico Normalizado, en donde se utiliza como disolvente de
extracción “cloroformo”.
El uso de disolventes altamente tóxicos para la salud humana, como benceno, cloroformo, y
el tiempo de exposición del personal que trabaja en los Laboratorios de ensayos
Fisicoquímicos, se relaciona directamente con el “Riesgo Químico” y el impacto ambiental
que genera estos agentes químicos. (PUBLICACIONES VERTICE.S.L, 2008)
El benceno es muy tóxico para los humanos, y hasta puede causar un grave daño hepático.
(Hart, Hart, & Craine, Química Orgánica, 1995). Está reconocido por la IARC (International
Agency for Research on Cáncer) como carcinógeno para el hombre (Barreno, García Oliver,
& Gutiérrez Montesinos, 2008).
La Agencia Internacional de Investigación sobre el Cáncer ha clasificado al cloroformo
como cancerígeno potencial. (Mosquera, Hidalgo, & Forjan, 2009)
Por lo antes indicado, es necesario investigar el uso de solventes verdes en el desarrollo de
métodos de ensayos, para la determinación de detergentes aniónicos en aguas limpias y
residuales.

2
Formulación del Problema
¿Es posible desarrollar un nuevo método analítico para determinar detergentes en aguas
limpias y residuales, que no sea toxico para el personal que realiza los análisis fisicoquímicos
y además sea amigable con el medio ambiente?
Objetivos
1.3.1 General
Desarrollar un método analítico para la determinación de detergentes aniónicos en aguas
limpias y residuales, que minimice el uso de disolventes tóxicos para la salud y para el
ambiente.
1.3.2 Específicos
Determinar: una mezcla óptima en volumen del d-limoneno con los diferentes
disolventes orgánicos; que facilite la extracción del analito, el rango de pH para la formación
del analito y la extracción de este, la influencia de la fuerza iónica en la formación del par
iónico, formado entre el detergente aniónico y el colorante cristal violeta y la longitud de
onda de máxima absorbancia para la especie química en estudio.
Construir la curva de calibración (Absorbancia en función de la concentración) para
determinar el rango lineal del analito (detergentes aniónicos).
Determinar los parámetros de calidad del método propuesto: límite de detección,
límite de cuantificación, linealidad, sensibilidad, precisión y exactitud.
Justificación de la Investigación
En el desarrollo de métodos analíticos los criterios cuantitativos y cualitativos como:
velocidad, factibilidad, habilidad y coste (Skoog, James Holler, & Nieman, 2001), son muy
importantes, pero no se debe minimizar las graves afecciones medio ambientales y los
riesgos para la salud humana por el uso de disolventes orgánicos volátiles (COV).
El personal que trabaja en los laboratorios de ensayos físico químico está expuestos a muchas
sustancias químicas consideradas toxicas para la salud, las mismas que caracterizan por tener
un límite de techo, además de su límite permitido de exposición (PEL) y el valor límite
umbral (TLV). El límite es una concentración en partes por millón (ppm) o miligramos por

3
metro cúbico (mg/m3), que no deben ser excedidos en un período específico de tiempo,
generalmente 15 minutos (Young, 2002). Esta exposición directa o indirecta de las personas
se ha convertido en un grave problema de salud pública puesto que se encuentra entre las
diez primeras causas de enfermedades profesionales con afección en diversos órganos y
sistemas del cuerpo humano como la piel, pulmones, sistema nervioso central, hígado
ocasionando en estos: dermatitis, síndrome orgánico cerebral, leucemia, cáncer y en ciertos
casos la muerte generando afectaciones de índole económico, familiar, laboral y social que
representa un obstáculo para el desarrollo del país.
Los disolventes orgánicos considerados tóxicos por la OMS, como: benceno, cloroformo,
están siendo reemplazados por los disolventes neotéricos que se obtienen de las materias
primas renovables, además presenta baja toxicidad, baja volatilidad, no son corrosivos ni
cancerígenos si utilizamos este tipo de disolventes podemos minimizar los riesgos químicos
laborales. (Sanchez & Angel, 2006).
La producción mundial de millones de toneladas y el consumo masivo de los tensioactivos
como producto de limpieza, en el hogar y a nivel industrial; los detergentes aniónicos es un
parámetro controlado por las normativas ambientales en las descargas líquidas; como
consecuencia de ello surge la necesidad de disponer de un método analítico amigable con el
medio ambiente.
En el organismo de acreditación SAE actualmente se encuentran registrado como
laboratorios de ensayos físico químico 46 sin contar con los laboratorios que realizan
actividades de docencia, razón más que suficiente para buscar una solución.
Limitaciones
Entre las limitaciones en el proceso de investigación fue de conseguir el disolvente, en este
caso del limoneno de grado reactivo, se procedió a trabajar con un reactivo grado técnico
para uso industrial.

4
CAPITULO II
MARCO TEÓRICO
Antecedentes de la Investigación
A principios del siglo XX los métodos clásicos fueron reemplazados por los métodos
instrumentales con la finalidad de resolver problemas analíticos. Con los métodos
instrumentales se aprovecha las propiedades físicas del analito como: conductividad, el
potencial del electrodo, la absorción y la emisión de la luz, la relación masa/carga, la
fluorescencia, técnicas cromatográficas y electroforéticas. Los fenómenos en que se
fundamenta los métodos instrumentales se conocieron mucho más antes. Su aplicación se
retrasó por falta de una instrumentación sencilla y fiable, gracias al desarrollo de la industria
electrónica e informática el desarrollo de los métodos instrumentales modernos ha
evolucionado paralelamente (Hernandez & Gonzales, 2002). Entre los métodos
instrumentales utilizados para la cuantificación del analito (detergente aniónico) se cita a
continuación.
2.1.1 Método 1: colorimétrico
Principio del método. En solución acuosa la molécula polar de los detergentes aniónicos
forma, con los colorantes catiónico, un complejo soluble en benceno, susceptible de una
determinación colorimétrica.
Procedimiento. Consiste en: colocar 300 ml de muestra, 10 ml de solución tampón de pH
1.0, más 1g de solución neutra de cristal de violeta y 30 ml de benceno, como disolvente de
extracción, (Hach, 1988)
2.1.2 Método 2: colorimétrico
Principio del método. En solución acuosa la molécula polar de los detergentes aniónicos
forma, con el azul de metileno (catiónico), un complejo soluble en cloroformo, susceptible
de una determinación colorimétrica.
Procedimiento. Utiliza cloroformo como disolvente de extracción, solución tampón de pH
10.5, solución neutra de azul de metileno. Efectuar las lecturas en el espectrofotómetro uv-
visble a 650 nm. (Rice, Baird, Eaton, & Clesceri, 2012)

5
2.1.3 Método 3: Absorción Atómica
Principio del método. Los detergentes aniónicos se asocian con la ortofenantrolina cúprica
que es extraíble con la metil-isobutil cetona en la que el cobre se determina por
espectrometría de Absorción atómica.
Procedimiento. En un embudo de decantación disolver 30 g de cloruro de sódico en un litro
de agua a analizar limpia. Acidificar con 10 ml de HCl 1N, 10 ml de solución de sulfato de
ortofenantrolina cúprica 0,025 M. añadir 25 ml y después 18 ml de MEK. Trasvasar a un
matraz y aforar de 50 ml con MEK. Nebulizar la solución en una llama aire-acetileno después
de cada muestra. Efectuar las lecturas a la longitud de onda de 324,7 nm. (Gagnon, 1979)
2.1.4 Método 4: Cromatografía Liquida de Alta Resolución (HPLC)
Principio del método
La determinación de LAS en muestras ambientales se realiza generalmente usando métodos
de cromatografía líquida con detección UV, la detección de fluorescencia, o la detección de
espectrometría de masas, que permite la identificación y determinación de LAS isómeros y
homólogos. Hay un número más limitado de métodos de cromatografía de gas, que puede
ser debido a la baja volatilidad de estos compuestos, siendo necesario el uso de reacciones
de derivatización del grupo sulfonato para obtener los compuestos más volátiles. La
electroforesis capilar con detección UV también se ha utilizado para la determinación de la
suma, homólogos e isómeros de LAS en productos para el hogar y muestras de aguas
residuales. Los métodos para la cuantificación de LAS en el suelo, en los lodos de aguas
residuales, en el sedimento, en los organismos biológicos, o en agua pueden ser reportados.
Sin embargo, estos métodos no se pueden aplicar directamente al análisis de plantas; se
necesitan medidas específicas de purificación. El principal problema para el análisis de los
contaminantes orgánicos en las plantas proviene de la complejidad de la matriz. Las plantas
tienen una estructura de tejido en particular, que dependen de la especie y la edad, y son muy
ricos en pigmentos, aceites esenciales, ácidos grasos, o alcoholes.
Procedimiento. Todos los productos químicos utilizados fueron de calidad analítica.
Metanol, acetonitrilo, agua, perclorato de sodio (NaClO4) y dodecilsulfato de sodio (SDS).
Los cartuchos de extracción de tamaño 20mmx80mm, arena de Fontainebleau (tamaño de
partícula 150-300um) para controlar la ebullición y en polvo (tamaño de partícula 60 a 100)
silicato de magnesio. Florisil para adsorber grasa se añadieron a la muestra en el cartucho de

6
extracción. La separación por HPLC se realizó con una columna de Inertsil ODS3 (C18) de
25 cm de largo de diámetro interno y 5 um de tamaño de partícula.
LAS que es un polvo tensioactivo comercial que contiene 80% de C10-C13 LAS. Esta
mezcla homóloga (LAS) comercial tiene la siguiente distribución de masas homólogo: C10
(14,3%), C11 (35,7%), C12 (30,8%) y C13 (19,2%). La solución estándar de la (1 g/L) se
preparó disolviendo 312,5 mg LAS en 250 ml de metanol / solución acuosa de SDS a 5 x10-
3mol/L (50/50, v/v). (Sablayrolles, Montrejaud, Silvestre, & Treilhou, 2009)
En todos los métodos citados, se utiliza reactivos (disolventes) que son altamente
carcinógenos, como el benceno y el cloroformo; metilisobutilcetona son irritable al sistema
respiratorio y el metanol, es una sustancia muy tóxica cuya ingestión puede causar ceguera
o la muerte. (Ferreiro Gomez, 2012)
Detergentes
2.2.1 Desarrollo Histórico
El ser humano con el transcurso del tiempo ha ido evolucionando, buscando aditivos que
mejoren la capacidad limpiadora del agua; una de las civilizaciones más antiguas como la
egipcia representaba mediante un símbolo la acción de lavar, con una persona sumergida en
el agua.
El origen del jabón ya como sal alcalina de un ácido graso, indudablemente se sitúa mucho
antes de la era cristiana. Los Sumerios 3000 a. C. ya comentaban de la fabricación y
describían las propiedades curativas de un “azufre jabonoso”. En Tello, Mesopotamia, se ha
encontrado, una lasca de arcilla del año 2500 a. C. en la que se relata la fabricación del jabón,
utilizando aceite y una cantidad precisa de hierba jabonosa. En un papiro egipcio
aproximadamente del año 1500 a. C. en un tratado médico, se indica, tratando aceites de
animales y plantas con ciertas sales (alcalinas) se obtenían un producto de tipo jabonoso útil
para combatir las enfermedades de la piel y también para el lavado. Existen referencias
bíblicas de ciertos productos líquidos alcalinos que poseían propiedades limpiadoras (Waite,
1984). Las tribus germanas de la época de César hervían cebo de cabra con potasa.
Efectuaban la misma reacción química que ahora lo realizan los fabricantes modernos de
jabones, a una escala industrial. (Wilkinson & More, 1990)

7
En España los musulmanes preparaban jabones a partir del aceite de oliva; sin embargo, en
las ciudades costeras del mediterráneo como Marsella, Venecia, Génova y otras donde se
desarrolló una importante industria jabonera por la abundancia del aceite de oliva. I). En
1971 el médico francés Nicolás Leblanc concluyó con el desarrollo de un método de
obtención del carbonato sódico, por tal razón es considerado como el fundador de la Química
Industrial. II) Entre 1813 y 1823 el Químico Michel Eugene–Chevreul profundizó sobre la
composición química de la grasa de origen natural y demostró que su formación era debido
a una reacción química de modo que los fabricantes del siglo XIX pudieron tener una idea
del proceso químico involucrado. Más tarde en 1861 el Químico Industrial Ernest Solvay
adquirió su primera patente para la fabricación de carbonato de sodio.
Durante los siglos XVIII y XIX la industria de los jabones sufrió un gran desarrollo
ampliando la oferta de productos con distintas formas y variedades como: jabones duros,
blandos, perfumados, etc. Debido a estos avances tecnológicos hubo un mayor consumo de
estos productos, ocasionando una mejora en la higiene personal y consecuentemente
contribuyendo al crecimiento exponencial de la población europea por la disminución de las
causas de mortalidad.
En 1878, en Alemania se elaboró un producto que además de tener jabón como uno de sus
componentes contenía silicato sódico se denominó Henkels-Bleichsoda puede ser
considerado como la primera formulación
Durante la primera guerra mundial era difícil la obtención de grasa en Alemania, ya que se
usaba con fines nutritivos, los científicos empezaron a estudiar la posibilidad de desarrollar
tensioactivos sintéticos para sustituir al jabón. En 1916 el Químico Fritz Gunter logró
sintetizar el primer jabón artificial se trataba del compuesto diisopropilnaftaleno sulfonato
sódico. En 1932 la Empresa Henkel comercializó alcoholes grasos como el oleico (C18:1) y
el esteárico (C18:0) procedentes del esperma de aceite de ballena, extendiéndose esta
comercialización a los alcoholes grasos como el láurico (C12:0) y el mirística (C14:0); por
presentar mejores propiedades. Un paso importante en el desarrollo de los detergentes fue el
uso de máquinas automáticas de lavado. En los años 50 se produjo un nuevo descubrimiento
en la línea de los detergentes, el alquílbenceno sulfonato ramificado del
inglés (BAB, Branched Alkylbenzene). Este detergente tenía un problema que no era
biodegradable, se dio una solución con la invención del alquilbenceno lineal del inglés

8
(LAB, Linear Alkylbenzene), a principios de los años 60; de esta forma apareció el sulfonato
de alquilbenceno lineal (LAS Linear Alkylbenzene Sulfonate), actualmente el detergente
sintético de mayor producción mundial. (Jiménez Díaz, 2009)
El consumo mundial anual de agentes activos de superficie o "tensioactivos" en 2006, se
estima que alcanzó los 13 millones de toneladas métricas, con la ruptura de las ventas
regionales tal como se representa en la Fig. 1
Los nuevos procesadores básicos de materias para extraer y refinar el petróleo crudo en
productos petroquímicos como los destilados de petróleo incluyendo parafinas, benceno y
otros aromáticos básicos; también se ha logrado extraer y convertir el gas natural en al etileno
y propileno. Los procesadores de productos oleo químicos extraer y purificar los aceites de
semilla de palma, soja, semilla de girasol, semilla de palma y de coco y los animales aportan
con el sebo para proporcionar aceites de triglicéridos con diferentes distribuciones de la
cadena. (Zoller & Paul, 2008).
2.2.2 Clasificación
La clasificación de los surfactantes se debe realizar atendiendo a la naturaleza de su grupo
hidrofílico. Así se puede clasificar en tensioactivos aniónicos, catiónicos, no iónicos y
anfóteros.
3
20
4 2
34 33
4
0
10
20
30
40
Americalatina
Asia delPacífico
ROW Canadá EstadosUnidos
Europa Japón
%
REGIÓN
PRODUCCION MUNDIAL DE TENSIOACTIVOS
Series1 Series2
Fig 1.- Porcentaje estimado de volumen de venta anual mundial de
agentes activos de superficie para 2006 por región, basado en un total de 13
millones de toneladas métricas.
Fuente: (Busines, 2006)

9
Surfactantes aniónicos
Son los tensoactivos más utilizados a escala mundial. La zona polar de este tipo de sustancias
está cargada negativamente, el contraión (sodio, potasio, hidrógeno o amonio) ejerce una
escasa influencia sobre las propiedades superficiales de estas sustancias. Este grupo polar
negativo suele ser los grupos sulfatos o sulfonatos.
En la Fig. 2.2 se representa la estructura química de un tensioactivo aniónico.
CH3 CH3
CH3 CH3
CH3
CH3
S OO
O-
CH3 CH3
S OO
O-
CH3 S
O
OO
-CH3
S
O
O
O-
a)
b)
c)
d)
Fig 2.- Ejemplos de tensioactivos aniónicos: a) Sulfonato de alquílbenceno lineal, b) Sulfonato de
1-n-alquilo, c) Sulfonato de alquilbenceno ramificado, d) Sulfonato de 4-(1-n-octil) benceno.
Fuente:. (Uri & Guy, 1999)
Surfactantes Catiónicos
Son compuestos orgánicos sintéticos que contiene en su estructura una parte polar hidrofílica
cargada positivamente y la parte hidrofóbica; entre las sustancias consideradas como
tensoactivos catiónicos tenemos: una sal de amonio cuaternario, amina o sal de fosfonio. Los
detergentes catiónicos no requieren protonarse para obtener carga positiva.
En la Fig.3 se representa la estructura química de sustancias tensioactivas catiónicas.

10
CH3
N+ H
Br-
CH3
N+
CH3
CH3CH3 Br
-
a)
b)
Fig. 3.- Ejemplos de Tensoactivos catiónicos: a) Bromuro de cetilpiridinio, b) Bromuro de
trimetilhexadecilamonio.
Fuente:. (Uri & Guy, 1999)
Surfactantes no iónicos
Sustancias que en disolución no originan iones (no cargas), pero si se solubiliza gracias al
carácter polar.
Estos productos son los mayoritarios en usos industriales. Un ejemplo se representa en la
Fig 4.
CH3
OOH
n
Fig 4.- Ejemplo de tensioactivo no iónico: Alcohol etoxilado.
Fuente:. (Uri & Guy, 1999)
Surfactantes anfóteros
Sustancias que pueden ionizarse positivamente o negativamente de acuerdo al medio que se
encuentre en especial al cambio de pH; pueden comportarse como tensioactivos aniónicos o
catiónicos, como ejemplo de este tipo de tensoactivos: las betaínas representado en la Fig.2.5

11
CH3
N+CH3
R
O
O-
NH
OH
R R
N
CH3
O
O-
CH3a) b)
Fig. 5.- Ejemplos de tensioactivos anfóteros. a) Alquilbetaína, b) Alquilamidopropilbetaína.
Fuente: (Uri & Guy, 1999)
2.2.3 Detergentes en aguas naturales y en aguas residuales
El potencial de los detergentes para contaminar el agua es alto debido a su uso extensivo
en distintos mercados, tanto de consumidores, como en ámbitos institucionales e
industriales. Se consumen anualmente más de cuatrocientas cincuenta mil toneladas en el
mercado doméstico de los Estados Unidos y algo más en Europa. La mayor parte de este
material, junto con otros ingredientes asociados con las formulaciones de los detergentes, se
vierte en las aguas residuales. (Baird, 2004)
2.2.4 Biodegradabilidad de los detergentes
Hasta los primeros años de la década de los 60 del siglo XX el ABS (alkilbenzene
sulfonate) era el surfactante más común usado en las formulaciones de detergentes. Sin
embargo, adolece de la desventaja de biodegradarse muy lentamente, debido a su estructura
en cadena ramificada. Una manifestación objetable de los detergentes no biodegradables lo
representó la capa de espuma que comenzó a aparecer en los vasos de agua potable en las
áreas donde el agua residual se reciclaba a través del suministro doméstico de agua. Lechos
espectaculares de espuma aparecieron cerca de las salidas del alcantarillado y en las plantas
de tratamiento de aguas residuales. Entre los otros efectos indeseables de los detergentes
persistentes en los procesos de tratamiento de aguas residuales que los llevan están la
disminución de la tensión superficial del agua, la desfloculación de los coloides; la flotación
de los sólidos; la emulsificación de grasas/aceites y la destrucción de bacterias útiles
(Weininger & Stermitz, 1988). Por consiguiente, el ABS está siendo reemplazado por un
surfactante biodegradable conocido como alquilsulfonato lineal LAS (linear alkyl sulfonate).
Por lo tanto, él LAS es mucho más biodegradable que el ABS, desafortunadamente, en países
como México, las empresas transnacionales que todavía conservan plantas productoras de
ABS en sus países de origen, exportan este producto por terceros países y lo siguen

12
manteniendo en la formulación de los detergentes en el mercado contaminando los cuerpos
o reservorios de agua de estos países. (Manahan, 2007)
Química Verde
La química verde fue adoptada como una propuesta novedosa para reducir y/o eliminar los
problemas ambientales derivados de actividades industriales. Según la US Environmental
Protection Agency (EPA), la química verde es el "uso de la química para la prevención de
la contaminación, y el diseño de productos químicos y procesos benéficos para el ambiente".
La química verde plantea 12 principios para conseguir sus objetivos.
1. Prevenir la creación de residuos. Resulta más útil evitar o reducir la producción de
desechos que tratarlos o limpiarlos tras su formación.
2. Maximizar la economía atómica. Los métodos sintéticos deben maximizar la
incorporación de cada material utilizado en el proceso.
3. Realizar síntesis química menos peligrosa. Consiste en elaborar procesos que
generen la mínima toxicidad e impacto ambiental.
4. Diseñar productos y compuestos menos peligrosos. Los productos químicos se
deben diseñar con una toxicidad mínima.
5. Utilizar disolventes y condiciones seguras de reacción. Las sustancias auxiliares de
los procesos químicos (disolventes, tampones, aditivos de separación, entre otros), han de
ser inocuas y reducirlas al mínimo.
6. Diseñar para la eficiencia energética. Debe minimizarse los requerimientos
energéticos para los procesos químicos, los cuales serán evaluados por su impacto
medioambiental y económico, y reducirlos al máximo, intentando llevar a cabo los métodos
de síntesis a temperatura y presión ambiente.
7. Utilizar materias primas renovables. Los materiales de partida utilizados deben
proceder de fuentes renovables, en la medida en que sea económica y técnicamente factible.
8. Evitar derivados químicos. La síntesis debe diseñarse con el uso mínimo de grupos
protectores para evitar pasos extras y reducir los desechos.
9. Utilizar catalizadores. Debe emplearse catalizadores lo más selectivos y
reutilizables posibles

13
10. Diseñar productos fácilmente degradables al final de su vida útil. Los productos
químicos han de ser diseñados de tal manera que al culminar su función no persistan en el
ambiente y puedan degradarse a derivados inertes o biodegradables.
11. Monitorear los procesos químicos en tiempo real para evitar la
contaminación. Debe crearse sistemas de control y monitorización continuos para prevenir
la producción de sustancias peligrosas durante los procesos.
12. Prevenir accidentes. Diseñar los procesos químicos, utilizando métodos y
sustancias que reduzcan los accidentes (emisiones, explosiones, incendios, entre otros), y
minimizar los daños cuando se produzca un accidente.
Los principios de la química verde fueron propuestos originalmente por Paul Anastas y John
Warner en su libro Green Chemistry, theory and practice (1998), y constituyen el pilar de la
química verde. La aplicación de estas estrategias en la implementación de procesos
innovadores, contribuirán a la sostenibilidad del Planeta en la sociedad, la economía y el
ambiente (Anastas, Kirchhoff, & Williamson, 2001).
Uno de los procesos que puede implementarse con base en la química verde, es la síntesis
química porque puede optimizarla mediante condiciones adecuadas, reduciendo los
requerimientos energéticos e incrementando la eficiencia de los procesos, con la catálisis y
el diseño de sustancias químicas más seguras. Por ejemplo: la oxidación de alcohol a grupo
carbonilo, genera una cantidad significativa de residuos peligrosos. En consecuencia, se ha
desarrollado un método alternativo que evita el uso de reactivos tóxicos, aplicando los
principios uno, tres y siete de la química verde. Por otra parte, Trost y sus colaboradores
demostraron cómo el uso de catalizadores maximiza la economía atómica (principio 2),
usando una variedad de catalizadores de paladio en reacciones de alquilación alílica. De
forma similar, las macrolactonas pueden obtenerse a partir del correspondiente ácido
carboxílico en un proceso catalizado por paladio, con un 100% de eficiencia atómica y a
temperatura ambiente, aplicando el principio 6. En forma análoga, los principio 4, 6 y 10, se
aplican en la síntesis catalítica de PPT (polímero poliaspartato térmico), que se utiliza en la
síntesis del ácido poliacrílico (Pájaro Castro & Olivero Verbel, 2011)
2.3.1 Solventes verdes
Los productos naturales, tales como hierbas aromáticas y especias, frutas y verduras, plantas
medicinales, micro y macroalgas, café y cacao y harinas, son mezclas complejas de

14
vitaminas, azúcares, proteínas y lípidos, fibras, aromas, aceites esenciales, pigmentos,
antioxidantes y otros compuestos orgánicos y minerales. Los análisis directos generalmente
no son posibles de lograr debido a la complejidad de las muestras de alimentos y el requisito
de muestras útiles en una forma líquida. Además, la aplicación directa de materias primas
no es posible porque en lugar de 1 g de aceite esencial utilizado para la aromatización de la
alimentación, cosmética, o industria de la perfumería, 1 kg de material aromático en bruto
será necesario (Kerton & Marriott, 2013).
Diferentes métodos pueden ser utilizados para la extracción de concentrado y valiosos
materiales, por ejemplo, la extracción de Soxhlet, maceración, la elución, y simultánea
destilación-extracción. Todas estas técnicas necesitan disolvente de petróleo para extraer los
biocompuestos. Las pérdidas de algunos compuestos, baja la eficiencia extracción, tiempo y
procedimientos (calentamiento prolongado y agitación, consumo energía, uso de grandes
volúmenes de disolventes, etc.). Estas deficiencias han llevado a la utilización de nuevas
técnicas sostenibles "verdes" y/o disolventes en la extracción, que implican típicamente
menos disolvente y energía, como por ejemplo por ultrasonido o extracción asistida por
microondas, o se refieren a disolvente alternativo, tal como extracción con fluido
supercrítico, agua subcrítica, la extracción y el uso de disolvente alternativo. La extracción
en condiciones extremas o no clásicos es actualmente una zona más dinámica y desarrollada
en la investigación aplicada y la industria. Alternativas a los procedimientos de extracción
convencionales o a los disolventes derivados del petróleo pueden aumentar la producción,
eficiencia y contribuir a la conservación del medio ambiente mediante la sustitución de la
utilización de disolventes de petróleo por solventes verdes y la reducción del consumo de la
energía fósil y la generación de sustancias peligrosas.
Con los crecientes precios de la energía y de la gasolina y la unidad para reducir el CO2 y
los compuestos orgánicos volátiles (COV) de las emisiones, la industria química y de
alimentos están en busca de nuevas tecnologías con el fin de reducir el consumo de energía
y disolventes, a cumplir con los requisitos legales en materia de emisiones, la seguridad del
producto / proceso, y el aumento de la calidad, así como la funcionalidad. La extracción con
disolventes de los productos naturales es uno de los más prometedores temas de innovación
que pueden contribuir al crecimiento sostenible de la química y de las industrias de
Alimentos. Las tecnologías de extracción existentes tienen una considerable tecnológica y
los cuellos de botella científicas para superar: a menudo requiere hasta un 50% de las

15
inversiones en una nueva planta y más de 70% de la energía total utilizada en el proceso y
menos de 50% de disolvente reciclado pierde en forma de emisiones de COV en los
alimentos, productos químicos finos y las industrias farmacéuticas. El solvente verde
utilizado para la extracción, tal como d-limoneno proporciona la base teórica necesaria y
algunos detalles (Barragan, 2010).
Limoneno: Origen, aplicaciones y Propiedades
El d-Limoneno es un hidrocarburo mono terpeno encontrado como el principal componente
del aceite esencial de cáscaras de cítricos (Fig. 6). Con una producción de más de 50 millones
de toneladas, la industria del zumo de naranja representa una fuente importante de d-
limoneno y una plataforma piloto de investigación desafiante para la valorización de los
subproductos. Se considera como GRAS (generalmente reconocido como seguro) de
material por la Administración de Alimentos y Fármacos de Estados Unidos y ha estado
jugando un papel importante en sabores y fragancias, así como agente de limpieza /
desengrasante en la industria y en aplicaciones domésticas. d-Limoneno se erige como un
sustituto valioso para los disolventes tradicionales, muchos de los cuales emiten
hidrocarburos aromáticos policíclicos (HAPs) o vapores de compuestos orgánicos volátiles
(COV). (Mira.B, Blasco.M, Berna.A, & Subirats.S, 1999)
CH3
CH3 CH2
Fig. 6.- Estructura química de d-limoneno
Fuente: (W.M.Haynes, 2013-2014)
Los disolventes que son comúnmente reemplazados con disolvente d-limoneno incluyen:
metiletil cetona, acetona, tolueno, éteres de glicol, y numerosos compuestos fluorados y

16
clorados. En la industria, el disolvente d-limoneno se mezcla típicamente con un agente
tensioactivo, produciendo una solución que contiene 5 a 15% de d-limoneno.
El principal inconveniente de la utilización de d-limoneno es su baja viscosidad y el mayor
consumo de energía relacionado con la recuperación de disolventes por evaporación debido
a su alto punto de ebullición (175°C), en comparación con n-hexano (69 °C). (Guenther.E,
2013)
Las propiedades químicas y físicas de d-limoneno en comparación con otros disolventes se
ilustran en la Tabla 1.
Tabla 1.- Propiedades relevantes de n-hexano, tolueno, diclorometano, benceno y d-limoneno
Propiedades n-Hexano Tolueno Diclorometano Benceno d-Limoneno
Fórmula Empírica C6H14 C6H5CH3 CH2Cl2 C6H6 C10H16
Peso Molecular (g/mol) 86.1 92.14 84.93 78.11 136.23
Punto de ebullición(°C) 68.7 110.6 40.0 80.1 175.5
Calor de vaporización
(KJ/kg. K)
334 351 28.6 396.0 353
Densidad 0.6603 0.8669 1,3250 0,879 0,8411
Toxicidad Si Si Si Si No
Impacto Ambiental Alto Alto Alto Alto Bajo
Fuente: (W. M. Haynes, 2013-2014)
Durante 130 años, Soxhlet ha sido la técnica más común para la recuperación de las grasas
y aceites procedentes de los vegetales; en la enseñanza y la investigación. Sin embargo, los
principales inconvenientes iniciada con Soxhlet como tiempo de extracción de largo, el

17
consumo de energía, y el uso de grandes cantidades de disolventes derivados del petróleo
han pedido un aumento de las preocupaciones ambientales. Por ejemplo, n-hexano, el
disolvente de elección para las grasas y aceites que utilizan la extracción de Soxhlet, está
clasificado en la parte superior de la lista de los disolventes peligrosos. Durante años,
muchos investigadores se han concentrado sus esfuerzos en la búsqueda de disolventes
alternativos. (Mohammad, 2012)
Serie eluotrópica
Una serie eluotrópica es una lista de disolventes ordenados según su poder de elución para
un adsorbente determinado. Esta serie es importante para determinar los
disolventes necesarios en la cromatografía de una mezcla de compuestos . Normalmente
comienzan con disolventes no-polares, como el n-hexano, y finalizan en disolventes polares
como el agua. El orden de disolventes en una serie eluotrópica depende a la vez de la fase
estacionaria así como del compuesto empleado para determinar el orden.
La fuerza de elución, fuerza eluyente, o fuerza eluotrópica, (εº), de un disolvente es una
medida de la energía de adsorción del disolvente tomando como referencia el sistema
pentano-sílice pura al que se asigna el valor 0,00. Por ejemplo, la serie de Snyder cuantifica
la fuerza de elución de cada disolvente en alúmina . Dicha fuerza indica la facilidad del
disolvente para formar enlaces de hidrógeno con las moléculas que queremos extraer, la cual
depende de su constante dieléctrica o de su momento dipolar. (Cazes, 2005)
2.4.1 Índice de polaridad de Snyder (P)
La capacidad de una molécula de interactuar en el medio se conoce como polaridad.
Polaridad de solventes puros. - Se define como la sumatoria del logaritmo de la constante de
distribución de los componentes presente.
P = ∑(logKD1 + logKD2 + logKD3 + ⋯ ) (2-1)
Mezcla de Solventes. - En general un solvente A que resulta demasiado débil, se mezcla con
otro solvente B que es demasiado fuerte se puede llegar a obtener una mezcla que sea útil
para lograr el objetivo mediante la siguiente ecuación se calcula la polaridad de una mezcla
de solventes.

18
P = XaPa + XbPb (2-2)
Donde Xa y Xb, representa las fracciones en volumen de los solventes y Pa, Pb la polaridad
de los solventes en estado puro. Los valores de polaridad y fuerza de elución se indica en la
siguiente Tabla 2.
Tabla 2.- Propiedades de los solventes orgánicos Fuerza de elución y polaridad.
Fuente: (Hamilton & Swell, 1982 Second Edition)
Método de extracción
2.5.1 Generalidades
Los métodos de extracción tienen mucho en común con los métodos de destilación. En la
destilación, la separación de los componentes se efectúa debido a la diferencia en las
presiones de vapor o volatilidad de los componentes. A una determinada temperatura y
presión, las concentraciones de equilibrio de un componente en la fase líquida, Cl y en la
fase vapor Cv se expresa mediante la ecuación (Lamarque & Zyjadlo, 2008)
K =Cl
Cv (0-3)
Solvente Fuerza Polar Índice de
Polaridad
Hexano 0,0 0,0
Dietil éter 1,4 2,9
Tetracloruro de carbono 0,0 1,7
Tolueno 0,7 2,3
Acetato de etilo 2,6 4,3
d-Limoneno 0,6
Benceno 0,5 3,0
Cloroformo 1,5 4,4
Acetona 5,1 5,4
Diclorometano 3,1 3,4
Acetonitrilo 8,8 6,2
Agua 15,3 9,0

19
En donde K es una constante de equilibrio. En un sistema de dos componentes, La K del
componente menos volátil es mayor que la unidad, y la K del componente más volátil es
menor que la unidad, por lo que en el proceso de evaporación se logra una separación parcial
de la mezcla inicial. La extracción es proceso análogo, en el que un soluto se distribuye entre
dos disolventes inmiscibles. Una ley similar define la relación de las concentraciones del
soluto en los dos disolventes, 1 y 2.
K =C1
C2
(2-4)
En donde K es el coeficiente de distribución o coeficiente de partición, un tipo especial de
constante de equilibrio que es prácticamente igual a las solubilidades relativas del soluto en
los dos disolventes. A menudo uno de los disolventes es agua y el otro es un disolvente
orgánico, con objeto de que las substancias inorgánicas iónicas, así como los compuestos
orgánicos polares se queden, en su mayor parte en la fase acuosa y los compuestos orgánicos
no polares pasen, en su mayor parte también a la fase orgánica. Esta es otra forma de decir:
“similar disuelve similar” En soluciones diluidas, en primera aproximación el coeficiente de
distribución es independiente de la concentración.
Extracción simple. Ley de distribución
Si el coeficiente de distribución es muy grande (>100), es probable que, con una sola
extracción, en un simple embudo de separación, sea suficiente para pasar un soluto de una
fase a otra. Sin embargo, se ha demostrado que es más efectivo emplear una cantidad dada
de disolvente de extracción en varias porciones pequeñas, que toda de una sola vez.
Extracción múltiple.
Después de la primera extracción cada una de las fases se puede poner en equilibrio
nuevamente con el disolvente contrario. Mediante una reciclización de las diferentes
fracciones intermedias se puede lograr, finalmente, una separación satisfactoria a costa de
considerables cantidades de disolvente y numerosas manipulaciones. El aparato de Craig
está diseñado para efectuar esta operación semiautomáticamente. Las relaciones
matemáticas que describen el proceso de Craig sirven para comprender muchas operaciones

20
en columna. El lineamiento de distribución de una substancia, a medida que avanza en este
aparato, se asemeja al de una separación por cromatografía (Pecsok & Shields, 1973).
Equilibrio Químico
Cuando una reacción química se realiza a una temperatura constante en un medio
homogéneo y transcurre de forma reversible la reacción química constituida por reactivos y
productos, se denomina Equilibrio Químico. Los equilibrios químicos transcurren a una
velocidad típica y está relacionada con el tiempo necesario para alcanzar el equilibrio. Todos
los equilibrios químicos son reacciones químicas, pero no todas las reacciones químicas son
equilibrios químicos.
Reactivos ↔ equilibrio ↔ productos
Para los análisis químicos interesan los equilibrios químicos que tienen lugar de forma
instantánea (Sanz Asencio, 2013).
La magnitud hacia el equilibrio determina, el cambio de energía libre entre los estados inicial
y final, y se representa por ∆G.
∆G = Gfinal − Ginicial (2-5)
La variación de energía libre en una reacción química cuando reactivos y productos están en
un estado normal o de referencia. Se denomina ∆G° y se calcula a partir de las energías libres
normales de formación.
∆G = ∆G°f(productos) − ∆G°f(reactivos) (2-6)
La termodinámica es el estudio de las características térmicas, eléctricas, químicas y
mecánicas; los estudios de termodinámica involucran muchas disciplinas, incluso la física,
la ingeniería y la química (Martí, Conde, Jimeno, & Mendez, 2008).
De las diversas ramas de la termodinámica, la más importante a la química es el estudio del
cambio en la energía durante una reacción química. Consideremos, por ejemplo, la reacción
de equilibrio general se muestra en la ecuación, que implica las especies A, B, C y D con los
coeficientes estequiométricos a, b, c y d.

21
aA + bB ⇋ cC + dD (2-7)
Dependiendo de las condiciones iniciales, la reacción se puede mover hacia la izquierda,
mover a la derecha, o estar en un estado de equilibrio. La comprensión de los factores que
determinar la posición final del equilibrio de las reacciones es uno de los objetivos de la
termodinámica química.
La energía libre de la reacción viene dada por función de la energía libre de Gibbs.
ΔG = ΔH − TΔS (2-8)
Donde T es la temperatura en grados Kelvin, y la 𝚫G, 𝚫H y 𝚫S son las diferencias en la
energía libre de Gibbs, la entalpía y la entropía entre los productos y los reactivos. El signo
de 𝚫G indica la dirección en la que una reacción se traslada hasta
llegar a su posición de equilibrio. Una reacción es termodinámicamente favorable cuando su
entalpía, 𝚫H disminuye su entropía 𝚫S aumenta. Sustituyendo las desigualdades 𝚫H <0
y 𝚫S> 0 en la ecuación muestra que una reacción es termodinámicamente favorable cuando
𝚫G es negativo; cuando es 𝚫G, positiva la reacción es desfavorable como está escrito
(aunque la reacción inversa es favorable). Una reacción en equilibrio tiene un 𝚫G de cero.
(Harvey, 2009).
Pares iónicos
El tratamiento por seguir para el estudio de extracción de pares iónicos es muy similar para
quelatos neutros. En este caso se trata de extraer una especie cargada, por ejemplo, el anión
A- , con una especie catiónica X+ que actúa como contra ion, formando un par iónico AX,
neutro e insoluble en la fase acuosa. La especie A- puede ser un anión orgánico o inorgánico
o bien un complejo o un quelato metálico cargado. La reacción de formación del par iónico
será.
A− + X+ ⇆ AX (2-9)

22
Este equilibrio está regido por la constante de asociación:
Kas =[AX]
[A−][X+]
(2-10)
En un sistema extractivo dado, este par iónico se distribuirá entre ambas fases según el
equilibrio siguiente:
XAa ⇆ XAo (2-11)
Cuya constante de distribución es:
KD = [XA]o
[XA]a
(2-12)
Considerando el proceso de formación y extracción del par iónico en forma global, el
equilibrio que se establece es el siguiente:
[𝐴−]𝑎 + [𝑋+]𝑎 ⇆ [𝑋𝐴]𝑜 (2-13)
Siendo la constante de extracción:
Kex = [XA]o
[A−]a[X+]a
(2-14)
Cuyo valor puede conocerse a partir de las constantes de equilibrio indicadas
anteriormente: multiplicando y dividiendo por [𝑋𝐴]𝑎 queda:
Kex = [XA]o
[A−]a[X+]a [XA]a
[XA]a= KasKD
(2-15)

23
Lo que indica que la extracción se favorece cuanto mayor es la constante de formación del
par iónico.
Para el interés de la investigación el par iónico debe ser más soluble en la fase orgánica
que en la fase acuosa, que sea capaz de extraer en un solo proceso.
La relación de distribución tiene la siguiente expresión:
D =[XA]o
[A−]a + [XA]a
(2-16)
En función de la constante de extracción queda:
D = Kex[X+]a = KasKD[X+]a (2-17)
Demostrándose que la extracción del par iónico también se favorece al aumentar la
concentración del contra ion en fase acuosa. En ciertos casos, en lugar de aumentar
excesivamente la concentración del contra ion, se varía la naturaleza de este.
La fase orgánica utilizada y, en especial, su capacidad para solvatar al par iónico tienen gran
influencia en la constante de extracción. (Valcarcel Cases & Gómez Hens, 1988).
Fuerza Iónica
Con el objeto de sistematizar los resultados para los coeficientes de actividad de sales
sencillas, tanto en estado puro como en presencia de otros electrolitos, introdujeron el
concepto de fuerza iónica (G.N. Lewis y M. Randall, 1912). Esta propiedad de una
disolución, representada por 𝜇𝑚 , se define como la semisuma de los términos obtenidos
multiplicando la molalidad de cada ion presente en disolución por el cuadrado de su valencia;
así.
μm =1
2∑ mjzj
2 (2-18)

24
Donde m representa la molalidad del ion j y zj es su valencia, efectuándose la suma para
todos los iones presentes en la disolución. (Glasstone, 1977)
Espectrometría de Absorción Ultravioleta – Visible
En el presente trabajo se tomó en cuenta la aplicación de la espectrofotometría de UV-visible
para la determinación de detergentes aniónicos en una variedad de muestras de agua. Sin
embargo, con el fin de aplicar espectrofotometría UV – visible apropiada y confiable, se
debe tener una cierta comprensión de los principios y prácticas sobre la que se basa esta
metodología.
2.9.1 Interacción de la luz con la materia
Los procesos espectroscópicos se basan en el hecho de que la radiación electromagnética
(EMR) interactúa con átomos y moléculas en formas discretas para producir perfiles de
absorción. Nuestra propia capacidad de percibir el color se debe a la actuación del ojo
humano como un detector para EMR. La propiedad de la REM que determina la gama de
color que se percibe es la longitud de onda. La parte del espectro electromagnético que se
conoce, como era de esperar que el ojo puede detectar, en la región visible. EMR puede ser
simplemente representada como una onda sinusoidal. Longitud de onda, λ, es la distancia
entre los picos o valles adyacentes. Nuestra capacidad para percibir el color depende de
muchos factores. Sin embargo, el mecanismo de interacción de EMR con la materia es de
gran importancia. Desde un punto de detección visual de vista, nuestra capacidad para
percibir diferentes colores depende del proceso óptico implicado, por ejemplo, si la luz es
absorbida o reflejada por el objeto observado. La longitud de onda, λ, de EMR puede ser
expresada como una función de su frecuencia, ν, y la velocidad de la luz, c, por la siguiente
ecuación simple:
ν = c
λ
(2-19)
Nuestros ojos no son uniformes con la respuesta a EMR en la región visible del espectro.
Ellos son los más sensibles en la región de 600 nm. La Figura 2 muestra la sensibilidad
relativa del ojo a la luz visible. Esta figura ilustra la importancia de la sensibilidad del
detector y el rango de longitud de onda para los detectores espectroscópicos. Sin embargo,

25
EMR se comporta como una partícula y como onda (la naturaleza dual de la luz); y la
longitud de onda de una partícula tal, un fotón, se relaciona con la energía por la ecuación.
E =hc
λ109
(2-20)
donde h es la constante de Planck (6.63 × 10-34 J s), c es la velocidad de la luz en el vacío
(2.998 × 108 ms-1), E es la energía del fotón y λ es la longitud de onda en nm. La región
visible del espectro electromagnético constituye, una pequeña parte, como puede verse en la
Fig.2.6
Es evidente que existe un enorme lapso de energías, de más de 18 órdenes de magnitud. La
ecuación que vincule la energía de longitud de onda es de fundamental importancia en la
espectroscopia.
2.9.2 Espectroscopía de absorción molecular uv-visible
La espectroscopia por absorción molecular se basa en la medición de la Transmitancia(T) o
de la Absorbancia(A) de soluciones que están en celdas transparentes que tienen una longitud
de trayectoria de b cm. Normalmente, la concentración de un analito absorbente se relaciona
en forma lineal con la absorbancia según la ley de Beer:
A = −logT = logPo
P= εbc
(2-21)
Fig. 7.- Espectro electromagnetic.
Fuente: Burgess Consultancy, ‘Rose Rae’, the Lendings, Startforth, Barnard Castle, Co
Durham, DL129AB, England

26
Por lo regular, la T y la A, no pueden medirse en el laboratorio porque la solución del analito
debe estar en un recipiente o cubeta transparente. La reflexión se presenta en las dos
interfaces aire/pared del recipiente y en las dos interfaces pared/solución. Fig. 6
Para compensar todos estos efectos, la potencia del haz transmitido por la solución del
analito se compara con la potencia del haz transmitido por una celda idéntica que contiene
sólo solvente. Entonces, la T y A experimental que se aproximan de manera notable a la T y
A verdadera se obtienen mediante las ecuaciones:
T = Psolución
Psolvente≈
P
Po (2-22)
A =logPsolvente
Psolución ≈ log
Po
P (2-23)
Los términos P y Po se refieren a la potencia de radiación, después de pasar a través de celdas
o cubetas que contienen al solvente y a las soluciones del analito.
Fig. 8.- Pérdidas por reflexión y dispersión.
Fuente: F. Settle, ed., Handbook of Instrumental Techniques for Analytical
Chemistry

27
2.9.3 Ley de Beer
La Ley de Beer se representa mediante la ecuación (2-21). La relación matemática se
define de la siguiente manera; considere el bloque de material absorbente (sólido, líquido o
gas) que se muestra en la Fig. 13.2.
Un haz de radiación monocromático paralelo de potencia Po choca de forma perpendicular
contra la superficie del bloque. Después de atravesar una longitud b de material, que contiene
n átomos, iones o moléculas absorbentes, su potencia disminuye hasta un valor P como
resultado de la absorción. Considérese una sección transversal del bloque con área S y
espesor infinitesimal dx. Esta sección contiene dn partículas absorbentes en la cual tendrá
lugar la captura del fotón y dará lugar a la absorción. El área total proyectada de estas
superficies se designa como dS; la relación entre el área de captura y el área total es dS/S,
esta relación representa la probabilidad de capturar fotones dentro de la sección.
La potencia del haz que entra en la sección, Px es proporcional al número de fotones por
unidad de área, y dPx representa la potencia absorbida en la sección.
La fracción absorbida es -dPx/Px, y esta relación es igual a la probabilidad media de captura.
Por consiguiente:
−dPx
Px=
dS
S
(2-24)
Fig. 9.- La radiación de inicial Po y la radiación de salida P luego de
atravesar la celda que contiene la solución con el analito.
Fuente: F. C. Strong, Anal. Chem.,1952, 24, p. 338.

28
Donde dn número de partículas y a es una constante de proporcionalidad que puede llamarse
sección transversal de captura.
− ∫dPx
Px= ∫
adn
S
n
0
P
Po ; al resolver la integral se tiene: −ln
P
Po=
an
S ;
Luego de transformar en logaritmos decimales se tiene:
logPo
P=
an
2,303S
(2-26)
Si el área S se expresa en función del volumen (V) dividido para la distancia de la celda b,
tenemos:
S =V
b
Por tanto, los valores numéricos se agrupan en una sola constante ɛ y la ley de Beer, se
representa mediante la siguiente ecuación.
logPo
P= ɛ. b. c = A
(2-27)
2.9.4 Aplicaciones de la Ley de Beer a mezclas
La Ley de Beer se puede aplicar a sustancias que contengan más un componente absorbente,
siempre que no haya interferencia entre las distintas especies. La absorbancia total para un
sistema de múltiples componentes se tiene:
dS = adn (2-25)

29
Atotal = ɛ1bc + ɛ2bc + ⋯ + ɛnbc (2-28)
Donde los subíndices 1, 2, y n son los componentes absorbentes.
2.9.5 Limitaciones de la Ley de Lambert-Beer
Se han encontrado pocas excepciones a la generalización de que la absorbancia está
relacionada en forma lineal con la longitud de la trayectoria. Por otra parte, se han encontrado
desviaciones frecuentes de la proporcionalidad directa entre la absorbancia medida y la
concentración cuando b es constante. Algunas de estas desviaciones, llamadas desviaciones
reales, son fundamentales y representan limitaciones propias de la ley. Otras resultan de la
forma en que se realizan las mediciones de absorbancia (desviaciones instrumentales) o son
consecuencia de cambios químicos que ocurren cuando se modifica la concentración
(desviaciones químicas).
Limitaciones reales de la Ley de Beer
La ley de Beer describe el comportamiento de absorción de medios que contienen
concentraciones de analito relativamente bajas; en este sentido es una ley restrictiva. A
concentraciones altas (casi siempre > 0.01 M), el grado de las interacciones soluto-solvente,
soluto-soluto, o los puentes de hidrógeno pueden afectar el ambiente del analito y su
capacidad de absorción. También surge desviaciones de la ley de Beer porque la absortividad
depende del índice de refracción del medio.
Desviaciones químicas aparentes
Se produce desviaciones aparentes de la Ley de Beer cuando un analito se disocia, se asocia
o reacciona con un solvente para originar un producto con un espectro de absorción diferente
al del analito.
Desviaciones instrumentales debidas a la radiación policromática
La Ley de Beer se cumple en forma rigurosa cuando las mediciones se realizan con radiación
monocromática. Las fuentes policromáticas que tienen una distribución continua de

30
longitudes de onda se usan junto con una red o filtro para aislar una banda más o menos
simétrica de longitudes de onda que rodean a la longitud de onda que se quiere usar.
Desviaciones instrumentales debidas a radiación parásitas
La radiación que emerge del monocromador está contaminada por pequeñas cantidades de
radiación dispersa o parásita llamada luz parásita, y se define como la que proviene de un
instrumento que esta fuera de la banda de longitud de onda seleccionada para la
determinación. Por lo general esta luz parásita se debe a la dispersión, reflexión desde las
superficies de redes, lentes o espejos, filtros y ventanas. La radiación parásita difiere de la
radiación principal; puede que no atraviese la muestra. (Sharpe, 1984)
Celdas desajustadas
Si la longitud de trayectoria de las celdas que contienen las soluciones del analito y del
blanco no son iguales ni tienen características ópticas equivalentes, habrá una ordenada al
origen k en la curva de calibración. Para que no ocurra este error se utilizan celdas
cuidadosamente ajustadas o se aplica un procedimiento de regresión lineal para calcular
tanto la pendiente como la ordenada al origen de la curva de calibración, siendo la mejor
estrategia. Otra manera de evitar el problema de las celdas desajustadas es usar sólo una
celda y mantenerla en la misma posición tanto en la medición del blanco como en la del
analito.
2.9.6 Efectos del Ruido instrumental
En los análisis espectrofotométricos, con frecuencia, la exactitud y la precisión están
limitados por las incertidumbres o ruidos asociados con los instrumentos. (Rothman, Crouch,
& Ingle, 1975)
Ruido instrumental como función de la transmitancia
Una medición espectrofotométrica involucra tres pasos: un ajuste de 0% de transmitancia,
un ajuste del 100% de transmitancia y una medida del porcentaje de T cuando la muestra se
sitúa en la trayectoria de la radiación. El ruido asociado con cada una de estas etapas se
combina para dar una incertidumbre neta al valor final de T.
Fuentes de ruido instrumental

31
En un estudio teórico y experimental Rothman, Crouch y Ingle describieron varias fuentes
de incertidumbre instrumentales y mostraron su efecto neto sobre la precisión de las
mediciones de la absorbancia o transmitancia. Estas incertidumbres se dividen en tres
categorías: Caso I, la precisión es independiente de T. Caso II, la precisión es directamente
proporcional a (T2 + T)1/2. Caso III, las incertidumbres son directamente proporcionales a
T.
Efecto de la anchura de la rendija en las mediciones de la Absorbancia
Para resolver espectros complejos se requieren anchuras de rendija angostas. Pero el
inconveniente es que al reducir la anchura de rendija en un factor de 10 hay una reducción
del poder radiante en un factor de 100 porque la energía radiante es proporcional al cuadrado
de la anchura de rendija. Existe una relación de desventaja entre la resolución y la relación
señal-ruido, a menudo se elige la relación promedio.
En general es aconsejable no estrechar la rendija más de lo que sería necesario para la
resolución del espectro.
Efecto de la radiación dispersada
En los extremos de la Longitud de Onda de un instrumento, cuando las medidas se realizan
a longitudes de onda extremas de un instrumento, los efectos de la radiación parásita pueden
ser incluso más grave y, en ocasiones puede aparecer bandas de absorción falsas.
A longitudes de onda por debajo de alrededor de los 380 nm, las ventanas, las celdas y el
prisma comienzan a absorber radiación, lo que reduce la energía que llega al transductor. La
señal de salida de la fuente desciende con rapidez en esta región y también disminuye la
sensibilidad del transductor. Por consiguiente, la señal total para el ajuste de 100 % T puede
ser tan bajo como 1 o 2 % de la señal correspondiente en la región comprendida entre 500 y
650 nm. La radiación dispersada está compuesta a menudo por longitudes de onda a las
cuales el instrumento es muy sensible. Por tanto, los efectos de la radiación parásita pueden
verse muy amplificadas, en algunos casos la señal de salida producida por la radiación
parásita podría exceder a la del haz que proviene del monocromador, en estas circunstancias
el componente de la T medida que se debe a la radiación parásita podría ser igual a la T
verdadera, o hasta podría excederla.

32
2.9.7 Instrumentación
En la actualidad existen cientos de instrumentos y modelos a elegir. Algunos son sencillos y
barato; otros son equipos complejos controladas por computadoras y caro. Con frecuencia
los instrumentos más sencillos son útiles en la región visible para mediciones cuantitativas
en una sola longitud de onda. Los instrumentos más complejos tienen la capacidad de realizar
barridos espectrales a resoluciones que se puede elegir, son capaces de medir en la región
visible y ultravioleta, compensar las fluctuaciones en la intensidad de la fuente y varias otras
características. (Jarnutowski, Ferraro, & Lankin, 1992)
Componentes de los instrumentos
Los instrumentos para medir la radiación ultravioleta y visible están compuestos por uno o
más de los siguientes componentes: fuentes, selectores de longitud de onda, recipientes para
la muestra, transductores de radiación y procesadores de señal-dispositivos de lectura.
Fuentes
Para las mediciones de absorción molecular es necesario disponer con una fuente continua,
cuya potencia radiante no cambie en forma brusca en un intervalo considerable de longitudes
de onda.
Lámparas de Deuterio e Hidrógeno. La excitación eléctrica del deuterio o
hidrogeno a baja presión produce un espectro continuo en la región ultravioleta. El
mecanismo por el cual se produce dicho espectro requiere la formación inicial de una especie
molecular excitada y luego su disociación para dar dos especies atómicas más un fotón
ultravioleta. La mayor parte de las lámparas modernas de este tipo contienen Deuterio y son
de bajo voltaje (40V). En la región UV (190-400 nm) produce un espectro continuo. A
longitudes mayores a 400 nm los espectros dejan de ser continuos, y están formado de líneas
de emisión y bandas que se superponen sobre el espectro continuo débil.
Lámparas de filamento de Tungsteno. La fuente más común de radiación visible
es la lámpara de filamento de tungsteno. La lámpara de filamento de tungsteno es útil para
la región de longitudes de onda comprendidas entre 350 y 2500 nm. En la región visible, la
energía de señal de salida de una lámpara de tungsteno varía aproximadamente con la cuarta
potencia del voltaje de operación. Las lámparas tungsteno/halógenos, también llamadas

33
lámparas de cuarzo-halógeno, contienen una cierta cantidad de yodo dentro de la envoltura
de cuarzo, en la que se aloja el filamento de tungsteno. El tiempo de vida de una lámpara de
filamento de tungsteno/halógeno es el doble que la de una lámpara normal, por esta razón se
encuentra en los equipos modernos este tipo de lámparas.
Diodos emisores de luz. Un diodo emisor de luz (LED) es un dispositivo de unión
pn que cuando tiene polarización directa produce energía radiante. Son muy comunes los
diodos fabricados con arseniuros de aluminio y galio (λm = 900 nm), fosfuro de arsénico y
galio (λm = 650 nm), fosfuro de galio (λm = 550 nm), nitruro de galio (λm = 465 nm) y nitruro
de galio e indio (λm = 450 nm). Las mezclas de estos compuestos se usan para desplazar el
máximo de longitud de onda a cualquier lugar en la región 375 a 1000 nm o más. Estos
diodos producen espectros continuos en un intervalo angosto de longitud de onda (20 a 50
nm). Estos diodos se pueden usar como fuentes semicromáticas o con filtros de interferencia
para hacer aún más angosta la salida del espectro. Puede trabajar en modo continuo o modo
pulsado.
Tienen la ventaja de poseer vidas útiles más largas y afectan menos al medio ambiente.
Lámparas de arco de xenón. Estas lámparas producen una radiación intensa al
pasar la corriente a través de la atmosfera de xenón. El espectro es continuo en un intervalo
comprendido entre casi 200, 1000 nm, con el máximo de intensidad alrededor de 500nm. En
algunos instrumentos las lámparas funcionan en forma intermitente mediante descargas
regulares que proceden de un condensador.
Recipientes para la muestra
Al igual que los instrumentos ópticos de un instrumento de Absorción, las celdas o cubetas
en las que se coloca la muestra y el disolvente debe ser de un material que deje pasar la
radiación de la región espectral de interés. Para trabajar en la región ultravioleta (<350 nm)
se requiere de recipientes de cuarzo o sílice fundido.
Los vidrios de silicatos se emplean en la región entre 350 y 2000 nm. También se puede
utilizar recipientes de plástico en la región visible.
Las mejores celdas tienen ventanas perpendiculares a la dirección del haz para reducir al
mínimo las perdidas por reflexión. La longitud de la trayectoria más común para los estudios
en las regiones UV y visible es de 1cm. También se puede encontrar cubetas 0,1 hasta 10

34
cm. Al utilizar cubetas cilíndricas se debe poner especial cuidado en repetir la posición de la
cubeta respecto al haz, debido a que puede causar errores importantes. La calidad de las
medidas de absorbancia depende en gran medida del uso y mantenimiento que se haga a las
cubetas. Las huellas dactilares, la grasa u otras señales sobre las paredes de la celda alteran
las características de transmisión. Por tanto, es indispensable una limpieza completa antes y
después de usarlas. Las cubetas ajustadas nunca deben secar mediante la acción de calor, en
un horno o sobre una llama ya que este tratamiento puede causar daños físicos o cambios en
la longitud de la trayectoria.
Tipos de Instrumentos
Entre los tipos de instrumentos espectroscópicos se citan los cuatro más generales: 1) de haz
sencillo, 2) de doble haz especial, 3) de doble haz temporal y 4) multicanal.
Instrumentos de haz sencillo
Un instrumento de haz sencillo consta de una lámpara de tungsteno o deuterio, un filtro o un
monocromador para seleccionar la longitud de onda, cubetas ajustadas que pueden
interponerse de manera alternada en el haz de la radiación, un transductor, un amplificador
y un dispositivo de lectura. Por lo regular, un instrumento de haz sencillo necesita de una
fuente de alimentación estabilizada. Entre los instrumentos de haz sencillo hay grandes
diferencias en cuanto a su complejidad y características de funcionamiento. El más sencillo
y barato consta de una bombilla de tungsteno conectada a una batería, un conjunto de filtros
de vidrios, tubos de ensayos en donde va la muestra, una celda fotovoltaica como transductor
y un pequeño medidor analógico como dispositivo de lectura. En el otro extremo están los
instrumentos complejos controlados mediante computadoras, que funcionan en un intervalo
de longitudes de onda de 200 a 1000 nm o más. Estos espectrofotómetros utilizan como
fuente intercambiable lámparas de tungsteno o deuterio, celda de sílice rectangulares y están
equipados con un monocromador de red de alta resolución y rendijas variables. Como
transductor se emplean tubos fotomultiplicadores y la señal de salida, a menudo se digitaliza,
se procesa y se almacena en una computadora.
Instrumentos de doble haz

35
Muchos fotómetros y espectrofotómetros modernos se basan en un diseño de doble haz. Los
instrumentos de doble haz tienen la ventaja de que compensan todo menos la mayoría de las
fluctuaciones cortas en la salida radiante de la fuente, así como la deriva en el transductor y
el amplificador. Además, permite el registro continuo de espectros de transmitancia o
absorbancia.
Instrumentos de varios canales
Un nuevo tipo de espectrofotómetro apareció en el mercado a principio de los años ochenta,
se basa en uno de los detectores en serie. Estos instrumentos se apegan al diseño de un solo
haz. Con los sistemas multicanal, el sistema dispersor es un espectrógrafo de red colocado
después de la celda de la muestra o referencia. El detector en serie se instala en el plano focal
del espectrógrafo, en donde choca la radiación dispersada.
Con los diseños de un solo haz, la corriente residual del acomodo se mide y se almacena en
la computadora.
Los instrumentos multicanal se pueden configurar como espectrofotómetros de doble haz
temporales. Los espectrómetros multicanal utilizan tanto fotodiodos en serie como
dispositivos de acoplamiento de carga que tienen la capacidad de un espectro completo en
apenas algunos milisegundos.
Un instrumento multicanal es una poderosa herramienta para estudios de productos
intermedios transitorios en reacciones moderadamente rápidas, para estudios cinéticos y para
las determinaciones cualitativas y cuantitativas de los componentes que salen de una
columna de cromatografía de líquido o de una columna de electroforesis capilar. (Skoog,
Holler, & Crouch, 2008)
Desarrollo de Métodos Analíticos
El desarrollo de un método analítico implica por un lado la adaptación de un método ya
existente realizando cambios menores para adecuarlo a una nueva aplicación. Por ejemplo,
un método para determinar tolueno en agua puede ser adaptado a partir de un método
establecido para el benceno en agua. La matriz es la misma, y los dos analitos tienen
propiedades muy similares.

36
Es probable que los mismos principios de separación, identificación, cuantificación que se
aplican al benceno también puedan ser aplicables al tolueno. Si, por el contrario, se requiere
un método para determinar el benceno en el suelo, la adaptación del método de benceno en
agua puede no ser la mejor opción.
La adaptación de algún otro método para la determinación de materia orgánica en el suelo
puede ser un mejor punto de partida.
Por otro lado, el químico analítico puede comenzar a esbozar algunas ideas y aplicar su
conocimiento y experiencia para idear un método adecuado. Esto implica claramente mucho
más trabajo y más dudas sobre la idoneidad del método final. No es inusual para el desarrollo
del método trabajar con varias ideas simultáneamente antes de elegir finalmente la más
adecuada. A pesar del esfuerzo invertido en el desarrollo del método, no hay ninguna
garantía de que el método vaya a funcionar adecuadamente durante la validación (o bajo
condiciones de rutina en un laboratorio particular). Al implicar a diferente personal en la
fase de desarrollo y de validación, se puede comprobar que las instrucciones (del
procedimiento de medición) se entienden y se aplican. (Morillas Bravo, 2016)
Selección de un Método Analítico
En la selección de un método en muy importante considerar el tiempo que requerirá el
análisis, lo sensible que debe ser el método y al intervalo de concentraciones al que debe
adaptarse, selectividad que se requiere, las propiedades físicas y químicas de la matriz de la
muestra y una consideración importante desde un punto de vista económico es el número de
muestras que hay que analizar; si es elevado, se puede invertir más tiempo y dinero en la
instrumentación, en el desarrollo del método y en la calibración. Además, si el número sea
muy elevado, debería elegirse un método que precisara del mínimo tiempo de dedicación del
operador a cada muestra. Por otro lado, si solo se han de analizar pocas muestras, la elección
adecuada suele ser la de un método más sencillo, aunque sea más largo y que requiera poco
o ningún tratamiento previo.
Calibración de métodos instrumentales
Una parte muy importante de todos los procedimientos analíticos es la calibración y
estandarización del proceso. La calibración determina la relación entre la respuesta analítica
y la concentración del analito. Por lo regular, se determina mediante el uso de normas

37
químicas. Casi todos los métodos analíticos requieren algún tipo de calibración según
normas químicas. (Skoog, Holler, & Crouch, 2008).
2.12.1 Comparación con estándares
Se describen dos tipos de métodos de comparación, la técnica de comparación directa y el
procedimiento de titulación.
Comparación directa
Algunos procedimientos analíticos requieren la comparación de una propiedad del analito (o
del producto de una reacción con el analito) con estándares o patrones tales que la propiedad
que se está probando concuerde de manera muy cercana con la del estándar. Por ejemplo, en
los primeros colorímetros, el color producido como resultado de una reacción química de un
analito se comparaba con el color producido por la reacción de estándares. Si la
concentración del estándar se variaba por dilución, por ejemplo, era posible obtener una
coincidencia de color casi exacta. La concentración del analito era entonces igual a la
concentración del estándar después de la dilución. Tal procedimiento se llama comparación
nula o método de isomación.
Titulaciones.
Están entre las más precisas de todos los procedimientos analíticos, en una titulación, el
analito reacciona con un reactivo estandarizado, la cantidad de titulante varía hasta que se
alcance la equivalencia química, según lo indica el cambio de color del indicador químico o
el cambio en una respuesta del instrumento.
2.12.2 Calibración de un estándar externo
Un patrón externo se prepara por separado de la muestra. En cambio, un estándar interno se
añade a la muestra. Los estándares externos se usan para calibrar instrumentos y

38
procedimientos cuando no hay efectos de interferencia de la matriz de componentes sobre la
disolución del analito. Lo ideal es usar tres o más de las disoluciones en el proceso de
calibración. No obstante, se puede contar en calibraciones de dos puntos en algunos análisis
de rutina. La calibración se consigue al obtener la señal de respuesta (absorbancia, altura del
pico, área del pico) en función de la concentración conocida del analito.
Una curva de calibración se prepara con los datos o ajustándoles una ecuación matemática
aceptable. El paso siguiente es la etapa de predicción, en la que se obtiene la señal de
respuesta y se usa para predecir la concentración desconocida del analito, a partir de la curva
de calibración con la ecuación de mejor ajuste. La concentración del analito en la muestra
original se calcula luego aplicando los factores de dilución.
Método de los mínimos cuadrados.
El método de los mínimos cuadrados se basa en dos suposiciones. La primera es que hay en
realidad una relación lineal entre la respuesta medida y, y la concentración x del analito
estándar.
La relación matemática que representa esta suposición se llama modelo de regresión, y se
podría representar con y = mx + b; donde b es la ordenada al origen o intersección con el eje
y, es decir, el valor de y cuando x es cero, y m es la pendiente de la recta. (Skoog, Holler, &
Crouch, 2008)
2.12.3 Características de desempeño de los instrumentos
Los parámetros de calidad se revelan en términos numéricos como se puede observar en la
Tabla 3.
Estos parámetros se usan en el estudio de los diferentes instrumentos y métodos
instrumentales.

39
Tabla 3.- Criterios numéricos para la seleccionar métodos analíticos
Criterio Parámetro de calidad
1 Precisión Desviación estándar absoluta
Desviación estándar, Varianza
Coeficiente de variación
2 Sesgo Error absoluto sistemático
Error sistemático relativo
3 Sensibilidad Sensibilidad de calibración
Sensibilidad de analítica
4 Límite de detección Blanco más tres veces la
desviación estándar
del blanco
5 Intervalo de
Concentración
Concentración entre el Límite de
Cuantificación (LOQ) y el Límite
de linealidad (LOL)
6 Selectividad Coeficiente de selectividad
Fuente: (Skoog, Holler, & Crouch, 2008).
Tabla 4.- Otras características a tener en cuenta en la elección del método
Características en la elección del método
1 Velocidad
2 Facilidad y comodidad
3 Habilidad del operador
4 Coste y disponibilidad del equipo
5 Coste por muestra
Fuente:(Skoog & Holler & Crouch, 2008)

40
Parámetros de calidad
2.13.1 Linealidad
Es un intervalo de concentraciones útil para un método analítico en el que la pendiente de la
recta obtenida permanece constante.
Dicho intervalo se extiende desde la concentración más pequeña de analito con la que puede
realizarse las medidas con una exactitud y precisión adecuada (Límite de cuantificación LQ)
hasta la concentración en la que la curva de calibración se desvía de la linealidad (límite de
linealidad LL).
Para que un método sea aplicable, debe tener un rango de, al menos, dos órdenes de
magnitud.
Es necesario verificar el rango comprobando que se obtienen una precisión y exactitud igual
de aceptable cuando el método se aplica a muestras con una concentración de analito en los
extremos del rango que cuando se aplica a muestras que contienen una concentración de
analito que se encuentra en el centro del rango. (Sierra, Perez, Gomez, & Morante, 2010).
Por ejemplo, si se postula una relación lineal:
y = a + bx (2-29)
Dos pares de valores (x1, y1) y (x2, y2) determinan las constantes en la ecuación. Esto resulta
satisfactorio, tomando en cuenta que las cantidades observadas no presentan ningún error.
En la práctica, hay errores en nuestras observaciones, y si se realizan algunas más, digamos
(x 3, y3), es posible obtener un punto que no se ajusta de manera exacta a la línea recta que
pasa por los dos puntos originales. Los métodos estadísticos permiten ajustar la “mejor” línea
a una serie de datos dada, en lugar de simplemente trazar una línea “a ojo”. El principio en
el que se basa el ajuste de la “mejor” línea es el de mínimos cuadrados, y se establece que,
si y es una función lineal de una variable independiente x, la posición más probable de una
recta 𝒚 = 𝒂 + 𝒃𝒙 es tal que la suma de los cuadrados de las desviaciones de todos los puntos
(xi, yi) respecto de la línea es un mínimo; las desviaciones se miden en la dirección del eje
y. Cabe destacar que el supuesto considerando consiste en que x está libre de errores (es la
asignada), o bien está sujeta solo a errores insignificantes, en tanto que, y es la cantidad

41
observada o medida, sujeta a errores que deben ser “eliminados” por el método de mínimos
cuadrados. La y observada es pues un valor aleatorio a partir de la población de valores de
y que corresponden a una x dada. Nuestro problema consiste en encontrar los valores de a y
b para el caso de la línea de “mejor ajuste”. Siendo a la ordenada al origen y b la pendiente
de la curva.
a =∑ x2 ∑ y−∑ x ∑ xy
n ∑ −(∑ x)2
x2
(2-30)
b =n ∑ xy−∑ x ∑ y
n ∑ x2−(∑ x)
2 (2-31)
El coeficiente de correlación se denota por r, cuando existe una correlación perfecta, esto
es, cuando todos los puntos quedan exactamente en cada una de las dos líneas de regresión,
las líneas coinciden; por tanto, sus pendientes son iguales.
Cuando el valor es cero, no existe correlación; cuando es igual a la unidad, la correlación es
perfecta, para calcular se utiliza la ecuación:
r =n ∑ xy − ∑ x ∑ y
√[n ∑ x2
− (∑ x)2][n ∑ y2 − (∑ y)2]
(2-32)
El coeficiente de correlación r debe quedar en el intervalo 0≤ |𝒓| ≤ 𝟏, pero, en la práctica,
debido a los errores aleatorios, 0<|r|<1. (Kennedy & Neville, 1982)
Como se ha visto, los valores de r obtenidos en análisis instrumental son normalmente muy
altos, de manera que un valor calculado, junto con la gráfica de calibrado, suele ser a menudo
suficiente para asegurar al analista que de hecho ha obtenido una relación lineal útil. Sin
embargo, en algunas circunstancias, se obtienen valores de r mucho más bajos. En estos
casos será necesario emplear un contraste estadístico adecuado para ver si el coeficiente de
correlación es realmente significativo, teniendo en cuenta el número de puntos, usados en su
cálculo. El método más simple de hacer esto es calcular el valor de tr, usando la siguiente
ecuación.

42
t = |r|√(n−2)
√(1−r2)
(2-33)
Para probar una correlación significativa, es decir Ho= correlación cero. El valor t calculada
se compara con el valor tabulado al nivel de significación deseado, utilizando un contraste t
de dos colas y (n-2) grados de libertad. La hipótesis nula en este caso es que no existe
correlación entre x e y. Si el valor calculado de t es mayor que el valor tabulado, se rechaza
la hipótesis nula y se concluye en tal caso que existe una correlación significativa. Como se
esperaba, cuanto más próximo este |r| de 1, es decir cuanto más acusada, se haga la relación
lineal, se obtiene valores más grandes de t. (Miller & Miller, 2002). Los errores aleatorios
en los valores de pendiente (b) y ordenada (a) en el origen son importantes. En primer lugar,
se debe calcular el estadístico Sx/y, que estima los errores aleatorios en la dirección de y.
Sy/x = √(y−ŷ)2
n−2
(2-34)
Una vez obtenido un valor de Sy/x, se puede calcular Sb y Sa las desviaciones estándar de la
pendiente (b) y ordenada en el origen (a). Estas vienen dadas por:
Desviación estándar de la pendiente:
Sb= Sy/x
√∑ (xi−x̅)2i
(2-35)
Desviación estándar de la ordenada en el origen:
Sb= Sy/x√∑ xi
2
n ∑ (xi−x⃐ )2
i
(2-36)
Así pues, los límites de confianza de la pendiente vienen dados por: b ± 𝒕(𝒏−𝟐) 𝑺𝒃 , donde t
se obtiene para un nivel de confianza deseado y (n-2) grados de libertad. Similarmente los

43
límites de confianza para la ordenada en el origen vienen dados por: a ± 𝒕(𝒏−𝟐) 𝑺𝒂. (Miller &
Miller, 2002).
2.13.2 Intervalo lineal
La definición del intervalo lineal de un método analítico, que va desde la concentración más
pequeña a la que se puede realizar en medidas cuantitativas (límite de cuantificación LOQ)
hasta la concentración a la que la curva de calibrado se desvía de la linealidad (límite de
linealidad LOL). Para las medidas cuantitativas se toma como límite inferior, en general, la
que corresponde a diez veces la desviación estándar de las medidas repetidas en un blanco o
10𝑠𝑏𝑙. En este punto la desviación estándar relativa es del orden de un 30 por 100 y
disminuye con rapidez cuando las concentraciones aumentan. En el límite de detección, la
desviación estándar relativa es del 100 por 100.
Para que un método analítico sea útil, debe tener un intervalo lineal de, al menos dos órdenes
de magnitud. Algunos métodos tienen un intervalo de concentración aplicable de cinco a seis
órdenes de magnitud.
2.13.3 Selectividad
La selectividad de un método indica el grado de ausencia de interferencias con otras especies
que contiene la matriz de la muestra. Desafortunadamente ningún método está totalmente
libre de interferencias y, con frecuencia hay que realizar diversas etapas para minimizar los
efectos. (Skoog, James Holler, & Nieman, 2001)
2.13.4 Sensibilidad
En general se acepta que la sensibilidad de un instrumento o de un método es una medida de
su capacidad de diferenciar pequeñas variaciones en la concentración del analito. Dos
factores limitan la sensibilidad: la pendiente de la curva de calibrado y la reproducibilidad o
precisión del sistema de medida. Entre dos métodos que tengan igual precisión, será más
sensible aquel cuya curva de calibrado tenga mayor pendiente.
La definición cuantitativa de sensibilidad, aceptada por la Unión 𝛾 Internacional de Química
Pura y Aplicada (IUPAC), es la sensibilidad de calibrado, que se define como la pendiente
de la curva de calibrado a la concentración objeto de estudio. La mayoría de las curvas de

44
calibrado que se usan en química analítica son lineales y se pueden representar mediante la
ecuación.
S = mc + Sbl (2-37)
En la que S es la señal medida, c es la concentración del analito, Sbl, es la señal instrumental
de un blanco y m es la pendiente de la línea recta. El valor de la 𝑆𝑏𝑙 , será la intersección de
la recta con el eje y. En dichas curvas la sensibilidad de calibrado es independiente de la
concentración c y es igual a m. La sensibilidad de calibrado como parámetro de calidad tiene
el inconveniente de no tener en cuenta la precisión de las mediadas individuales.
Mandel y Stiehler consideraron la necesidad de incluir la precisión en un tratamiento
matemático coherente para la sensibilidad y proponen la siguiente definición para la
sensibilidad analítica,𝜸.
γ =m
SS (2-38)
Aquí, m es de nuevo la pendiente de la curva de calibrado y 𝑆𝑆 es la desviación estándar de
las medidas.
La sensibilidad analítica tiene la ventaja de ser relativamente insensible a los factores de
amplificación. Por ejemplo, al aumentar la ganancia de un instrumento por un factor de
cinco, el valor de m se incrementará en cinco veces. Sin embargo, este aumento vendrá
acompañado, en general, del correspondiente aumento en SS, y por tanto la sensibilidad
analítica se mantendrá prácticamente constante. La ventaja de la sensibilidad analítica radica
en su independencia de las unidades de medida de medida de S.
Una desventaja de la sensibilidad analítica es que generalmente depende de la concentración,
ya que 𝑆𝑆 puede variar con ella.
Es la medida de la capacidad del procedimiento analítico que permanece inalterado por
variaciones pequeñas, pero deliberadas en los parámetros del método y proporciona una
indicación de su confiabilidad durante su uso normal.

45
2.13.5 Límite de detección
La definición cualitativa más aceptada para el límite de detección es la mínima concentración
o la mínima masa de analito que se puede detectar para un nivel de confianza dado. Este
límite depende de la relación entre la magnitud de la señal analítica y el valor de las
fluctuaciones estadísticas de la señal del blanco. Por tanto, a no ser que la señal analítica sea
mayor que la del blanco, en un múltiplo k de la variación del blanco debida a errores
aleatorios, no será posible detectar con certeza esta señal. Así, al aproximarse al límite de
detección, la señal analítica y su desviación estándar se aproximan a la señal del blanco Sbl
y a su desviación estándar Sbl. Por tanto, la mínima señal analítica distinguible Sm se
considera que es igual a la suma de la señal media del blanco 𝑆̅bl más un múltiplo k de la
desviación estándar del mismo.
Esto es:
Sm = Sbl + kSbl (2-39)
Experimentalmente, se puede demostrar 𝑺𝒎 se puede determinar realizando 20 o 30 medidas
del blanco, preferiblemente durante un amplio período de tiempo.
A continuación, los datos se tratan estadísticamente para obtener 𝑆𝑏𝑙 y 𝒔𝒃𝒍 . Finalmente, la
pendiente de la Ecuación (2.32) y 𝑺𝒎 se utilizan para calcular LD que se define como límite
de detección, y cuya ecuación es:
LD = 𝐒𝐦−𝐒𝐛𝐥
𝐦
(2-40)
Como ha indicado Ingle, para determinar el valor de k en la Ecuación (2-34) se han usado
numerosas alternativas, relacionadas correctas o incorrectamente con los estadísticos t y z.
Káiser argumenta que un valor razonable para la constante es k= 3. Considera que es
incorrecto suponer una distribución estrictamente normal de los resultados a partir de las
medias del blanco, y que cuando k= 3, el nivel de confianza de detección será de un 95 por
100 en la mayoría de los casos.

46
Así mismo considera poco ventajoso usar un valor mayor de k y por tanto un mayor nivel de
confianza. Long y Winefordner, en un estudio sobre límites de detección, también
recomiendan la utilización de k=3.
Límite de detección (LD) Se define el límite de detección como la magnitud mínima que
puede detectarse en un ensayo, pero no cuantificarse con un valor exacto. Por tanto, se puede
decir que el límite de detección, en nuestros análisis químicos por FRX, es aquella
concentración que proporciona una señal en el instrumento significativamente diferente de
la señal de una muestra en blanco (o señal de fondo).
De los distintos métodos para obtener el límite de detección se ha optado por calcular la
cantidad promedio de analito que proporciona una señal igual a la señal del blanco (yn) más
tres veces la desviación estándar del blanco (Sbl). Siendo n el número de veces que se repite
la medida.
LD = yn + 3Sbl (2-41)
Otra forma de calcular el límite de detección (LD) para un nivel de confianza del 95 %, se
basa en estimar la mínima señal analítica distinguible (Sd) como la señal media del blanco
analítico (Sbl), procedente de al menos 20 medidas, más un múltiplo 3 de la desviación
estándar del blanco (sdbl).
A una señal Sd le corresponde una concentración que es el LD, lo que se consigue
sustituyendo en la recta de calibrado. Si la señal del blanco es cero, coincide con el valor de
la ordenada en el origen de la recta de calibrado, la expresión para el cálculo del LD quedaría.
(Sierra, Perez, Gomez, & Morante, 2010)
Sd = Sbl + 3sdbl (2-42)

47
2.13.6 Límite de Cuantificación (LC)
Se define el límite de cuantificación como la magnitud mínima que puede determinarse con
un nivel aceptable de exactitud. De los distintos métodos para obtener el límite de
cuantificación se ha optado por calcular la cantidad promedio de analito que proporciona
una señal igual a la señal del blanco (Yn) más diez veces la desviación estándar del blanco
(Sbl). (Sevilla, 2010)
LC = Yn + 10Sbl (2-44)
El LC se considera como el límite inferior del rango lineal. Su cálculo se realiza a partir de
la señal mínima cuantificable (Sq) que es la señal media del blanco analítico (Sbl), procedente
de al menos 20 medidas, más un múltiplo 10 de la desviación stándar del blanco (sdbl).
Sq = Sbl + 3sdbl (2-45)
A una señal Sq le corresponde una concentración que es el LC. Si la señal del blanco es cero,
coincide con el valor de la ordenada en el origen de la recta de calibrado, la expresión para
el cálculo del LC con la siguiente ecuación. (Sierra, Perez, Gomez, & Morante, 2010)
LC = (10sdbl)/m (2-46)
2.13.7 Precisión
La precisión de los datos analíticos se define como el grado de concordancia entre los datos
que se han obtenido. Además, indica la medida del error aleatorio de un análisis.
LD =3sdbl
m
(2-43)

48
Tabla 5.- Parámetros de calidad para la precisión de los métodos analíticos.
Términos Definición
Desviación estándar absoluta, (s)
s = √∑ (Xi − X̅)N
i=1
N − 1
Desviación estándar relativa (RSD) RSD = s
X̅
Coeficiente de variación, (CV) CV = s
X̅ × 100%
Varianza s2
�̅� = 𝐦𝐞𝐝𝐢𝐚 𝐝𝐞 𝐍 𝐦𝐞𝐝𝐢𝐝𝐚𝐬 = ∑ 𝐗𝐢𝐍
𝐢=𝟏
𝐍 ,Xi= valor numérico de la iésima medida.
Fuente: (Skoog&Holler&Crouch, 2008)
La precisión del método se puede definir como el grado de concordancia entre los resultados
obtenidos cuando un método se aplica, repetidamente y desde el principio, sobre distintas
porciones representativas desde una misma muestra. La precisión mide el error aleatorio de
un método. La precisión del método puede estimarse de varias formas: (Sierra Alonso &
Perez Quintanilla, 2010)
Repetibilidad
"Se refiere a una estimación de precisión obtenida a partir de mediciones repetidas
realizadas en un laboratorio por un solo analista que utiliza el mismo equipo en una escala
de tiempo breve (se denominan" condiciones de repetibilidad "). Da una indicación de la
variación a corto plazo en los resultados de la medición y se usa típicamente para estimar la
diferencia probable entre los resultados de medición repetidos obtenidos en un solo lote de
análisis. La precisión en las condiciones de repetibilidad, a menudo denominada
simplemente "repetibilidad", también se denomina precisión dentro o fuera del ensayo.

49
Precisión Intermedia
La precisión intermedia se refiere a una estimación de precisión obtenida a partir de
mediciones repetidas realizadas en un solo laboratorio en condiciones más variables que las
condiciones de repetibilidad. Idealmente, las condiciones de precisión intermedias deberían
reflejar, en la medida de lo posible, las condiciones de uso rutinario del método (por ejemplo,
mediciones efectuadas en días diferentes por diferentes analistas utilizando diferentes
equipos en el mismo laboratorio). No siempre es posible variar todos estos factores dentro
de un laboratorio; Las condiciones en las que se obtiene una estimación de la precisión
intermedia deben ser siempre registradas. La ISO 5725-3 sugiere una nomenclatura para
registrar las condiciones.9 La "precisión intermedia" también se conoce como
"reproducibilidad dentro del laboratorio".
Reproducibilidad.
La reproducibilidad definida por la ISO 3534 se refiere a una estimación de precisión
obtenida a partir de mediciones repetidas realizadas en diferentes laboratorios por diferentes
analistas utilizando diferentes equipos. Por lo tanto, debe ser evaluado realizando un estudio
interlaboratorio.
Obsérvese que el término «reproducibilidad» también se ha utilizado más generalmente para
describir cualquier condición de medición distinta de la repetición; Por lo tanto, es a menudo
útil calificar el término (por ejemplo, "reproducibilidad del laboratorio inter-6"). (Ellison,
Barwick, & Duguid Farrant, 2009)
2.13.8 Diseño de Factores Anidados: Una variación del tema
El diseño estándar factorial, tiene dos características notables: cada nivel de cada factor
ocurre con todos los niveles de los demás factores, y es posible examinar la interacción entre
ellos.
En cierto tipo de estudios, los niveles de un factor, B, no serán idénticos en todos los niveles
de otro factor, A.
Cada nivel del factor A contendrá diferentes niveles del factor B.

50
Se dice que los niveles del factor B, están anidados dentro de los niveles del factor A.
En este caso, recibe el nombre de diseños de factor anidado, también se conoce como
diseños jerárquicos. (Kuehl, 2001)
Análisis simple de la varianza
Supóngase k grupos muéstrales, de tamaño respectivos p, todos iguales, siendo n el número
total de elementos muéstrales (n=kp), supuesto que cada grupo muestral procede de una
población normal y que todas las poblaciones normales tienen la misma varianza
(desconocida).
A cada valor muestral Xij se le asigna dos subíndices ij (i-ésimo grupo, j-ésimo valor), donde
1≤ i≤ k y 1≤ j ≤ p. Las observaciones se ordenan, normalmente, en forma de tablas:
Tabla 6.- Para el Análisis Simple de la Varianza
Grupo muestral
Observaciones 1 2 … I … k-1 k
1 x11
2
…
J xij
…
p-1
P xkp
Fuente: (Kuehl, 2001)

51
Las medias de cada grupo muestral están definidas por:
�̅� =∑ ∑ 𝑥𝑖𝑗
𝑝𝑗=1
𝑘𝑖=1
𝑛
(2-47)
La media general es:
x̅ =∑ px̅i
ki=1
n=
∑ x̅i ki=1
k
(2-48)
Tabla 7.- Análisis de Varianza
Análisis simple de la varianza
Origen de la varianza Grados de
libertad (v)
Sumas de diferencias cuadráticas
(SDC)
Diferencias
cuadráticas m
(DCM=SDC/v)
Entre grupos(between) v1=k-1 SDCB=∑ p(xi − x)2ki=1 DCMB=SDCB/k-1
Dentro del
grupo(within)
v2= n-k SDCW=∑ ∑ (xij − xi)2p
j=1ki=1 DCMW=SDCW/n-k
Total v=n-1
(=v1-v2)
SDCT=∑ ∑ (xij − x)2p
j=1ki=1
(=SDCB+SDCW)
DCMT=SDCT/n-1
Fuente: (Kuehl, 2001)
Las diferencias cuadráticas medias (DMC) son las respectivas varianzas, y cabrá esperar que
sean aproximadamente iguales si todos los grupos proceden de una misma población.
Para aceptar los grupos estudiados como no diferentes, obtener el valor estimado de F:

52
F =DCMB
DCMW
(2-49)
Y compararlo con el correspondiente valor de F tabulado (para el ⍺ prefijado, v1=k-1 y v2=
n-k).
Si Fc < Ftabulado, podremos decir que no existen diferencias significativas entre los distintos
grupos muestrales.
En el caso, contrario, de que existen diferencias significativas (al nivel ⍺ prefijado) entre los
grupos muestrales, se deberá considerar que al menos dos de las medias de grupo son
distintas entre sí.
La desviación estándar de repetibilidad (𝑠𝑟) es
sr = √DCMW (2-50)
La desviación estándar de reproducibilidad (𝒔𝑹) es:
sR=√sr
2 + sL2 (2-51)
Donde
sL 2 =
DCMB− DCMW
p
(2-52)
Siendo el denominador (p) igual al número de observaciones que se realizan cada día (en
cada nivel) (cuando es el caso de un diseño experimental homogéneo de factores (totalmente
anidados) (como el que se ha planteado)).

53
Si por efectos aleatorios, 𝑠𝐿2 < 0, debe asumirse 𝑠𝐿
2 = 0, normalmente debería cumplirse que
DCMB > DCMW y en caso contrario deberían existir razones que lo justificaran. (Feinberg,
1995).
Límite de repetibilidad (Lr). Es el valor menor o igual esperado con una probabilidad del
95%, de la diferencia absoluta entre dos resultados de ensayo obtenidos en condiciones de
repetibilidad.
El límite de repetibilidad está dado por la siguiente ecuación
Lr = t∞ × √2 × σr (2-53)
Donde, t∞ es el valor de t student de dos colas para n= ∞, para una confianza dada
(normalmente se usa un nivel de confianza del 95%, cuando el valor de t es de 1.96), y 𝜎𝑟 o
sr es la desviación estándar medida en condiciones de repetibilidad.
Límite de reproducibilidad (LR). Valor menor o igual esperado con una probabilidad del
95%, de la diferencia absoluta entre dos resultados de ensayo obtenidos en condiciones de
reproducibilidad.
El límite de reproducibilidad está dado por la siguiente ecuación
LR = t∞ × √2 × σR (2-54)
Donde, t∞ es el valor de t student de dos colas para n= ∞, para una confianza dada
(normalmente se usa un nivel de confianza del 95%, cuando el valor de t es de 1.96), y 𝜎𝑅 o
sR es la desviación estándar medida en condiciones de reproducibilidad. (Eurachem, 1998).
Se realiza mínimo tres repeticiones por nivel de concentración (Taverniers, De Loose, &
Bockstaele, 2004)
Evaluación estadística de las estimaciones de precisión
Comparación con un valor de precisión específico
Si la desviación estándar obtenida del estudio de precisión excede marginalmente el valor
establecido durante la especificación del requisito de medición, puede utilizarse una prueba

54
F para determinar si la estimación de precisión es significativamente mayor que el valor
objetivo:
F =s2
σo2
(2-55)
Donde s es la estimación de precisión obtenida del estudio de validación y σo es la precisión
objetivo (expresada como una desviación estándar). Para obtener el valor crítico apropiado
para F, se supone que σo tiene grados infinitos de libertad. (Ellison, Barwick, & Duguid
Farrant, 2009)
2.13.9 Exactitud.
La exactitud es el grado de concordancia entre el resultado de una determinación o la media
de los resultados y el valor verdadero (Gallego, Garcinuño, & Morcillo, 2013)
Sesgo
El sesgo mide el error sistemático, o determinado, de un método analítico. El sesgo se define
mediante la ecuación:
Sesgo = μ − 𝒳𝔱 (2-55)
Donde μ es la media de la población de la concentración de un analito en una muestra cuya
concentración verdadera es 𝓧𝖙 . Para determinar la exactitud hay que analizar uno o varios
materiales de referencia cuya concentración de analito es conocida. Sin embargo, los
resultados de dichos análisis también tendrán tanto errores aleatorios como errores
sistemáticos, pero si se realiza un número suficiente de determinaciones, se puede determinar
el valor de la media, para un nivel de confianza dado. Se puede suponer que la media de 20
o 30 análisis replicados es una buena estimación de la media de la población µ. Cualquier
diferencia entre esta media y la concentración del analito indicada en el material de
referencia se puede atribuir al sesgo.

55
En general, al desarrollar un método analítico, todos los esfuerzos se dirigen hacia la
identificación de las causas del sesgo y a su eliminación o corrección mediante el uso de
blancos y el calibrado del instrumento.
La exactitud describe si el resultado experimental es correcto. Estrictamente hablando, el
único tipo de medida que puede ser completamente exacto es aquel que consiste en el
recuento de objetos. Todos los demás tipos de medidas contienen errores y aportan sólo una
aproximación del valor verdadero.
La exactitud es un término relativo, en el sentido de que un método es exacto o inexacto
dependiendo en gran medida de las necesidades del científico y de las dificultades del
problema analítico.
Por ejemplo, un método analítico con el cual se obtiene resultados que se encuentran
comprendidos entre ±10 por 100, o una parte por billón, de la cantidad correcta de mercurio
presente en una muestra de tejido de pescado que contiene 10 partes por billón, del metal se
consideraría de una exactitud razonable. Por el contrario, un procedimiento con el que se
obtienen resultados comprendidos entre ± 10 por 100 de la cantidad correcta de mercurio
presente en un mineral que contiene el 20 por 100 del metal, normalmente se juzgaría como
inaceptable por inexacto.
La exactitud se expresa en términos de errores absolutos y relativos.
El error absoluto (E a)
De la media (o promedio) 𝑥 del análisis de un pequeño conjunto de replicados se expresa
mediante la relación:
Ea = x̅ − xt (2-56)
Donde xt es el valor aceptado (como verdadero) de la cantidad medida. Con frecuencia, es
útil expresar la exactitud en términos de error relativo (er), donde:
error relativo =x−xt
xt× 100% (2-57)

56
Como se ve, es habitual expresar el error relativo como un porcentaje (%); en otros casos el
cociente se multiplica por 1000 para dar el error en tanto por mil (‰).Observe que ambos
errores, absoluto y relativo, llevan un signo positivo para indicar que el resultado medido es
mayor que el valor verdadero y un signo negativo en el caso contrario (Skoog, James Holler,
& Nieman, 2001).
Z-score
Es un valor calculado que indica cuantas desviaciones estándares, un determinado valor está
alejado de su correspondiente media.
El z-score se calcula, restando el resultado del control de la media correspondiente y luego
dividiendo esta diferencia por la desviación estándar observada, para el mismo material de
control.
Ejemplo: µ = media; x = resultado del control; s = desviación estándar observada.
z = (x − μ)/s (2-58)
Un z-score de 2.4 indica que el valor de control observado está 2.4 desviaciones estándar,
alejadas de la media, pero que aún no excede el límite 3s. (Saez Ramires & Gómez
Cambronero, 2006)
Z-score por parte de los organizadores La interpretación de la puntuación es la siguiente:
Satisfactorio si |z| < 2,
Cuestionable si 2 < |z| < 3,
Insatisfactorio si |z| > 3
Recuperación (R)
Es la fracción de la sustancia agregada a la muestra (muestra fortificada) antes del análisis,
al ser analizadas muestras fortificadas y sin fortificar.

57
La recuperación permite ver el rendimiento de un método analítico en cuanto al proceso de
extracción y la cantidad del analito existente en la muestra original. Por lo cual, la
recuperación esta intrínsecamente relacionada a las características de la matriz de la muestra.
Se recomienda realizar a lo menos 6 mediciones de cada uno en lo posible en tres niveles.
Se debe considerar al elegir estos niveles el rango de la curva de calibración del método, el
LOD y el LMP establecido. De manera que los niveles seleccionados permitan entregar la
mejor información posible respecto a la capacidad de recuperación del método, en cuanto a
estos valores críticos. Se calcula de la siguiente manera:
R = (Ce − Co
Ca)
(2-59)
Siendo:
R= Recuperación
Ce = es la concentración de analito de la muestra enriquecida.
Co= es la concentración de analito medida en la muestra sin adicionar.
Ca= es la concentración de analito adicionado a la muestra enriquecida.
Se puede igualmente expresar en porcentaje de recuperación (%R): se calcula de la
siguiente manera:
%R = [R]x100 (2-60)
En caso de evaluar la recuperación, se deberá realizar prueba t, en la cual el tcal<tcrit:
tcal =[100 − %R]
s√n
(2-61)
Donde:
tcal = t observado o calculado
R = Recuperación
s = Desviación estándar de las lecturas del porcentaje de recuperación

58
n = N.º de lecturas o valores observados
Si el tcalc > tcrit (hay diferencia estadísticamente significativa), los resultados reportados
deberán ser corregidos.
Evaluación de la Exactitud
La precisión describe la variación de los resultados obtenidos a partir del análisis de
repetición de una muestra, pero no da ninguna información acerca de cuán cerca están los
resultados de la concentración verdadera del analito en la muestra. Es posible que un método
de prueba produzca resultados que estén en estrecho acuerdo entre sí, pero que sean
consistentemente más bajos (o más altos) de lo que deberían ser. La fidelidad se define como
«La proximidad del acuerdo entre la expectativa de un resultado de la prueba o un resultado
de la medición y un valor verdadero». La veracidad se expresa normalmente en términos de
sesgo. La definición incluye una nota que, en la práctica, un valor de referencia aceptado se
sustituye por el valor verdadero. ¿La exactitud puede evaluarse comparando la media de los
resultados de la medición 𝑥 con un valor de referencia aceptado (µo).
Evaluar el sesgo implica realizar un análisis repetido de un material adecuado que contiene
una cantidad conocida del analito (este es el valor de referencia, µo). La exactitud es la
diferencia entre el promedio de los resultados de la prueba y el valor de referencia:
E = x̅ - µo (2-62)
La exactitud también se expresa frecuentemente como un porcentaje:
%E = x̅ − μo
μox(100)
(2-63)
Y como proporción (en particular al evaluar la «recuperación»):
%R =x̅
μox(100)
(2-64)

59
Uno de los problemas al planear un estudio de la exactitud del método es la selección de un
valor de referencia adecuado. Hay una serie de opciones:
Un material de referencia certificado (CRM)
Muestras con prueba de punta
Un método de referencia.
Las estimaciones de exactitud se evalúan estadísticamente utilizando las pruebas t de
Student.
Comparación con un valor de referencia
Una vez obtenida una estimación de la exactitud, por ejemplo, utilizando la ecuación (2-62),
se puede realizarse una prueba t de Student para determinar si la exactitud observada es
estadísticamente significativa. La estadística de prueba se calcula utilizando la ecuación
t =(x − μ)
s√n
(2-65)
Donde n es el número de mediciones de repetición utilizadas para obtener x y s es la
desviación estándar de los n resultados de medición. (Ellison, Barwick, & Duguid Farrant,
2009)
Hipótesis De Investigación
Ho: No es factible desarrollar un método analítico seguro y ecológico, usando d-limoneno
como disolvente de extracción de la especie química (analito) y el cumplimiento de los
criterios de los parámetros de calidad.
Ha: Es factible desarrollar un método analítico seguro y ecológico, usando d-limoneno como
disolvente de extracción de la especie química (analito) y el cumplimiento de los criterios
de los parámetros de calidad.

60
Variables
Las variables aleatorias continuas toman valores sobre un intervalo o una colección de
intervalos, que surgen de un proceso de medición. La información se obtiene de la medida
de volúmenes y de los instrumentos de medidas. Todas las operaciones citadas implican
medidas y estas tienen un cierto grado de imprecisión. (Araya Alpizar, 2004)
Tabla 8.- Variables Independiente y dependiente
Independiente Dependiente Respuesta
Concentración de analito
(detergente)
Señal analítica Absorbancia
Método analítico Precisión Desviación estándar absoluta y relativa.
Varianza
Exactitud Sesgo
Porcentaje de recuperación
Z score
Robustez Repetibilidad
Reproducibilidad
Límite de detección Concentración límite detectable
Límite de cuantificación Concentración límite medible
Intervalo de trabajo Rango de concentración aplicable
Fuente: Salomón Chacha.

61
CAPITULO III
METODOLOGIA
Diseño metodológico
3.1.1 Tipo de Investigación
Método científico, (inductivo deductivo) enfoque cualitativo, cuantitativo y mixto y métodos
particulares si los hay, por ejemplo, documental, histórico, etc.
En el proceso de la investigación se siguió los lineamientos del método científico; y la parte
experimental forma parte de este. El desarrollo de la investigación se basa en los
fundamentos teóricos de la Química y complementa en la determinación de las variables
independientes mediante ensayos de laboratorios, para cumplir con los objetivos y la
hipótesis planteada. (Figueroa. M & Ramírez. S, 2014)
Población y Muestra
3.2.1 Población
Solución madre patrón de detergente (lauril sulfato de sodio) de 0,1g/L
3.2.2 Muestra
Los estándares preparados a partir de la solución madre, muestras de aguas residuales
de la industria textil y un material de referencia certificado.
3.2.3 Diseño muestral
Preparación de la muestra. Las muestras utilizadas en el proceso de desarrollo del método,
los estándares de diferentes concentraciones fueron preparadas con un patrón de “sodium
lauryl sufate”, una vez puesto a punto el método se realizó los ensayos con muestras de aguas
residuales, en particular de las Industrias textiles.

62
Diseño experimental
Para el diseño experimental se ha divido en dos partes la investigación: La puesta a punto
del Método Propuesto y la determinación de los Parámetros de Calidad.
3.3.1 Puesta a punto del método analítico propuesto
Consiste en ajustar y afinar las distintas variables del método propuesto, como la longitud
de onda, la concentración del colorante empleado, el disolvente (mezcla) de extracción, el
efecto de la fuerza iónica, el efecto del pH, el tiempo de extracción de la especie formada, el
tiempo de estabilidad del par iónico e intervalo lineal.
3.3.2 Parámetros de Calidad
Determinación del límite de detección y cuantificación
Se preparó 20 muestras de blanco, la misma que fue analizado individualmente según el
método propuesto y se determinó la desviación estándar de las 20 lecturas. (3.2.1). El límite
de detección (LD). Se determinó mediante la ecuación (3-11). El límite de cuantificación
(LQ). Se determinó mediante la ecuación (3-12).
Determinación de la linealidad
Se realizó 5 curvas de calibración con 6 estándares de diferentes concentraciones (0,10; 0,20;
0,30; 0,40; 0,50 y 0,60) ppm. Con los valores de absorbancias en el eje de las ordenadas y
las concentraciones en el eje de las abscisas se trazó la gráfica: Absorbancia en función de
la concentración (detergente). En el gráfico se puede observar la pendiente (b), la
intersección u ordenada al origen (a) y el coeficiente de correlación (r). El coeficiente de
correlación fue analizado por medio de una prueba “t de student”, para determinar si la
correlación es significativa o no, además se plantea la hipótesis nula, Ho: no existe
correlación entre y/x y la hipótesis alternativa, Ha: existe correlación entre y/x.
Estudios de repetibilidad y reproducibilidad
El estudio de la precisión se realizó a través del análisis simple de varianza (ANOVA de
factores totalmente anidados y homogéneos), se calculó las desviaciones estándar de
repetibilidad (sr) y de reproducibilidad (sR) para cada uno de los niveles. Este parámetro se
determinó con los 6 estándares de diferente concentración de detergente (0.1, 0.2, 0.3, 0.4,

63
0.5 y 0.6) ppm, cada estándar se medió 4 veces durante 5 días (5 semanas), en el mismo
equipo y con dos analistas.
Estudios de la exactitud
La exactitud se determinó mediante el porcentaje de recuperación de un material de
referencia (estándar) preparado a partir de la solución madre; realizando 10 determinaciones
en el nivel medio.
Diseño estadístico
Tabla 9.- Parámetros considerados para el diseño Experimental
Diseño Estadístico
Parámetros Diseño Experimental Recolección de datos
Precisión Diseño de factores Tabla de ANOVA, 4 repeticiones, por 5
anidados días para cada nivel
Exactitud Prueba t de medias Tabla de cuatro columnas con 10
Repeticiones
Robustez Diseños de factores
anidados
Tabla de ANOVA, 4 repeticiones, por 5
días para cada nivel
Límite de
detección
Curvas de correlación Tablas de correlación
regresión lineal Abs- Concentración. 6 estándares y
por 5 días
Límite de
Cuantificación
Curvas de correlación Tablas de correlación
regresión lineal Abs- Concentración. 6 estándares y
por 5 días
Intervalo de
trabajo
Curvas de correlación Tablas de correlación
regresión lineal Abs- Concentración. 6 estándares y 5 días
Fuente: Salomón Chacha

64
Técnicas analíticas empleadas en el trabajo de investigación
Método Espectrofotométrico de referencia
5540 C. Surfactantes amónicos como SAAM.
a) Definición y principio: Las sustancias activas para el azul de metileno (SAAM) llevan
a cabo la transferencia del azul de metileno, un tinte catiónico, de una solución acuosa
a un líquido orgánico inmiscible, hasta equilibrio. Esto ocurre a través de la formación
de un par iónico entre el anión SAAM y el catión azul de metileno. La intensidad del
color azul resultante en la fase orgánica es una medida de la SAAM. Los surfactantes
aniónicos se encuentran entre las más destacadas de las muchas sustancias, naturales y
sintéticas, que muestran actividad al azul de metileno. El método SAAM es útil
para valorar el contenido de surfactante aniónico de las aguas limpias y residuales. Este
método es relativamente simple y preciso. Comprende tres extracciones sucesivas en
cloroformo (CHCl3) a partir de medio acuoso ácido que contenga azul de metileno en
exceso, seguidas de lavado por contracorriente con agua, y la determinación del color
azul en el CHCl3 por espectrofotometría a 652 nm. El método es aplicable a
concentraciones de SAAM inferiores a unos 0,025 mg/l.
b) Respuesta de surfactante aniónico: Los jabones no responden al método SAAM.
Los utilizados en, o como, detergentes, son sales básicas de ácidos grasos de C10-20. [RCO2]-
Na+, y aunque de naturaleza aniónica se ionizan tan débilmente que no se forma un par iónico
extraíble bajo las condiciones de la prueba. Los surfactantes aniónicos no jabones utilizados
habitualmente en las formulaciones de los detergentes responden con gran fuerza.
Incluyen sobre todo surfactantes del tipo sulfonato [RSO3]- Na+, del tipo éster de sulfato
[ROSO3]-Na+ y no iónicos sulfatados [REnOSO3]-Na+. Son recuperados casi por completo
por una extracción simple en CHCl3.
El sulfonato de alquílbenceno lineal (SAL) es el surfactante aniónico más utilizado y se
emplea para estandarizar el método SAAM. El SAL no es un compuesto sencillo, sino que
puede constar de algunos o todos los 26 isómeros y homólogos con estructura
[R’C6H4SO3], donde R’ es un grupo alquilo secundario lineal cuya longitud oscila entre 10
y 14 átomos de carbono.
Los surfactantes de tipo sulfonato y sulfato responden juntos en el análisis SAAM, pero
pueden diferenciarse por otros medios. El tipo sulfato se descompone por hidrólisis acida; la
reducción resultante de las SAAM corresponde al contenido de surfactante sulfato original

65
mientras que las SAAM restantes corresponden a los surfactantes sulfonato. El
alquilbenceno sulfonato puede ser identificado y cuantificado por espectrometría infrarroja
después de la purificación. El SAL puede distinguirse de otros surfactantes alquilbenceno
sulfonato por métodos infrarrojos; puede ser identificado de forma inequívoca y su
composición detallada isómero-homólogo determinada por cromatografía de gases-
desulfonación.
c) Interferencias: Se producen interferencias positivas por todas las demás especies de
SAAM presentes; si se busca una determinación directa de cualquier especie individual de
SAAM, tal como el SAL, todos los demás interfieren. Sustancias tales como sulfonatos,
sulfatos, carboxilatos y fenoles orgánicos, y tiocianatos, cianatos, nitratos y cloruros
inorgánicos pueden también transferir más o menos azul de metileno a la fase cloroformo.
Cuanto más pobre la extracción de sus pares iónicos, más efectiva es la etapa de lavado en
contracorriente acuosa para extraer estas interferencias positivas; la interferencia del cloruro
es eliminada casi por completo y la del nitrato también lo es en gran medida por la
contracorriente. Debido a la variada extracción de SAAM no surfactantes, desviaciones en
la proporción de CHCl3 y en el procedimiento de lavado en contra corriente pueden inducir
diferencias significativas en las SAAM totales observadas, aunque la recuperación de los
surfactantes de tipo sulfonato y sulfato serán sustancialmente completas en todos los casos.
Interferencias negativas pueden ser consecuencia de la presencia de surfactantes catiónicos
y otros materiales catiónicos, tales como aminas, porque compiten con el azul de metileno
en la formación de pares iónicos. La materia en partículas puede producir interferencia
negativa por la adsorción de las SAAM. Aunque alguna SAAM adsorbida puede
desadsorberse y emparejarse durante las extracciones de CHCl3, la recuperación puede ser
incompleta y variable. Redúzcanse al mínimo las interferencias debidas a los materiales no
surfactantes por cancelación, si es necesario (sección 5540B). Elimínense los surfactantes
catiónicos que interfieren y otros materiales catiónicos utilizando una resina de intercambio
catiónico bajo condiciones adecuadas Manipúlese la adsorción de las SAAM por partículas,
preferiblemente filtrando y analizando las insolubles. Con o sin filtración, las SAAM
adsorbidas pueden ser desadsorbidas por hidrólisis acida; sin embargo, la SAAM originada
en cualquier surfactante de tipo éster sulfato presente es destruida a la vez. Los sulfuros, a
menudo presentes en las aguas residuales sin tratar o primariamente tratadas, pueden
reaccionar con el azul de metileno para formar un producto de reducción incoloro, haciendo
imposible el análisis. Elimínese esta interferencia mediante oxidación previa con peróxido
de hidrógeno.

66
d) Peso molecular: Los resultados de la prueba parecerán diferentes si se expresan en
términos de peso y no de cantidades molares. Cantidades equimolares de dos surfactantes
aniónicos con diferentes pesos moleculares darán colores sustancialmente iguales en la capa
de CHCl3, aunque las cantidades por peso pueden diferir de forma significativa. Si los
resultados van a expresarse por peso, como suele solicitarse, debe conocerse el peso
molecular promedio del surfactante medido o usarse una curva de calibración construida con
ese compuesto concreto.
Dado que dicha información detallada suele faltar, infórmese de los resultados en términos
de una curva de calibración estándar, por ejemplo «0,65 mg SAAM/l (calculado como SAL,
mol peso 318)».
e) Cantidad detectable mínima: Alrededor de 10 µg de SAAM (calculada como SAL).
f) Aplicación: El método SAAM ha sido aplicado con éxito a muestras de agua potable. En
las aguas residuales, los residuos industriales, y el lodo, numerosos materiales que suelen
estar presentes pueden interferir gravemente si se intenta la determinación directa de las
SAAM. La mayoría de las interferencias de fase acuosa de no surfactantes pueden ser
eliminadas por cancelación.
2. Instrumental
a) Equipo calorimétrico: Se utilizó el siguiente equipo.
1) Espectrofotómetro: A la longitud de onda de 652 nm, que proporcione un trayecto de
luz de 1 cm o más largo.
2) Fotómetro de filtro: Que proporcione un trayecto de luz de 1 cm o más largo y está
equipado con un filtro de color rojo que muestre una transmitancia máxima próxima a 652
nm.
b) Embudos de separación: 500 ml, preferiblemente con tapones y llaves de paso de TFE
inerte.
3. Reactivos
a) Solución SAL de reserva: Pésese una cantidad de material de referencia*igual a 1,00 g de
SAL en una base activa del 100 por 100. Disuélvase en agua y dilúyase hasta 1.000 ml; 1,00
ml = 1,00 mg SAL. Guárdese en un refrigerador para minimizar la biodegradación. Si es
necesario, prepárese cada semana.
b) Solución SAL patrón: Dilúyanse 10,00 ml de la solución anterior hasta 1.000 ml con agua;
1,00 ml = 10,0 µg SAL. Prepárese a diario.

67
c) Solución indicadora de fenolftaleína, alcohólica.
d) Hidróxido de sodio, NaOH, 1N.
e) Ácido sulfúrico, H2SO4, 1N y 6N.
f) Cloroformo, CHCl3; PRECAUCIÓN: El cloroformo es tóxico y un probable carcinógeno.
Adóptense las precauciones apropiadas contra la inhalación y la exposición a la piel.
Fig. 10.- Espectrofotómetro Jasco V-630.
Fuente. JASCO Analytical Instruments
g) Reactivo azul de metileno: Disuélvanse 100 mg de azul de metileno † en 100 ml de agua.
Pásense 30 ml a un matraz de 1.000 ml. Añádanse 500 ml de agua, 41 ml de H2SO4
6N, y 50 g de fosfato de sodio, monobásico, monohidratado, NaH2PO4 • H2O. Agítese hasta
que se disuelva. Dilúyase hasta 1.000 ml.
h) Solución de lavado: Añádanse 41 ml de H2SO4 6N a 500 ml de agua en un matraz de
1.000 ml. Añádanse 50 g de NaH2PO4 • HO y agítese hasta que se disuelva. Dilúyase a
1.000 ml.
i) Metanol, CH3OH. PRECAUCIÓN: Los vapores de metanol son inflamables y tóxicos;
tómense las precauciones adecuadas.
j) Peróxido de hidrógeno, H2O2, al 30 por 100.
k) Lana de vidrio: Extráigase previamente con CHCl3 hasta eliminar las interferencias.
l) Agua, destilada o desionizada, libre de SAAM. Utilícese para hacer todos los reactivos y
diluciones.
4. Procedimiento

68
a) Preparación de la curva de calibración: Prepárese una serie de 10 embudos de
separación con 0, 1,00, 3,00, 5,00, 7,00, 9,00, 11,00, 13,00, 15,00 y 20,00 ml de solución
SAL patrón. Añádase suficiente agua para completar el volumen total de 100 ml en cada
embudo.
b) Tamaño de la muestra: Para el análisis directo de las aguas limpias y residuales,
selecciónese el volumen de muestra sobre la base de la concentración de SAAM esperada:
Tabla 10.- Tamaño de la muestra en función de la concentración de SAAM.
Concentración de SAAM esperada
mg/L
Muestra tomada Ml
0,025 – 0,080 400
0,08 – 0,40 250
0,40 – 2,0 100
Fuente: Métodos Normalizados APHA
Si la concentración esperada de SAAM es superior a 2 mg/l, dilúyase la muestra que
contenga 40 a 200 µg de SAAM hasta 100 ml con agua.
Para analizar las muestras purificadas por cancelación, disuélvase el residuo de cancelación
(sección 5540B.4e) en 10 a 20 ml de metanol, pésese cuantitativamente la cantidad entera (o
una porción apropiada si se esperan más de 200 µg de SAAM) a un volumen de entre 25 y
50 ml de agua, evapórese sin hervir hasta que haya desaparecido el metanol, añadiendo agua
si es necesario para evitar que se seque, y dilúyase hasta unos 100 ml con agua.
c) Tratamiento con peróxido: Si es necesario evitar la decoloración del azul de metileno
por sulfuros, añádanse unas pocas gotas de H2O2 al 30 por 100.
d) Apareamiento iónico y extracción:
1) Añádase muestra a un embudo de separación. Alcalinícese añadiendo gotas de NaOH 1N,
utilizando indicador de fenolftaleína. Elimínese el color rosa añadiendo gotas de H2SO4 1N.
2) Añádanse 10 ml de CHCl3 y 25 ml de azul de metileno. Agítese el embudo vigorosamente
durante 30 segundos y déjese que se separen las fases. Alternativamente, colóquese una barra
de agitación magnética en el embudo de separación; túmbese el embudo sobre su lateral en
un agitador magnético y ajústese la velocidad de agitación para producir un movimiento de
sacudimiento. Excesiva agitación puede hacer que emulsione.

69
Para romper las emulsiones persistentes añádase un volumen pequeño de alcohol
isopropílico (< 10 ml); añádase el mismo volumen de alcohol isopropílico a todos los
patrones. Algunas muestras requieren un período más largo de separación de fases que otras.
Antes de drenar la capa de CHCl3, muévase suavemente y déjese reposar.
3) Sepárese la capa de CHCl3 en un segundo embudo de separación. enjuáguese el tubo
producido por el primer embudo de separación con una pequeña cantidad de CHCl3. Repítase
la extracción dos veces más, utilizando 10 ml de CHCl3 cada vez. Si el color azul de la fase
acuosa se debilita o desaparece, deséchese y repítase, utilizando una muestra más pequeña.
4) Combínense todos los extractos de CHCl4 en el segundo embudo de separación. Añádanse
50 ml de solución de lavado y agítese vigorosamente durante 30 segundos. No se forman
emulsiones en esta etapa. Déjese reposar, muévase y extraiga la capa de CHCl3 a través del
embudo que contenga un tapón de lana de vidrio en un matraz volumétrico de 100 ml; el
filtrado debe ser claro. Extráigase dos veces la solución de lavado con 10 ml de CHCl3 cada
uno y añádase al matraz a través de la lana de vidrio. Recójanse los lavados en el matraz
volumétrico, dilúyase hasta la marca con CHCl3, y mézclese bien.
e) Determinación: Determínese la absorbancia a 652 nm frente a un blanco de CHCl3
5. Cálculo
A partir de la curva de calibración (apartado 4a) léanse los microgramos de SAL aparente
que corresponden a la absorbancia medida.
𝑚𝑔 𝑆𝐴𝐴𝑀
𝐿=
𝑢𝑔𝑆𝐴𝐿 𝑎𝑝𝑎𝑟𝑒𝑛𝑡𝑒
𝑚𝑙 𝑑𝑒 𝑚𝑢𝑒𝑠𝑡𝑟𝑎 𝑜𝑟𝑖𝑔𝑖𝑛𝑎𝑙
Informar como «SAAM, calculada como SAL, mol peso».
6. Precisión y sesgo
En 110 laboratorios se analizó una muestra que contenía 270 µg de SAL/l en agua destilada
con una desviación estándar relativa del 14,8 por 100 y un error relativo de 10,6 por 100.
(SAL, surfactante aniónico lineal)
Una muestra de agua potable a la que se añadió 480 µg de SAL/l fue analizada en 110
laboratorios con una desviación estándar relativa del 9,9 por 100 y un error relativo del 1,3
por 100. Una muestra de agua de río con2,94 mg de SAL/l añadida fue analizada en 110
laboratorios con una desviación estándar relativa del 9,1 por 100 y un error relativo del 1,4
por 100. (Clesceri, Greenberg, & Trussell, 1992)

70
Método Espectrofotométrico Propuesto
Principio
Los detergentes aniónicos en solución acuosa, con un colorante catiónico (cristal de violeta)
forma par-iónico soluble en solventes apolares (benceno, cloroformo, cloro-benceno)
(Esquivel & Leal, 2004), en este caso particular en “d-limoneno”.
Reactivos y Materiales
-Solución tampón tipo sulfato pH 2.
-Solución de cristal violeta en cloruro de sodio (0,5% p/p).
-Limoneno (85% de pureza).
- Solución madre patrón de detergente (lauril sulfato de sodio) de 0,1g/L: detergente aniónico
100 mg; agua destilada aforo a 1000 mL.
- Solución hija patrón de detergente de 10 mg/L. Diluir 100 mL de la solución madre a 1000
mL con agua destilada.
- Espectrofotómetro uv-visible HACH DR/4000
- Celdas de cuarzo, de vidrio de 25 ml o celdas de plástico.
- Material de vidrio: embudos de separación de 500 ml, pipetas, matraces aforados de 25,
50, 100 y 1000 ml de capacidad, embudo simple para líquidos, probetas de 10, 25 y 500 ml,
matraz Erlenmeyer y vasos de precipitación.
-Balanza analítica

71
Fig. 11.- Espectrofotómetro HACH DR/4000.
Fuente: Hach Company
Preparación de la curva de calibrado
En una serie de embudos de separación de 500 ml, numerados, se agregó los reactivos
sucesivamente de acuerdo a la tabla 12. Con agitación fuerte y uniforme durante 1minuto,
se agregó el solvente (limoneno), y agitación suave durante 30 segundos, evitando que se
forme una emulsión. Se dejó en reposo por 10 minutos, luego se procedió a separar la fase
orgánica, filtrar en papel filtro impregnado de limoneno y una mota de algodón y recoger los
extractos de limoneno en matraces aforados de 25 ml o en viales de vidrio de 30 ml. Se
realizó la lectura de los 6 estándares en el espectrofotómetro a la longitud de onda de 605,4
nm. Con las absorbancias obtenidas se construyó la curva de calibrado.
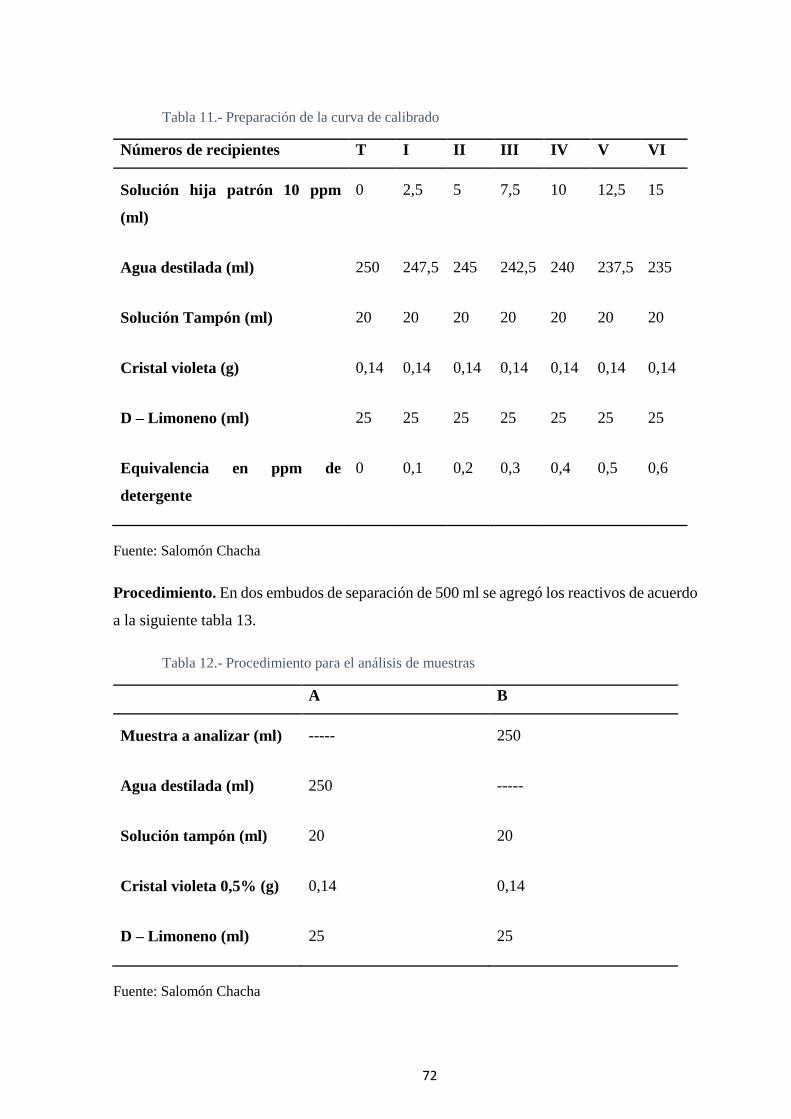
72
Tabla 11.- Preparación de la curva de calibrado
Números de recipientes T I II III IV V VI
Solución hija patrón 10 ppm
(ml)
0 2,5 5 7,5 10 12,5 15
Agua destilada (ml) 250 247,5 245 242,5 240 237,5 235
Solución Tampón (ml) 20 20 20 20 20 20 20
Cristal violeta (g) 0,14 0,14 0,14 0,14 0,14 0,14 0,14
D – Limoneno (ml) 25 25 25 25 25 25 25
Equivalencia en ppm de
detergente
0 0,1 0,2 0,3 0,4 0,5 0,6
Fuente: Salomón Chacha
Procedimiento. En dos embudos de separación de 500 ml se agregó los reactivos de acuerdo
a la siguiente tabla 13.
Tabla 12.- Procedimiento para el análisis de muestras
A B
Muestra a analizar (ml) ----- 250
Agua destilada (ml) 250 -----
Solución tampón (ml) 20 20
Cristal violeta 0,5% (g) 0,14 0,14
D – Limoneno (ml) 25 25
Fuente: Salomón Chacha

73
Para cada embudo de separación, se aplicó la extracción como se detalla en la preparación
de la curva de calibrado y las respectivas lecturas de la muestra en el espectrofotómetrometro
uv-visible a la longitud de onda de 605,4 nm. Siempre la primera lectura será del testigo,
luego se procedió con la muestra problema. Se obtuvo los resultados. Para una muestra
líquida de 250 ml, la curva da directamente el contenido de detergente aniónico, expresado
en miligramos por litro de agua (residual o potable).
Técnicas e Instrumentos de Recolección de Datos
3.6.1 Técnica:
Observación y análisis
3.6.2 Instrumento:
Tablas Excel:
Tabla de Resultados en Absorbancia de la Extracción del par iónico con
mezclas de disolventes.
Tabla de Resultados en absorbancia de la Extracción del colorante catiónico
sin el analito con mezclas de disolventes.
Tabla de Resultados en absorbancia de la especie extraída con L-X en
diferentes proporciones.
Tabla de Resultados en Absorbancia del Efecto a diferentes valores de pH en
la formación y extracción del par iónico.
Tabla de Resultados en absorbancia del Efecto de la fuerza Iónica en la
formación y extracción del par iónico.
Tabla de Resultados en Absorbancia de la concentración optima del CV.
Tabla de Resultados en Absorbancia para la determinación del intervalo
dinámico.
Tabla de Resultados corregidos para elegir el intervalo dinámico.
Tabla de Resultados en Absorbancia y la concentración en (ppm) obtenidos
a partir de las lecturas de 20 blancos.
Tabla de Resultados en Absorbancia de las lecturas de los estándares de
diferentes concentraciones en 5 días para la determinación de la linealidad.

74
Tabla de Resultados para la determinación de parámetros estadísticos
básicos.
Tabla de Resultados de la Función respuesta instrumental y análisis de las
curvas de calibración.
Tabla de resultados de la curva global y de los limites superior e inferior.
Tabla de resultados de la función respuesta instrumental de la curva global.
Tabla de resultados de las pendientes de las curvas de calibración.
Tabla de resultados de la desviación estándar de la repetibilidad en los 6
niveles.
Tabla de resultados de la desviación estándar de la reproducibilidad en los 6
niveles.
Tabla de resultados de la F calculada en los 6 niveles
Tabla de resultados del porcentaje de recuperación para evaluar la exactitud.
3.6.3 Descripción de instrumentos
Por el tipo de investigación el instrumento de recolección de datos fue el programa Excel
para la elaboración de las tablas; este programa contiene todas las herramientas para
obtener los resultados confiables y los cálculos exactos. Se trabajó con grandes cantidades
de datos en la cual intervienen todas las variables, facilita la interpretación de los resultados
y se ahorra tiempo en los cálculos.
Los datos colocados en las tablas Excel son las lecturas obtenidas del equipo
espectrofotómetro uv-visible.
El documento final de la investigación está organizado siguiendo los formatos y esquemas
estándar de la institución (UCE) y normas internacionales como la APA.
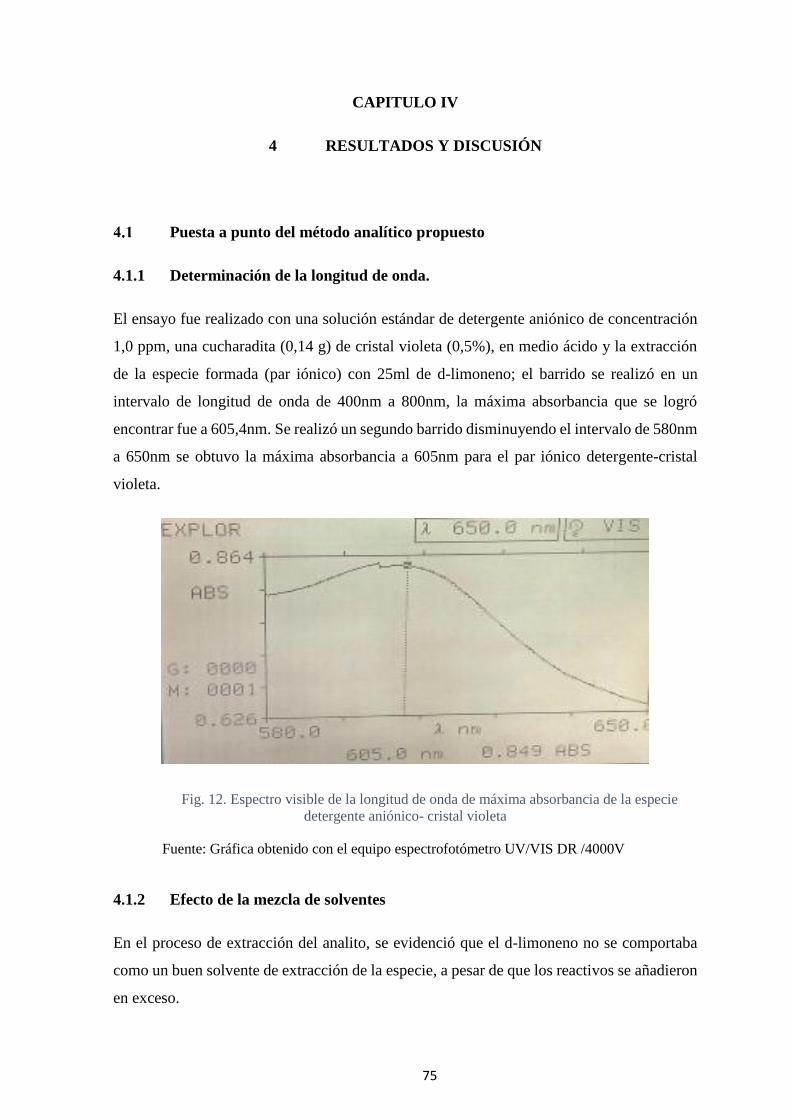
75
CAPITULO IV
4 RESULTADOS Y DISCUSIÓN
Puesta a punto del método analítico propuesto
4.1.1 Determinación de la longitud de onda.
El ensayo fue realizado con una solución estándar de detergente aniónico de concentración
1,0 ppm, una cucharadita (0,14 g) de cristal violeta (0,5%), en medio ácido y la extracción
de la especie formada (par iónico) con 25ml de d-limoneno; el barrido se realizó en un
intervalo de longitud de onda de 400nm a 800nm, la máxima absorbancia que se logró
encontrar fue a 605,4nm. Se realizó un segundo barrido disminuyendo el intervalo de 580nm
a 650nm se obtuvo la máxima absorbancia a 605nm para el par iónico detergente-cristal
violeta.
Fig. 12. Espectro visible de la longitud de onda de máxima absorbancia de la especie
detergente aniónico- cristal violeta
Fuente: Gráfica obtenido con el equipo espectrofotómetro UV/VIS DR /4000V
4.1.2 Efecto de la mezcla de solventes
En el proceso de extracción del analito, se evidenció que el d-limoneno no se comportaba
como un buen solvente de extracción de la especie, a pesar de que los reactivos se añadieron
en exceso.

76
Para superar este inconveniente se realizó las extracciones con mezclas de d-limoneno y
diversos solventes tomando en cuenta la serie eluotrófica. Para este ensayo se utilizó estándar
de 0,5ppm de detergente, cristal violeta (0,5%), d-limoneno y solventes: alcohol amílico,
metanol, etanol, hexano, etileno, tolueno y xileno. La proporción de las mezclas fueron 20
ml de d-limoneno y 5 ml de otros solventes, de acuerdo con la polaridad de estos.
El extracto se procedió a leer en el espectrofotómetro a 605nm y se obtuvo los siguientes
resultados en absorbancia (A).
La simbología que se utilizó fue la siguiente: Limoneno: L, Alcohol Amílico: AA, Metanol:
Me, Etanol: Et, Hexano H, Etileno: E, Tolueno: T y Xileno: X
Tabla 13.- Resultados en Absorbancia de la Extracción del par iónico con mezclas de disolventes
N Mezcla de Solventes Absorbancia
1 L 0,000
2 L-AA 0,656
3 L-Me 0,525
4 L-Et 0,568
5 L-H 0,016
6 L-E 0,198
7 L-T 0,120
8 L-X 0,183
Fuente: Salomón Chacha
Con los resultados de la tabla 13, se trazó una gráfica absorbancia en función de las mezclas
de diferentes solventes.
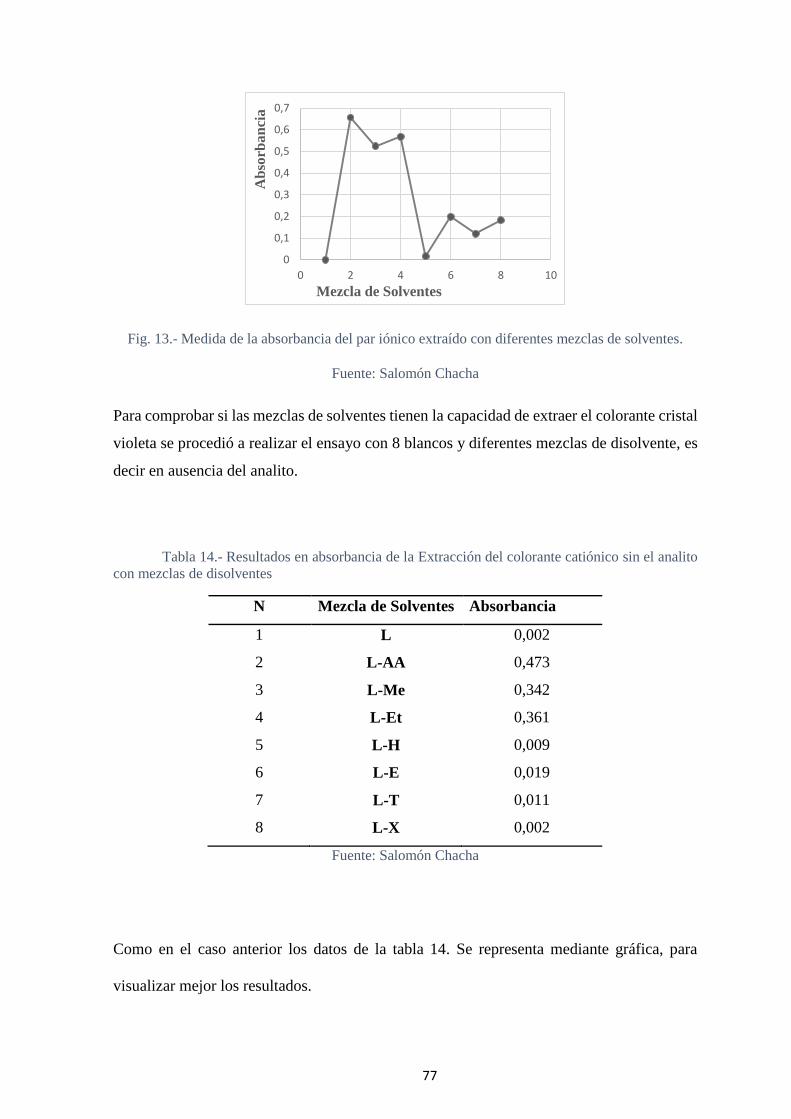
77
Fig. 13.- Medida de la absorbancia del par iónico extraído con diferentes mezclas de solventes.
Fuente: Salomón Chacha
Para comprobar si las mezclas de solventes tienen la capacidad de extraer el colorante cristal
violeta se procedió a realizar el ensayo con 8 blancos y diferentes mezclas de disolvente, es
decir en ausencia del analito.
Tabla 14.- Resultados en absorbancia de la Extracción del colorante catiónico sin el analito
con mezclas de disolventes
N Mezcla de Solventes Absorbancia
1 L 0,002
2 L-AA 0,473
3 L-Me 0,342
4 L-Et 0,361
5 L-H 0,009
6 L-E 0,019
7 L-T 0,011
8 L-X 0,002
Fuente: Salomón Chacha
Como en el caso anterior los datos de la tabla 14. Se representa mediante gráfica, para
visualizar mejor los resultados.
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0 2 4 6 8 10
Ab
sorb
an
cia
Mezcla de Solventes
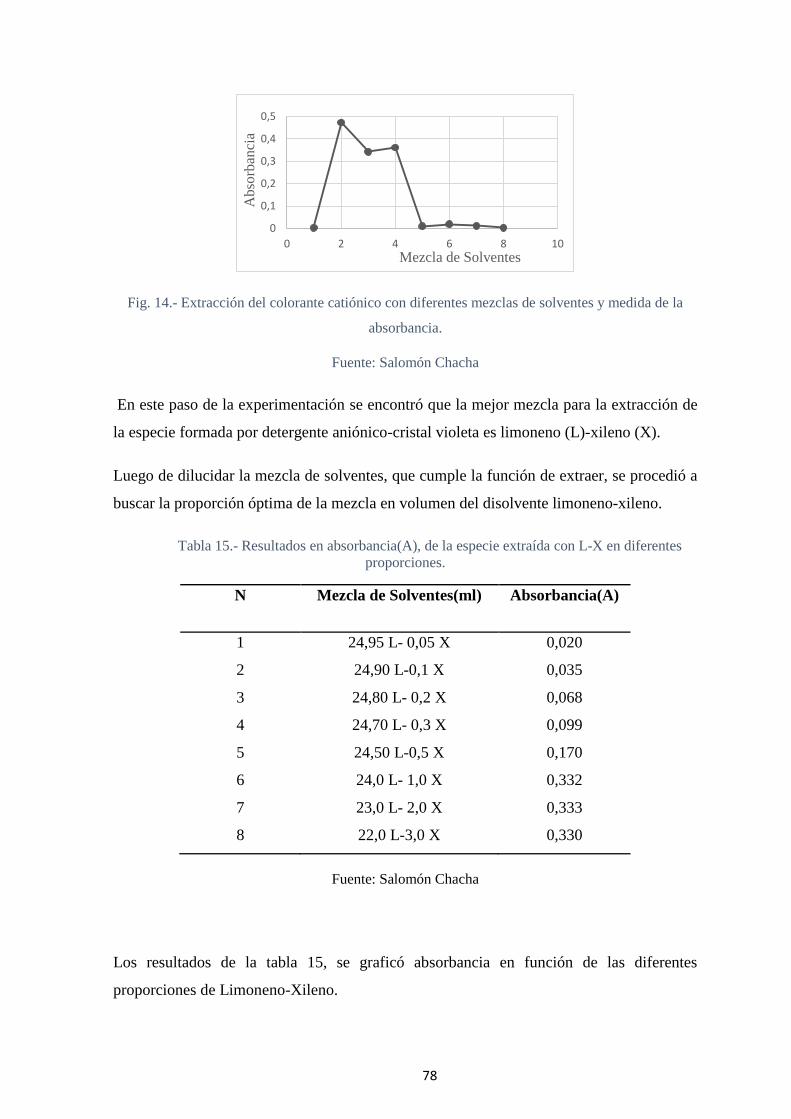
78
Fig. 14.- Extracción del colorante catiónico con diferentes mezclas de solventes y medida de la
absorbancia.
Fuente: Salomón Chacha
En este paso de la experimentación se encontró que la mejor mezcla para la extracción de
la especie formada por detergente aniónico-cristal violeta es limoneno (L)-xileno (X).
Luego de dilucidar la mezcla de solventes, que cumple la función de extraer, se procedió a
buscar la proporción óptima de la mezcla en volumen del disolvente limoneno-xileno.
Tabla 15.- Resultados en absorbancia(A), de la especie extraída con L-X en diferentes
proporciones.
N Mezcla de Solventes(ml) Absorbancia(A)
1 24,95 L- 0,05 X 0,020
2 24,90 L-0,1 X 0,035
3 24,80 L- 0,2 X 0,068
4 24,70 L- 0,3 X 0,099
5 24,50 L-0,5 X 0,170
6 24,0 L- 1,0 X 0,332
7 23,0 L- 2,0 X 0,333
8 22,0 L-3,0 X 0,330
Fuente: Salomón Chacha
Los resultados de la tabla 15, se graficó absorbancia en función de las diferentes
proporciones de Limoneno-Xileno.
0
0,1
0,2
0,3
0,4
0,5
0 2 4 6 8 10A
bso
rban
cia
Mezcla de Solventes
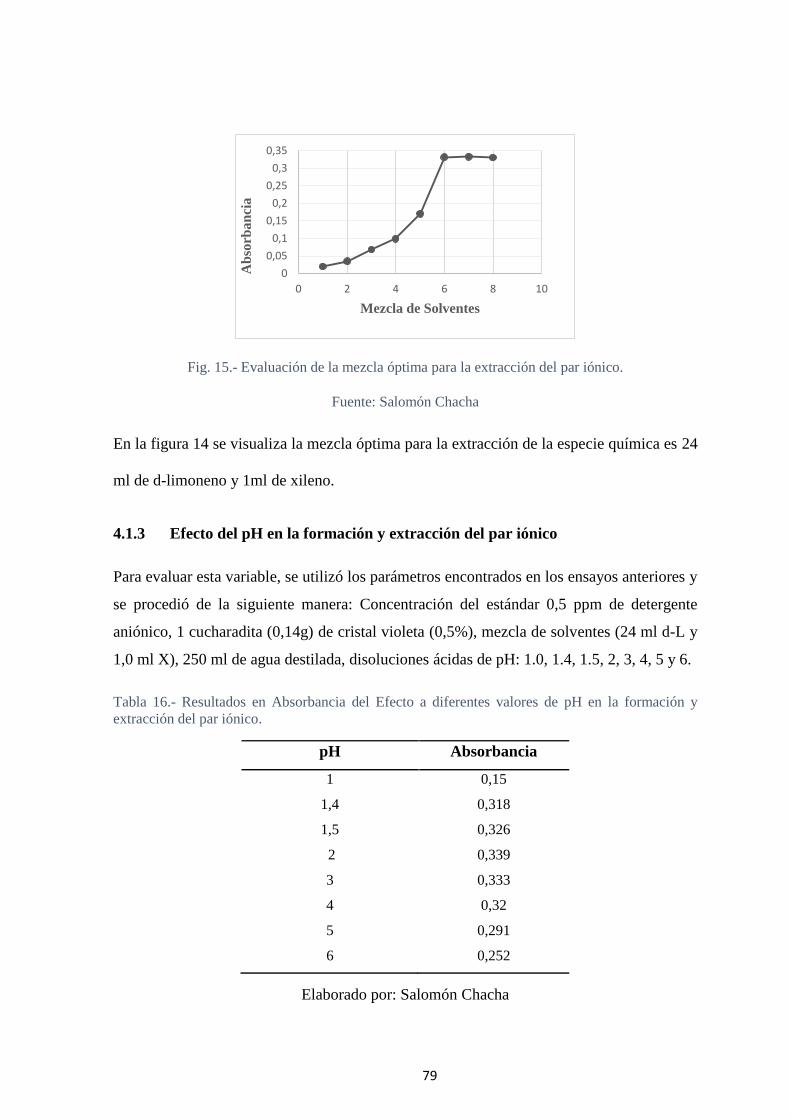
79
Fig. 15.- Evaluación de la mezcla óptima para la extracción del par iónico.
Fuente: Salomón Chacha
En la figura 14 se visualiza la mezcla óptima para la extracción de la especie química es 24
ml de d-limoneno y 1ml de xileno.
4.1.3 Efecto del pH en la formación y extracción del par iónico
Para evaluar esta variable, se utilizó los parámetros encontrados en los ensayos anteriores y
se procedió de la siguiente manera: Concentración del estándar 0,5 ppm de detergente
aniónico, 1 cucharadita (0,14g) de cristal violeta (0,5%), mezcla de solventes (24 ml d-L y
1,0 ml X), 250 ml de agua destilada, disoluciones ácidas de pH: 1.0, 1.4, 1.5, 2, 3, 4, 5 y 6.
Tabla 16.- Resultados en Absorbancia del Efecto a diferentes valores de pH en la formación y
extracción del par iónico.
pH Absorbancia
1 0,15
1,4 0,318
1,5 0,326
2 0,339
3 0,333
4 0,32
5 0,291
6 0,252
Elaborado por: Salomón Chacha
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0 2 4 6 8 10
Ab
sorb
an
cia
Mezcla de Solventes
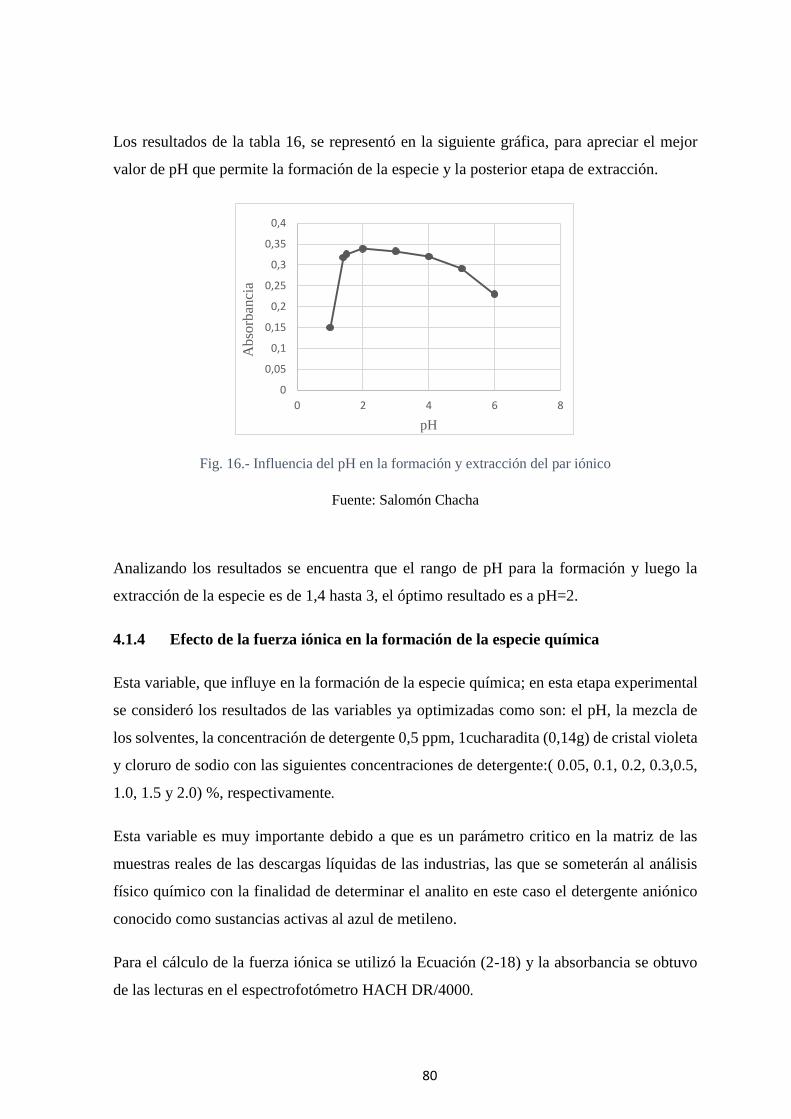
80
Los resultados de la tabla 16, se representó en la siguiente gráfica, para apreciar el mejor
valor de pH que permite la formación de la especie y la posterior etapa de extracción.
Fig. 16.- Influencia del pH en la formación y extracción del par iónico
Fuente: Salomón Chacha
Analizando los resultados se encuentra que el rango de pH para la formación y luego la
extracción de la especie es de 1,4 hasta 3, el óptimo resultado es a pH=2.
4.1.4 Efecto de la fuerza iónica en la formación de la especie química
Esta variable, que influye en la formación de la especie química; en esta etapa experimental
se consideró los resultados de las variables ya optimizadas como son: el pH, la mezcla de
los solventes, la concentración de detergente 0,5 ppm, 1cucharadita (0,14g) de cristal violeta
y cloruro de sodio con las siguientes concentraciones de detergente:( 0.05, 0.1, 0.2, 0.3,0.5,
1.0, 1.5 y 2.0) %, respectivamente.
Esta variable es muy importante debido a que es un parámetro critico en la matriz de las
muestras reales de las descargas líquidas de las industrias, las que se someterán al análisis
físico químico con la finalidad de determinar el analito en este caso el detergente aniónico
conocido como sustancias activas al azul de metileno.
Para el cálculo de la fuerza iónica se utilizó la Ecuación (2-18) y la absorbancia se obtuvo
de las lecturas en el espectrofotómetro HACH DR/4000.
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0,4
0 2 4 6 8
Ab
sorb
anci
a
pH
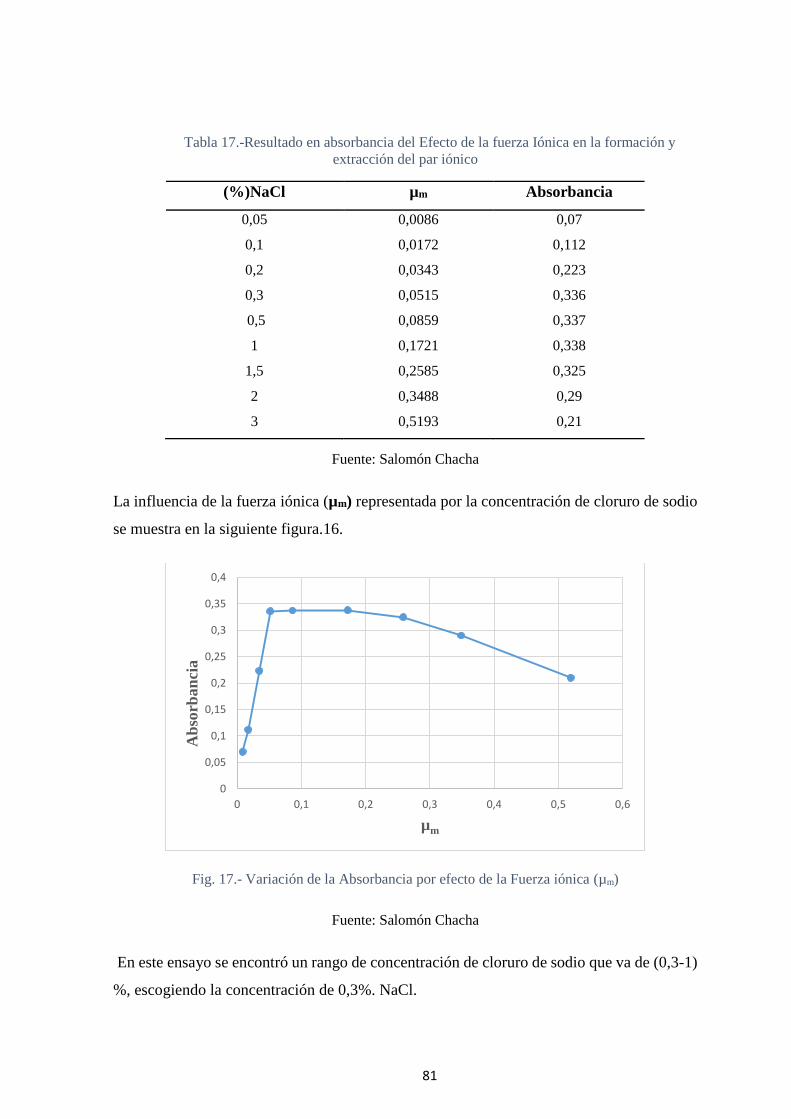
81
Tabla 17.-Resultado en absorbancia del Efecto de la fuerza Iónica en la formación y
extracción del par iónico
(%)NaCl µm Absorbancia
0,05 0,0086 0,07
0,1 0,0172 0,112
0,2 0,0343 0,223
0,3 0,0515 0,336
0,5 0,0859 0,337
1 0,1721 0,338
1,5 0,2585 0,325
2 0,3488 0,29
3 0,5193 0,21
Fuente: Salomón Chacha
La influencia de la fuerza iónica (µm) representada por la concentración de cloruro de sodio
se muestra en la siguiente figura.16.
Fig. 17.- Variación de la Absorbancia por efecto de la Fuerza iónica (µm)
Fuente: Salomón Chacha
En este ensayo se encontró un rango de concentración de cloruro de sodio que va de (0,3-1)
%, escogiendo la concentración de 0,3%. NaCl.
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0,4
0 0,1 0,2 0,3 0,4 0,5 0,6
Ab
sorb
an
cia
µm
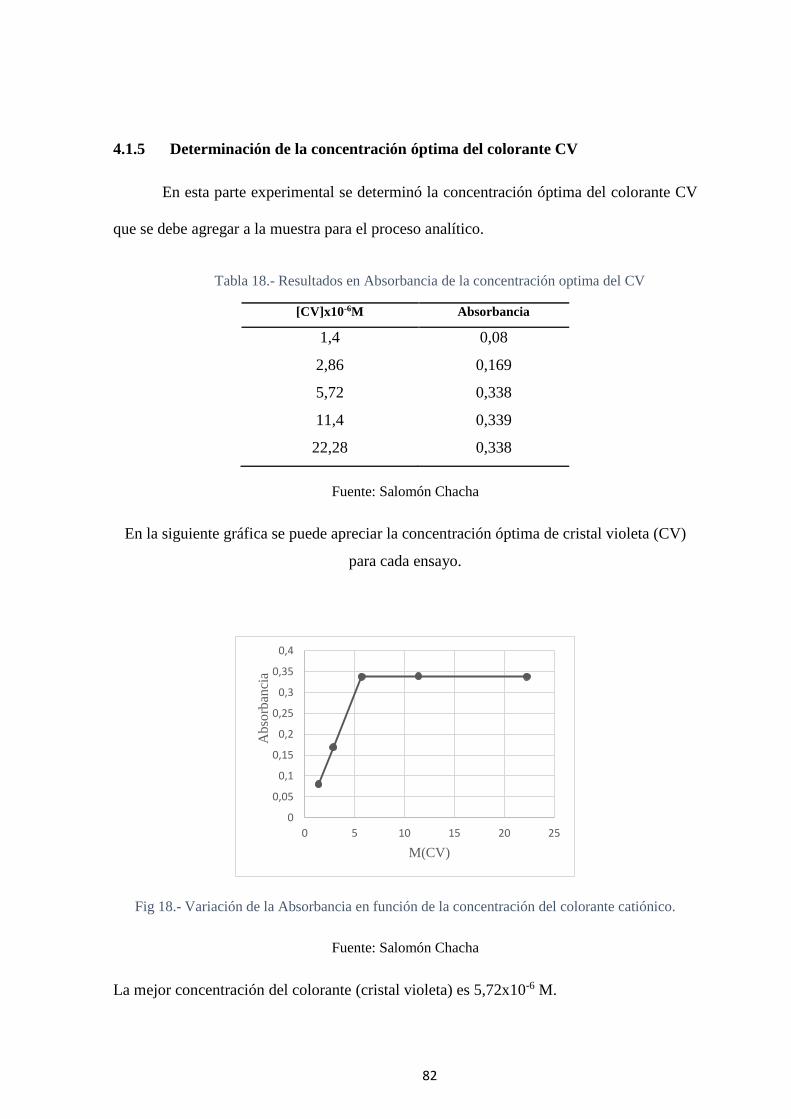
82
4.1.5 Determinación de la concentración óptima del colorante CV
En esta parte experimental se determinó la concentración óptima del colorante CV
que se debe agregar a la muestra para el proceso analítico.
Tabla 18.- Resultados en Absorbancia de la concentración optima del CV
[CV]x10-6M Absorbancia
1,4 0,08
2,86 0,169
5,72 0,338
11,4 0,339
22,28 0,338
Fuente: Salomón Chacha
En la siguiente gráfica se puede apreciar la concentración óptima de cristal violeta (CV)
para cada ensayo.
Fig 18.- Variación de la Absorbancia en función de la concentración del colorante catiónico.
Fuente: Salomón Chacha
La mejor concentración del colorante (cristal violeta) es 5,72x10-6 M.
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0,4
0 5 10 15 20 25
Abso
rban
cia
M(CV)
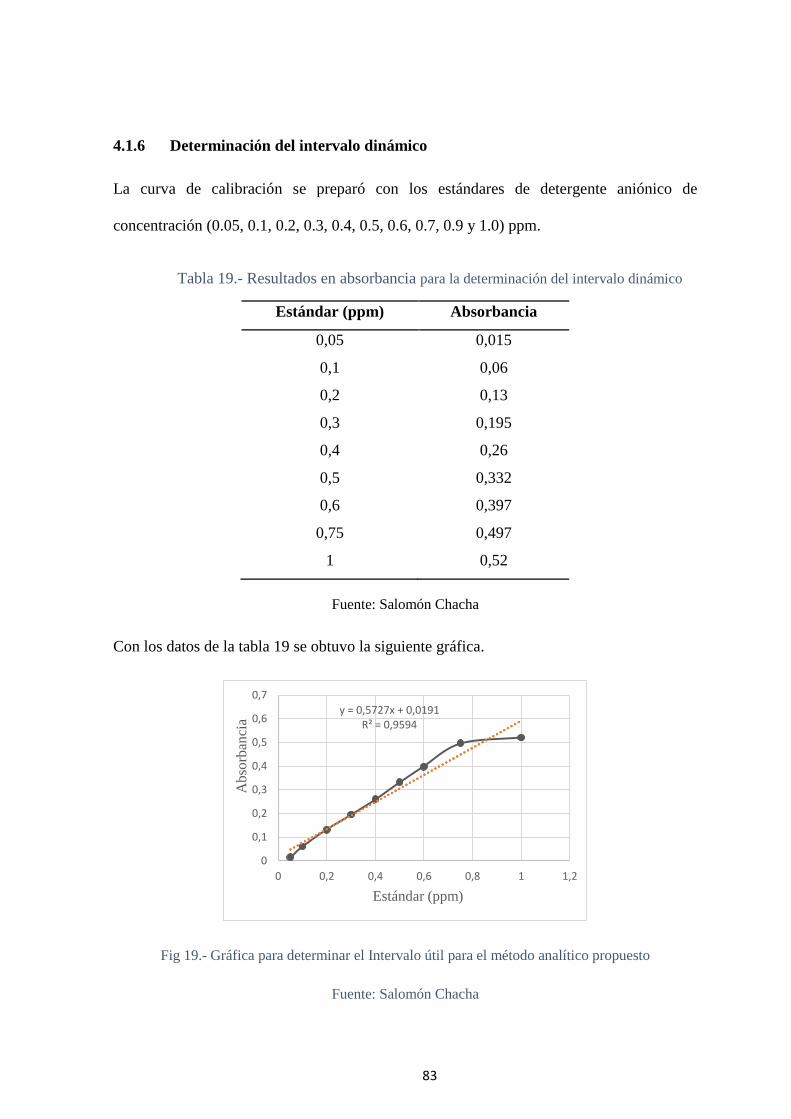
83
4.1.6 Determinación del intervalo dinámico
La curva de calibración se preparó con los estándares de detergente aniónico de
concentración (0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.9 y 1.0) ppm.
Tabla 19.- Resultados en absorbancia para la determinación del intervalo dinámico
Estándar (ppm) Absorbancia
0,05 0,015
0,1 0,06
0,2 0,13
0,3 0,195
0,4 0,26
0,5 0,332
0,6 0,397
0,75 0,497
1 0,52
Fuente: Salomón Chacha
Con los datos de la tabla 19 se obtuvo la siguiente gráfica.
Fig 19.- Gráfica para determinar el Intervalo útil para el método analítico propuesto
Fuente: Salomón Chacha
y = 0,5727x + 0,0191R² = 0,9594
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0 0,2 0,4 0,6 0,8 1 1,2
Abso
rban
cia
Estándar (ppm)
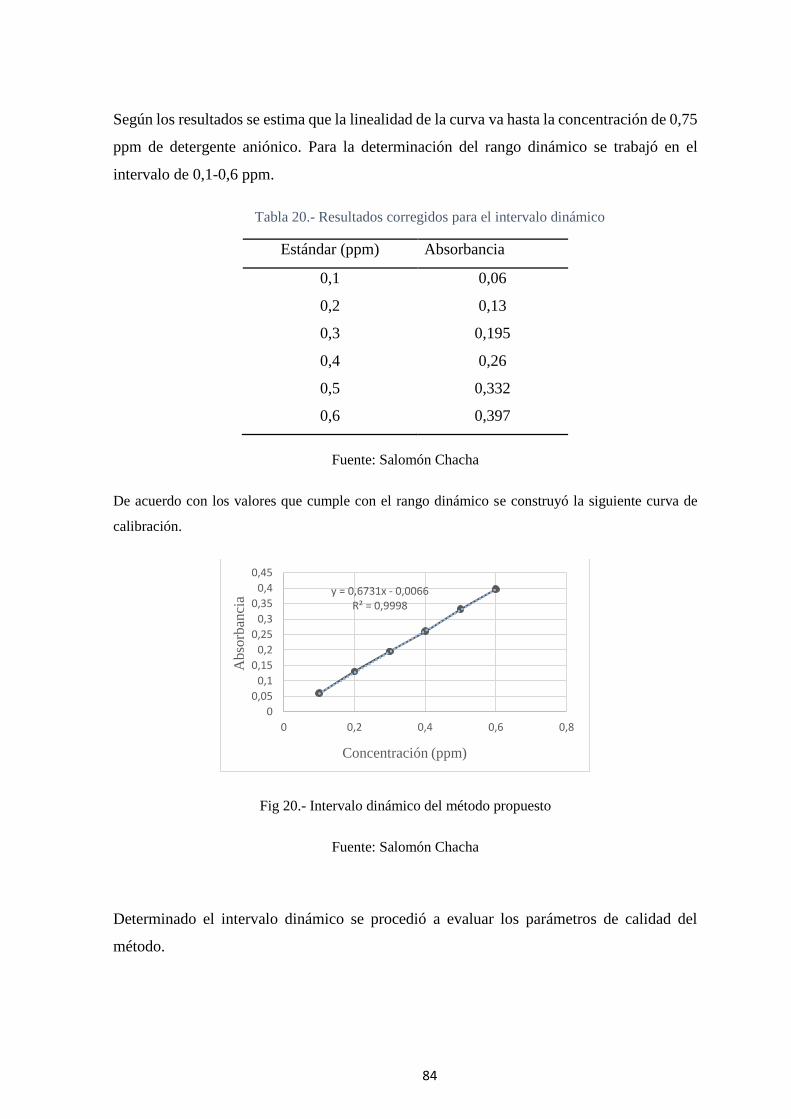
84
Según los resultados se estima que la linealidad de la curva va hasta la concentración de 0,75
ppm de detergente aniónico. Para la determinación del rango dinámico se trabajó en el
intervalo de 0,1-0,6 ppm.
Tabla 20.- Resultados corregidos para el intervalo dinámico
Estándar (ppm) Absorbancia
0,1 0,06
0,2 0,13
0,3 0,195
0,4 0,26
0,5 0,332
0,6 0,397
Fuente: Salomón Chacha
De acuerdo con los valores que cumple con el rango dinámico se construyó la siguiente curva de
calibración.
Fig 20.- Intervalo dinámico del método propuesto
Fuente: Salomón Chacha
Determinado el intervalo dinámico se procedió a evaluar los parámetros de calidad del
método.
y = 0,6731x - 0,0066R² = 0,9998
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0,4
0,45
0 0,2 0,4 0,6 0,8
Abso
rban
cia
Concentración (ppm)
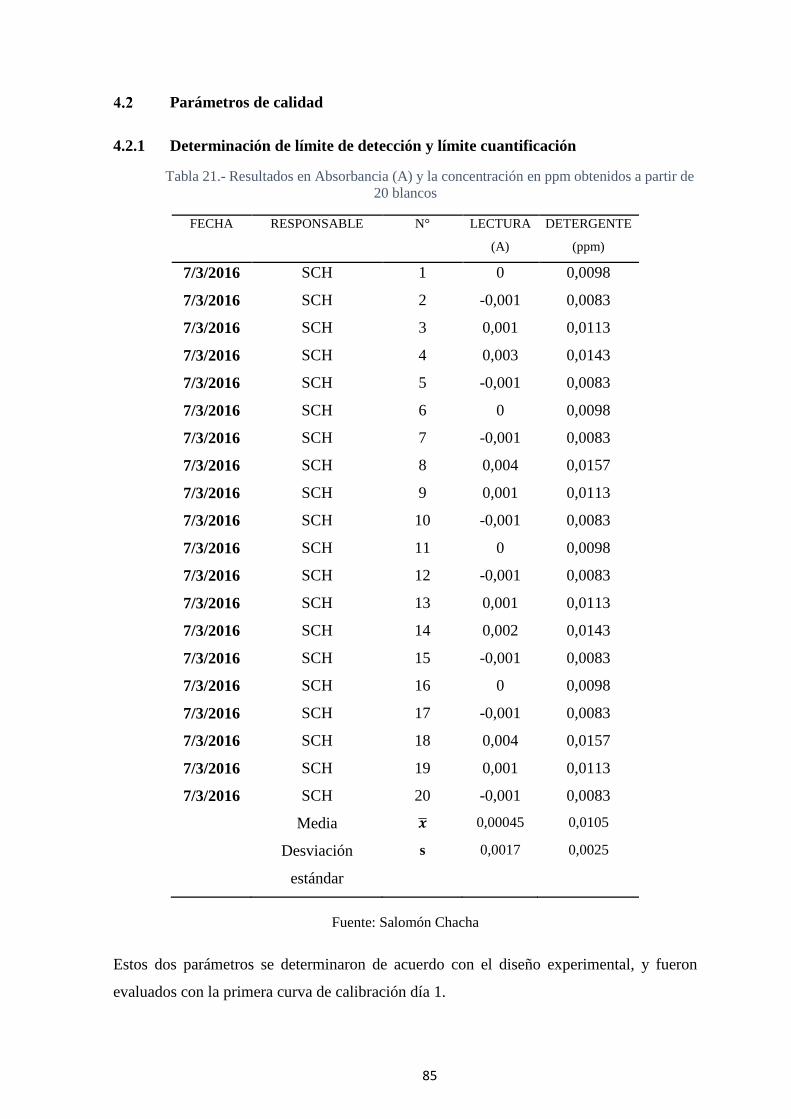
85
Parámetros de calidad
4.2.1 Determinación de límite de detección y límite cuantificación
Tabla 21.- Resultados en Absorbancia (A) y la concentración en ppm obtenidos a partir de
20 blancos
FECHA RESPONSABLE N° LECTURA
(A)
DETERGENTE
(ppm)
7/3/2016 SCH 1 0 0,0098
7/3/2016 SCH 2 -0,001 0,0083
7/3/2016 SCH 3 0,001 0,0113
7/3/2016 SCH 4 0,003 0,0143
7/3/2016 SCH 5 -0,001 0,0083
7/3/2016 SCH 6 0 0,0098
7/3/2016 SCH 7 -0,001 0,0083
7/3/2016 SCH 8 0,004 0,0157
7/3/2016 SCH 9 0,001 0,0113
7/3/2016 SCH 10 -0,001 0,0083
7/3/2016 SCH 11 0 0,0098
7/3/2016 SCH 12 -0,001 0,0083
7/3/2016 SCH 13 0,001 0,0113
7/3/2016 SCH 14 0,002 0,0143
7/3/2016 SCH 15 -0,001 0,0083
7/3/2016 SCH 16 0 0,0098
7/3/2016 SCH 17 -0,001 0,0083
7/3/2016 SCH 18 0,004 0,0157
7/3/2016 SCH 19 0,001 0,0113
7/3/2016 SCH 20 -0,001 0,0083
Media �̅� 0,00045 0,0105
Desviación
estándar
s 0,0017 0,0025
Fuente: Salomón Chacha
Estos dos parámetros se determinaron de acuerdo con el diseño experimental, y fueron
evaluados con la primera curva de calibración día 1.
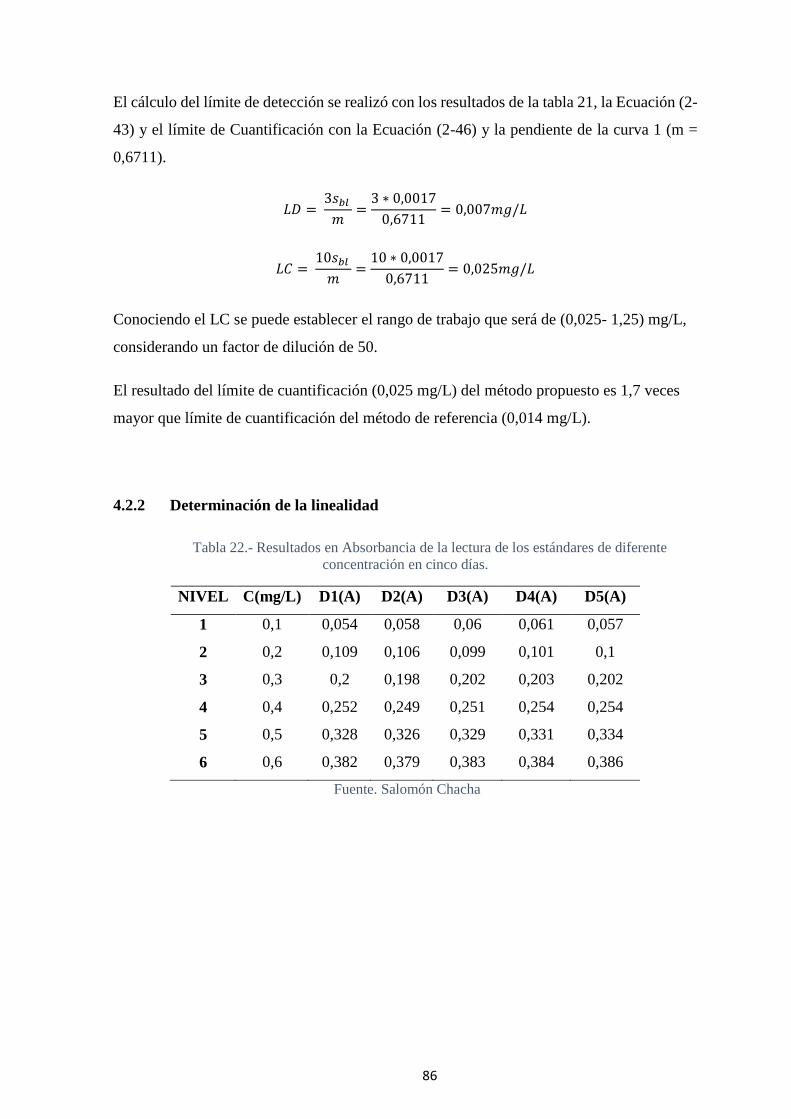
86
El cálculo del límite de detección se realizó con los resultados de la tabla 21, la Ecuación (2-
43) y el límite de Cuantificación con la Ecuación (2-46) y la pendiente de la curva 1 (m =
0,6711).
𝐿𝐷 = 3𝑠𝑏𝑙
𝑚=
3 ∗ 0,0017
0,6711= 0,007𝑚𝑔/𝐿
𝐿𝐶 = 10𝑠𝑏𝑙
𝑚=
10 ∗ 0,0017
0,6711= 0,025𝑚𝑔/𝐿
Conociendo el LC se puede establecer el rango de trabajo que será de (0,025- 1,25) mg/L,
considerando un factor de dilución de 50.
El resultado del límite de cuantificación (0,025 mg/L) del método propuesto es 1,7 veces
mayor que límite de cuantificación del método de referencia (0,014 mg/L).
4.2.2 Determinación de la linealidad
Tabla 22.- Resultados en Absorbancia de la lectura de los estándares de diferente
concentración en cinco días.
NIVEL C(mg/L) D1(A) D2(A) D3(A) D4(A) D5(A)
1 0,1 0,054 0,058 0,06 0,061 0,057
2 0,2 0,109 0,106 0,099 0,101 0,1
3 0,3 0,2 0,198 0,202 0,203 0,202
4 0,4 0,252 0,249 0,251 0,254 0,254
5 0,5 0,328 0,326 0,329 0,331 0,334
6 0,6 0,382 0,379 0,383 0,384 0,386
Fuente. Salomón Chacha
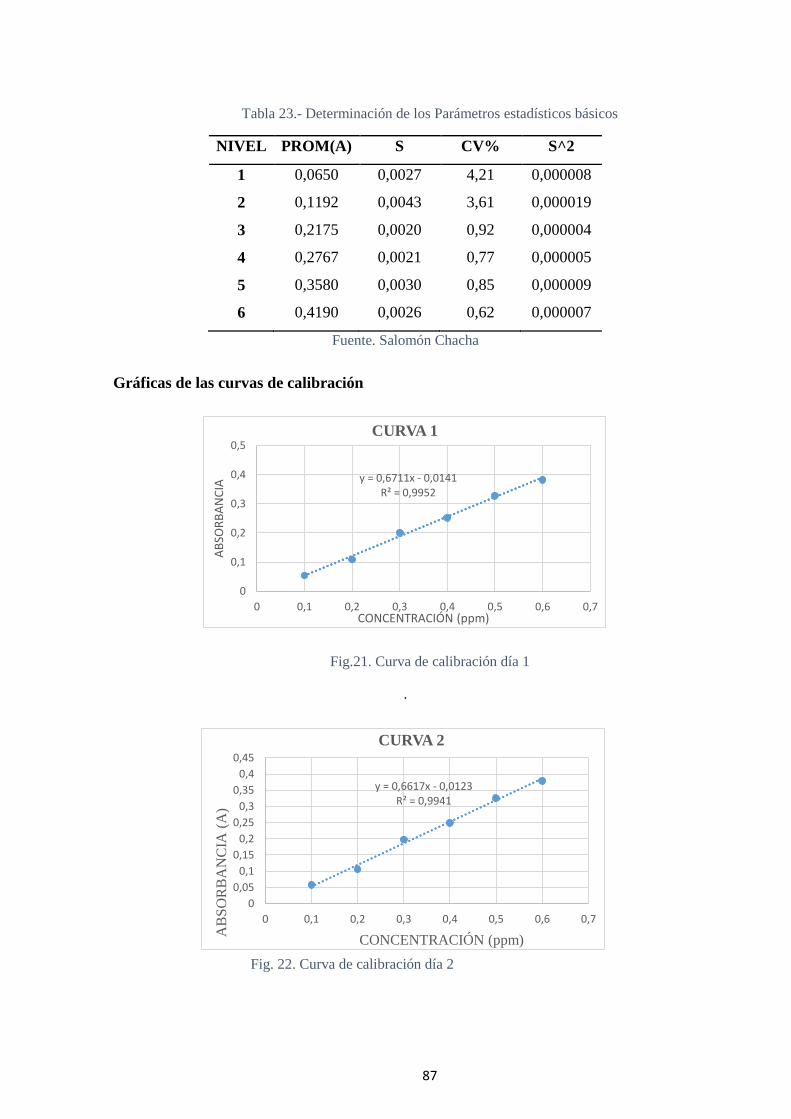
87
Tabla 23.- Determinación de los Parámetros estadísticos básicos
NIVEL PROM(A) S CV% S^2
1 0,0650 0,0027 4,21 0,000008
2 0,1192 0,0043 3,61 0,000019
3 0,2175 0,0020 0,92 0,000004
4 0,2767 0,0021 0,77 0,000005
5 0,3580 0,0030 0,85 0,000009
6 0,4190 0,0026 0,62 0,000007
Fuente. Salomón Chacha
Gráficas de las curvas de calibración
Fig.21. Curva de calibración día 1
.
Fig 21.- Curva de calibración día 2.
Fuente: Salomón Chacha
y = 0,6711x - 0,0141R² = 0,9952
0
0,1
0,2
0,3
0,4
0,5
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7
AB
SOR
BA
NC
IA
CONCENTRACIÓN (ppm)
CURVA 1
y = 0,6617x - 0,0123R² = 0,9941
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0,4
0,45
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7
AB
SO
RB
AN
CIA
(A
)
CONCENTRACIÓN (ppm)
CURVA 2
Fig. 22. Curva de calibración día 2
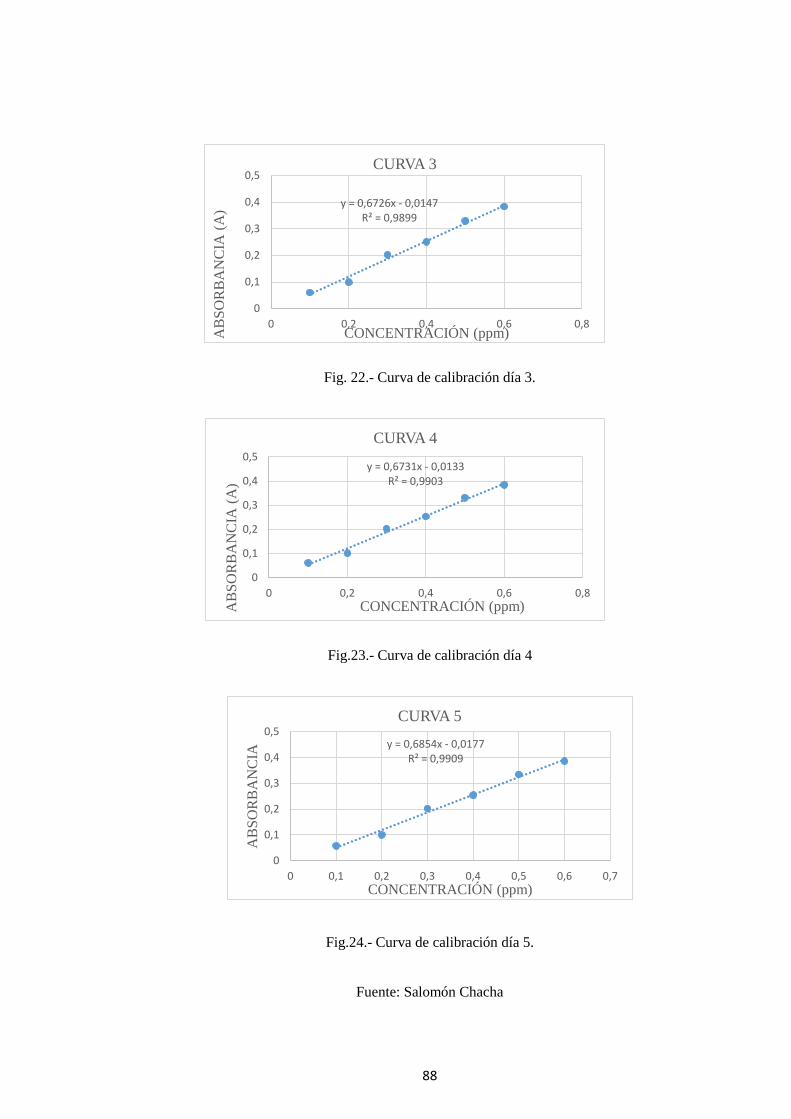
88
Fig. 22.- Curva de calibración día 3.
Fig.23.- Curva de calibración día 4
Fig.24.- Curva de calibración día 5.
Fuente: Salomón Chacha
y = 0,6726x - 0,0147R² = 0,9899
0
0,1
0,2
0,3
0,4
0,5
0 0,2 0,4 0,6 0,8
AB
SO
RB
AN
CIA
(A
)
CONCENTRACIÓN (ppm)
CURVA 3
y = 0,6731x - 0,0133R² = 0,9903
0
0,1
0,2
0,3
0,4
0,5
0 0,2 0,4 0,6 0,8
AB
SO
RB
AN
CIA
(A
)
CONCENTRACIÓN (ppm)
CURVA 4
y = 0,6854x - 0,0177R² = 0,9909
0
0,1
0,2
0,3
0,4
0,5
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7
AB
SO
RB
AN
CIA
CONCENTRACIÓN (ppm)
CURVA 5
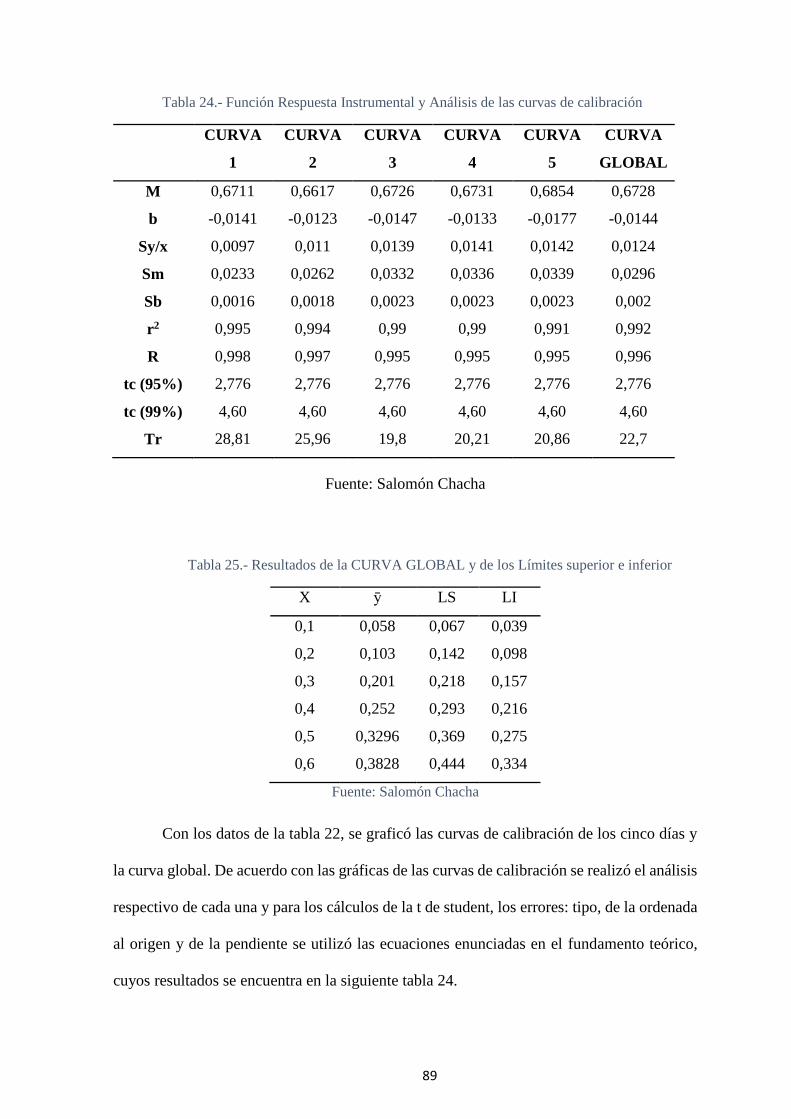
89
Tabla 24.- Función Respuesta Instrumental y Análisis de las curvas de calibración
CURVA
1
CURVA
2
CURVA
3
CURVA
4
CURVA
5
CURVA
GLOBAL
M 0,6711 0,6617 0,6726 0,6731 0,6854 0,6728
b -0,0141 -0,0123 -0,0147 -0,0133 -0,0177 -0,0144
Sy/x 0,0097 0,011 0,0139 0,0141 0,0142 0,0124
Sm 0,0233 0,0262 0,0332 0,0336 0,0339 0,0296
Sb 0,0016 0,0018 0,0023 0,0023 0,0023 0,002
r2 0,995 0,994 0,99 0,99 0,991 0,992
R 0,998 0,997 0,995 0,995 0,995 0,996
tc (95%) 2,776 2,776 2,776 2,776 2,776 2,776
tc (99%) 4,60 4,60 4,60 4,60 4,60 4,60
Tr 28,81 25,96 19,8 20,21 20,86 22,7
Fuente: Salomón Chacha
Tabla 25.- Resultados de la CURVA GLOBAL y de los Límites superior e inferior
X ӯ LS LI
0,1 0,058 0,067 0,039
0,2 0,103 0,142 0,098
0,3 0,201 0,218 0,157
0,4 0,252 0,293 0,216
0,5 0,3296 0,369 0,275
0,6 0,3828 0,444 0,334
Fuente: Salomón Chacha
Con los datos de la tabla 22, se graficó las curvas de calibración de los cinco días y
la curva global. De acuerdo con las gráficas de las curvas de calibración se realizó el análisis
respectivo de cada una y para los cálculos de la t de student, los errores: tipo, de la ordenada
al origen y de la pendiente se utilizó las ecuaciones enunciadas en el fundamento teórico,
cuyos resultados se encuentra en la siguiente tabla 24.
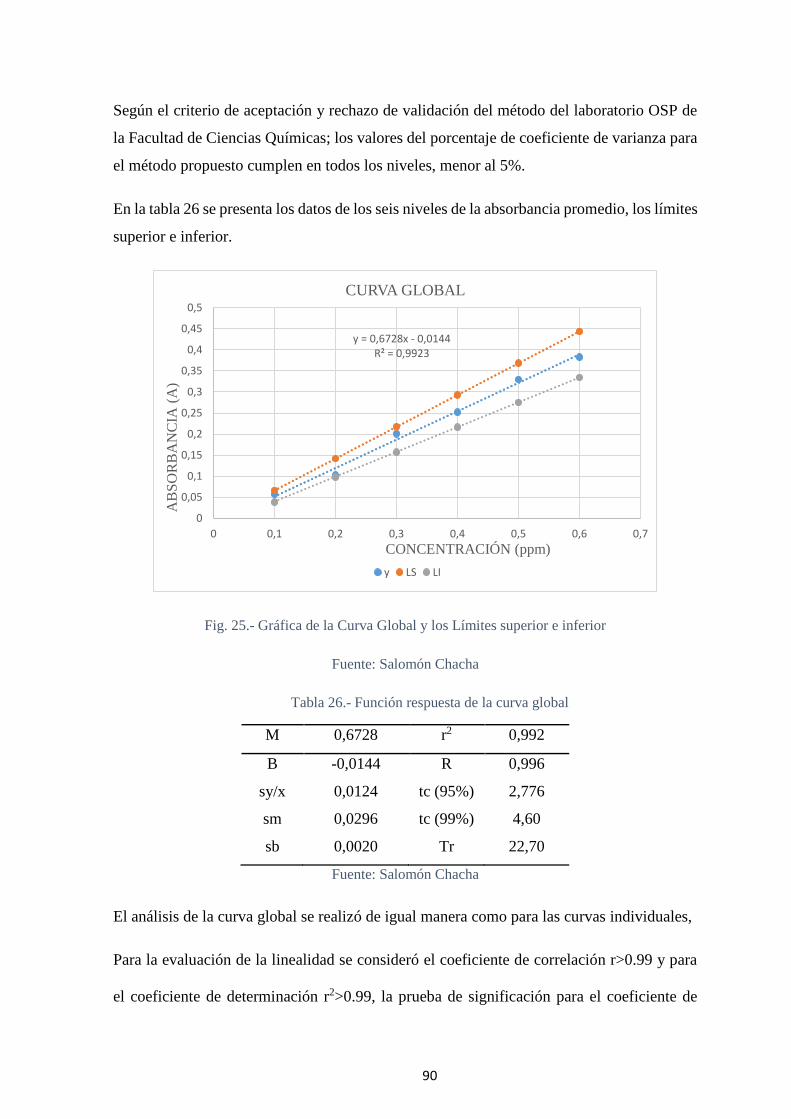
90
Según el criterio de aceptación y rechazo de validación del método del laboratorio OSP de
la Facultad de Ciencias Químicas; los valores del porcentaje de coeficiente de varianza para
el método propuesto cumplen en todos los niveles, menor al 5%.
En la tabla 26 se presenta los datos de los seis niveles de la absorbancia promedio, los límites
superior e inferior.
Fig. 25.- Gráfica de la Curva Global y los Límites superior e inferior
Fuente: Salomón Chacha
Tabla 26.- Función respuesta de la curva global
M 0,6728 r2 0,992
B -0,0144 R 0,996
sy/x 0,0124 tc (95%) 2,776
sm 0,0296 tc (99%) 4,60
sb 0,0020 Tr 22,70
Fuente: Salomón Chacha
El análisis de la curva global se realizó de igual manera como para las curvas individuales,
Para la evaluación de la linealidad se consideró el coeficiente de correlación r>0.99 y para
el coeficiente de determinación r2>0.99, la prueba de significación para el coeficiente de
y = 0,6728x - 0,0144R² = 0,9923
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0,4
0,45
0,5
0 0,1 0,2 0,3 0,4 0,5 0,6 0,7
AB
SO
RB
AN
CIA
(A
)
CONCENTRACIÓN (ppm)
CURVA GLOBAL
y LS LI

91
correlación se calculó con la ecuación (2-33), para las cinco curvas de calibración y para la
curva global, cuyos datos están en las tablas 24 y 26. Los valores críticos tabulados de tc
obtenido de la tabla para n=4,𝛼 = 0,05 𝑦 0,01 para un contraste de 2 colas (Anexo Tabla
A2), son menores en los dos casos al 95% y 99% para el valor calculado de tr; por tal razón
la prueba es significativa y se rechaza la hipótesis nula.
Sensibilidad
Tabla 27.- Resultados de las pendientes de las curvas de calibración
Curvas Pendiente(A.l/mg)
1 0,6711
2 0,6617
3 0,6726
4 0,6731
5 0,6854
GLOBAL 0,6728
Fuente: Salomón Chacha
La sensibilidad del método se determinó por medio de la pendiente de las curvas de
calibración, incluyendo la pendiente de la curva de calibración global, los valores se
representan en la tabla 27.
La curva de calibración realizado el quinto día representa la más sensible para el método
propuesto, debido a que la pendiente es mayor a las demás curvas de calibración.
Precisión
El estudio de la precisión se realizó a través del análisis simple de varianza (ANOVA de
factores totalmente anidados y homogéneos), las desviaciones estándar de repetibilidad (sr)
y de reproducibilidad (sR), considerado como precisión intermedia; para cada uno de los
niveles de ensayo. Los resultados experimentales se ubican en la siguiente tabla.28.
Repetibilidad. Se procedió de acuerdo con el diseño experimental, a continuación, en la
tabla 28. se presenta los resultados.
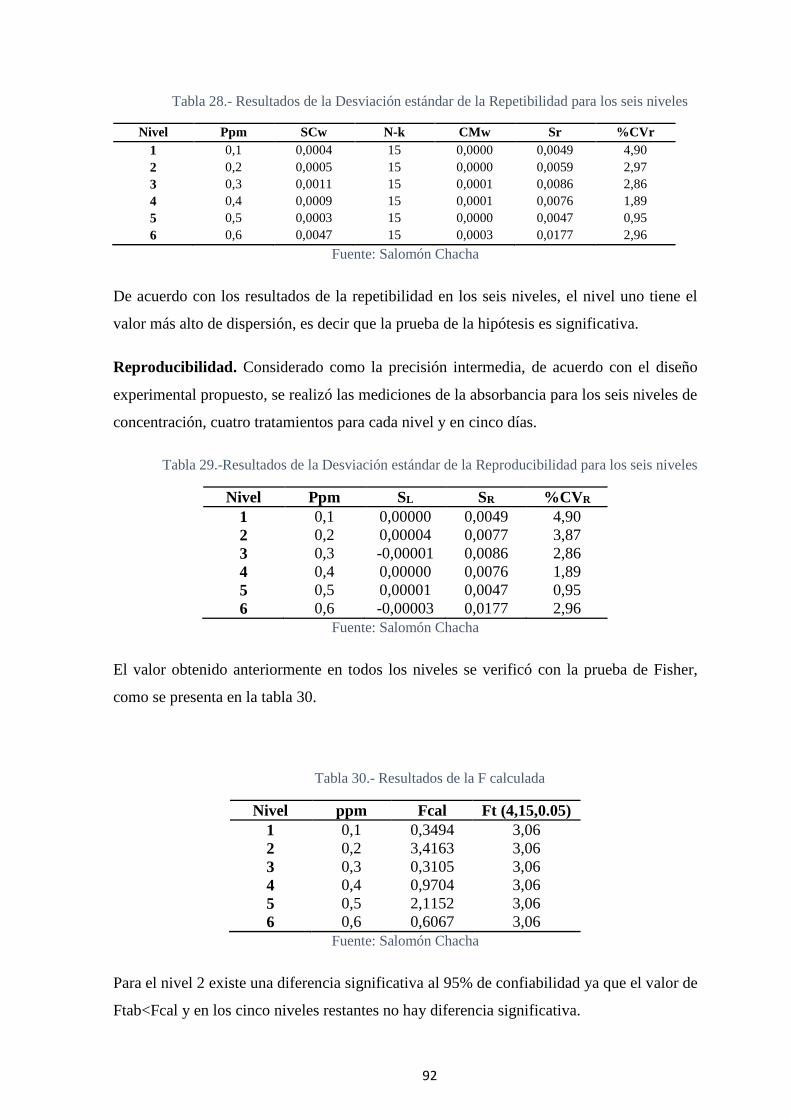
92
Tabla 28.- Resultados de la Desviación estándar de la Repetibilidad para los seis niveles
Nivel Ppm SCw N-k CMw Sr %CVr
1 0,1 0,0004 15 0,0000 0,0049 4,90
2 0,2 0,0005 15 0,0000 0,0059 2,97
3 0,3 0,0011 15 0,0001 0,0086 2,86
4 0,4 0,0009 15 0,0001 0,0076 1,89
5 0,5 0,0003 15 0,0000 0,0047 0,95
6 0,6 0,0047 15 0,0003 0,0177 2,96
Fuente: Salomón Chacha
De acuerdo con los resultados de la repetibilidad en los seis niveles, el nivel uno tiene el
valor más alto de dispersión, es decir que la prueba de la hipótesis es significativa.
Reproducibilidad. Considerado como la precisión intermedia, de acuerdo con el diseño
experimental propuesto, se realizó las mediciones de la absorbancia para los seis niveles de
concentración, cuatro tratamientos para cada nivel y en cinco días.
Tabla 29.-Resultados de la Desviación estándar de la Reproducibilidad para los seis niveles
Nivel Ppm SL SR %CVR
1 0,1 0,00000 0,0049 4,90
2 0,2 0,00004 0,0077 3,87
3 0,3 -0,00001 0,0086 2,86
4 0,4 0,00000 0,0076 1,89
5 0,5 0,00001 0,0047 0,95
6 0,6 -0,00003 0,0177 2,96
Fuente: Salomón Chacha
El valor obtenido anteriormente en todos los niveles se verificó con la prueba de Fisher,
como se presenta en la tabla 30.
Tabla 30.- Resultados de la F calculada
Nivel ppm Fcal Ft (4,15,0.05)
1 0,1 0,3494 3,06
2 0,2 3,4163 3,06
3 0,3 0,3105 3,06
4 0,4 0,9704 3,06
5 0,5 2,1152 3,06
6 0,6 0,6067 3,06
Fuente: Salomón Chacha
Para el nivel 2 existe una diferencia significativa al 95% de confiabilidad ya que el valor de
Ftab<Fcal y en los cinco niveles restantes no hay diferencia significativa.
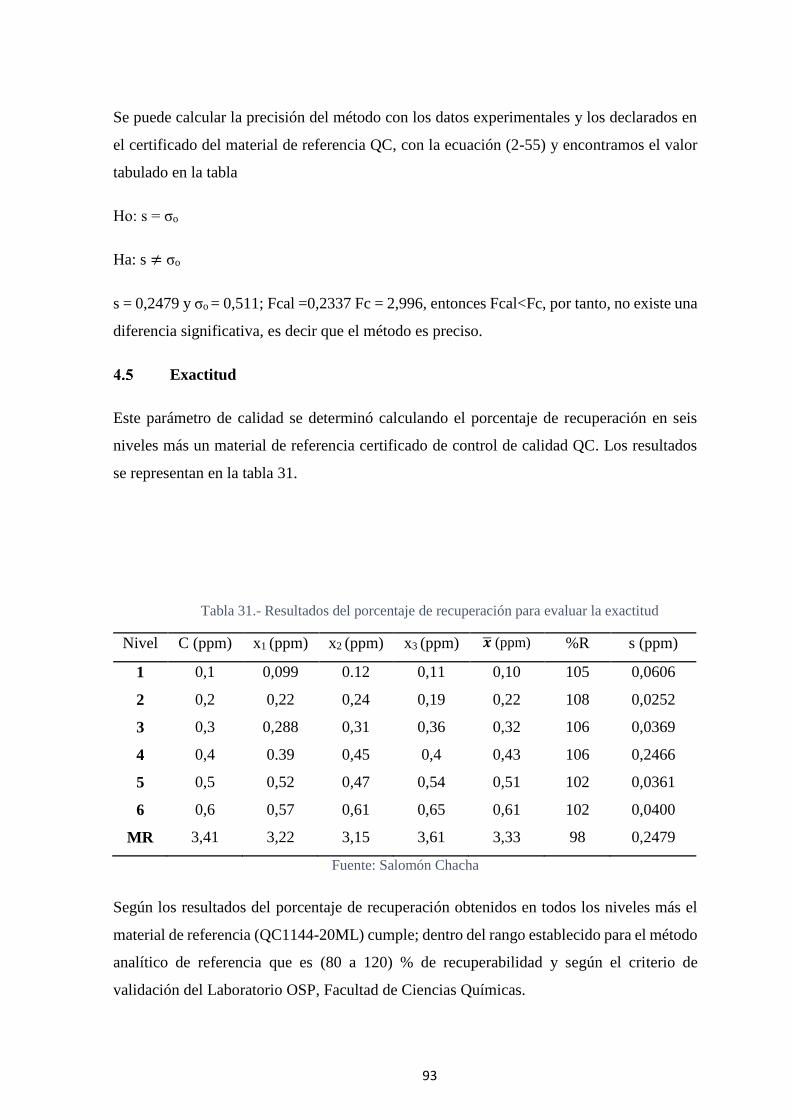
93
Se puede calcular la precisión del método con los datos experimentales y los declarados en
el certificado del material de referencia QC, con la ecuación (2-55) y encontramos el valor
tabulado en la tabla
Ho: s = σo
Ha: s ≠ σo
s = 0,2479 y σo = 0,511; Fcal =0,2337 Fc = 2,996, entonces Fcal<Fc, por tanto, no existe una
diferencia significativa, es decir que el método es preciso.
Exactitud
Este parámetro de calidad se determinó calculando el porcentaje de recuperación en seis
niveles más un material de referencia certificado de control de calidad QC. Los resultados
se representan en la tabla 31.
Tabla 31.- Resultados del porcentaje de recuperación para evaluar la exactitud
Nivel C (ppm) x1 (ppm) x2 (ppm) x3 (ppm) �̅� (ppm) %R s (ppm)
1 0,1 0,099 0.12 0,11 0,10 105 0,0606
2 0,2 0,22 0,24 0,19 0,22 108 0,0252
3 0,3 0,288 0,31 0,36 0,32 106 0,0369
4 0,4 0.39 0,45 0,4 0,43 106 0,2466
5 0,5 0,52 0,47 0,54 0,51 102 0,0361
6 0,6 0,57 0,61 0,65 0,61 102 0,0400
MR 3,41 3,22 3,15 3,61 3,33 98 0,2479
Fuente: Salomón Chacha
Según los resultados del porcentaje de recuperación obtenidos en todos los niveles más el
material de referencia (QC1144-20ML) cumple; dentro del rango establecido para el método
analítico de referencia que es (80 a 120) % de recuperabilidad y según el criterio de
validación del Laboratorio OSP, Facultad de Ciencias Químicas.

94
Otra forma de determinar la exactitud del método es con los datos experimentales de un
MRC, QC1144 y el valor declarado en el certificado, según la ecuación (2-64) que representa
al porcentaje de recuperación.
�̅� = 3,33 µo = 3,41
%R=97,65; como en el caso anterior el porcentaje de recuperación está dentro de los criterios
de aceptación del laboratorio OSP, Facultad de Ciencias Químicas.
La prueba estadística se realizó utilizando la ecuación (2-65), se planteó la siguiente
hipótesis: Ho= Que el método no está sujeto a errores sistemáticos
se calcula con los siguientes datos: �̅� = 3,33; µo = 3,41; n=3 y s= 0,2479
tcal= -0,558, tc = 3,18 entonces tc>tcal, la Ho se acepta, es decir que no hay evidencia de errores
sistemáticos.

95
CAPITULO V
CONCLUSIONES Y RECOMENDACIONES
Conclusiones
El proyecto de investigación plasmado en este documento ha llegado a las siguientes
conclusiones.
En la puesta a punto del método se encontró:
La mezcla apropiada para la extracción de la especie química que contiene al
analito es el limoneno con el xileno, la proporción optima es de 24:1, en
volumen.
La máxima absorbancia que alcanza la especie química es a una longitud de
onda de 605 nm.
El rango de pH adecuado para la formación y extracción de la especie
química es de (1,4-2) unidades de pH.
La influencia de la fuerza iónica para favorecer la formación y extracción de
la especie química está en el rango de concentración de (0,33-1,0) % (p/v) de
NaCl.
La concentración mínima del colorante catiónico es de 5,72x10-6 M.
Con respecto a los parámetros de calidad:
El intervalo de trabajo se determinó a partir del Límite de Cuantificación (0,025-
1,25) mg/L.
En la evaluación de la curva de calibración global, mediante la prueba de t-
Student que determina la correlación entre la respuesta y la concentración; que el
modelo es lineal. El coeficiente de correlación r2>0,99
La sensibilidad del método que tiene relación con la pendiente es 0,6728 L/mg.
La exactitud tiene un rango de recuperación de 98-108 %.
El método es selectivo porque se trabajó con muestras sintéticas o patrones, es
decir no se apreció interferencias en las lecturas.

96
Por medio del análisis de varianza de determinó la precisión con una desviación
estándar de repetibilidad de un rango de 0,0047-0,0177 mg/L y una desviación
estándar de reproducibilidad que está en el mismo rango.
El límite de detección es de 0,007 mg/L
El límite de cuantificación es de 0,025mg/L
En base a los resultados de las variables; mezcla de solvente, pH, fuerza iónica y los
parámetros de calidad se concluye que el método alternativo ha sido posible desarrollar.
Recomendaciones
Entre las recomendaciones al desarrollar métodos analíticos se citan las siguientes:
Utilizar sustancias químicamente puras, los productos comerciales están compuestas
por mezclas de isómeros (Tensioactivos).
Los equipos de medida como espectrofotómetros, balanzas, pipetas deben estar
calibrados y con su respectivo cronograma de mantenimiento, además el material
de vidrio limpio y en buen estado.
Si en la muestra de aguas residuales existe sustancias oxidantes como: Cl2, KMnO4,
K2Cr2O7, o sustancias reductoras como los sulfuros, sulfitos, se debe hacer un
tratamiento previo antes de someter al ensayo.
La agitación no debe ser fuerte para evitar formar una emulsión
Si la muestra de agua residual contiene altos contenido de minerales se procederá a
la dilución.
Las muestras con materia sólida en suspensión o microrganismos se debe practicar
una extracción previa.
El método desarrollado se recomienda someter a una validación para que los
resultados sean confiables.

97
6 BIBLIOGRAFIA
(JCGM), C. C. (4 de Febrero de 2009). http://www.sim-
metrologia.org.br/docs/span_VIM.pdf. Recuperado el 25 de Enero de 2014, de
http://www.metrologia.cl/link.cgi/Noticias/11.
Anastas, P., Kirchhoff, M., & Williamson, T. (2001). Catalysis as a foundational pillar of
green chemistry. CENTER FOR GREEN CHEMISTRY & GREEN
ENGINEERING AT YALE, 3-13.
Araya Alpizar, C. M. (2004). Estadistica para Laboratorista Químico (Primera ed.). San José
de Costa Rica: De la Universidad de Universidad de Costa Rica. Ciudad Universitaria
Rodrigo Facio.
Baird, C. (2004). Química Ambiental. Barcelona: REVERTÉ, S.A.
Barragan, O. L. (2010). Desarrollo salud Humana y Amenazas Ambientales. La Plata: de la
Universidad nacional de la Plata .
Barreno, A. M., García Oliver, A., & Gutiérrez Montesinos, A. (2008). EXPOSICIÓN
LABORAL A DISOLVENTES (Primera ed.). Madrid, España: Ambarpack.
Obtenido de
http://www.cancerceroeneltrabajo.ccoo.es/comunes/recursos/99924/pub44957_Exp
osicion_laboral_a_disolventes.pdf
Busines, G. S. (2006). Surface Active Agents. Global Industry Analysts, 10-15.
Cazes, J. (2005). Encyclopedia of chromatography. Florida Atlantic University USA.:
Taylor & Francis Group.
Clesceri, L., Greenberg, A., & Trussell, R. (1992). MÉTODOS NORMALIZADOS Para el
Análisis de Aguas Potables y Residuales. Madrid: Díaz de Santos.S.A.
Colin, B. (2001). Química Ambiental. Barcelona: REVERTÉ, S.A.
Duffau, B. (2010). Validación de métodos y determinación de la incertidumbre de
Medición:"Aspectos generales sobre la validación de Métodos". Instituto de Salud
Pública de Chile, 50-53.
Ellison, S., Barwick, V., & Duguid Farrant, T. (2009). Practical Statistics for the Analytical.
Cambrige: The Royal Society of Chemistry.
Esquivel, E., & Leal, L. (2004). Cromatografía en Fase Reversa. Métodos Físicos químicos
en Biotecnología, 20-25.

98
Eurachem. (1998). The fitness for purpose of analytical methods : a laboratory guide to
method validation and related topics. Teddington: Laboratory of the Government
Chemist.
Feinberg, M. (1995). Basics of interlaboratory studies: the trends in the new ISO 5725
standard edition. Published journal article available from ScienceDirect, Part 2.
Ferreiro Gomez, R. (2012). Larousse Diccionario esencial Química. México: ISBN 84-8332-
658-2 (Larousse Editorial, S.L.).
Figueroa. M, A., & Ramírez. S, H. (2014). INTRODUCCIÓN A LA METODOLOGÍA
EXPERIMENTAL. Universidad de Guadalajara: Pearson Educación de México S.A
de C.V.
Gagnon, M. J. (19 de julio de 1979). NOTE ON A RAPID AND SENSITIVE METHOD
FOR THE DETERMINATION OF ANIONIC DETERGENTS IN NATURAL
WATERS AT THE ppb NIVEL. Water Research, 13, 53-56.
Gallego, A., Garcinuño, R., & Morcillo, M. (2013). Expeimentación en Química Analítica.
Madrid: www.uned.es/publicaciones.
Gary D, C. (2009). Química Analítica (Sexta edición ed.). México, México: McGraw Hill /
INTERAMERICANA EDITORES S.A.
Glasstone, S. (1977). TERMODINÁMICA PARA QUÍMICOS. Madrid: Aguilar S.A.
Ediciones.
Guenther.E. (2013). The Essential Oil. History-Origin in Plants-Production-Analysis. Vol1.
New York: R.E.Kreiger Publishing.
Hach, C. (1988). Procedures Manual. Loveland, Colorado, USA.
Hamilton, R., & Swell, R. (1982 Second Edition). INTRODUCCTION TO HIGH
PERFORMANCE LIQUID CHROMATOGRAPHY . London New York: Chapman
and HallMethuen Inc.
Hart, H. (1995). Química organica. México: McGRAW-HILL.
Hart, H., Hart, D. J., & Craine, L. E. (1995). Química Orgánica. México: McGRAW_HILL
INTERAMERICANA DE MÉXICO,S.A.de C.V.
Harvey, D. (2009). Analytical Chemistry. Illinois, U.S.A: MacGrow-Hill Companies.
Hernandez Sanchez, C., & Rubio Armendaris, L. (2011). TRIHALOMETANOS EN
AGUAS DE CONSUMO HUMANO. REVISTA DE TOXICOLOGIA, 109-113.

99
Hernandez, L., & Gonzales, C. (2002). INTRODUCCIÓN AL ANALISIS
INSTRUMENTAL (Primera ed.). Barcelona, España: Ariel S.A.
Jarnutowski, R., Ferraro, J., & Lankin, D. (1992). Spectroscopy. Chemical Analytical, 22.
Jiménez Díaz, I. (27 de Febrero de 2009). DESARROLLO DE METODOLOGÍAS
ANALÍTICAS PARA LA DETERMINACIÓN DE TENSIOACTIVOS Y SUS
PRODUCTOS DE DEGRADACIÓN MEDIANTE DIFERENTES TÉNICAS
SEPARATIVAS. Granada, España, España: Editorial de la Universidad de Granada.
Kennedy, J. B., & Neville, A. M. (1982). Estadistica para Ciencias e Ingeniería.
México,D.F.: HARLA, S.A.
Kerton, F. M., & Marriott, R. (2013). Alternative Solvents for Green Chemistry . Cambrige:
United kingdom.
Kikuchi, M., Tokai, A., & Yoshida, T. (1986). DETERMINATION OF TRACE LEVELS
OF LINEAR ALKYLBENZENESULFONATES IN THE MARINE
ENVIRONMENT BY HIGH-PERFORMANCE LIQUID CHROMATOGRAPHY.
WATER RESEARCH, 643-650.
Kuehl, R. O. (2001). Diseño de Experimentos. México: International Thomson editores, S.A.
Lamarque, A., & Zyjadlo, J. (2008). Fundamentos teórico-práctico de Química Orgánica.
Córdoba, Argentina: ENCUENTRO GRUPO EDITOR.
Manahan, S. E. (2007). Introducción a la Quimica Ambiental. Barcelona: Reverté,S.A.
Martí, B., Conde, L., Jimeno, A., & Mendez, H. (2008). QUIMICA ANALITICA
CUALITATIVA. Madrid: Thomson Editores Spain.
Martinez Toledo, A., & Cueva Díaz, M. d. (Marzo 2011). PRODUCCION DE BTX EN
MEXICO:USOS, TOXICOLOGIA Y ANALISIS. TLATEMOANI Revista
Académica de Investigación, 1-7.
Miller, J. N., & Miller, J. C. (2002). Estadistica y Quimiometría para Química Analítica.
Madrid: Prentice Hall.
Miller, J., & Miller, J. (2002). ESTADÍSTICA Y QUIMIOMETRÍA PARA QUÍMICA
ANALÍTICA. Madrid: PEARSON EDUCACIÓN.
Mira.B, Blasco.M, Berna.A, & Subirats.S. (01 de Enero de 1999). Supercritical CO2
extraction of essential oil from orange peel. Effect of operation conditions on the
extract composition. The Journal Supercritical Fluids, 14, 95-104.
Mohammad, A. (2012). Green Solvent I. Aligarh, India: Springer Dordrecht Heidelberg New
York London.

100
Morillas Bravo, P. P. (15 de 02 de 2016). (www.eurolab.org.es. Obtenido de
www.eurolab.org.es
Mosquera, M., Hidalgo, J., & Forjan, E. (2009). Evaluación del contenido en trihalometanos
en aguas de consumo de municipios pertenecientes a una misma zona de
Abastecimiento. Higiene y Sanidad Ambiental, 9, 404-411. Recuperado el 05 de
enero de 2015, de nievesmos@yahoo
Pájaro Castro, P., & Olivero Verbel, J. (19 de Diciembre de 2011). QUIMICA VERDE:
NUEVO RETO. Scielo, 21(2). Obtenido de
http://www.scielo.org.co/scielo.php?script=sci_arttext&pid=S0124-
81702011000200009
Pecsok, R. L., & Shields, L. D. (1973). Métodos Modernos de Análisis Químico. México:
Limusa,S,A.
PUBLICACIONES VERTICE.S.L. (2008). GESTION
MEDIOAMBIENTAL:MANIPULACION DE RESIDUOS Y PRODUCTOS
QUIMICOS. Málaga, ESPAÑA: VERTICE .S.L.
Ramis Ramos, G., & García Alvarez, M. C. (2001). Quimiometría. Madrid: Síntesis,S.A.
Rice, E. W., Baird, R. B., Eaton, A. D., & Clesceri, L. S. (2012). Standard Methods. For the
Examination of Water and Wastewater. Baltimore,Maryland: American,Public
Health Association;American Water work,Association;Water Environment
Federation.
Rothman, L., Crouch, S., & Ingle, J. (1975). Theoretical and experimental investigation of
factors affecting precision in molecular absorption spectrophotometry. Analytical
Chemistry, 1226-1233.
Rouessac, F., & Ruessac, A. (2003). Análisis Químico (Quinta ed.). Madrid, España:
McGRAW-HILL/INTERAMERICANA DE ESPAÑA, S.A. Recuperado el 23 de 12
de 2014
Sablayrolles, C., Montrejaud, M., Silvestre, J., & Treilhou, M. (2009). Trace Determination
of Linear Alkylbenzene Sulfonates: Application in Artificially Polluted Soil—
Carrots System. International Journal of Analytical Chemistry, 6.
Saez Ramires, S., & Gómez Cambronero, L. G. (2006). Sistema de mejora continua de la
calidad en el laboratorio: teoría y práctica. Valencia: Universitat de Valencia.
Sanchez, M., & Angel, M. (2006). Prevención de Riesgos Laborales Básico.
Antakira,Málaga: Antakira Grafic.
Sanz Asencio, J. (2013). Equilibrios Químicos. Madrid: Visión Libros.

101
Sevilla, U. d. (Agosto de 2010).
http://investigacion.us.es/docs/web/files/ag10_err_ld_lc_frx.pdf. Recuperado el 05
de 2013, de Av.Reina Mercedes,N° 4-B 41012 Sevilla España
Sharpe, M. (1984). Stray light in UV-VIS spectrophotometers. Analytical Chemistry, 339-
356.
Sierra Alonso, I., & Perez Quintanilla, D. (2010). Análisis Instrumental. Coruña:
Gesbiblo.S.L.
Sierra, I., Perez, D., Gomez, S., & Morante, S. (2010). Analisis Instrumental. Coruña:
Gesbiblio,S.L.
Skoog, D. A., Holler, J. E., & Crouch, S. R. (2008). PRINCIPIOS DE ANÁLISIS
INSTRUMENTAL. México: Cengage Learning Editores S.A.
Skoog, D. A., James Holler, F., & Nieman, T. A. (2001). PRINCIPIOS DE ANALISIS
INSTRUMENTAL. Madrid: McGRAW-HILL/INTERAMERICANA DE
ESPAÑA, S. A. U.
Skoog, D. A., James Holler, F., & Nieman, T. A. (2001). PRINCIPIOS DE ANALISIS
INSTRUMENTAL. Madrid, ESPAÑA: McGRAW-HILL/INTERAMERICANA
DE ESPAÑA, S.A.U.
Skoog, D. A., James Holler, F., & Nieman, T. A. (2001). Principios de Análisis instrumental
(Quinto ed.). Madrid: McGRAW-HILL/INTERAMERICANA DE ESPAÑA,S.A.U.
Skoog, D. A., West, D. M., & Holler, F. J. (2010). Fundamentos de Química Analítica.
Barcelona, España: Reverté,S.A.
Skoog, D., Holler, F., & Crouch, S. (2008). Principios de Analisis Instrumental. En D.
Skoog, F. Holler, & S. Crouch, Espectroscopía Molecular (págs. 335-354). México
D.F: Cenage Learning.
Solano, C., & Jorge. (2004). Prácticas recomendadas para determinar y reportar la
incertidumbre de las mediciones en Química Análitica. San José: Editorial de la
Universidad de Costa Rica.
Taverniers, I., De Loose, M., & Bockstaele, V. (2004). Trends in quality in the analytical
Laboratory II.Analytical method Validation and quality assurance. Trend in
Analytical Chemistry, 23(8), 535-552.
Uri, Z., & Guy, B. (1999). HANDBOOK OF DETERGENTS (Vol. 82). New York: Marcel
Dekker,Inc. Recuperado el Agosto de 2015, de http;//www.dekker.com
Valcarcel Cases, M., & Gómez Hens, A. (1988). TECNICAS ANALITICAS DE
SEPARACION. Barcelona, ESPAÑA: EDITORIAL REVERTÉ S.A.

102
W.M.Haynes. (2013-2014). HANDBOOK OF CHEMISTRY AND PHYSICS. London
New York: Taylor & Francis Group.
Waite, T. (1984). Principles of Water Qualify. Florida, USA: Academic Press Inc.
Weininger, S. J., & Stermitz, F. R. (1988). Química Orgánica. Barcelona: REVERTÉ, S.A.
Wilkinson, J., & More, R. (1990). Cosmetología de Harry. Madrid: Ediciones DIAZ
SANTOS, S.A.
Young, J. A. (2002). SEGURIDAD EN LOS LABORATORIOS QUIMICOS
ACADEMICOS VOLUMEN I. Washington : Una Publicación de la Sociedad
Americana de Química.
Zoller, U., & Paul, S. (2008). HANDBOOK OF DETERGENTS: PRODUCTIONS. Brokins
U.S.A: CRC PRESS Taylor & Fracis Group.
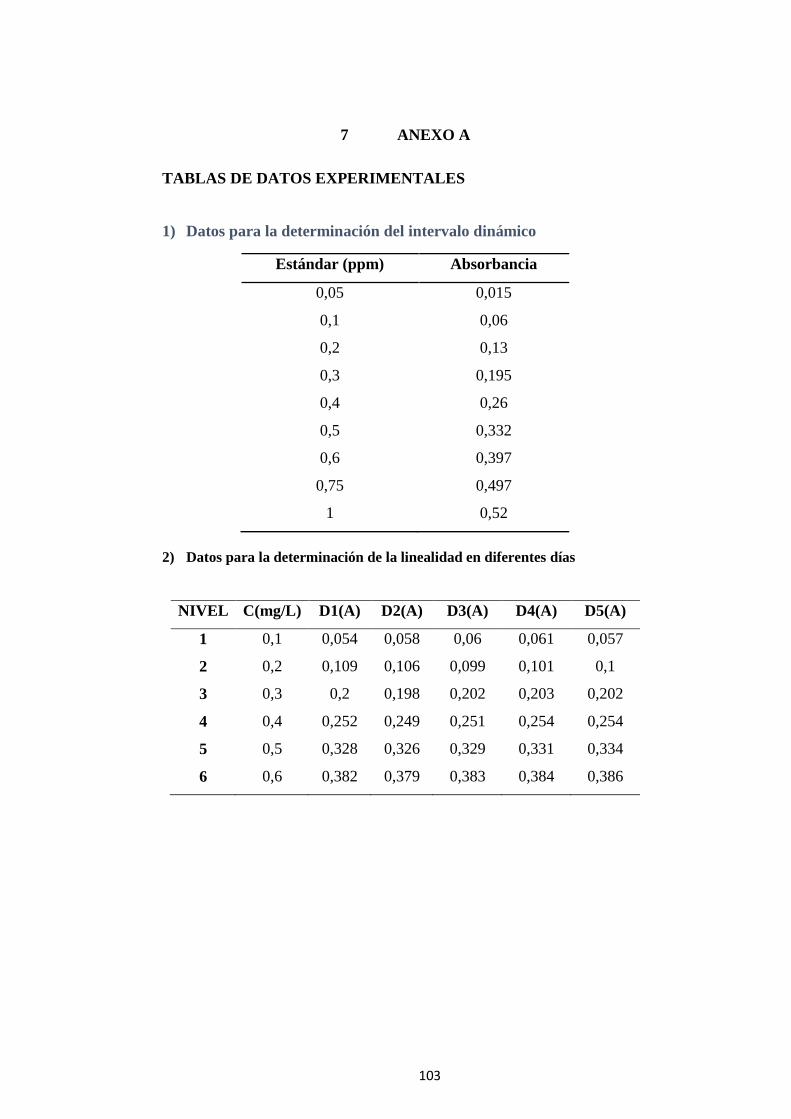
103
7 ANEXO A
TABLAS DE DATOS EXPERIMENTALES
1) Datos para la determinación del intervalo dinámico
Estándar (ppm) Absorbancia
0,05 0,015
0,1 0,06
0,2 0,13
0,3 0,195
0,4 0,26
0,5 0,332
0,6 0,397
0,75 0,497
1 0,52
2) Datos para la determinación de la linealidad en diferentes días
NIVEL C(mg/L) D1(A) D2(A) D3(A) D4(A) D5(A)
1 0,1 0,054 0,058 0,06 0,061 0,057
2 0,2 0,109 0,106 0,099 0,101 0,1
3 0,3 0,2 0,198 0,202 0,203 0,202
4 0,4 0,252 0,249 0,251 0,254 0,254
5 0,5 0,328 0,326 0,329 0,331 0,334
6 0,6 0,382 0,379 0,383 0,384 0,386
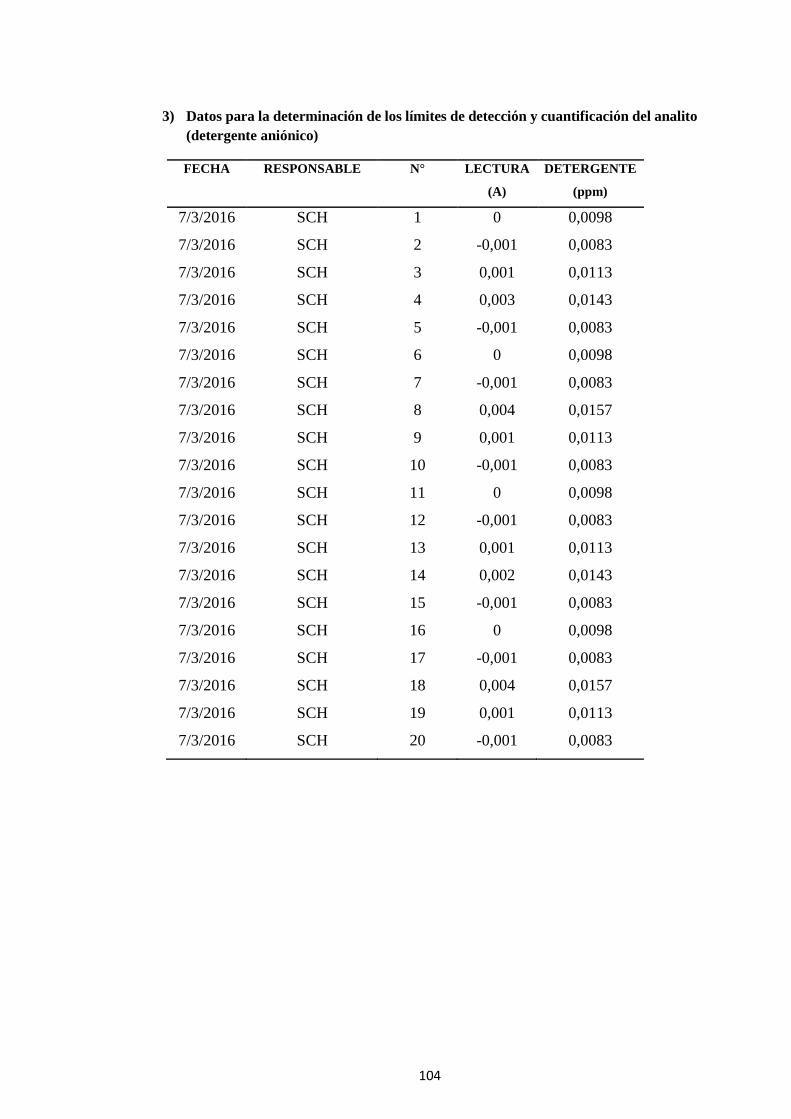
104
3) Datos para la determinación de los límites de detección y cuantificación del analito
(detergente aniónico)
FECHA RESPONSABLE N° LECTURA
(A)
DETERGENTE
(ppm)
7/3/2016 SCH 1 0 0,0098
7/3/2016 SCH 2 -0,001 0,0083
7/3/2016 SCH 3 0,001 0,0113
7/3/2016 SCH 4 0,003 0,0143
7/3/2016 SCH 5 -0,001 0,0083
7/3/2016 SCH 6 0 0,0098
7/3/2016 SCH 7 -0,001 0,0083
7/3/2016 SCH 8 0,004 0,0157
7/3/2016 SCH 9 0,001 0,0113
7/3/2016 SCH 10 -0,001 0,0083
7/3/2016 SCH 11 0 0,0098
7/3/2016 SCH 12 -0,001 0,0083
7/3/2016 SCH 13 0,001 0,0113
7/3/2016 SCH 14 0,002 0,0143
7/3/2016 SCH 15 -0,001 0,0083
7/3/2016 SCH 16 0 0,0098
7/3/2016 SCH 17 -0,001 0,0083
7/3/2016 SCH 18 0,004 0,0157
7/3/2016 SCH 19 0,001 0,0113
7/3/2016 SCH 20 -0,001 0,0083
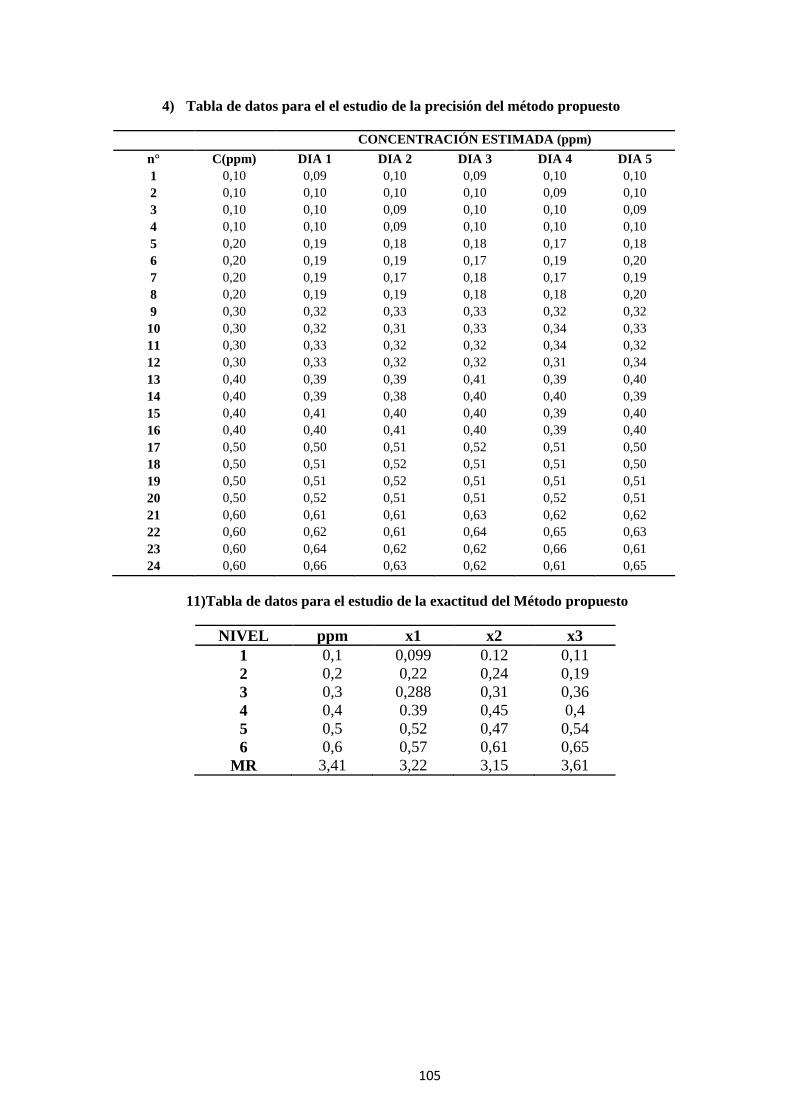
105
4) Tabla de datos para el el estudio de la precisión del método propuesto
CONCENTRACIÓN ESTIMADA (ppm)
n° C(ppm) DIA 1 DIA 2 DIA 3 DIA 4 DIA 5
1 0,10 0,09 0,10 0,09 0,10 0,10
2 0,10 0,10 0,10 0,10 0,09 0,10
3 0,10 0,10 0,09 0,10 0,10 0,09
4 0,10 0,10 0,09 0,10 0,10 0,10
5 0,20 0,19 0,18 0,18 0,17 0,18
6 0,20 0,19 0,19 0,17 0,19 0,20
7 0,20 0,19 0,17 0,18 0,17 0,19
8 0,20 0,19 0,19 0,18 0,18 0,20
9 0,30 0,32 0,33 0,33 0,32 0,32
10 0,30 0,32 0,31 0,33 0,34 0,33
11 0,30 0,33 0,32 0,32 0,34 0,32
12 0,30 0,33 0,32 0,32 0,31 0,34
13 0,40 0,39 0,39 0,41 0,39 0,40
14 0,40 0,39 0,38 0,40 0,40 0,39
15 0,40 0,41 0,40 0,40 0,39 0,40
16 0,40 0,40 0,41 0,40 0,39 0,40
17 0,50 0,50 0,51 0,52 0,51 0,50
18 0,50 0,51 0,52 0,51 0,51 0,50
19 0,50 0,51 0,52 0,51 0,51 0,51
20 0,50 0,52 0,51 0,51 0,52 0,51
21 0,60 0,61 0,61 0,63 0,62 0,62
22 0,60 0,62 0,61 0,64 0,65 0,63
23 0,60 0,64 0,62 0,62 0,66 0,61
24 0,60 0,66 0,63 0,62 0,61 0,65
11)Tabla de datos para el estudio de la exactitud del Método propuesto
NIVEL ppm x1 x2 x3
1 0,1 0,099 0.12 0,11
2 0,2 0,22 0,24 0,19
3 0,3 0,288 0,31 0,36
4 0,4 0.39 0,45 0,4
5 0,5 0,52 0,47 0,54
6 0,6 0,57 0,61 0,65
MR 3,41 3,22 3,15 3,61
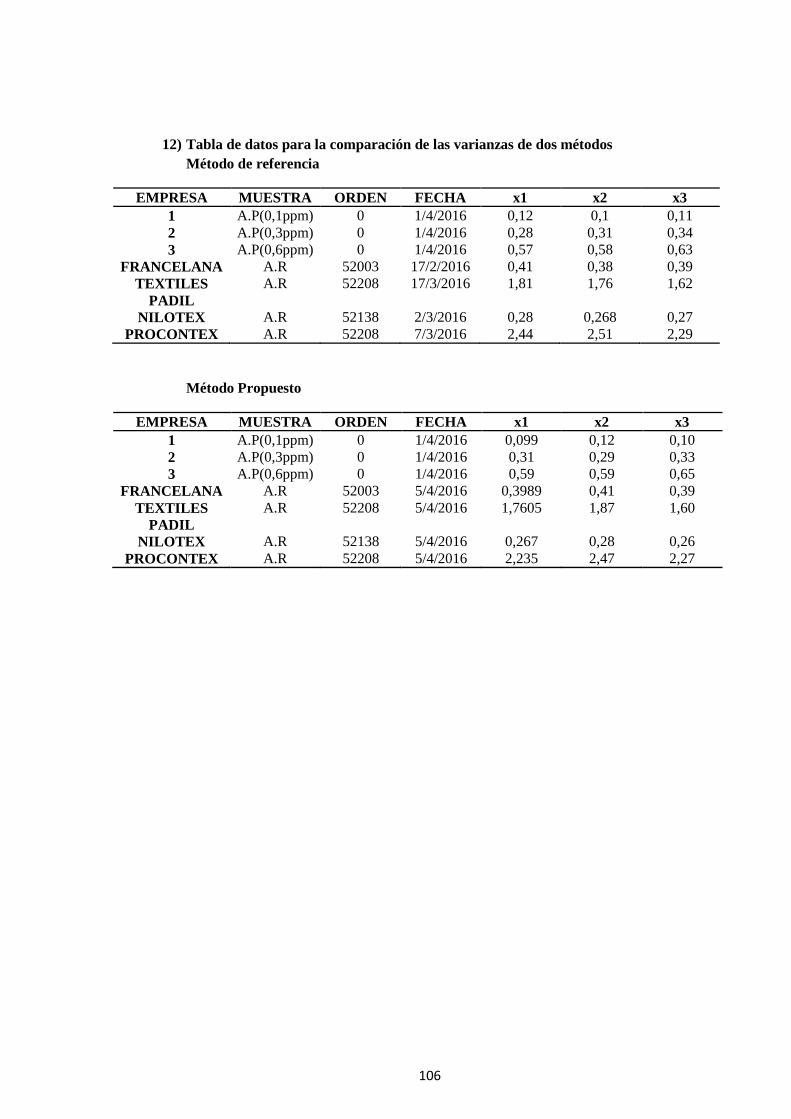
106
12) Tabla de datos para la comparación de las varianzas de dos métodos
Método de referencia
EMPRESA MUESTRA ORDEN FECHA x1 x2 x3
1 A.P(0,1ppm) 0 1/4/2016 0,12 0,1 0,11
2 A.P(0,3ppm) 0 1/4/2016 0,28 0,31 0,34
3 A.P(0,6ppm) 0 1/4/2016 0,57 0,58 0,63
FRANCELANA A.R 52003 17/2/2016 0,41 0,38 0,39
TEXTILES
PADIL
A.R 52208 17/3/2016 1,81 1,76 1,62
NILOTEX A.R 52138 2/3/2016 0,28 0,268 0,27
PROCONTEX A.R 52208 7/3/2016 2,44 2,51 2,29
Método Propuesto
EMPRESA MUESTRA ORDEN FECHA x1 x2 x3
1 A.P(0,1ppm) 0 1/4/2016 0,099 0,12 0,10
2 A.P(0,3ppm) 0 1/4/2016 0,31 0,29 0,33
3 A.P(0,6ppm) 0 1/4/2016 0,59 0,59 0,65
FRANCELANA A.R 52003 5/4/2016 0,3989 0,41 0,39
TEXTILES
PADIL
A.R 52208 5/4/2016 1,7605 1,87 1,60
NILOTEX A.R 52138 5/4/2016 0,267 0,28 0,26
PROCONTEX A.R 52208 5/4/2016 2,235 2,47 2,27

107
ANEXO B
TABLAS ESTADISTICAS
Fuente: (Miller & Miller, 2002)
108

109
Fuente: (Miller & Miller, 2002)

110
ANEXO C
1. CERTIFICADO DE MANTENIMIENTO DE LOS
ESPECTROFOTÓMETROS
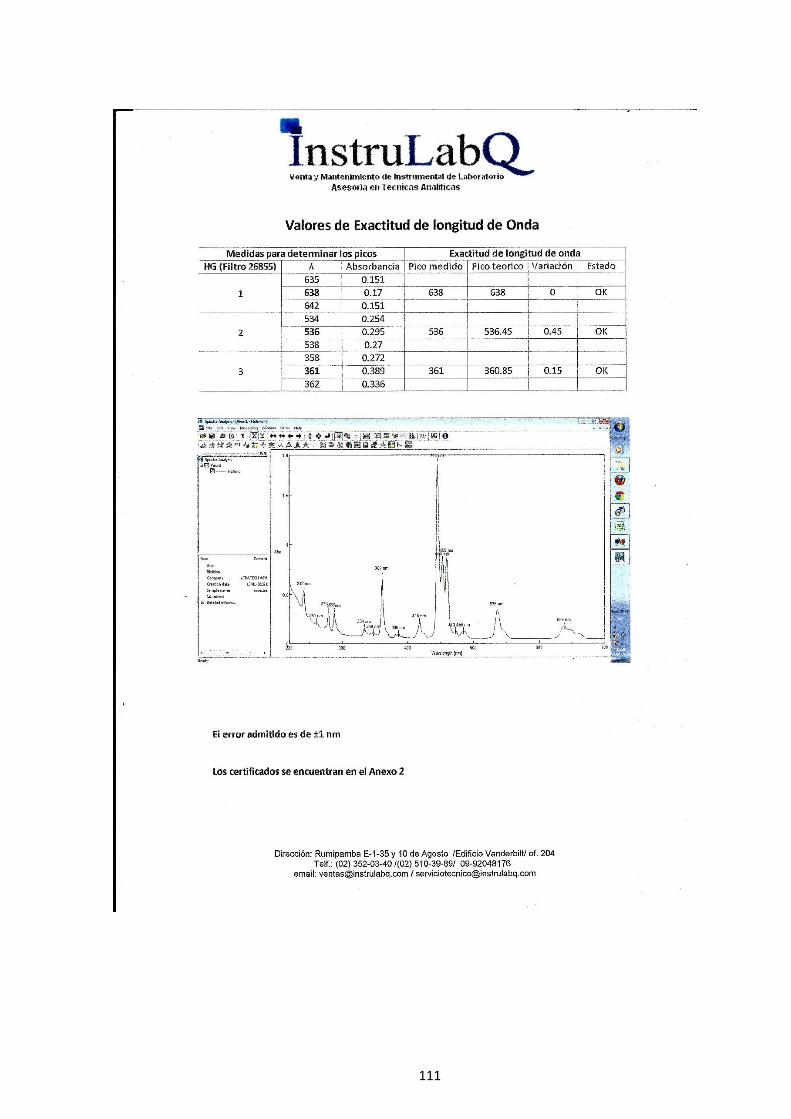
111

112
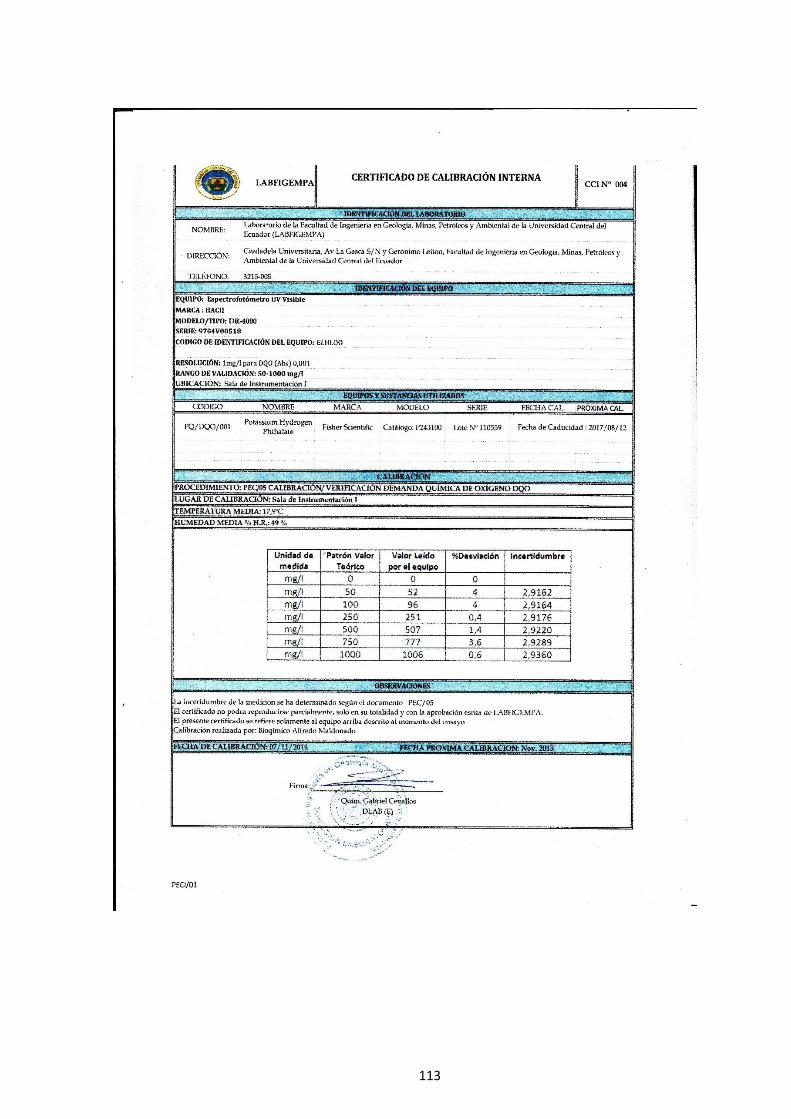
113

114

115
2.- CERTIFICADO DE LOS REACTIVOS

116

117





























