Embed Size (px)
Citation preview

UNIVERSIDAD DE CHILE FACULTAD DE CIENCIAS FÍSICAS Y MATEMÁTICAS DEPARTAMENTO DE INGENIERIA QUÍMICA Y BIOTEGNOLOGÍA
ESTUDIO DE REACTIVIDAD DE COMPLEJOS DE COORDINACIÓN EN REACCIONES DE POLIMERIZACIÓN DE OLEFINAS
TESIS PARA OPTAR AL GRADO DE DOCTOR EN QUÍMICA
CAROLINA ESTHER VALDEBENITO CALABRÁN
PROFESOR GUIA: RAÚL QUIJADA ABARCA RENE ROJAS GUERRERO
MIEMBROS DE LA COMISION: CARLOS DIAZ VALENZUELA
FRANCISCO JAVIER GIL LLAMBIAS MAURICIO ISAAC CASANOVA
EDUARDO SOTO BUSTAMANTE
SANTIAGO DE CHILE ENERO 2010

A mis Padres y a todos los que hicieron posible este sueño…

iii
AGRADECIMIENTOS
En primer lugar quiero agradecer a mis profesores guías, Dr. Raúl Quijada y Dr.
René Rojas por su dirección y por el apoyo recibido a lo largo de la realización de esta
Tesis. Agradezco a los profesores de mi comisión: Dr. Carlos Díaz, Dr. Francisco Javier
Gil, Dr. Mauricio Isaac y Dr. Eduardo Soto, por sus correcciones y recomendaciones
durante el desarrollo de esta Tesis.
Quiero dar gracias a la profesora María Teresa Garland y Andrés Ibañez de la
Universidad de Chile por los análisis de rayos-X. Al profesor Daniel Serafini de la
Universidad de Santiago de Chile, por las facilidades en los análisis DSC. Agradezco al
profesor Dr. Gerhard Erker de la Univerisidad Westfälische Wilhelms-Universität por
haberme recibido en su laboratorio durante la realización de la estadía de investigación en
Alemania.
También quiero agradecer a mis compañeros del Laboratorio de Ingeniería de
Polímeros: Vivianne D., Brian P., Christian C. y Humberto P por su colaboración y
consejos entregados. Agradezco especialmente a Juan B. por su ayuda y por estar ahí en
los buenos y malos momentos. A Paulina Q. por los análisis de GPC y DSC y a Lorena G.
por su colaboración en la caracterización de los polímeros. Y muy en especial a mis
compañeras de estudios Paula Z., María Eulalia V. y Carla F. por las alegrías y penas
compartidas en este largo camino.
Quiero extender un sincero agradecimiento a los profesores: Mauricio Valderrama,
Verónica Arancibia y Raúl Contreras de la Pontificia Universidad Católica de Chile, por
acogerme en el laboratorio como una más de sus alumnos. A todos los que forman parte de
este laboratorio y muy en especial a mis compañeros: Abigail S., Javiera U., Rodrigo C.,
Alan C., Manuel E. y John H. por su constante colaboración en el desarrollo de esta tesis.
Finalmente agradezco a mis padres y a mi hermano por darme la fuerza y
tranquilidad necesaria para terminar esta tesis. A mis amigos por su incondicional entrega
y a todos los que forman parte de mi gran familia, gracias por estar ahí en cada momento.
La realización de esta Tesis Doctoral ha sido posible gracias a la Comisión Nacional
de Investigación Científica y Tecnológica (CONICYT) por la beca de estudios de Post
grado y de termino de tesis doctoral. Además de los proyectos AT- 24071084,
FONDECYT 11060384 y FONDAP 11980002.

iv
TABLA DE CONTENIDOS
INDICE DE FIGURAS……………………………………………………….. INDICE DE TABLAS ………………………………………………………… LISTA DE ABREVIATURAS ……………………………………………….. RESUMEN………………………………………………………...................... SUMMARY …………………………………………………….........................
vii xi xii xiii xv
CAPITULO I INTRODUCCION
1.1 Antecedentes Generales……………………………………………… 1 1.2 Catalizadores Ziegler-Natta………………………………………….. 4 1.3 Catalizadores Metalocénicos…………………………………………. 10 1.4 Catalizadores Post-Metalocénicos…………………………………… 15 1.4.1 Complejos con Metales de Transición Tempranos (Zr, Ti)………….. 20 1.4.2 Complejos con Metales de Transición Avanzados (Fe, Ni, Pd)……... 27 1.5 Hipótesis…………………………………………………………… 40 1.6 Objetivos………………………………………………………….... 40 1.6.1 Objetivo General…………………………………………………....... 40 1.6.2 Objetivos Específicos………………………………………………… 40 CAPITULO 2 PARTE EXPERIMENTAL
2.1 Método General de Síntesis………………………………………...... 42 2.2 Síntesis de Ligandos y Complejos…………………………………… 44 2.3 Caracterización de Ligandos y Complejos…………………………… 46 2.4 Polimerización de Etileno……………………………………………. 47 2.5 Caracterización de los Polímeros…………………………………….. 49 CAPITULO 3 RESULTADOS Y DISCUSIÓN
3.1 Síntesis y Caracterización de Ligandos……………………………… 51 3.1.1 Ligandos β-cetoiminas y β-diiminas…………………………………. 51 3.1.2 Ligandos β-cetoiminas y β-diiminas Sustituidos…………………….. 58 3.1.3 Ligandos N-(imidoilamidinas) y Acetamidinicos………………........ 62 3.1.3.1 Ligandos N(imidoilamidinico) con Sustituyentes Alquilados……….. 66 3.1.3.2 Ligandos N(imidoilamidinico) con Anillos Sustituidos con Flúor y
Nitrógenos. ……………………………………………………….......
72 3.2 Síntesis de Complejos de Titanio y Circonio………………………… 83 3.2.1 Síntesis y Caracterización de Complejos de Ti(IV) y Zr(IV) con L1 y
L5……………………………………………………………………
83

v
3.2.2 Síntesis y Caracterización de Complejos de Zr(IV) con L6………….. 86 3.3 Síntesis y Caracterización de Complejos de Níquel………………..... 92 3.3.1 Complejos con ligandos N-(imidoilamidinicos) con Sustituyentes
Alquilicos…………………………………………………………...
92 3.3.2 Complejos con ligandos N-(imidoilamidinicos) Sustituidos con Flúor
y Nitrógenos…………………………………………………………..
97 3.4 Reactividad de Complejos en Polimerizaciones de Etileno………….. 104 3.4.1 Complejos de Circonio con Ligandos L1 y L5……………………...... 104 3.4.2 Complejos de Titanio con Ligandos L1 y L5…………………………. 106 3.4.3 Complejos de Circonio con Ligandos L6…………………………….. 107 3.4.4 Complejos de Níquel…………………………………………………. 109 3.4.4.1 Complejos de Níquel con ligandos N-(imidoilamidinicos) Sustituidos
con Flúor……………………………………………………………...
113 3.4.4.2 Complejos de Níquel con ligandos N-(imidoilamidinicos) Sustituidos
con Nitrógenos………………………………………………………..
117 CAPÍTULO 4 CONCLUSIONES
126
CAPÍTULO 5 REFERENCIAS
128
ANEXO 1 Síntesis de Ligandos………………………………………………….. 139 Preparación de ligandos Iminicos……………………………………. 138 Ligandos Acetamidinicos……………………………………………. 143 Preparación de Sales de Ligandos Iminicos……………………......... 152 Preparación de Complejos…………………………………………… 153 Síntesis de Complejos de Circonio y Titanio………………………… 152 Síntesis de Complejos de Níquel…………………………………...... 156 INDICE DE FIGURAS Figura 1. Estructura de polietilenos. a) Polietileno de Baja Densidad
(PEBD) b) Polietileno Lineal de Alta Densidad(PEAD), c) Polietileno Lineal de Baja Densidad (PELBD)……………..
2
Figura 2. Microestructura de Polipropileno…………………………… 3 Figura 3. Estructura Cristalina TiCl3…………………………………… 5
Figura 4. Sistema Catalítico Ziegler-Natta Típico. A) Formación Especie Activa, b) Mecanismo de Propagación de la Cadena
Figura 5. Mecanismo de Terminación de Cadena, Eliminación β-hidrógeno……………………………………………………
5 6
Figura 6. Mecanismos de Terminación de Cadena…………………… 7 Figura 7. Diferentes Estructuras Posibles de MAO: 1 lineal; 2 y 3
cíclicos; 4 cluster y 5 red tridimensional………………….. 11

vi
Figura 8. Mecanismo de Complejación, Metilación y Activación con MAO…………………………………………...
12
Figura 9. Mecanismo de Desactivación y Reactivación de Complejos Metalocénicos………………………………………………..
13
Figura 10. Activación General de Complejos Metalocénicos y Propagación de la Cadena Polimérica………………………..
14
Figura 11. Tipos de Ligandos R = 2,6(i-Pr)2C6H3; 2,6Me2C6H3; 2-t-BuC6H4……………………………………………………….
16
Figura 12. Ruta de Síntesis de Ligandos β-diiminas…………………… 18 Figura 13. Ligandos Aniónicos a) β-diiminicos, b) β-dicetoiminas, c)
Base de Schiff……………………………………………….. 19
Figura 14. Modos de Coordinación de Ligandos Bidentados a Metales de Transición…………………………………………………
19
Figura 15. Complejos Post-metalocénicos con Metales de Transición Avanzados……………………………………………………
20
Figura 16. Sistema Catalítico Post-Metalocénicos……………………… 21 Figura 17. Catalizador Post-metalocénicos FI…………………………. 22 Figura 18. Configuraciones Posibles de Sistemas Catalíticos Post-
metalocénico…………………………………………………. 23
Figura 19. Variaciones en Ligandos N,O de Complejos Post-metalocénicos………………………………………………..
24
Figura 20. Catalizador con Sustituyentes Halógenos, G. Coates……….. 25 Figura 21. Sistemas Catalíticos de W. Keim para la Oligomerización de
Etileno……………………………………………………….. 28
Figura 22. Ligandos N,N utilizados en Complejos de Níquel(II) y Paladio(II)…………………………………………………… 29
Figura 23. a) Formación Sitio Activo; b) Propagación de Cadena; c) Terminación de Cadena………………………………………
32
Figura 24. Sitios de Coordinación Axial (Ax) y Ecuatorial (Ec) de Centros Metálicos y su Potencial Interacción Estérica con Sustituyentes Voluminosos (R)………………………………
32
Figura 25. Mecanismo Propuesto para Chain Walking y la Obtención de Polímeros Lineales y Ramificados…………………………...
34
Figura 26. Ligandos a) N,O; b) N,N; c) P,P; d) N,N,N………………… 34 Figura 27. Polimerización de Propileno con Catalizador de Ni(II) de
Simetría C2 y Centros quirales…………………………….. 36
Figura 28. Complejos Post-metalocénicos con Heteroátomos en su Estructura……………………………………………………..
37
Figura 29. Complejos Post-metalocénicos de Níquel Neutros Capaces de Polimerizar Polietileno……………………………………….
38
Figura 30. Activación de Sistemas Post-metalocénicos………………… 39 Figura 31. Cámara Seca MBRAUN…………………………………….. 42 Figura 32. Diagrama y Línea de Polimerización de Etileno…………….. 48 Figura 33. Síntesis de β-cetoiminas……………………………………. 51

vii
Figura 34. Síntesis de 4-(1-fenil-etilamino)-pent-3-en-2ona (L 1). 52 Figura 35. Espectro 1H-RMN de Ligando L 1…………………………… 53 Figura 36. Espectro 13C-RMN de Ligando L 1…………………………… 54 Figura 37 Estructuras de Ligandos L 2 y L 3…………………………… 54 Figura 38. Síntesis de Ligando L 4………………………………………. 56 Figura 39. Espectro 1H-RMN de Ligando L 4........................................... 57 Figura 40. Incorporación de Funcionalidad Ciano……………………… 58
Figura 41. Ligandos β-cetoiminas y β-dicetoiminas con Funcionalidad Adicional……………………………………………………..
59
Figura 42. Comparación de Espectro 1H-RMN de L 1 y L 5……………... 60 Figura 43. Espectro FT-IR de Ligando L 5………………………………. 61 Figura 44. Síntesis de Ligandos N-(imidoilamidinas)…………………... 62
Figura 45. Síntesis de N-(2,6-diisopropilfenil)acetamida (Isómeros P1cis y P1trans)………………………………………………………
63
Figura 46. Síntesis de Cloruro de N-(2,6-diisopropilfenil)acetimidoilo (P2)……………………………………………………………
63
Figura 47. Espectro 1H-RMN del Compuesto P2……………………….. 64 Figura 48. Síntesis de los Isómeros de L 8………………………………. 66
Figura 49. Espectro 1H-RMN de Isómeros L 8, M = Isómero Mayoritario…………………………………………………...
67
Figura 50. Formación de Isómeros L 8a y L 8b…………………………... 68 Figura 51. Estructura ORTEP de L 8a y L 8b……………………………... 68 Figura 52. Síntesis de Ligando L 9………………………………………. 70 Figura 53. Estructura ORTEP de L 9…………………….……………….. 70 Figura 54. Síntesis de Ligando Fluorado L 10……………………………. 72 Figura 55. Estructura ORTEP de L 10……………………………………. 73 Figura 56. Síntesis de Ligandos L 11 y L 12………………………………. 75 Figura 57. Estructura ORTEP de L 11 …………………………………… 75 Figura 58. Comparación Espectros 1H-NMR Ligandos L 10 y L 12……….. 77 Figura 59. Síntesis de Ligandos L 13……………………………………... 78 Figura 60. Espectro 1H-RMN de Ligando L 13………………………….. 79 Figura 61. Síntesis de Ligando L 14……………………………………… 80 Figura 62. Espectro 1H-RMN del Ligando L 14…………………………. 81 Figura 63. Espectro 13C-RMN del Ligando L 14………………………… 82 Figura 64. Formación de Sal de Litio de Ligandos L 1 y L 5…………….. 83 Figura 65. Síntesis de Complejos de Circonio y Titanio………………… 84 Figura 66. Síntesis de Complejo de Circonio con L 6……………………. 87
Figura 67. Síntesis de Complejo de Circonio con Precursor (NMe2)2ZrCl2…………………………………………………
88
Figura 68. Espectro 1H-RMN para Complejo 6…………………………. 89 Figura 69. Síntesis de Complejo 7……………………………………….. 90 Figura 70. Estructura ORTEP de Complejo 7 ………………………….. 91 Figura 71. Obtención de Mezcla Isomérica de Complejos de Níquel 8… 93

viii
Figura 72. Estructura ORTEP de 8a y 8b ……………………………….. 94 Figura 73. Síntesis de Complejo de Níquel más Simétrico 9……………. 96 Figura 74. Estructura ORTEP de complejo 9……..……………………... 97 Figura 75. Síntesis de Complejos Níquel 10 y 11……………………….. 98 Figura 76. Estructura ORTEP de Complejo 12………………………….. 99 Figura 77. Síntesis de Complejo 13……………………………………… 100 Figura 78. Complejo de Níquel Esperado……………………………….. 101 Figura 79. Síntesis de Complejo de Níquel con Ligandos Monodentado.. 102 Figura 80. Estructura ORTEP de 14 …………………………………….. 102 Figura 81. Complejos 1 y 2 Utilizados en la Polimerización de Etileno… 104 Figura 82. Sistemas 5 y 6 Utilizados para la Polimerización de Etileno… 107
Figura 83. Catalizadores de Níquel Utilizados para Polimerización de Etileno………………………………………………………...
110
Figura 84. Sistema Activo para Catalizador de Níquel………………….. 111
Figura 85. Análisis de GPC para Polietilenos: (a) Catalizador 8a + 8b y (b) Catalizador 9, Tabla 3.4.4.1, Relativo al Poliestireno Estándar 135 °C………………………………………………
113
Figura 86. Catalizadores de Níquel con Ligandos Fluorados……………. 113
Figura 87. Influencia de Sustituyentes Fluorados en Complejos de Níquel…………………………………………………………
115
Figura 88. Análisis de GPC para Polietilenos: (a) Catalizador 10 y (b) Deconvolución para catalizador 10 a 60 ºC…………………..
116
Figura 89. Análisis de GPC para Polietilenos Obtenido con Catalizador 12……………………………………………………………..
117
Figura 90. Complejos de Níquel con Ligandos Piridínicos……………… 117 Figura 91. Sistema de Activación para Sistemas con Anillo Piridínico…. 118
Figura 92. Análisis de GPC para Polietilenos Obtenido con Catalizador 13……………………………………………………………...
119
Figura 93. Espectro de Masa para Oligómero Obtenido con Complejo 14……………………………………………………………...
121
Figura 94. Patrón de Fragmentación Correspondiente a 1-octadeceno….. 122 Figura 95. Catalizadores de Circonio de Referencia…………………….. 123 Figura 96. Catalizadores de Níquel de Referencia………………………. 124

ix
INDICE DE TABLAS Tabla 1. Resumen de Ligandos Sintetizados…………………………. 44 Tabla 2. Resumen de Complejos Sintetizados……………………… 45
Tabla 3. Resumen de Ligandos N-(imidoilamidinas) y Acetamidinicos Síntetizados………………………………
65
Tabla 4. Distancias de enlace (Å) y ángulos (º) de Ligandos (N)-imidoilamidina………………………………………………
69
Tabla 5. Distancias de enlaces (Å) y ángulos (º) de L 9……………………………………………………………..
71
Tabla 6. Distancias de enlace (Å) y ángulos (º) para Ligandos L 10 y L 11…………………………………………………………..
73
Tabla 7. Distancias de enlace (Å) y ángulos (º) para Complejo 7………………………………………………………………
91
Tabla 8. Distancias de enlace (Å) y ángulos (º) para los Complejos de 8a, 8b y 9……………………………………………………
95
Tabla 9. Distancias de enlace (Å) y ángulos (º) para los Complejo 12.. 100 Tabla 10. Distancias de enlace (Å) y ángulos (º) para los Complejo 14.. 103
Tabla 11. Resultados de Polimerización con Complejos de Circonio 1 y 2……………………………………………………………
105
Tabla 12. Resultados de Polimerización con Complejos de Titanio 3 y 4……………………………………………………………..
106
Tabla 13. Resultados de Polimerización de Etileno con Complejos de Circonio 5 y 6……………………………………………….
108
Tabla 14. Resultados de Polimerización con Complejos de Níquel N-imidoilamidina……………………………………………….
110
Tabla 15. Resultados de Polimerización de Etileno con Complejos de 10 y 12……………………………………………………….
114
Tabla 16. Resultados de Polimerización de Etileno con Complejos de 13 y 14……………………………………………………….
118
Tabla 17. Rango de eficiencia de un catalizador en base a su actividad.. 122 Tabla 18. Resumen de Actividad Catalítica de Complejos de Circonio.. 123
Tabla 19. Resumen de Actividad Catalítica de Complejos de Níquel. 124

x
LISTA DE ABREVIATURAS PE Polietileno PP Polipropileno PDI Indice de Polidispersidad Mw Peso molecular en peso Mn Peso Molecular en número MAO Metilaluminoxano DME 2-metoxietil éter Me Metilo Bn Bencilo COD Bis(1,5-ciclooctadieno) i-Pr Isopropilo t-But ter-butilo Ph Fenilo CN Ciano Et3N Trietilamina THF Tetrahidrofurano DCM Diclorometano CDCl3 Cloroformo deuterado TMS Tetrametilsilano Hz Hertz. ppm Partes por millón. J Constante de acoplamiento s Singulete d Doblete t Triplete p Pentuplete m Multiplete IR Infrarrojo DSC Calorimetría diferencial de barrido GPC Cromatografía de permeación en gel TCB 2,2,4-triclorobenceno BHT 2,6-di-t-butil-4-metilfenol RMN Resonancia magnética nuclear [η] Viscosidad intrínseca RT Temperatura ambiente Tf Temperatura de fusión

xi
RESUMEN
El desarrollo de nuevos y mejores sistemas catalíticos para la polimerización de
olefinas depende entre, otras factores de la naturaleza, número y características de los
ligandos que se coordinan al centro metálico. Entre los metales de transición más utilizados
para la síntesis de estos sistemas se encuentran Níquel, Circonio y Titanio. La utilización
de cada uno de ellos depende de los requerimientos específicos en las propiedades de los
polímeros que se desea obtener.
Esta investigación contempla el desarrollo de nuevos ligandos para la obtención de
complejos con metales de transición que sean capaces de llevar a cabo reacciones de
polimerización de etileno. Los ligandos sintetizados son del tipo bidentados N,N - N,O,
específicamente β-cetoiminas, β-diiminas, N-(imidoilamidinicos) y acetamidinicos. Estos
han sido obtenidos con buenos rendimientos y caracterizados completamente. Las rutas de
síntesis utilizadas para su obtención esto resultan ser una muy buena alternativa para la
preparación de derivados que contengan es su estructura grupos funcionales o
heteroátomos.
La introducción de sustituyentes es de gran interés para estudiar los factores
estéricos y electrónicos de los ligandos sobre centros metálicos y su influencia en la
reactividad en reacciones de polimerización de olefinas. En el caso de ligandos β-
cetoiminas y β-diiminas el grupo funcional incorporado corresponde al ciano (CN). Para los
ligandos N-imidoilamidinicos sintetizados, la diferencia radica en los grupos sustituyentes
de los anillos fenílicos que conforman al ligando. Es así que se tienen sustituyentes como
metilos, isopropilos, átomos de flúor, o simplemente presentan un anillo piridínico en su
estructura. Con la misma ruta de síntesis utilizada para la obtención de los ligandos N-
imidoilamidinicos es posible obtener los acetamidinico, los cuales también presentan
sustituyentes adicionales en su estructura. Se realiza una discusión completa acerca de su
síntesis y caracterización de este tipo de ligandos ha sido realizada a lo largo de este trabajo
de tesis.

xii
Con los ligandos β-cetoiminas y β-diiminas se han sintetizado complejos tanto de
titanio como de circonio, los cuales resultaron ser activos para la polimerización de etileno
en presencia de metilaluminoxano (MAO) con actividades catalíticas moderadas. Las
actividades catalíticas resultaron ser mayores cuando el centro metálico corresponde a
circonio (290 Kg/mol Zr*h*bar) en comparación a titanio (90 Kg/mol Ti*h*bar). Por otra
parte, la incorporación de la funcionalidad CN aumenta la actividad catalítica en un factor
de 3 para los complejos de circonio, en idénticas condiciones de reacción. Los polímeros
obtenidos con ambos metales poseen alto peso molecular (> 300 (Kg/mol)) y corresponden
a polietilenos lineales, con temperaturas de fusión sobre 134 ºC.
Tanto los ligandos N-(imidoilamidinicos) y acetamidinicos fueron utilizados para la
formación de complejos de níquel por la reacción directa con el precursor níquel (II)
dibromo 2-metoxietil éter. Todos ellos han sido bien caracterizados y han resultado ser
activos para la polimerización de etileno en presencia de MAO. Las actividades catalíticas,
de los sistemas que contienen solo grupos alquilos, son moderadas (547 Kg/mol Ni*h) y
los polietilenos obtenidos poseen alto peso molecular (~ 160 (Kg/mol)) con una
distribución de peso molecular monomodal y puntos de fusión de 134 ºC. Sin embargo,
dependiendo de las condiciones de reacción estas características pueden variar, lográndose
distribuciones bimodales con menores pesos moleculares (~ 87 (Kg/mol)).
Para los sistemas que presentan heteroátomos en su estructura, las actividades
catalíticas encontradas también son moderadas y dependen de las condiciones de reacción,
como temperatura y presión. Por otra parte, la distribución de pesos moleculares también se
ve afectada por las diferentes condiciones de reacción, sin embargo en todos los casos se
obtienen polietilenos de alto peso molecular con temperaturas de fusión características de
polímeros lineales (~ 134 ºC).

xiii
ABSTRACT
The development of new and better catalytic systems for olefin polymerization
depends, among other factors, on the nature, number and characteristics of the ligands that
are coordinated to the metal center. The transition metals typically used for the synthesis of
these systems are nickel, zirconium and titanium. The use of each depends on the specific
characteristics and properties desired in the polymers that will be produced.
This research is focused on the development of new ligands for the synthesis of
transition metal complexes capable of being catalytically active toward ethylene
polymerization reactions. The ligands synthesized are of the N,N- N,O- bidentated type,
specifically β-ketoimines, β-diimines, N-(imidoylamidines) and acetamidines. These
compounds were obtained in good yields and each fully characterized. The synthetic routes
used for their preparation proved to be a very good alternative for the synthesis of derivate
compounds that contain functional groups or heteroatoms in their structure.
The introduction of substituent groups is of great interest for the study of the
influence of steric and electronic factors in the metallic centers and in the reactivity towards
olefin polymerization reactions. In the case of β-ketoimine and β-diimine ligands, the cyano
group (CN) is added. For the synthesized N-imidoylamidine ligands, the difference lies in
the substitution present in the ligand’s aromatic rings. Therefore, methyl and isopropyl
groups as well as fluoride atoms were used, among others, as substitutent groups, or
simply, a pyridine ring is used. With the same synthetic pathway used for the preparation of
the N-imidoylamidine ligands, it was possible to obtain the acetamidine compounds, which
contain additional substituents as well. A complete discussion on the synthesis and
characterization of these type of ligands was performed along this thesis work.
Using the β-ketoimine and β-diimine ligands, zirconium and titanium organometalic
complexes were synthesized. Those compounds showed moderate activity towards ethylene
polymerization in the presence of methylaluminoxane (MAO). The obtained activities were
higher when zirconium was used as metallic centr (290 Kg/mol Zr*h*bar) instead of
titanium (90 Kg/mol Ti*h*bar). When the CN functionality was introduced, the catalytic
activity increased by a factor of 3 in the case of zirconium compounds, under the same
reaction conditions. The obtained polymers with both zirconium and titanium complexes

xiv
were linear polyethylenes with high molecular weight (> 300 Kg/mol) and fusion
temperatures over 134 ºC.
The N-imidoylamidine, and the acetamidine ligands were used to synthesize the
corresponding nickel compounds by reacting them directly with dibromo(2,2’-
dimethoxyethylether)nickel(II). These compounds were fully characterized and proved to
be catalytically active towards ethylene polymerization in the presence of MAO. The
catalytic activities were moderate (547 kg/mol Ni*h) for the systems containing only alkyl
groups, and the polymers obtained had high molecular weights (~ 160 kg/mol), monomodal
molecular weight distributions and melting temperatures of 134°C. However, it was
possible to obtain bimodal distributions and lower molecular weights (~ 87 Kg/mol),
depending on the reaction conditions used.
In the case of the systems that contained heteroatoms in their structure, the catalytic
activities were found to be moderate and depend on the reaction conditions, such
temperature and pressure. Additionally, the molecular weight distribution was also affected
by the reaction conditions, however, in all the studied cases, high molecular weight
polyethylenes were obtained, with melting temperatures similar to those of linear polymers
(~ 134 ºC).

1
CAPÍTULO 1
INTRODUCCIÓN
1.1 Antecedentes Generales
Las poliolefinas constituyen la familia de polímeros con uno de los mayores
desarrollos en el mundo, que incluye materiales de gran producción llamados
commodities, como polietileno (PE), polipropileno (PP), y otros elastómeros como:
etileno–propileno (EPR), poli-1-buteno o etileno-polipropileno-dieno (EPDM). Esto
ha originado grandes avances científicos y tecnológicos en el diseño de nuevos
catalizadores y procesos, provocando que la velocidad de crecimiento de estos sea
exponencial en comparación a otros materiales de este volumen de producción
llegando a ser cerca de un 60% de todos los plásticos comercializados[1].
Dos son las características principales por las cuales las poliolefinas se
distinguen de otros polímeros, siendo una de ellas la relación costo-rendimiento,
donde éstas tienen un costo de producción y procesamiento menor que otros
plásticos. La segunda, está relacionada a las propiedades que estas poseen como, alta
rigidez y buena resistencia tanto química como física. Esto hace que sean materiales
útiles en la elaboración de diferentes productos como piezas de automóviles, fibras o
films para empaques.
Como se comentó estos avances en la síntesis de poliolefinas están
estrechamente relacionados con el progreso de los sistemas catalíticos que las
producen. Se sabe que las primeras poliolefinas sintéticas fueron obtenidas a fines de
los años 1890 por Hinderman, y colaboradores vía descomposición de diazometano.
Luego en los años 1930 Imperial Chemical Industries (ICI), E. W. Fawcett y R.O.
Gibson descubrieron que era posible obtener polietileno altamente ramificado,
conocido como Polietileno de Baja Densidad (PEBD, Figura 1, a). Este se obtiene
vía polimerización de radicalar libre, en condiciones de alta presión de etileno (1000
a 3000 bar) y temperatura (150 a 350 °C). Michael Perrin de ICI continúa estudiando
esta reacción con el propósito de optimizar la formación de este tipo de material.
Removiendo benzaldehído de la mezcla de reacción y agregando deliberadamente
peróxidos, Perrin logra las condiciones de reacción que le permiten sintetizar

2
polietileno de manera reproducible y en buen rendimiento. Este proceso fue
patentado en 1937 y PEBD comienza a ser producido comercialmente 1939[2].
n( )n( ) CH3 CH3
CH3
CH3 CH3
CH3
CH3
CH3
CH3
CH3
CH3
n( )m
R
CH3 CH3
Figura 1. Estructura de polietilenos. a) Polietileno de Baja Densidad (PEBD) b)
Polietileno Lineal de Alta Densidad(PEAD), c) Polietileno Lineal de Baja Densidad
(PELBD).
El descubrimiento y comercialización de polietileno de alta densidad
(PEAD), ocurre a comienzos de los años 1940, por C. S. Marvel en Dupont y F.
Mayo en U. S. Rubber Company (Figura 1, b). En 1943, M. Fischer en BASF,
sintetiza PEAD usando polvo de aluminio y tetrafluoruro titanio. Una patente por
este descubrimiento fue hecha en 1943, y comienza un periodo de interés comercial
en la producción de PEAD. En 1945, P. Hogan, R. Banks, G. Bailey, y E. Reid de
Phillips Petroleum Company producen PEBD a través de un sistema catalítico
compuesto de óxido de níquel, soportado en alúmina-sílice. Más tarde, Hogan y
Banks adicionan sal de cromo a este sistema con el propósito de producir PEAD,
logrando patentar este proceso en 1953[3].
En el año 1953 Karl Ziegler y colaboradores[4] descubrieron accidentalmente
que trazas de níquel catalizaban reacciones de oligomerización de etileno en
presencia de compuestos de alquil-aluminio. A partir de estos descubrimientos
comienza una nueva era de generaciones de catalizadores, donde Ziegler estudio la
acción de variadas mezclas entre metales de transición y alquil-aluminios,
concluyendo que los metales del grupo 4 (Ti, Zr, Hf) al 6 (Cr, Mo, W) polimerizaban

3
etileno a presiones y temperaturas moderadas (10-20 atm; 50-75 ºC) en presencia de
un cocatalizador que es el alquilaluminio. Los sistemas que contenían titanio
resultaron ser los que presentaban las actividades catalíticas más altas y se obtenían
polímeros de alto peso moleculares. Otra característica importante de este nuevo tipo
de sistema es que producen polietilenos lineales los cuales no presentan
ramificaciones en su estructura llamándose polietilenos de alta densidad (PEAD).
Paralelamente, Giulio Natta y colaboradores, estudiaban este tipo de
catalizadores en reacciones de polimerización con α-olefinas, donde verificaron la
posibilidad de producir polímeros estereoregulares[5, 6]. Uno de los polímeros
altamente utilizado industrialmente y que presenta una alta estereoregularidad es el
polipropileno, con tres tipos diferentes de microestructura (atáctico, isotáctico y
sindiotáctico). Polipropileno atáctico, es el que tiene una orientación aleatoria de los
grupos metilos a lo largo de la cadena polimérica, dando como resultados materiales
amorfos (ceras o aceites). El polipropileno isotáctico, es aquel en el cual los grupos
metilos se encuentran orientados para un mismo lado a lo largo de la cadena
polimérica. Por último en el caso del polipropileno sindiotáctico muestra que la
orientación de los grupos metilos es alternada a lo largo de la cadena (Figura 2). Los
polímeros isostáticos y sindiotácticos presentan una gran cristalinidad a diferencia de
los atácticos que poseen una estructura amorfa. Esta regularidad se ve reflejada en su
alto punto de fusión (165 ºC), cristalinidad, alta dureza y resistencia a la
deformación.
Figura 2. Microestructura de Polipropileno.

4
Debido a estos descubrimientos K. Ziegler y G.Natta recibieron el premio
Nobel en 1963. Los sistemas catalíticos desarrollados son conocidos como
catalizadores Ziegler-Natta, y actualmente contituyen la base de la produccion
industrial de polietilenos lineales de alta densidad, polipropilenos isotácticos y sus
copolímeros.
Otro sistema catalítico de coordinación para la polimerización de olefinas que
es descubierto en esta época, son los catalizadores del tipo Phillips, llamados así por
ser descubiertos por dicha compañia, Phillips Petroleum Co. Estos corresponden a
catalizadores a base de CrO3 soportado sobre sílica (SiO2) el cual es activado a altas
temperaturas[7]. Si bien los catalizadores de Phillips poseen actividades catalíticas
menores (2000 Kg PE/mol Cr*h*atm) a las mostradas por los catalizadores Ziegler-
Natta, su importancia radica en que con ellos es posible de obtener polietileno de alta
densidad (Figura 1, b) y polietilenos lineales de baja densidad (PELBD, Figura 1, c).
El PELBD corresponde a un polímero con ramificaciones muy cortas y uniformes, el
cual se obtiene por reacciones de copolimeriación de etileno con α-olefinas como 1-
buteno, 1-penteno o 1-hexeno (condiciones 40 atm, 80-100 ºC). El PELBD es un
copolímero importante en la producción de films flexibles y materiales de embalaje,
debido a que posee características como buena resistencia a la tracción, o al impacto
a baja temperatura.
1.2 Catalizadores Ziegler-Natta
Como se ha mencionado uno de los sistemas catalíticos más importantes lo
constituyen los catalizadores Ziegler-Natta. Los precursores cataliticos de dichos
sistemas requieren de la reacción de un alquilmetal (o hidruro metálico) con una sal
de metal de transición en atmósfera inerte. Los alquilmetal se basan en metales del
grupo I a III (Al, Zn, Mg, Be, Li, Ga), mientras que los metales de transición que
forman las sales son Ti, V, Cr, Co ó Ni. Un sistema catalítico típico (par
catalizador/cocatalizador) lo constituyen TiCl3 y Al(C2H5)2Cl. En estos sistemas, la

5
especie catalítica se forma por una reacción de alquilación sobre TiCl4, seguido por
una reducción por eliminación del grupo alquilo a un estado trivalente, (TiCl3).
Figura 3. Estructura Cristalina TiCl3
El TiCl3 cocristalizado con AlCl3, es posteriormente alquilado para formar el
centro activo capaz de coordinar al monomero (interaccion π entre el monómero y el
orbital d vacio del titanio que se encuentra en la parte externa del cristal). (Figura 4,
(a)). En el mecanismo de propagación se considera que el monómero forma un
complejo de coordinación con el catalizador y es insertado en el enlace polarizado
Ti-C del extremo creciente de la cadena polimérica. Un mecanismo propuesto para la
propagación de las especies se muestra en el Figura 4, (b).
Figura 4. Sistema Catalítico Ziegler-Natta Típico. A) Formación Especie Activa, b)
Mecanismo de Propagación de la Cadena.

6
En este el monómero se coordina en el orbital vacante y se inserta a la cadena
polimérica, con la regeneración del orbital vacante en una orientación diferente. La
adición continuada conduce a la formación de un polímero.
Con los catalizadores Ziegler-Natta es posible de obtener polímeros
isostáticos. La naturaleza de la superficie del catalizador parece ser importante para
conferir estereoregularidad en estos pasos. La propagación probablemente tiene lugar
en centros activos de la superficie del cristal del compuesto de metal de transición.
La estructura química y cristalinidad del catalizador determina la orientación del
monómero que se adiciona a la cadena creciente. La fuerza impulsora para la
propagación resulta de este modo de interacción entre el monómero y los ligandos
del metal de transición en el sitio activo. La polimerización en este caso se verá
influenciada por varios parámetros, como lo son la relación Al/Ti, temperatura o la
concentración de los reactivos.
Por otra parte existen mecanismos mediante los cuales las cadenas
poliméricas terminan, estas reacciones se denominan de transferencia o de término.
Una de ellas es la eliminación β-hidrógeno, la cual consiste en que el centro
metálico del catalizador atrae un átomo de hidrógeno del carbono β de la cadena de
polímero en crecimiento, formando un enlace M-H y dejando una cadena polimérica
terminada con una insaturación. (Figura 5).
Figura 5. Mecanismo de Terminación de Cadena, Eliminación β-hidrógeno
Otra de las reacciones de terminación de cadena es la transferencia al
monómero. En este caso existe una eliminación del hidrogeno del carbono β de la

7
cadena en crecimiento, el cual reacciona con el monómero que se está insertando al
centro activo, con la formación de un enlace M-R (Figura 6, a). Cuando la cadena de
polímero en crecimiento unida a un centro activo interacciona con el grupo metilo de
una molécula de cocatalizador (RmAl), puede ocurrir la terminación por transferencia
al cocatalizador. En esta se forma un enlace Al-C en el centro activo y una cadena
polimérica unida a un átomo de Al, la cual por hidrólisis forma el polímero final
(Figura 6, b). Dependiendo de las características del sistema catalítico es cuál de las
reacciones de transferencia mencionadas anteriormente la que ocurrirá.
Figura 6. Mecanismos de Terminación de Cadena
El desarrollo de los catalizadores Ziegler-Natta ha sufrido una constante
evolución, desde 1955 se pueden considerar la existencia de cuatro generaciones de
catalizadores basados en titanio. Los primeros catalizadores llamados convencionales
o de primera generación presentan bajas actividades catalíticas (1000g de polímero/g
de Ti) así como baja estereoespecificidad. En el caso de propeno, se obtiene
principalmente polipropileno atáctico, sin ninguna morfología determinada. Por otra
parte, en estos sistemas solo los átomos de Ti de la superficie se encuentran
disponibles para interactuar con el cocatalizador y crear sitios activos. Debido a que
el cristal de TiCl3 posee diferentes tipos de caras o entornos alrededor de los titanios
activos, es que existen múltiples sitios que dan origen a polímeros de variados pesos
moleculares. Esto hace que los catalizadores Ziegler-Natta se conozcan como sistema
multi-sitio que produce polímeros con distribución de peso molecular amplio. Los
esfuerzos posteriores se focalizaron en la búsqueda de sistemas donde la mayor parte
de los atomos de titanio participaran en la formación de centros activos (buzqueda de
sistemas más eficientes), estudiándose así tres caminos posibles: la reducción del

8
tamaño de las partículas de catalizador, dispersar la sal de titanio sobre una superficie
o utilizar sales solubles.
La segunda generación de catalizadores contempla la modificación química,
como introducción de donores internos y externos. Estas variaciones hacen posible
aumentar la actividad catalítica, controlar la estereoregularidad de los polímeros, así
como su peso molecular.
La tercera generación introduce variaciones físicas a los catalizadores, como
soportar la sal de titanio en sólidos inorgánicos como SiO2, Al2O3 o MgCl2. El
motivo de la utilización de estos soportes como el MgCl2 es porque el radio iónico
del Mg2+ (0,066 nm) es muy parecido al del Ti4+ (0,068 nm). Este sistema mostró alta
actividad para polietileno y polipropileno, sin embargo para este último la
estereoespecificidad fue menor. Este problema se resolvio con la adición de bases de
Lewis, las cuales hacen posible obtener sistemas con alta actividad catalítica y
estereoespecificidad. Cuando la base se adiciona al soporte MgCl2 o al TiCl3 se
denomina donor interno. Si la base se combinada con el cocatalizador se le denomina
donor externo. Un tipo de donor interno utilizado industrialmente es el benzoato de
etilo, mientras que como donor externo es utilizado el metil-p-tolueno. Es decir, la
tercera generación se compone de sales de titanio soportadas sobre MgCl2, las cuales
son modificadas por diferentes tipos de donores. La alta eficiencia catalítica y alta
estereoespecificidad hace innecesaria la etapa de remoción del polímero atáctico
desde el polímero obtenido, siendo esta una ventaja en comparación a los sistemas
anteriores. La última generación de catalizadores Ziegler-Natta aparece en los años
setenta y consiste en soportar el TiCl4 sobre partículas esféricas de MgCl2, con lo que
se puede controlar la forma y tamaño de la partícula de polímero. Además esto hace
posible aumentar aún más la actividad catalítica, la estereoregularidad (PP
isostáctico) y tener un control morfológico del polímero obtenido. Por otra parte la
introducción de un nuevo tipo de donores como el 1,3 dieter como componente
interno hace posible la obtención de polipropileno isostático sin la necesidad de
bases externas, siendo utilizada esta metodología para la obtención de este tipo de
polímero industrialmente. Una de las últimas modificaciones realizadas a esta
generación de catalizadores es la introducción de alquilsilanos como donores

9
externos, la diferencia esencial de este tipo de sistemas es la amplia distribución de
pesos moleculares que pueden ser obtenidas (PDI = 10-15). A continuación se
resumen las cuatro generaciones de catalizadores Ziegler-Natta y sus principales
modificaciones.
1ª Generación - TiCl3/ AlClEt2
2ª Generación - TiCl3/AlClEt2/ Donores internos y externos.
3ª Generación - TiCl4/AlClEt2/Base de Lewis interna/MgCl2/ Base de Lewis
externa. (Soporte)
4ª Generación - TiCl4/AlClEt2/Base de Lewis interna/MgCl2(esférico)/Base
de Lewis externa. (Morfología)
Los mecanismos de reacción de las polimerizaciones de olefinas, llevados a
cabo por catalizadores Ziegler-Natta resultan ser complejos debido a su naturaleza
heterogénea. Esto provocó que en los años 1980 los estudios sobre desarrollo de
sistemas catalíticos estuviera dirigido a la búsqueda de sistemas homogéneos para
llevar a cabo la polimerización de olefinas.
Es así que se comenzó con el estudio de sistemas catalíticos bien definidos
con el fin de elucidar los mecanismos de polimerización que pudieran ser extensivos
a sistemas heterogéneos. Estos sistemas se conocen como metalocenos y fueron de
fundamental importancia para el progreso y conocimiento de sistemas homogéneos.
Estos condujeron a nuevos polímeros y principalmente a copolímeros con
propiedades nunca antes observadas, que no pueden ser obtenidos con la tecnología
Ziegler-Natta tradicional.

10
1.3. Catalizadores Metalocénicos
Los catalizadores metalocenos son complejos organometálicos formados por
metales de transición del grupo IV (Ti, Zr, Hf) unidos a dos anillos aromáticos como
por ejemplo ciclopentadienilo (Cp), indenilo (Ind) a través de interacciones tipo π y a
ligandos como iones haluros o metilos mediante enlaces σ.
Los complejos metalocenos son catalíticamente inactivos en reacciones de
polimerización. La formación de la especie activa necesita de un ácido de Lewis que
actúe como cocatalizador, y sea capaz de formar un centro metálico catiónico y un
enlace metal carbono. Los primeros catalizadores homogéneos a base de metalocenos
(Cp2TiCl2/AlR2Cl; R = alquilo), descrito por Breslow y Natta en 1955, fue en la
polimerización de etileno dando bajas actividades (2,5-10 Kg PE/(mol
Ti*h*atm))[8]. Posteriormente Kaminsky y Sinn descubrieron que la hidrólisis
controlada de trimetilaluminio produce un ácido de Lewis denominado
Metilaluminoxano (MAO), el cual en presencia del metaloceno genera un sistema
catalítico 10.000 veces más activo comparado a los que se forman con otros
alquilaluminios[9].
Los alquilaluminoxanos son preparados por variados métodos, el más
utilizado corresponde a la hidrólisis controlada de respectivo alquil alumnio. Esta
una reacción es altamente exotérmica y se debe llevar a cabo a baja temperatura con
solventes inertes como tolueno[10]. Ecuación 1.
4Al(CH3)3 + 3H2O Al4O3(CH3)6 + 6CH4 Ec.1
Esta reacción es la simplificación de la hidrólisis que ocurre en múltiples
pasos. Se conoce que la hidrólisis procede por la formación de un complejo
alquilaluminio-agua, el cual elimina metano para formar un complejo de hidróxido
de dimetilaluminio. Esta rápida asociación da dímeros u oligómeros en solución. Así
la estructura del MAO es compleja y ha sido investigada mediante medidas
crioscópicas, reacciones de hidrólisis, espectroscopia infrarroja, ultravioleta y de
resonancia magnética nuclear (RMN)[11]. El peso molecular determinado por
medidas crioscópicas es de 1000 a 1500 g/mol. Trabajos de Sinn[11] y Barron[12]

11
han promovido detalles sobre la estructura del MAO, donde los diferentes
oligómeros tiene una unidad como la mostrada en la Ecuación 1.
Esta unidad se puede asociar dando como resultado átomos de aluminio
insaturados. Hay átomos de aluminio tri- y tetra- coordinados, donde los trivalentes
muestran una gran acides de Lewis. Algunas de las estructuras propuestas para el
MAO se muestran en la Figura 7. Existen estructuras más complejos como la
producida por la unión de cuatro unidades Al4O3(CH3)6, donde se puede formar una
estructura conocida como cluster de MAO.
Figura 7. Diferentes Estructuras Posibles de MAO: 1 lineal; 2 y 3 cíclicos; 4 cluster
y 5 red tridimensional[13].
Los sistemas metalocenos/MAO, en particular de circonio presenta
actividades mayores que su análogo con AlR3 y superiores a las obtenidas con los
catalizadores Ziegler-Natta soportados en MgCl2. Sin embargo para que la activación
con MAO sea efectiva, son necesarias altas concentraciones de este compuesto,
típicamente son usadas razones Al/Zr en el rango de 103-104. Lo anterior implica
necesariamente que no todas las especies presentes en el MAO tienen la acidez
necesaria para generar a la especie activa o bien no participan en la activación del
centro metálico.
Uno de los componentes fundamentales para acceder a altas actividades
catalíticas con catalizadores de sitio único es el cocatalizador. Una de las funciones

12
del cocatalizador es la formación del par ionico con el metaloceno. Otra de las
funciones, es la alquilación del complejo metalocénico halogenado. En el primer
paso el compuesto monometil es formado rápidamente y en exceso de MAO da la
especie dialquilada. Para que el sitio formado sea activo es necesario al menos retirar
un grupo alquilo del metaloceno[14-16]. El rol más importante del MAO como
cocatalizador en la polimerización de olefinas en sistemas metalocénicos es la
alquilación del metal de transición y la producción del complejo alquil catión del tipo
Cp2MR+ como especie catalíticamente activa[17]. La estructura y función del MAO
puede ser representada por los múltiples equilibrios, sin embargo las reacciones que
existen entre este y un circonoceno se resume en la Figura 8.
Complejación
L2ZrCl2 + MAO L2ZrCl2*MAO
Metilación
L2ZrCl2*MAO L2Zr(CH3)Cl + Al
Cl
H3C
O
L2Zr(CH3)Cl*MAO L2Zr(CH3)2 + MAO*Cl2
Activación
L2Zr(CH3)Cl + MAO L2Zr(CH3)Cl*MAO
[L2ZrCH3]+ [MAO*Cl]-
Figura 8. Mecanismo de Complejación, Metilación y Activación Creados con MAO.

13
Otros alquilaluminio como etilaluminoxano o isobutilaluminoxano han sido
utilizado como cocatalizador en lugar de MAO, sin embargo los sistemas con estos
cocatalizadores han mostrado una menor actividad catalítica.
Existen asociada a este proceso reacciones como la desactivación la cual
consiste en la transferencia de hidrógeno-α, del catión L2ZrCH3+ al MAO,
produciendo un sistema inactivo con la consecuente producción de metano. (Figura
9). Cuando ocurre la condensación del metaloceno con el MAO se forma las especies
Zr-CH2-Al o Zr-CH2-Zr. Estos compuestos son inactivos y la presencia de estos es
una de las razones por las cuales los complejos metalocénicos se desactivan[18].
Desactivación
L2Zr C
H
H
H + Al
Cl
H3C
O L2Zr CH2 Al
Cl
O + CH4
Reactivación
L2ZrH2C Al
CH3
O + Al
H3C
H3C
O [L2ZrCH3]
+
O Al
CH3
H2C Al O
CH3
MAO-
Figura 9. Mecanismo de Desactivación y Reactivación de Complejos Metalocénicos
El proceso de formación de la especie activa se puede resumir en dos etapas.
La primera de ellas es la metilación del metal por el MAO y la segunda es la
generación de un sitio de coordinación libre donde se generara además una carga
positiva en el metal. La formación de polímero ocurre en dos etapas: primero la
coordinación del monómero al metal y segundo su inserción en la cadena en
crecimiento.

14
Figura 10. Activación General de Complejos Metalocénicos y Propagación de la
Cadena Polimérica[19].
Los pasos de terminación de cadena son los que determinan el peso molecular
de un polímero. Parece ser que el proceso dominante en la terminación es la
eliminación β-hidrógeno, con la consecuente formación de un hidruro metálico que
podría reiniciar el crecimiento de una nueva cadena. Sin embargo los otros tipos de
terminación anteriormente descritos también pueden tener lugar en este tipo de
sistema.
Existen diferencias importantes entre catalizadores metalocénicos y Ziegler-
Natta; una de ellas es que los primeros muestran un solo tipo de sitio activo y
producen polímeros con distribuciones de peso molecular estrechas (Mw/Mn ≈ 2), en
cambio los sistemas Ziegler-Natta poseen distribuciones de pesos moleculares anchas
(Mw/Mn ≈ 3-10), ya que son sistemas multi-sitio. Por otra parte el peso molecular
también puede ser controlado por modificaciones estructurales en los anillos
ciclopentadienilo, donde un mayor impedimento estérico dificulta la eliminación-β,
aumentando así el peso molecular.
Desde los primeros estudios realizados con los catalizadores metalocénicos
hasta la fecha, se han desarrollado numerosas modificaciones en los sustituyentes

15
aromáticos, así como entre los fragmentos Cp, a través de puentes (-CH2CH2-, -
CMe2-, SiMe2-). Estas variaciones han hecho posible la obtención de nuevos
polímeros donde se puede controlar el peso molecular; permitiendo obtener desde
oligómeros hasta polímeros con un muy alto peso molecular[20]. Por otra parte la
introducción de puentes hace posible la obtención de complejos capaces de dar
polímeros estereoregulares. Aquellos sistemas que presentan una simetría del tipo C2
frente a monómeros proquirales, hacen la inserción siempre por la misma cara con la
idea de minimizar las interacciones estereoquímicas del grupo metilo del propileno
con el centro metálico. La estereoquímica de la inserción se mantiene a lo largo de la
propagación de la cadena lo que forma un polipropileno isotáctico[21]. Si la simetría
del catalizador es del tipo Cs, la inserción de la olefina es alternada de un lado a otra
desde un plano, obteniéndose polipropileno sindiotáctico[22]
1.4. Catalizadores Post-Metalocénicos
La comercialización tanto de polietileno como de polipropileno sintetizado
mediante metalocenos ha sido posible, sin embargo el costo asociado a la síntesis de
estos sistemas, ha sido elevado[23]. Por otra parte estos sistemas poseen desventajas
como una menor tolerancia a la presencia de grupos funcionales o impurezas polares,
menor estabilidad térmica o fotoquímica. Esto ha derivado en la búsqueda de nuevos
complejos, con metales de transición del grupo IV (Zr, Ti o Hf) o de grupos
posteriores como Ni, Co, Fe, etc con una variedad de ligandos diferentes, dando
origen a la nueva generación de catalizadores llamados Post-metalocénicos[24-27].
Estos sistemas al ser activados con ácidos de Lewis muestran altas actividades
catalíticas. Por otra parte con estos sistemas es posible obtener variados tipos de
polímeros como polietilenos altamente ramificados, copolímeros de etilenos con
monómeros polares[28-31], polímeros con polidispersidad ~ 1,0[32-35], poli(1-
hexeno) de alto peso molecular[36-38], copolímeros y polímeros en bloque[39, 40],
etc., los cuales no han sido posibles de obtener con los catalizadores convencionales
(Ziegler-Natta, Metalocénicos).

16
En la búsqueda de sistemas catalíticos que permitan acceder a nuevos tipos de
polímeros y copolímeros, a significado que desde mediados de los años 90´ el foco
de las investigaciones en el área de catálisis de olefinas este centrado en el desarrollo
y aplicación de catalizadores basados en metales de transición del grupo IV con
ligandos bidentados y tridentados. En la Figura 11 se incluyen algunos ligandos que
han resultados ser muy eficientes como parte de estos nuevos sistemas[41-44].
Particularmente porque a través de estos se puede modular propiedades estéricas,
electrónicas, de simetría y estabilidad, de los compuestos de coordinación
resultantes.
Figura 11. Tipos de Ligandos R = 2,6(i-Pr)2C6H3; 2,6Me2C6H3; 2-t-BuC6H4
Estos ligandos puedes ser obtenido mediante reacciones químicas clásicas, así
por ejemplo de la Figura 1.4.1 los ligandos a, b, c y d, pueden ser preparados por
reacciones de condensación de una anilina particular (sustituyente R), con una cetona
adecuada, en razones estequiométricas especificas, para dar ligandos mono o
disustituidos[45, 46].

17
En el caso de ligandos iminicos-heterociclos, la condensación se realiza con
heterociclos de aldehídos. Los ligandos tridentados como bis(imino)bipiridina
(Figura 11 f) es obtenido por una oxidación selectiva para dar 6,6-diformil-2,2-
bipiridina, seguida por la condensación de la anilina apropiada.
En este contexto la química de los ligandos β-dicetoiminas
([{N(Ar)C(Me)} 2CH]-, nacnac)[46] ha sido muy estudiada dada la amplia variedad
de anilinas disponibles comercialmente y que permiten acceder a un gran número de
estos ligandos con diferentes características estéricas y electrónicas. Esta última
basada en la incorporación de átomos de flúor a los sustituyentes, los cuales,
influencian notoriamente las propiedades del compuesto[47-49]. Otra ventaja que
presenta este tipo de ligandos, es el alto rendimiento en su síntesis.
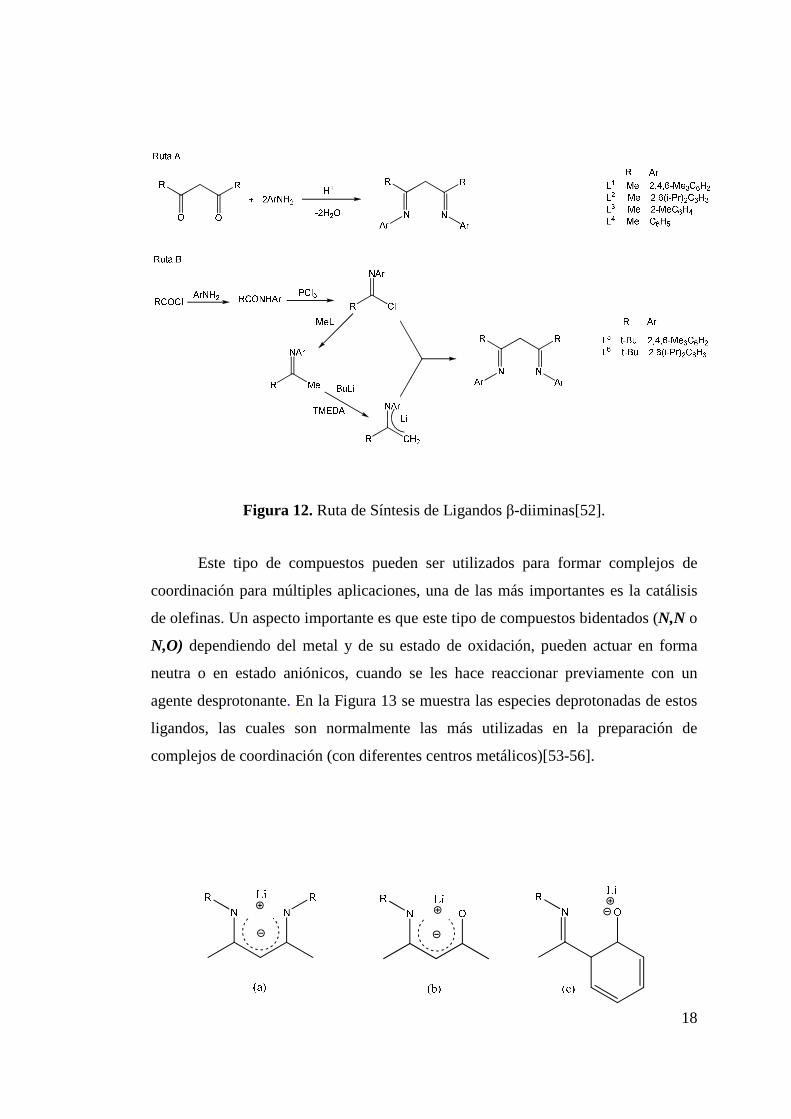
En la Figura 12 se muestran dos rutas de síntesis para la obtención de
ligandos β-diiminicos. La ruta más conveniente (ruta A), corresponde a la reacción
de condensación de una cetona con una amina aromática y es catalizada por un
ácido[50, 51]. La segunda; ruta B, es especialmente útil para iminas muy
voluminosas, donde la condensación es muy lenta o prácticamente no ocurre. La ruta
B incluso es aplicable a la síntesis de ligandos asimétricos, donde los sustituyentes de
la imina son distintos.
18
Figura 12. Ruta de Síntesis de Ligandos β-diiminas[52].
Este tipo de compuestos pueden ser utilizados para formar complejos de
coordinación para múltiples aplicaciones, una de las más importantes es la catálisis
de olefinas. Un aspecto importante es que este tipo de compuestos bidentados (N,N o
N,O) dependiendo del metal y de su estado de oxidación, pueden actuar en forma
neutra o en estado aniónicos, cuando se les hace reaccionar previamente con un
agente desprotonante. En la Figura 13 se muestra las especies deprotonadas de estos
ligandos, las cuales son normalmente las más utilizadas en la preparación de
complejos de coordinación (con diferentes centros metálicos)[53-56].

19
Figura 13. Ligandos Anionicos a) β-diiminicos, b) β-dicetoiminas, c) base de Schiff
El modo de coordinación de los ligandos al centro metálico, está determinada
por la densidad de carga en torno de los sitios básicos del ligando (neutra o aniónica),
pero también por la acidez y estabilidad del metal con el cual se le desee coordinar.
Algunas de las formas de coordinación más frecuentes se resumen en la Figura 14. N
N
M
N
N
M
N
N
M
NH2
NH2
M
N N MM
N
N
M
M
Figura 14. Modos de coordinación de Ligandos bidentados a Metales de Transición.
Dependiendo del tipo de metal que se utilice y las características del ligando
(neutro o aniónico) es el tipo de coordinación que será más probable. En el caso de
metales de transición tempranos, lo más común es ver dos ligandos aniónicos
coordinados a un centro metálico, en cambio en sistemas de Ni, Pd y Fe,
normalmente solo un ligando se coordina y se puede encontrar en forma neutro o
monoaniónica.

20
1.4.1. Complejos con Metales de Transición Tempranos (Zr, Ti)
Características de los ligandos, como las mencionadas anteriormente, junto
con el enorme interés industrial por el desarrollo de sistemas catalíticos para
polimerización de olefinas más simples y eficientes, son la base para la síntesis de los
sistemas catalíticos con metales de transición como circonio y titanio. En la Figura
15 se muestran diferentes complejos post-metalocénicos con metales de transición
tempranos. En estos se pueden observar que los ligandos se coordinan al metal de
forma monoaniónicos. En la mayoría de los casos existen dos ligandos por metal lo
cual está estrechamente relacionado con la compensación de carga del catión
utilizado (ligandos y halógenos). Este tipo de compuesto en general son preparados a
partir de la sal del metal de transición (MCl4, MCl4*DME, MCl2Me2, etc) con la sal
del ligando respectivo o por reacción de los ligandos protonados con precursores de
metales de transición como MBn4 o M(NMe)4. Estas son reacciones directas (una
etapa) y se llevan a cabo en condiciones de temperaturas moderadas o bajas.
Afortunadamente ambos métodos han resultado ser muy efectivos para acceder a
complejos de estos metales. Esto complejos han mostrado ser mucho más estables,
(estables al aire por algunas horas) comparados con los metalocenos.
Figura 15. Complejos Post-metalocénicos con Metales de Transición Tempranos[55].

21
Complejos como los señalados en la Figura 15 muestran excelente
comportamiento para la polimerización de olefinas, con actividades catalíticas del
orden de 6000 KgPE/molM*h*bar, considerándose alta para complejos que no
poseen anillos ciclopentadienilo en su estructura (catalizadores metalocénicos.)
Algunos de estos sistemas mostraron una relación lineal entre tiempo de reacción y
peso molecular “polimerización viva”. Esto significa que en la polimerización no
existe la terminación de cadena. Esto hace que las cadenas de polímero sean todas de
un mismo tamaño obteniéndose distribuciones de peso molecular cercano a uno (1,0-
1,15). En este caso el crecimiento de la cadena polímero se interrumpe por la adición
de un agente externo que forma una especie no reactiva o por dejar de suministrar el
monómero a polimerizar[56].
Figura 16 Sistema Catalítico Post-Metalocénicos[57-60].
Estudios computacionales han utilizado como modelos sistemas metalocenos
con metales del grupo IV, mostrando que la polimerización de etileno es un proceso
que involucra un cambio electrónico entre el metal y el ligando que forman parte del
complejo. En los sistemas post metalocénicos este efecto también se aprecia,
haciendo que el ligando juegue un rol predominante en la polimerización debido a la
flexibilidad de su estructura (cambios electrónicos y estéricos), la cual influye en el
sistema final[59].
En general los sistemas catalíticos para
polimerización de olefinas están
compuestos por; un metal de transición
central, ligandos enlazados a este, una
cadena polimérica creciente, una olefina
coordinada y un cocatalizador (Figura
16)
M = Metal L = Ligando P = Cadena Polimérica

22
Ejemplos de esta influencia lo muestran los sistemas reportados por Fujita y
colaboradores, los cuales son llamados catalizadores FI. Esta catálisis FI consiste en
metales de transición del grupo IV unido a un par de ligandos monoaniónicos no
simétricos del tipo fenoxi-iminas (N,O) [59-62]. Un ejemplo específico lo constituye
un sistema como el mostrado en la Figura 17.
Distancia de Enlace (A)
Ti-O 1,852(4)
Ti-N 2,236(4)
Ti-Cl 2,305(2)
Angúlos de Enlace (º)
O-Ti-O 171,6(2)
N-Ti-N 76,4(2)
Cl-Ti-Cl 103,10(8)
Figura 17. Catalizador Post-metalocénicos FI.
La estructura molecular de este tipo de sistemas (fenoxi-imina)2MCl2 (M =
Ti, Zr, Hf) han sido ampliamente estudiados mediante RMN y difracción de Rayos-X
(resolución estructural) llegando a la conclusión que para este tipo de sistema existen
cinco configuraciones posibles (Figura 18). Esto es producto de las diferentes
disposiciones espaciales que adoptan los ligandos (uno en relación al otro) al unirse
al metal. Normalmente poseen una geometría octaédrica en torno al centro metálico,
con simetría C2. La estructura precisa de la especie activa con MAO, para este tipo
de sistemas no ha sido aun elucidada, sin embargo estudios de DFT sugieren que la
especie catalíticamente activa es la que posee una geometría octaédrica con un
arreglo, tran-O, cis-N, cis-cadena polimerica/olefina coordinada, en el caso que los
sustituyentes voluminosos están en la cercanía del oxigeno[61].

23
Figura 18. Configuraciones Posibles de Sistemas Catalíticos Post-metalocénico.
Adicionalmente la geometría C2 de la especie activa tiene la capacidad de ser
estereoselectiva en polimerizaciones de α-olefinas. Esta relación ya había sido
observada para los complejos metalocénicos[62].
Estructuralmente si se hace una comparación entre los complejos de Ti y Zr,
que estén unidos a iguales ligandos fenoxi-imina, se puede notar que el átomo de
titanio se encuentra más protegido (estéricamente más impedido) en comparación al
complejo de circonio. Esto se puede apreciar en las menores distancias de enlace Ti-
O, Ti-N y Ti-Cl y al menor ángulo O-Ti-O, producto del menor radio iónico Ti+4
(0,68 Å) comparado con Zr+4 (0,86 Å) aunque los dos complejos tengan la misma
conformación. Este efecto hace que las actividades catalíticas de los complejos de
circonio sean mayores, ya que se encuentra más disponible para la coordinación con
los monómeros de las olefinas[61].
Los catalizadores FI generalmente muestran altas actividades catalíticas,
comparables o incluso mayores que las encontradas con sistemas metalocenicos. Un
ejemplo es el complejo bis[N-(3-tert-butilsalicilidine)anilinato]Circonio (IV) dicloro
que presenta una actividad de 519 (Kg-PE/mmol Zr*h) la cual es 20 veces más alta
que la encontrada para el sistema Cp2ZrCl2/MAO (27 Kg-PE/mmol-Zr*h) bajo las

24
mismas condiciones de reacción[63]. La clave de los catalizadores FI es que sus
propiedades estéricas y electrónicas pueden ser variadas fácilmente. Estudios de
actividades catalíticas de complejos de titanio con ligandos fenoxi-imina con
diferentes sustituyentes muestra que mientras más voluminosos es R1 afecta tanto la
actividad catalítica como el peso molecular de los polímeros obtenidos (Figura 19).
Así por ejemplo, el incremento en el tamaño de R1 hidrógeno a metilo incrementa la
actividad catalítica y el peso molecular del polietileno en un factor de 6, si se le
compara con un sistema donde el sustituyente es solo hidrógeno. Este aumento es
más pronunciado cuando los sustituyentes son t-butilo o trimetilsilano. Este mismo
efecto de R1 se ha visto en los complejos de circonio con los correspondientes
ligandos fenoxi-imina.
Figura 19. Variaciones en Ligandos N,O de Complejos Post-metalocénicos.
El incremento en la actividad catalítica al introducir sustituyentes
voluminosos, se atribuye a que estos estarían jugando un rol esencial en la separación
entre la especie activa catiónica y el cocatalizador aniónico. Esta separación produce
un mayor espacio para llevar a cabo la polimerización de olefina. Por otra parte, el
aumento en el peso molecular esta atribuido a la mayor congestión producida por el
sustituyente voluminoso, que hace que disminuya la terminación de cadena.
Los incrementos en el volumen de R2, produce disminuciones tanto en la
actividad catalítica como en el peso molecular. Este efecto puede estar asociado a la
mayor congestión que crea el sustituyente alrededor de centro activo, debido a su
Complejo R1 R2 R3
1 H H H
2 Me H H
3 t-Bu Me H

25
disposición espacial que obstaculiza el acceso de etileno al centro activo. El
sustituyente R3 tiene efecto en la actividad catalítica, pero casi no influye en el peso
molecular del polímero obtenido, sin embargo la influencia no está bien clara debido
a que su posición espacial está muy lejos del centro activo [60].
Paralelamente G. Coates, desarrollo una variedad de complejos de titanio
para la polimerización de olefinas, enfocándose principalmente en la influencia que
tenia la variación de entornos estéricos de los ligandos sobre el sistema catalítico[64-
66]. (Figura 20)
Figura 20. Catalizador con Sustituyentes Halógenos, G. Coates[67].
Una de las variables a estudiar fue la influencia de la incorporación de
halógenos en los ligandos sobre la actividad catalítica y las características de los
polímeros obtenidos. El complejo que posee anillo aromático sin sustituyentes (1),
cuando es activado con MAO es capaz de producir polipropileno sindiotáctico (94
%) de bajo peso molecular (Mn = 9910, Mw/Mn= 2,14). La incorporación de anillos
fluorados al sistema provoca un cambio en la densidad del titanio haciéndolo más
positivo lo involucra un aumento en la actividad catalítica, en al menos un orden de
magnitud. Por otra parte, este sistema (2) produce una mayor estereoespecificidad del
polipropileno obtenido (99 %). Otra de las características del polímero obtenido con
estos sistemas es la distribución de peso molecular que es típica de un sistema de
polimerización viva (Mw/Mn = 1,1). La capacidad de ser un sistema living hace
posible que con este sistema se obtengan copolímeros en bloque polietileno,
polipropileno. Esto se logra con el catalizador 2 en presencia de MAO a 0 ºC,

26
obteniéndose polipropileno sindiotáctico (Mn = 38400; Mw/Mn = 1,1). Luego la
adición de etileno al reactor produce polímero de alto peso molecular (Mn = 145100;
Mw/Mn = 1,2) dando como resultado el primer sistema capaz de sintetizar polímero
dibloque sindio-poli(propileno)-block-poli(etileno-co-propileno)[67] (Figura 20)
Estos sistemas corroboran las tendencias y resultados mostrados por los
complejos de Fujita, donde los factores estéricos y electrónicos de los ligandos tienen
influencia sobre la reactividad de los complejos y las características de los polímeros
obtenidos.
Sin embargo todos los sistemas hasta ahora mostrados con metales de
transición temprana (Ti, Zr, Hf) tiene en común su alta oxofilicidad, esto implica
que las reacciones de polimerización pueden ser envenenadas con la presencia de
olefinas funcionalizadas. Las polimerizaciones con este tipo de olefinas solo pueden
llevarse a cabo si la funcionalidad esta protegidas (sustratos específicos) o si en el
medio de reacción existe un alto contenido de ácidos de Lewis (protección por
complejación)[68, 69]. El costo de estos múltiples pasos dificulta la competitividad
de estos sistemas hacia la comercialización de los productos obtenidos con ellos.
Siendo este el motivo por el cual este tipo de copolímero (etileno/monómero
funcionalizados) y polímeros de etileno de baja densidad aun se siguen realizando
mediante polimerización de radical libre[70]. Como una alternativa para la obtención
de copolímeros funcionalizados, (mediante polimerización por coordinación) es que
se ha investigado la utilización de complejos de metales de transición como Fe, Ni,
Pd, etc.

27
1.4.2. Complejos con Metales de Transición Tardíos (Fe, Ni, Pd)
Los centros metálicos mencionados anteriormente tienen características
comunes, una de ellas es que poseen orbitales d semi llenos y bajos potenciales de
ionización, esto último hace que existan por ejemplo los cationes Ni(II) y Pd(II) con
configuración d8 y el Fe (II) con configuración d6. La configuración electrónica de
estos metales hace posible que su geometría molecular sea del tipo tetraédrico o
cuadrada plana dependiendo del tipo de ligandos que posea. La diferencia más
notable que presenta cada una de estas geometrías es que definen las propiedades
magnéticas de los complejos. Cuando el níquel adquiere una configuración cuadrada
plana el sistema es diamagnético, en cambio si la geometría es tetraédrica es
paramagnético.
Las características electrónicas antes mencionadas, hacen que los metales de
transición como Ni(II), Pd(II) y Fe(II) sean más bien ácidos blandos (clasificación de
Pearson) provocando que su reactividad hacia ligandos polidentados del tipo P,P;
P,N; P,O; N,N; N,O; N,N,N; P,N,N, entre otros, sea suficiente para poder formar
complejos[71-79]. Los ligandos que contienen fósforo en su estructura, han tenido
gran relevancia en el desarrollo y uso de compuestos organometálicos en reacciones
catalíticas. Algunos ejemplos son las reacciones de acoplamiento C-C, de
alquilación, hidrogenación y en reacciones de Heck[80-86]. En esta última los
complejos con ligandos nitrogenados funcionan generalmente mejor que los a base
de fósforo.
Algunas de las reacciones de acoplamiento C-C, utilizando catalizadores de
níquel pueden ocurrir con un alto grado de selectividad, obteniéndose oligómeros o
polímeros de etileno. La obtención de uno u otro depende de las características del
ligando que este coordinado al metal.
Uno de los primeros sistemas reportados para la oligomerización de etileno
fue por W. Keim en 1994 (Figura 21, a). Este es un complejo de níquel con ligandos
aniónico P,O que se obtiene por la reacción directa del ligando con Ni(COD)2 (COD
= ciclooctadieno) mediante adición oxidativa con PhBr en presencia de la sal de
ligando y de PPh3 en tolueno[87].

28
Figura 21. Sistemas Catalíticos Post-metalocénicos, a) W. Keim b) J. Claverie c) Grubbs.
En tolueno el complejo formado es capaz de oligomerizar etileno sin la
presencia de un cocatalizador, formando productos lineales con un contenido de alfa
olefina de 98 % predominantemente C6 - C26. La producción de oligómeros con este
tipo de sistema, es la base para el proceso Shell Higher Olefin Process (SHOP).
Si el solvente es cambiado de tolueno a hexano el sistema es capaz de formar
polietileno. Este corresponde a un polietileno más bien lineal, con solo algunas
ramificaciones del tipo CH3, con terminación del tipo vinílica y peso molecular Mw =
9310 g/mol.
Como se menciono anteriormente, la obtención de polímeros de bajo o alto
peso molecular está estrechamente relacionada con las características del ligando. En
el caso de ligandos poco sustituidos la eliminación de hidrógeno-β compite con la
inserción de monómero, dando como resultados oligómeros. Con el fin de encontrar
sistemas capaces de producir polímeros de mayor peso molecular o de copolímerizar
etileno con otros monómeros, es que se han modificado los ligandos que coordinan al
metal. Un ejemplo lo constituyen los ligandos utilizados en los complejos de Keim,
donde el átomo de fosforo es reemplazado por nitrógeno (Figura 21, b). La
coordinación de este ligando a níquel, produce un complejo que no necesita la
presencia de un cocatalizador para llevar a cabo la polimerización de etileno, por otra
parte este sistema es capaz de formar policetonas por la copolimerización de etileno
con monóxido de carbono[88]. Otro tipo de ligando es el reportado por Grubss, el

29
cual consiste en un ligando monoaniónico N,O que forma con el níquel un anillo de
seis miembros (Figura 21, c). Este sistema es altamente activo para la polimerización
de etileno, sin la necesidad de un cocatalizador, dando como resultado un polímero
lineal con muy pocas ramificaciones[89].
Basado en los estudios de Keim, principalmente Brookhart y colaboradores
reportaron complejos diiminicos de níquel y paladio (II), los que corresponden a
especies metálicas dibromadas, con un ligando bidentado neutro N,N como los
mostrados en la Figura 22. Los diferentes sustituyentes de las iminas dan origen a
sistemas con variadas actividades catalíticas y que permiten acceder a materiales de
un amplio rango de pesos moleculares y microestructura[39, 90-93].
Figura 22. Ligandos N,N utilizados en Complejos de Níquel(II) y Paladio(II)
Más específicamente estos complejos resultaron ser activos para la
polimerización de etileno, propileno y 1-hexeno, en presencia de MAO. El sistema
de níquel con ligandos como (a) de Figura 22 mostró altas actividades catalíticas,
cercana a las de los complejos metalocénicos (11000 KgPE/(mol Ni*h), produciendo
polietilenos de alto peso molecular (650000 g/mol), y estrecha distribución de peso
molecular. Por otra parte este complejo es capaz de polimerizar 1-hexeno amorfo con
una actividad de 176 Kg/(mol Ni*h)[94, 95].

30
En el caso de los complejos de paladio fue posible de obtener polietileno con
ramificaciones distribuidas aleatoriamente a lo largo de la cadena polimérica (103
ramificaciones/1000 átomos de carbono). Al contrario de esto, con los complejos de
níquel se obtuvo un polímero altamente lineal, con solo algunas ramificaciones del
tipo CH3. La cantidad de ramificaciones en este caso es función de la temperatura, la
presión de etileno y la estructura del catalizador. Es así que se obtiene una mayor
cantidad de ramificación cuando se aumenta la temperatura, dando como resultado
una menor temperatura de fusión del polímero obtenido. Las altas presiones
disminuyen la cantidad de ramificaciones en el polímero, sin variar las actividades
catalíticas o el peso molecular.
El complejo de níquel con el ligando e) Figura 22 en condiciones de bajas
temperatura (23 ºC) produce polipropileno con una estrecha distribución de peso
molecular (Mw/Mn 1,4-1,6). Esta distribución sugiere que la transferencia de cadena
es menor a bajas temperatura, cuando la reacción se realiza a -10 ºC, se pueden
lograr distribuciones hasta de Mw/Mn ~ 1,13. Esto sugiere que con estos complejos,
también es posible suprimir la terminación de cadena (polimerización viva).
Por otra parte, estos complejos son capaces de copolimerizar etileno con
monómeros polares que contengan grupos éter, ester o ácidos en su estructura. Los
complejos catiónicos de paladio son capaces de copolimerizar etileno con olefinas
como acrilatos, dando productos de gran interés comercial, de altos pesos
moleculares y con distribución aleatoria del comonómero. Otros comonómeros
utilizados han sido los ácidos carboxílicos, sustituyentes epóxidos y derivados de
acrilatos[30]. Los complejos de níquel copolimerizan etileno con acrilatos, dando
polímeros altamente lineales con una menor incorporación del comonómero si se le
compara con paladio. En el caso de níquel los comonómeros polares utilizados han
sido metil vinil cetonas, CO, aldehídos, entre otros. [40, 72, 96, 97]

31
Sin embargo una de las características más significativas es el entorno
estérico de los ligandos diiminicos ya que determinan los diferentes procesos que
llevan a cabo la polimerización, como lo son la propagación, ramificación y
transferencia de cadena.
Al igual que en los sistemas catalíticos de Ziegler-Natta y metalocénicos, en
estos sistemas, la polimerización de olefinas ocurre debido a la presencia de un
centro metálico alquil catiónico (especie catalíticamente activa). En los sistemas de
níquel o paladio mostrados anteriormente, la formación de la especia activa se lleva a
cabo utilizando un cocatalizador, siendo generalmente MAO. Este al igual que en los
sistemas metalocénicos tiene la función de metilar el complejo y formar un par
ionico, cuyo sitio de coordinación es el responsable de coordinar la olefina (Figura
23, a). En el ciclo de propagación de cadena, el primer paso consiste en la inserción
de la olefina formando una especie alquil catiónica y la inserción del siguiente
monómero vuelve al sistema a su estado inicial para que siga creciendo la cadena
polimérica (Figura 23, b). La terminación de cadena ocurre generalmente es por
eliminación de hidrogeno-β (Figura 23, c).

32
Figura 23. a) Formación sitio activo; b) Propagación de Cadena; c) Terminación de
Cadena.
Brookhart ha señalado que el mecanismo de polimerización está relacionado
con los sustituyentes voluminosos de los ligandos. La inserción de los monómeros
hacia el centro metálico ocurre solo por los sitios ecuatoriales del complejo, en
cambio la terminación de cadena es debido a una coordinación en parte axial del
sistema catalítico. Esto implica que la presencia de sustituyentes voluminosos (R = i-
Pr, t-But) estarían bloqueando los sitios axiales del centro metálico, debido a que los
anillos de las α-diiminas se encuentran en una posición ortogonal al plano formado
por el metal y el ligando (Figura 24). Esto provoca que la terminación de cadena sea
menor a la propagación produciéndose polímeros de alto peso molecular[92].
N N
R
R
R
R
Ni
PEc Ec
Ax
Ax
(MAOBr2)
R : Me, i-Pr, t-But
Figura 24. Sitios de Coordinación Axial (Ax) y Ecuatorial (Ec) de Centros Metálicos
y su Potencial Interacción Estérica con Sustituyentes Voluminosos (R).
Por el contrario la reducción del volumen de los sustituyentes (R = H) hace
que la transferencia de cadena se vea incrementada, formándose oligómeros de
etileno, desde C4 hasta C26 con selectividades superiores al 94% de oligómeros

33
lineales [90]. Estos sistemas presentan una alta actividad catalítica, si se les compara
con los catalizadores neutros SHOP que utilizan ligandos bidentados P,O, se puede
ver que son 15 veces más activos. También tienen mayor estabilidad, no presentan
perdida en la actividad catalítica, incluso en periodos mayores a tres horas. Cambios
en las condiciones de reacción también provocan variaciones en la proporción de
oligómeros obtenidos, en general cuando la temperatura de reacción se incrementa,
las reacciones de transferencia de cadena se ven favorecidas, con respecto a la
propagación
Otra de las características de los polímeros que puede ser obtenida, variando
los ligandos en los diferentes complejos de níquel o paladio, es la alta ramificación
de cadena (100 ramificaciones/1000 átomos de carbono). Esta ha sido incluso
superior a la observada para polietilenos de baja densidad. El proceso mediante el
cual se explica la formación de ramificaciones ocurre durante la etapa propagación
de la cadena polimérica, e involucra una isomerización del sitio activo dentro de la
cadena polimérica este proceso es conocido como Chain Walking[98-100] (Figura
25). Por lo tanto, dependiendo de la posición donde el centro activo se encuentre, es
donde se insertara el siguiente monómero.
En este tipo de catalizadores la eliminación por hidrogeno-β y la
isomerización (chain walking) compiten con el proceso de propagación de la cadena.
El grado de ramificación con este tipo de sistemas depende de variables como la
temperatura, presión y tipo de ligando. Por ejemplo, el grado de ramificación
aumenta con la temperatura de reacción y disminuye con un aumento en la presión y
al disminuir el volumen de los sustituyentes de los ligandos α-diiminas.
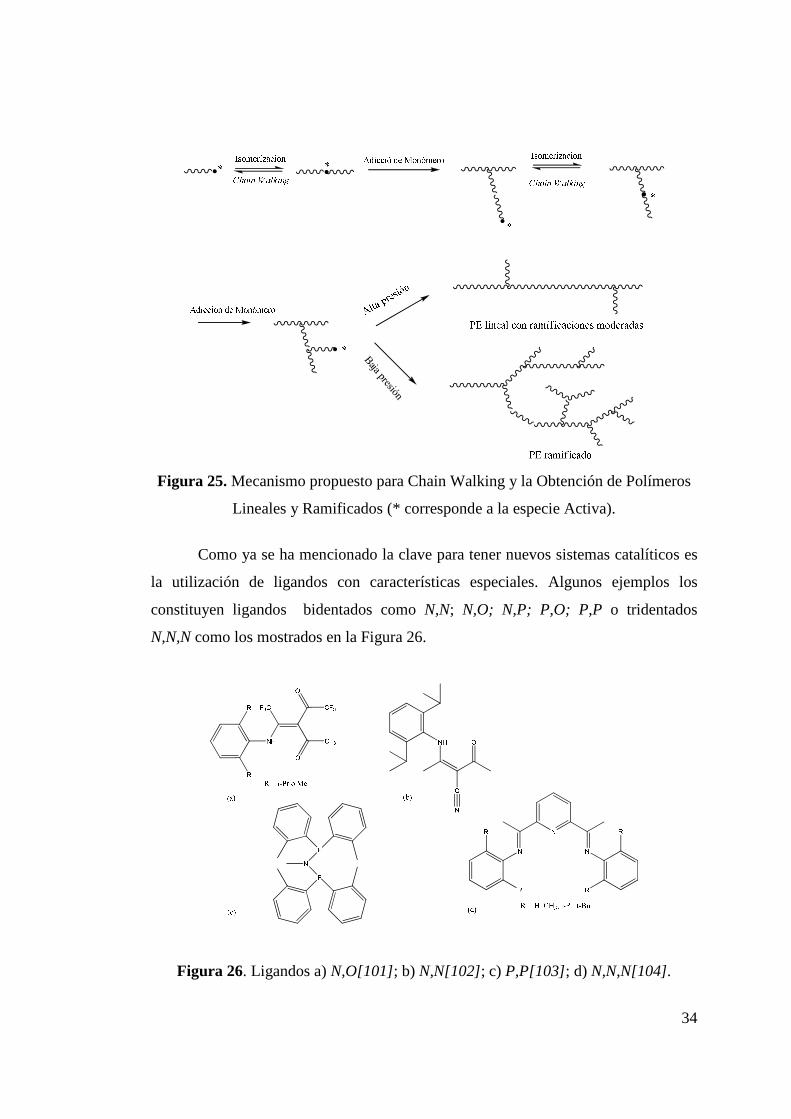
34
Baja
presión
Figura 25. Mecanismo propuesto para Chain Walking y la Obtención de Polímeros
Lineales y Ramificados (* corresponde a la especie Activa).
Como ya se ha mencionado la clave para tener nuevos sistemas catalíticos es
la utilización de ligandos con características especiales. Algunos ejemplos los
constituyen ligandos bidentados como N,N; N,O; N,P; P,O; P,P o tridentados
N,N,N como los mostrados en la Figura 26.
Figura 26. Ligandos a) N,O[101]; b) N,N[102]; c) P,P[103]; d) N,N,N[104].

35
Otro ejemplo lo constituyen los complejos de níquel con ligandos β-diiminas,
[ArN=C-(Me)CH2C(Me)=NAr]NiBr2 que en su coordinación al centro metálico
forma anillos de 6 miembros. Estos sistemas, si bien poseen los mismos sustituyentes
(Ar) que los sistemas α-diminicos ([ArN=C-(Me)C(Me)=NAr]NiBr2), la actividad
catalítica es muy diferente, siendo mayor en el caso de anillos de cinco miembros. El
polímero obtenido también difiere notablemente, aquellos obtenidos de sistemas
basados en β-diiminas, son mas cristalinos (más lineal) y de mayores pesos
moleculares. Estas diferencias pueden estar asociadas con centros metálicos más
protegidos, debido a la disposición que adoptan en una configuración hexa-
coordinada del ligando al centro metálico[105].
Las variedades de ligandos que pueden ser sintetizados y utilizados para la
formación de complejos de níquel (II) son muy numerosas. En el 2005, G.
Coates[106] y colaboradores reportaron un nuevo complejo α-diminico (N,N) que en
su estructura posee centros quirales, Figura 27. Este complejo en presencia de MAO
es activo para la polimerización de polipropileno. Adicionalmente con este sistema,
controlando la temperatura de reacción es posible acceder a propilenos
isoespecificos. A bajas temperatura se obtiene PP isotáctico cuya propiedad se pierde
con el incremento de ésta. De este modo mediante el control de la temperatura de
reacción se accede a polipropilenos en bloque (atáctico-isotáctico), con
características muy particulares.

36
Figura 27. Polimerización de Propileno con Catalizador de Ni(II) de Simetría C2 y
Centros quirales.
Como hemos visto las características de los ligandos (estéricos y electrónico)
y consecutivamente su interacción con los centros metálicos determinan la actividad
y características de los polímeros obtenidos. Recientemente se han reportado
complejos de Fe(II) y Co(II) con ligandos piridiniminicos, en los cuales también la
variación de los sustituyentes en las posiciones orto y para de los anillos de las
iminas, influencian la reactividad del centro metálico y definen las propiedades del
polímero obtenido[107]. Por ejemplo, la introducción de sustituyentes halogenados
pueden variar la densidad de carga del centro metálico haciéndolo más positivo y por
consiguiente mas reactivo hacia la polimerización de etileno.
En el caso de níquel también se han informado sistemas catalíticos en los
cuales se ha comprobado que la introducción de diferentes tipos de halógenos en la
estructura del ligando, provocan variaciones en la reactividad del sistema[108-113].
Por ejemplo el cambio de un protón a un CH3 en la posición para del anillo de las α-
diiminas, provoca un decrecimiento en la actividad catalítica y peso molecular del
polímero obtenido (desde 3610 a 3300 KgPE/mol Ni*h, Mw de 386 a 337 Kg/mol).
Cuando se introduce en esta posición un grupo atractor de electrones (Cl o Br) se ha
observado que aumenta tanto la actividad catalítica y en el peso molecular[114].

37
El hecho que la variación en la estructura de los ligandos produzca enormes
cambios en las actividades catalíticas y en las propiedades de los polímeros ha
significado un auge en la síntesis de complejos que contengan en su estructura
heteroátomos o grupos funcionales. Un ejemplo es mostrado en la Figura 28.
Figura 28. Complejos Post-metalocénicos con Heteroátomos en su
Estructura.
La introducción de este tipo de ligando con donores axiales, es una nueva
estrategia del diseño de catalizadores para la polimerización de etileno. La
introducción de la piridina produce una interacción axial con el metal y
eventualmente también con el cocatalizador, de modo que juegan un rol importante
para impedir la terminación de cadena y así producir polietileno de alto peso
molecular. Este efecto se ve tanto en los sistemas de níquel como de paladio, donde
se obtiene polímeros de alto peso molecular (108 Kg/mol para Ni y 880 Kg/mol para
Pd) y altamente lineales (137 ºC para Ni y 134 ºC para Pd)[115].
Otra estrategia basada en la modificación de ligandos incluye la
incorporación de grupos en la posición beta, como β-diazo, β-piridil, β-halógeno y β-
ciano, entre otros, a través de los cuales se puede variar la reactividad del centro
metálico[116-118]. La introducción de estas funcionalidades tiene como objeto
permitir por ejemplo un segundo tipo de interacción (del tipo exocíclica), sobre el
complejo. Mediante estas funcionalidades es posible cambiar la densidad electrónica
del centro metálico o más importante aún, permitir la heterogenización de estos
complejos sobre sólidos inorgánicos y de este modo incrementar las opciones de
estos sistemas hacia su aplicación en sistemas industriales. Un ejemplo lo
constituyen el complejo de níquel con ligando voluminosos α-ceto-β-diimina con

38
sustituyentes 2,6-diisopropilo, el cual posee un carbonilo como funcionalidad
adicional del tipo exocíclica. Esta funcionalidad permite una interacción con los
ácidos de Lewis que se utilizan para activar el sistema catalítico, incrementando la
electrofilicidad del centro metálico. Esto hace que la actividad catalítica se vea
incrementada desde 8 kg PE/mol Ni x h (sistema sin carbonilo) hasta 5800 kg
PE/mol Ni x h (sistema con carbonilo)[119].
Otros sistemas que han logrado gran impacto en el diseño de sistemas a base
de metales de transición avanzados, particularmente por su eficiencia en
copolimerización de etileno y monómeros polares, son aquellos en que la especie
activa es neutra. La principal diferencia entre los dos tipos de sistemas, catiónicos y
neutros se encuentra en el tipo de activación. Como ya fue mostrado, en el caso de
complejos como los de Brookhart donde el sistema es dihalogenado se necesita de la
presencia de un cocatalizador que cumpla la función de alquilar y dejar la vacancia
libre donde se insertara la olefina. En el caso de los complejos neutros de níquel (II),
algunos no requieren cocatalizador y son activados por la presencia del monómero
que desplaza un ligando neutro mono-coordinado al centro metálico (“single
component”)[120-122]. Un ejemplo es el complejo α-iminocarboxamida-Ni(η1-
CH2Ph)(PMe3) el cual es activo en presencia de Ni(COD)2 siendo capaz de producir
polietilenos con distribución de peso molecular ~ 1.0 [121].
Figura 29. Complejos Post-metalocenicos de Níquel Neutros Capaz de
Polimerizar Polietileno.

39
En este tipo de complejos, las funcionalidades adicionales, puede ser útil
también para activar el centro metálico. Por ejemplo Bazán[123] y colaboradores
mostraron que la adición de B(C6F5)3 o Al-(C6F5)3 a sistemas de níquel neutros
producen complejos zwitterionicos, los cuales son activos para dimerizar,
oligomerizar y polimerizar etileno (Figura 30, a). Más recientemente Rojas[102] y
colaboradores informaron que la adición de B(C6F5)3 a complejos que presentan una
funcionalidad adicional ciano, pueden ser activados a través de este grupo con ácidos
de Lewis (activación remota) provocando una redistribución de la densidad
electrónica removiéndola desde el níquel generando así una espacie activa para la
polimerización de etileno. (Figura 30, b)
Figura 30. Activación de Sistemas Post-metalocénicos.
En resumen se puede decir que los diferentes tipos de sistemas catalíticos de
níquel pueden ser catiónica o neutros, los cuales tienen metodologías de activación
diferentes. En el caso de los complejos catiónicos se requiere de un cocatalizador
para formar la especie activa (enlace metal-carbono y vacancia). En cambio en el
sistema neutro solo se requiere de un agente que desplace a un ligante neutro para
que se pueda introducir la olefina. Sin embargo en ambos tipos de sistemas puede
existir la presencia de grupos funcionales adicionales en el ligando, que permita la
activación remota del centro metálico, permitiendo que la actividad catalítica se vea
incrementada[122, 124].

40
1.5 Hipótesis
Se espera que utilizando un centro metálico de conocida reactividad, como
Zr, Ti y Ni, con nuevos ligandos del tipo (N,O) y (N,N) generen compuestos de
coordinación activos para la polimerización de olefinas. Las características
especificas del ligando y la presencia de grupos funcionales permitirán modular la
densidad electrónica y consecuentemente la reactividad del metal hacia la
polimerización.
1.6 Objetivos
1.6.1. Objetivo General
• Sintetizar complejos de coordinación con metales de transición, como Ni, Ti
y Zr, con diferentes tipos de ligandos, capaces de polimerizar etileno en
sistemas homogéneos.
1.6.2. Objetivos Específicos
• Sintetizar nuevos ligandos bidentados (N,O y N,N) con funcionalidad
adicional X = -CN, Piridina entre otras.
• Preparar y caracterizar los complejos de formula general X-(A,N)NiBr2 y (X-
(A,N))nMCl2 (M = Ti y Zr) (n = 1 o 2).

41
• Estudiar el comportamiento de estos complejos como catalizadores en
reacciones de polimerización de etileno con o sin la presencia de
cocatalizador (ácido de Lewis).
• Caracterizar los polímeros obtenidos determinando su peso molecular y
temperatura de fusión.

42
CAPÍTULO 2 PARTE EXPERIMENTAL
2.1. Método General de Síntesis.
Para la síntesis de los ligandos se utilizó técnicas Schlenk, solventes anhidros
y atmósfera de nitrógeno. La síntesis de los complejos se efectuó en una cámara seca
MBRAUN Glove Box, single unit Systems modelo 07-283, bajo atmósfera inerte de
nitrógeno, empleando solventes anhidros (Figura 31).
Figura 31. Cámara Seca MBRAUN
El secado y purificado de los solventes fue el mismo en todos los casos. Este
consiste en colocar el solvente a purificar en un balón de tres bocas con sodio
metálico y benzofenona.
� Benzofenona, procedencia Aldrich (Milwaukee, WI, USA).
� Sodio Metálico, procedencia Fluka Chemie AG (Buchs, Switzerland), grado
de pureza PA.
La mezcla se coloca bajo reflujo en ambiente de nitrógeno, por un tiempo de
2 horas mínimo, cuando el solvente se torna de color azul oscuro se encuentra listo
para la destilación y su utilización. Este procedimiento se realizó en los solventes:
� Tolueno

43
� Tetrahidrofurano (THF)
� Hexano
� Pentano
En el caso de Diclorometano el secado es con P2O5, destilandoce luego al vacio. Para la síntesis de los ligandos y complejos se utilizó reactivos de partida de
procedencia comercial entre los que se encuentran:
� 2,4-pentanodiona (99%, MW 100,12, Aldrich)
� R-(+)-α-metilbencilamina (ChiraSelect > 99,0%, MW 121,18, Aldrich)
� 2,6-diisopropilanilina (90% tech., MW 177,29, Aldrich)
� 2,6-dimetilanilina (99%, MW 121,18, Aldrich)
� 2,3,4,5,6-pentafluoroanilina (99%, MW 183,08, Aldrich)
� 4-aminopiridina (99 %, MW 94,12, Aldrich)
� ácido p-toluensulfónico (98,5%, MW 190,22, Aldrich)
� n-Butillitio (1,6M en hexano, MW 64,06, Aldrich)
� p-toluensulfonilciano (95% tech., MW 181,22, Aldrich)
� piridina (99%, MW 79,1, Aldrich)
� pentacloruro de fosforo (95% tech., MW 208,24, Aldrich)
� trietilamina (99% tech., MW101,19, Riedel-deHaen)
� tetracloruro de circonio (ZrCl4) (99,5%, Aldrich)
� tetracloruro de titanio (TiCl4) (1.0 M En diclorometano, MW 189,71, Aldrich)
� Precursor de níquel [{O(CH2CH2OCH3)2}NiBr 2] (MW 352,71, Aldrich)
2.2. Síntesis de Ligandos y Complejos
En las siguientes dos tablas se muestran en resumen los ligandos (Tabla 1) y
complejos (Tabla 2) sintetizados en esta tesis. La ruta de obtención de cada uno de
ellos se muestra en detalle en el Anexo 1. En este anexo también podrán ser
encontrados los análisis de espectroscopia de resonancia magnética nuclear (1H, 13C, 19F), de infrarrojo, espectros de masa y análisis de difracción de Rayos-X, realizados
a cada uno de estos productos que corroboran su obtención.

44
Tabla 1. Resumen de Ligandos Sintetizados. Ligando
L1 4-(1-fenil-etilamino)-pent-3-en-2ona.
L2 4-(2,6-diisopropilfenilimino)-pentano-2ona.
L3 2-(2,6-diisopropilfenilamina)-4-(2,6-diisopropilfenilimina)-pent-2-eno.
L4 5-(1-feniletilimino)-2,2,6,6-tetrametilhepta-3-ona
L5 2-acetil-3-(1-fenil-etilimino)but-2-enenitrilo
L6 2-acetil-3-(2,6-diisopropilfenilimino)-but-2-enenitrilo.
L7 3-(2,6-diisopropilfenilamino)-2-1-(2,6-diisopropilfenilimino)etil)but-2-enenitrilo.
L8a N’-(2,6-diisopropilfenil)-N-[1-(2,6-diisopropilfenilimino)-etil]-N-(2,6-dimetil)-acetamidina
L8b N-(2,6-diisopropilfenil)-N’-[1-(2,6-diisopropilfenilimino)-etil]-N-(2,6-dimetil)-acetamidina
L9 N,N´-bis-(2,6-dimeil-fenil)-N-[1-(2,6-dimetil-fenilimino)etil)]-acetamidina.
L10 N-(2,6-diisopropil-fenil)-N-[1-(2,6-diisopropil-fenilimino)-etil]-N-(2,3,4,5,6-petafluoro-fenil)-acetamidina
L11 N-1-(2,6-diisopropilfenil)-N´-(2,3,4,5,6-perfluorofenil)acetamidina
L12 N-(2,6-diisopropilfenil)-N-[1-(2,6-diisopropylfenilimino)-etil]-N´(2,3,4,5,6-pentafluorofenil)acetamidina
L13 N-(2,6-diisopopilfenil)-N-1,(2,6-diisopropilfenilimino)-N-(piridin-4-il) acetamidina
L14 N-(1-(4-iminopiridin-1(4H)-yl)etilideno)-2,6-diisopropylanilina
Tabla 2. Resumen de Complejos Sintetizados.
Complejo
1 [ZrCl4{Bis-[4-(1-fenil-etilamino)-pent-3-en-2ona]}] 2 [ZrCl4{Bis-[2-acetil-3-(1-fenil-etilimino)-but-2-
enenitrilo]}]. 3 [TiCl4{Bis-[4-(1-fenil-etilamino)-pent-3-en-2ona]}] 4 [TiCl4{Bis-[2-acetil-3-(1-fenil-etilimino)but-2-
enenitrilo]}] 5 ZrCl4[Bis(3-(2,6-diisopropilfenilimino)-2-1-(2,6-
diisopropilfenilimino) etil)but-2-enenitrilo)] 6 [ZrCl2{Tri[2-acetil-3-(2,6-diisopropilfenilimino)-but-2-
enenitilo]}] 7 [Zr(NMe2)2{Bis(2-acetil-3-(2,6-diisopropilfenilimino)-but-
2-enenitrilo)}]

45
10 [NiBr2{N’-(2,6-diisopropil-fenil)-N-[1-(2,6-diisopropil-fenilimino)-etil]-N-(2,6-dimetil)-acetamidina}]
11 [NiBr2{N-(2,6-diisopropil-fenil)-N’-[1-(2,6-diisopropil-fenilimino)-etil]-N’-(2,6-dimetil)-acetamidina}]
12 [NiBr2{N,N´-bis-(2,6-dimetil-fenil)-N-[1-(2,6-dimetil-fenilimino)etil)]-acetamidina}]
13 [NiBr2{N-1-(2,6-diisopropilfenil)-N´-(2,3,4,5,6-perfluorofenil)acetamidina}].
14 [NiBr2{N-(2,6-diisopropil-fenil)-N-[1-(2,6-diisopropil-fenilimino)-etil]-N-(2,3,4,5,6-petafluoro-fenil)-acetamidina}].
15 [NiBr2{N-(2,6-diisopropilfenil)-N-[1-(2,6-diisopropylfenilimino)-etil]-N´(2,3,4,5,6-pentafluorofenil)acetamidina}].
16 [NiBr2{N-(1-(4-iminopiridin-1(4H)-yl)etilideno)-2,6-diisopropylanilina}].
17 [NiBr2{N-(2,6-diisopopilfenil)-N-1,(2,6-diisopropilfenilimino)-N-(piridin-4-il) acetamidina}].

46
2.3. Caracterización de Ligandos y Complejos.
Los espectros de resonancia magnética nuclear (RMN) se realizaron en un
espectrómetro BRUKER, modelo AVANCE AC-400, utilizando cloroformo
(CDCl3), benceno y THF como solventes, todos del tipo deuterado, los cuales
contienen TMS (1%) como sustancia de referencia interna para protones y carbono.
Los desplazamientos químicos se expresan en partes por millón (ppm) respecto al
TMS y las constantes de acoplamiento (J) en Hertz (Hz). Para la multiplicidad de las
señales se emplean las siguientes abreviaturas: singulete (s), doblete (d), triplete (t),
cuadruplete (c), pentuplete (p) y multiplete (m).
Los espectros de infrarrojo (FT-IR) fueron realizados en un espectrofotómetro
con transformada de Fourier Bruker Vector-22, utilizando KBr como dispersante y
fueron registrados en el rango 4000 – 600 cm-1.
Los espectros de masa fueron realizados en un espectrómetro de masas de
tipo electrospray-trampa iónica ESQUIRE 4000 ESI-IT (Bruker Daltonics). La
metodología de inyección fue en forma directa c.a. 200 µL de la solución preparada
utilizando una bomba de jeringa (Cole-Parmer, IL-USA) a un flujo de 2,5 µL/min. La
nebulización se realizó utilizando los siguientes parámetros: temperatura de
nebulización, 300 ºC; presión de nebulización, 10 psi; flujo de gas nebulizador, 5
L/min. Se examinó la adquisición de espectros MS en polaridad positiva y negativa.
Los análisis de difracción de Rayos X fueron realizados en un difractor de
plataforma Bruker CCD. Se empleó el programa SMART para recolección de datos y
determinar los parámetros de celda unitaria. Los datos obtenidos fueron procesados
utilizando los programas SAINT y SADABS para obtener el archivo de datos de
reflexiones. Cálculos posteriores fueron realizados empleando el programa
SHELXTL. La estructura fue resuelta empleando métodos directos, y refinada en F2
empleando métodos de mínimos cuadrados de matriz completa.

47
2.4 Polimerización de Etileno.
Los reactivos utilizados en la etapa de polimerización de etileno son:
-Solventes: Grado de pureza técnico:
� Acetona
� Acido Clorhídrico
� Alcohol Etílico
� Alcohol Metílico
Grado de pureza PA:
� Tolueno, procedencia J. T. Baker (Xalostoc, México) previamente secado.
-Gases
� Etileno, procedencia Petroquim S.A. (Chile), grado petroquímico.
� Nitrógeno, procedencia Aga S.A (Chile), grado de purificación 99,95 %.
-Cocatalizador
� Metilaluminoxano, procedencia Witco Gmbh (Bergkamem, Germany), grado
de pureza PA, concentración 1,66 mol/L (10 %) de aluminio en solución de tolueno.
Las polimerizaciones se realizaron en un reactor autoclave marca Parr, de 600
mL con tolueno como solvente. Este cuenta con un manómetro, una termocupla y un
sistema de agitación mecánica. Además, se encuentra conectado a un baño
termorregulado, con el cual se controla la temperatura de reacción. En la Figura 32,
se muestra un diagrama de la línea de polimerización de etileno.

48
Figura 32. Diagrama y Línea de Polimerización de Etileno.
Antes de cada polimerización, el material utilizado en la reacción, se
mantiene en la estufa a 100 ºC, aproximadamente por 2 horas. Tanto el catalizador
como el cocatalizador MAO son sacados de la cámara de atmósfera inerte como una
solución en tolueno. Estos son adicionados al reactor, donde se controla la
temperatura de reacción, la presión y la agitación mecánica.
En el caso de complejos de níquel las reacciones se realizan en 100 mL de
tolueno a temperaturas entre 40 – 70 ºC, presión entre 2-4 bar y agitación constante
de 1000 rpm. Para los complejos de circonio y titanio se utilizan 240 ml de tolueno y
las temperaturas entre 45 y 70 ºC a 2 bar de presión. Para ambos casos el tiempo de
reacción es de 30 min con una relación Al/M de 2000 (M = Ni, Zr, Ti).
Las reacciones se detienen cerrando la válvula de etileno y desactivando el
catalizador y exceso de MAO con una mezcla Metanol/HCl (10 %). El polímero
obtenido es filtrado, lavado y secado hasta peso constante.

49
2.5 Caracterización de los Polímeros.
La temperatura de fusión se determinó por calorimetría diferencial de barrido
(DSC) utilizando un calorímetro DSC290 Modulated TA Instruments, a una
velocidad de calentamiento de 10 ºC/min. Los datos colectados fueron tomados del
segundo ciclo.
El peso molecular (Mw) y el indice de polidispersion de los polímeros se
determinaron por cromatografia de permeacion de geles en un cromatografo Waters
Alliance GPC2000 provisto con un detector de indice de refracción diferencial
(model 150C). Se utilizaron un conjunto de 3 columnas Styragel HT ( HT6E, HT5 y
HT3) que permite determinar pesos entre 500 y 1x107 g/mol. Como fase movil se
utilizao 2,2,4-triclorobenceno (TCB), al cual se le adiciono 2,6-di-t-butil-4-
metilfenol (BHT) como antioxidante (0,025%) .La concentracion de las muestras fue
de 2.5 mg/mL y el flujo de analisis fue 1 mL/min la temperatura fue de 135 ºC. La
calibracion universal fue realizada con estandares de poliestireno con estrecha
distribucion de pesos moleculares.
� 1,2,4 triclorobenceno, procedencia Aldrich, grado pureza PA.
Para los polímeros obtenidos con los complejos de circonio, el peso
molecular fue determinado mediante la viscosidad intrínseca [η]. Ésta fue
determinada usando un viscosímetro capilar tipo Ubbelohde. Las muestras fueron
disueltas en decalina aditivada con BHT 0,1 %, y medidas a 135 ºC de temperatura.
� Decalina, procedencia Fluka Chemie AG (Buchs, Switzerland), grado de
pureza PA, se aditiva con antioxidante BHT al 0,1%.
El valor de [η] es estimado usando el método de un solo punto de
concentración en la ecuación de Martin corregida. Para ello se utiliza una única
solución de polímero con una concentración de entre 0,18 - 0,20 g/dL.
(t-k/t)/(to – k/to) = c·[η]·exp(k5[η] c) +1 Ecuación 2
donde,

50
k5 : Constante del solvente, 0,32 para la decalina
k : B/A (constante de calibración de viscosímetro)
to : tiempo de escurrimiento del solvente
t : tiempo de escurrimiento de solución de polímero
c : concentración de solución, calculada mediante la Ecuación 3.
c = (M·100)/(V·1,107) Ecuación 3
donde,
M : masa de la muestra
V : volumen de decalina a 20 ºC
1,107 : factor de corrección de volumen.
El viscosímetro fue calibrado con agua destilada, a varias temperaturas,
obteniéndose así las constantes A y B. La viscosidad intrínseca está relacionada con
el peso molecular por la ecuación de Kuhn- Mark-Houwink-Sakurada Ecuación 4.
[η] = K·Mvα Ecuación 4.
donde:
Mv : peso molecular viscosimétrico medio
K y α : constantes empíricas que dependen de cada sistema particular; polímero,
solvente y temperatura. Estas se determinan a partir de una representación doble
logarítmica de la viscosidad intrínseca frente al peso molecular.
La determinación de fracción oligomérica se realizó mediante cromatografía
de gases acoplada a espectrometría de masas. El cromatógrafo consta de una
columna Angilent, HP-5MS, 30m x 0,250 mm, el carrier posee un flujo constante: 1
ml/min inyectado por metodología splitless. El análisis de masa se realiza con un
espectrómetro Modelo MAT 95XP, Thermo Finnigan. Analizador de doble enfoque,
sector magnético y eléctrico. La ionización es por impacto de electrones, EI, 70 eV.

51
CAPÍTULO 3
RESULTADOS Y DISCUSIÓN
En este capítulo se muestran los resultados obtenidos a lo largo del trabajo de
tesis y se discuten los aspectos más importantes acerca de síntesis y caracterización
de ligandos, su utilización en la preparación de complejos con metales de transición
(Ti, Zr y Ni) y el estudio de reactividad de estos en reacciones de polimerización de
etileno.
Los resultados se han dividido en cuatro partes: la primera corresponde a la
síntesis y caracterización de ligandos del tipo: β-cetoiminas, β-diiminas, N-
(imidoilamidinicos) y acetamidinicos. En la segunda parte se describe la síntesis y
caracterización de complejos de circonio y titanio. La tercera parte corresponde a la
síntesis y caracterización de complejos de níquel con los ligandos anteriormente
descritos y por último la cuarta parte resume los estudios de reactividad de algunos
de los complejos sintetizados, en reacciones de polimerización de etileno.
3.1 Síntesis y Caracterización de Ligandos
La síntesis y caracterización de ligandos se divide en tres partes: i) β-
cetoiminas y β-diiminas, ii) β-cetoiminas y β-diiminas sustituidas y iii) N-
(imidoilamidinicos) y acetamidinicos.
3.1.1. Ligandos β-cetoiminas y β-diiminas
Las rutas de síntesis para la generación de grupos iminas consisten en la
condensación de una anilina en una cetona de acuerdo al siguiente mecanismo
(Figura 33).
OO
+NH
R
HNO
R
Benceno H+
-H2O
Figura 33. Síntesis de β-cetoiminas.

52
Estas reacciones son catalizada por ácido (fuerte como HCl o débiles por
ejemplo acido p-toluensulfónico) utilizando benceno como solvente o etanol para
aquellas donde se requiere HCl, en un sistema Dean Starck para extraer el agua
formada en la reacción y así desplazar el equilibrio hacia la formación de producto
Siguiendo este procedimiento fueron preparados los siguientes compuestos:
� 4-(1-fenil-etilamino)-pent-3-en-2ona. L 1
� 4-(2,6-diisopropilfenilimino)-pentano-2ona. L 2
� 2-(2,6-diisopropilfenilamina)-4-(2,6-diisopropilfenilimina)-pent-2-eno. L 3
� 5-(1-feniletilimino)-2,2,6,6-tetrametilhepta-3-ona. L 4
4-(1-fenil-etilamino)-pent-3-en-2ona (L 1), se obtiene por la reacción de 3,3
dimetil-pentano-2,4-diona y (R)-(+)-α-metilbencilamina en una razón equimolar,
utilizando benceno como solvente y ácido p-toluensulfónico como catalizador
(Figura 34)
O O
+H2N
*
O HNBenceno, H+
*
(L 1)-H2O
Figura 34. Síntesis de 4-(1-fenil-etilamino)-pent-3-en-2ona (L 1).
El compuesto L 1 se obtiene después de purificación mediante cromatografía
(éter/hexano) como un aceite amarillo en un rendimiento de 82 %. El espectro de 1H-
RMN (Figura 35) mostró la formación de un único isómero, donde la multiplicidad
(pentuplete, 4,96 ppm) del protón enlazado al carbono quiral indica que existe un
acoplamiento con los protones del CH3 y con el protón de la amina. Además en la
Figura 35 se puede apreciar dos singletes en 2,01 y 1,75 ppm correspondientes a las
CH3 terminales del grupo cetónico (CH3-CO) y imínico (CH3CN) respectivamente.
Estos aparecen a campo más bajo que los protones del CH3 unido al carbono quiral
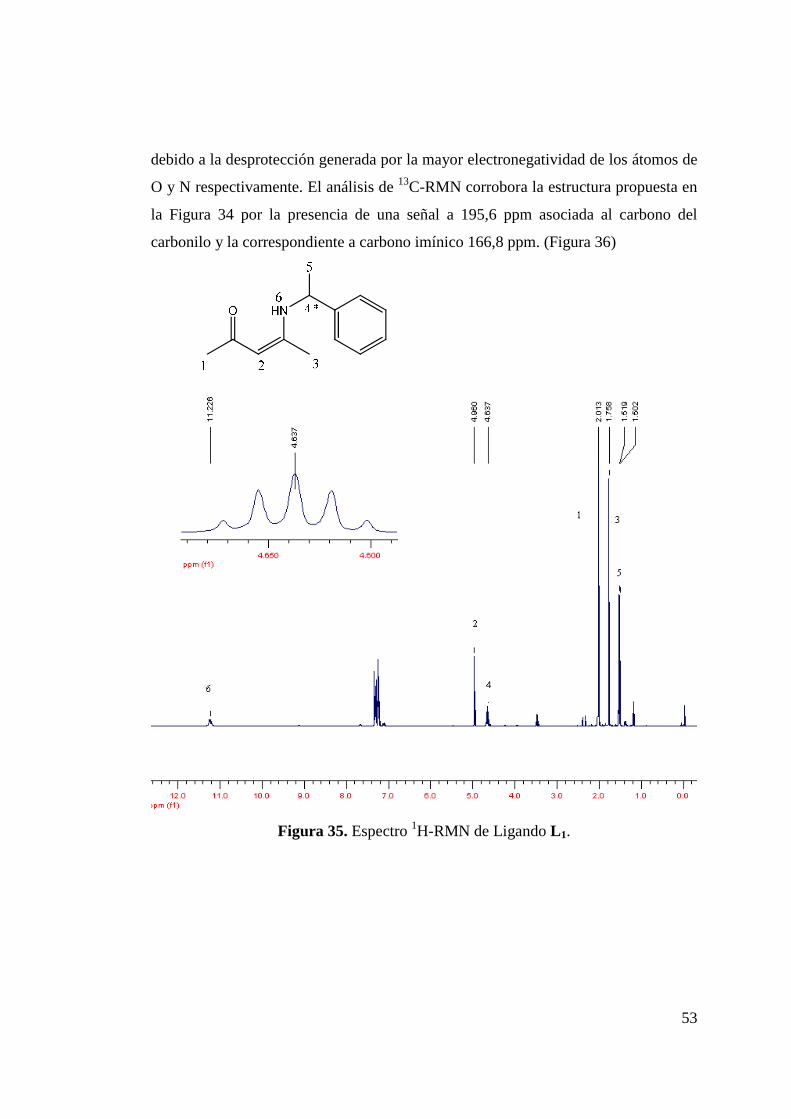
53
debido a la desprotección generada por la mayor electronegatividad de los átomos de
O y N respectivamente. El análisis de 13C-RMN corrobora la estructura propuesta en
la Figura 34 por la presencia de una señal a 195,6 ppm asociada al carbono del
carbonilo y la correspondiente a carbono imínico 166,8 ppm. (Figura 36)
Figura 35. Espectro 1H-RMN de Ligando L 1.
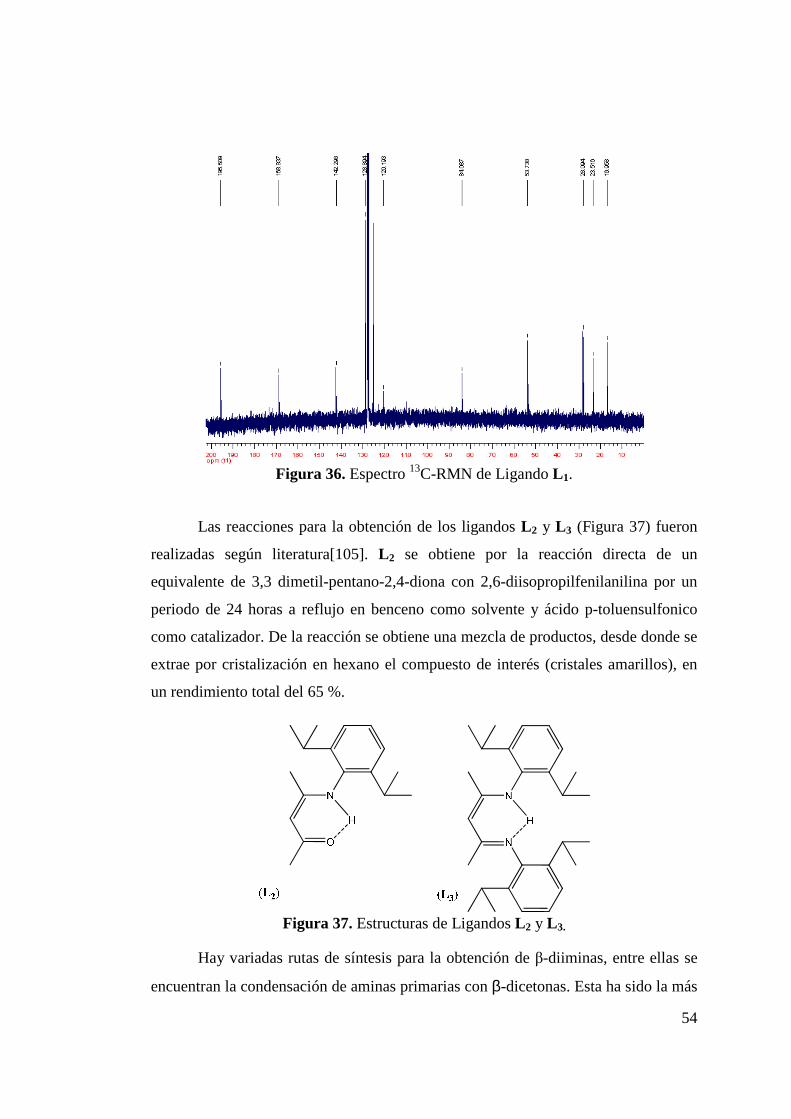
54
Figura 36. Espectro 13C-RMN de Ligando L 1.
Las reacciones para la obtención de los ligandos L 2 y L 3 (Figura 37) fueron
realizadas según literatura[105]. L 2 se obtiene por la reacción directa de un
equivalente de 3,3 dimetil-pentano-2,4-diona con 2,6-diisopropilfenilanilina por un
periodo de 24 horas a reflujo en benceno como solvente y ácido p-toluensulfonico
como catalizador. De la reacción se obtiene una mezcla de productos, desde donde se
extrae por cristalización en hexano el compuesto de interés (cristales amarillos), en
un rendimiento total del 65 %.
Figura 37. Estructuras de Ligandos L 2 y L 3.
Hay variadas rutas de síntesis para la obtención de β-diiminas, entre ellas se
encuentran la condensación de aminas primarias con β-dicetonas. Esta ha sido la más

55
utilizada debido a las ventajas que presenta; donde se destacan, los buenos
rendimientos, la utilización de reactivos convenientes económicamente o fáciles de
sintetizar en grandes cantidades. La conversión desde β-dicetonas a β-diimina
mediante condensación puede realizarse por etapas; en la primera se obtiene la
enaminocetona y luego por una segunda condensación se obtiene la β-diiminas[125].
Este procedimiento es adecuado especialmente en la preparación de β-diimina
asimétrica y donde ambos materiales de partida contienen sustituyentes poco
voluminosos. Para sistemas más voluminosos existen rutas de síntesis como las
mostradas en la introducción[42].
La síntesis de L 3, se lleva a cabo por un camino distinto al utilizado con L 2,
esto debido a que la segunda conversión del grupo ceto a imina resulta ser menos
favorables por factores estéricos. Esta reacción se lleva a cabo utilizando como
catalizador ácido clorhídrico concentrado, en relación equimolar, etanol como
solvente, y reflujo por un periodo de 72 h [105]. El producto es obtenido después de
recristalización en metanol en un 71 % de rendimiento, la caracterización se realiza
mediante 1H-RMN y 13C-RMN.
Con el propósito de estudiar ligandos del tipo N,O, que presenten un mayor
impedimento estérico en torno a los átomos nitrógeno y oxigeno, es que se diseñó la
síntesis del ligando 5-(1-feniletilimino)-2,2,6,6-tetrametilhepta-3-ona (L 4). Siguiendo
la metodología utilizada en la Figura 3.1.1.1, esta nueva reacción no ocurre. Es así
que diferentes rutas de síntesis fueron probadas: reacción catalizada con ácido
clorhídrico[126], formación de iminas catalizadas por TiCl4 [127-130], sin obtenerse
el ligando deseado. Este ligando finalmente fue obtenido utilizando microondas
como fuente de energía y arcilla (MK-10) como catalizador (Figura 38)

56
*
Figura 38. Síntesis de Ligando L4.
La mejor conversión (50 %), determinada cualitativamente mediante
cromatografía en capa fina, se obtiene después de 1 h de reacción. No obstante, la
purificación utilizando columna cromatográfica (hexano/éter) no resultó efectiva y
sólo se logra un rendimiento de 20 %. La caracterización por 1H-RMN muestra que
el producto obtenido corresponde a L 4. La existencia del pentuplete a 5,17 ppm
(Figura 39) indica una conectividad atómica similar a la mostrada previamente en
compuesto L 1.
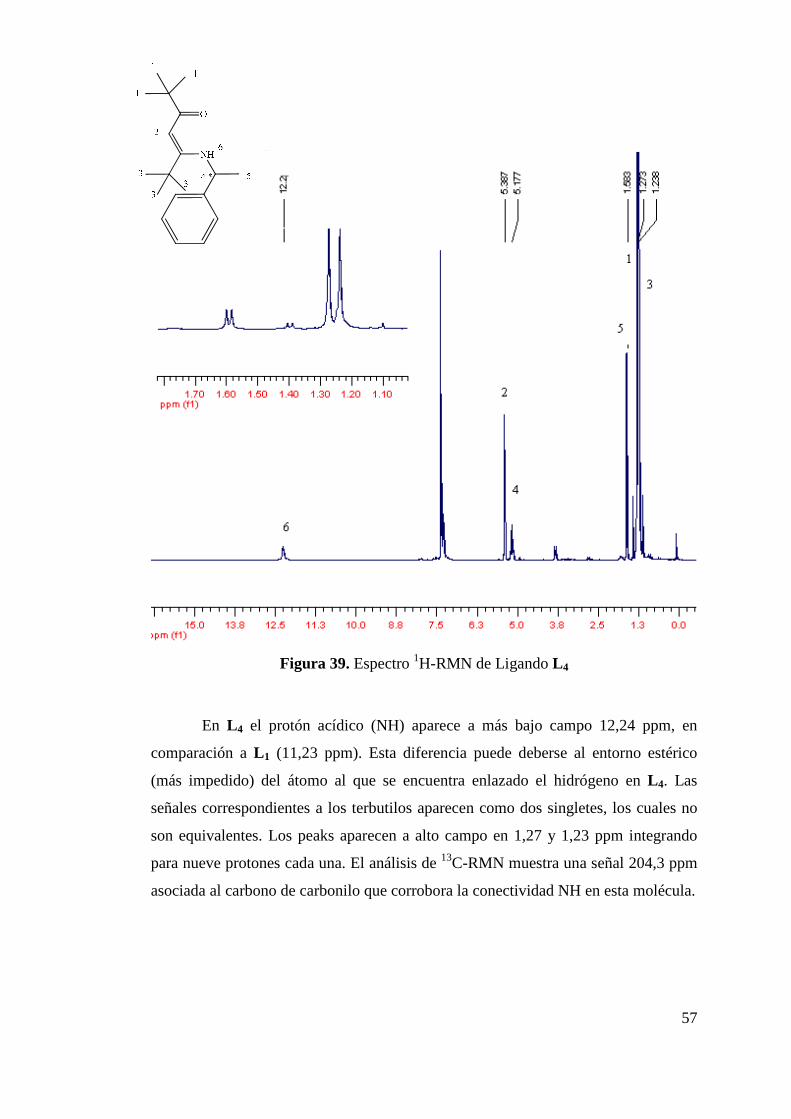
57
Figura 39. Espectro 1H-RMN de Ligando L 4
En L 4 el protón acídico (NH) aparece a más bajo campo 12,24 ppm, en
comparación a L 1 (11,23 ppm). Esta diferencia puede deberse al entorno estérico
(más impedido) del átomo al que se encuentra enlazado el hidrógeno en L 4. Las
señales correspondientes a los terbutilos aparecen como dos singletes, los cuales no
son equivalentes. Los peaks aparecen a alto campo en 1,27 y 1,23 ppm integrando
para nueve protones cada una. El análisis de 13C-RMN muestra una señal 204,3 ppm
asociada al carbono de carbonilo que corrobora la conectividad NH en esta molécula.

58
La sustitución de un hidrógeno en el carbono alfa de las β-cetoiminas y β-
diiminas, es una ruta conveniente por la cual se puede introducir una funcionalidad
adicional en este tipo de ligandos. Entre ellas se encuentran: anillos aromáticos,
carbonilos, cianos, nitrilos, entre otros. Estos sustituyentes pueden variar el entorno
estérico y electrónico de los nitrógenos y oxígenos del ligando, haciéndolos más o
menos reactivos en reacciones posteriores.
3.1.2. Ligandos β-cetoiminas y β-diiminas sustituidos.
En compuestos como L 2 y L 3, se realizaron reacciones de sustitución para
incorporar un grupo ciano (CN), este debido a: sus características básicas asociada al
par de electrones libres en el nitrógeno, su hibridación (sp) que puede ser parte de
una conjugación mayor en el ligando y también porque su posición espacial no
permite que este actué como un ligando tridentado.
Figura 40. Incorporación de Funcionalidad Ciano.
Como se muestra en la Figura 40, la incorporación del grupo CN en el
carbono beta imínico, fue realizada a través de una sustitución electrofílica. Esta se
realiza en dos etapas consecutivas i) desprotonación del ligando con n-butillitio,
formando la sal respectiva y ii) reacción insitu de sustitución electrofílica con p-
toluensulfonilciano.

59
Siguiendo este procedimiento fueron preparados los siguientes ligandos:
NH
O
CN
(L5)
N
O
H
N
N
HCNCN
(L6) (L7)
*
Figura 41. Ligandos β-cetoiminas y β-diimina con Funcionalidad Adicional.
� 2-acetil-3-(1-fenil-etilimino)-but-2-enenitrilo. L 5
� 2-acetil-3-(2,6-diisopropilfenilimino)-but-2-enenitrilo. L 6
� 3-(2,6-diisopropilfenilamino)-2-1-(2,6-diisopropilfenilimino)etil)-but-2-
enenitrilo. L 7
Los productos fueron caracterizados mediante 1H-RMN, 13C-RMN y FT-IR.
En el caso de L 5 el espectro 1H-RMN (Figura 42, Espectro rojo) muestra la
desaparición de la señal en 4,96 ppm correspondiente al singlete CH del ligando L 1
(Figura 42, Espectro azul) indicando la sustitución de éste, por el grupo CN.
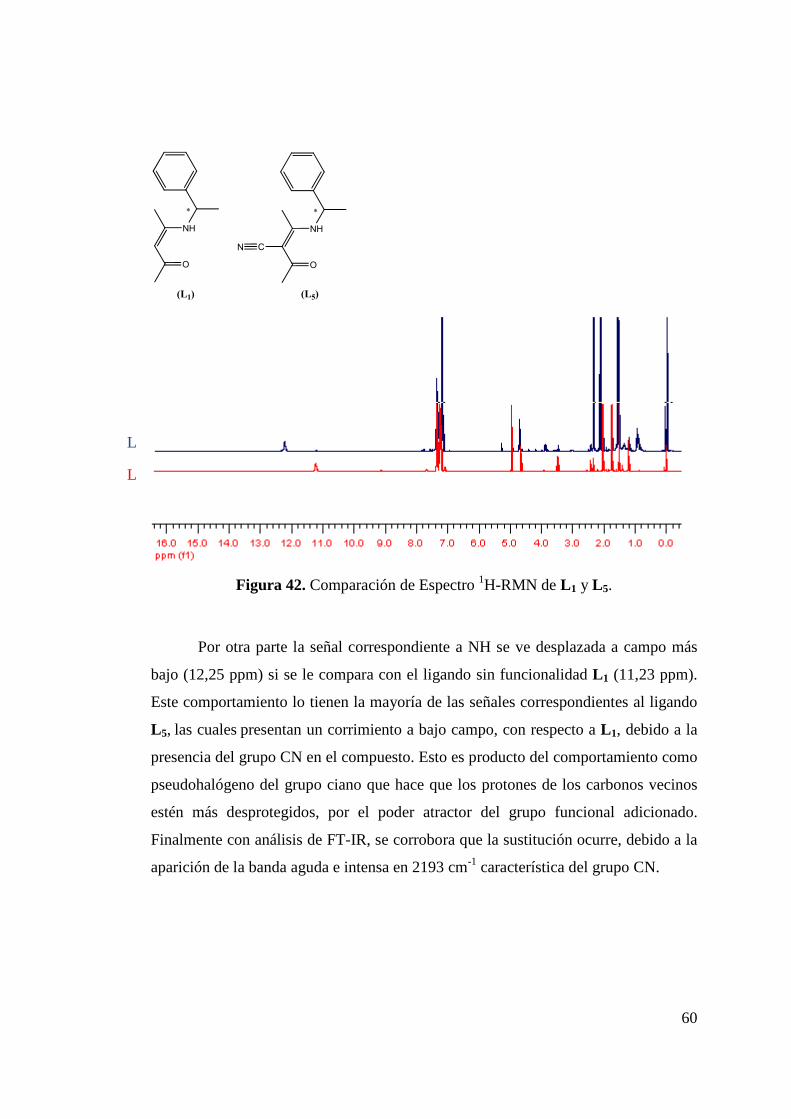
60
NH
O
(L1)
*
NH
O
CN
(L5)
*
Figura 42. Comparación de Espectro 1H-RMN de L 1 y L 5.
Por otra parte la señal correspondiente a NH se ve desplazada a campo más
bajo (12,25 ppm) si se le compara con el ligando sin funcionalidad L 1 (11,23 ppm).
Este comportamiento lo tienen la mayoría de las señales correspondientes al ligando
L 5, las cuales presentan un corrimiento a bajo campo, con respecto a L 1, debido a la
presencia del grupo CN en el compuesto. Esto es producto del comportamiento como
pseudohalógeno del grupo ciano que hace que los protones de los carbonos vecinos
estén más desprotegidos, por el poder atractor del grupo funcional adicionado.
Finalmente con análisis de FT-IR, se corrobora que la sustitución ocurre, debido a la
aparición de la banda aguda e intensa en 2193 cm-1 característica del grupo CN.
L
L
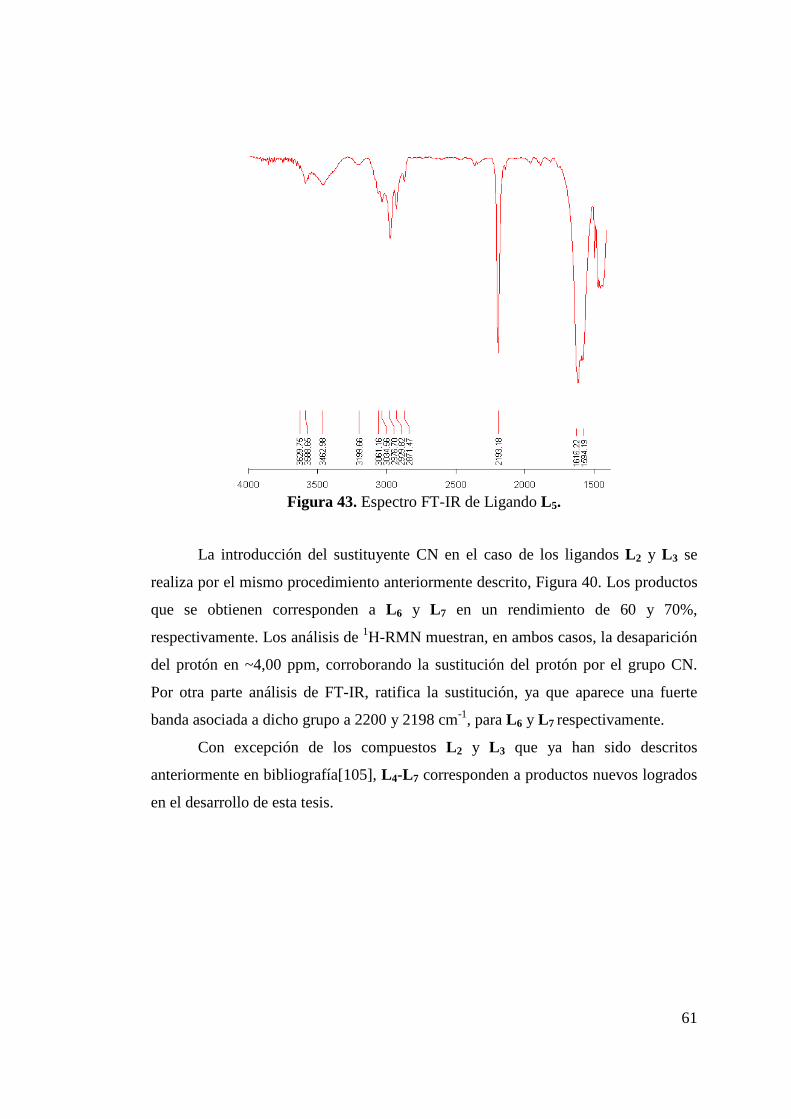
61
Figura 43. Espectro FT-IR de Ligando L 5.
La introducción del sustituyente CN en el caso de los ligandos L 2 y L 3 se
realiza por el mismo procedimiento anteriormente descrito, Figura 40. Los productos
que se obtienen corresponden a L 6 y L 7 en un rendimiento de 60 y 70%,
respectivamente. Los análisis de 1H-RMN muestran, en ambos casos, la desaparición
del protón en ~4,00 ppm, corroborando la sustitución del protón por el grupo CN.
Por otra parte análisis de FT-IR, ratifica la sustitución, ya que aparece una fuerte
banda asociada a dicho grupo a 2200 y 2198 cm-1, para L 6 y L 7 respectivamente.
Con excepción de los compuestos L 2 y L 3 que ya han sido descritos
anteriormente en bibliografía[105], L4-L 7 corresponden a productos nuevos logrados
en el desarrollo de esta tesis.

62
3.1.3 Ligandos N-(imidoilamidinas) y Acetamidinicos
La necesidad de nuevos ligandos polidentados que puedan ser usados en la
síntesis de eficientes y novedosos compuestos organometálicos, ha provocado un
enorme avance en el diseño y síntesis de ligandos.
En esta tesis, los esfuerzos se han centrado en el desarrollo de nuevos
ligandos bidentados (N,N) con sustituyentes voluminosos en torno de las iminas y
que contengan sustituyentes en la posición α de éstas. Este tipo de ligandos es
conocido como N-(imidoilamidina).
El método general de síntesis se muestra en la Figura 44 y consiste en la
reacción del precursor N-(2,6-diisopropilfenil)acetimidoilo y una anilina, en
presencia de trietilamina (NEt3)[131].
Figura 44. Síntesis de Ligandos N-(imidoilamidinas).
Para la obtención del precursores N-(2,6-diisopropilfenil)acetimidoilo es
necesario sintetizar la N-(2,6-diisopropilfenil)acetamida (P1)[132]. Esta se prepara
por reacción directa de cloruro de acetilo y 2,6-diisopropilanilina, como lo muestra la
Figura 45.

63
NH2
Cl
OPiridina
Reflujo, 3 h.+NH
O
O NH
+
(P1cis) (P1trans)
Figura 45. Síntesis de N-(2,6-diisopropilfenil)acetamida (Isómeros P1cis y P1trans)
Después de recristalización en metanol, P1 es obtenido como cristales blancos
en un 85 % de rendimiento. La caracterización mediante 1H-RMN muestra la
presencia de dos isómeros geométricos, que difieren en la orientación del grupo
carbonilo y el sustituyente 2,6-diisopropilfenil con respecto al enlace C–N.
Al observar los valores de las integrales de los pentupletes correspondientes a
los protones CH del isopropilo, se observa que en la mezcla, los dos isómeros se
encuentran en una proporción trans-cis 1:2, donde el isómero mayoritario es el Pcis.
Por otra parte el espectro FT-IR muestra las bandas características esperadas
para el producto, que corresponden al grupo amido secundario (υNH) en 3022 cm-1 y
el grupo amido secundario (υC=O) en 1649 cm-1.
Con la obtención de N-(2,6-diisopropilfenil)acetamida es posible sintetizar el
cloruro de N-(2,6-diisopropilfenil)acetimidoilo (P2), por la reacción directa de P1 y
pentacloruro de fósforo (PCl5), mediante un procedimiento descrito en la
literatura[131, 133]. Esta se realiza en condiciones anhidras, empleando benceno
como solvente y reflujando la mezcla por 3 horas (Figura 46).
Figura 46. Síntesis de Cloruro de N-(2,6-diisopropilfenil)acetimidoilo (P2).
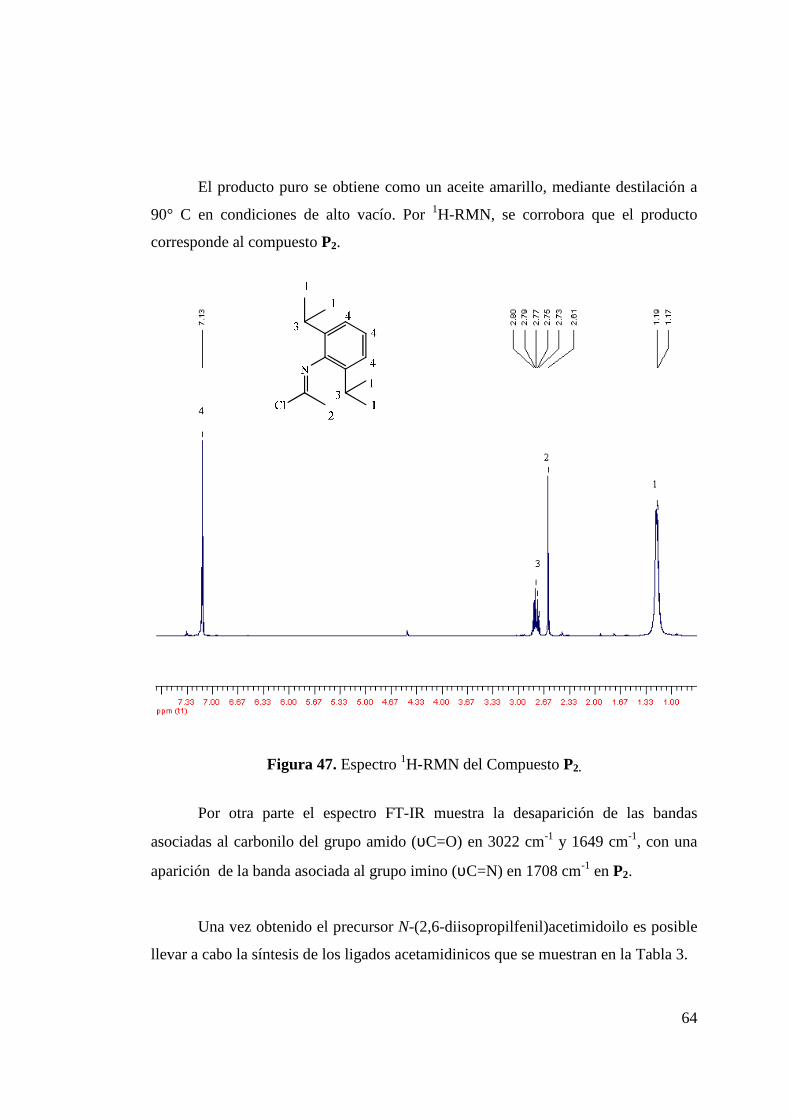
64
El producto puro se obtiene como un aceite amarillo, mediante destilación a
90° C en condiciones de alto vacío. Por 1H-RMN, se corrobora que el producto
corresponde al compuesto P2.
Figura 47. Espectro 1H-RMN del Compuesto P2.
Por otra parte el espectro FT-IR muestra la desaparición de las bandas
asociadas al carbonilo del grupo amido (υC=O) en 3022 cm-1 y 1649 cm-1, con una
aparición de la banda asociada al grupo imino (υC=N) en 1708 cm-1 en P2.
Una vez obtenido el precursor N-(2,6-diisopropilfenil)acetimidoilo es posible
llevar a cabo la síntesis de los ligados acetamidinicos que se muestran en la Tabla 3.

65
Tabla 3. Resumen de Ligandos N-(imidoilamidinas) y Acetamidinicos Síntetizados. Ligando Estructura
� N’-(2,6-diisopropilfenil)-N-[1-(2,6-diisopropilfenilimino)-etil]-N-(2,6-dimetil)acetamidina.
L 8a
� N-N´-bis(2,6-diisopropilfenil)-N-
[1-(2,6-dimetilfenilimino)-etil]acetamidina.
L 8b
� N,N´-bis-(2,6-dimeil-fenil)-N-[1-
(2,6-dimetil-fenilimino)etil)]acetamidina.
L 9
� N-(2,6-diisopropil-fenil)-N-[1-
(2,6-diisopropil-fenilimino)-etil]-N-(2,3,4,5,6-petafluoro-fenil)acetamidina.
L 10
� N-1-(2,6-diisopropilfenil)-N´-
(2,3,4,5,6-perfluorofenil)acetamidina. L 11 H
N N
F
FF
F
F � N,N´-bis(2,6-diisopropilfenil)-N-
(1-(2,3,4,5,6-pentafluorofenilimino)etil)acetamidina.
L 12
� N-(2,6-diisopropil-fenil)-N-1,(2,6-
diisopropilfenilimino)-N-(piridin-4-il) acetamidina.
L 13
� N-(1-(4-iminopiridin-1(4H)-
yl)etilideno)-2,6-diisopropylanilina. L 14
N
H3C
N NH

66
Los ligandos mostrados en la Tabla 3.1.3.1 se pueden separar en dos
categorías: N(imidoilamidinico), donde se incluyen los que tiene sustituyentes
alquílicos en los anillos (L 8 y L 9) y los que poseen anillos sustituidos con flúor o
nitrógeno (L 10, L12 y L 13). La segunda categoría corresponde a intermediarios del
tipo acetamidinicos (L 11 y L 14).
3.1.3.1. Ligandos N(imidoilamidinico) con Sustituyentes Alquílicos.
El primer ligando sintetizado corresponde a L 8, el cual se obtiene por la
reacción de dos equivalentes de cloruro de (2,6-diisopropilfenil)acetoimidoil con 2,6-
dimetilanilina en presencia de Et3N utilizando tolueno como solvente. El producto de
la reacción es un sólido blanco obtenido después de purificación por cristalización en
metanol en un rendimiento de 85 % (Figura 48). Los análisis 1H y 13C NMR
muestran una mezcla de isómeros para L 8, los que corresponden a 2,6-
MeC6H3N(C(Me)NC6H3-2,6-i-Pr)2 (L 8a) y 2,6-i-PrC6H3N(C(Me)NC6H3-2,6-i-
Pr)(C(Me)NC6H3- 2,6-Me) (L 8b).
Figura 48. Síntesis de los Isómeros de L 8.
La razón de los isómeros es 10:1 y es determina por la relación de las
integrales asociadas a los grupos isopropilos de los anillos aromáticos. En el isómero
simétrico L 8a la señal aparece en 2,92 ppm, mientras que el isómero menor y menos
simétrico L 8b tiene dos señales de los grupos isopropilos (3,41 y 2,86 ppm).
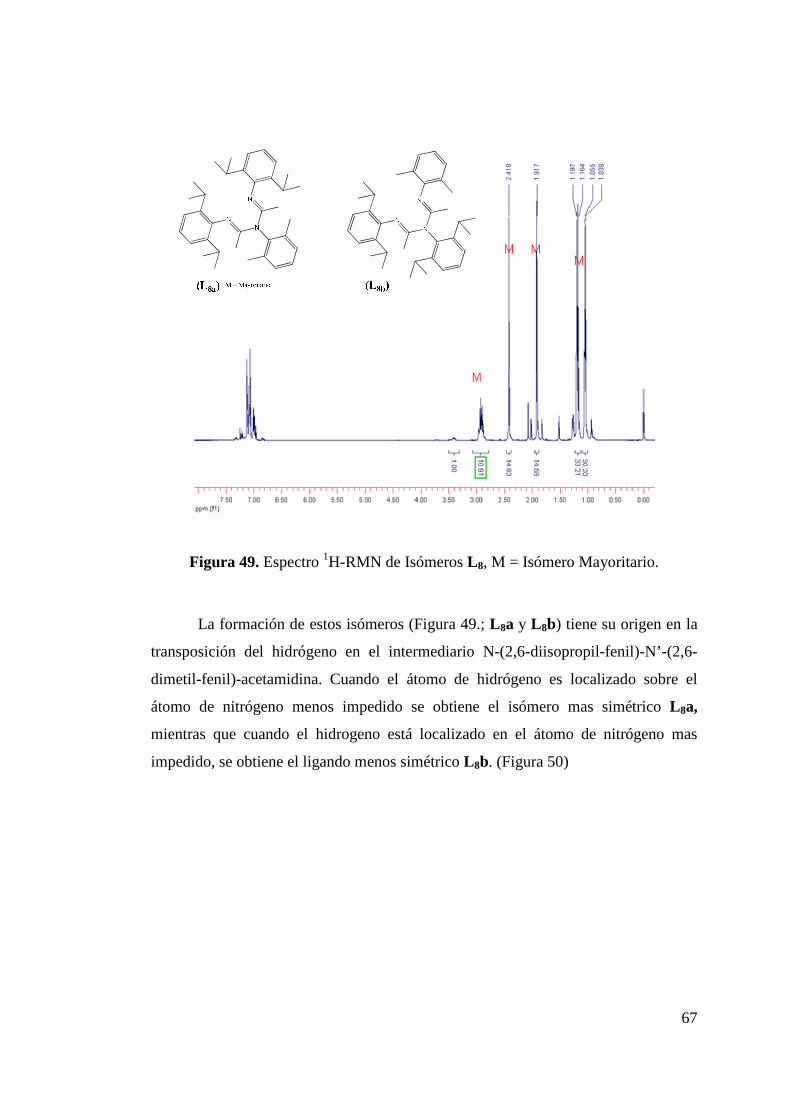
67
Figura 49. Espectro 1H-RMN de Isómeros L 8, M = Isómero Mayoritario.
La formación de estos isómeros (Figura 49.; L 8a y L 8b) tiene su origen en la
transposición del hidrógeno en el intermediario N-(2,6-diisopropil-fenil)-N’-(2,6-
dimetil-fenil)-acetamidina. Cuando el átomo de hidrógeno es localizado sobre el
átomo de nitrógeno menos impedido se obtiene el isómero mas simétrico L 8a,
mientras que cuando el hidrogeno está localizado en el átomo de nitrógeno mas
impedido, se obtiene el ligando menos simétrico L 8b. (Figura 50)
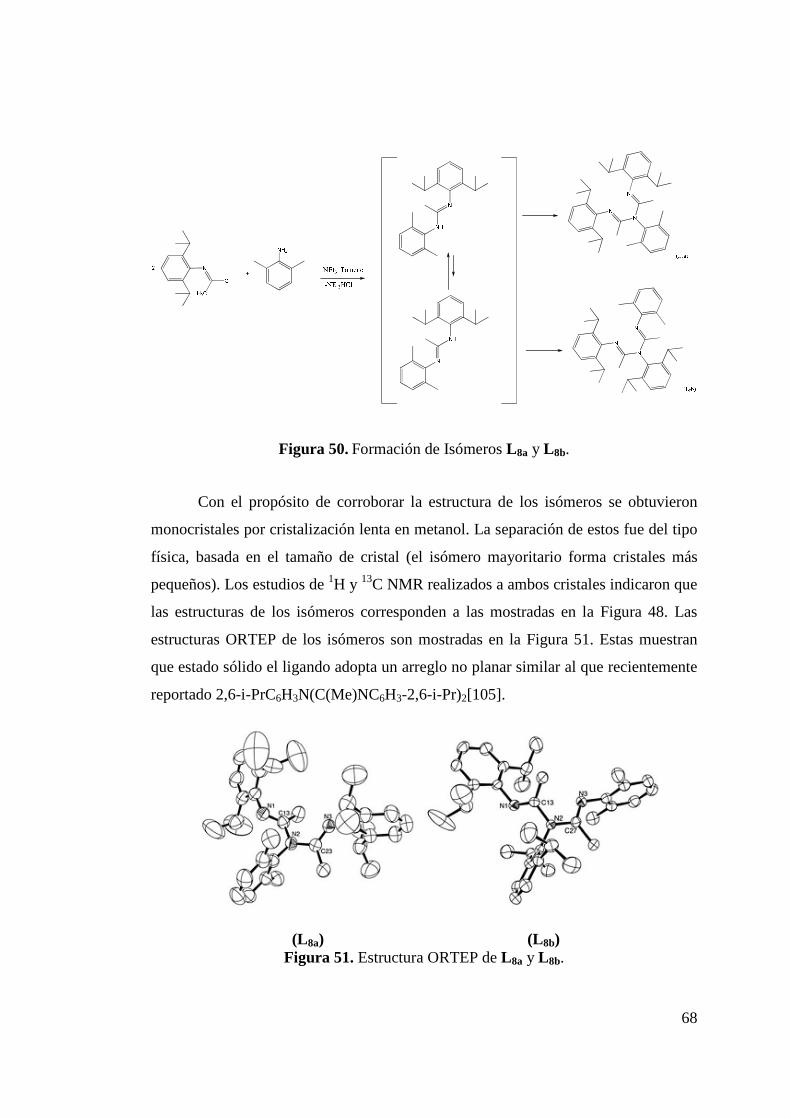
68
Figura 50. Formación de Isómeros L 8a y L 8b.
Con el propósito de corroborar la estructura de los isómeros se obtuvieron
monocristales por cristalización lenta en metanol. La separación de estos fue del tipo
física, basada en el tamaño de cristal (el isómero mayoritario forma cristales más
pequeños). Los estudios de 1H y 13C NMR realizados a ambos cristales indicaron que
las estructuras de los isómeros corresponden a las mostradas en la Figura 48. Las
estructuras ORTEP de los isómeros son mostradas en la Figura 51. Estas muestran
que estado sólido el ligando adopta un arreglo no planar similar al que recientemente
reportado 2,6-i-PrC6H3N(C(Me)NC6H3-2,6-i-Pr)2[105].
(L8a) (L8b)
Figura 51. Estructura ORTEP de L 8a y L 8b.
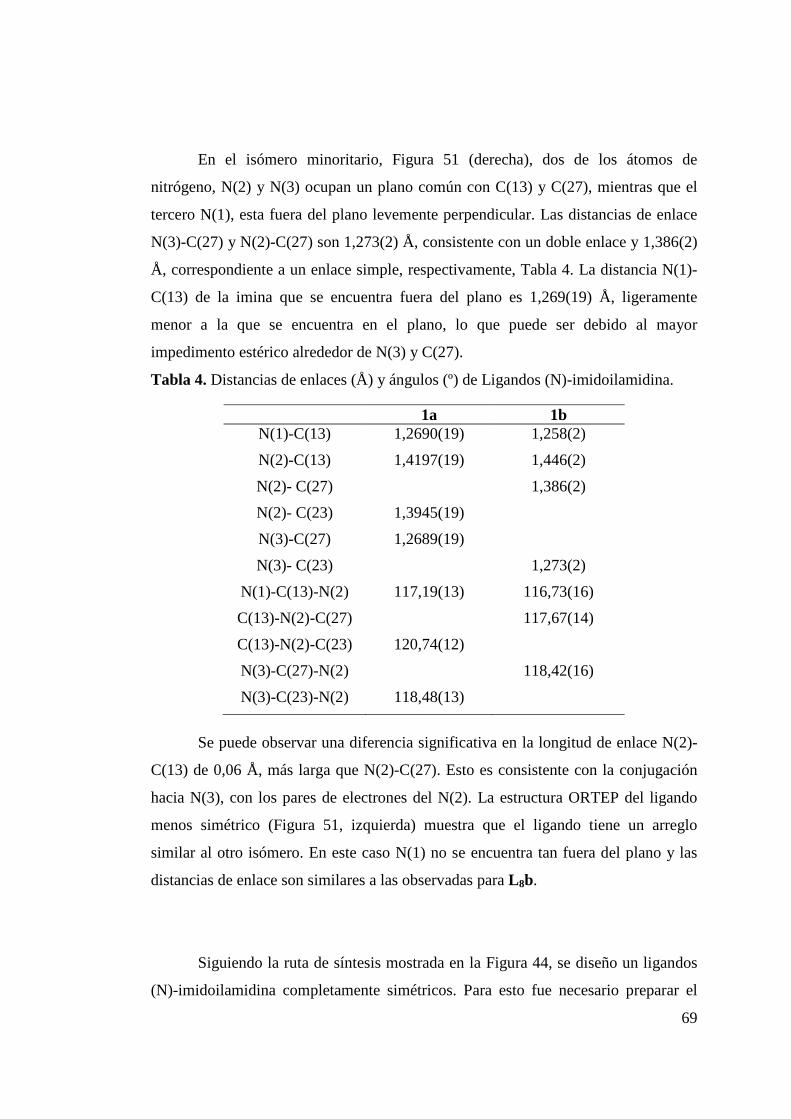
69
En el isómero minoritario, Figura 51 (derecha), dos de los átomos de
nitrógeno, N(2) y N(3) ocupan un plano común con C(13) y C(27), mientras que el
tercero N(1), esta fuera del plano levemente perpendicular. Las distancias de enlace
N(3)-C(27) y N(2)-C(27) son 1,273(2) Å, consistente con un doble enlace y 1,386(2)
Å, correspondiente a un enlace simple, respectivamente, Tabla 4. La distancia N(1)-
C(13) de la imina que se encuentra fuera del plano es 1,269(19) Å, ligeramente
menor a la que se encuentra en el plano, lo que puede ser debido al mayor
impedimento estérico alrededor de N(3) y C(27).
Tabla 4. Distancias de enlaces (Å) y ángulos (º) de Ligandos (N)-imidoilamidina.
1a 1b N(1)-C(13) 1,2690(19) 1,258(2)
N(2)-C(13) 1,4197(19) 1,446(2)
N(2)- C(27) 1,386(2)
N(2)- C(23) 1,3945(19)
N(3)-C(27) 1,2689(19)
N(3)- C(23) 1,273(2)
N(1)-C(13)-N(2) 117,19(13) 116,73(16)
C(13)-N(2)-C(27) 117,67(14)
C(13)-N(2)-C(23) 120,74(12)
N(3)-C(27)-N(2) 118,42(16)
N(3)-C(23)-N(2) 118,48(13)
Se puede observar una diferencia significativa en la longitud de enlace N(2)-
C(13) de 0,06 Å, más larga que N(2)-C(27). Esto es consistente con la conjugación
hacia N(3), con los pares de electrones del N(2). La estructura ORTEP del ligando
menos simétrico (Figura 51, izquierda) muestra que el ligando tiene un arreglo
similar al otro isómero. En este caso N(1) no se encuentra tan fuera del plano y las
distancias de enlace son similares a las observadas para L 8b.
Siguiendo la ruta de síntesis mostrada en la Figura 44, se diseño un ligandos
(N)-imidoilamidina completamente simétricos. Para esto fue necesario preparar el

70
precursor 2,6-dimetilfenilimidoil, por la reacción de 2,6-dimetilanilina con PCl5 en
condiciones de reacción similares a P2. Una vez obtenido se hace la reacción de éste
con un equivalente de 2,6-dimetilanilina dando como producto el N,N´-bis-(2,6-
dimetil-fenil)-N-[1-(2,6-dimetil-fenilimino)etil)]-acetamidina (L 9) (Figura 52)
N
Cl
H3C
2
+
NH2
NEt3, Tolueno
-NEt3HClN
N
N
(L 9) Figura 52. Síntesis de Ligando L 9.
El ligando es purificado por cristalización en etanol obteniéndose en un 87 %
de rendimiento. Análisis de 1H y 13C NMR muestran como se esperaba la presencia
de un solo isómero. Señales características incluyen: 2,46 ppm (s, 6H, Ph-CH3), 2,19
(s, 12H, Ph-CH3), 1,95 (s, 6H, (C=N)CH3). Monocristales son obtenidos por
evaporación lenta desde etanol, Figura 53.
Figura 53. Estructura ORTEP de L9

71
Tabla 5. Distancias de enlaces (Å) y ángulos (º) de L9.
L 9
N(1)-C(19) 1,400(2)
N(1)-C(9) 1,422(2)
N(2)- C(9) 1,265(2)
C(19)-N(3) 1,273(2)
N(2)-C(9)-N(1) 117,05(14)
C(9)-N(1)-C(19) 120,40(13)
N(1)-C(19)-N(3) 118,35(14)
La configuración o conectividad atómica corrobora lo predicho por 1H-RMN,
adicionalmente muestra que en estado sólido solo N(1) y N(3) se encuentran en un
plano. El tercer nitrógeno N(2) se ubica fuera de este y en dirección opuesta. Las
distancias de enlaces N(1)-C(19) y N(1)-C(9) son 1,400(2) y 1,422(2) Å,
respectivamente típicas de enlaces simples. Y en el caso de N(2)- C(9) y C(19)-N(3)
las distancia son 1,265(2) y 1,273(2) Å, respectivamente, típicas de enlace doble,
indicando una ausencia de conjugación en el ligando.
Estudios previos han mostrado que la introducción de grupos atractores de
electrones o heteroátomos en los ligandos que forman parte de sistemas cataliticos,
influyen en la reactividad de estos hacia la polimerización de olefinas. En este
trabajo es de interés estudiar efectos estéricos y electrónicos de los ligandos sobre
centros metálicos. Las rutas de síntesis mostradas anteriormente resultan ser una muy
buena alternativa para la preparación de ligandos con este tipo de grupos en su
estructura.

72
3.1.3.2. Ligandos N(imidoilamidinico) con Anillos Sustituidos con Flúor y
Nitrógenos.
Uno de los sistemas que se desea obtener es el N-(2,6-diisopropil-fenil)-N-[1-
(2,6-diisopropil-fenilimino)-etil]-N-(2,3,4,5,6-petafluoro-fenil)-acetamidina (L 10). La
ruta de síntesis se muestra en la Figura 54 y consiste en la adición de dos
equivalentes de (2,6-diisopropilfenil)acetimidoil a 2,3,4,5,6-pentafluoroanilina en
presencia de Et3N, en tolueno, bajo reflujo por 6 h.
N
N
N
F
F
F
F
F
N
Cl
H3C
FF
NH2
F F
F
+
2 NEt3, Tolueno
-NEt3HCl
(L10)
Figura 54 Síntesis de Ligando Fluorado L 10.
El ligando L 10 se obtiene después de recristalización en metanol, con un
rendimiento de 55 %. Los espectros de 1H, 13C y 19F-NMR del compuesto aislado,
son consistentes con la presencia de un único isómero a diferencia de lo que ocurre
con los ligandos de esta misma familia anteriormente descritos L 8a y L8b.
Algunas señales características del espectro de 1H NMR, son: 2,84 (p, 4H, 3JHH = 6,8 Hz, CH-iPr) y 2,00 (s, 6H, CH3) ppm. Las señalas que corroboran la
presencia un solo anillo fluorado se obtiene del espectro de 19F NMR en: -147,29, -
156,66, -160,35 ppm que corresponden a los cinco flúor del anillo. Monocristales de
L 10 han sido obtenidos por evaporación lenta de metanol. Representación de la
conectividad atómica de L 10 (ORTEP) se muestra en la Figura 55.

73
En el ligando L 10, los grupos fenilos adoptan un arreglo perpendicular al plano
formado por los átomos de nitrógenos, similar a 2,6-MeC6H3N(C(Me)NC6H3-2,6-i-
Pr)2. Sin embargo si se observa la estructura ORTEP podemos notar que los átomos
N(1), N(2) y N(3) ocupan un plano común, con una mínima distorsión a diferencia
de lo encontrado para L 8. En la Tabla 6 se muestran las principales longitudes de
enlaces y ángulos para el ligando L 10.
Tabla 6. Distancia de enlace (Å) y ángulo (º) para ligandos L 10 y L 11.
L10 L 11
C(1)-N(1) 1,267(3) N(1)-C(24) 1,407(3)
C(1)-C(3) 1,483(3) N(1)-C(7) 1,297(3)
C(1)-N(2) 1,422(3) N(2)- C(7) 1,348(3)
C(2)-N(2) 1,422(3) N(2)- C(32) 1,431(3)
C(2)-C(4) 1,498(4) C(7)- C(31) 1,500(4)
C(2)-N(3) 1,262(3) N(1)-C(30)-N(2) 120,1(2)
N(1)-C(1)-N(2) 114,4(2) N(1)-C(30)-C(31) 124,6(2)
N(1)-C(1)-C(3) 126,2(2) C(30)-N(1)-C(24) 118,5(2)
N(3)-C(2)-N(2) 119,0(2) C(30)-N(2)-C(32) 124,31(19)
N(2)-C(2)-C(4) 114,1(2)
C(2)-N(2)-C(2) 126,81(18)
Figura 55. Estructura ORTEP de L 10.

74
Las longitudes de enlace C(1)-N(1) y C(2)-N(3) son 1,267(3) y 1,262(3) Å
respectivamente, característicos de enlace doble. En el caso de C(1)-N(2) y C(2)-
N(2) son típicas de enlace simple 1,422(3) Å corroborando la estructura propuesta
para L 10.
Como se mostró en la Figura 50 la formación de los ligandos N-
(imidoilamidinas) poseen un intermediario, el cual posee una transposición del
protón de la imina entre los nitrógenos, lo que explica la obtención de isómeros. En
este caso la transposición del protón no ocurre, sino que se encuentra en el nitrógeno
que contiene al anillo fluorado haciéndolo mas acídico. La presencia de la base
podría estar definiendo la formación del producto final; es decir, la reacción sobre el
segundo equivalente de cloruro de imidoil estaría siendo una reacción nucleofílica de
la carga negativa del ligando (ubicada favorablemente en el átomo de nitrógeno que
contiene al anillo fluorado) sobre el carbono carbonílico del precursor P2. Esto
explicaría la ausencia del segundo isómero.
Con el propósito de entender los factores que afectan la selectivita de este tipo
de reacciones, es que se decidió realizar la síntesis anterior por etapas para aislar el
intermediario. La combinación de una razón estequiométricas de (2,6-
diisopropilfenil)acetimidoil y 2,3,4,5,6-pentafluoroanilina lleva a la formación de
dicho compuesto. (Figura 56, (i)). Esta reacción se lleva a cabo en tolueno como
solvente y se mantiene bajo reflujo por 6 h. El producto de la reacción se obtiene
como un sólido blanco en un 70 % de rendimiento después de recristalización en
metanol.

75
Figura 56. Síntesis de Ligandos L 11 y L 12.
Los análisis de 1H, 13C y 19F NMR muestran la presencia de un solo isómero
que corresponde a N-(2,6-diisopropilfenil)-N-(perfluorofenil)acetamidina L11,
señales características de los espectros de protón y flúor son: 1H-NMR 5,95 (s, 1H,
NH), 3,22 (p, 2H, CH-iPr), 2,04(s, 3H, CH3), 1,29 (d, 6H, CH3-iPr), 1,23 (d, 6H,
CH3-iPr); 19F NMR -146,60, -153,99, -162,61 ppm respectivamente.
Monocristales del ligando acetamidinico L 11 fueron obtenidos por
evaporación lenta del solvente de una solución de este en metanol. (Figura 57)
En estado sólido los dos átomos de nitrógenos y el carbono C(7) se
encuentran en un plano, con una geometría del tipo trigonal alrededor del átomo de
carbono. Las longitudes de enlace N(1)-C(7) y N(2)-C(7) son 1,297(3) y 1,348(3) Å
respectivamente. Estas longitudes muestran el carácter doble y simple, de estos
enlaces y consecutivamente son indicativos de que el átomo de hidrogeno se localiza
en el nitrógeno N(2). Adicionalmente, el ligando co-cristaliza con una molécula de
metanol. La posición en la cual se encuentra la molécula de metanol, corrobora que
Figura 57. Estructura ORTEP de L 11.

76
el protón esta en el átomo de nitrógeno que posee isopropilos en el anillo fenílico, ya
que forman enlace por puente de hidrógeno.
Una vez obtenido L 11, la segunda etapa de esta síntesis (Figura 56. (ii)),
consiste en la adición de un equivalente de 2,6-diisopropilfenilimidoil a un
equivalente de L 11, utilizando tolueno como solvente y dejando la reacción bajo
reflujo por 6 h. Esta reacción se lleva a cabo en ausencia de base (NEt3). De la
reacción se obtiene el segundo isómero N-(2,6-diisopropilfenil)-N-[1-(2,6-
diisopropylfenilimino)-etil]-N´(2,3,4,5,6-pentafluorofenil)acetamidina (L 12), como
un sólido de color blanco en un 60 % de rendimiento después de cristalización en
metanol.
La caracterización del producto mediante 1H, 13C y 19F NMR, indica la
presencia de un solo isómero, pero diferente al obtenido por la reacción directa
mostrada en la Figura 54. Entre algunas de las señales características de este nuevo
compuesto se incluyen las correspondientes a los isopropilos 3,26 (p, 2H, 3JHH = 6,8
Hz, CH-iPr) y 2,79 (p, 2H, 3JHH = 6,8 Hz, CH-iPr), que a diferencia de L 11 no son
equivalentes.
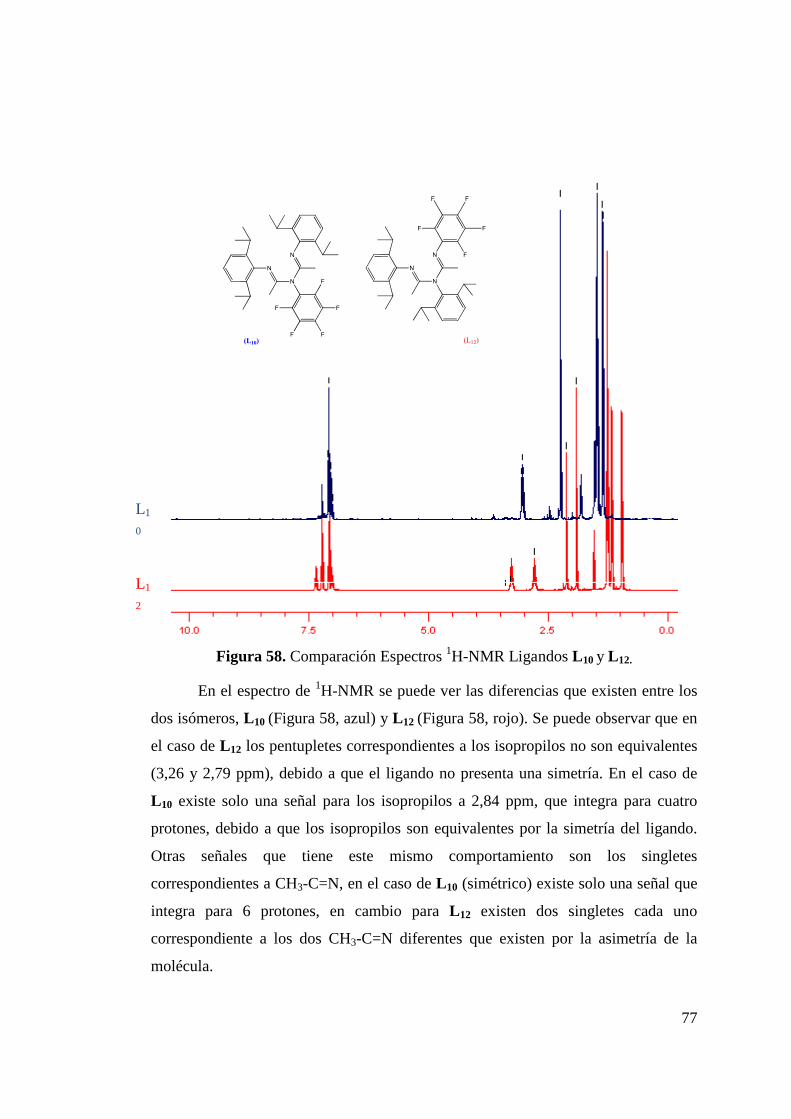
77
Figura 58. Comparación Espectros 1H-NMR Ligandos L10 y L12.
En el espectro de 1H-NMR se puede ver las diferencias que existen entre los
dos isómeros, L 10 (Figura 58, azul) y L 12 (Figura 58, rojo). Se puede observar que en
el caso de L 12 los pentupletes correspondientes a los isopropilos no son equivalentes
(3,26 y 2,79 ppm), debido a que el ligando no presenta una simetría. En el caso de
L 10 existe solo una señal para los isopropilos a 2,84 ppm, que integra para cuatro
protones, debido a que los isopropilos son equivalentes por la simetría del ligando.
Otras señales que tiene este mismo comportamiento son los singletes
correspondientes a CH3-C=N, en el caso de L 10 (simétrico) existe solo una señal que
integra para 6 protones, en cambio para L 12 existen dos singletes cada uno
correspondiente a los dos CH3-C=N diferentes que existen por la asimetría de la
molécula.
L1
0
L1
2
N
N
N F
F
F
F
F
(L12)
N
N
N
F
F
F
F
F(L10)

78
Los resultados mostrados anteriormente, indican que mediante este proceso
de síntesis es posible acceder a nuevos ligandos, particularmente a aquellos que
contengan en su estructura grupos funcionales y que en esta tesis son de interés para
la preparación de catalizadores. Ejemplos de estas funcionalidades además son
piridinas, nitrilos, carbonilos etc.
Siguiendo la metodología descrita para la síntesis de ligandos N-
(imidoilamidinas), se hace la reacción de 2,6-diisopropilfenilimidoil con 4-
aminopiridina en presencia de NEt3 utilizando tolueno como solvente. Esto con el fin
de obtener un ligando sustituido en el nitrógeno alfa de la imina con un anillo que
presente un átomo libre de nitrógeno.
Figura 59. Síntesis de Ligandos L 13.
En este caso el producto crudo de la reacción muestra por 1H-NMR la
presencia de más de un compuesto, sin embargo solo es posible aislar el producto L 13
después de columna cromatográfica (éter/hexano) en un 50 % de rendimiento. En la
Figura 60 se muestra el espectro del ligando L 13.
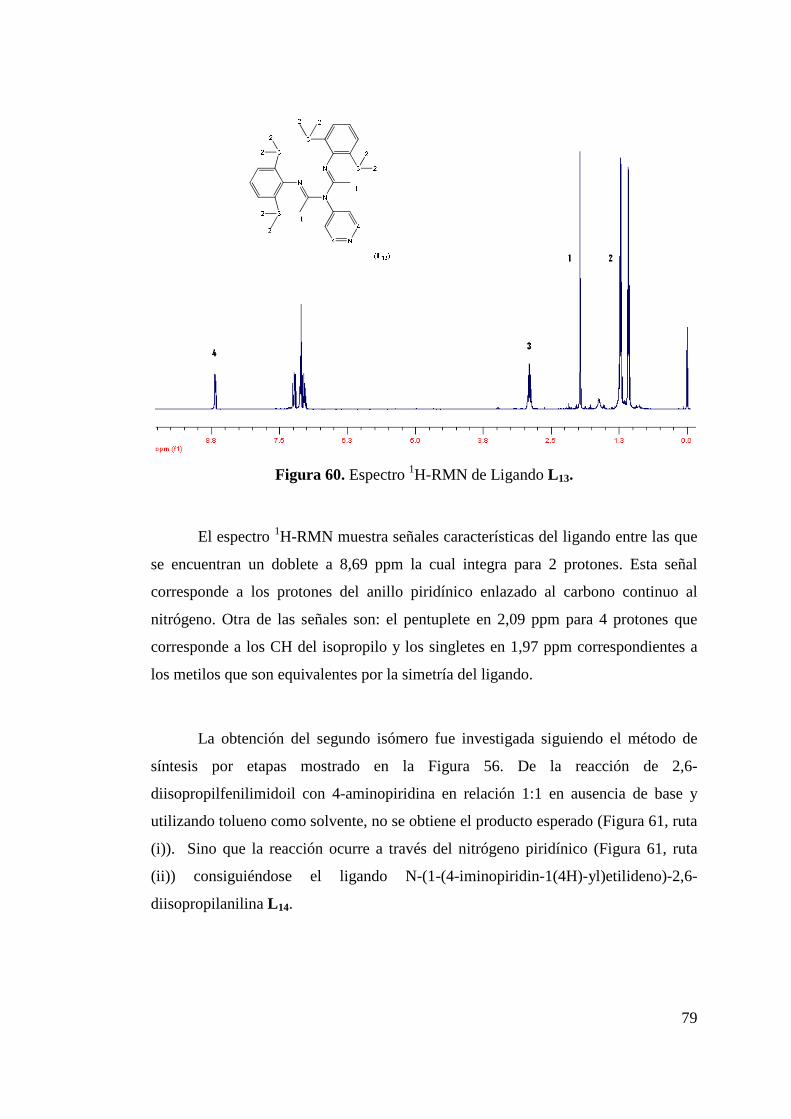
79
Figura 60. Espectro 1H-RMN de Ligando L13.
El espectro 1H-RMN muestra señales características del ligando entre las que
se encuentran un doblete a 8,69 ppm la cual integra para 2 protones. Esta señal
corresponde a los protones del anillo piridínico enlazado al carbono continuo al
nitrógeno. Otra de las señales son: el pentuplete en 2,09 ppm para 4 protones que
corresponde a los CH del isopropilo y los singletes en 1,97 ppm correspondientes a
los metilos que son equivalentes por la simetría del ligando.
La obtención del segundo isómero fue investigada siguiendo el método de
síntesis por etapas mostrado en la Figura 56. De la reacción de 2,6-
diisopropilfenilimidoil con 4-aminopiridina en relación 1:1 en ausencia de base y
utilizando tolueno como solvente, no se obtiene el producto esperado (Figura 61, ruta
(i)). Sino que la reacción ocurre a través del nitrógeno piridínico (Figura 61, ruta
(ii)) consiguiéndose el ligando N-(1-(4-iminopiridin-1(4H)-yl)etilideno)-2,6-
diisopropilanilina L 14.

80
Figura 61. Síntesis de Ligando L14
El producto de esta reacción, indica que el mayor carácter nucleófilo del
nitrógeno piridínico respecto al de la anilina, (Kb piridina = 2,3*10-9; anilina =
4,2*10-10, respectivamente) hace más favorable que la reacción de 4-aminopiridina
con el precursor 2,6-diisopropilfenilimidoil ocurra a través de un ataque nucleofílico
del nitrógeno del anillo hacia el átomo de carbono de grupo iminico deficiente en
electrones.
El producto de esta reacción es obtenido, después de cristalización en
metanol, como un sólido amarillo en un 60 % de rendimiento. En la Figura 62 se
muestra el espectro de 1H-RMN que corrobora la existencia de un único isómero,
correspondiente a L 14.
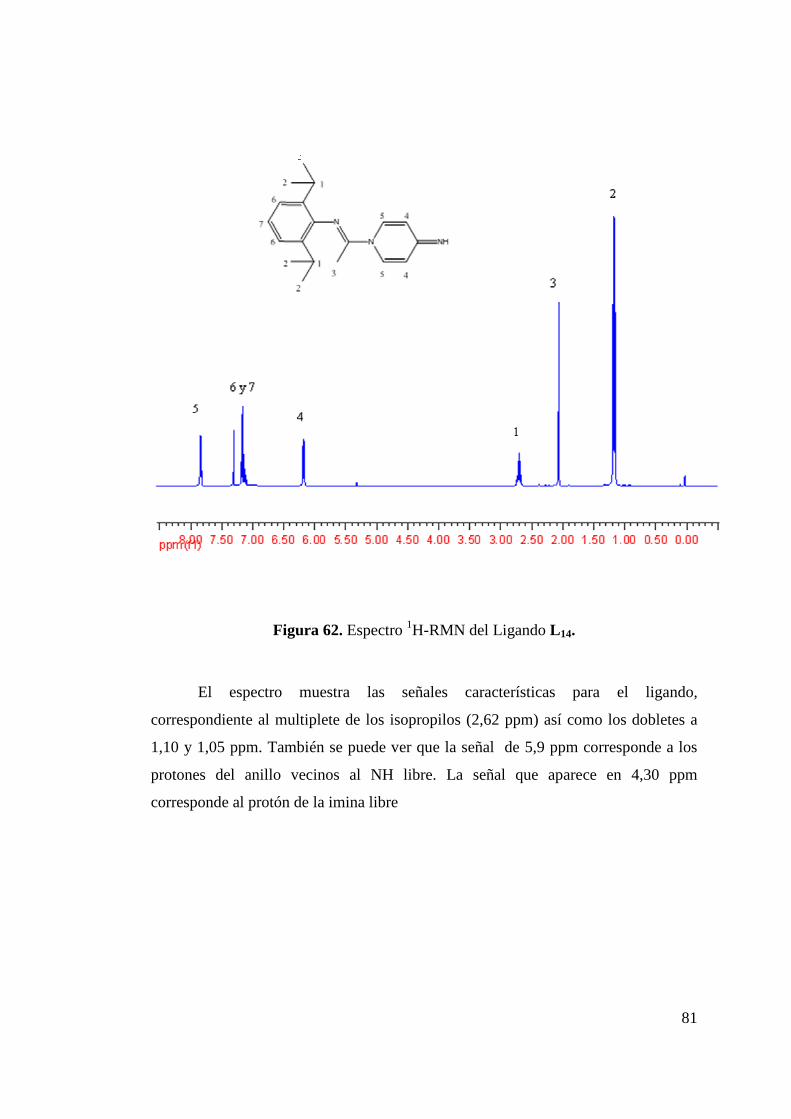
81
Figura 62. Espectro 1H-RMN del Ligando L14.
El espectro muestra las señales características para el ligando,
correspondiente al multiplete de los isopropilos (2,62 ppm) así como los dobletes a
1,10 y 1,05 ppm. También se puede ver que la señal de 5,9 ppm corresponde a los
protones del anillo vecinos al NH libre. La señal que aparece en 4,30 ppm
corresponde al protón de la imina libre
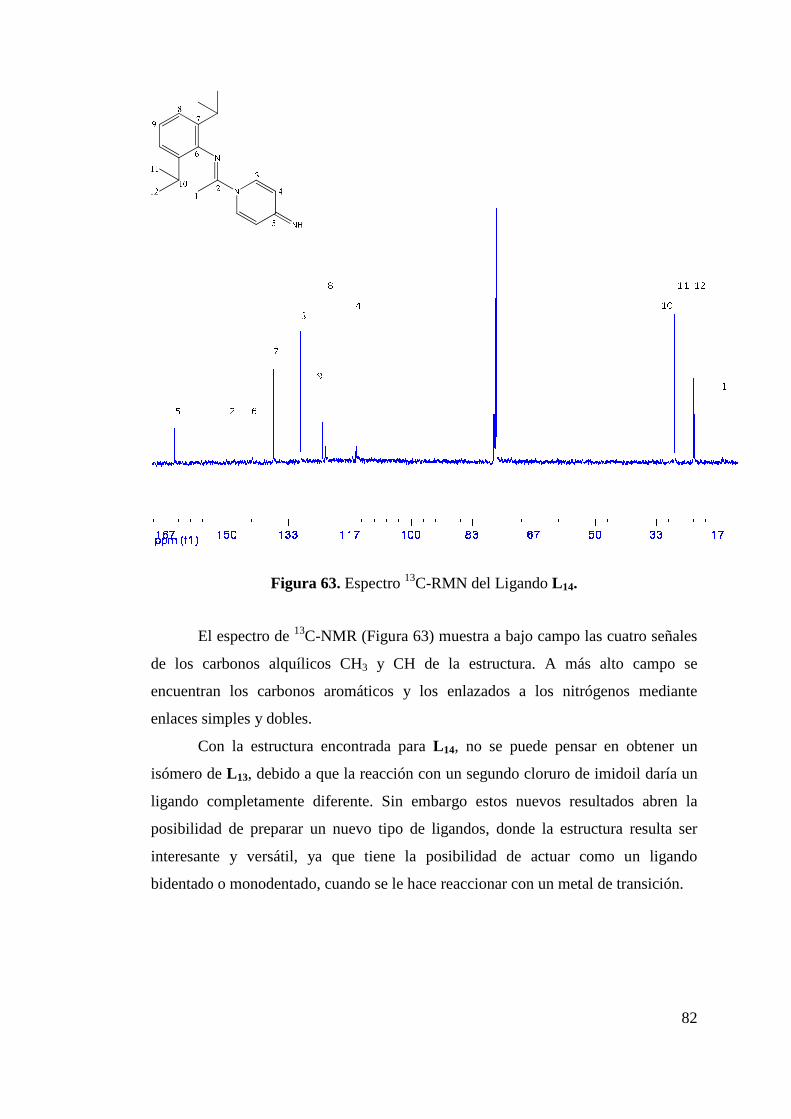
82
Figura 63. Espectro 13C-RMN del Ligando L14.
El espectro de 13C-NMR (Figura 63) muestra a bajo campo las cuatro señales
de los carbonos alquílicos CH3 y CH de la estructura. A más alto campo se
encuentran los carbonos aromáticos y los enlazados a los nitrógenos mediante
enlaces simples y dobles.
Con la estructura encontrada para L 14, no se puede pensar en obtener un
isómero de L 13, debido a que la reacción con un segundo cloruro de imidoil daría un
ligando completamente diferente. Sin embargo estos nuevos resultados abren la
posibilidad de preparar un nuevo tipo de ligandos, donde la estructura resulta ser
interesante y versátil, ya que tiene la posibilidad de actuar como un ligando
bidentado o monodentado, cuando se le hace reaccionar con un metal de transición.

83
3.2. Síntesis de Complejos de Titanio y Circonio.
En esta sección se describe la síntesis y caracterización de complejos de
Zr(IV) y Ti(IV). Los resultados se dividen en dos partes: la primera muestra los
complejos de titanio y circonio con ligandos que poseen un carbono quiral (4-(1-
fenil-etilamino)-pent-3-en-2ona (L 1) y 2-acetil-3-(1-fenil-etilimino)but-2-enenitrilo
(L 5). La segunda parte muestra la síntesis del complejo de circonio con el ligando
L 6.
3.2.1 Síntesis y Caracterización de Complejos de Ti(IV) y Zr(IV) con L 1 y L5
La síntesis de estos complejos se lleva a cabo por la reacción directa de los
precursores de metales de transición con la sal de litio de los ligandos. La obtención
de la sal de litio en ambos casos es por la reacción del ligando con n-butillitio en
THF a -78 º C. (Figura 64)
Figura 64. Formación de Sal de Litio de Ligandos L 1 y L 5.
El análisis mediante 1H-RMN muestra que para ambos ligandos desaparece el
protón acídico de ~ 12 ppm, lo que corrobora la obtención de las sales de litio.

84
Una vez obtenida la sal del ligando se hace la reacción para la obtención del
complejo en THF con una razón estequiometrica 2:1 sal del ligando, precursor del
metal MCl4 (M = Ti o Zr). (Figura 65)
O
N
Li +MCl4
2
*
ONH
M
O
Cl
Cl HN
O
N
M
Cl Cl
O
N
Cl
Cl
THF22 ºC12 h
a)
b)
X
X
X
1) M = Zr; X= H2) M = Zr; X= CN3) M = Ti; X = H4) M = Ti; X = CN
*
*
*
*
X
Disustituido
Cl
M
O
Cl
Cl
N
Cl H
X
*
Monosustituido
ó
X
Figura 65. Síntesis de Complejos de Circonio y Titanio.
Los complejos ZrCl4{Bis-[4-(1-fenil-etilamino)-pent-3-en-2ona]} (1) y
ZrCl4{Bis-[2-acetil-3-(1-fenil-etilimino)-but-2-enenitrilo]} (2) se obtienen después
de 12 h de reacción a temperatura ambiente, seguido por eliminación del solvente y
precipitación con éter. En ambos casos el producto obtenido es un sólido amarillo y
los rendimientos de las reacciones son 72 y 69 % respectivamente.
Los análisis de 1H-RMN de los complejos de circonio 1 y 2 muestran que la
reacción no procede como se esperaba Figura 65, ruta a). Los espectros muestran que
el ligando se vuelve a protonar, lo que se observa con la aparición de la señal a
campo bajo del protón acídico ~12 ppm. Esto se observa en ambos casos, es decir es
independiente de la sal de ligando que se utilice (L 1 y L5). Si se compara el

85
corrimiento de estas señales con la de los ligandos puros, se observa un corrimiento
hacia bajo campo en ambos complejos. Algunos de las señales que muestran este
efecto en el sistema que tiene la funcionalidad CN (2) corresponden a las del protón
acídico. Este en el ligando L 5 aparece en 12,25 ppm, en cambio cuando forma parte
del complejo esta señal aparece en 12,45 ppm. La otra señal característica que sufre
esta variación es la que corresponde al pentuplete de los isopropilos, la cual aparece
en 4,17 ppm en el ligando y se corre a 4,68 ppm cuando L 5 forma parte del complejo
2. Por otra parte los análisis de FT-IR también muestran una variación entre los
espectros del ligando y del complejo, donde la banda correspondiente a CN del
ligando ve desplazada respecto al complejo desde 2198 a 2207 cm-1, debido al
cambio en el entorno que posee al estar coordinado al centro metalico, estos hechos
corroboran la coordinación del ligando al metal.
Como el ligando presenta una alta solubilidad en éter, en el procedimiento
experimental de síntesis del complejo se lava para extraer con este solvente el
ligando que pudiera quedar sin reaccionar. Esto con el fin de asegurarse que el sólido
obtenido corresponde al ligando enlazado al centro metálico y no es solo ligando
libre.
Al igual que en los complejos de circonio, los complejos de titanio TiCl4{Bis-
[4-(1-fenil-etilamino)-pent-3-en-2ona]} (3) y TiCl4{Bis-[2-acetil-3-(1-fenil-
etilimino)but-2-enenitrilo]} (4), poseen coordinados los ligandos de forma protonada.
Esto se observa por la aparición del protón acídico en 11,57 y 12,27 ppm en los
complejos 3 y 4 respectivamente. El espectro de FT-IR muestra la señal característica
para la funcionalidad CN en 2201 cm-1. Estos hechos corroboran la formación del
complejo de titanio, al igual que en el caso de circonio
Tanto los complejos de Ti(IV) como de Zr(IV) son inestable al aire, por este
motivo no fue posible realizar una caracterización mediante análisis elemental o
espectrometría de masa, solo se realizaron análisis en atmósfera inerte, los cuales no
son suficientes para decir cuál es el tipo de coordinación del ligando al centro
metálico. Se realizaron innumerables esfuerzos para obtener monocristales de estos

86
complejos, lamentablemente esto no fue posible, lo que impide determinar la
conectividad en estos complejos. Es así que se proponen dos estructuras posibles,
una con dos ligandos protonados unido al centro metálico y la otra en que solo un
ligando de forma protonada este coordinado.
Estos resultados son muy novedosos, ya que en general los informados sobre
síntesis de complejos de circonio y titanio, con ligandos bidentados (N,O), como los
sintetizados en esta tesis, muestran que dos ligandos se coordinan al metal de
transición de forma monoaniónica, desplazando dos átomos de Cl. Esto es debido a
que la coordinación del tipo bidentada está más favorecida por el efecto quelato
cuando los ligandos son monoaniónicos.
Con el propósito de evitar la reprotonación del ligando, las reacciones se
realizaron en condiciones extremas de ausencia de oxigeno y humedad, tanto del
sistema de reacción como de los solventes. Sin embargo en todos los casos se vuelve
a obtener los compuestos donde la especie protonada se encuentra enlazado al centro
metálico. Otra ruta de síntesis consistió en la utilización del precursor DMEZrCl4,
que por tener un ligando bidentado enlazado al metal, es más estable y menos
susceptible a la descomposición por trazas de H2O generando HCl, el cual puede ser
la fuente de reprotonación de la sal del ligando.
Esta reacción se realizó en tolueno o THF. Sin embargo no se pudo evitar la
protonación del ligando, obteniéndose el mismo producto que con el precursor
anterior.
3.2.2 Síntesis y Caracterización de Complejos de Zr(IV) con L 6
En la preparación de complejos de circonio, se utilizó como alternativa a los
ligandos que poseen un grupo quiral en su estructura, el ligando 2-acetil-3-(2,6-
diisopropilfenilimino)-but-2enenitrilo (L 6). Este ligando contiene grupos alquilicos
mas voluminosos en su estructura y se espera que estas características aumente la
solubilidad de su sal y los productos finales de esta reacción, favoreciendo su
purificación y caracterización.

87
Para la síntesis del complejo es necesario obtener la sal de litio de L6. Esta se
obtiene por la reacción del ligando con n-butillitio a -78 ºC en THF. Una vez
obtenida, ésta se hace reaccionar con el precursor DMEZrCl4 en THF, por 12 h a
temperatura ambiente, en una razón estequiometrica 2:1. (Figura 66)
Figura 66. Síntesis de Complejo de Circonio con L6
Nuevamente independiente de las condiciones de reacción como solvente,
temperatura, tiempo de reacción, etc el espectro 1H-NMR muestra que el complejo
obtenido es ZrCl4[Bis(3-(2,6-diisopropilfenilimino)-2-1-(2,6-diisopropilfenilimino)
etil)but-2-enenitrilo)]n (n = numero de ligandos) (5) donde el ligando se encuentra de
forma protonada. Si se compara el espectro de 1H-RMN del ligando con el producto
de esta reacción se observa un corrimiento en las principales señales. Los más
representativos corresponde al peak de los isopropilos que en el complejo aparecen
en 2,74 ppm y en el ligando a 3,79 ppm, es decir a campo más alto debido a la
coordinación del ligando al centro metálico.
En el espectro de FT-IR también se observa la protonación del ligando por la
señal correspondiente a NH en 3676 cm-1. Además, la banda correspondiente al
grupo CN muestra un desplazamiento desde 2200 cm-1 (ligando) a 2184 cm-1
(complejo). Todos estos resultados estarían corroborando la unión del ligando en su
forma protonada al centro metálico de circonio. Sin embargo al igual que en los
complejos anteriores no se ha podido determinar el número de ligandos coordinados
al metal, por la inestabilidad del sistema al aire, lo que impide otras técnicas de
caracterización. De este sistema tampoco fue posible obtener la estructura cristalina.

88
En resumen, se puede decir que utilizando precursores como ZrCl4 y
DMEZrCl4 en la obtención de complejos de circonio, no se obtienen los productos
esperados (ligando desprotonado coordinado al metal). En todos los casos se obtiene
un complejo donde el ligando está unido al centro metálico de forma protonada y en
una relación no determinada.
Teniendo en consideración los resultados anteriores, una alternativa viable en
este tipo de síntesis corresponde a la utilización de precursores de circonio capaces
de reaccionar con el ligando protonado, como lo son (NMe2)2ZrCl2 y Zr(NMe2)4.
La primera reacción consiste en agregar el precursor (NMe2)2ZrCl2 a una
solución de L 6 en tolueno. La reacción se deja bajo agitación a temperatura ambiente
durante 12h. (Figura 67)
Figura 67. Síntesis de Complejo de Circonio con Precursor (NMe2)2ZrCl2.
El producto ZrCl2{Tri[2-acetil-3-(2,6-diisopropilfenilimino)-but-2enenitilo
(6) fue obtenido después de recristalización en pentano es un sólido amarillo en un
60 %. El espectro de 1H-NMR muestra la coordinación del ligando al centro
metálico, sin embargo si observamos en la Figura 68, podemos ver que existe un
ligando protonado (L 6) formando parte del complejo y dos ligandos en forma
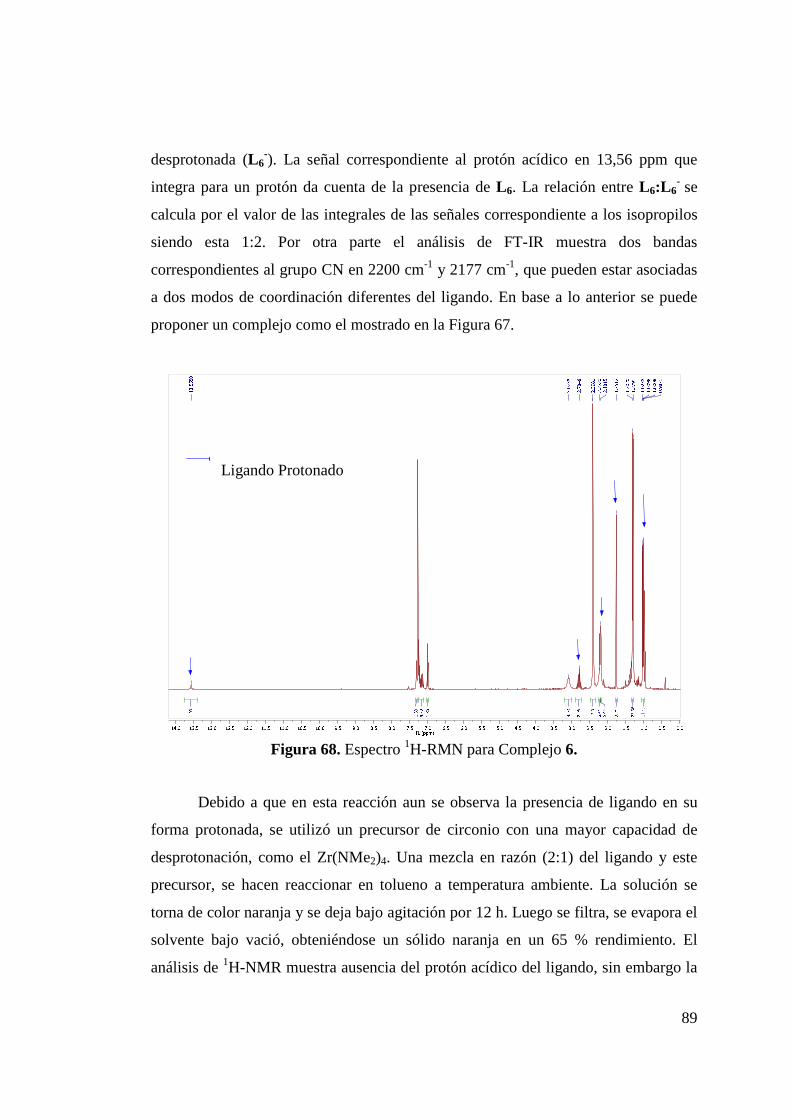
89
desprotonada (L 6-). La señal correspondiente al protón acídico en 13,56 ppm que
integra para un protón da cuenta de la presencia de L 6. La relación entre L 6:L 6- se
calcula por el valor de las integrales de las señales correspondiente a los isopropilos
siendo esta 1:2. Por otra parte el análisis de FT-IR muestra dos bandas
correspondientes al grupo CN en 2200 cm-1 y 2177 cm-1, que pueden estar asociadas
a dos modos de coordinación diferentes del ligando. En base a lo anterior se puede
proponer un complejo como el mostrado en la Figura 67.
Figura 68. Espectro 1H-RMN para Complejo 6.
Debido a que en esta reacción aun se observa la presencia de ligando en su
forma protonada, se utilizó un precursor de circonio con una mayor capacidad de
desprotonación, como el Zr(NMe2)4. Una mezcla en razón (2:1) del ligando y este
precursor, se hacen reaccionar en tolueno a temperatura ambiente. La solución se
torna de color naranja y se deja bajo agitación por 12 h. Luego se filtra, se evapora el
solvente bajo vació, obteniéndose un sólido naranja en un 65 % rendimiento. El
análisis de 1H-NMR muestra ausencia del protón acídico del ligando, sin embargo la
Ligando Protonado

90
complejidad del resto del espectro hace dificil una asignación clara de los demás
protones del ligando.
El espectro de FT-IR muestra la señal característica del grupo CN en 2200
cm-1 que indica la presencia del ligando en el producto. Esfuerzos para purificar
dicho producto resultaron infructuosos debido a su alta solubilidad.
De esta reacción también fue separada una pequeña fracción (10% del
rendimiento teórico de la reacción), que precipita de la solución madre. En este
producto, el ligando se encuentra desprotonado. La asignación de los peak e
integración de las señales corresponden al complejo Zr(NMe2)2{Bis(2-acetil-3-(2,6-
diisopropilfenilimino)-but-2enenitrilo) (7).
Figura 69. Síntesis de Complejo 7.
Monocristales del complejo 7 se han obtenido por difusión de
tolueno/pentano a -30 ºC. Estos son resueltos mediante difracción de Rayos-X y en la
Figura 69 se muestra la conectividad encontrada para este complejo.

91
La estructura molecular del complejo 7 muestra que este posee una simetría
del tipo C2, donde el metal tiene una coordinación del tipo octaédrica ligeramente
distorsionada. Se puede ver que la disposición espacial de los átomos de nitrógeno es
del tipo trans (N(1)-Zr-N(1a) 157,16(7)º) así como la de los átomos de oxigeno es del
tipo cis (O(1)-Zr-O(1a) 81,27(8)º) al igual que los grupos dimetilamino (N(3)-Zr-
N(3a) 98,55(10)º). Las distancias y ángulos de enlace se resumen en la Tabla 7.
Tabla 7. Distancia de enlace (Å) y ángulo (º) para Complejo 7.
5
Zr-O(1) 2,1415(18)
Zr-N(1) 2,359(2)
Zr-N(3) 2,059(2)
O(1)-C(4) 1,270(3)
C(3)-C(4) 1,391(4)
N(1)-C(2) 1,450(4)
O(1)-Zr-N(3) 88,91(9)
N(1)-Zr-O(1) 77,58(1)
N(1a)-Zr-N(1) 157,16(7)
N(3)-Zr-O(1a) 169,02(8)
N(3a)-Zr-O(1) 171,26(9)
N(3)-Zr-N(3a) 98,55(10)
Figura 70. Estructura ORTEP de Complejo 7.

92
El bajo rendimiento en la obtención de este complejo hace imposible la etapa
de alquilación, para su posterior utilización como complejo en la polimerización de
etileno.
3.3. Síntesis y Caracterización de Complejos de Níquel.
En esta sección se presentan los resultados sobre la síntesis y caracterización
de complejos de níquel a partir de los ligandos mostraros en la sección 3.1.2.
3.3.1. Complejos con ligandos N-(imidoilamidinicos) con Sustituyentes
Alquílicos.
A partir de los compuestos N-(imidoilamidinicos) L 8(a,b), se prepararon los
complejos de níquel [NiBr2{N’-(2,6-diisopropil-fenil)-N-[1-(2,6-diisopropil-
fenilimino)-etil]-N-(2,6-dimetil)-acetamidina}] (8a) y [NiBr2{N-(2,6-diisopropil-
fenil)-N-[1-(2,6-diisopropil-fenilimino)-etil]-N’-(2,6-dimetil)-acetamidina}] (8b).
Estos fueron obtenidos de la reacción de níquel dibromo 2-metoxietil éter,
(NiBr2[O-(C2H4OMe)2]) con la mezcla de isómeros L 8a y L 8b. La reacción se lleva a
cabo en THF a temperatura ambiente por un periodo de 4 días. (Figura 71)
O
O
O
NiBr2
THF
4 dias
N
N
NNi
BrBr
N
N
NNi
BrBr
(L8a) + (L8b) +
Isómeros
+
(8a)
(8b)

93
Figura 71. Obtención de Mezcla Isomérica de Complejos de Níquel 8.
El producto de esta reacción es estable al aire y es obtenido después de
filtración, lavado con éter y recristalización en THF-éter, como un sólido cristalino
de color violeta en un 70 % de rendimiento.
Monocristales de este producto fueron obtenidos de la misma mezcla de
solventes. Mediante microscopio se observaron dos tipos de cristales (tamaño y
formas diferentes) que fueron aislados y estudiados por difracción. En la Figura 72 se
pueden observar las estructuras cristalinas de ambos cristales que corresponden a los
dos isómeros formados en la reacción.
La estructura molecular para ambos isómeros muestra un anillo de seis miembros
con enlaces N,N al centro metálico. Además la geometría observada es del tipo
tetraédrica ligeramente distorsionada alrededor del níquel. En el complejo 8a (más
simétrico) el átomo Br(2) es casi perpendicular al plano formado por el anillo. Se
puede ver que los ángulos corresponden a N(1)-Ni-Br(2) es 101,32(9)° mientras que
N(1)-Ni-Br(1) es 122,18(9)°. Las distancias de enlace N(3)-C(27), N(1)-C(13) y
N(2)-C(27), N(2)-C(13) son 1,293(5), 1,279(5) y 1,392(5), 1,405(5),

94
respectivamente. Las dos primeras corresponden a enlace doble y las otras a
enlaces simples. (Tabla 8)
8a 8b
Figura 72. Estructura ORTEP de 8a y 8b.
El segundo isómero 8b muestra distancias de enlaces similares N(3)-C(23),
N(1)-C(9) y N(2)-C(23), N(2)-C(9); 1,283(3), 1,287(3) y 1,411(3), 1,402(3)
respectivamente. Si observamos las distancias N-C de la amina del complejo 8b
(N(2)-C(23), N(2)-C(9)) podemos ver que son similares, lo cual es diferente a lo
observado para estos mismos enlaces para el ligando L 8b. La similitud en distancias
de enlace de las aminas es debido a la coordinación de ligando al centro metálico de
níquel.
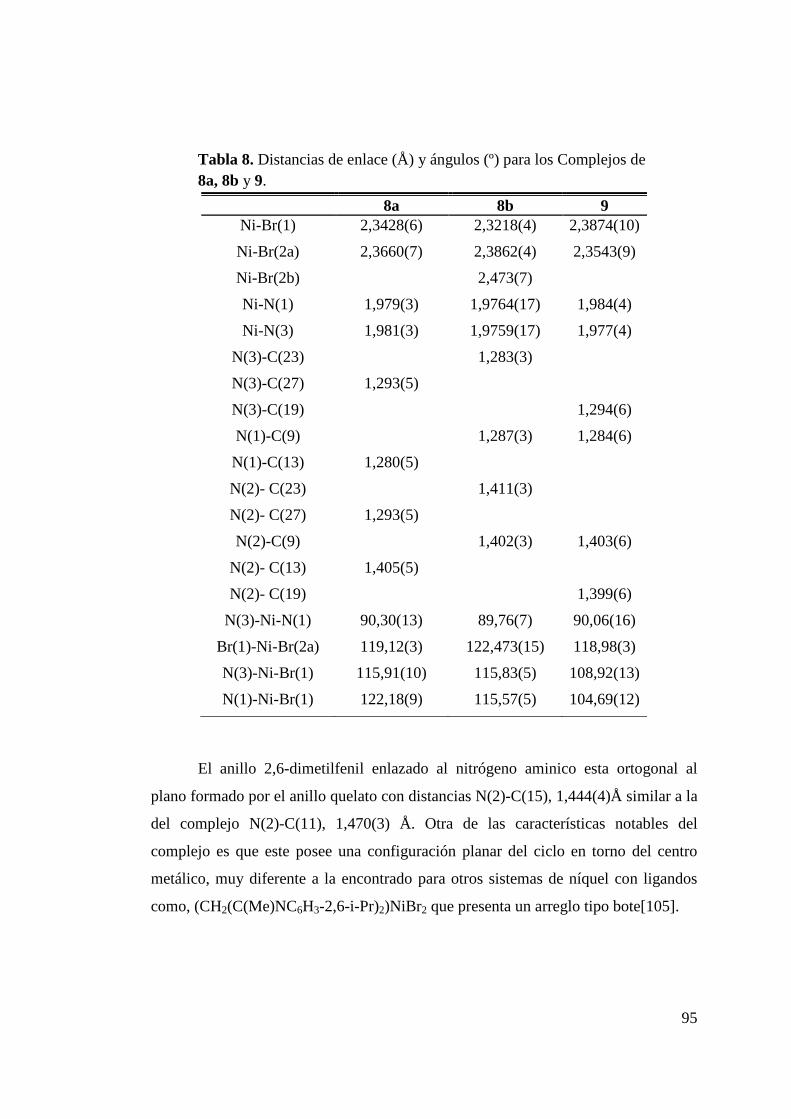
95
Tabla 8. Distancias de enlace (Å) y ángulos (º) para los Complejos de 8a, 8b y 9.
8a 8b 9 Ni-Br(1) 2,3428(6) 2,3218(4) 2,3874(10)
Ni-Br(2a) 2,3660(7) 2,3862(4) 2,3543(9)
Ni-Br(2b) 2,473(7)
Ni-N(1) 1,979(3) 1,9764(17) 1,984(4)
Ni-N(3) 1,981(3) 1,9759(17) 1,977(4)
N(3)-C(23)
N(3)-C(27)
N(3)-C(19)
1,293(5)
1,283(3)
1,294(6)
N(1)-C(9)
N(1)-C(13)
1,280(5)
1,287(3) 1,284(6)
N(2)- C(23)
N(2)- C(27)
1,293(5)
1,411(3)
N(2)-C(9)
N(2)- C(13)
N(2)- C(19)
1,405(5)
1,402(3) 1,403(6)
1,399(6)
N(3)-Ni-N(1) 90,30(13) 89,76(7) 90,06(16)
Br(1)-Ni-Br(2a) 119,12(3) 122,473(15) 118,98(3)
N(3)-Ni-Br(1) 115,91(10) 115,83(5) 108,92(13)
N(1)-Ni-Br(1) 122,18(9) 115,57(5) 104,69(12)
El anillo 2,6-dimetilfenil enlazado al nitrógeno aminico esta ortogonal al
plano formado por el anillo quelato con distancias N(2)-C(15), 1,444(4)Å similar a la
del complejo N(2)-C(11), 1,470(3) Å. Otra de las características notables del
complejo es que este posee una configuración planar del ciclo en torno del centro
metálico, muy diferente a la encontrado para otros sistemas de níquel con ligandos
como, (CH2(C(Me)NC6H3-2,6-i-Pr)2)NiBr2 que presenta un arreglo tipo bote[105].

96
La síntesis del complejo de níquel menos impedidos y mas simétricos se
realiza por la adición del precursor de níquel dibromo 2-metoxietil éter al ligando L 9,
utilizando DCM como solvente, a 75 ºC durante 48 h (Figura 73). El complejo se
obtiene después de filtración, evaporación de solvente y cristalización desde DCM.
Figura 73. Síntesis de Complejo de Níquel más Simétrico 9.
Monocrsitales del complejo 9 se obtuvieron por difusión de éter a DCM a
temperatura ambiente. La resolución estructural obtenida de los resultados de las
mediciones de Rayos-X son mostrados en la Figura 74. La estructura molecular
muestra un anillo de seis miembros con el átomo metálico. La reducción en el
volumen estérico alrededor del átomo de níquel da como resultado un complejo de
geometría tetraédrica menos distorsionada si se le compara con los complejos más
impedidos 8a y 8b.
Las distancias de enlaces de las iminas N(1)-C(9) y N(3)-C(19) son 1,284(6)
y 1,294(6) respectivamente, similares a las distancias para los complejos 8a y 8b.
(Tabla 8).

97
En la sección de síntesis de ligandos se mostraron compuestos del tipo N-
(imidoilamidinas) que contienen anillos fluorados o heteroátomos en su estructura.
La presencia de grupos funcionales, dadorores o atractores de electrones así como la
de heteroátomos, normalmente son determinantes en su reactividad o en la capacidad
de coordinación de estos ligandos hacia metales. En esta sección se muestran los
resultados del estudio de como la presencia de sustituyentes electrón deficientes o
grupos funcionales en el ligando afectan en la síntesis de complejos de níquel.
3.3.2. Complejos con Ligandos N-(imidoilamidinicos) Sustituidos con Flúor y
Nitrógenos.
Uno de los sistemas sintetizado es el [NiBr2{N-1-(2,6-diisopropilfenil)-N´-
(2,3,4,5,6-perfluorofenil)acetamidina}] (10). Este se obtiene por la adición de L 10 al
precursor níquel dibromo 2-metoxietil éter en THF. La reacción se deja bajo
agitación por 48 h a 60 ºC. Otro de los sistemas obtenidos es el [NiBr2{N-1-(2,6-
diisopropilfenil)-N´-(2,3,4,5,6-perfluorofenil)acetamidina}] (12), esto es por la
reacción del ligando L 12 con el mismo precursor y en las mismas condiciones de
reacción (Figura 75).
Figura 74. Estructura ORTEP de complejo 9.

98
Figura 75. Síntesis de Complejos Níquel 10 y 11.
Estas reacciones tienen un rendimiento de 55 % y 60 % para los complejos 10
y 12 respectivamente. En ambos casos se obtienen como sólidos de color morado
después de ser lavados con éter. Los dos complejos resultaron ser estables al aire lo
que permitió una mejor caracterización de ellos. Análisis de 1H y 13C NMR no se
pudieron realizar debido a las características paramagnéticas de los complejos, lo
cual dificulta la asignación de las señales obtenidas. El análisis de espectroscopia de
masa indica que el compuesto 10 posee una masa de 765,1 g/mol lo que concuerda
con el compuesto menos un átomo de bromo (M+-Br).

99
En el caso del complejo 12 se obtuvieron monocristales por evaporación lenta
de DCM. La estructura ORTEP se muestra en la Figura 76.
Figura 76. Estructura ORTEP de Complejo 12.
La estructura molecular muestra la formación de un anillo de seis miembros,
donde el ligando N,N está coordinado al níquel dibromo como era esperado. El
complejo muestra un arreglo del tipo tetraédrico distorsionado, donde el átomo Br(2)
es casi perpendicular al plano del anillo, lo que se ve reflejado en los ángulos N(1)-
Ni-Br(2) (99,99(7) º) mientras que el ángulo N(1)-Ni-Br(1) es (113,48(7) º). Las
distancias de enlace para N(3)-C(2), N(3)-C(4) y N(1)-C(2), N(2)-C(4) son 1,397(4),
1,406(4) y 1,294(4), 1,289(4), indicando la configuración de enlace doble y simple
respectivamente. (Tabla 9)

100
Tabla 9. Distancias de enlace (Å) y ángulos (º) para los Complejo 12.
Distancias Ángulos
N(1)-C(2) 1,294(4) N(1)-Ni-N(5) 88,06(10)
N(3)-C(2) 1,397(4) N(5)-Ni-Br(1) 122,27(7)
N(3)- C(4) 1,406(4) N(1)-Ni-Br(1) 113,48(7)
N(5)- C(4) 1,289(4) N(1)-Ni-Br(2) 99,99(7)
Ni- N(1) 1,977(3) N(5)-Ni-Br(2) 103,75(7)
Ni- N(5) 1,966(2) C(2)-N(3)-C(4) 126,3(2)
Ni- Br(1) 2,3111(5)
Ni- Br(2) 2,3681(5)
Siguiendo la misma metodología a partir de L 13 se preparó el complejo
[NiBr2{N-(1-(4-iminopiridin-1(4H)-yl)etilideno)-2,6-diisopropylanilina}] (13)
(Figura 77). El producto es obtenido como un sólido azul después de ser lavado con
éter en un 73% de rendimiento, después de recristalización. El complejo al igual que
los otros sistemas de níquel reportados en esta tesis resulta ser estable al aire.
Figura 77. Síntesis de Complejo 13

101
No fue posible obtener monocristal para ser estudiados mediante Rayos-X,
sin embargo el análisis de espectroscopia de masa indica que el compuesto posee una
masa de 715,3 g/mol, lo que concuerda con el complejo propuesto en la Figura 77.
En la preparación de L 12, se obtuvo el intermediario L 11, el cual también se
utilizó para la formación de complejos. Era de esperar que el ligando se coordinara
de forma bidentada al níquel, formando un complejo de cuatro miembros[134]
(Figura 78). Sin embargo la reacción de L 11 con el precursor de níquel (II) dibromo
2-metoxietil éter no produce el complejo esperado, incluso cuando se cambian las
condiciones de reacción como solvente, temperatura o tiempo de reacción.
Figura 78. Complejo de Níquel Esperado.
El nuevo ligando L 14, también fue utilizado para la síntesis de complejos de
níquel. En este caso como el ligando puede actuar como un ligando monodentado la
reacción se hizo en una relación estequiometrica 2:1 (ligando: precursor). Esta fue
realizada en DCM, a temperatura ambiente por 4 h. Después de evaporación de
solvente y lavado con éter se obtiene un sólido de color verde oscuro en un
rendimiento de 85%. (Figura 79)
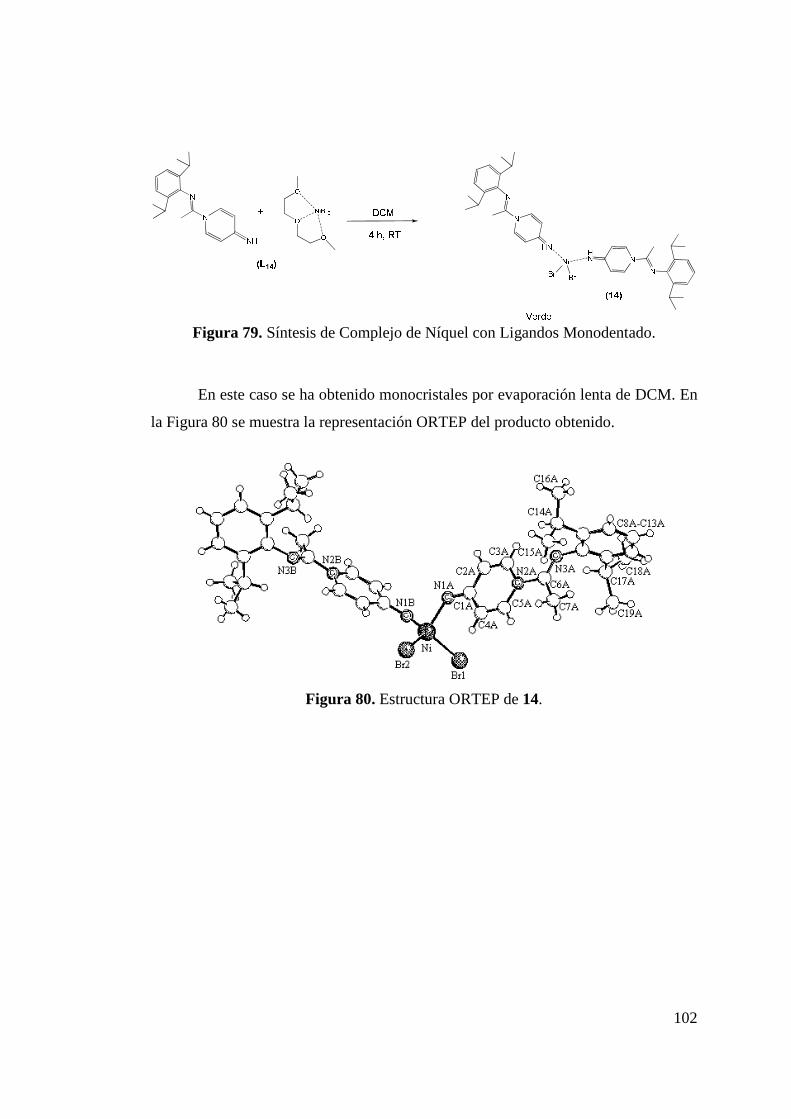
102
Figura 79. Síntesis de Complejo de Níquel con Ligandos Monodentado.
En este caso se ha obtenido monocristales por evaporación lenta de DCM. En
la Figura 80 se muestra la representación ORTEP del producto obtenido.
Figura 80. Estructura ORTEP de 14.
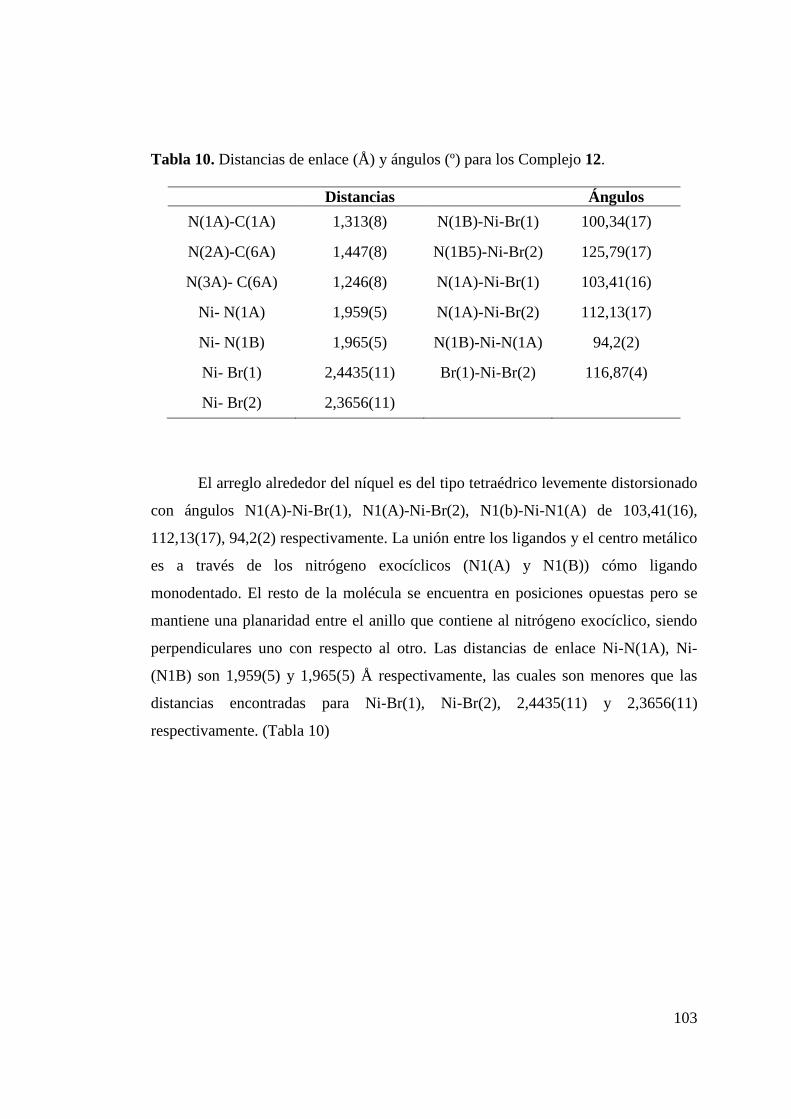
103
Tabla 10. Distancias de enlace (Å) y ángulos (º) para los Complejo 12.
Distancias Ángulos
N(1A)-C(1A) 1,313(8) N(1B)-Ni-Br(1) 100,34(17)
N(2A)-C(6A) 1,447(8) N(1B5)-Ni-Br(2) 125,79(17)
N(3A)- C(6A) 1,246(8) N(1A)-Ni-Br(1) 103,41(16)
Ni- N(1A) 1,959(5) N(1A)-Ni-Br(2) 112,13(17)
Ni- N(1B) 1,965(5) N(1B)-Ni-N(1A) 94,2(2)
Ni- Br(1) 2,4435(11) Br(1)-Ni-Br(2) 116,87(4)
Ni- Br(2) 2,3656(11)
El arreglo alrededor del níquel es del tipo tetraédrico levemente distorsionado
con ángulos N1(A)-Ni-Br(1), N1(A)-Ni-Br(2), N1(b)-Ni-N1(A) de 103,41(16),
112,13(17), 94,2(2) respectivamente. La unión entre los ligandos y el centro metálico
es a través de los nitrógeno exocíclicos (N1(A) y N1(B)) cómo ligando
monodentado. El resto de la molécula se encuentra en posiciones opuestas pero se
mantiene una planaridad entre el anillo que contiene al nitrógeno exocíclico, siendo
perpendiculares uno con respecto al otro. Las distancias de enlace Ni-N(1A), Ni-
(N1B) son 1,959(5) y 1,965(5) Å respectivamente, las cuales son menores que las
distancias encontradas para Ni-Br(1), Ni-Br(2), 2,4435(11) y 2,3656(11)
respectivamente. (Tabla 10)

104
3.4 Reactividad de Complejos en Polimerizaciones de Etileno.
En esta sección se presentan los resultados obtenidos en las reacciones de
polimerización de etileno utilizando algunos de los complejos presentados
anteriormente. Los complejos que fueron utilizados son aquellos que mostraron
buenos rendimientos en la etapa de síntesis.
La presentación de los resultados será dividida en dos partes: la primera
muestra las reacciones de polimerización con complejos de Titanio y Circonio en
presencia de cocatalizador MAO. La segunda parte muestra los resultados obtenidos
en las polimerizaciones con complejos de Níquel. Para ambos tipos de catalizadores
se estudió la influencia de parámetros de reacción como: temperatura, la presión del
monómero, razón Al/Metal, en la actividad catalítica del sistema y características del
polímero obtenido (peso molecular y temperatura de fusión).
3.4.1 Complejos de Circonio con ligandos L1 y L 5
El estudio de reactividad de complejos de circonio se realiza con los
complejos 1 y 2 (Esquema) los cuales tienen como diferencia la presencia de una
funcionalidad adicional CN en el ligando de características quirales.
*
Figura 81. Complejos 1 y 2 utilizados en la Polimerización de Etileno.
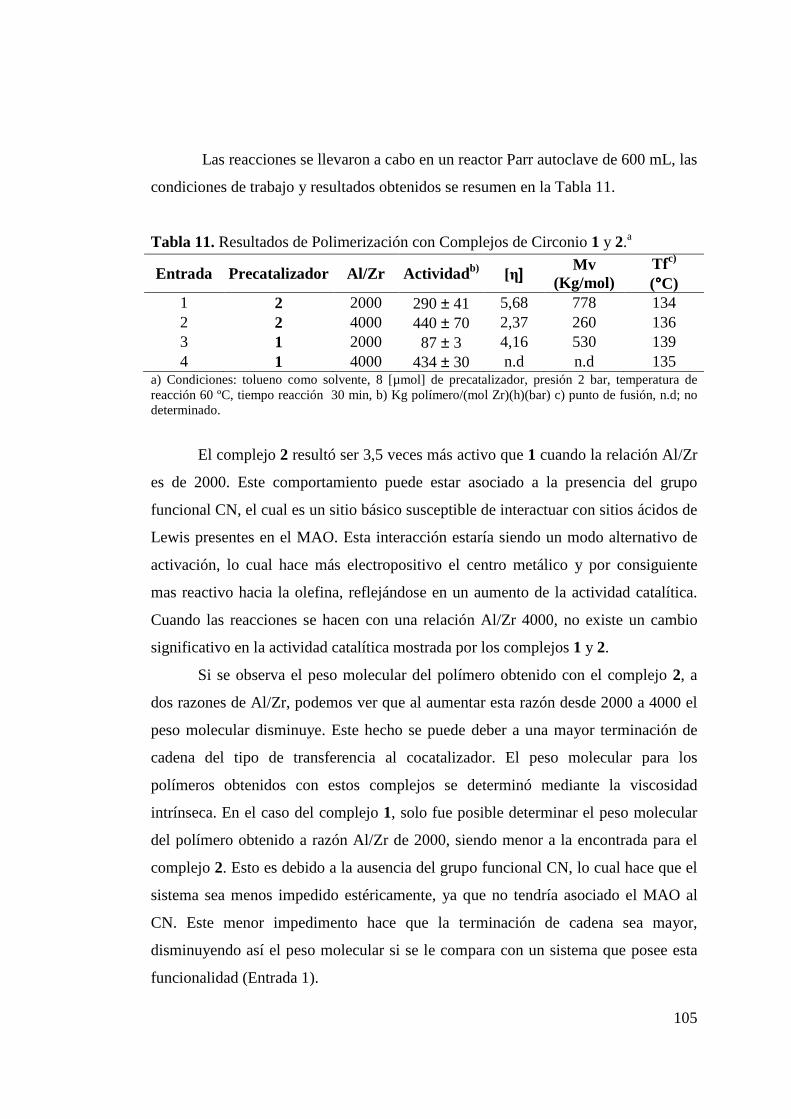
105
Las reacciones se llevaron a cabo en un reactor Parr autoclave de 600 mL, las
condiciones de trabajo y resultados obtenidos se resumen en la Tabla 11.
Tabla 11. Resultados de Polimerización con Complejos de Circonio 1 y 2.a
Entrada Precatalizador Al/Zr Actividad b) [η]]]] Mv (Kg/mol)
Tf c) (°°°°C)
1 2 2000 290 ± 41 5,68 778 134 2 2 4000 440 ± 70 2,37 260 136 3 1 2000 87 ± 3 4,16 530 139 4 1 4000 434 ± 30 n.d n.d 135
a) Condiciones: tolueno como solvente, 8 [µmol] de precatalizador, presión 2 bar, temperatura de reacción 60 ºC, tiempo reacción 30 min, b) Kg polímero/(mol Zr)(h)(bar) c) punto de fusión, n.d; no determinado.
El complejo 2 resultó ser 3,5 veces más activo que 1 cuando la relación Al/Zr
es de 2000. Este comportamiento puede estar asociado a la presencia del grupo
funcional CN, el cual es un sitio básico susceptible de interactuar con sitios ácidos de
Lewis presentes en el MAO. Esta interacción estaría siendo un modo alternativo de
activación, lo cual hace más electropositivo el centro metálico y por consiguiente
mas reactivo hacia la olefina, reflejándose en un aumento de la actividad catalítica.
Cuando las reacciones se hacen con una relación Al/Zr 4000, no existe un cambio
significativo en la actividad catalítica mostrada por los complejos 1 y 2.
Si se observa el peso molecular del polímero obtenido con el complejo 2, a
dos razones de Al/Zr, podemos ver que al aumentar esta razón desde 2000 a 4000 el
peso molecular disminuye. Este hecho se puede deber a una mayor terminación de
cadena del tipo de transferencia al cocatalizador. El peso molecular para los
polímeros obtenidos con estos complejos se determinó mediante la viscosidad
intrínseca. En el caso del complejo 1, solo fue posible determinar el peso molecular
del polímero obtenido a razón Al/Zr de 2000, siendo menor a la encontrada para el
complejo 2. Esto es debido a la ausencia del grupo funcional CN, lo cual hace que el
sistema sea menos impedido estéricamente, ya que no tendría asociado el MAO al
CN. Este menor impedimento hace que la terminación de cadena sea mayor,
disminuyendo así el peso molecular si se le compara con un sistema que posee esta
funcionalidad (Entrada 1).

106
A los polímeros obtenidos se les determinó la temperatura de fusión,
mediante calorimetría diferencial de barrido, las muestras fueron calentadas de 0 °C a
160 °C a 10 °C /min, obteniéndose el peak de fusión del segundo ciclo de
calentamiento. Los polímeros obtenidos con los complejos 1 y 2 corresponden a
polietilenos lineales, con temperaturas de fusión sobre 134 ºC.
3.4.2 Complejos de Titanio con ligandos L1 y L 5
En el mismo sistema de polimerización, se probó la reactividad de los
complejos 3 y 4 en la polimerización de etileno (Figura 81). Las condiciones de
reacción son las mismas que para circonio y se resumen en la Tabla 12.
Tabla 12. Resultados de Polimerización con Complejos de Titanio 3 y 4a).
Entrada Precatalizador Actividadb) [η]]]] Mv (Kg/mol)
Tf c) (°°°°C)
1 3 40 ± 8 4,3 280 138 2 4 90 ± 18 n.d n.d 136
a) Condiciones: tolueno como solvente, 8 [µmol] de precatalizador, MAO como cocatalizador a una razón de Al/Ti de 2000, presión 2 bar, temperatura de reacción 60 ºC, tiempo reacción 30 min, b) Kg polímero/(mol Ti)(h)(bar) c) punto de fusión, n.d; no determinado.
El complejo 4 resultó ser 2 veces más activo que 3 en las mismas condiciones
de reacción, observándose un comportamiento similar al mostrado por los complejos
de circonio. Esto se explica de igual manera, debido a la interacción del grupo
funcional CN, con el cocatalizador MAO, que provoca un cambio en la densidad
electrónica del titanio dejándolo más catiónico, siendo así mas reactivo hacia el doble
enlace de etileno y por consiguiente aumenta la actividad catalítica. Los polímeros
obtenidos usando el sistema catalítico 3/MAO, muestran un peso molecular de
280(Kg/mol), lo que se considera como alto peso molecular. Sin embargo, no fue
posible la determinación de este parámetro para los polímeros obtenidos con 4/MAO,
debido a su baja solubilidad en decalina aditivada, este hecho se puede relacionar con

107
un alto peso molecular, que impide la solubilidad en estas condiciones. Las
temperaturas de fusión indican que el polímero es altamente lineal, en ambos casos.
Si comparamos los complejos de circonio y titanio que contiene ligandos con
funcionalidad adicional CN, en igual condiciones de reacción (Entrada 1 Tabla 11;
Entrada 2, Tabla 12) podemos ver que se obtiene mayores actividades catalíticas con
los sistemas de circonio. Esto está estrechamente relacionado con las características
de los metales, donde el radio iónico de Zr+4 (0,86 Å) es mayor que el del Ti+4 (0,68
Å), haciendo que este más expuesto para la coordinación de la olefina, aumentando
así la actividad catalítica[61].
3.4.3. Complejos de Circonio con ligandos L6
Se realizaron estudios de polimerización con los complejos de circonio con
ligandos 2-acetil-3-(2,6-diisopropilfenilimino)-but-2-enenitrilo. Los dos complejos
utilizados son aquellos que mostraron un buen rendimiento en la etapa de síntesis y
que han sido bien caracterizados (Figura 82).
Figura 82. Sistemas 5 y 6 Utilizados para la Polimerización de Etileno.
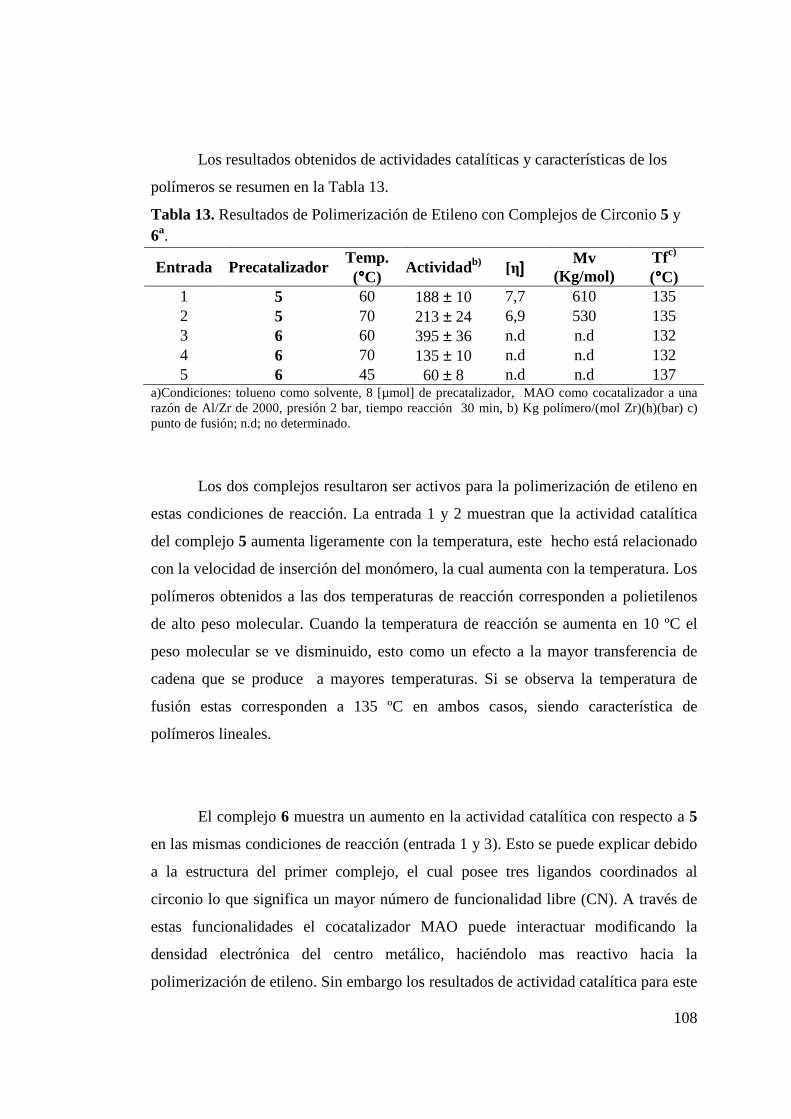
108
Los resultados obtenidos de actividades catalíticas y características de los
polímeros se resumen en la Tabla 13.
Tabla 13. Resultados de Polimerización de Etileno con Complejos de Circonio 5 y 6a.
Entrada Precatalizador Temp. (°°°°C)
Actividadb) [η]]]] Mv (Kg/mol)
Tf c) (°°°°C)
1 5 60 188 ± 10 7,7 610 135 2 5 70 213 ± 24 6,9 530 135 3 6 60 395 ± 36 n.d n.d 132 4 6 70 135 ± 10 n.d n.d 132 5 6 45 60 ± 8 n.d n.d 137
a)Condiciones: tolueno como solvente, 8 [µmol] de precatalizador, MAO como cocatalizador a una razón de Al/Zr de 2000, presión 2 bar, tiempo reacción 30 min, b) Kg polímero/(mol Zr)(h)(bar) c) punto de fusión; n.d; no determinado.
Los dos complejos resultaron ser activos para la polimerización de etileno en
estas condiciones de reacción. La entrada 1 y 2 muestran que la actividad catalítica
del complejo 5 aumenta ligeramente con la temperatura, este hecho está relacionado
con la velocidad de inserción del monómero, la cual aumenta con la temperatura. Los
polímeros obtenidos a las dos temperaturas de reacción corresponden a polietilenos
de alto peso molecular. Cuando la temperatura de reacción se aumenta en 10 ºC el
peso molecular se ve disminuido, esto como un efecto a la mayor transferencia de
cadena que se produce a mayores temperaturas. Si se observa la temperatura de
fusión estas corresponden a 135 ºC en ambos casos, siendo característica de
polímeros lineales.
El complejo 6 muestra un aumento en la actividad catalítica con respecto a 5
en las mismas condiciones de reacción (entrada 1 y 3). Esto se puede explicar debido
a la estructura del primer complejo, el cual posee tres ligandos coordinados al
circonio lo que significa un mayor número de funcionalidad libre (CN). A través de
estas funcionalidades el cocatalizador MAO puede interactuar modificando la
densidad electrónica del centro metálico, haciéndolo mas reactivo hacia la
polimerización de etileno. Sin embargo los resultados de actividad catalítica para este

109
sistema (entrada 3-5) no muestran una tendencia clara respecto a la temperatura, a 70
ºC se aprecia una disminución en la actividad catalítica, lo que puede estar asociado a
la descomposición del catalizador. El peso molecular de los polímeros obtenidos por
este sistema no fue posible de determinar mediante la viscosidad intrínseca debido a
la poca solubilidad del polímero en la decalina aditivada, lo que está directamente
relacionado con el alto peso molecular de los polímeros. Las temperaturas de fusión
son de 132 ºC a altas temperatura, cuando la temperatura de reacción disminuye a 45
ºC la temperatura de fusión es de 137 ºC lo que puede estar relacionado con una
mayor cristalinidad del polímero que hace que la temperatura de fusión aumente.
3.4.4. Complejos de Níquel
En esta parte se muestran los resultados obtenidos con los complejos
de níquel que contiene ligandos N-(imidoilamidinicos) que poseen en su estructura
sustituyentes alquílicos. Con el propósito de determinar, cual es la influencia en la
reactividad que produce la incorporación de un átomo de nitrógeno (amína terciaria)
en la posición alfa al grupo imíno, se incluye en la tabla de polimerización los datos
de reactividad del complejo [ArN=(C-Me)CCH2(Me)=NAr]NiBr2[105] (A). Este
complejo ha sido sintetizado y probado en la polimerización de etileno en las mismas
condiciones que los nuevos complejos sintetizados en esta tesis. Las características
estructurales de este compuesto lo hacen un buen referente para observar la
influencia sobre la reactividad que puede tener la incorporación de heteroátomos en
la estructura de este tipo de complejos.

110
Figura 83. Catalizadores de Níquel Utilizados para Polimerización de Etileno.
Todos estos complejos tienen una característica común y es que son estables
al aire, lo que ayuda a la menor desactivación de los sistemas, provocada por
humedad u otras impurezas del sistema de reacción.
En principio se realizaron estudios de polimerización de etileno con la mezcla
de isómeros (8a + 8b) y el complejo 9. Los resultados de actividad catalítica, peso
molecular y temperatura de fusión se resumen en la Tabla 14.
Tabla 14. Resultados de Polimerización con Complejos de Níquel N-
imidoilamidinaa.
Entrada Precatalizador Temp. (°°°°C)
Presión (bar)
Actividad b M w
(Kg/mol) Mw/Mn
Tf c (°°°°C)
1 A* 60 4 8 ± 3 422 2,6 -
2 8a+8b 60 2 141 ± 10 160 1,9 132
3 8a+8b 60 4 547 ± 22 169 1,7 132
4 8a+8b 70 4 540 ± 19 87 21
1,7, 1,04**
136
5 9 60 2 45 ± 13 98 1,8 138
6 9 60 4 95 ± 17 112 1,9 136 a) Condiciones: tolueno como solvente, 8 [µmol] de precatalizador, MAO como cocatalizador a una razón de Al/Zr de 2000 en 100 mL de tolueno, tiempo de reacción 30 min b) Actividad = Kg polímero/(mol Ni)(h) c) punto de fusión ºC. A* precatalizador de referencia** distribución bimodal.
Los complejos 8a + 8b y 9 en presencia de MAO son activos para la
polimerizar de etileno bajo diferentes condiciones de reacción. La entrada 2 y 3
muestra los resultados obtenidos a diferentes presiones de reacción para la mezcla de

111
precatalizadores 8a + 8b, al igual que las entradas 5 y 6 para el sistema 9/MAO. Si se
comparan estas entradas se puede observar que en ambos sistemas el consumo de
etileno se incrementa al aumentar la presión. Este comportamiento está asociado a la
mayor concentración de etileno que hace que la inserción sea mayor.
Si comparamos los resultados obtenidos con el sistema 9/MAO y 8a +
8b/MAO, entrada 2, 5 y 3, 6, respectivamente podemos ver que la actividad catalítica
disminuye al reducir el tamaño de los sustituyentes en el anillo fenílico de los
ligandos N-imidoilamidina alrededor del níquel.
Figura 84. Sistema Activo para Catalizador de Níquel.
Esto puede deberse a que al disminuir el tamaño de los sustituyentes R
(Figura 84) el contraión (MAO-) puede acercarse más al centro metálico
estabilizando mejor la carga y consecutivamente reduce la reactividad del níquel.
Este efecto sigue la misma tendencia de sistemas diiminicos de níquel anteriormente
reportados[92].
Si comparamos la reactividad de la entrada 1 (catalizador de referencia) con
los nuevos sistemas, podemos observar que la presencia del nitrógeno en la posición
alfa al grupo imínico, provoca un marcado aumento en la actividad catalítica. Este
efecto puede estar asociado a una coordinación entre el átomo de nitrógeno (base) y

112
el cocatalizador MAO (ácido) la que influencia la reactividad del centro metálico, al
variar su densidad electrónica.
A los polímeros obtenidos se les determinó la temperatura de fusión,
mediante calorimetría diferencial de barrido, las muestras fueron calentadas de 0 °C a
160 °C a 10 °C /min, obteniéndose el peak de fusión del segundo ciclo de
calentamiento. Los polímeros logrados con la mezcla de isómeros y con el complejo
9 corresponden a polietilenos lineales, con temperaturas de fusión de 132 ºC y 137
ºC respectivamente.
Por otra parte, mediante cromatografía permeación en gel se determinó el
peso molecular de los polímeros obtenidos. Si se comparan las entradas 2 y 5 con 3 y
6 se puede ver que el peso molecular del polímero resultante decrece cuando se
reduce el tamaño de los sustituyentes en los ligando imidoilamidina. Esto es debido a
que el menor impedimento estérico deja más libre las posiciones axiales del
complejo, siendo más factible que ocurran los procesos de terminación de cadena al
de propagación, que solo ocurre por las posiciones ecuatoriales[92]. Los dos sistemas
presentan las mismas tendencia en los pesos molecular al incrementar la presión
desde 2 a 4 bar, Figura 85 (a) líneas roja y negra para catalizador 8a + 8b y Figura 85
(b) para catalizador 9. Este comportamiento puede estar asociado a un mayor índice
de propagación a mayor concentración de etileno. En estas condiciones la
distribución de peso molecular del polietileno se mantuvo constante. Cuando la
temperatura de reacción se aumenta a 70 ºC, el sistema que está compuesto por
isómeros posee una distribución de peso molecular bimodal con máximos en 98
Kg/mol y 21 Kg/mol. La fracción de más alto peso molecular corresponde a la
producida por la mezcla de isómeros, siendo menor que la mostrada en la entrada 2
debido a que la temperatura de reacción afecta los procesos de propagación de la
cadena polimérica, favoreciendo la terminación[135]. La deconvolución de la grafica
Figura 85 (a), línea azul, muestra que el nuevo sitio de reacción se comporta como un
catalizador de polimerización viva (PDI = 1,04). Por otra parte como era esperado se
aprecia que el PDIs (Mw/Mn) en promedio es 1,8, lo cual indica que los sistemas son
catalizadores de sitio único.
113
(a) (b)
Figura 85. Análisis de GPC para Polietilenos: (a) Precatalizador 8a + 8b y (b) Precatalizador 9, Tabla 3.4.4.1, Relativo al Poliestireno como Estándar 135 °C.
3.4.4.1. Complejos de Níquel con ligandos N-(imidoilamidinicos) Sustituidos con
Flúor
El comportamiento catalítico de los complejos que poseen anillos aromáticos
sustituidos con átomos de flúor también fue estudiado. Los resultados obtenidos se
resumen en la Tabla 15.
Figura 86. Catalizadores de Níquel con Ligandos Fluorados.
Tabla 15. Resultados de Polimerización de Etileno con Complejos de 10 y 12a

114
Entrada Precatalizador Temp. (°°°°C)
Actividad b M w
c (Kg/mol)
Mw/Mn Tf d
(°°°°C) 1 10 60 387 ± 35 162 2,1 131 2 10 70 145 ± 7 122 2,5 134 3 12 60 468 ± 51 84 1,6 134 4 12 70 70 ± 9 184 3,1 135
a)Las polimerizaciones fueron realizadas en reactor autoclave de 600 mL con 8 µmol de precatalizador una razón Al/Ni 2000 en 100 mL de tolueno, presión 4 bar, tiempo de reacción es de 30 min b) Actividad = Kg polímero/(mol Ni)(h) c) pero molecular determinado mediante GPC utilizando poliestireno como estándar d) punto de fusión ºC.
Se puede observar que los complejos 10 y 12 en presencia de MAO resultan
activos para la polimerización de etileno, obteniéndose mayores actividades a 60 ºC.
Si comparamos el sistema 10/MAO (entrada 1), que posee el anillo fenílico
sustituido en el nitrógeno alfa del grupo imínico con la mezcla de isómeros 8a + 8b
(entrada 2, Tabla 8) se observa que el primero presenta menor actividades catalíticas,
sin embargo sigue siendo mayor que el sistema de referencia (entrada 1 Tabla 8).
Este es un comportamiento contrario al esperado, observándose que los sustituyentes
no contribuyen al cambio en la densidad electrónica del centro metálico,
provablemente debido a que no existe una conjugación completa del ligando. El
efecto de los sustituyentes de flúor se estaría reflejando en el nitrógeno que no
coordina al metal, dejando menos disponible el par electrónico libre, dificultando su
interacción con el MAO y consecuentemente no aumentando la actividad catalítica.
Figura 87. Influencia de Sustituyentes Fluorados en Complejos de Níquel.

115
En el caso del sistema 12/MAO (entrada 3), se ve que la actividad catalítica
es mayor, este efecto está influenciado por la presencia del anillo fluorado en uno de
los nitrógenos adyacente al centro metálico, provocando un efecto de atractor de
densidad electrónica, dejando al átomo de níquel más positivo haciéndolo mas
reactivo hacia el monómero de etileno. Sin embargo el aumento en 10 ºC en la
temperatura de reacción provoca una disminución en la actividad catalítica en los dos
sistemas (10 y 12), posiblemente por descomposición de la especia activa.
El peso molecular de los polímeros obtenidos con el complejo 10 es alto,
viéndose que el aumento en 10 º C hace que el peso molecular disminuya producto
de la mayor transferencia de cadena. El complejo 12 muestra un peso molecular
menor (entrada 3) si se le compara con 10 en las mismas condiciones de reacción
(entrada 1). Esto puede estar asociado a un menor impedimento estérico alrededor
del centro metálico, ya que una de las iminas solo tiene un anillo sustituido con flúor
(12) lo que facilita la terminación de cadena, en comparación al complejo 10 que
posee grupos isopropilos que retardan la terminación de cadena. Si observamos las
curvas de GPC (Figura 88), podemos ver que en el caso del complejos 10, cuando la
reacción se realiza a 60 ºC, existe una distribución de peso molecular que muestra
tres máximos. Esto estaría asociado a que el sistema es capaz de producir polímeros
de tres pesos moleculares diferentes, siendo el de mayor proporción el presentado en
la Tabla 15. Sin embargo las otras dos fracciones corresponden a polímeros de 13
Kg/mol y 407 Kg/mol, lo cual es posible de determinar por la deconvolución de las
graficas.
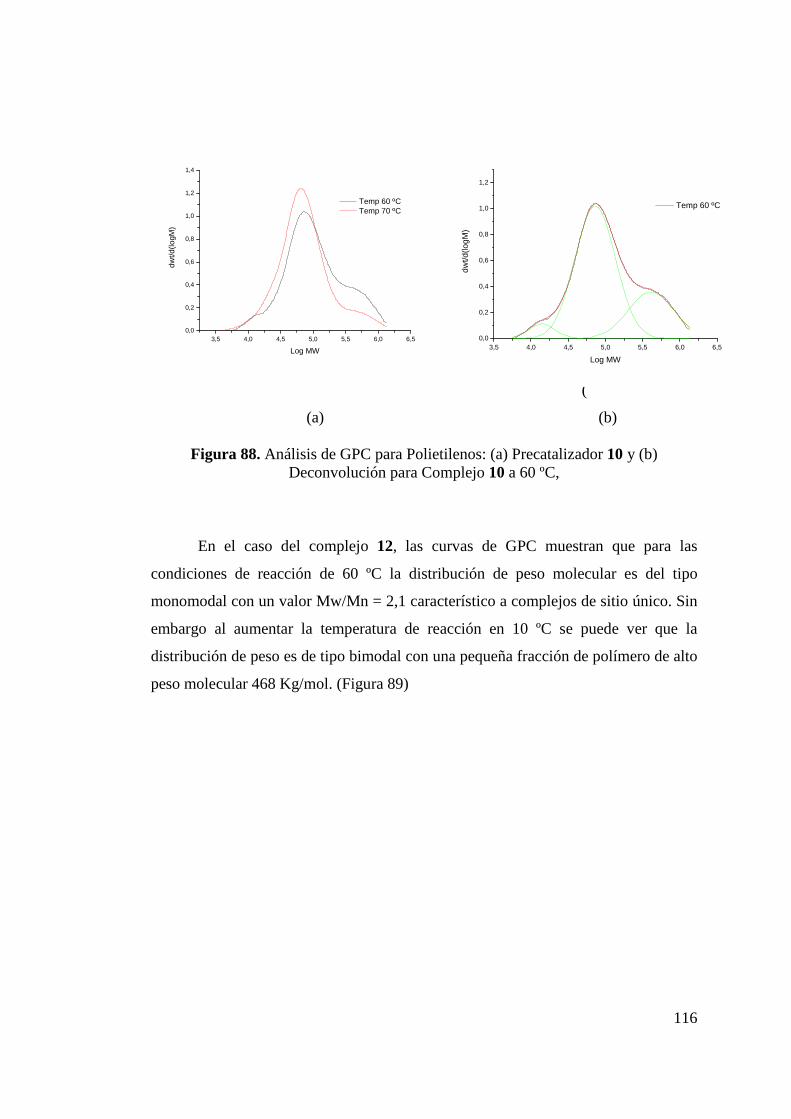
116
3,5 4,0 4,5 5,0 5,5 6,0 6,50,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
Temp 60 ºC Temp 70 ºC
dwt/d
(logM
)
Log MW
(
(a) (b)
Figura 88. Análisis de GPC para Polietilenos: (a) Precatalizador 10 y (b) Deconvolución para Complejo 10 a 60 ºC,
En el caso del complejo 12, las curvas de GPC muestran que para las
condiciones de reacción de 60 ºC la distribución de peso molecular es del tipo
monomodal con un valor Mw/Mn = 2,1 característico a complejos de sitio único. Sin
embargo al aumentar la temperatura de reacción en 10 ºC se puede ver que la
distribución de peso es de tipo bimodal con una pequeña fracción de polímero de alto
peso molecular 468 Kg/mol. (Figura 89)
3,5 4,0 4,5 5,0 5,5 6,0 6,50,0
0,2
0,4
0,6
0,8
1,0
1,2
Temp 60 ºC
dwt/d
(logM
)
Log MW
117
Figura 89. Análisis de GPC para polietilenos Obtenido con Precatalizador 12.
3.4.4.2. Complejos de Níquel con ligandos N-(imidoilamidinicos) Sustituidos
con Nitrógenos.
Los complejos que poseen un anillo piridínico también fueron testeados en
reacciones de polimerización de etileno (Figura 90). Los resultados de actividades
catalíticas y temperatura de fusión son mostrados en la Tabla 16.
N
N
HN
N
NHNNi
BrBr
(14)
N
N
N
N
(13)
Ni
Br
Br
Figura 90. Complejos de Níquel con Ligandos Piridínicos.

118
Tabla 16. Resultados de Polimerización de Etileno con Complejos de 13 y 14a.
Entrada Precatalizador Temp. (°°°°C)
Actividad b M w
c (Kg/mol)
Mw/Mn Tf d (°°°°C)
1 13 60 68 ± 4 102 1,8 126
2 13 70 110 ± 14 98 2,0 129
3 13- B(C6F5)3 60 260 ± 46 402 1,6 131
4 14 20 200 ± 16 C18 - -
5 14 60 468 ± 2 89 1,6 132
6 14 70 178 ± 24 80 1,8 132 a)Las polimerizaciones fueron realizadas en reactor autoclave de 600 ml con 8 µmol de precatalizador una razón Al/Ni 2000 en 100 mL de tolueno. El tiempo de reacción es de 30 min b) Kg polímero/(mol Ni)(h) c) punto de fusión ºC.
Si observamos los resultados obtenidos por el sistema 13/MAO (entradas 1 y
2) podemos ver que éste posee una menor actividad catalítica que los sistemas 8a +
8b/MAO que no presentan heteroátomos en su estructura. Como se muestra en el
Figura 91, el complejo 13 posee un nitrógeno piridinico libre, el cual debería
interactuar con el cocatalizador MAO, generando una especie zwitterionica, más
activa hacia la polimerización de etileno.
Figura 91. Sistema de Activación para Sistemas con Anillo Piridínico.
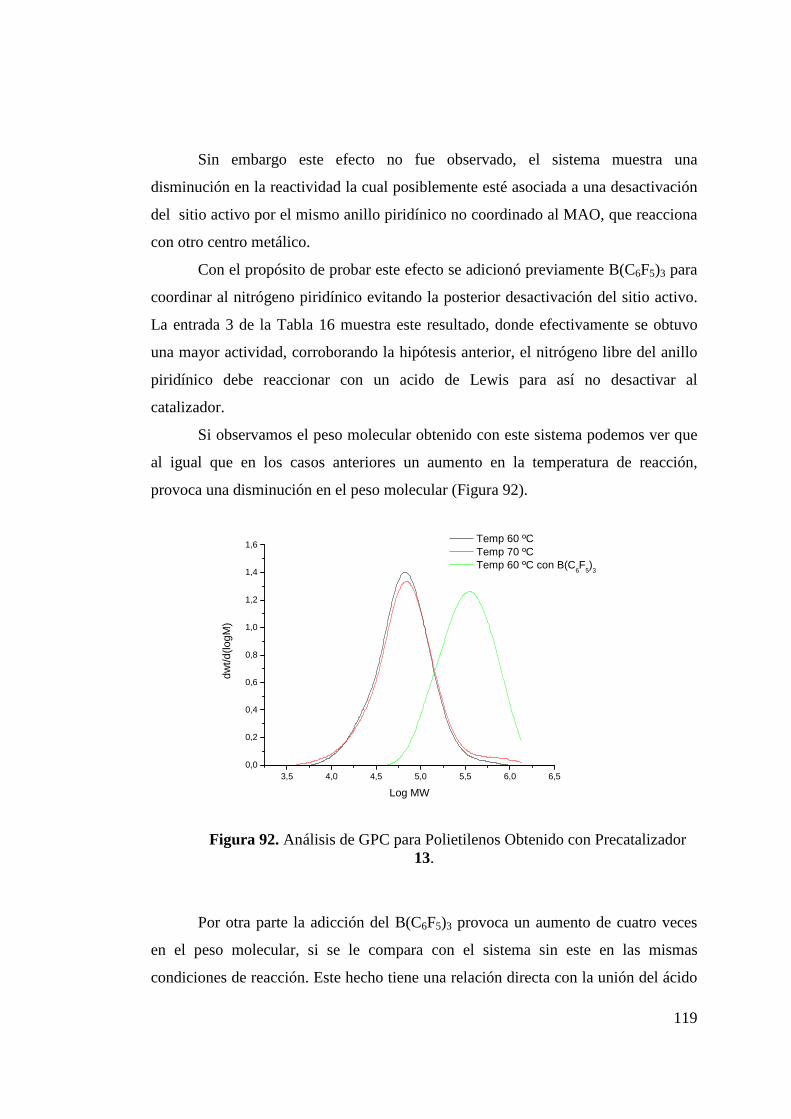
119
Sin embargo este efecto no fue observado, el sistema muestra una
disminución en la reactividad la cual posiblemente esté asociada a una desactivación
del sitio activo por el mismo anillo piridínico no coordinado al MAO, que reacciona
con otro centro metálico.
Con el propósito de probar este efecto se adicionó previamente B(C6F5)3 para
coordinar al nitrógeno piridínico evitando la posterior desactivación del sitio activo.
La entrada 3 de la Tabla 16 muestra este resultado, donde efectivamente se obtuvo
una mayor actividad, corroborando la hipótesis anterior, el nitrógeno libre del anillo
piridínico debe reaccionar con un acido de Lewis para así no desactivar al
catalizador.
Si observamos el peso molecular obtenido con este sistema podemos ver que
al igual que en los casos anteriores un aumento en la temperatura de reacción,
provoca una disminución en el peso molecular (Figura 92).
3,5 4,0 4,5 5,0 5,5 6,0 6,50,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6 Temp 60 ºC Temp 70 ºC Temp 60 ºC con B(C
6F
5)3
dwt/d
(logM
)
Log MW
Figura 92. Análisis de GPC para Polietilenos Obtenido con Precatalizador 13.
Por otra parte la adicción del B(C6F5)3 provoca un aumento de cuatro veces
en el peso molecular, si se le compara con el sistema sin este en las mismas
condiciones de reacción. Este hecho tiene una relación directa con la unión del ácido

120
de Lewis en los nitrógenos con pares de electrones libres, que hace que el volumen
estérico del sistema sea mayor, provocando una disminución en la transferencia de
cadena y por consiguiente aumentando el peso molecular. Por otra parte se puede ver
en todos los casos la distribución de peso molecular es del tipo monomodal, lo que
indica un solo tipo de sitio activo y con un Mw/Mn ~ 2,0. Los polímeros obtenidos
con este sistema mostraron en promedio un punto de fusión de 127 ºC, siendo algo
menor a lo encontrado con los otros sistemas, lo que indica un cierto grado de
ramificación del polímero obtenido.
Como se señalo en la sección de síntesis de compuestos de níquel, el
complejo 14 corresponde a una clase distinta a los demás discutidos en esta tesis. En
este caso el centro metálico, enlazado a ligandos monodentados, se encuentra
estéricamente menos impedido. No hay muchos ejemplos que muestren que sistemas
similares presenten reactividad considerable en reacciones de polimerización de
etileno. Los existentes y que presentan un centro metálico estéricamente disponible,
siguen siendo bidentados y en general oligomerízan etileno. Por esta razón se decidió
estudiar el comportamiento catalítico de este sistema.
El sistema 14/MAO resulto ser activo para polimerizar etileno a 60 ºC,
mostrando una actividad de 458 Kg/molNi*h y un peso molecular de 89 Kg/mol
siendo más bajo que los obtenidos con los complejos de níquel con ligandos
bidentados. El polietileno obtenido cuando la temperatura de la reacción es de 70 ºC,
es de menor peso molecular que el obtenido a 60 º C. Este hecho se debe a que la
temperatura de reacción afecta los procesos de propagación de la cadena polimérica,
favoreciendo la terminación.
Para el catalizador 14 no está claro la conectividad molecular de la especie
activa; en general y como se mostró anteriormente polímeros de alto peso molecular
son obtenidos de sistemas donde el centro metálico esta estéricamente muy
impedido. En esa dirección y teniendo en consideración que el complejo 14 presenta
grupos imínicos libres, es posible que en el medio de reacción se generen especies
más impedidas estéricamente por la coordinación de moléculas de MAO en dichos
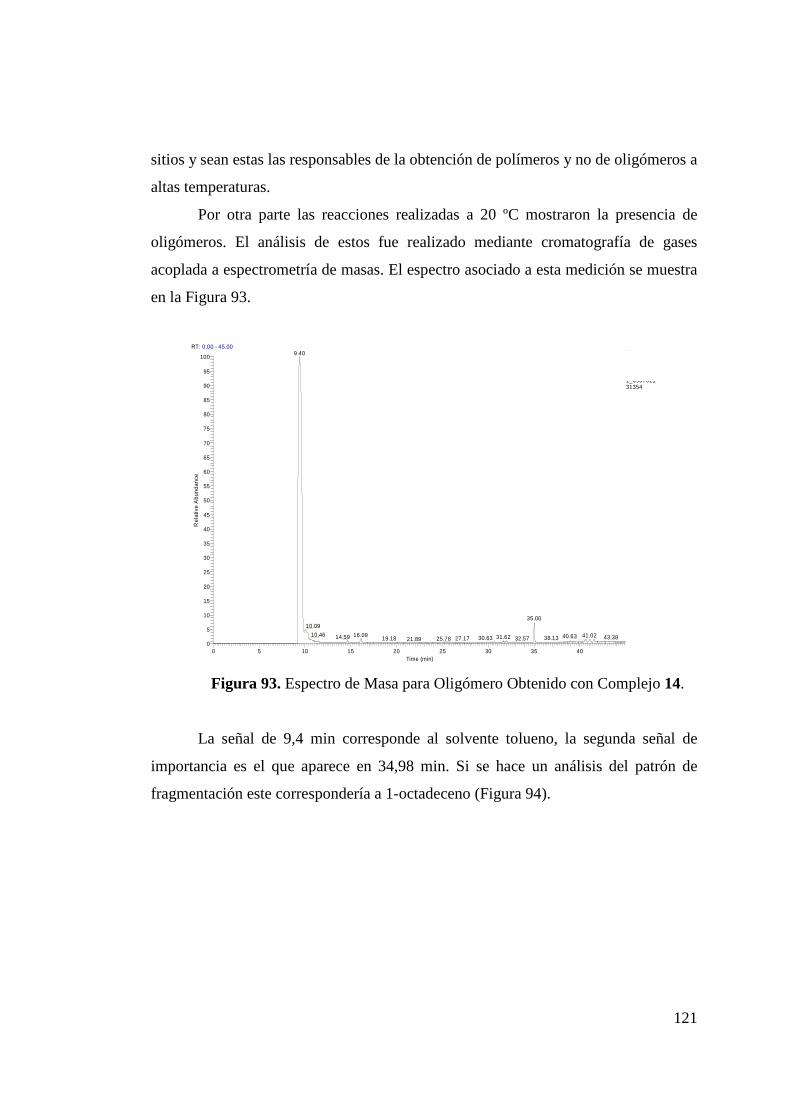
121
sitios y sean estas las responsables de la obtención de polímeros y no de oligómeros a
altas temperaturas.
Por otra parte las reacciones realizadas a 20 ºC mostraron la presencia de
oligómeros. El análisis de estos fue realizado mediante cromatografía de gases
acoplada a espectrometría de masas. El espectro asociado a esta medición se muestra
en la Figura 93.
RT: 0.00 - 45.00
0 5 10 15 20 25 30 35 40Time (min)
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
Rel
ativ
e A
bund
ance
9.40
35.0010.09
10.46 16.09 41.0240.6314.59 31.62 43.3838.1330.63 32.5719.18 27.1725.7821.89
NL:1.90E6TIC F: MS rene_mu1_1_090701131354
Figura 93. Espectro de Masa para Oligómero Obtenido con Complejo 14.
La señal de 9,4 min corresponde al solvente tolueno, la segunda señal de
importancia es el que aparece en 34,98 min. Si se hace un análisis del patrón de
fragmentación este correspondería a 1-octadeceno (Figura 94).
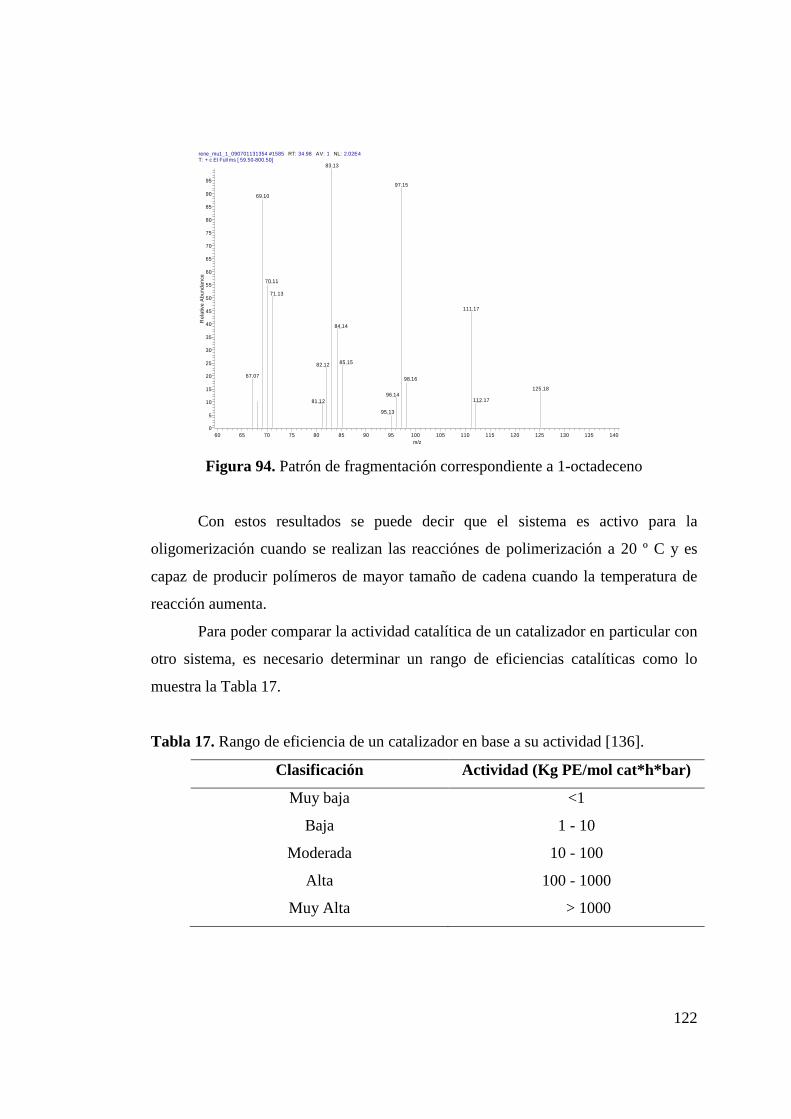
122
rene_mu1_1_090701131354 #1585 RT: 34.98 AV: 1 NL: 2.02E4T: + c EI Full ms [ 59.50-800.50]
60 65 70 75 80 85 90 95 100 105 110 115 120 125 130 135 140m/z
0
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
Rel
ativ
e A
bund
ance
83.13
97.15
69.10
70.11
71.13
111.17
84.14
85.1582.12
67.07 98.16
125.1896.14
112.1781.12
95.13
Figura 94. Patrón de fragmentación correspondiente a 1-octadeceno
Con estos resultados se puede decir que el sistema es activo para la
oligomerización cuando se realizan las reacciónes de polimerización a 20 º C y es
capaz de producir polímeros de mayor tamaño de cadena cuando la temperatura de
reacción aumenta.
Para poder comparar la actividad catalítica de un catalizador en particular con
otro sistema, es necesario determinar un rango de eficiencias catalíticas como lo
muestra la Tabla 17.
Tabla 17. Rango de eficiencia de un catalizador en base a su actividad [136].
Clasificación Actividad (Kg PE/mol cat*h*bar)
Muy baja <1
Baja 1 - 10
Moderada 10 - 100
Alta 100 - 1000
Muy Alta > 1000

123
Las actividades catalíticas obtenidas por los sistemas de circonio
pueden ser comparadas con sistemas metalocénicos y post-metalocénicos
(Figura 95), según el rango de eficiencia catalítica como se muestra en la
Tabla 18.
ZrCl
Cl
Zr ClCl
A B C
Figura 95. Catalizadores de Circonio de Referencia.
Tabla 18. Resumen de Actividad Catalítica de Complejos de Circonio.
Catalizador Actividad
(Kg PE/mol cat*h*bar)
Eficiencia M w
(Kg/mol) Mw/Mn
A Metalocénicos
3600 Muy alta 208 2,1
B Metalocénicos
33300 Muy alta 120 1,9
C 51900 Muy alta 100 -
1 87 Moderada 530 -
2 290 Alta 778 -
5 188 Alta 610 -
6 395 Alta - -
Los complejos metalocénicos y post-metalocénicos mostrados poseen
actividades catalíticas en un rango de eficiencia muy alta, en cambio los
complejos de circonio sintetizados en esta tesis solo logran llegar a una
eficiencia catalítica alta. Sin embargo con estos nuevos sistemas es posible de
obtener polímeros de mayor peso molecular (> 500 Kg/mol) si se compara

124
con los catalizadores metalocénicos. En resumen, se puede decir que la
variación en la estructura del ligando, juega un rol tanto en la determinación
de la actividad catalítica como en las características del peso molecular de los
polímeros obtenidos.
En el caso de los complejos de níquel esta comparación también
puede ser llevada a cabo, si se compara con dos complejos de níquel
dibromados (Figura 96) que ya han sido informados en la literatura.
A B
Figura 96. Catalizadores de Níquel de Referencia.
Tabla 19. Resumen de Actividad Catalítica de Complejos de Níquel.
Catalizador Actividad (Kg PE/mol cat*h)
Eficiencia M w
(Kg/mol) Mw/Mn
A 8 Baja 422 2,6
B 5800 Muy Alta 380 -
8a + 8b 547 Alta 169 1,7
9 95 Moderada 112 1,9
10 387 Alta 162 2,1
12 468 Alta 84 1,6
13 68 Moderada 102 1,8
Los complejos de níquel informados en la literatura, dependiendo de
las características estructurales es la actividad catalítica que presentan. En el
caso de A, se puede ver que esta es baja si se le compara con el sistema B que
N
N
O Ni
Br
Br
Ar
Ar

125
posee incorporado una funcionalidad adicional que presenta una actividad
muy alta. Los complejos sintetizados en esta tesis muestran este mismo
comportamiento, dependiendo de la estructura del catalizador es la actividad
catalítica que se obtiene, sin embargo en todos los casos las actividades
catalíticas son moderadas o altas. Esto indica que los complejos de níquel
sintetizados en esta tesis son comparables a los ya existentes hasta la fecha.

126
CAPÍTULO 4
CONCLUSIONES
De los resultados obtenidos a lo largo de la tesis se puede concluir que:
� A través de reacciones de condensación de una anilina con una cetona,
catalizadas por acido, fue posible sintetizar exitosamente nuevos ligandos
bidentados N,O y N,N (tipo β-cetoimina y β-diiminoamina). Las variaciones
en la estructura de los ligandos está determinada por la naturaleza de la amina
utilizada, así como de los sustituyentes que puedan ser introducidos. Es así
que ha sido posible obtener ligandos con grupos quirales en su estructura o
con una funcionalidad ciano.
� Las rutas de síntesis estudiadas a partir del cloruro de imidoil resultaron
altamente eficiente para la síntesis de ligandos N-imidoilamidinicos y
acetamidinicos. El control de las diferentes rutas de síntesis hace posible la
obtención de ligandos que presenten diferentes isómeros, que contengan
grupos atractores de electrones o heteroátomos en la estructura.
� En la síntesis de compuestos de circonio a partir de precursores ZrCl4 y
DMEZrCl4 y sales de litio de los ligandos, siempre se obtiene el ligando
reprotonado coordinado al centro metálico. La utilización de precursores
desprotonantes tales como (NMe2)2ZrCl2 y (NMe2)4Zr ayudan a la formación
de complejos donde el ligando se encuentra desprotonado. No obstante la
selectividad de la reacciones es muy bajo, obteniéndose siempre una mezcla
de compuestos.
� La reacción del los ligandos N-imidoilamidinicos y acetamidinicos con el
precursor [{O(CH3CH2OCH3)2}NiBr 2] generó en todos los casos un complejo

127
neutro de níquel. Los complejos obtenidos presentan una estructura del tipo
tetraédrica con un ligante bidentado y dos bromuros, de características
paramagnéticas.
� Los complejos de circonio tetra y diclorados en presencia de MAO resultaron
ser activos para la polimerización de etileno. Los polímeros obtenidos de
estas reacciones corresponden a polietilenos de alto peso molecular con
puntos de fusiones cercanos a 135 ºC, característicos de polietilenos lineales.
� Los complejos de níquel con ligandos bidentados N-imidoilamidinicos son
activos para la polimerización de etileno en presencia de MAO. Las
reactividades en general son moderadas comparadas a aquellas reportadas en
literatura para sistemas similares. Las propiedades de los polímeros siguen las
tendencias esperadas en relación al entorno estérico del centro metálico,
centro metálico más impedido generan polímeros de mayor peso molecular.
En general los polietilenos obtenidos son de alto peso moleculare, con
temperaturas de fusión de 135 ºC, característicos de polietilenos lineales
� La incorporación de heteroátomos en la estructura de los complejos no mostró
una influencia positiva en la reactividad del catalizador, probablemente
debido a una ausencia de conjugación directa hacia el centro metálico. Por
otra parte la existencia de nitrógenos libres en el complejo provoca una
descativación en el sistema la que se ve reducida al proteger el heteroátomo.
� [NiBr2N-(1-(4-iminopiridin-1(4H)-yl)etilideno)-2,6-diisopropilanilina] es un
nuevo tipo de compuesto que presenta ligandos que en presencia de MAO y
controlando las condiciones de reacción se puede acceder a oligómeros y
polímero de mayor peso molecular.

128
CAPITULO 5
REFERENCIAS
1. GALLI P., VECELLIO G., Polyolefins: The most promising large-volume materials
for the 21st century. Journal of Polymer Science Part A: Polymer Chemistry. 42(3):396-415,2004.
2. FAWCETT E., GIBSON R., PERRIN M., WILLIAMS E., British Patent. 471:590,1937.
3. BAILEY G., REID J., U. S. Patents 2. 381 198,1945. 4. ZIEGLER K., HOLZKAMP E., BRELL H., Angewandte Chemie International
Edition. 67 541,1955. 5. NATTA G., Angewandte Chemie International Edition. 68 393,1956. 6. NATTA G., Angewandte Chemie International Edition. 76 553,1964. 7. CLARK A., HOGAN J.P., BANKS R.L., LANNING W.C., Marlex Catalyst
Systems. Industrial & Engineering Chemistry. 48(7):1152-1155,2002. 8. BRESLOW D., NEWBURG N., bis-(cyclopentadienyl)-Titanium dihloride
alkylaluminum complex as catalysts for the polymerization of ethylene. Journal of the American Chemical Society. 79(18):5072-5073,2002.
9. SINN H., KAMINSKY W., VOLLMER H., RÜDIGER W., Lebende Polymere; bei Ziegler-Katalysatoren extremer Produktivität. Angewandte Chemie. 92(5):396-402,1980.
10. STORR A., JONES K., LAUBENGAYER A., The partial hydrolysis of ethylalane compounds. Journal of the American Chemical Society. 90(12):3173-3177,2002.
11. SINN H., Proposal for structure and effect of methylalumoxane based on mass balance and phase separation experiments. Macromolecules Symposia. 97:27,1995.
12. KOIDE Y., BOTT S., BARRON A., Alumoxanes as cocatalysts in the palladium-catalyzed copolymerization of carbon monoxide and ethylene: genesis of a structure activity relationship. Organometallics. 15(9):2213-2226,1996.
13. PASYNKIEWICZ S., Alumoxanes: Synthesis, structures, complexes and reactions. Polyhedron. 9(2-3):429-453,1990.
14. EISCH J., POMBRIK S., ZHENG G., Organometallic compounds of Group III. Active sites for ethylene polymerization with titanium(IV) catalysts in homogeneous media: multinuclear NMR study of ion-pair equilibria and their relation to catalyst activity. Organometallics. 12(10):3856-3863,2002.
15. TAIT P., KAMINSKY W., SINN H., Transition metals and organometallics as catalysts for olefin polymerization. Springer, Berlin,1988.
16. JORDAN R., DASHER W., ECHOLS S., Reactive cationic dicyclopentadienyl zirconium(IV) complexes. Journal of the American Chemical Society. 108(7):1718-1719,2002.
17. TRITTO I., SACCHI M., LI S., NMR study of the reactions in CpMeCl/AlMe and CpTiMeCl/methylalumoxane systems, catalysts for olefin polymerization. Macromolecular Rapid Communications. 15(3):217-223,1994.
18. KAMINSKY W., STEIGER R., Polymerization of olefins with homogeneous zirconocene/alumoxane catalysts. Polyhedron. 7(22-23):2375-2381,1988.
19. BABUSHKIN D., SEMIKOLENOVA N., ZAKHAROV V., TALSI E., Mechanism of dimethylzirconocene activation with methylaluminoxane: NMR monitoring of intermediates at high Al/Zr ratios. Macromolecular Chemistry and Physics. 201(5):558-567,2000.
20. QUIJADA R., GUEVARA J., GALLAND G., RABAGLIATI F., LOPEZ-MAJADA J., Synthesis and properties coming from the copolymerization of propene

129
with [alpha]-olefins using different metallocene catalysts. Polymer. 46(5):1567-1574,2005.
21. MANSEL S., RIEF U., PROSENC M., KIRSTEN R., BRINTZINGER H., ansa-Metallocene derivatives, zirconocene complexes with a spirosilane bridge: syntheses, crystal structures and properties as olefin polymerization catalysts. Journal of Organometallic Chemistry. 512(1-2):225-236,1996.
22. HERZOG T., ZUBRIS D., BERCAW J., A new class of zirconocene catalysts for the syndiospecific polymerization of propylene and its modification for varying polypropylene from isotactic to syndiotactic. Journal of the American Chemical Society. 118(47):11988-11989,1996.
23. RESCONI I., FRITZE C., Editor Pasquini, N; 2ª Edicción. Handbook in Polypropylene 2005.
24. GIBSON V., SPITZMESSER S., Advances in Non-Metallocene Olefin Polymerization Catalysis. Chemical Reviews. 103(1):283-316,2002.
25. SAITO J., MITANI M., MATSUI S., TOHI Y., MAKIO H., NAKANO T., TANAKA H., KASHIWA N., FUJITA T., A New Titanium Complex Having Two Phenoxy-Imine Chelate Ligands for Ethylene Polymerization. Macromolecular Chemistry and Physics. 203(1):59-65,2002.
26. KIMMO H., BARBRO L., MIKA P., MARKKU L., Ethylene polymerizations with novel tantalum(V) aminopyridinato complex/MAO systems. Macromolecular Rapid Communications. 18(8):635-638,1997.
27. STEPHAN D., GUERIN F., SPENCE R., KOCH L., GAO X., et al., Remarkably Active Non-Metallocene Ethylene Polymerization Catalysts. Organometallics. 18(11):2046-2048,1999.
28. KOCHI T., NODA S., YOSHIMURA K., NOZAKI K., Formation of Linear Copolymers of Ethylene and Acrylonitrile Catalyzed by Phosphine Sulfonate Palladium Complexes. Journal of the American Chemical Society. 129(29):8948-8949,2007.
29. GUIRONNET D., ROESLE P., RUÌNZI T., GOÌTTKER-SCHNETMANN I., MECKING S., Insertion Polymerization of Acrylate. Journal of the American Chemical Society. 131(2):422-423,2008.
30. MECKING S., JOHNSON L., WANG L., BROOKHART M., Mechanistic Studies of the Palladium-Catalyzed Copolymerization of Ethylene and alfa-Olefins with Methyl Acrylate. Journal of the American Chemical Society. 120(5):888-899,1998.
31. POPENEY C., GUAN Z., A Mechanistic Investigation on Copolymerization of Ethylene with Polar Monomers Using a Cyclophane-Based Pd(II) alfa-Diimine Catalyst. Journal of the American Chemical Society. 131(34):12384-12393,2009.
32. HAGIHARA H., SHIONO T., IKEDA T., Living Polymerization of Propene and 1-Hexene with the [t-BuNSiMe2Flu]TiMe2/B(C6F5)3 Catalyst. Macromolecules. 31:3184,1998.
33. DOMSKI G., ROSE J., COATES G., BOLIG A., BROOKHART M., Living alkene polymerization: New methods for the precision synthesis of polyolefins. Progress in Polymer Science. 32(1):30-92,2007.
34. TALARICO G., BUSICO V., CAVALLO L., Living Propene Polymerization with Bis(phenoxyimine) Group 4 Metal Catalysts: New Strategies and Old Concepts. Organometallics. 23(25):5989-5993,2004.
35. KILLIAN C., TEMPEL D., JOHNSON L., BROOKHART M., Living Polymerization of alfa-Olefins Using NiII alfa±-Diimine Catalysts. Synthesis of New Block Polymers Based on alfa-Olefins. Journal of the American Chemical Society. 118(46):11664-11665,1996.

130
36. SAITO J., MITANI M., MATSUI S., KASHIWA N., FUJITA T., Polymerization of 1-hexene with bis[N-(3-tert-butylsalicylidene)phenylaminato]titanium(IV) dichloride using 1-3Al/Ph-3CB(C6F5)4 as a cocatalyst. Macromolecular Rapid Communications. 21(18):1333-1336,2000.
37. SCHROCK R., BAUMANN R., REID S., GOODMAN J., STUMPF R., DAVIS W., Synthesis of Titanium, Zirconium, and Hafnium Complexes that Contain Diamido Donor Ligands of the Type [(t-BuN-o-C6H4)2O]2- and an Evaluation of Activated Versions for the Polymerization of 1-Hexene. Organometallics. 18(18):3649-3670,1999.
38. GOODMAN J., SCHROCK R., Kinetic Investigation of the Polymerization of 1-Hexene by the {[t-BuNON]ZrMe}[B(C6F5)4] Initiator. Organometallics. 20(24):5205-5211,2001.
39. JOHNSON L., KILLIAN C., BROOKHART M., New Pd(II)- and Ni(II)-Based Catalysts for Polymerization of Ethylene and alpha.-Olefins. Journal of the American Chemical Society. 117(23):6414-6415,2002.
40. JOHNSON L., MECKING S., BROOKHART M., Copolymerization of Ethylene and Propylene with Functionalized Vinyl Monomers by Palladium(II) Catalysts. Journal of the American Chemical Society. 118(1):267-268,1996.
41. GIBSON V., SPITZMESSER S., Advances in Non-Metallocene Olefin Polymerization Catalysis. Chemical Reviews. 103:283,2003.
42. BOURGET-MERLE L., LAPPERT M., SEVERN J., The Chemistry of alfa-Diketiminatometal Complexes. Chemical Reviews. 102:3031,2002.
43. ITTEL S., JOHNSON L., BROOKHART M., Late-Metal Catalysts for Ethylene Homo- and Copolymerization. Chemical Reviews. 100:1169,2000.
44. YANG X., SUN X., HAN F., LIU B., TANG T., WANG Z., GAO M., XIE Z., BU S., One-Pot Screening of Titanium Catalysts for Ethylene Polymerization. Organometallics. 27:4618,2008.
45. PARKS J., HOLM R., Synthesis, solution stereochemistry, and electron delocalization properties of bis(beta.-iminoamino)nickel(II) complexes. Inorganic Chemistry. 7(7):1408-1416,2002.
46. MINDIOLA D., Nacnac, Are You Still There? The Evolution of beta-Diketiminate Complexes of Nickel. Angewandte Chemie International Edition. 48(34):6198-6200,2009.
47. GUANGYONG X., CHANGTAO Q., Dramatic electronic effect of fluoro substituents on the olefin polymerization activity of mono beta-diiminato titanium complexes. Journal of Polymer Science Part A: Polymer Chemistry. 46(1):211-217,2008.
48. ANNUNZIATA L., PAPPALARDO D., TEDESCO C., PELLECCHIA C., Bis[(amidomethyl)pyridine] Zirconium(IV) Complexes: Synthesis, Characterization, and Activity as Olefin Polymerization Catalysts. Organometallics. 28(3):688-697,2009.
49. GAGIEVA S., SUKHOVA T., SAVINOV D., OPTOV V., BRAVAYA N., BELOKON Y., BULYCHEV B., New fluorine-containing bissalicylidenimine-titanium complexes for olefin polymerization. Journal of Applied Polymer Science. 95(5):1040-1049,2005.
50. PARKS J., WAGNER B., HOLM R., Three-dimensional macrocyclic encapsulation reactions. II. Synthesis and properties of nonoctahedral clathro chelates derived from tris(2-aldoximo-6-pyridyl)phosphine and boron trifluoride or tetrafluoroborate. Inorganic Chemistry. 10(11):2472-2478,2002.

131
51. LI X., DAI K., YE W., PAN L., LI Y., New Titanium Complexes with Two Enaminoketonato Chelate Ligands: Syntheses, Structures, and Olefin Polymerization Activities. Organometallics. 23(6):1223-1230,2004.
52. BUDZELAAR P., VAN OORT A., ORPEN A., alfa-Diiminato Complexes of VIII and TiIII Formation and Structure of Stable Paramagnetic Dialkylmetal Compounds. European Journal of Inorganic Chemistry. 10:1485-1494,1998.
53. BINYUAN L., CHUNYING T., LI Z., WEIDONG Y., WENJUN Z., Copolymerization of carbon dioxide and cyclohexene oxide with zinc/beta-ketoiminato complexes: Investigating the influence of the electron structure of zinc/beta-ketoiminato complexes on the catalytic activity and resultant copolymer properties. Journal of Polymer Science Part A: Polymer Chemistry. 44(21):6243-6251,2006.
54. LESIKAR L., GUSHWA A., RICHARDS A., Synthesis, characterization, and steric hindrance comparisons of selected transition and main group metal b-ketoiminato complexes. Journal of Organometallic Chemistry. 693 3245,2008.
55. YOSHIDA Y., MATSUI S., FUJITA T., Bis(pyrrolide-imine) Ti complexes with MAO: A new family of high performance catalysts for olefin polymerization. Journal of Organometallic Chemistry. 690(20):4382-4397,2005.
56. MITANI M., FURUYAMA R., MOHRI J., SAITO J., ISHII S., TERAO H., NAKANO T., TANAKA H., FUJITA T., Syndiospecific Living Propylene Polymerization Catalyzed by Titanium Complexes Having Fluorine-Containing Phenoxy-Imine Chelate Ligands. Journal of the American Chemical Society. 125:4293,2003.
57. MATSUI S., MITANI M., SAITO J., TOHI Y., MAKIO H., TANAKA H., FUJITA T., Post-Metallocenes: A New Bis(salicylaldiminato) Zirconium Complex for Ethylene Polymerization. Chemistry Letters:1263,1999.
58. MATSUI S., MITANI M., SAITO J., MATSUKAWA N., TANAKA H., NAKANO T., FUJITA T., Post-Metallocenes: Catalytic Perfomance of New Bis(salicylaldiminato) Zirconium Complexes for Ethylene Polymerization. Chemistry Letters:554,2000.
59. MATSUI S., FUJITA T., FI Catalysts: super active new ethylene polymerization catalysts. Catalysis Today. 66(1):63-73,2001.
60. FURUYAMA R., SAITO J., ISHII S.-I., MITANI M., MATSUI S., et al., Ethylene and propylene polymerization behavior of a series of bis(phenoxy-imine)titanium complexes. Journal of Molecular Catalysis A: Chemical. 200(1-2):31-42,2003.
61. HARUYUKI M., NORIO K., TERUNORI F., FI Catalysts: A New Family of High Performance Catalysts for Olefin Polymerization. Advanced Synthesis & Catalysis. 344(5):477-493,2002.
62. HANS H.B., DAVID F., ROLF M., BERNHARD R., ROBERT M.W., Stereospecific Olefin Polymerization with Chiral Metallocene Catalysts. Angewandte Chemie International Edition in English. 34(11):1143-1170,1995.
63. MATSUI S., MITANI M., SAITO J., TOHI Y., MAKIO H., et al., A Family of Zirconium Complexes Having Two Phenoxy-Imine Chelate Ligands for Olefin Polymerization. Journal of the American Chemical Society. 123(28):6847-6856,2001.
64. MASON A., COATES G., New Phenoxyketimine Titanium Complexes: Combining Isotacticity and Living Behavior in Propylene Polymerization. Journal of the American Chemical Society. 126:16326,2004.

132
65. HUSTAD P., COATES G., Insertion/Isomerization Polymerization of 1,5-Hexadiene: Synthesis of Functional Propylene Copolymers and Block Copolymers. Journal of the American Chemical Society. 124(39):11578-11579,2002.
66. TIAN J., G. C., Development of a Diversity-Based Approach for the Discovery of Stereoselective Polymerization Catalysts: Identification of a Catalyst for the Synthesis of Syndiotactic Polypropylene. Angewandte Chemie. 39(20):3626-3629,2000.
67. TIAN J., HUSTAD P., COATES G., A New Catalyst for Highly Syndiospecific Living Olefin Polymerization: Homopolymers and Block Copolymers from Ethylene and Propylene. Journal of the American Chemical Society. 123(21):5134-5135,2001.
68. MARQUES M., CORREIA S., ASCENSO J., RIBEIRO A., GOMES P., DIAS A., FOSTER P., RAUSCH M., CHIEN J., Polymerization with TMA-protected polar vinyl comonomers. Catalyzed by group 4 metal complexes with 5-type ligands. Journal of Polymer Science Part A: Polymer Chemistry. 37(14):2457-2469,1999.
69. XIAOFAN Z., SHANGTAO C., HUAYI L., ZHICHENG Z., YINGYING L., CHUNHONG W., YOULIANG H., Copolymerizations of ethylene and polar comonomers with bis(phenoxyketimine) group IV complexes: Effects of the central metal properties. Journal of Polymer Science Part A: Polymer Chemistry. 45(1):59-68,2007.
70. MARK H., In Encyclopedia of Polymer Science and Engineering. Eds.; Wiley-Interscience: New York,1987.
71. TANABIKI M., SUNADA Y., NAGASHIMA H., A Bis-[C(trimethylsilyl)-N-arylimino]dimethylsilane, Me3Si(CNAr)SiMe2(CNAr)SiMe3 (Ar = 2,6-xylyl), as a New alfa-Diimine Ligand. Organometallics. 26(24):6055-6058,2007.
72. LI X., LI Y., LI Y., CHEN Y., HU N., Copolymerization of Ethylene with Methyl Methacrylate with Neutral Nickel(II) Complexes Bearing alfa-Ketoiminato Chelate Ligands. Organometallics. 24(10):2502-2510,2005.
73. GUAN Z., MARSHALL W., Synthesis of New Phosphine Imine Ligands and Their Effects on the Thermal Stability of Late-Transition-Metal Olefin Polymerization Catalysts. Organometallics. 21(17):3580-3586,2002.
74. DAUGULIS O., BROOKHART M., Polymerization of Ethylene with Cationic Palladium and Nickel Catalysts Containing Bulky Nonenolizable Imine Phosphine Ligands. Organometallics. 21(26):5926-5934,2002.
75. WANG H., ZHANG J., MENG X., JIN G., Nickel (II) complexes with [beta]-enaminoketonato chelate ligands: Synthesis, solid-structure characterization and reactivity toward the addition polymerization of norbornene. Journal of Organometallic Chemistry. 691(6):1275-1281,2006.
76. ALBERS I., ÁLVAREZ E., CÁMPORA J., MAYA C., PALMA P., SÁNCHEZ L., PASSAGLIA E., Cationic [eta]3-benzyl nickel compounds with diphosphine ligands as catalyst precursors for ethylene oligomerization/polymerization: influence of the diphosphine bite angle. Journal of Organometallic Chemistry. 689(4):833-839,2004.
77. KEIM W., KILLAT S., NOBILE C., SURANNA G., ENGLERT U., WANG R., MECKING S., SCHRÖDER D., Synthesis, characterisation and catalytic activity of Pd(II) and Ni(II) complexes with new cyclic [alpha]-diphenylphosphino-ketoimines. Crystal structure of 2,6-diisopropyl-N-(2-diphenylphosphino-cyclopentylidene)aniline and of 2,6-diisopropyl-N-(2-diphenylphosphino-cyclohexylidene)aniline. Journal of Organometallic Chemistry. 662(1-2):150-171,2002.

133
78. KAWAGUCHI H., MATSUO T., Aryloxide-based multidentate ligands for early transition metals and f-element metals. Journal of Organometallic Chemistry. 689(24):4228-4243,2004.
79. SHULTZ C., LEDFORD J.O., DESIMONE J., BROOKHART M., Kinetic Studies of Migratory Insertion Reactions at the (1,3-Bis(diphenylphosphino)propane)Pd(II) Center and Their Relationship to the Alternating Copolymerization of Ethylene and Carbon Monoxide. Journal of the American Chemical Society. 122(27):6351-6356,2000.
80. MARKIES B., KRUIS D., RIETVELD M., VERKERK K., BOERSMA J., KOOIJMAN H., LAKIN M., SPEK A., VAN KOTEN G., Alkene and Carbon Monoxide Insertion Reactions of Nitrogen-Coordinated Monoorganopalladium(II) Complexes: The Stepwise Construction of Alternating Copolymers of CO and Alkenes on a Palladium(II) Center. Journal of the American Chemical Society. 117:5263,1995.
81. DRENT E., VAN BROEKHOVEN J., DOYLE M., Efficient palladium catalysts for the copolymerization of carbon monoxide with olefins to produce perfectly alternating polyketones. Journal of Organometallic Chemistry. 417(1-2):235-251,1991.
82. AYUSMAN S., ZHAOZHONG J., Palladium(II)-catalyzed alternating copolymerization and terpolymerization of carbon monoxide with .alpha.-olefins: formation of syndiotactic copolymers as well as terpolymers with both syndiotactic and atactic segments. Macromolecules. 26(5):911-915,2002.
83. CARSTEN B., Bis(4,5-dihydrooxazolyl) Derivatives in Asymmetric Catalysis. Angewandte Chemie International Edition in English. 30(5):542-543,1991.
84. LEUTENEGGER U., UMBRICHT G., FAHRNI C., VON MATT P., PFALTZ A., 5-aza-semicorrins: A new class of bidentate nitrogen ligands for enantioselective catalysis. Tetrahedron. 48(11):2143-2156,1992.
85. VAN ASSELT R., ELSEVIER C., Palladium Complexes Containing Rigid Bidentate Nitrogen Ligands as Catalysts for carbon---carbon bond Formation. Tetrahedron. 50(2):323-334,1991.
86. TOGNI A., VENANZI L., Nitrogen Donors in Organometallic Chemistry and Homogeneous Catalysis. Angewandte Chemie International Edition in English. 33(5):497-526,1994.
87. KEIM W., SCHULZ R., Chelate Control in the Nickel-complex Catalysed Homogeneous Oligomerization of Ethylene. Journal of Molecular Catalysis. 92(1):21-33,1994.
88. SOULA R., BROYER J.P., LLAURO M.F., TOMOV A., SPITZ R., CLAVERIE J., DRUJON X., MALINGE J., SAUDEMONT T., Very Active Neutral P,O-Chelated Nickel Catalysts for Ethylene Polymerization. Macromolecules. 34(8):2438-2442,2001.
89. WANG C., FRIEDRICH S., YOUNKIN T.R., LI R.T., GRUBBS R.H., BANSLEBEN D.A., DAY M.W., Neutral Nickel(II)-Based Catalysts for Ethylene Polymerization. Organometallics. 17(15):3149-3151,1998.
90. KILLIAN C., JOHNSON L., BROOKHART M., Preparation of Linear alpha±-Olefins Using Cationic Nickel(II) alpha±-Diimine Catalysts. Organometallics. 16(10):2005-2007,1997.
91. LEATHERMAN M., SVEJDA S., JOHNSON L., BROOKHART M., Mechanistic Studies of Nickel(II) Alkyl Agostic Cations and Alkyl Ethylene Complexes: Investigations of Chain Propagation and Isomerization in (alpha±-diimine)Ni(II)-Catalyzed Ethylene Polymerization. Journal of the American Chemical Society. 125(10):3068-3081,2003.

134
92. DENG L., WOO T., CAVALLO L., MARGL P., ZIEGLER T., The Role of Bulky Substituents in Brookhart-Type Ni(II) Diimine Catalyzed Olefin Polymerization: A Combined Density Functional Theory and Molecular Mechanics Study. Journal of the American Chemical Society. 119(26):6177-6186,1997.
93. JENKINS J., BROOKHART M., A Mechanistic Investigation of the Polymerization of Ethylene Catalyzed by Neutral Ni(II) Complexes Derived from Bulky Anilinotropone Ligands. Journal of the American Chemical Society. 126(18):5827-5842,2004.
94. YANG X., STERN C., MARKS T., Cationic Zirconocene Olefin Polymerization Catalysts Based on the Organo-Lewis Acid Tris(pentafluorophenyl)borane. A Synthetic,Structural, Solution Dynamic, and Polymerization Catalytic Study. Journal of the American Chemical Society. 116(22):10015-10031,2002.
95. CROWTHER D., BAENZIGER N., JORDAN R., Group 4 metal Dicarbollide Chemistry. Synthesis, Structures, and Reactivity of Electrophilic Alkyl Complexes (Cp*)(C2B9H11)M(R), M = Hf, Zr. Journal of the American Chemical Society. 113(4):1455-1457,2002.
96. SZABO M., GALEA N., MICHALAK A., YANG S., GROUX L., PIERS W., ZIEGLER T., Copolymerization of Ethylene with Polar Monomers: Chain Propagation and Side Reactions. A DFT Theoretical Study Using Zwitterionic Ni(II) and Pd(II) Catalysts. Journal of the American Chemical Society. 127(42):14692-14703,2005.
97. FERNANDES S., SOARES A., LEMOS F., LEMOS M., MANO J., MALDANIS R., RAUSCH M., CHIEN J., MARQUES M., Copolymerization of Ethylene/Unsaturated Alcohols Using Nickel Catalysts: Effect of the Ligand on the Activity and Comonomer Incorporation. Journal of Organometallic Chemistry. 690(4):895-909,2005.
98. GUAN Z., COTTS P., MCCORD E., MCLAIN S., Chain Walking: A New Strategy to Control Polymer Topology. science. 283:2059,1999.
99. GUAN Z., Control of Polymer Topology through Late-Transition-Metal Catalysis. Journal of Polymer Science: Part A: Polymer Chemistry,. 41:3680,2003.
100. GUAN Z., Control of Polymer Topology by Chain-Walking Catalysts. chemical european journal. 8:3087,2002.
101. ZHANG J., KE Z., BAO F., LONG J., GAO H., ZHU F., WU Q., Ethylene Polymerization and Oligomerization Catalyzed by Bulky [beta]-diketiminato Ni(II) and [beta]-diimine Ni(II) Complexes/Methylaluminoxane Systems. Journal of Molecular Catalysis A: Chemical. 249(1-2):31-39,2006.
102. BOARDMAN B., VALDERRAMA J., MUÑOZ F., WU G., BAZAN G., ROJAS R., Remote Activation of Nickel Complexes by Coordination of B(C6F5)3 to an Exocyclic Carbonitrile Functionality. Organometallics. 27(8):1671-1674,2008.
103. COOLEY N., GREEN S., WASS D., HESLOP K., ORPEN A., PRINGLE P., Nickel Ethylene Polymerization Catalysts Based on Phosphorus Ligands. Organometallics. 20(23):4769-4771,2001.
104. BRYLIAKOV K., SEMIKOLENOVA N., ZAKHAROV V., TALSI E., Active Intermediates of Ethylene Polymerization over 2,6-Bis(imino)pyridyl Iron Complex Activated with Aluminum Trialkyls and Methylaluminoxane. Organometallics. 23(22):5375-5378,2004.
105. FELDMAN J., MCLAIN S., PARTHASARATHY A., MARSHALL W., CALABRESE J., ARTHUR S., Electrophilic Metal Precursors and a alpha-Diimine Ligand for Nickel(II)- and Palladium(II)-Catalyzed Ethylene Polymerization. Organometallics. 16(8):1514-1516,1997.

135
106. CHERIAN A., ROSE J., LOBKOVSKY E., COATES G., A C2-Symmetric, Living alpha±-Diimine Ni(II) Catalyst: Regioblock Copolymers from Propylene. Journal of the American Chemical Society. 127(40):13770-13771,2005.
107. LIU J., ZHENG Y., LI Y., PAN L., LI Y., HU N., Fe(II) and Co(II) Pyridinebisimine Complexes Bearing Different Substituents on ortho- and para-position of Imines: Synthesis, Characterization and Behavior of Ethylene polymerization. Journal of Organometallic Chemistry. 690(5):1233-1239,2005.
108. HELLDÖRFER M., MILIUS W., ALT H., The Influence of Halogen Substituents at the Ligand Framework of ([alpha]-diimine)Nickel(II) Catalyst Precursors on their Behavior in Ethylene Oligomerization and Polymerization. Journal of Molecular Catalysis A: Chemical. 197(1-2):1-13,2003.
109. HELLDÖRFER M., BACKHAUS J., MILIUS W., ALT H., ([alpha]-Diimine)Nickel(II) Complexes Containing Chloro Substituted Ligands as Catalyst Precursors for the Oligomerization and Polymerization of Ethylene. Journal of Molecular Catalysis A: Chemical. 193(1-2):59-70,2003.
110. ZHANG T., GUO D., JIE S., SUN W., LI T., YANG X., Influence of Electronic Effect on Catalytic Activity of Salicylaldiminato Nickel(II) Complexes. Journal of Polymer Science Part A: Polymer Chemistry. 42(19):4765-4774,2004.
111. WOO T., ZIEGLER T., The Influence of Electronic and Steric Factors on Chain Branching in Ethylene Polymerization by Brookhart-type Ni(II) diimine Catalysts: a Combined Density Functional Theory and Molecular Mechanics Study. Journal of Organometallic Chemistry. 591(1-2):204-213,1999.
112. SONG C., TANG L., LI Y., LI X., CHEN J., LI Y., Preparation of Linear alpha-olefins to High-Molecular Weight Polyethylenes Using Cationic alpha-diimine Nickel(II) Complexes Containing Chloro-substituted Ligands. Journal of Polymer Science Part A: Polymer Chemistry. 44(6):1964-1974,2006.
113. YAOFENG C., CHANGTAO S., JIE S., Fluoro-Substituted 2,6-Bis(imino)pyridyl Iron and Cobalt Complexes: High-Activity Ethylene Oligomerization Catalysts. Organometallics. 22(6):1231-1236,2003.
114. LIU J., LI T., LI J., HU N., Ethylene Polymerization by (alpha-diimine)nickel(II) Complexes Bearing Different Substituents on para-position of Imines Activated with MMAO. Journal of Applied Polymer Science. 109(2):700-707,2008.
115. LEUNG D., ZILLER J., GUAN Z., Axial Donating Ligands: A New Strategy for Late Transition Metal Olefin Polymerization Catalysis. Journal of the American Chemical Society. 130(24):7538-7539,2008.
116. MACEDO F., GWENGO C., LINDEMAN C., SMITH M., GARDINIER J., beta-Diketonate, beta-Ketoiminate, and beta-Diiminate Complexes of Difluoroboron. European Journal of Inorganic Chemistry. 2008(20):3200-3211,2008.
117. NIEUWENHUYZEN M., SCHOBERT R., HAMPEL F., HOOPS S., Reactions of Dicarbonyltitanocenes with 2-diazo-1,3-diketones: O-,N- versus O-,O- Chelation and Self-assembly of a Novel Heteroleptic Ti5O6-cage Compound. Inorganica Chimica Acta. 304(1):118-121,2000.
118. ISAKOVA V., BAIDINA I., MOROZOVA N., IGUMENOV I., [gamma]-Halogenated Iridium(III) Acetylacetonates. Polyhedron. 19(9):1097-1103,2000.
119. AZOULAY J., ROJAS R., SERRANO A., OHTAKI H., GALLAND G., WU G., BAZAN G., Nickel alpha-Keto-beta-Diimine Initiators for Olefin Polymerization. Angewandte Chemie International Edition. 48(6):1089-1092,2009.
120. ROJAS R., GALLAND G., WU G., BAZAN G., Single-Component alpha±-Iminocarboxamide Nickel Ethylene Polymerization and Copolymerization Initiators. Organometallics. 26(22):5339-5345,2007.

136
121. AZOULAY J., ITIGAKI K., WU G., BAZAN G., Influence of Steric and Electronic Perturbations on the Polymerization Activities of alpha±-Iminocarboxamide Nickel Complexes. Organometallics. 27(10):2273-2280,2008.
122. LEE B., BAZAN G., VELA J., KOMON Z., BU X., alpha±-Iminocarboxamidato Nickel(II) Ethylene Polymerization Catalysts. Journal of the American Chemical Society. 123(22):5352-5353,2001.
123. ROJAS R., WASILKE J., WU G., ZILLER J., BAZAN G., alpha±-Iminocarboxamide Nickel Complexes: Synthesis and Uses in Ethylene Polymerization. Organometallics. 24(23):5644-5653,2005.
124. KOMON Z., BU X., BAZAN G., Synthesis of Butene- Ethylene and Hexene-Butene-Ethylene Copolymers from Ethylene via Tandem Action of Well-Defined Homogeneous Catalysts. Journal of the American Chemical Society. 122(8):1830-1831,2000.
125. BOURGET-MERLE L., LAPPERT M.F., SEVERN J.R., The Chemistry of β-Diketiminatometal Complexes. Chemical Reviews. 102(9):3031-3066,2002.
126. CARLSON R.N., A, Acta Chemica Scandinavica. 38:49,1984. 127. ROLF C.U.L., LARS HANSSON, Efficient Synthesis if Imines by a Modified
Titanium Tetrachloride Procedure. Acta Chemica Scandinavica. 46:1211,1992. 128. CARLSON R.N., A; , Improved Titanium Tetrachloride Procedure for Enamine
Synthesis. II. Scope of the Reaction. Acta Chemica Scandinavica. 38:49,1984. 129. MUZART S.B.A.F.H.A.J., Access to Enimines Via Titanium Tetrachloride-
Promoted Condensation of Amines With Arylenones Synthetic Communications. 31:39,2001.
130. YUANHE GAO Q.Z., AND JIAXI XU*, A Convenient and Effective Method for Synthesizing β-Amino-,β-Unsaturated Esters and Ketones Synthetic Communications. 35:909,2004.
131. STEPHAN J.D.M.A.D.W., Neutral and cationic aluminium complexes of a sterically demanding N-imidoylamidine ligand. Dalton Transactions:2089,2006.
132. RENÉ T. BOERÉ V.K.A.G.W., Synthesis of some very bulky N,N9-disubstituted amidines and initial studies of their coordination chemistry. Dalton Transactions. 24:4147.
133. PETER H.M.B., OORT A.B.V., ORPEN A.G., alfa-Diiminato Complexes of VIII and TiIII Formation and Structure of Stable Paramagnetic Dialkylmetal Compounds. European Journal of Inorganic Chemistry. 1998(10):1485-1494,1998.
134. LIU F.-S., GAO H.-Y., SONG K.-M., ZHAO Y., LONG J.-M., ZHANG L., ZHU F.-M., WU Q., Neutral nickel complexes chelating monoamidinate ligands: Syntheses, characterizations and catalytic properties toward ethylene oligomerization and norbornene polymerization. Polyhedron. 28(4):673-678,2009.
135. JINGYU L. YANGUO L., YUESHENG L., NINGHAI H., Ethylene polymerization by (alpha-diimine)nickel(II) complexes bearing different substituents on para-position of imines activated with MMAO. Journal of Applied Polymer Science. 109(2):700-707,2008.

137
ANEXO 1 (Parte Experimental)
SINTESIS DE LIGANDOS
Preparación de Ligandos iminicos.
(i) Síntesis de 4-(1-fenil-etilamino)-pent-3-en-2ona (L1).
La síntesis fue llevada a cabo mediante la adición de 2,4-pentadiona (1,65 g,
165 mmol) a (R)-(+)-α-metilbencilamina (2 g, 165 mmol) disuelta en 50 mL de
benceno en presencia de ácido p-toluensulfonico como catalizador. La mezcla fue
agitada por un periodo de 12 h bajo reflujo, el solvente es removido bajo vacío
obteniéndose un aceite café. El producto es purificado mediante cromatografía
obteniéndose en un 82 % de rendimiento. 1H-NMR (399,95 MHz, d6-benceno, 300 K): δ = 11,23 (s, 1H, NH), 7,18-6,99 (m,
5H, H-Ph ), 4,96 (s, 1H, CH), 4,63 (p, 1H, 3JHH = 6,8 Hz, *CH-Ph), 2,01 (s, 3H,
CH3), 1,75 (s, 3H, CH3), 1,51 (d, 3H, 3JHH = 6,8 Hz, CH3-CH). 13C-NMR (399,95
MHz, d6-benceno, 300 K): δ = 195,61; 166,84; 142,29; 128,77; 126,99; 120,19;
84,09; 53,74; 28,09; 23,51; 16,95. FT-IR (KBr, cm-1): 3442(s), 2971(d), 2927(d),
2871(d), 1610(s)
(ii) Síntesis de 4-(2,6-diisopropilfenilimino)-pentano-2ona (L2).
El ligando es obtenido por la adición de 2,6-diisopropilanilina (7,1 g, 40,0
mmol) a 2,4-pentanodiona (8,1 g, 80,9 mmol) en benceno, seguido de ácido p-
toluensulfonico como catalizador. La reacción es reflujada por un día a 110 ºC, el
solvente es removido bajo vacío obteniéndose un sólido naranjo. El producto es
purificado por cristalización en hexano consiguiéndose un sólido blanco en un 72 %
de rendimiento. 1H-RMN (399,95 MHz, CDCl3, 300 K): δ = 12,24 (s, 1H, NH), 7,31 (t, 1H, 3JHH =
7,7 Hz, Hp-Ph), 7,18 (d, 2H, 3JHH = 7,7 Hz, Hm-Ph), 4,72 (s, 1H, Hβ), 3,07 (sep, 2H, 3JHH = 6,8 Hz, CH-iPr), 2,12 (s, 3H, CH3), 1,63 (s, 3H, CH3), 1,22 (d, 6H, 3JHH = 6,8
Hz, CH3-iPr), 1,16 (d, 6H, 3JHH = 6,8 Hz, CH3-iPr). 13C-RMN (399,95 MHz, CDCl3,

138
300 K): δ = 195,9; 163,3; 146,3; 133,5; 128,2; 123,6; 95,6; 29,1; 28,5; 24,6; 22,7;
19,2.
(iii) Síntesis de 2-(2,6-diisopropilfenilamina)-4-(2,6-diisopropilfenilimina)-pent-
2-eno (L3).
La síntesis de L 3 fue realizada siguiendo el procedimiento descrito en
literatura tal como se menciona a continuación. A una mezcla de 2,4-pentadiona (3 g,
29 mmol) y 2,6-diisopropilanilina (11 g, 62 mmol) en etanol, se le adiciona HCl 32%
(4 g). La reacción es colocada bajo reflujo durante 72 h con agitación constante. A la
solución resultante se le remueve el solvente bajo vacío, se extrae con CH2Cl2 y se
lava con una solución saturada de NaCO3 y luego tres veces con agua. La fase
orgánica se seca sobre Na2SO4, se evapora el solvente obteniéndose un aceite
amarillo oscuro. El producto es obtenido después de recristalización como un sólido
blanco en un 71 % de rendimiento. 1H-NMR (399,95 MHz, CDCl3, 300 K): δ = 12,11 (s, 1H, NH), 7,13 (t, 6H, H-Ph ),
4,87 (s, 1H, CH), 3,16 (sep, 4H, 3JHH = 6,9 Hz, CH-iPr), 1,71 (s, 6H, CH3), 1,22 (d,
12H, 3JHH = 6,8 Hz, CH3), 1,12 (d, 12H, 3JHH = 6,8 Hz, CH3). 13C-NMR (399,95
MHz, CDCl3, 300 K): δ = 161,19; 142,49; 140,59; 125,03; 123,04; 93,27; 28,24;
24,26; 23,25; 20,78.
(iv) Síntesis 5-(1-feniletilimino)-2,2,6,6-tetrametilhepta-3-ona (L4).
Este ligando fue preparado utilizando microondas como fuente de energía. En
un tubo de reacción se coloca 2,2,6,6-tetrametilheptadiona (0,3 g, 1,63 mmol), R-(+)-
metilbencilamina (0,79 g, 6,5 mmol) utilizándose arcilla MK-10 (250 mg) como
catalizador. Las condiciones de reacción son 1 h a 200 ºC y una potencia de 300
MW. El producto es purificado mediante columna cromatográfica (hexano/éter),
obteniéndose un rendimiento de 20 %. 1H-NMR (399,95 MHz, CDCl3, 300 K): δ = 12,23 (s, 1H, NH), 7,34-7,20 (m, 5H, H-
Ph ), 5,38 (s, 1H, CH), 5,17 ( p, 1H, 3JHH = 6,4 Hz, *CH-Ph), 1,58 (d, 3H, 3JHH = 6,4
Hz, CH3-CH), 1,27 ( s, 9H, CH3-tBu), 1,23 ( s, 9H, CH3-tBu). 13C NMR (399,95
MHz, CDCl3, 300 K): δ = 204,25, 172,79, 144,74, 128,66, 126,82, 125,54, 86,96,
54,36, 41,95, 36,21, 29,5, 28,24, 26,19.

139
(v) Síntesis de 2-acetil-3-(1-fenil-etilimino)but-2-enenitrilo (L 5).
Este ligando fue preparado por desprotonación de 4-(1-fenil-etilamino)-pent-
3-en-2ona (0,5 g, 2,5 mmol) con n-butillitio (1,6 M en hexano, 1,8 ml, 3 mmol) en
THF a -78 ºC. La reacción se deja alcanzar temperatura ambiente y se agita por 3 h.
La solución de color amarilla es nuevamente enfriada a -78 ºC, y se le adiciona una
suspensión de p-toluensulfonilciano (0,54 g, 3 mmol) en THF, tornándose más
oscura. La solución resultante es agitada por 12 h, luego el solvente es removido bajo
vació, el producto extraído con CH2Cl2 y lavado con una solución de NaCl saturada.
La fase orgánica es separada y secada sobre Na2SO4. La solución es filtrada y el
solvente removido bajo vacío colectándose un aceite amarillo. El producto puro es
obtenido en un 60% de rendimiento. 1H-NMR (399,95 MHz, CDCl3, 300 K): 12,25 (s, 1H, NH), 7,36 -7,19 (m, 5H, H-Ph
), 4,17 (p, 1H, 3JHH = 6,8 Hz, *CH-Ph), 2,33 (s, 3H, CH3), 2,12 (s, 3H, CH3), 1,58 (d,
3H, 3JHH = 6,8 Hz, CH3-CH). 13C-NMR (399,95 MHz, CDCl3, 300 K): δ = 196,33;
170,02; 142,01; 129,27; 128,05; 125,46; 95,22; 83,71; 54,55; 28,61; 24,27; 17,97.
FT-IR (KBr, cm-1): 3629(s), 2969(d), 2193(s), 1617(s), 1283(s).
(vi) Síntesis de 2-acetil-3-(2,6-diisopropilfenilimino)-but-2-enenitrilo (L 6).
A una solución de 4-(2,6-diisopropilfenilimino)-pentano-2ona (1,47 g, 5,7
mmol) se le adiciona n-butillitio (1,6 M en hexano, 24,5 ml, 7,2 mmol) en THF a -78
ºC. La reacción se deja agitando a temperatura ambiente por 3 h, obteniéndose la sal
de litio. La suspensión resultante es nuevamente enfriada a -78 º C y se le agrega una
suspensión de p-toluensulfonilciano (1,12 g, 6,2 mmol) en THF. La solución
resultante se deja agitando por 12 hr. El solvente es removido bajo vacio, el producto
es disuelto en CH2Cl2, para luego ser lavado con una solución de NaCl saturada. La
fase orgánica es separada y secada sobre Na2SO4. La solución es filtrada y el
solvente removido bajo vacío colectándose un aceite café. El producto puro es
obtenido como cristales amarillos, en un 60 % de rendimiento después de
recristalización de hexano. 1H NMR (399,95 MHz, d6-benceno, 298 K): δ = 13,55 (s, 1H, NH), 7,04 (t, 1H, 3JHH
= 8,0 Hz, Hp-Ph), 6,90 (d, 2H, 3JHH = 8,0 Hz, Hm-Ph) 2,80 (sep, 2H, 3JHH = 8,0 Hz,

140
CH-iPr), 2,39 (s, 3H, CH3), 1,75 (s, 3H, CH3), 1,00 (dd, 12H, 3JHH = 8,0 Hz, CH3-
iPr). 13C NMR (399.95 MHz, d6-benceno, 298 K): δ = 196,82; 170,90; 145,49;
132,27; 129,41; 124,16; 120,09; 84,67; 28,89; 28,29; 24,16; 22,38; 17,84. FT-IR
(KBr, cm-1): 3474(d), 2963(s), 2929(d), 2868(d), 2200(s), 1625(s), 1275(s), 806(s).
(vi) Síntesis 3-(2,6-diisopropilfenilamino)-2-1-(2,6-diisopropilfenilimino)etil)but-
2-enenitrilo (L7)
A una solución de 2-(2,6-diisopropilfenilamina)-4-(2,6 diisopropilfenilimina)-
pent-2-eno (1,6 g, 3,8 mmol) se le adiciona n-butillitio (1,6 M en hexano, 2,6 mL, 4,2
mmol) en THF a -78 ºC. La reacción es llevada a temperatura ambiente y se agita
por 3 h. La solución es nuevamente enfriada a -78 ºC, y se adiciona una suspensión
de p-toluensulfonilciano (0,7 g, 3,8 mmol) en THF. La solución resultante es agitada
por 12 h, el solvente es removido bajo vacio y el producto disuelto en CH2Cl2. La
solución se lava con una solución saturada de NaCl, la fase orgánica es separada y
secada sobre Na2SO4. El solvente es removido bajo vacío, el producto es purificado
mediante recristalización en hexano obteniéndose un sólido amarillo en un 70 % de
rendimiento.
1H-NMR (399,95 MHz, CDCl3, 300 K): 13,99 (s, 1H, NH), 7,43-7,20 (m, 6H, H-Ph),
3,11 (sep, 4H, 3JHH = 6,8 Hz, CH-iPr), 2,05 (s, 6H, CH3), 1,20 (d, 12H, 3JHH = 6,8 Hz,
CH3-iPr) 1,12 (d, 12H, 3JHH = 6,8 Hz, CH3-iPr). 13C-NMR (399,95 MHz, CDCl3, 300
K): δ = 196,33; 142,01; 129,27; 128,05; 125,46; 83,71; 54,55; 28,61; 24,27; 17,97.
FT-IR (KBr, cm-1): 3441(s), 2961(s), 2198(s), 1610(s)

141
Ligandos Acetamidinicos
(i) Síntesis de N-(2,6-diisopropilfenil)acetamida (P1).
A una solución de 2,6-diisopropilanilina (10 g, 56, 4 mmol) en 100 mL de
piridina seca se le agrega lentamente cloruro de acetilo (4 mL, 56,4 mmol) a -20 ºC.
La solución pasa de transparente a amarilla con la formación de un precipitado
blanco. La mezcla se deja bajo reflujo por 3 h, luego la piridina es removida bajo
vacío y el residuo disuelto en 100 mL de diclorometano. La solución es lavada tres
veces con 100 ml de agua, secada sobre MgSO4, y filtrada. El solvente es removido
bajo vacio, obteniéndose un sólido amarillo. El producto fue aislado y purificado
mediante recristalización en tolueno, obteniéndose un sólido blanco en un 85 % de
rendimiento. El espectro 1H-RMN mostró la presencia de dos isómeros. 1H-RMN (399,95 MHz, CDCl3, 300 K): Isómero mayoritario (P1cis): δ = 7,35-7,15
(m, 3H, H-Ph); 6,72 (s, 1H, NH); 3,06 (sep, 2H, 3JHH = 8 Hz, CH-iPr), 2,20 (s, 3H,
CH3), 1,18 (d, 12H, 3JHH = 8 Hz, CH3-iPr). Isómero minoritario (P1trans): δ = 7,35-
7,15 (m, 3H, H-Ph), 6,84 (s, 1H, NH), 3,18 (sept, 2H, 3JHH = 8 Hz, CH-iPr), 1,70 (s,
3H, CH3), 1,22 (d, 6H, 3JHH = 8 Hz, CH3-iPr), 1,15 (d, 6H, JHH = 8 Hz, CH3-iPr). 13C-NMR (399,95 MHz, CDCl3, 300 K): δ = 196,33; 142,01; 129,27; 128,05;
125,46; 83,71; 54,55; 28,61; 24,27; 17,97.
(ii) Síntesis de cloruro de N-(2,6-diisopropilfenil)acetoimidoilo (P2).
Se disuelve pentacloruro de fósforo (10,9 g, 52,3 mmol) en 50 mL de
benceno a 30 ºC. Tras disolver totalmente la sal, la solución se enfría a temperatura
ambiente y se le adiciona N-(2,6-diisopropilfenil)acetamida (10,5 g, 46,5 mmol). La
solución se colocó bajo reflujo durante 3 h, tornándose de color amarillo intenso. El
benceno y las impurezas volátiles fueron eliminadas mediante destilación a presión
reducida (30-40 °C), obteniéndose un aceite café-rojizo. El compuesto fue aislado y
purificado de éste aceite mediante destilación a presión reducida (90-95 ºC en alto
vacío). El producto obtenido finalmente es un aceite amarillo en un 82% de
rendimiento.

142
1H-RMN (399,95 MHz, CDCl3, 300 K): δ = 7,26 –7,28 (m, 3H, H-Ph), 2,90 (sep,
2H, 3JHH = 8 Hz, CH-iPr), 2,73 (s, 3H, CH3), 1,31 (s, 12H, CH3-iPr).
(iii) Síntesis de N’-(2,6-diisopropilfenil)-N-[1-(2,6-diisopropilfenilimino)-etil ]-N-
(2,6-dimetil)acetamidina (L8)
(2,6-diisopropilfenil)acetoimidoilo (2 g, 8,4 mmol) es adicionado a una
solución de 2,6-dimetilanilina (0,51 g, 4,2 mmol), trietilamina (0,85 g, 8,4 mmol) en
60 ml de tolueno. La mezcla de reacción es llevada hasta reflujo por 3 h. Pasando de
transparente a opalescente, ésta es lavada tres veces con agua y secada bajo Na2SO4.
El solvente es removido bajo vacio, obteniéndose un sólido amarillo. Cristalización
desde etanol da como producto un sólido blanco en 85 % de rendimiento. El espectro 1H-RMN muestra la presencia de dos isómeros en una relación 10:1 N’-(2,6-
diisopropilfenil)-N-[1-(2,6-diisopropil-fenilimino) -etil]-N-(2,6-
dimetil)acetamidina (L 8a) y N-(2,6-diisopropil-fenil)-N´-[1-(2,6-diisopropil-
fenilimino)-etil ]-N’-(2,6-dimetil)acetamidina (L 8b) respectivamente. 1H-NMR (399,95 MHz, CDCl3, 300 K): Isómero Mayoritario L 8a, δ = 7,26-7,00 (m,
9H, H-Ph ), 2,92 (sep, 4H, 3JHH = 6,8 Hz, CH-iPr), 2,42 (s, 6H, CH3), 1,92 (s, 6H,
CH3), 1,19 (d, 12H, 3JHH = 6.8 Hz, CH3-iPr), 1,06 (d, 12H, 3JHH = 6,8 Hz, CH3-iPr).
Isómero MinoritarioL 8b, δ 7,34-6,84 (m, 6H, H-Ph ), 3,41 (sep, 2H, 3JHH = 6,8 Hz,
CH-iPr), 2,86 (sep, 2H, 3JHH = 6,8 Hz CH-iPr), 2,08 (s, 6H, CH3), 2,01 (s, 3H, CH3),
1,82 (s, 3H, CH3), 1,28 (d, 6H, 3JHH = 6,4 Hz, CH3-iPr), 1,27 (d, 6H, 3JHH = 6,4 Hz,
CH3-iPr), 1,18 (d, 6H, 3JHH = 6,8 Hz. CH3-iPr), 0,94 (d, 6H, 3JHH = 6,8 Hz, CH3-iPr). 13C-NMR (399,95 MHz, CDCl3, 300 K): Isómero Mayoritario L 8a, 156,47; 147,86;
137,32, 137,22; 128,38; 127,23; 122,87; 122,78; 28,10; 24,43; 22,94; 19,18; 18,40;
Isómero Minoritario L 8b, 158,00; 147,89; 137,00; 128,58; 127,88; 127,25; 124,06;
122,86; 122,82; 29,06; 27,94; 24,67; 24,45; 23,53; 22,58; 18,15.

143
(iv) Síntesis de N,N´-bis-(2,6-dimeil-fenil)-N-[1-(2,6-dimetil-
fenilimino)etil)]acetamidina (L 9).
A una solución de 2,6-dimetilanilina (0,38 g, 3,2 mmol), trietilamina (0,64 g,
6,4 mmol) en 60 mL de tolueno se adicionó (2,6-dimetilfenil)acetoimidoilo (1 g, 6,4
mmol). La reacción es agitada bajo reflujo durante 3 h, cambiando de transparente a
opalescente. La solución es llevada a temperatura ambiente, lavada tres veces con
agua y secada sobre Na2SO4. El solvente es removido bajo vacio, obteniéndose un
sólido amarillo. Este es purificado mediante cristalización en etanol dando un sólido
blanco en un 87 % de rendimiento. 1H-NMR (399,95 MHz, CDCl3, 300 K): δ 7,18 (m, 3H, H-Ph), 7,04 (d, 4H, 3JHH =
7,6 Hz, H-Ph), 6,87 (t, 2H, 3JHH = 7,6 Hz, H-Ph) 2,46 (s, 6H, CH3-Ph), 2,19 (s, 12H,
CH3-Ph), 1,95 (s, 6H, (C=N)CH3). 13C-NMR (399,95 MHz, CDCl3, 300 K): 156,78;
147,53; 141,01; 137,08; 128,58; 127,99; 127,87; 127,05; 122,31; 18,96; 18,59;
18,52.
(v) Síntesis de N-(2,6-diisopropil-fenil)-N-[1-(2,6-diisopropil-fenilimino)-etil]-N-
(2,3,4,5,6-petafluoro-fenil)acetamidina (L10).
(2,6-diisopropilfenil)acetoimidoil (2 g, 8,4 mmol) fue adicionado a una
solución de 2,3,4,5,6-pentafluoroanilina (0,77 g, 4,2 mmol), trietilamina (0,85 g, 8,4
mmol) en 60 mL de tolueno. La mezcla de reacción es agitada bajo reflujo por 6 h.
La solución fue lavada tres veces con agua y secada bajo Na2SO4. El solvente es
removido bajo vacío obteniéndose un sólido amarillo. La cristalización en tolueno da
como resultado un sólido blanco en un 55 % de rendimiento. 1H-NMR (399,95 MHz, CDCl3, 300 K): δ 7,11-7,02 (m, 6H, H-Ph), 2,84 (sep, 4H, 3JHH = 6,8 Hz, CH-iPr), 2,00 (s, 6H, CH3), 1,21 (d, 12H, 3JHH = 7,2 Hz, CH3-iPr),
1,08 (d, 12H, 3JHH = 6,8 Hz, CH3-iPr). 13C-NMR (399,95 MHz, CDCl3, 300 K):
155,99; 144,03; 137,26; 123,81; 123,26; 29,92; 28,36; 23,44; 22,89; 18,99. 19F-NMR
(200 MHz, CDCl3, 300 K): δ = -147,29, -156,66, -160,35. FT-IR (KBr, cm-1):
3402(s), 2968(s), 2929(m), 2869(m), 1638(s). EI-MS(+) : m/z = 586,32

144
(vi) N-1-(2,6-diisopropilfenil)-N´-(2,3,4,5,6-perfluorofenil)acetamidina (L11).
A una solución de (2,6-dimetilfenil)acetoimidoil (1 g, 4,2 mmol) en tolueno
fue adicionado 2,3,4,5,6-pentafluoroanilina (0,77 g, 4,2 mmol). La reacción se deja
bajo reflujo durante 6 h. La solución resultante posee un precipitado el que se colecta
por filtración, correspondiendo a L 11·HCl. El sólido es disuelto en 30 mL de
diclorometano y tratado tres veces con una solución acuosa al 25% de NH3. La
solución es secada bajo Na2SO4 y el solvente es removido bajo vacio obteniéndose
un aceite amarillo. El producto es obtenido como cristales blancos después de
purificación por cristalización desde metanol en un 70 % de rendimiento. 1H-NMR (399,95 MHz, CDCl3, 300 K): δ 7,33-7,20 (m, 3H, H-Ph), 5,95 (s, 1H,
NH), 3,22 (sep, 2H, CH-iPr), 2,04 (s, 3H, CH3), 1,29 (d, 6H, 3JHH = 6,4 Hz, CH3-iPr),
1,23 (d, 6H, 3JHH = 6,8 Hz, CH3-iPr). 13C-NMR (399,95 MHz, CDCl3, 300 K,):,
159,79; 146,96; 132,26; 128,94; 124,12; 123,85; 29,04; 28,63; 24,53; 24,43; 23,45;
23,05; 18,50. 19F-NMR (200 MHz, CDCl3, 300 K): δ = -146,60; -153,99; -162,61.
FT-IR (KBr, cm-1): 3402(s), 3061(w), 2968(s), 2929(m), 2869(m), 1638(s). EI-
MS(+): m/z = 385,16.
(vii) Síntesis de N,N´-bis(2,6-diisopropilfenil)-N-(1-(2,3,4,5,6-
pentafluorofenilimino)etil)acetamidina (L12).
A una solución de N-1-(2,6-diisopropilfenil)-N´-(2,3,4,5,6-
perfluorofenil)acetamidina (1,0 g, 2,6 mmol) en tolueno se le agrega un equivalente
de (2,6-diisopropilfenil)acetoimidoilo (0,62 g, 2,6 mmol) a temperatura ambiente. La
mezcla resultante se deja bajo reflujo por 6 h, luego esta es filtrada para colectar el
precipitado formado, éste se disuelve en diclorometano y es tratado con una solución
acuosa de NH3 al 25 %. La solución se seca en Na2SO4 y el solvente es removido
bajo vacio obteniéndose un sólido amarillo. Este es purificado por cristalización en
etanol dando como producto un sólido blanco en un 60 % de rendimiento. 1H-NMR (399,95 MHz, CDCl3, 300 K): δ = 7,38-7,22 (m, 3H, H-Ph), 7,09-6,99 (m,
3H, H-Ph), 3,26 (sep, 2H, 3JHH = 6,8 Hz, CH-iPr), 2,79 (sep, 2H, 3JHH = 6,8 Hz, CH-
iPr), 2,12 (s, 3H, CH3), 1,91 (s, 3H, CH3), 1,26 (dd, 12H, 3JHH = 6,8 Hz, CH3-iPr),
1,17 (d, 12H, 3JHH = 6,8 Hz, CH3-iPr), 0,96(d, 12H, 3JHH = 6,8 Hz, CH3-iPr). 13C-

145
NMR (399,95 MHz, CDCl3, 300 K): 163,25; 157,78; 147,59; 144,25; 137,09;
136,79; 129,34; 124,53; 123,49; 123,14; 29,12; 28,20; 24,69; 24,41; 23,62; 22,66;
20,23; 19,52. 19F- NMR (200 MHz, CDCl3, 300 K): δ = -146,16; -157,56; -158,64.
EI-MS(+): m/z = 586,2
(viii) Síntesis de N-(2,6-diisopropilfenil)-N-1-(2,6-diisopropilfenilimino)-N-
(piridin-4-il)acetamidina (L 13).
A una solución de 4-aminopiridina (0,20 g, 2,1mmol) y trietilamina (0,42 g,
4,2 mmol) en 60 ml de tolueno se le adicionó (2,6-dimetilfenil)acetimidoil (1g, 4,2
mmol). La reacción se dejo bajo reflujo y agitación durante 6 h. La solución
resultante fue lavada tres veces con agua y secada bajo Na2SO4. El solvente es
removido bajo vacio obteniéndose un sólido café. El producto de la reacción es
purificado mediante columna cromatográfica (éter/hexano) obteniéndose un sólido
amarillo después de evaporación de los solventes en un 50 % de rendimiento. 1H-NMR (399,95 MHz, CDCl3, 300 K): δ 8,69 (d, 2H, 3JHH = 6,8 Hz, H-piridínico),
7,25 (d, 2H, 3JHH = 6,8 Hz,H-piridínico), 7,12 (d, 4H, 3JHH = 6,8 Hz, p-H-Ph), 7,04
(m, 2H, 3JHH = 6,8 Hz, m-H-Ph), 2,90 (sep, 4H, 3JHH = 6,8 Hz, CH-iPr), 1,97 (s, 6H,
CH3), 1,23 (d, 12H, 3JHH = 6,8 Hz, CH3-iPr), 1,09 (d, 12H, 3JHH = 6,8 Hz, CH3-iPr). 13C-NMR (399,95 MHz, CDCl3, 300 K): 157,82; 151,31; 151,25; 144,64; 137,15;
123,86; 123,69; 123,23; 28,44; 23,68; 23,03; 19,77. FT-IR (KBr, cm-1): 2959(s),
2930(d), 2867(d), 1652(s), 1239(s).
(ix) N-(1-(4-iminopiridin-1(4H)-yl)etilideno)-2,6-diisopropylanilina (L 14).
A una solución de (2,6-dimetilfenil)acetimidoil (1 g, 4,2 mmol) en tolueno se
le adiciona lentamente la 4-aminopiridina (0,40 g, 4,2 mmol). La reacción se deja
bajo reflujo por 6 h. Se filtra, obteniéndose un sólido que corresponde a L 14·HCl. El
producto es disuelto en 30 mL de diclorometano y tratado tres veces con una
solución acuosa al 25% de NH3, secado bajo Na2SO4 y filtrado. El solvente es
removido bajo vacio dando como producto un aceite café. La cristalización desde
metanol da como resultado un sólido amarillo en un 60 % de rendimiento.

146
1H-NMR (399,95 MHz, CDCl3, 300 K): δ 7,27 (d, 2H, 3JHH = 8 Hz, H-piridínico),
7,25-7,10 (m, 3H, 3JHH = 8 Hz, H-Ph), 5,92 (d, 2H, 3JHH = 8 Hz, H-PhN), 2,62 (sep,
2H, 3JHH = 6,8 Hz, CH-iPr), 1,23 (s, 3H, CH3), 1,10 (d, 6H, 3JHH = 6,8 Hz, CH3-iPr),
1,05 (d, 6H, 3JHH = 6,8 Hz, CH3-iPr). 13C-NMR (399,95 MHz, CDCl3, 300 K):
164,16; 150,27; 143,25; 137,32; 130,36; 124,28; 123,44; 114,90; 28,57; 23,4; 23,00;
15,32. FT-IR (KBr, cm-1): 3423(d), 2960(s), 2924(d), 2867(d), 1651(s), 1288(s),
1074(s), 849(s). EI-MS(+): m/z = 296,21.
Algunos de los ligandos anteriormente descritos fueron caracterizados
mediante Rayos-X. En las siguientes tablas se resumen los valores de refinamiento
de las estructuras cristalinas encontradas.
Tabla i. Valores de refinamiento estructura cristalina de ligandos L 8a y L 8b.
L 8a L 8b
formula empírica C36H49 N3 C36H49 N3 Peso molecular 523,78 523,78 temperatura(K) 150 (2) 150(2) Longitud de onda(Å) 0,71073 0,71073 Sistema cristalino Monoclinic Triclinic Grupo espacial P2(1)/c P-1 Dimensiones celda unitaria(Å, deg)
a = 10,8098(4) b = 13,8640(5) c = 22,6187(7) α= 90 β= 95,2900(10)° γ = 90
a = 10,2414(11) b = 10,6912(12) c = 16,2338(18) α= 82,350(2) β= 74,429(2) γ = 73,118(2)
Volumen(Å3) 3375,4(2) 1635,5(3) Z 4 2 densidad (calculado)Mg/m3
1.031 1.064
Coeficiente absorción(mm-1)
0,060 0,062
F(000) 1144 572 Tamaño cristal(mm3) 0,59 x 0,55 x 0,46 0,19 x 0,09 x 0,08 θ rango(deg) 1,89 to 27,90° 1,99 to 27,77 Rango índice -14 ≤ h ≤ 13
-18 ≤ k ≤ 18 -29 ≤ l ≤ 28
-10 ≤ h ≤ 113 -11≤ k ≤ 13 -21 ≤ l ≤ 21

147
Reflexiones colectadas
27470 7932
Reflexiones independientes
7647 [R(int) = 0.0229]
6711 [R(int) = 0.0573]
Complete desde θ = 26.48 ° (%) y 28.52° (%)
100,0 96,9
datos / restricciones / parámetros
7647 / 3 / 367 6711/ 0 / 364
F2 1,065 0,696 final R índices [I>2σ(I)] R1 = 0,0668, wR2
= 0,2067 R1 = 0,0548, wR2 = 0,0824
R índices (todos los datos)
R1 = 0.0909, wR2 = 0,2297
R1 = 0,1252, wR2 = 0,1310
Peaks mas grande e.Å-3 0,337 y -0,221 0,262 y -0,226
Tabla ii. Valores de refinamiento estructura cristalina de ligandos L 9.
L 9
formula empírica C28H33 N3 Peso molecular 411,57 temperatura(K) 150 (2) Longitud de onda(Å) 0,71073 Sistema cristalino Tetragonal Grupo espacial I4(1)/a Dimensiones celda unitaria(Å, deg)
a = 28,775(3) b = 28,775(3) c = 12,127(2) α= 90 β= 90 γ = 90
Volumen(Å3) 10042(2) Z 16 densidad (calculado)Mg/m3 1,089 Coeficiente absorción(mm-1) 0,064 F(000) 3552 Tamaño cristal(mm3) 0,40x 0,23x 0,19 θ rango(deg) 1,82 a 27,91 Rango índice -37 ≤ h ≤ 37
-35 ≤ k ≤ 36 -15 ≤ l ≤ 15
Reflexiones colectadas 35683 Reflexiones independientes 5728 [R(int) =
0,0676]

148
Complete desde θ = 26.48 ° (%) y 28.52° (%)
100,0
datos / restricciones / parámetros 5728/ 0 / 288 F2 1,027 final R índices [I>2σ(I)] R1 = 0,0560,
wR2 = 0,1135 R índices (todos los datos) R1 = 0,0884,
wR2 = 0,1287 Peaks mas grande e.Å-3 0,287 and -0,273
Tabla iii . Valores de refinamiento estructura cristalina de ligandos L 10 y L 11.
L10 L 11
formula empírica C34H40F5N3 C21H24F5N2O Peso molecular 585,69 415,42 temperatura(K) 298(2) 298(2) Longitud de onda(Å) 0,71073 0,71073 Sistema cristalino Monoclínico Monoclínico Grupo espacial P2(1)/n P2(1)/n Dimensiones celda unitaria(Å, deg)
a = 15,5920(15) b = 11,4499(12) c = 18,0860(18) α= 90 β= 93,195(3) γ = 90
a = 9,5228(10) b = 13,9902(15) c = 16,7103(18) α= 90 β= 94,239(2) γ = 90
Volumen(Å3) 3223,8(6) 2220,2(4) Z 4 4 densidad (calculado)Mg/m3 1,207 1.243 Coeficiente absorción(mm-1) 0,091 0.106 F(000) 1240 868 Tamaño cristal(mm3) 0,35x0,3x0,1 0,61 x 0,49 x 0,38 θ rango(deg) 1,68 to 26,46 1,90 to 27,63 Rango índice -19 ≤ h ≤ 19
-14 ≤ k ≤ 13 -20 ≤ l ≤ 22
-12 ≤ h ≤ 12 -17≤ k ≤ 17 -21 ≤ l ≤ 20
Reflexiones colectadas 25127 14924 Reflexiones independientes 6512 [R(int) =
0,0578] 4746 [R(int) = 0,0485]
Complete desde θ = 26.48 ° (%) y 28.52° (%)
97,6 98,3
datos / restricciones / 6512 / 0 / 401 4746 / 0 / 269

149
parámetros F2 1,381 1,014 final R índices [I>2σ(I)] R1 = 0,0758, wR2
= 0,1516 R1 = 0,0810, wR2 = 0,2351
R índices (todos los datos) R1 = 0,1495, wR2 = 0,1764
R1 = 0,1274, wR2 = 0,2762
Peaks mas grande e.Å-3 0,276 y -0,239 0,513 y -0,219
Definitions: R1= ∑||Fo|-|Fc||] / ∑[|Fo|, wR2= [∑(Fo2-Fc
2)2] / ∑[w(Fo2)2]]1/2, GOF=
[∑[w(Fo2-Fc
2)2]/(n-p)]1/2.

150
Preparación de Sales de Ligandos iminicos.
(i) Síntesis de sal de litio-4-(1-metil-1-fenil-etilamino)-pent-3-en-2ona.
A una solución de 4-(1-metil-1-fenil-etilamino)-pent-3-en-2ona, (1 g, 4,9
mmol), disuelta en THF se le agrega n-butillitio (1,6 M en hexano, 3,06 mL, 4,9
mmol) a -78 ºC. La solución se mantiene bajo agitación por 2 h. La sal precipita en
THF y se colecta por filtración, para ser lavada con éter. El producto es un sólido
amarillo, con un 90 % de rendimiento. 1H NMR (399,95 MHz, d6-benceno, 298 K): δ 7,15-6,99 (m, 5H, H-Ph), 4,52 (p, 2H,
3JHH = 6,8 Hz, CH-iPr), 2,23 (s, 3H, CH3), 2,01 (s, 3H, CH3), 1,43-1,40 (dd, 12H, 3JHH = 6,8 Hz, CH3-iPr)
(ii) Síntesis de sal de litio-2-acetil-3-(1-fenil-etilimino)-but-2-enenitrilo.
La obtención de esta sal es mediante la adición de n-butillitio (1,6 M en
hexano, 364 mg, 1,30 mmol) a una solución en THF de 2-acetil-3-(1-fenil-
etilimino)but-2-enenitrilo (300 mg, 1,30 mmol) a -78 ºC. La solución es agitada por 3
h, obteniéndose un precipitado blanco, el cual es filtrado y lavado con pentano. El
producto corresponde a un sólido blanco con un rendimiento de 90 %. 1H-NMR (399,95 MHz, d8-tetrahydrofuran, 300 K): δ = 7,16-6,87 (m, 5H, H-Ph),
2,80 (sep, 1H, 3JHH = 6,4 Hz, CH-iPr), 2,07 (s, 3H, CH3), 1,69 (s, 3H, CH3), 1,01 (d,
3H, 3JHH = 6,4 Hz, CH3-iPr). FT-IR (KBr, cm-1): 3504,2(s), 2965,7(s), 2932(d),
2279,7(s), 1615,1(s), 1329,9(s), 812,2(s).
(iii) Síntesis de sal de litio-2-acetil-3-(2,6-diisopropilfenilimino)-but-2enenitrilo.
La sal es preparada por la adición de n-butillitio (1,6 M en hexano, 0,55 mL,
0,8 mmol) a una solución de 2-acetil-3-(2,6-diisopropilfenilimino)-but-2enenitrilo
(0,25 g, 0,8 mmol) en tolueno a -78 ºC. La solución alcanza temperatura ambiente y
es agitada por 3 h. El solvente es removido bajo vacío y el producto es lavado tres
veces con pentano, obteniéndose como un sólido blanco en un 90 % de rendimiento. 1H-NMR (300 MHz, d8-tetrahydrofuran, 298 K): δ 7,08-6,87 (m, 3H, H-Ph), 2,80
(sep, 2H, 3JHH = 6,8 Hz, CH-iPr), 2,07 (s, 3H, CH3), 1,69 (s, 3H, CH3), 1,01-0,97 (dd,

151
12H, 3JHH = 6,8 Hz, CH3-iPr). FT-IR (KBr, cm-1): 2981(s), 2928(s), 2192(s), 1604(s),
1238(s).
PREPARACION DE COMPLEJOS
Síntesis de Complejos de Circonio y Titanio
(i) Síntesis de [ZrCl4{Bis-[4-(1-fenil-etilamino)-pent-3-en-2ona]}] (1).
A la sal, Potasio-4-(1-metil-1-fenil-etilamino)-pent-3-en-2ona (300 mg, 1,2
mmol) disuelto en THF se le añade ZrCl4 (150 mg, 0,64 mmol) como una suspensión
en este mismo solvente, en una razón estequiométrica 2:1. La reacción fue realizada
a temperatura ambiente y se mantiene bajo agitación por 12 h. La solución resultante
de color rojo se filtra, se evapora el solvente, se extrae tres veces con éter. El
producto se obtiene como un sólido amarillo oscuro en un 72 % de rendimiento.
FT-IR (KBr, cm-1): 2953(s), 2923(s), 1462(s), 1377(s)
(ii) Síntesis de [ZrCl4{Bis-[2-acetil-3-(1-fenil-etilimino)but-2-enenitrilo (2).
El complejo se obtiene por reacción de la sal de litio-2-acetil-3-(1-fenil-
etilimino)-butilonitrilo (67 mg, 0,28 mmol) y de ZrCl4 (54 mg, 0,14 mmol) disueltos
en THF. La mezcla se deja bajo agitación por 12 h. La solución resultante de color
naranjo se filtra, se evapora el solvente y se extrae tres veces con éter. El producto es
un sólido amarillo, con un rendimiento de 69 %. 1H-NMR (399,95 MHz, d8-tetrahydrofuran, 300 K): 12,25 (s, 1H, NH), 7,22-7,06
(m, 5H, H-Ph), 4,68 (p, 1H, 3JHH = 13,6 Hz, CH-Ph), 2,11 (s, 3H, CH3), 1,96 ( s, 3H,
CH3), 1,36 (d, 3H, 3JHH= 13,6 Hz, CH3-CH). 13C-NMR (399,95 MHz, d8-
tetrahydrofuran, 300 K): 187,38; 167,07; 146,83; 128,82; 127,08; 126,35; 126,82;
67,19; 59,41; 28,63; 24,96; 19.64. FT-IR (KBr, cm-1): 2925(s), 2854(s), 2207(s),
1610(s), 1461(s), 1377(s)

152
(iii) Síntesis de [TiCl4{Bis-[4-(1-fenil-etilamino)-pent-3-en-2ona]}] (3).
A Potasio-4-(1-Metil-1-fenil-etilamino)-pent-3-en-2ona (1 g, 4,9 mmol)
disuelto en THF se le agrega TiCl4 (1,0 M en tolueno, 2,5 mL, 2,5 mmol) en razón
estequiométrica 2:1. La solución se torna de color rojo con la aparición de
precipitado. Esta es filtrada, lavada con pentano y secada bajo vació. El producto es
un sólido de color café oscuro y se obtiene en un rendimiento de 70%. 1H-NMR (399,95 MHz, d6-benceno, 300 K): δ = 11,57 (s, 1H, NH), 6,98-7,68 (m,
5H, H-Ph), 4,95 (s, 1H, CH), 5,14 (p, 1H, 3JHH = 14 Hz, CH-Ph), 1,66 (d, 3H, 3JHH =
14 Hz, CH3-CH), 1,55 (s, 3H, CH3), 1,29 (s, 3H, CH3).
(iv) Síntesis de [TiCl4{Bis-[2-acetil-3-(1-fenil-etilimino)but-2-enenitrilo]}] (4).
A la sal de litio-2-acetil-3-(1-fenil-etilimino)-butilonitrilo (0,35 g, 1,3 mmol)
disuelta en THF se le agrega TiCl4 (1,0 M en tolueno, 1,2 mL, 0,67 mmol). La
reacción se deja bajo agitación por 12h, obteniéndose un precipitado de color café. El
precipitado es colectado, lavado con pentano y secado bajo vacio. El producto se
obtiene como un sólido café con un rendimiento de 65%. 1H-NMR (399,95 MHz, THF, 300 K): δ = 12,27 (s, 1H, NH), 7,28-7,04 (m, 5H, H-
Ph), 4,64 (p, 1H, 3JHH = 6,2 Hz, CH-Ph), 1,38 (s, 3H, CH3), 1,00 (s, 3H, CH3), 0,78
(d, 3H, 3JHH = 6,2 Hz, CH3-CH)
(v) Síntesis de [ZrCl4{Bis-[2-acetil-3-(2,6-diisopropilfenilimino)-but-2-
enenitrilo]}] (5)
A una solución de la sal de litio de 2-acetil-3-(2,6-diisopropilfenilimino)-but-
2enenitrilo (100 mg, 0,34 mmol) en THF se le adiciona el precursor DMEZrCl4 (0,56
mg, 0,17 mmol) en una razón estequiométrica 2:1. La solución de color amarillo se
deja bajo agitación por 12 h a temperatura ambiente. Ésta se filtra, se evapora el
solvente y se lava tres veces con pentano. El producto es un sólido de color amarillo,
con un rendimiento de 60 %. 1H-NMR (300 MHz, d8-tetrahidrofurano, 300 K): δ 12,93(s, 1H, NH) 7,34-7,17 (m,
3H, H-Ph), 2,74 (m, 2H, 3JHH = 6,8 Hz, CH-Ph), 2,21 (s, 3H, CH3), 1,87 (s, 3H, CH3),

153
1.13-1,02 (dd, 12H, 3JHH = 6,8 Hz, CH3-Ph). FT-IR (KBr, cm-1): 3676(s), 2963(s),
2928(s), 2184(s), 1599(s).
(vi) Síntesis de [ZrCl2{Tri[2-acetil-3-(2,6-diisopropilfenilimino)-but-2enenitilo]}]
(6).
El complejo se obtiene por la adición del ligando 2-acetil-3-(2,6-
diisopropilfenilimino)-but-2enenitrilo (70 mg, 0,24 mmol) a (NMe2)2ZrCl2 (47 mg,
0,12 mmol) en THF. La solución se deja bajo agitación por 16 h, se evapora el
solvente y el producto es lavado tres veces con pentano. Se obtiene un sólido
amarillo en un rendimiento de 60 %. 1H-NMR (300 MHz, d8-tetrahydrofuran, 300 K): 13.56 (s, 1H, NH); 2.78 (sep, 2H,
CH-iPr); 2.21 (s, 3H, CH3); 1.75 (s, 3H, CH3), 1.02-0.98 (dd, 12H, CH3-iPr) y dos
ligandos en forma desprotonada 3.07 (m, 4H, CH-iPr) ; 2.39 (s, 12H, CH3); 1.3-1.28
(dd, 12H, CH3-iPr). FT-IR (KBr): 3695(d), 2961(s), 2927(s), 2200(s), 2177(s),
1622(s), 1571(s).
(vii) Síntesis de [Zr(NMe2)2{Bis(2-acetil-3-(2,6-diisopropilfenilimino)-but-
2enenitrilo)}] (7)
A una solución de 2-acetil-3-(2,6-diisopropilfenilimino)-but-2enenitrilo (100
mg, 0,34 mmol) en tolueno se le adiciona el precursor (NMe2)4Zr (0,47 mg, 0,17
mmol) en una razón estequiométrica 2:1. La solución de color naranjo se deja bajo
agitación por 12 h a temperatura ambiente. Ésta se filtra recolectándose una pequeña
fracción (10 % del total) que corresponde a un precipitado de color amarillo. A la
fracción soluble se le evapora el solvente y se obtiene un sólido color naranja en un
rendimiento de 60 %. La complejidad del espectro dificulta la asignación de las
señales del producto naranja.

154
Síntesis de Complejos de Níquel
(i) Síntesis de [NiBr2{N’-(2,6-diisopropil-fenil)-N-[1-(2,6-diisopropil-f enilimino)-
etil]-N-(2,6-dimetil)-acetamidina}] (8a) y [NiBr 2{ N-(2,6-diisopropil-fenil)-N-[1-
(2,6-diisopropil-fenilimino)-etil ]-N’-(2,6-dimetil)-acetamidina}] (8b)
Un equivalente de níquel dibromo 2-metoxietil éter (0,041 g, 1,34 mmol) es
adicionado a una solución de la mezcla de isómeros L 8a y L 8b, (0,07 g, 1,34 mmol)
en THF. La mezcla es agitada a temperatura ambiente por 4 días. La reacción
muestra un cambio progresivo en el color desde verde hasta morado. La solución
resultante es filtrada, el solvente es evaporado y el sólido es extraído con éter. El
producto se obtiene como un sólido de color morado en un 70 % de rendimiento. Su
caracterización mediante 1H y 13C RMN no es posible debido a las características
paramagnéticas del compuesto.
FT-IR (KBr, cm-1): 3439(m), 3063(d), 3021(d), 2961(s), 2925(m), 2867(m), 1644(s).
EI-MS (+): m/z = 662,31 (M+ - Br).
(ii) Síntesis de [NiBr2{N,N´-bis-(2,6-dimetil-fenil)-N-[1-(2,6-dimetil-
fenilimino)etil)]-acetamidina}] (9)
Un equivalente de níquel dibromo 2-metoxietil éter (0,070 g, 2,3 mmol) fue
adicionado a una solución de N,N´-bis-(2,6-dimetil-fenil)-N-[1-(2,6-dimetil-
fenilimino)etil)]-acetamidina, (0,10 g, 2,43 mmol) en DCM. La mezcla fue agitada
por 48 h a temperatura ambiente. La reacción muestra un cambio progresivo de color
desde verde a morado. La solución resultante es filtrada, se evapora el solvente y el
sólido es extraído con éter. El producto obtenido es un sólido de color morado con un
rendimiento de 86%. Análisis de 1H y 13C RMN no fueron posibles de realizar
debido a la característica paramagnética del producto.
FT-IR (KBr Pellet, cm-1): 3422(s), 3060(d), 2962(s), 2926(m), 2867(m), 1632(s).
EI-MS(+): m/z = 629,10.

155
(iii) Síntesis de [NiBr2{N-(2,6-diisopropil-fenil)-N-[1-(2,6-diisopropil-fenilimino)-
etil]-N-(2,3,4,5,6-petafluoro-fenil)acetamidina}] (10).
Un equivalente de níquel dibromo 2-metoxietil éter (0,09 g, 2,56 mmol) fue
adicionado a una solución de L 10 (0,15 g, 2,56 mmol) en DCM. La mezcla fue
agitada a 60 ºC por 48 h. La solución muestra un progresivo cambio de verde a
morado. La solución resultante fue filtrada, el solvente evaporado y el sólido extraído
con éter. El producto un sólido morado con un 70 % de rendimiento. Caracterización
mediante NMR no fue posible por las características paramagnéticas del producto.
EI-MS(+): m/z = 765,1 (M+-Br)
(iv) Síntesis de [NiBr2{N,N´-bis(2,6-diisopropilfenil)-N-(1-(2,3,4,5,6-
pentafluorofenilimino)etil)acetamidina}] (12).
Un equivalente de níquel dibromo 2-metoxietil éter (0,09 g, 2,56 mmol) fue
adicionado a una solución de L 12 (0,15 g, 2,56 mmol) en DCM. La mezcla fue
agitada a 60 ºC por 48 h. La solución resultante de color morada fue filtrada, el
solvente evaporado y el sólido extraído con éter. El producto un sólido morado con
un 77 % de rendimiento.
FT-IR (KBr Pellet, cm-1): 3047 (d), 2968(s), 2928(s), 2870(d), 1635(s), 1300(s).
(v) Síntesis de [NiBr2{N-(2,6-diisopopilfenil)-N-1,(2,6-diisopropilfenilimino)-N-
(piridin-4-il) acetamidina}] (13).
Un equivalente de níquel dibromo 2-metoxietil éter (0,11 g, 3,10 mmol) fue
adicionado a una solución de L 13 (0,15 g, 3,10 mmol) en DCM. La reacción se deja
bajo agitación por 4 h a temperatura ambiente, mostrando un cambio desde verde
hasta azul. La solución resultante se filtra, el solvente es evaporado y el sólido
extraído con éter. El producto es un sólido azul con un rendimiento de 73%.
EI-MS(+): m/z = 715,3

156
(vi) Síntesis de [NiBr2 {N-(1-(4-iminopiridin-1(4H)-yl)etilideno)-2,6-
diisopropylanilina}] (14).
Un equivalente de níquel dibromo 2-metoxietil éter (0,11 g, 3,10 mmol) fue
adicionado a una solución de L 14, (0,15 g, 3,10 mmol) en DCM. La mezcla fue
agitada a temperatura ambiente por 4 h. Esta muestra un rápido cambio desde naranja
hasta verde. La solución resultante es filtrada, evaporado el solvente, extraída con
éter. El producto un sólido verde en un rendimiento del 85%. Análisis de Rayos-X
confirman la obtención del producto esperado.
Algunos de los complejos anteriormente descritos fueron caracterizados
mediante Rayos-X. En las siguientes tablas se resumen los valores de refinamiento
de las estructuras cristalinas encontradas.
Tabla iv. Valores de refinamiento estructura cristalina de Complejos 8a y 8b.
8a 8b formula empírica C36H49 N3 Br2 Ni C36H49 N3 Br2 Ni Peso molecular 742,31 742,31 temperatura(K) 150 (2) 150(2) Longitud de onda(Å) 0,71073 0,71073 Sistema cristalino Triclinic Monoclinic Grupo espacial P-1 P2(1)/n Dimensiones celda unitaria(Å, deg)
a = 10,0112(9) b = 11,1804(10) c = 16,9769(15) α= 90,3390(10) β= 99,7510(10) γ = 108,0690(10)
a = 11,2211(8) b = 17,8539(13) c = 18,2779(14) α= 90 β= 102,3690(10) γ = 90
Volumen(Å3) 1776,9(3) 3576,8(5) Z 2 4 densidad (calculado)Mg/m3 1,387 1,378 Coeficiente absorción(mm-1) 2,822 2,804 F(000) 768 1536 Tamaño cristal(mm3) 0,36x 0,32x 0,05 0,64 x 0,41 x 0,35
θ rango(deg) 1,92 to 27,69 1,96 to 27,83
157
Rango índice -12 ≤ h ≤ 13 -14 ≤ k ≤ 14 -22 ≤ l ≤ 21
-14 ≤ h ≤ 14 -23 ≤ k ≤ 23 -23 ≤ l ≤ 23
Reflexiones colectadas 17808 25610
Reflexiones independientes 7596[R(int)= 0,0298]
7905[R(int)= 0,0245]
Complete desde θ = 26.48 ° (%) y 28.52° (%)
98,8 99,9
datos / restricciones / parámetros
7596 / 0 / 392 7905/ 0 / 395
F2 1,059 1,059 final R índices [I>2σ(I)] R1 = 0,0521, wR2 =
0,1290 R1 = 0,0335, wR2 = 0,0800
R índices (todos los datos) R1 = 0,0718, wR2 = 0,1386
R1 = 0,0443, wR2 = 0,0838
Peaks mas grande e.Å-3 1,457 y -0,602 0,860 y -0,415
Tabla v. Valores de refinamiento estructura cristalina de Complejo 9.
9 formula empírica C28H33 N3NiBr2x CCl2H2 Peso molecular 715,03 temperatura(K) 150 (2) Longitud de onda(Å) 0,71073 Sistema cristalino Monoclinic Grupo espacial P2(1)/c Dimensiones celda unitaria(Å, deg)
a = 13,631(2) b =13,0323(19) c = 18,203(3) α= 90 β= 107,422(2) γ = 90
Volumen(Å3) 3085,3(8) Z 4 densidad (calculado)Mg/m3 1,539 Coeficiente absorción(mm-1) 3,415 F(000) 1448 Tamaño cristal(mm3) 0,25x 0,25 x 0,09 θ rango(deg) 1,57 to 26,55
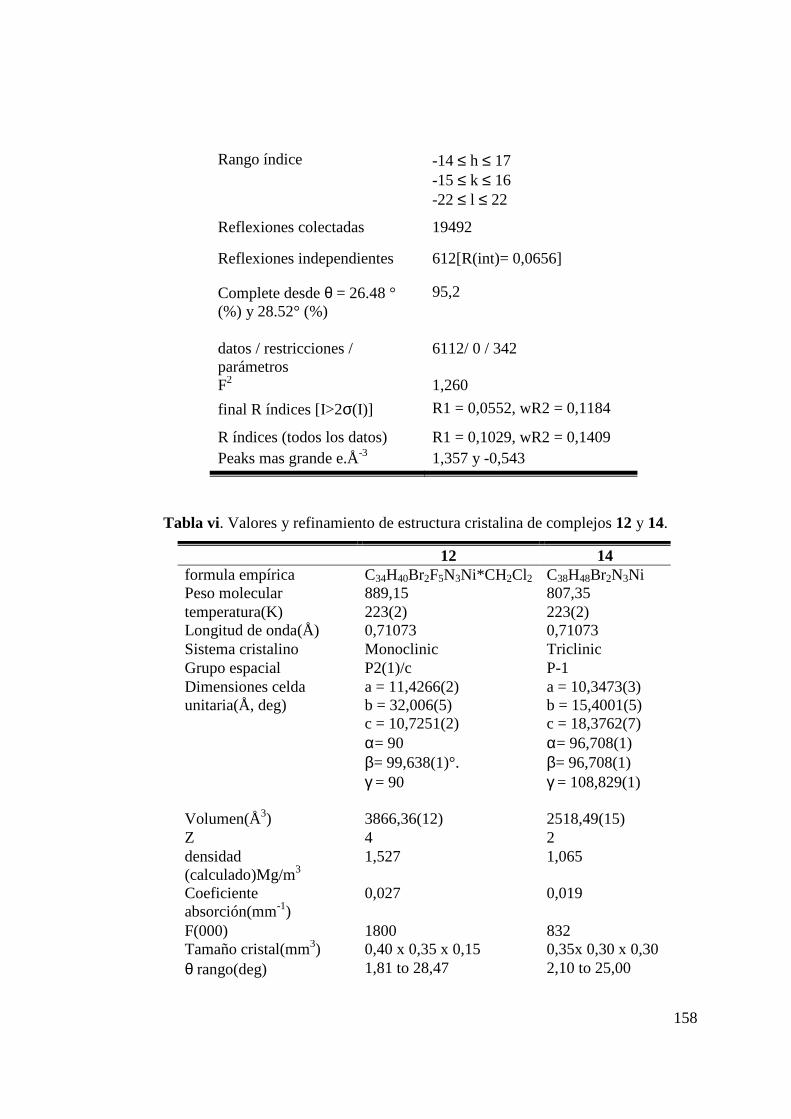
158
Rango índice -14 ≤ h ≤ 17 -15 ≤ k ≤ 16 -22 ≤ l ≤ 22
Reflexiones colectadas 19492
Reflexiones independientes 612[R(int)= 0,0656]
Complete desde θ = 26.48 ° (%) y 28.52° (%)
95,2
datos / restricciones / parámetros
6112/ 0 / 342
F2 1,260
final R índices [I>2σ(I)] R1 = 0,0552, wR2 = 0,1184
R índices (todos los datos) R1 = 0,1029, wR2 = 0,1409 Peaks mas grande e.Å-3 1,357 y -0,543
Tabla vi. Valores y refinamiento de estructura cristalina de complejos 12 y 14.
12 14 formula empírica C34H40Br2F5N3Ni*CH2Cl2 C38H48Br2N3Ni Peso molecular 889,15 807,35 temperatura(K) 223(2) 223(2) Longitud de onda(Å) 0,71073 0,71073 Sistema cristalino Monoclinic Triclinic Grupo espacial P2(1)/c P-1 Dimensiones celda unitaria(Å, deg)
a = 11,4266(2) b = 32,006(5) c = 10,7251(2) α= 90 β= 99,638(1)°. γ = 90
a = 10,3473(3) b = 15,4001(5) c = 18,3762(7) α= 96,708(1) β= 96,708(1) γ = 108,829(1)
Volumen(Å3) 3866,36(12) 2518,49(15) Z 4 2 densidad (calculado)Mg/m3
1,527 1,065
Coeficiente absorción(mm-1)
0,027 0,019
F(000) 1800 832 Tamaño cristal(mm3) 0,40 x 0,35 x 0,15 0,35x 0,30 x 0,30 θ rango(deg) 1,81 to 28,47 2,10 to 25,00
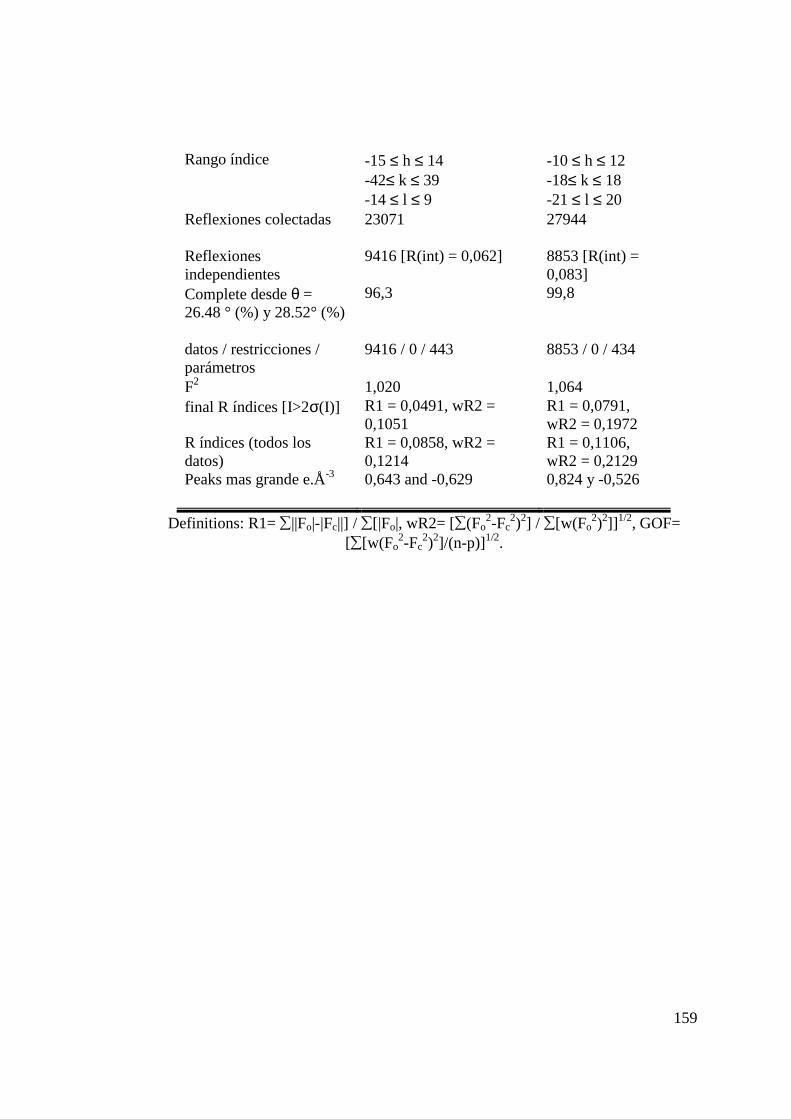
159
Rango índice -15 ≤ h ≤ 14 -42≤ k ≤ 39 -14 ≤ l ≤ 9
-10 ≤ h ≤ 12 -18≤ k ≤ 18 -21 ≤ l ≤ 20
Reflexiones colectadas
23071 27944
Reflexiones independientes
9416 [R(int) = 0,062] 8853 [R(int) = 0,083]
Complete desde θ = 26.48 ° (%) y 28.52° (%)
96,3 99,8
datos / restricciones / parámetros
9416 / 0 / 443 8853 / 0 / 434
F2 1,020 1,064 final R índices [I>2σ(I)] R1 = 0,0491, wR2 =
0,1051 R1 = 0,0791, wR2 = 0,1972
R índices (todos los datos)
R1 = 0,0858, wR2 = 0,1214
R1 = 0,1106, wR2 = 0,2129
Peaks mas grande e.Å-3 0,643 and -0,629 0,824 y -0,526
Definitions: R1= ∑||Fo|-|Fc||] / ∑[|Fo|, wR2= [∑(Fo2-Fc
2)2] / ∑[w(Fo2)2]]1/2, GOF=
[∑[w(Fo2-Fc
2)2]/(n-p)]1/2.
![Complejos de hierro no hémico con nitroxilo y sus ... · RMN 1H. Los estudios de reactividad de [FeII(CN) 5HNO] ... 3–/2–, ya que su actividad farmacológica podría estar](https://img.pdfslide.es/doc/110x75/5edf5aedad6a402d666ab4a1/complejos-de-hierro-no-hmico-con-nitroxilo-y-sus-rmn-1h-los-estudios-de.jpg)




























