
CENTRO DE INVESTIGACIÓN CIENTÍFICA DE
YUCATÁN
Posgrado en Energía Renovable
Síntesis y caracterización de materiales híbridos a
base de óxido de grafeno/polímeros intrínsecamente
conductores para su empleo en supercondensadores
Tesis que presenta:
Paola Navid García Hernández
En opción al título de
Maestro en Ciencias en Energía Renovable
Mérida, Yucatán a Noviembre de 2013



VI
Agradecimientos
A la beca CONACYT Nacional número 290649.
A la beca Mixta de CONACYT número 290674, durante la estancia en el extranjero.
A la beca de movilidad otorgada por el CICY, para la estancia en el extranjero.
Al proyecto de Ciencia Básica 2011 número 166356, con título “Síntesis y caracterización
de materiales híbridos a base de polímeros electroconductores y grafeno para su
potencial aplicación en sistemas de almacenamiento de energía”.
A mi directora de tesis la Dra. Daniella E. Pacheco Catalán por el apoyo brindado, la
orientación y la confianza que me tuvo para poder sacar adelante éste proyecto. Sobre
todo por la paciencia que tuvo hacia mí y motivarme en cada momento para poder así
aprender cada día más. Por fomentar en el equipo de trabajo un ambiente de
compañerismo y amistad.
A mis tutores y revisores de tesis; Dra. Mascha A. Smit, Dr. José Antonio Azamar Barrios,
Dr. Enrique Morales Bergas y Dr. Jorge Alonso Uribe Calderón; por el tiempo y
correcciones durante el proceso de la tesis.
Al Instituto de Ciencia y Tecnología de Polímeros (ICTP), España, por sus instalaciones y
equipos facilitados durante mi estancia. Al Dr. Enrique, Carmen Del Río Bueno, María
Carmen Ojeda García y Justyna Chojnack por los conocimientos que me brindaron y la
amabilidad con la que me trataron durante mi estancia en Madrid.
A Justyna Chojnack y José David Gómez Varga del ICTP por las caracterizaciones
realizadas de difracción de rayos-X, TGA, micrografías SEM de alta resolución.

VII
A M. en C. Jorge Arturo Domínguez Maldonado por las caracterizaciones de FTIR y Q. I.
Tanit Toledano Thompson por las micrografías SEM realizadas. A José Martín Baas
López por las espectroscopias Raman y su apoyo técnico al igual que a Isaí Carrera León.
A Julia González Montilla por su apoyo administrativo durante el transcurso de la
maestría y la amistad que me brindó.
A mis compañeros y amigos de la Unidad de Energía Renovable, que han hecho más
llevadero el camino recorrido durante la maestría, que se convirtieron en una familia para
mí. Por permitirme compartir y aprender con ellos estos años, por las experiencias que
hemos tenido juntos y nos hicieron ser más unidos.
A mis hermanos de proyecto, Lupita, Martín y José Manuel.
A todo el equipo de las Mandarinas Mecánicas con quienes pasé momentos divertidos,
disfrutando de la convivencia en otro ámbito aparte del académico.
A Madaleni que en poco tiempo se convirtió en una gran amiga que me ha brindado su
apoyo.
A esa personita que supo darme sus consejos cuando más lo necesitaba.
A mis padres Martín e Imelda por confiar en mí en todo momento, por darme la
oportunidad de seguir adelante, por apoyarme en las decisiones que he tomado. A mi
hermanito Alejandro y mi hermanita Karen por ser mi compañía a lo largo de mi vida,
porque siempre sigan sus sueños, porque lleguen tan lejos como ellos lo deseen y
puedan sobre llevar cualquier obstáculo que se les presente. A mis padres y hermanos
que a pesar de la distancia siempre estuvieron al pendiente de mí; por entender y tomar
de la mejor manera el que no estuviera con ellos en casa, porque éste esfuerzo no sólo
fue mío, sino de ellos también.

VIII
A MIS PADRES POR SU INFINITO AMOR
A MIS HERMANOS POR SU AMOR Y COMPRENSIÓN
A MI MEJOR AMIGA POR SU APOYO Y AMISTAD

IX

X
TABLA DE CONTENIDO
RESUMEN ..................................................................................................................................... XXII
ABSTRACT .................................................................................................................................. XXIV
INTRODUCCIÓN .............................................................................................................................. 1
Hipótesis ........................................................................................................................................ 3
Objetivo general ........................................................................................................................... 3
Objetivos específicos ................................................................................................................... 3
CAPÍTULO 1: ANTECEDENTES .................................................................................................. 5
1.1.- Supercondensadores .......................................................................................................... 5
1.1.1.- Introducción ................................................................................................................... 5
1.1.2.- Supercondensadores de doble capa ........................................................................ 9
1.1.2.1.- Principio de la doble capa electroquímica ...................................................... 10
1.1.3.- Supercondensadores pseudocapacitivos............................................................... 13
1.1.3.1.- Principio de los supercondensadores redox ................................................... 13
1.1.4.- Materiales para electrodos de supercondensadores de doble capa ................. 16
1.1.5.- Materiales para electrodos de supercondensadores pseudocapacitivos .......... 17
1.1.6- Supercondensadores de materiales híbridos ......................................................... 18
1.2.- Grafeno ............................................................................................................................... 19
1.2.1.- Métodos de obtención ............................................................................................... 21
1.3.- Polímeros intrínsecamente conductores ....................................................................... 23
1.3.1.- Proceso de dopado .................................................................................................... 23
1.3.2.- Métodos de síntesis de polímeros intrínsecamente conductores ...................... 25
1.3.3.- Mecanismos de polimerización ................................................................................ 27
1.3.3.1.- Polimerización por radicales libres ................................................................... 27
1.3.3.2.- Polimerización iónica .......................................................................................... 29
1.3.4.- Poli(pirrol) .................................................................................................................... 30
1.3.5.- Poli(3,4-etilenodioxitiofeno) ...................................................................................... 31

XI
1.4.- Materiales híbridos a base de grafeno/polímero intrínsecamente conductor .......... 34
1.5.- Técnicas de caracterización del material híbrido ......................................................... 35
1.5.1.- Microscopia electrónica de barrido (SEM) ............................................................. 35
1.5.2.- Espectroscopia Raman ............................................................................................. 35
1.5.3.- Difracción de Rayos X ............................................................................................... 36
1.5.4.- Análisis termogravimétrico ........................................................................................ 38
1.5.5.- Espectroscopia infrarroja con transformada de Fourier ....................................... 39
1.5.6.- Voltamperometría cíclica .......................................................................................... 39
1.5.7.- Ciclos galvanostáticos de carga-descarga ............................................................. 42
1.5.8.- Espectroscopia de impedancia electroquímica ..................................................... 43
CAPÍTULO 2: METODOLOGÍA .................................................................................................... 45
2.1.- Síntesis de emulsión inversa de nanotubos de polímeros intrínsecamente
conductores ................................................................................................................................. 45
2.2.- Síntesis de óxido de grafito.............................................................................................. 47
2.3.- Síntesis del material híbrido ............................................................................................ 48
2.4.- Caracterización de los materiales ................................................................................... 51
2.4.1.- Microscopía electrónica de barrido ......................................................................... 51
2.4.2.- Espectroscopia Raman ............................................................................................. 51
2.4.3.- Difracción de Rayos X ............................................................................................... 51
2.4.4.- Análisis termogravimétrico ........................................................................................ 52
2.4.5.- Espectroscopia infrarroja con transformada de Fourier ....................................... 52
2.4.6.- Caracterización electroquímica ................................................................................ 52
CAPÍTULO 3: RESULTADOS Y DISCUSIONES ...................................................................... 55
3.1.- Microscopía electrónica de barrido ................................................................................. 55
3.2.- Espectroscopia Raman .................................................................................................... 57
3.3.- Difracción de Rayos X ...................................................................................................... 61
3.4.- Análisis Termogravimétrico.............................................................................................. 64
3.5.- Análisis de FTIR ................................................................................................................ 66
3.6.- Caracterización electroquímica ....................................................................................... 70

XII
3.6.1.- Voltamperometría cíclica .......................................................................................... 70
3.6.2.- Ciclos galvanostáticos de carga-descarga ............................................................. 75
3.6.3.- Espectroscopia de impedancia electroquímica ..................................................... 79
CONCLUSIONES ........................................................................................................................... 85
REFERENCIAS .............................................................................................................................. 87

XIII

XIV
ÍNDICE DE FIGURAS
Figura 1.1.- Comparación de la densidad de potencia vs densidad de energía de diversos
dispositivos para almacenamiento de energía eléctrica. ........................................................... 6
Figura 1.2.- Esquematización de los diferentes tipos de condensadores y sus
componentes. .................................................................................................................................... 7
Figura 1.3.- Clasificación de los supercondensadores de acuerdo al proceso de
almacenamiento que emplean. ...................................................................................................... 8
Figura 1.4.- Representación de los procesos de carga y descarga en un condensador
electroquímico de doble capa......................................................................................................... 9
Figura 1.5.- Modelo de Helmholtz; a) organización rígida de los iones; b) variación del
potencial electrostático, ϕ, con distancia x, desde el electrodo; c) variación de la
capacitancia, Cd, con un potencial aplicado. .............................................................................. 11
Figura 1.6.- Esquematización de los modelos de Bockris correspondientes; a) distribución
de los iones y las moléculas del solvente, representa una molécula de agua; b) variación
del potencial electrostático, ϕ, con distancia, x, del electrodo de acuerdo con el potencial
aplicado. ........................................................................................................................................... 13
Figura 1.7.- Formas alotrópicas del carbono. ............................................................................ 19
Figura 1.8.- Proceso de carga de un electrodo de polímero intrínsecamente conductor; a)
P-Dopaje, b) N-Dopaje. ................................................................................................................. 25
Figura 1.9.- Proceso de término de crecimiento de la cadena por medio de dos cadenas
cortas. ............................................................................................................................................... 28
Figura 1.10.- Proceso de término de crecimiento de la cadena por medio de un átomo de
hidrógeno. ........................................................................................................................................ 28
Figura 1.11.- Mecanismo de polimerización catiónica.............................................................. 29
Figura 1.12.- Mecanismo de polimerización aniónica. ............................................................. 30
Figura 1.13.- Esquema de la estructura química del monómero de pirrol y la unidad
repetitiva del poli(pirrol). ................................................................................................................ 30
Figura 1.14.- Estructura del a) poli(pirrol) reducido, b) polarón y c) bipolarón. .................... 31
Figura 1.15.- Esquema de la estructura química del monómero EDOT y de la unidad
repetitiva del PEDOT. .................................................................................................................... 32

XV
Figura 1.16.- Estructura del a) poli(3,4-etilenedioxitiofeno), b) polarón y c) bipolarón. ....... 33
Figura 1.17.- Ejemplo de gráficas por espectroscopia Raman del grafeno. ......................... 36
Figura 1.18.- Difracción de rayos X en un cristal. ..................................................................... 37
Figura 1.19.- Esquema de una curva TG con una sola etapa de descomposición. ............ 38
Figura 1.20.- Señales de potencial de excitación utilizadas en voltamperometría. ............. 40
Figura 1.21.- Una celda típica electroquímica utilizada en voltamperometría consiste en un
electrodo de trabajo, un contra electrodo, y un electrodo de referencia. La celda puede ser
burbujeada con un gas inerte para remover el O2 disuelto. .................................................... 41
Figura 1.22.- Voltamperometría cíclica característica de los procesos de carga/descarga
en un condensador electroquímico. ............................................................................................ 41
Figura 1.23.- Esquema representativo de ciclos de carga y descarga a corriente
constante. ........................................................................................................................................ 43
Figura 1.24.- Representación esquemática del gráfico de impedancia Nyquis. .................. 44
Figura 2.1.- Estructura del AOT. .................................................................................................. 46
Figura 2.2.- Esquema representativo del óxido de grafito funcionalizado con la terminación
amino. ............................................................................................................................................... 49
Figura 2.3.- Esquema representativo de los materiales híbridos sintetizados. .................... 50
Figura 3.1.- Micrografías obtenidas por FESEM: a. nanotubos de poli(3,4-
etilenodioxitiofeno), b. nanotubos de poli(pirrol), c. óxido de grafito-amino, d. óxido de
grafito/PEDOT y e. óxido de grafito/PPy. ................................................................................... 56
Figura 3.2.- Micrografías de los materiales híbridos; a. GraphO-PEDOT y b. GraphO-PPy.
........................................................................................................................................................... 57
Figura 3.3.- Comparación del espectro Raman del GO-PEDOT, GO-amino, Óxido de
grafito y PEDOT. ............................................................................................................................. 59
Figura 3.4.- Comparación del espectro Raman del GO-PPy, GO-amino, Óxido de grafito y
PPy. .................................................................................................................................................. 60
Figura 3.5.- Patrón de difracción de rayos X del PEDOT, óxido de grafito, GraphO-PEDOT
y del grafito. ..................................................................................................................................... 62
Figura 3.6.- Patrones de difracción de rayos X del PPy, óxido de grafito, GraphO-PPy y
grafito................................................................................................................................................ 63
Figura 3.7.- TGA de los materiales sintetizados en atmósfera de nitrógeno. ....................... 66

XVI
Figura 3.8.- Espectros de FTIR correspondientes al óxido de grafito, GraphO-amino,
GraphO-PEDOT, PEDOT; obtenidos por ATR. ......................................................................... 68
Figura 3.9.- Espectros FTIR del óxido de grafito, GO-amino, PPy y GO-PPy; obtenidos por
ATR. .................................................................................................................................................. 69
Figura 3.10.- a) Voltamperometrías cíclicas a diferentes velocidades de barrido para a)
óxido de grafito, b) PEDOT y c) GraphO-PEDOT; en 2M H2SO4. .......................................... 72
Figura 3.11.- a) Voltamperometrías cíclicas a diferentes velocidades de barrido para a)
óxido de grafito, b) PPy y c) GraphO-PPy; en 2M H2SO4. ....................................................... 73
Figura 3.12.- Capacitancia específica del óxido de grafito, PEDOT y GO-PEDOT a), y del
óxido de grafito, PPy y del GO-PPy; velocidad de barrio 10 mV/s. ........................................ 75
Figura 3.13.- Ciclos galvanostáticos de los materiales sintetizados a 2 mA, en 2M H2SO4.
........................................................................................................................................................... 77
Figura 3.14.- Diagramas Nyquist de los materiales sintetizados, en 2M H2SO4. ................. 80
Figura 3.15.- Circuitos equivalentes: a) PEDOT y PPy; b) GraphO, GraphO-PEDOT y
GraphO-PPy. ................................................................................................................................... 81

XVII

XVIII
ÍNDICE DE TABLAS
Tabla 1.1.- Parámetros de un sistema de almacenamiento de energía para 1 mega Joule
(MJ) ensamblado utilizando los componentes de cada uno de los tres tipos de
condensadores. ................................................................................................................................ 7
Tabla 1.2.- Materiales basados en carbono empleados para electrodos en
supercondensadores de doble capa. .......................................................................................... 16
Tabla 1.3.- Materiales empleados para electrodos en supercondensadores
pseudocapacitivos. ......................................................................................................................... 17
Tabla 1.4.- Valores de capacitancia específica reportadas de materiales híbridos basados
en los materiales de interés. ......................................................................................................... 35
Tabla 3.1.- Bandas D, G y la relación ID/IG. ................................................................................ 61
Tabla 3.2.- Parámetros XRD de los materiales sintetizados. .................................................. 64
Tabla 3.3.- Valores de capacitancia calculados por voltamperometría cíclica. .................... 74
Tabla 3.4.- Valores de ERS y eficiencia obtenidos por las curvas galvanostáticas. ........... 78
Tabla 3.5.- Valores de capacitancia obtenidos por ciclos galvanostáticos y
voltamperometría cíclica. .............................................................................................................. 79
Tabla 3.6.- Valores obtenidos de ajustes a partir de los circuitos equivalentes. .................. 82
Tabla 3.7.- Comparación de las ERS de los materiales sintetizados obtenidas por
impedancia y por ciclos galvanostáticos. .................................................................................... 82

XIX

XX
TABLA DE ABREVIATURAS
AOT Bis(2-etilhexil)-sulfosuccinato sódico
CPE Elemento de fase constante
DTG Primera derivada de la curva TG
EDLC Condensador electroquímico de doble capa
EDOT 3,4-etilenodioxitiofeno
EDR Resistencia distribuida equivalente
EIS Espectroscopia de impedancia electroquímica
ESR Resistencia en serie equivalente
FESEM Microscopio electrónico de barrido de alta resolución
FTIR Espectroscopia infrarroja con transformada de Fourier
GEIS Espectroscopia de impedancia electroquímica galvánica
GraphO Óxido de grafito
GraphO-amino Óxido de grafito con terminación amino
GraphO-PEDOT Óxido de grafito/poli(3,4-etilenodioxitiofeno)
GraphO-PPy Óxido de grafito/poli(pirrol)
ICP Polímero intrínsecamente conductor
IHP Plano interior de Helmholtz
Lc Tamaño medio del cristal
MLG Grafeno multicapas
OG Óxido de grafeno
OHP Plano exterior de Helmholtz
PEDOT Poli(3,4-etilenodioxitiofeno)
PPy Poli(pirrol)
Py Pirrol
SCE Electrodo de referencia de calomelanos
SEM Microscopio electrónico de barrido
TGA Análisis termogravimétrico
VC Voltamperometría cíclica
XRD Difracción de rayos X

XXI

XXII
RESUMEN
En los supercondensadores, el material de los electrodos es un factor primordial en el
almacenamiento de energía, ya que deben de tener una gran área superficial. Por lo cual
en esta investigación, se sintetizan materiales híbridos a partir de óxido de grafito con
polímeros intrínsecamente conductores, poli(pirrol) y poli(3,4-etilenodioxitiofeno); con el fin
de aumentar la capacidad de almacenar energía de estos materiales híbridos, en
comparación al óxido de grafito. Se empleó la polimerización de microemulsión inversa
para obtener nanotubos de los polímeros, así como el método de Brodie para obtener el
óxido de grafito. Las hojas de óxido de grafito se funcionalizaron y se ancló el polímeros
intrínsecamente conductores.
Se confirmó la obtención del óxido de grafito multicapas mediante pruebas de difracción
de rayos X (XRD) al material obtenido de la síntesis del método de Brodie. La morfología
de nanotubos de los polímeros conductores se corroboró mediante microscopía
electrónica de barrido (SEM). Se alcanzaron capacitancias específicas en un electrolito
acuoso 2M H2SO4 a 5 mV/s de 19 F/g para el óxido de grafito/poli(3,4-etilenodioxitiofeno)
y 18 F/g para el óxido de grafito/poli(pirrol). Comparando éstos valores con la capacitancia
específica del óxido de grafito de 9 F/g, se concluye que existe un aumento en la
capacitancia específica de los materiales híbridos, con lo que podrían ser aplicados como
electrodos en supercondensadores.

XXIII

XXIV
ABSTRACT
The nature of the electrode materials is a crucial factor for its performance in energy
storage applications; those materials should have a large specific surface area. In this
research, hybrid materials from graphite oxide and intrinsically conductive polymer (i.e.
poly(pyrrole) and poly(3,4-ethylenedioxythiophene) were synthesized, the resulting
materials are considered to have improve energy storage capacity compared with graphite
oxide. Polymer nanotubes were synthesized using reverse microemulsion polymerization
and graphite oxide was prepared using the Brodies’s method. Oxide graphite sheets were
functionalized and intrinsically conducting polymer was anchored.
X-ray diffraction (XRD) results confirmed the production of multi-layer graphite oxide. The
morphology of the polymer nanotubes were determined by scanning electron microscopy
(SEM). A specific capacitance of 19 F/g for graphite oxide/poly(3,4-
ethylenedioxythiophene and 18 F/g for graphite oxide/poly(pyrrole) in 2M H2SO4 aqueous
electrolyte were archived. Graphite oxide specific capacitance was 9 F/g, the hybrid
material show an increase in the specific capacitance compared with graphite oxide, so
that could be applied as electrodes in supercapacitors.

XXV

INTRODUCCIÓN
1
INTRODUCCIÓN
Las tecnologías de almacenamiento de energía proporcionan valiosos beneficios a las
fuentes de energías, ya sea fósil o renovable; tanto para mejorar la estabilidad, la calidad
de energía y la confiabilidad del suministro [1]. Los sistemas de almacenamiento de
energía eléctrica pueden ser divididos en dos categorías principales: las baterías y los
condensadores electroquímicos. Las baterías almacenan energía en forma de reacciones
químicas, en donde existe movimiento de carga y de materia entre los electrodos;
mientras que los condensadores electroquímicos almacenan energía directamente como
carga de manera electrostática, o sea, que no se llevan a cabo reacciones farádicas [2-4].
Las diferencias fundamentales entre las baterías y los condensadores electroquímicos
radican en la ciclabilidad de carga-descarga, en el tiempo de reacción, y en la energía y
potencia que se entrega en la salida. Las baterías almacenan más energía por unidad de
masa que los condensadores electroquímicos, debido a que se llevan a cabo reacciones
farádicas en los electrodos, lo que provoca cambios en la estructura moléculas de los
electrodos afectando su estabilidad después de algunos cientos de ciclos de carga-
descarga [5].
A diferencia de las baterías y las celdas de combustible en las que la energía recolectada
se almacena en los enlaces químicos, los supercondensadores utilizan la superficie de
sus electrodos para almacenar la carga electrostáticamente mediante procesos no-
faradaicos [6, 7]. La capacitancia de estos materiales está en el rango de decenas de
faradios por gramo de material activo; a diferencia de los condensadores dieléctricos
tradicionales, la cual está en el rango de microfaradios. No obstante, la energía
almacenada en los supercondensadores es directamente proporcional a la capacidad de
carga que pueden almacenar sus electrodos [6, 8-10]. Los materiales utilizados como
electrodos de supercondensadores deben tener una área superficial grande, superior a
los 1000 m2/g [11].
Con el objetivo de aumentar la capacidad de almacenar energía de los
supercondensadores, en éste trabajo se sintetizaron materiales híbridos a base de óxido

INTRODUCCIÓN
2
de grafito/poli(pirrol) y óxido de grafito/poli(3,4-etilenodioxitiofeno). El óxido de grafito se
sintetizó mediante el método de Brodie [12], y los polímeros intrínsecamente conductores,
poli(pirrol) y el poli(3,4-etilenodioxitiofeno) empleando una polimerización de emulsión
inversa para obtener nanotubos de ambos polímeros. Posteriormente, se ancló el
polímero sobre el óxido de grafito con ayuda de grupos funcionales (grupo amino); se
espera que con la inserción de los nanotubos entre las capas de óxido de grafito exista
una mayor separación entre las hojas del óxido de grafito para así impedir que se
reaglomeren.
Los materiales obtenidos (los polímeros intrínsecamente conductores, el óxido de grafito y
los materiales híbridos sintetizados) fueron caracterizados morfológica (SEM), físico-
química (TGA, FTIR, XRD y Raman) y electroquímicamente (VC, galvanometría de carga
y descarga, y EIS).
En el capítulo uno se expone los antecedentes de los supercondensadores así como sus
características dependiendo de la forma de almacenar la energía, las propiedades y
métodos de obtención del óxido de grafito y de los polímeros intrínsecamente
conductores. Además, se describen las diferentes técnicas que se utilizaron para
caracterizar los materiales obtenidos en el presente trabajo.
En el capítulo dos se describe la metodología que se empleó para la obtención del óxido
de grafito, de los polímeros intrínsecamente conductores y el procedimiento empleado
para el anclaje de los nanotubos del polímero intrínsecamente conductor en la superficie
del óxido de grafito.
El capítulo tres comprende la parte de la discusión de los resultados obtenidos de las
diferentes técnicas de caracterización.
Por último se presentan las conclusiones del trabajo con los resultados obtenidos en la
caracterización de los distintos materiales.

INTRODUCCIÓN
3
Hipótesis
La inclusión de nanotubos de poli(pirrol) o poli(3,4-etilenodioxitiofeno) entre las láminas de
óxido de grafito, mediante la inserción de un grupo funcional amino con terminación de
anillo aromático como promotor de la cadena polimérica, permitirá la obtención de un
material hibrido que reduzca la resistencia equivalente del óxido de grafito, aumente los
valores de capacitancia específica y el tiempo de los ciclos de carga y descarga, para
utilizarse como electrodos en supercondensadores.
Objetivo general
Síntesis y caracterización de nanotubos de poli(3,4-etilenodioxitiofeno) y poli(pirrol),
anclados sobre óxido de grafito, para su posible aplicación como material para electrodos
en supercondensadores.
Objetivos específicos
Obtención de óxido de grafito mediante una oxidación química del grafito por el
método de Brodie.
Síntesis de nanotubos de los polímeros intrínsecamente conductores de poli(3,4-
etilenodioxitiofeno) y poli(pirrol) anclados sobre el óxido de grafito, para la
obtención de materiales híbridos.
Caracterización morfológica, fisicoquímica y electroquímica de los materiales
individuales y los materiales híbridos por medio de microscopía electrónica de
barrido (SEM), espectroscopia infrarroja con transformada de Fourier (FTIR),
difracción de rayos X (XRD), análisis termogravimétrico (TGA), espectroscopia
Raman, voltamperometrías cíclicas, ciclos galvanostáticos de carga-descarga y
espectroscopia de impedancia electroquímica.

INTRODUCCIÓN
4

CAPÍTULO 1: ANTECEDENTES
5
CAPÍTULO 1
ANTECEDENTES
1.1.- Supercondensadores
1.1.1.- Introducción
Los condensadores electroquímicos, también llamados supercondensadores o
ultracondensadores, son capaces de entregar mayor densidad de potencia que las
baterías y mayor densidad de energía que los condensadores dieléctricos; además
cuentan con un rango amplio de temperatura operativa, un intervalo bajo de auto-
descarga, vida cíclica larga y eficiencia alta en su desempeño en el almacenamiento de la
energía [8, 13-15]. Son usados principalmente en aplicaciones que requieren impulsos de
energía durante periodos cortos de tiempo [10, 14, 16, 17].
En la figura 1.1, denominada diagrama de Ragone, se presentan los dispositivos típicos
de almacenamiento y conversión de energía, en términos de su energía específica y la
potencia específica [18]. Un supercondensador se puede considerar como un dispositivo
intermedio entre un condensador tradicional y una batería, en base a la energía específica
y potencia específica; los supercondensadores cubren varios órdenes de magnitud de la
brecha que existe entre ambos sistemas [7, 18]. Como ya se mencionó previamente, las
diferencias fundamentales entre las baterías y los condensadores electroquímicos radican
en la ciclabilidad de carga-descarga, en el tiempo de reacción, y en la energía y potencia
que se entrega en la salida.

CAPÍTULO 1: ANTECEDENTES
6
Figura 1.1.- Comparación de la densidad de potencia vs densidad de energía de diversos
dispositivos para almacenamiento de energía eléctrica [14, 18, 19].
Los condensadores se clasifican en: electrostáticos, electrolíticos y electroquímicos (ver
figura 1.2) [4, 20]. Los condensadores electrostáticos consisten de dos electrodos
metálicos separados por un dieléctrico; almacenan la carga de una manera física y no
química. Un condensador electrolítico es similar en construcción a un condensador
electrostático, sin embargo, difiere en el proceso de almacenamiento, ya que se tiene un
electrolito en contacto directo con los electrodos metálicos; el mecanismo de
almacenamiento es químico. La densidad de energía es alrededor de 10 veces mayor que
la de los condensadores electroestáticos. En general, los condensadores electroquímicos
también utilizan soluciones electrolíticas, pero poseen electrodos altamente porosos, por
lo que tienen una capacidad mayor de almacenar energía por unidad de volumen que los
condensadores electrostáticos y electrolíticos. Tienen alta capacitancia específica de
aproximadamente 100 veces más que la de un condensador electrolítico [4, 20].

CAPÍTULO 1: ANTECEDENTES
7
Figura 1.2.- Esquematización de los diferentes tipos de condensadores y sus componentes [20].
Los condensadores electroquímicos presentan algunas ventajas frente a los otros dos
tipos de condensadores; entre ellas destaca que para almacenar la misma cantidad de
energía los sistemas electroquímicos presentan una menor masa y también un menor
volumen a comparación de los condensadores electrostáticos y electrolíticos (ver tabla
1.1).
Tabla 1.1.- Parámetros de un sistema de almacenamiento de energía para 1 mega Joule (MJ)
ensamblado utilizando los componentes de cada uno de los tres tipos de condensadores [4].
Tipo de
condensador
Masa de un
sistema de 1MJ
(kg)
Volumen de un
sistema de 1MJ
(m3)
Costo de un
sistema de 1MJ
(k US $)
Tiempo de
respuesta (s)
Electrostático 200000 140 700 10-9
Electrolítico 10000 2.2 300 10-4
Electroquímico 100 0.1 15 1

CAPÍTULO 1: ANTECEDENTES
8
En los últimos años, los supercondensadores han sido aplicados en diferentes
dispositivos que van desde teléfonos celulares hasta vehículos eléctricos híbridos [21].
Los supercondensadores son aplicados principalmente en dispositivos que requieren una
gran cantidad de carga entregada en un tiempo muy corto como cámaras digitales y
herramientas eléctricas, o de manera híbrida con baterías recargables como las baterías
de plomo-ácido en vehículos de sistemas de transmisión eléctrica [15, 21, 22].
Los dos primeros supercondensadores comerciales aparecieron en el mercado en 1978
(Gold Capacitors de Panasonic/Matsushita) y en 1980 (Supercap de NEC/Tokin). Éstos
productos proveían una tensión nominal en un rango de 2.3 a 6 V y valores de
capacitancia de 10-2 F hasta varios Faradios [18].
Debido a los diferentes mecanismos para almacenar la energía, los supercondensadores
se clasifican como condensadores electroquímicos de doble capa (EDLC) y
supercondensadores redox (ver figura 1.3) [23, 24].
Figura 1.3.- Clasificación de los supercondensadores de acuerdo al proceso de almacenamiento
que emplean [19].

CAPÍTULO 1: ANTECEDENTES
9
1.1.2.- Supercondensadores de doble capa
Los condensadores electroquímicos de doble capa usualmente están formados por dos
electrodos de carbono y un electrolito soportado en una matriz porosa interpuesta entre
los electrodos (ver figura 1.2) [10, 15, 19, 25].
Los supercondensadores de doble capa son dispositivos de almacenamiento de energía
electroquímicos donde las cargas se almacenan en la interface entre el electrodo y el
electrolito en la doble capa electroquímica, también llamada capa de Helmholtz [4, 10, 13,
25-27]. Los iones del electrolito son adsorbidos electrostáticamente sobre la superficie de
los electrodos porosos sin que ocurran reacciones farádicas [13, 27].
Durante el proceso de descarga, la solución electrolítica comprende cationes cargados
positivamente y aniones con carga negativa. Una vez que el dispositivo está cargado, los
cationes son atraídos a la superficie del electrodo negativo y los aniones, por el electrodo
positivo; formado de ésta manera una doble capa (ver figura 1.4) [4, 19].
Figura 1.4.- Representación de los procesos de carga y descarga en un condensador
electroquímico de doble capa [2].
Tal como para capacitores dieléctricos, la capacitancia, expresada en Faradios (F), se
define como la relación entre la carga total en coulombs (Q) en cada electrodo y el
diferencial de potencial (V) aplicado [3, 4, 19, 28]:
V
QC
[1.1]

CAPÍTULO 1: ANTECEDENTES
10
Ya que un supercondensador de doble capa consta de dos electrodos, con una
capacitancia de doble capa electroquímica en cada uno de ellos, la capacidad del
supercondensador está dada por [18, 29]:
21sup
111
DLDLdorercondensa CCC
[1.2]
donde CDL1 y CDL2 son las capacitancias de los dos electrodos del supercondensador.
La energía E almacenada en un condensador es directamente proporcional a su
capacitancia y al cuadrado del potencial a través de los electrodos [14, 18, 23, 29, 30]:
2
2
1CVE
[1.3]
La potencia específica es limitada por la resistencia en serie equivalente, que a su vez
está determinada principalmente por la conductividad de la capa activa y el separador, así
como la resistencia de contacto en la placa final. Las baterías, la potencia máxima del
supercondensador se obtiene cuando la resistencia externa es igual a la resistencia
interna. El máximo de energía eléctrica utilizable está dada por [18, 23, 29]:
R
UP
4
2
max [1.4]
donde R es la resistencia interna del condensador y U es el potencial de operación.
1.1.2.1.- Principio de la doble capa electroquímica
Las reacciones electroquímicas tienen lugar en la interfase entre un conductor electrónico
(electrodo), y un conductor iónico (electrolito) [31]. En este dispositivo la energía eléctrica
almacenada se basa en la separación de especies cargadas en una doble capa eléctrica
a través de la interface electrodo/electrolito; esto da lugar a una región llamada doble

CAPÍTULO 1: ANTECEDENTES
11
capa. Se han propuesto muchos modelos para explicar el comportamiento de
transferencia de carga que se lleva a cabo en el electrolito; a continuación se presentan
dos de los modelos fundamentales; el modelo de Helmholtz y el modelo de agua-dipolo.
El primero fue un modelo preliminar de doble capa, mientras que el segundo, que es el
que actualmente se emplea, describe de una manera más completa las fases que se
presentan durante el proceso [32].
a) Modelo de Helmholtz o de doble capa rígida
El modelo propone la existencia de una capa de electrones en la superficie del electrodo y
una monocapa de iones en el electrolito, con igual magnitud y signo opuesto; por lo que
se le da la designación de doble capa, en donde las interacciones no se extienden más
allá en la solución, dando lugar así a una alta capacitancia en la interfase (ver figura 1.5)
[31, 33, 34].
Figura 1.5.- Modelo de Helmholtz; a) organización rígida de los iones; b) variación del potencial
electrostático, ϕ, con distancia x, desde el electrodo; c) variación de la capacitancia, Cd, con un
potencial aplicado [33].

CAPÍTULO 1: ANTECEDENTES
12
La capacitancia diferencial es definida por:
E
σC
[1.5]
donde σ es la densidad de carga y E es el potencial eléctrico [3, 31, 33]. De acuerdo al
modelo de Helmholtz la capacitancia diferencial es dada por:
24π
εCH
[1.6]
CH es la capacitancia diferencial predicha por el modelo de Helmholtz, y es un valor
constante que depende de la constante dieléctrica (ε) y de la separación de la capa de
carga (δ) [28, 33, 35]. Por lo tanto CH incrementa cuando decrece la separación entre la
superficie del electrodo y la capa iónica del electrolito [28].
Entre los principales inconvenientes de este modelo se encuentra el hecho de que no
toma en cuenta las interacciones que se producen adicionalmente a las del electrodo y la
capa de especies adsorbidas, y tampoco considera la dependencia de la concentración
del electrolito [33].
b) Modelo de Bockris, Devanathan y Müller o de agua-dipolo
En este modelo se toma en cuenta la interacción que existe entre los disolventes
dipolares, como el agua o líquidos orgánicos, y el electrodo; ya que la concentración del
solvente es siempre mayor a la concentración del soluto (ver figura 1.6) [33, 35]. El plano
interior de Helmholtz (IHP) comprende todas las especies que están específicamente
adsorbidas en la superficie del electrodo. El plano exterior de Helmholtz (OHP) está
compuesto de los iones que están más cercanos a la superficie del electrodo, solvatados,
pero no están específicamente adsorbidos [33, 35]. La variación del potencial con la
distancia, a través de la interface electrodo/electrolito revela una fuerte caída entre el

CAPÍTULO 1: ANTECEDENTES
13
electrodo y IHP, luego un pequeño aumento entre IHP y OHP [35]. En la actualidad, éste
modelo es el que se utiliza para definir el comportamiento de la doble capa.
Figura 1.6.- Esquematización de los modelos de Bockris correspondientes; a) distribución de los
iones y las moléculas del solvente, representa una molécula de agua; b) variación del
potencial electrostático, ϕ, con distancia, x, del electrodo de acuerdo con el potencial aplicado [33].
1.1.3.- Supercondensadores pseudocapacitivos
Un supercondensador pseudocapacitivo almacena la energía por dos mecanismos;
además de la doble capa electroquímica, su principal mecanismo de almacenamiento de
carga es farádicamente mediante reacciones tipo redox, adsorción de iones o
intercalación en un mecanismo denominado pseudocapacitancia. Ésta última depende
tanto del tipo de material así como del electrolito; involucra a todo el material activo del
electrodo en sí y no únicamente la superficie de éste. Se ha observado que la
pseudocapacitancia contribuye al aumento de la capacitancia y la densidad de energía de
los condensadores electroquímicos [14].
1.1.3.1.- Principio de los supercondensadores redox
El mecanismo de carga y descarga en los supercondensadores implica la transferencia de
carga eléctrica entre las fases [27]; esto se lleva a cabo en los electrodos cuando al
aplicar un potencial se inducen corrientes farádicas en reacciones como adsorción de
iones o de la oxidación-reducción de materiales electroactivos [15, 34].

CAPÍTULO 1: ANTECEDENTES
14
a) Reacciones redox
A partir de las reacciones de oxidación-reducción que se originan en el electrodo se
produce una transferencia de cargas, indicadas como Oad + ne- → Red [15].
Considerando que el proceso redox se produce en un potencial casi constante en un
dispositivo el potencial del electrodo varía proporcionalmente a la carga utilizada dq en un
pseudocondensador, como se muestra en la siguiente fórmula [14]:
C*dUdq [1.7]
El potencial de reducción del electrodo es dado por la ecuación de Nernst:
1ln0
zF
RTEE
[1.8]
donde E0 es el potencial estándar, R es la constante de los gases, T es la temperatura
absoluta, F es la constante de Faraday, z es la carga eléctrica del ion considerado, y ℜ es
definido como ([ox]/ ([ox] + [red]), donde los corchetes denotan la concentración de las
especies [30].
a) Adsorción de iones
Entre los fenómenos que ocurren durante la pseudocapacitancia se encuentra la
adsorción de iones (quimisorción); en muchos de los casos solamente un tipo de ion es
específicamente adsorbido, el anión (excepto en el caso de electrolitos orgánicos). El
depósito de los iones provenientes del electrolito, para formar una monocapa sobre la
superficie del electrodo, es un proceso reversible que se traduce en una transferencia de
carga de Faraday [30].

CAPÍTULO 1: ANTECEDENTES
15
El proceso de adsorción/desorción que se lleva a cabo en el electrodo se puede expresar
de la siguiente manera [15, 30]:
adsV
c ASeSAAA
1 [1.9]
donde A es la especie iónica, S es el sustrato, c es la concentración de los iones
depositables, 1-θA es el área fraccional de la superficie libre disponible para adsorción, θA,
y V es el potencial del electrodo. En este proceso se involucra el movimiento de iones y
electrones dentro del material.
Por ejemplo, la ecuación de reacción de la superficie para el Pt en solución acida,
quedaría de la siguiente forma [36]:
OHPtHeOHPt
upadaerficie oc
adsK
potencialV
c
breerficie liHH 2
sup
3
sup
1
[1.10]
Si se asume que los sitios están ocupados al azar en una red fija, se puede determinar la
cobertura a partir de la ecuación de adsorción de Langmuir [30]:
RT
VFKc
θ
θ
A
A exp1
[1.11]
K es la constante de equilibrio electroquímico. Un cambio en la cobertura de dθ es
directamente proporcional a la carga dq [30]:
dθqdq 1 [1.12]

CAPÍTULO 1: ANTECEDENTES
16
donde q1 es la cantidad de carga necesaria para formar o disolver una monocapa
completa. Puesto que θ es una función de V, lo que da una relación pseudocapacitiva [30,
36, 37]:
2
1
exp1
exp
RT
VFKc
RT
VFKc
RT
FqCφ
[1.13]
1.1.4.- Materiales para electrodos de supercondensadores de doble capa
Los supercondensadores basados en la doble capa electroquímica requieren electrodos
con una área superficial grande y resistencia electrónica baja [29]. El carbono, en varias
modificaciones, es el material más utilizado para los electrodos (ver tabla 1.2). Las
razones para usar carbono son muchas como bajo costo, área superficial grande,
disponibilidad, entre otras [18].
Entre los diferentes tipos de carbonos que se han utilizado como electrodos se
encuentran: nanofibras de carbono, grafito nanoestructurado, carbono activado, tela de
carbono, nanotubos de carbono, carbono mesoporoso nanoestructurado, compuestos de
carbono mesoporoso y carbono poroso de precursores termoplásticos [20, 38].
Tabla 1.2.- Materiales basados en carbono empleados para electrodos en supercondensadores de
doble capa [15].
Material electrodo Electrolito Voltaje de
trabajo (V)
Capacitancia
específica (F/g) Ref
Carbono Activado (AC) Et4NBF4 + PC (1M) 1.5 40 [39]
Grafito Et4NBF4 + PC (1M) 3.0 12 [40]
Aerogeles de carbono Et4NBF4 + PC (1.5M) 3.0 160 [41]
Carbono mesoporoso KOH (30 wt%) 0.9 180 [42]
Carbono meso/macroporoso KOH (6M) 0.8 130 [43]
Tela de carbono activado KOH (6M) 1.0 208 [44]

CAPÍTULO 1: ANTECEDENTES
17
Nanotubos de carbono
(CNTs) de pared simple EMITFSI 2.3 50 [45]
CNTs de pared múltiple TEMANF4 + PC (1.96M) 2.5 13 [46]
CNTs/poli(pirrol)/MnO2 Na2SO4 (1M) 0.9 281 [47]
1.1.5.- Materiales para electrodos de supercondensadores pseudocapacitivos
Los electrodos de los supercondensadores con pseudocapacitancia emplean óxidos de
metales de transición o polímeros intrínsecamente conductores (tabla 1.3) [15, 21, 38].
Los óxidos metálicos presentan una gran capacitancia específica a bajas resistencias [20].
Varios óxidos metálicos, tales como RuO2, IrO2 y NiOx, Ni(OH)2, MnO2, Co2O3, TiO2 y V2O5
MoO, han sido estudiados como materiales para los electrodos [24].
La pseudocapacitancia de polímeros intrínsecamente conductores surge por su rápido y
reversible proceso de oxidación y reducción. Los polímeros p-dopados son los más
utilizados ya que son más estables frente a la degradación que los polímeros n-dopables
[15]; Los oxido-reducción provocan estrés en la cadena principal produciendo cambios en
la estructura en especial para los polímeros n-dopados.
Tabla 1.3.- Materiales empleados para electrodos en supercondensadores pseudocapacitivos [15].
Material electrodo Electrolito Voltaje de
trabajo (V)
Capacitancia
específica (F/g) Ref
Óxidos de
metales de
transición
RuO2 • H2O H2SO4 (0.5M) 1.0 650 [48]
Nanoláminas de
ácido ruténico/Au H2SO4 (0.5M) 1.2 620 [49]
H0.2RuO2.1 • nH2O H2SO4 (0.5M) 1.2 390 [50]
RuO2/carbono Hidrogel PVA 0.8 1000 [51]
Ru1-yCryO2/TiO2
Amorfo KOH (1M) 0.9 1272 [52]
MnO2 K2SO4 (0.5M) 0.8 261 [53]
MnO2/AC K2SO4 (0.65M) 2.2 29 [54]
SnO2/aerogel H2SO4 (1M) 1.0 68 [55]

CAPÍTULO 1: ANTECEDENTES
18
carbono
Ni(OH)2 KOH (3 %) 0.8 578 [56]
Ni(OH)2/AC KOH (6M) 0.8 194 [57]
Óxidos de cobalto-
níquel/CNTs KOH (1M) 1.0 569 [58]
Mischmetal base-
Níquel/AC BMIM-PF6 1.0 357 [59]
Mo2N/Ta2O5 H2SO4 (3.5M) 0.8 106 [60]
WX/carbono H2SO4 (1M) 0.9 477 [61]
MnFe2O4 LiPF6 (1M)
+EC/EMC 2.5 126 [62]
TiN KOH (1M) 0.2 238 [63]
V2O5 KCl (2M) 0.7 262 [64]
Polímeros
intrínsecamente
conductores
Poli(3-metiltiofeno) PYR14TFSI 3.6 25 [65]
Poli(3-
metiltiofeno)/MnO2 Na2SO4 (1M) 1.0 381 [66]
Poli(pirrol)/AC Pirrol (0.5M) +
β-NSA (0.5M) 0.9 345 [67]
Poli(anilina)/MnO2 Na2SO4 (0.1M) 1.2 715 [68]
Poli(anilina)/AC KOH (6M) 0.9 588 [69]
1.1.6- Supercondensadores de materiales híbridos
Los supercondensadores híbridos intentan aprovechar las ventajas y disminuir las
desventajas de los EDLCs y los pseudocondensadores, mejorando así sus características
de rendimiento. El empleo de los procesos farádicos y no farádicos para almacenar la
carga ha hecho que se alcancen energías y densidades de potencia superiores que el de
los EDLCs y pseudocondensadores de manera individual [19, 38].
Los electrodos compuestos integran materiales basados en carbono, con materiales
poliméricos intrínsecamente conductores u óxidos de metales de transición; los
mecanismos de almacenamiento de carga son de forma física o química en el electrodo.

CAPÍTULO 1: ANTECEDENTES
19
Los materiales basados en carbono facilitan una doble capa de carga y también una área
superficial grande que incrementa así el contacto entre los materiales pseudocapacitivos
depositados y el electrolito [19]. Los materiales pseudocapacitivos son capaces de
aumentar aún más la capacidad del electrodo compuesto a través de reacciones farádicas
[19].
En los últimos años, ha habido un gran interés en el grafeno el cual se ha convertido en
un material prometedor en el almacenamiento de energía debido a su gran superficie y
conductividad eléctrica [70]. Polímeros intrínsecamente conductores (ICP), como el
poli(pirrol) (PPy), poli(anilina) (PANI) y poli(3,4-etilenodioxitiofeno) (PEDOT), poseen una
buena conductividad eléctrica y pseudocapacitancias grandes, estas propiedades hacen
de estos materiales una buena opción para ser utilizados en supercondensadores [71].
1.2.- Grafeno
El grafeno consiste en una red 2D hexagonal de átomos de carbono cuasi plana con
pequeñas ondulaciones, dando la apariencia de una panal de abejas [72-76]. El grafeno
es el componente básico de todos los demás elementos grafíticos; puede ser apilado
formando grafito y diamante (3D), enrollado para formar nanotubos (1D), envueltos para
formar fulerenos (0D) (ver figura 1.7) [72, 73, 77].
Figura 1.7.- Formas alotrópicas del carbono [73, 75].

CAPÍTULO 1: ANTECEDENTES
20
Las propiedades electrónicas del grafeno se deben a la alta calidad de su red cristalina en
2D; esto implica una densidad de defectos excepcionalmente baja, que normalmente
actúan como centros de dispersión que inhiben el transporte de carga eléctrica [77].
Algo importante a cerca del transporte de carga en el grafeno es su ambipolaridad. Esto
significa que el grafeno tiene la capacidad de cambiar entre el uso de portadores positivos
(huecos) y negativos (electrones) sobre la marcha en función de la señal de entrada, en
donde sus movilidades pueden exceder 15000 cm2/Vs incluso bajo condiciones
ambientales, pudiendo éstas llegar hasta ≈100000 cm2/Vs si se eliminan las impurezas
que generan dispersión [73, 76, 77].
Entre las propiedades más destacadas de este material están una conductividad térmica
grande (~5000 W/mK) y resistencia eléctrica baja (50 x10-6 Ωcm), elasticidad y dureza
altas, resistencia a las radiaciones ionizantes, bajo peso, efecto de Joule bajo (se calienta
menos al conducir electrones) y consumo de electricidad bajo [73, 74, 78, 79]. El área
superficial teórica es reportada aproximadamente entre 2630 m2/g, superando a los
nanotubos monocapa y el grafito, cuyas áreas superficiales han reportado ser ~1315 m2/g
y ~10 m2/g, respectivamente [16, 79].
En la actualidad ha existido confusión sobre hasta qué punto un material puede ser
considerado una capa de grafeno, por lo que la revista Carbon (volumen 65, 2013) publicó
una primera nomenclatura para los carbonos de dos dimensiones propuesta por un comité
de investigadores, con la finalidad de homogenizar la descripción de éstos materiales [80].
A continuación se presenta la nomenclatura para designar a éstos materiales 2D:
Grafeno.- Una sola hoja de un solo átomo de espesor estructurada de manera
hexagonal, átomos con enlace sp2 que no son una parte integral de un material de
carbono.
Grafeno bicapa, grafeno tricapa.- Material laminar 2D, que consiste en 2 o 3 capas
de grafeno apiladas bien definidas.

CAPÍTULO 1: ANTECEDENTES
21
Grafeno multicapas (MLG).- Material 2D similar a una lamina que consta de un
número pequeño contable de capas de grafeno apiladas, de alrededor de 2 a 10
capas.
Óxido de grafeno (GO).- Grafeno modificado químicamente, preparado por la
oxidación y la exfoliación que se acompaña de una modificación oxidativa del
plano basal.
Óxido de grafito.- Sólido sintetizado por la oxidación de grafito a través de
procesos que funcionalizan los planos basales y así aumentar la distancia entre
las capas. El óxido de grafito puede ser exfoliado en solución para formar óxido de
grafeno (monocapa) o parcialmente exfoliado para obtener óxido de grafeno de
pocas capas.
1.2.1.- Métodos de obtención
Diversas propuestas han sido desarrolladas para producir una o varias capas de grafeno,
entre las que se encuentran la exfoliación mecánica de grafito, crecimiento epitaxial,
exfoliación química, la reducción química de los óxidos de grafeno y la deposición química
de vapor [16, 74, 81, 82].
a) Exfoliación micromecánica
En esta técnica, la superficie de un cristal de grafito se le somete a un raspado fino, de
arriba hacia abajo, mediante el empleo de cualquier objeto de superficie sólida, o bien, al
desescamamiento repetido utilizando cinta adhesiva con el propósito de extraer hojuelas
extremadamente delgadas unidas a estos objetos [73, 83].
b) Exfoliación química
Consiste en insertar moléculas o átomos en una masa de grafito, de tal manera que los
planos de grafeno puedan ser separados por las moléculas o átomos insertados.
Después, mediante una reacción química, se separan los átomos o moléculas que se

CAPÍTULO 1: ANTECEDENTES
22
insertaron obteniéndose un sedimento consistente de residuos y hojas de grafeno [73,
77].
c) Reducción química del óxido de grafito
La reducción del óxido de grafito es una vía prometedora para lograr la producción en
masa de grafeno a través de tres pasos: la oxidación del grafito, la exfoliación del óxido de
grafito (obteniendo óxido de grafeno), y la reducción del óxido de grafeno [77, 81]. El
método de Brodie fue el primer método reportado para obtener óxido de grafito, en 1859;
este método se basa en la oxidación del grafito mediante la adición de ácido nítrico
fumante y clorato de potasio [12, 84]. Por otra parte, Hummers y Offeman desarrollaron un
método de oxidación al hacer reaccionar al grafito con una mezcla de permanganato de
potasio (KMnO4) y ácido sulfúrico concentrado (H2SO4) [79, 81, 85, 86].
Una variedad de métodos térmicos y mecánicos pueden ser utilizados para exfoliar el
óxido de grafeno a una capa individual de óxido de grafeno (GO), siendo la agitación de
éste en un solvente y/o el uso de ultrasonidos son más comunes [81]. Por último, el óxido
de grafeno puede reducirse mediante hidracina u otros agentes reductores, obteniendo
así el grafeno [81].
d) Crecimiento epitaxial y depósito químico de vapor
De Heer fue pionero en reportar el método epitaxial, donde el grafeno resulta de la
reducción a altas temperaturas del carburo de silicio. El proceso es relativamente sencillo,
el silicón desorbe alrededor de los 1000 °C en ultra vacío [77, 83]. El segundo método es
el depósito químico de vapor (CVC) de vapor de grafeno en las películas de metales de
transición. Éste procedimiento se basa en la saturación de carbono en presencia de un
metal de transición por medio de la exposición de un gas de hidrocarburos a altas
temperaturas. A menudo, las películas de níquel se utilizan con gas metano. Al enfriarse
el sustrato, la solubilidad del carbono en el metal de transición decrece y una delgada
capa de carbono se precipita desde la superficie [77, 83, 87].

CAPÍTULO 1: ANTECEDENTES
23
1.3.- Polímeros intrínsecamente conductores
Los polímeros intrínsecamente conductores (ICP) son polímeros que conducen electrones
a través de su estructura [88-91]. Consisten básicamente en una cadena carbonatada
muy larga que presenta una conjugación muy extendida; ya que está formado por
moléculas que repiten su estructura en forma periódica. Son moléculas que presentan una
distribución de dobles enlaces C = C alternados a lo largo de la cadena [90, 91]. La
diferencia de éstos polímeros con los otros tipos de polímeros, se deriva de un proceso de
dopaje, en donde el polímero tiene un esqueleto conjugado en el cual el sistema-π se
deslocaliza, dando así la formación de complejos de transferencia de carga de los
donantes de electrones, tal es el caso del sodio o potasio, o por aceptores de electrones
como el I2 o FeCl3, por ejemplo [89, 92]. La capacidad de almacenar carga en la superficie
y en el interior del material proporciona una gran capacidad de almacenamiento de carga
por unidad de superficie de dicho material [91].
Para realizar la síntesis de éstos materiales se puede emplear tanto la vía química o
electroquímica [34]. Ejemplos de polímeros intrínsecamente conductores que han sido
comúnmente utilizados en la fabricación de nanocompuestos son la poli(anilina) (PANI),
poli(pirrol) (PPy), poli(3,4-etilenodioxitiofeno), entre otros [93].
Los polímeros intrínsecamente conductores ofrecen tres propiedades importantes para los
supercondensadores: capacitancia específica alta, debido a la participación de toda la
masa del polímero en el proceso de carga, por ejemplo para el PEDOT se han reportado
110 F/g [94] y 200 F/g [95], y para el PPy 130 F/g [94]; alta conductividad en el estado
dopado, la cual se encuentra en el rango de los metales y semiconductores (2 x104 – x10-8
S/cm); y rápida cinética de transferencia de electrones de carga y descarga [21].
1.3.1.- Proceso de dopado
El dopaje tipo-p conlleva al oxidación parcial del sistema-π, con lo que la eliminación de
electrones-π de la conjugación nos da una carga positiva neta; mientras que el dopaje
tipo-n se refiere a la reducción parcial del sistema-π de la cadena principal del polímero,
dando una carga negativa neta [96]. La manera en la cual los polímeros intrínsecamente

CAPÍTULO 1: ANTECEDENTES
24
conductores almacenan y liberan la carga es a través de procesos oxido-reducción.
Cuando se produce la oxidación o dopaje, los iones son transferidos a la estructura del
polímero; mientras que durante la reducción o desdopaje, los iones son liberados de
nuevo en la solución (ver figura 1.8) [97]. Cuando se produce un flujo de electrones y
movimiento de iones por un proceso de carga; el polímero p-dopado se carga
positivamente lo que produce movimiento de los aniones en el electrolito hacia el polímero
para así compensar la carga; en el caso del polímero n-dopado se caga negativamente,
mientras que los cationes se mueven del electrolito hacia el polímero. Durante la
descarga, los polímeros p-dopados y n-dopados vuelven a su estado neutral, y los iones
regresan a la solución.

CAPÍTULO 1: ANTECEDENTES
25
Figura 1.8.- Proceso de carga de un electrodo de polímero intrínsecamente conductor; a) P-
Dopaje, b) N-Dopaje [4, 97].
1.3.2.- Métodos de síntesis de polímeros intrínsecamente conductores
Los polímeros intrínsecamente conductores pueden ser sintetizados principalmente por
vía química o electroquímica [11, 59]. En el primer caso, una solución del monómero es
oxidada con un agente oxidante cuyo potencial de oxidación corresponde al potencial de
oxidación del monómero, formando un precipitado del polímero intrínsecamente conductor
[11, 59], mientras que por medio del método electroquímico, se genera una película de
polímero intrínsecamente conductor sobre un sustrato metálico en una celda
electroquímica que contiene el monómero, el solvente y la molécula dopante; la

CAPÍTULO 1: ANTECEDENTES
26
polimerización ocurre por oxidación a través de una corriente eléctrica [59]. Los
principales métodos de obtención de polímeros intrínsecamente conductores son los que
se presentan a continuación.
Síntesis directa.- Método desarrollado por Shirakawa en 1977, en donde la
pared interna de un recipiente de vidrio se recubre con un catalizador Ziegler-
Natta, posteriormente, mediante el paso de una corriente de acetileno da lugar
a una película brillante de poli(acetileno), debido a un exceso de catalizador
[90, 91].
Oxidación química.- En éste caso, a una disolución se añade un oxidante cuyo
potencial corresponde al potencial de oxidación del monómero, un ejemplo de
éste serían: las sales de Fe3+ en disoluciones de pirrol, posteriormente se
forma un precipitado negro, el cual corresponde al poli(pirrol) [90, 91].
Oxidación electroquímica.- En éste caso el proceso es heterogéneo y se
produce sobre el ánodo de una celda electroquímica que contiene un
disolvente y una sal. El flujo de un potencia o corriente anódica a través del
electrodo de trabajo, generando así iones en la solución por medio de los
cuales se produce una película polimérica en del ánodo [90, 91].
Precursores.- Un polímero precursor, generalmente soluble, es aplicado en la
superficie deseada. Por calentamiento se descompone dando una molécula
gaseosa y un polímero intrínsecamente conductor insoluble [90, 91].
Hay otros métodos para realizar la polimerización pero son menos empleados
como la polimerización fotoiniciada, la polimerización por condensación, la
síntesis por emulsión inversa.

CAPÍTULO 1: ANTECEDENTES
27
1.3.3.- Mecanismos de polimerización
La polimerización implica la apertura del doble enlace del monómero, la cual se da en tres
pasos principales, iniciación, propagación y terminación. Dependiendo de la naturaleza
del iniciador, se puede llevar a cabo una apertura homolítica en donde se forman los
radicales libres desarrollando así una polimerización por radicales libres (química o
electroquímicamente), o una apertura hererolítica, mediante moléculas iónicas o
polarizadas en la cual se forman las especies iónicas, llevándose a cabo así una
polimerización catiónica o una polimerización aniónica [98].
1.3.3.1.- Polimerización por radicales libres
Mediante éste mecanismo, la iniciación se origina en el radical libre, ya sea por medio de
una colisión térmica, por radiación electromagnética o por medio de una descomposición
del iniciador. Posteriormente, este radical se adiciona a un monómero para romper ese
doble enlace del carbono. En la ecuación 1.14, se describe el proceso de iniciador.
RMMR
nRI
a
d
k
k
[1.14]
donde I es el iniciador, R• el radical libre formado, M el monómero, kd la constante de
descomposición del iniciador y ka la constante de iniciación del iniciador. Lo importante
durante la etapa la iniciación es que la actividad radical esté localizada en el átomo de
carbono del monómero [98-100]. Durante el proceso de propagación la cadena polimérica
va creciendo por el extremo en donde se encuentra el monómero radical:
1n
k
n
3
k
2
2
k
1
MMM
MMM
MMM
p
p
p
[1.15]

CAPÍTULO 1: ANTECEDENTES
28
siendo en éste caso kp la constante de propagación, Mn se refiere a n número de
monómeros presentes en la cadena polimérica, mientras que Mn+1 hace referencia a la
adición de un monómero más a la cadena polimérica que le precede [98, 100].
En el caso del término del crecimiento de la cadena polimérica, ésta se puede presentar
principalmente de dos maneras. La primera se presenta por medio de la combinación de
dos cadenas más cortas, que se encuentran en crecimiento, y de éste modo formar una
sola cadena (ver figura 1.9).
Figura 1.9.- Proceso de término de crecimiento de la cadena por medio de dos cadenas cortas.
La segunda forma de término, es por medio de la separación de un átomo de hidrógeno
en una cadena que se encuentra en crecimiento. Ésta reacción se conoce como
desproporción (ver figura 1.10) [98-100].
Figura 1.10.- Proceso de término de crecimiento de la cadena por medio de un átomo de
hidrógeno.
Debido a esto, se forman dos tipos de cadenas, una con el extremo saturado y otra con el
extremo insaturado. Generalmente, en una reacción de polimerización, se llevan a cabo
ambos tipos de terminaciones en diferente proporción, todo depende del monómero con el
que se trabaje y las condiciones de polimerización utilizadas [98-100].

CAPÍTULO 1: ANTECEDENTES
29
1.3.3.2.- Polimerización iónica
Éste tipo de polimerización se divide en dos categorías: catiónica y aniónica, dependiendo
del tipo de iniciador que se emplee. No todos los polímeros pueden ser polimerizados por
vía iónica, mucho tiene que ver la estructura del monómero [100].
a) Polimerización catiónica
Éste mecanismo de polimerización, se inicia mediante compuestos ácidos, en donde
actúa el iniciador, en el doble enlace del monómero, formando un catión, como se
muestra en el siguiente esquema (ver figura 1.11):
Figura 1.11.- Mecanismo de polimerización catiónica.
En éste caso, la velocidad de polimerización es muy sensible a la presencia de
impurezas. Para la polimerización se requiere de un catalizador el cual actuará como el
donador de electrones en la reacción. El proceso de propagación se lleva a cabo cuando
el par iónico iniciador empieza a crecer por medio de la incorporación del monómero. La
polimerización se puede terminar de dos formas: por la expulsión de un protón para
generar un alqueno o la combinación con un contraión [98-101].
b) Polimerización aniónica
En éste caso, el mecanismo puede ser iniciado por medio de una base o por medio de
compuestos organometálicos. El proceso de iniciación se representa a continuación
(figura 1.12), en donde se inicia la polimerización por transferencia de un electrón al doble
enlace del monómero [102]:

CAPÍTULO 1: ANTECEDENTES
30
Figura 1.12.- Mecanismo de polimerización aniónica.
En éste método de polimerización es difícil realizar la reacción de terminación, ya que en
la propagación se consume por completo el monómero quedando los aniones terminales
activos aún. Las terminales de éstos polímeros se pueden desactivar añadiendo
sustancias que reaccionen con los carbaniones, tales como el agua, ácido o alcohol [98-
101].
1.3.4.- Poli(pirrol)
El poli(pirrol) (PPy) es una polisal que puede ser producida en forma de polvo,
recubrimiento o películas, es intrínsecamente conductor, y altamente estable [89, 103] El
PPy puede ser preparado ya sea por oxidación química o electroquímica del monómero
pirrol (ver figura 1.13) [104, 105].
Figura 1.13.- Esquema de la estructura química del monómero de pirrol y la unidad repetitiva del
poli(pirrol) [88].
El polímero intrínsecamente conductor denominado poli(pirrol) es sintetizado fácilmente
utilizando uno de amplia gama de electrolitos acuosos u orgánicos [89]. Entre los
polímeros intrínsecamente conductores conocidos hasta la fecha, los basados en PPy han
atraído un interés especial debido a su conductividad alta (101 – 103 S/cm), su facilidad y
flexibilidad en la preparación, alta estabilidad térmica y buenas propiedades mecánicas
[103, 106].
La conductividad eléctrica del PPy es el producto de dos factores importantes, el número
de portadores (electrones o huecos) y la movilidad de los portadores de carga (polarón y

CAPÍTULO 1: ANTECEDENTES
31
bipolarón, ver figura 1.14). Las cargas positivas creadas en la estructura del polímero
(polarones) son los portadores de carga para la conducción eléctrica [106].
Figura 1.14.- Estructura del a) poli(pirrol) reducido, b) polarón y c) bipolarón [107].
El proceso de oxido reducción del poli(pirrol) implica cambios en el peso del polímero, así
como en las transiciones de electrones [103]. La conductividad eléctrica en el PPy implica
el movimiento de los portadores de carga positiva y/o electrones a lo largo de las cadenas
de los polímeros y el salto de éstos portadores entre las cadenas [108]. Actualmente, se
conoce que el PPy puede ser sintetizado tanto en medio acuoso como en medio orgánico,
y que la naturaleza del medio electrolítico, así como su pH, afectan las propiedades
eléctricas y mecánicas del polímero [109].
1.3.5.- Poli(3,4-etilenodioxitiofeno)
El poli(3,4-etilenedioxitiofeno) (PEDOT) es un derivado del poli(tiofeno). Es uno de los
más importantes polímeros intrínsecamente conductores debido a sus propiedades
eléctricas superiores y su alta estabilidad térmica, PEDOT es también uno de los pocos
politiofenos que pueden ser sintetizados por medio de la polimerización química oxidativa
simple, sin utilizar algún catalizador [104].

CAPÍTULO 1: ANTECEDENTES
32
Durante la segunda mitad de la década de 1980, los científicos de los laboratorios de
investigación de Bayer AG en Alemania desarrollaron un nuevo derivado del poli(tiofeno),
que tiene una estructura como se muestra a continuación (ver figura 1.15) [110].
Figura 1.15.- Esquema de la estructura química del monómero EDOT y de la unidad repetitiva del
PEDOT [88].
Este polímero intrínsecamente conductor, a menudo abreviado como PEDT o PEDOT, fue
inicialmente desarrollado para proporcionar un polímero intrínsecamente conductor
soluble que careciera de la presencia de acoplamientos indeseables α,β- y β,β en la
estructura del polímero. Sin embargo se encontró que era un polímero insoluble. Además
de una conductividad muy alta de alrededor de 300 S/cm, se encontró que el PEDOT es
casi transparente en capas delgadas y muestra una alta estabilidad en estado oxidado
(ver figura 1.16) [110].

CAPÍTULO 1: ANTECEDENTES
33
Figura 1.16.- Estructura del a) poli(3,4-etilenedioxitiofeno), b) polarón y c) bipolarón [111].
El problema de solubilidad se resolvió posteriormente mediante el uso de un polielectrolíto
soluble en agua, poli(estiren sulfónico) (PSS), esto dio lugar a una combinación soluble en
agua [110].
La síntesis de derivados PEDOT se pueden dividir en tres diferentes tipos de reacciones
de polimerización [110]:
Polimerización química oxidativa a base de los monómeros EDOT.
Polimerización electroquímica a base de los monómeros EDOT
Polimerización en presencia de metales de transición mediante el acoplamiento de
dihalos derivados del EDOT.

CAPÍTULO 1: ANTECEDENTES
34
La polimerización química de los derivados del EDOT se puede realizar empleando varios
métodos y oxidantes. El método clásico emplea agentes oxidantes como FeCl3 o Fe(OT)3,
teniendo como resultado un compuesto insoluble, el PEDOT [110].
Se han llevado a cabo investigaciones para utilizar al PEDOT como material en los
electrodos para supercondensadores, utilizando varios tipos de electrolitos debido a que
posee características interesantes, entre ellas una buena estabilidad química,
propiedades de gran flexibilidad y alta conductividad eléctrica reportada de 1 a 10 S/cm
[112, 113].
1.4.- Materiales híbridos a base de grafeno/polímero intrínsecamente conductor
Anteriormente a éste trabajo se han realizado trabajos en los cuales se ha empleado el
grafeno o el óxido de grafeno acoplado a polímeros intrínsecamente conductores; en
aplicaciones de supercondensadores.
Por ejemplo, en el trabajo de Y. Han, L. Hao, et al. [114], se reporta la síntesis de un
material compuesto de óxido de grafito/PPy mediante una polimerización in situ utilizando
persulfato de amonio como iniciador, en presencia de óxido de grafeno, usando HCl como
dopante. El material obtenido presentó una capacitancia específica de 197 F/g a una
densidad de corriente de 500 mA/g, en un electrolito de 1M H2SO4, en una celda de tres
electrodos; calomel y platino; como electrodo de trabajo se depositó el material sobre
grafito.
F. Alvi, M. K. Ram, et al. [71] sintetizaron un material híbrido de grafeno/poli(3,4-
etilenodioxitiofeno) por medio de una técnica de polimerización química oxidativa, se
realizó la caracterización electroquímica a los supercondensadores en dos diferentes
electrolitos, H2SO4 y HCl, a una concentración de 2M; se encontró un mayor valor de
capacitancia específica en HCl de 374 F/g, valor obtenido en la descarga.
Se han realizado otras investigaciones en las cuales se han mostrado valores elevados de
capacitancia de materiales con grafeno (ver tabla 1.4), lo que los hace prometedores
como electrodos de supercondensadores. Los valores que se reportan en éste trabajo son

CAPÍTULO 1: ANTECEDENTES
35
solamente del material en sí, o sea, sin ser ensamblados como electrodos en un
supercapacitor.
Tabla 1.4.- Valores de capacitancia específica reportadas de materiales híbridos basados en los
materiales de interés.
Material Capacitancia específica (F/g)
Grafeno-PEDOT [18] 390
Grafeno-PPy nanocompuesto [115] 240
Óxido de grafito/PPy [114] 197
1.5.- Técnicas de caracterización del material híbrido
1.5.1.- Microscopia electrónica de barrido (SEM)
El microscopio electrónico de barrido utiliza un haz de electrones de alta energía para
generar una variedad de señales en la superficie de muestras sólidas [116]. Las señales
que se derivan de las interacciones de electrones de la muestra, dan información sobre la
muestra, incluyendo la morfología, la composición química y la orientación de los
materiales que componen la muestra [117]. En la mayoría de las aplicaciones, los datos
se recogen sobre un área seleccionada de la superficie de la muestra, y una imagen de
dos dimensiones se general mostrando las variaciones espaciales en estas propiedades
[117].
1.5.2.- Espectroscopia Raman
Los espectros Raman se obtienen al irradiar una muestra con una fuente láser de
radiación monocromática visible o infrarroja. Durante la irradiación, se registra el espectro
de la radiación dispersada un cierto ángulo con un espectrómetro. Su detección y medida
resulta más difícil que el espectro infrarrojo [18]. Los instrumentos para la espectroscopia
Raman moderna consta de tres componentes: una fuente láser, un sistema de iluminación
de la muestra y un espectrómetro. Por su alta intensidad el laser es la fuente más común

CAPÍTULO 1: ANTECEDENTES
36
utilizada para la espectroscopia Raman. Entre las cinco fuentes más utilizadas se
encuentran el ion argón, ion criptón, helio/neón, láser de diodos y Nd/YAG [116].
Así mismo, ésta técnica puede ofrecer una manera rápida y eficaz para la caracterización
de la estructura y la calidad del grafeno (ver figura 1.17).
Figura 1.17.- Ejemplo de gráficas por espectroscopia Raman del grafeno [118].
La banda G representa una doble degeneración del modo fonón en el centro de la zona
Brillouin, lo que se debe a la presencia de redes de carbono sp2, mientras que la banda D
es apreciable cuando existe la presencia de defectos inducidos en la estructura del grafito,
que se pueden deber a los grupos funcionales [119].
1.5.3.- Difracción de Rayos X
La interacción entre el vector eléctrico de la radiación X y los electrones de la materia que
atraviesa da lugar a una dispersión, donde existen interferencias (constructivas y
destructivas) entre los rayos dispersados; el resultado es la difracción [116, 120]. Cuando
un haz de rayos X incide sobre la superficie de un cristal formando un ángulo θ, una
porción del haz es dispersada por la capa de átomos de la superficie; la porción no
dispersada del haz penetra en la segunda capa de átomos. El efecto acumulativo de esta
dispersión producida por los centros regularmente espaciados de cristal es la difracción
del haz (ver figura 1.18) [116].

CAPÍTULO 1: ANTECEDENTES
37
Figura 1.18.- Difracción de rayos X en un cristal [120].
La diferencia de las trayectorias de los rayos reflejados de los planos adyacentes es 2d
sen θ, en donde θ es medido del plano. Cuando la diferencia entre las ondas que inciden
y las que se difractan es un múltiplo entero de la longitud de onda de los rayos X se puede
plantear la siguiente ecuación, conocida como ley de Bragg [120, 121]:
nλθd sen2 [1.16]
donde λ es la longitud de onda del haz, n es un número entero; como el valor máximo de
sen θ es 1, el valor mínimo de d (para n=1) es la mitad de la longitud de onda de los rayos
X [120].
El segmento de rayos X que es absorbido por el material, aumenta a medida que
disminuye la energía. Debido a esto, se puede identificar la composición de la muestra,
los elementos tienen valores específicos de energía; éstos son conocidos como bordes de
absorción, los cuales se producen por el aumento brusco de la cantidad energía
adsorbida. Esto se puede atribuir al hecho de que este aumento corresponde a la
expulsión de un electrón del elemento [120].
Para estimar el tamaño de los cristales en el material, perpendicular al plano, mediante la
anchura del pico de difracción medido se emplea la ecuación de Scherrer [122, 123]:
Bc
θB
kλL
cos [1.17]

CAPÍTULO 1: ANTECEDENTES
38
donde k es una constante que depende de la forma de los cristales, λ es la longitud de
onda del haz, B es la mitad de la anchura total de su intensidad máxima y θB es el ángulo
de dispersión de B [124].
1.5.4.- Análisis termogravimétrico
La termogravimetría (TG) estudia el cambio o pérdida de masa en una muestra como una
función del tiempo, empleando ya sea una temperatura determinada o un rango de
temperatura, y una velocidad de calentamiento [125, 126]. Cuando en las curvas de TG se
presenta una sola etapa de descomposición, hay dos temperaturas características (figura
1.19); la temperatura inicial Ti de descomposición y la temperatura final Tf; donde Ti es la
temperatura más baja a la que el inicio de un cambio de masa puede ser detectado por
termo equilibrio en condiciones de operación particulares y Tf es la temperatura final en la
que se lleva a cabo la descomposición por completo [126].
Figura 1.19.- Esquema de una curva TG con una sola etapa de descomposición [126].
En ocasiones existen superposiciones de las reacciones las cuales son difíciles de
determinar; por lo que para poder determinar cada contribución, se realiza la primera
derivada de la curva TG (DTG). En la DTG, la variación de la masa con respecto de la
temperatura (dm/dT) se representa gráficamente en función a la temperatura; para un
intervalo de masa constante, dm/dt es cero. Un pico en la curva DTG se produce cuando
la tasa de variación de la masa es un máximo. Las gráficas de la DTG se caracterizan por
el pico de temperatura máxima (Tmax) y la temperatura de inicio del pico (Te) [126].

CAPÍTULO 1: ANTECEDENTES
39
1.5.5.- Espectroscopia infrarroja con transformada de Fourier
Es una técnica experimental que se emplea para obtener información sobre la estructura
de moléculas. Las moléculas absorben la luz infrarroja en las frecuencias que coinciden
con la energía de las vibraciones moleculares, lo que proporciona una huella digital de la
estructura molecular. Por lo que a ésta técnica también se le conoce como espectroscopia
vibracional. El patrón de vibraciones es único para una molécula dada, y la intensidad de
la absorción está relacionada con la cantidad de absorbente. En la región de radiación
infrarroja, cada grupo tiene diferentes patrones de vibración tales como: estiramiento,
flexión, oscilación, entre otras. Por lo tanto, la espectroscopia infrarroja permite la
determinación de los componentes o grupos de átomos que absorben en el infrarrojo a
frecuenticas específicas [127].
1.5.6.- Voltamperometría cíclica
El principio de la técnica de voltamperometría se basa en aplicar una excitación de
potencial variable a una celda electroquímica, lo cual provoca una respuesta de intensidad
de corriente [116, 120]; en la figura 1.20 se pueden observar cuatro señales de excitación
que comúnmente se utilizan en voltamperometría [116].
Los parámetros importantes en un voltamperograma cíclico son el potencial de pico
catódico, el potencial de pico anódico, la corriente de pico catódica y la corriente de pico
anódica [3, 116].

CAPÍTULO 1: ANTECEDENTES
40
Figura 1.20.- Señales de potencial de excitación utilizadas en voltamperometría [116].
Para la realización de la caracterización electroquímica, se requiere de una celda
electroquímica, la cual generalmente puede ser de 2, 3 o hasta 4 electrodos. Sin
embargo, generalmente para la caracterización del material se utiliza la celda
electroquímica de tres electrodos, la cual consiste del electrodo de trabajo en donde se
oxida o reduce el analito; el electrodo de referencia que mantiene su potencial constante
durante el experimento, para así conocer cuál es el potencial aplicado al electrodo de
trabajo, y del contra electrodo, utilizado para crear un circuito cerrado (ver figura 1.21)
[120].

CAPÍTULO 1: ANTECEDENTES
41
Figura 1.21.- Una celda típica electroquímica utilizada en voltamperometría consiste en un
electrodo de trabajo, un contra electrodo, y un electrodo de referencia. La celda puede ser
burbujeada con un gas inerte para remover el O2 disuelto [120].
Esta técnica permite conocer tanto el comportamiento capacitivo del material, así como
determinar el valor de capacitancia. Un comportamiento ideal de capacitancia de doble
capa de un material para electrodo es expresado como un rectángulo en una
voltamperometría, pero en supercondensadores de doble capa reales el rectángulo toma
una forma de paralelogramo con picos irregulares [32]. En la figura 1.22 se puede
observar las diferentes respuestas que se pueden obtener en una voltamperometría de un
condensador electroquímico.
Figura 1.22.- Voltamperometría cíclica característica de los procesos de carga/descarga en un
condensador electroquímico [32].

CAPÍTULO 1: ANTECEDENTES
42
1.5.7.- Ciclos galvanostáticos de carga-descarga
En éste caso, el electrodo de trabajo se somete a una corriente constante (carga o
descarga). El voltaje generado en función del tiempo se registran entre valores mínimos y
máximos [14]. Los datos que éstas mediciones determinan son la capacitancia del
electrodo en función del voltaje y el tiempo, cambio del potencial en función del estado de
carga, la reversibilidad termodinámica, la ciclabilidad o estabilidad al ciclado, y la
estimación de la caída óhmica [14].
En el caso de las curvas de carga-descarga no lineal, la capacitancia implica que se están
llevando a cabo procesos de doble capa y reacciones farádicas; ya que un
comportamiento capacitivo puro, conduce a los mismos tiempos de carga y descarga, si
no, esto es una indicación de que se produjo una reacción irreversible farádica [14, 128].
A partir de las curvas de carga-descarga se puede calcular la capacitancia específica del
condensador (Csp) (ver figura 1.23), mediante la siguiente ecuación:
mΔV
It
mΔV
QC d
sp22
[1.18]
donde Q representa la carga (C), ΔV2 es el rango de potencia al cual se lleva a cabo la
descarga (V), m es la masa del material activo en el supercondensador (g), I es la
corriente de carga-descarga (A) y td es el tiempo de descarga del dispositivo [129].
También se puede calcular la resistencia interna en serie (ERS), utilizando la caída de
potencia que se presenta por la resistencia del supercondensador, ΔV1 o IR; y la corriente
de carga-descarga, I.
I
VESR
2
1 [1.19]

CAPÍTULO 1: ANTECEDENTES
43
Figura 1.23.- Esquema representativo de ciclos de carga y descarga a corriente constante.
Para conocer la eficiencia que el dispositivo brinda, se emplea la siguiente expresión:
100xt
tEf
c
d [1.20]
en donde t es el tiempo de carga del supercondensador [129].
1.5.8.- Espectroscopia de impedancia electroquímica
Es un método de evaluación del rendimiento de un componente en el dominio de la
frecuencia. En el diagrama de Nyquist de la figura 1.24 se compara un condensador ideal
y un condensador electroquímico simplificado, ambos con la misma ESR (Resistencia en
Serie Equivalente a 1 kHz) [18].
El condensador ideal presenta una línea vertical mientras el condensador electroquímico
comienza con una línea de impedancia de 45° y se acerca a una línea casi vertical sólo en
las frecuencias bajas. La pendiente no vertical de la impedancia de cualquier condensador
electroquímico real puede ser fácilmente reproducido en cualquier modelo de ecuaciones
mediante el remplazo de la expresión de la capacitancia con la expresión de un elemento
de fase constante (CPE) [18].

CAPÍTULO 1: ANTECEDENTES
44
La región de 45° (región Warburg) es una consecuencia de la resistencia distribuida:
capacitancia dentro de un electrodo poroso. A frecuencias más altas la resistencia así
como la capacitancia de los electrodos porosos decrecen, porque sólo parte de la capa
porosa activa es accesible a altas frecuencias. El condensador electroquímico por lo tanto
puede ser representado por un condensador ideal con un incremento en la resistencia en
serie equivalente (ESR) por la resistencia distribuida equivalente (EDR) [18, 130].
Figura 1.24.- Representación esquemática del gráfico de impedancia Nyquis [18].

CAPÍTULO 2: DISEÑO METODOLÓGICO
45
CAPÍTULO 2
METODOLOGÍA
A continuación se describen los procedimientos de síntesis empleados en éste trabajo
para la obtención de los polímeros intrínsecamente conductores, el óxido de grafito y los
materiales híbridos. La síntesis de los polímeros intrínsecamente conductores estuvo
enfocada en la obtención de nanotubos, que al ser estructuras más alineadas nos
permitiría separar las capas del óxido de grafito y aumentar así la capacidad de
almacenamiento de carga eléctrica [131]. El óxido de grafito se obtuvo empleando el
método de Brodie [84, 132].
Una vez obtenido el óxido de grafito se llevó a cabo un proceso de funcionalización
empleando 6((1H-pirrol-2-carbonilo)oxi)hexano-1-amino, el cual posee en uno de sus
extremos un grupo amino y en el otro un anillo aromático pirrol. El grupo amino es capaz
de reaccionar selectivamente en medio básico con los grupos epoxi superficiales de las
láminas de óxido de grafito, quedando unido el grupo funcional a la superficie del óxido de
grafito. Posteriormente, en una segunda etapa, es posible emplear el anillo de pirrol
terminal como unidad monomérica en la polimerización, con lo que el polímero estaría
unido covalentemente a la superficie del material de carbono.
2.1.- Síntesis de emulsión inversa de nanotubos de polímeros intrínsecamente
conductores
Se emplearon dos monómeros diferentes, pirrol y 3,4-etilenodioxitiofeno, que durante el
proceso de polimerización oxidativa se anclarán a la superficie del óxido de grafito
funcionalizado a través del anillo aromático de pirrol, dando lugar en cada caso a los
materiales híbridos, óxido de grafito/poli(pirrol) y óxido de grafito/poli(3,4-
etilenodioxitiofeno). En ambos casos el proceso de polimerización se llevó a cabo en
emulsión; se forman micelas de forma cilíndrica a partir de la interacción de un
tensoactivo, disuelto en un disolvente orgánico, en este caso: bis(2-etilhexil)-
sulfosuccinato sódico (AOT) (ver figura 2.1), y una solución acuosa de FeCl3, que actuará
como oxidante en la polimerización del monómero. Una vez incorporado el monómero, es

CAPÍTULO 2: DISEÑO METODOLÓGICO
46
de esperar que el proceso de reacción dé lugar a la formación de micro/nanotubos del
polímero.
Como monómeros se emplearon pirrol (Aldrich, 98 % grado reactivo) y 3,4-
etilenodioxitiofeno (Bayer AG., grado reactivo). El proceso de polimerización se realizó de
acuerdo al procedimiento descrito por E. Morales y J.L. Acosta [133], adecuado a las
condiciones a las que se trabaja en nuestro laboratorio. Tanto el pirrol como el 3,4-
etilenodioxitiofeno fueron destilados a vacío y almacenados en refrigeración.
Figura 2.1.- Estructura del AOT.
Se preparó la solución para la polimerización utilizando 18 gramos de bis(2-etilhexil)
sulfosuccinato sódico (AOT; Aldrich, 98 % grado reactivo) al cual se le agregó 80 ml de
hexano (LAB-SCAN Analytical Sciences, grado reactivo); una vez disuelto el AOT se dejó
reaccionar aproximadamente durante 24 horas. Luego se agregó a la solución 2 ml de
FeCl3 (Aldrich, 97 %) 9 M disuelto en agua; posteriormente se dejó reposar por 48 horas
para favorecer la formación de las micelas; se obtiene una solución espesa amarilla
brillante. Transcurridas las 48 horas se agregó 1 g de monómero para comenzar el
proceso de polimerización, se dejó reposar sin ningún tipo de perturbación.
Tras 24 horas de reacción se extrajo el polímero de la solución filtrándolo, empleando una
membrana de Nylon® de 0.45 μm. Se lavó con abundante agua destilada y etanol para
arrastrar todos los residuos orgánicos del interior del polímero. El polímero que se obtuvo
se almacenó en un vial de vidrio para colocarlo en el tubo de secado a vacío a una
temperatura de 80 °C durante 24 horas.

CAPÍTULO 2: DISEÑO METODOLÓGICO
47
2.2.- Síntesis de óxido de grafito
El óxido de grafito se obtuvo mediante la oxidación química del grafito empleando el
método descrito por Brodie [84], que consiste en la oxidación química del grafito en
presencia de ácido nítrico fumante y clorato de potasio a baja temperatura. De manera
complementaría a ésta metodología, se utilizó además la de M. Hernández, et al. [132],
tomando de ésta metodología la temperatura de la reacción así como el tiempo de
reacción. Estos métodos fueron adecuados a las condiciones a las que se trabaja en
nuestro laboratorio.
La reacción de oxidación del grafito se llevó a cabo a una temperatura de 0°C
recirculando etanol mediante el criostato; ya que ésta síntesis es exotérmica; por lo cual,
el adecuado manejo de los reactivos es de suma importancia. En el reactor de vidrio que
se utilizó para llevar a cabo la reacción, se agregaron 5 g de grafito en hojuela; de manera
paulatina y por las paredes del reactor, se agregaron 100 ml de ácido nítrico fumante
(Sigma Aldrich, ≥99.5 % grado reactivo); en toda la reacción se empleó la agitación; y
observando que la temperatura no aumentara en la solución.
Una vez que se estabilizó la temperatura a 0 °C nuevamente, se comenzó a agregar 40 g
de clorato de potasio (Fluka, 99.0 % grado reactivo), éste pasó se realizó cautelosamente,
ya que el ácido puede reaccionar violentamente con el clorato de potasio, además de los
gases que se expiden de la reacción, óxido nítrico y dióxido de nitrógeno; se debe
emplear bata, lentes de seguridad, máscara para gases y vapores, y guantes de
neopreno. El reactivo se agregó poco a poco; éste proceso tuvo una duración aproximada
de 90 minutos. Ya que se terminó de agregar por completo el clorato de potasio, se dejó
reaccionar la solución durante 21 horas con agitación continua, a una temperatura de 0°C.
Para extraer la solución del reactor se empleó un vaso de precipitado con un 1 litro de
agua destilada, el cual se colocó en un baño de hielo para mantener fría el agua. La
solución se pipeteó del reactor y con precaución se colocó la solución en el agua destilada
fría, se debe tener cuidado con los gases que ésta reacción desprende (óxido nítrico y
dióxido de nitrógeno); además, al ser la reacción exotérmica, el agua se calienta al
agregar la solución. Al terminar de extraer la solución, se procedió a realizar el filtrado a

CAPÍTULO 2: DISEÑO METODOLÓGICO
48
vacío para obtener el óxido de grafito, se lavar para retirar los restos de la reacción para
neutralizar el producto; el filtrado se llevó a cabo empleando una membrana de Nylon ®.
Posteriormente se secó a una temperatura de 80°C a vacío.
2.3.- Síntesis del material híbrido
Para obtener el material híbrido, se funcionalizó el óxido de grafito (GraphO) obtenido con
un compuesto con terminación amino (GraphO-amino), posteriormente se llevó a cabo el
anclaje con el polímero. Para la primera funcionalización se utilizó como referencia la
metodología de H. Yang; C. Shan, et al. [134], mientras que para la síntesis del híbrido, se
llevó a cabo el mismo procedimiento que se describió anteriormente para la
polimerización en la sección 2.1, sólo que en este caso en presencia del óxido de grafito.
a) Adición del grupo funcional amino en el óxido de grafito
Para la introducción del grupo funcional amino, se preparó una suspensión de óxido de
grafito, agregando 280 mg de óxido de grafito a 560 ml de agua desionizada para
posteriormente con ayuda de una punta ultrasónica realizar una dispersión durante 3
horas. A la suspensión obtenida se le agregaron 560 mg de 6-((1H-pirrol-2-
carbonilo)oxi)hexano-1-aminio, que es el compuesto con terminación amino que se
empleará para el anclaje con el grupo epoxi del óxido de grafito (ver figura 2.2).
Posteriormente, se agregaron 560 mg de hidróxido de potasio (Sigma Aldrich, KOH ≥85
%) a la suspensión, para luego dejarla durante 30 minutos en el baño ultrasónico;
posteriormente se colocó la solución en una placa de agitación y se dejó en agitación
vigorosa a una temperatura de 80 °C durante 24 horas. Una vez terminada la reacción, el
material se filtró y lavó con agua desionizada y etanol, empleando una bomba de vacío y
una membrana de Nylon® de 0.45 μm, hasta alcanzar un pH neutro; por último el material
obtenido se secó a 80 °C en la estufa por 24 horas.

CAPÍTULO 2: DISEÑO METODOLÓGICO
49
Figura 2.2.- Esquema representativo del óxido de grafito funcionalizado con la terminación amino.
b) Polimerización por emulsión inversa en presencia del óxido de grafito con
terminación amino
Se tomaron 130 mg del GraphO-amino, y se hizo una suspensión en 4 ml de hexano, la
cual se colocó durante 2 horas en la punta ultrasónica para lograr una mejor dispersión.
Se hizo una hibridación con la intención de tener en el peso total del material un 10 % del
polímero en peso. Después de dispersar el material, se procedió a agregar 234 mg de

CAPÍTULO 2: DISEÑO METODOLÓGICO
50
AOT, se le dio una agitación leve y después se dejó reaccionar por 24 horas sin agitación;
posteriormente se le incorporaron 39 mg de FeCl3 disuelto en agua desionizada a la
solución, luego se dejó reposar por 48 horas. Transcurrido el tiempo, se añadieron 13 mg
del monómero.
Después de 24 horas de polimerización en reposo, se extrajo el material para filtrarlo y
lavarlo con agua desionizada y etanol; hasta alcanzar un pH 7. Al terminar, el material se
colocó en la estufa a 80 °C durante 24 horas. Este procedimiento tuvo como objetivo
pbtener un material híbrido en polvo, ya sea óxido de grafito/PEDOT (GraphO-PEDOT) u
óxido de grafito/PPy (GraphO/PPy) acorde la a figura 2.3.
Figura 2.3.- Esquema representativo de los materiales híbridos sintetizados.

CAPÍTULO 2: DISEÑO METODOLÓGICO
51
2.4.- Caracterización de los materiales
Con la finalidad de conocer la contribución de cada componente del material híbrido,
primero se evaluaron, por medio de diferentes técnicas de caracterización que a
continuación se mencionan, a los polímeros intrínsecamente conductores; posteriormente
tanto al óxido de grafito como al óxido de grafito con el compuesto amino, y por último a
los materiales híbridos basados en óxido de grafito y polímeros intrínsecamente
conductores.
2.4.1.- Microscopía electrónica de barrido
Se empleó el microscopio electrónico de barrido HITACHI modelo SU8000, las
micrografías se hicieron a vacío con una intensidad de haz de 1.0 kV. A los polímeros se
le realizaron análisis SEM para observar la morfología y corroborar la obtención de
nanotubos. Las magnificaciones utilizadas fueron de 30.0K y 8.0K para ambos polímeros,
poli(pirrol) y poli(3,4-etilenodioxitiofeno). En el caso del óxido de grafito con terminación
amino y los dos materiales híbridos se usaron magnificaciones de 10.0K, 30.0K y 50.0K;
para lograr examinar la morfología de las hojas de óxido de grafito y el cambio que ocurrió
en ésta al realizar la hibridación con los polímeros.
2.4.2.- Espectroscopia Raman
Se utilizó un Espectrómetro Raman HoloSpec™ VPT system de Kaiser optical systems,
inc.; con una longitud de onda de excitación de 632.8 nm. El rango de frecuencias
empleado fue de 300 a 1900 cm-1.
2.4.3.- Difracción de Rayos X
Los difractogramas de Rayos X de las diferentes muestras se obtuvieron entre 10 y 70°
(2θ) mediante un barrido por pasos de tamaño 0.02° (2θ) y 1 s de tiempo de contaje,
empleando un difractómetro D-8 Bruker. La radiación utilizada fue Cu-K α. Los ensayos se
realizaron sobre pastillas obtenidas por comprensión en frío a una presión de 500 kg en el
caso de los dos polímeros y de 200 kg para el resto de los materiales.

CAPÍTULO 2: DISEÑO METODOLÓGICO
52
2.4.4.- Análisis termogravimétrico
El estudio de estabilidad térmica (TGA) de los diferentes materiales sintetizados se llevó a
cabo empleando un equipo TA Q500. Los ensayos se realizaron en atmósfera de
nitrógeno en el rango de temperatura comprendido entre 35 y 900 °C, en el caso del óxido
de grafito el rango de temperatura fue de entre 35 y 800 °C; la velocidad de calentamiento
fue de 10 °C/min y el flujo de nitrógeno empleado de 50 ml/min.
2.4.5.- Espectroscopia infrarroja con transformada de Fourier
El estudio de FTIR, se llevó a cabo utilizando un FTIR Thermo Scientific Nicolet 8700,
mediante la técnica de reflexión total atenuada (ATR), en un intervalo de frecuencias de
entre 4000 a 650 cm-1.
2.4.6.- Caracterización electroquímica
La caracterización electroquímica de los polímeros sintetizados se realizó empleando una
celda de tres electrodos; utilizando como electrodo de referencia de calomelanos (SCE),
una barra de grafito como contraelectrodo y como electrodo de trabajo tela de carbón
(ElectroChem, Inc. EC-CC1-060) de 1cm2 de área sobre la que se depositó una tinta que
contenía el material a caracterizar. Como electrolito se empleó una solución acuosa de
2M H2SO4. El electrodo de trabajo se compone de una tela de carbón, que previamente a
la deposición de la tinta se le dio tratamiento con 2M H2SO4 durante 24 horas,
posteriormente se lavó con agua desionizada y se secó a 100 °C durante 24 horas. La
tinta que se empleó para la caracterización contenía 7 mg del material, 20 μl de Nafion®
117 y 50 μl de alcohol isopropílico; para el caso del PPy, PEDOT, GraphO-PEDOT y
GraphO-PPy se utilizó la misma proporción; las tintas fueron puestas en un baño
ultrasónico durante 30 min para lograr homogenizar y posteriormente se colocó sobre la
tela, mediante una micropipeta gota por gota.
Para realizar estas pruebas se utilizó un potenciostato marca BioLogic Science
Instruments. En la caracterización por voltamperometría cíclica se realizaron 8 ciclos con
una ventana de potencial de -0.2 a 1 V vs SCE para GraphO-PEDOT y PEDOT, mientras

CAPÍTULO 2: DISEÑO METODOLÓGICO
53
que PPy y GraphO-PPy, la ventana de potencial que se utilizó fue de -0.3 a 1 V vs SCE.
En ambos casos, las velocidades de barrido fueron de 5, 10, 20, 40, 60, y 80 mV/s. En los
ciclos galvanostáticos, se utilizó una ventana de potencial entre 0 y 1 V vs SCE, a
densidades de corriente de 2, 5 y 10 mA.
En la evaluación del óxido de grafito se empleó una celda de tres electrodos; utilizando
como electrodo de referencia un electrodo saturado de calomel, SCE y un contraelectrodo
de grafito, en un electrolito de 2M H2SO4. Para el electrodo de trabajo se empleó tela de
carbón de 1cm2 de área en la cual se depositó en uno de los lados una pasta de óxido de
grafito, que contenía 7 mg del óxido, 20 μl de Nafion® y 50 μl de alcohol isopropílico. Se
utilizó un potenciostato marca BioLogic Science Instruments. Para la voltamperometría
cíclica se realizaron 8 ciclos con dos ventana de potencial, la primera de -0.2 a 1 V vs
SCE y la otra de -0.3 a 1 V vs SCE, esto con el fin de poder hacer la comparación con los
materiales híbridos; a velocidades de barrido de 5, 10, 20, 40, 60, y 80 mV/s. En los ciclos
galvanostáticos, se utilizó un potencial entre 0 y 1 V vs SCE, a densidades de corriente de
2, 5 y 10 mA.

CAPÍTULO 2: DISEÑO METODOLÓGICO
54

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
55
CAPÍTULO 3
RESULTADOS Y DISCUSIONES
En éste capítulo se presentan los resultados que se han obtenido a lo largo del trabajo;
enfocado en la síntesis de los polímeros intrínsecamente conductores, del óxido de grafito
y de los materiales híbridos óxido de grafito/polímero intrínsecamente conductor; por vía
química, por emulsión inversa.
3.1.- Microscopía electrónica de barrido
En la figura 3.1 (a–e) se presentan las micrografías SEM de los materiales sintetizados
PEDOT, PPy, GraphO-amino, GraphO-PEDOT y GraphO-PPy. Para calcular el tamaño de
los nanotubos se hicieron 20 medidas sobre las micrografías de los polímeros, utilizando
el programa ImageJ 1.47v para Windows, calibrándolo con la regleta de cada unas de las
micrografías SEM; para posteriormente calcular el promedio de los nanotubos.
En la figura 3.1a se observan los nanotubos de poli(3,4-etilenodioxitiofeno), que dan un
aspecto de tubos delgados rugosos entrelazados como hebras de polímero,
aproximadamente 0.24 μm de diámetro en promedio, los cuales forman tubos más
gruesos de 0.94 μm en promedio aproximadamente; además de que también existen
lugares con acumulación de polímero sin una morfología definida, aglomeraciones. Los
nanotubos de poli(pirrol) se muestran en la micrografía b, donde su morfología es más
sólida en comparación con la de los nanotubos de PEDOT, o sea, los tubos son más
definidos y fáciles de identificar, de aproximadamente 0.5 μm de diámetro en promedio.
La aglomeración presente en los polímeros se debe a que la polimerización se llevó a
cabo mediante síntesis química; con las medidas de los tubos de polímero, se sabe que
no se obtuvieron nanotubos, ya que las dimensiones están en el orden de las micras.

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
56
Figura 3.1.- Micrografías obtenidas por FESEM: a. nanotubos de poli(3,4-etilenodioxitiofeno), b.
nanotubos de poli(pirrol), c. óxido de grafito-amino, d. óxido de grafito/PEDOT y e. óxido de
grafito/PPy.
La micrografía de la figura 3.1 c es el óxido de grafito-amino, en donde se nota la
presencia de hojas delgadas con bordes irregulares apiladas desordenadamente; en la
superficie de éstas hojas se encuentran algunas irregularidades, dando la percepción de
una “sábana arrugada”. La figura 3.1 d y e, son los dos materiales híbridos sintetizados, el

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
57
GraphO/PEDOT y el GraphO/PPy, respectivamente. Con las micrografías del
GraphO/PEDOT se puede observar la formación de partículas aglomeradas,
correspondientes al polímero en el material híbrido (círculos rojos); así como también se
observan una mayor rugosidad en toda la superficie de las láminas debida al polímero
(figura 3.2 a).
Figura 3.2.- Micrografías de los materiales híbridos; a. GraphO-PEDOT y b. GraphO-PPy.
Las micrografías del GraphO-PPy (figura 3.2 b) muestran la presencia de polímero sobre
la superficie del GraphO-amino, en forma de pequeñas aglomeraciones, con diámetros
promedio aproximados de 26 nm, también es notorio que se formó una especie de capa o
costra de PPy que cubre la superficie de las hojas, ya que éstas tiene ahora un aspecto
más rugoso a comparación del material sin el polímero.
3.2.- Espectroscopia Raman
En la figura 3.3 se presentan los espectros de los materiales relacionados al híbrido
GraphO-PEDOT. En el espectro del PEDOT se observan tres picos, el primero a 1367 cm-
1 que se atribuye al estiramiento del enlace C–C, el más intenso de ellos a 1426 cm-1
atribuido a la tensión de deformación asimétrica de los enlaces C=C del anillo tiofeno y el

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
58
último a 1509 cm-1 atribuido a la tensión de deformación asimétrica en el plano de los
enlaces C=C del anillo tiofeno. Adicionalmente aparece otro pico a 990 cm-1, poco notorio,
que se debe a la deformación del anillo oxietileno [135-138].
En la misma figura, el espectro del óxido de grafito muestra dos picos característicos a
1329 y 1588 cm-1, que corresponden a la banda D y G, respectivamente; en donde la
intensidad de la banda D es notablemente mayor que la banda G. La banda G se atribuye
a la vibración en el plano de átomos de carbono sp2 y a un fonón de centro de zona
Brillouin doblemente degenerado con simetría E2g [118]; la banda D corresponde al modo
de respiración de los fonones en la simetría del punto k de la simetría A1g, la cual se
relacionan con diversos defectos: desorden del ángulo de enlace, desorden de la longitud
de enlace, hibridación, átomos de carbono funcionalizados, entre otros [139, 140]. Con lo
que se puede comprobar que se obtuvo un óxido de grafito desordenado por la intensidad
que presenta la banda D del material.
El GraphO-amino también muestra las bandas D y G a 1316 y 1588 cm-1, siendo éstas de
menor intensidad que las del óxido de grafito; sin embargo, la banda D aún presenta
mayor intensidad que la banda G [75, 79]. En el material híbrido de GraphO-PEDOT se
encuentran cinco picos correspondientes tanto al PEDOT a 573, 990, 1366, 1426 y 1515
cm-1, como a los dos picos del GraphO-amino a 1324 y 1592 cm-1, donde la banda D
sigue siendo más intensa que la banda G.
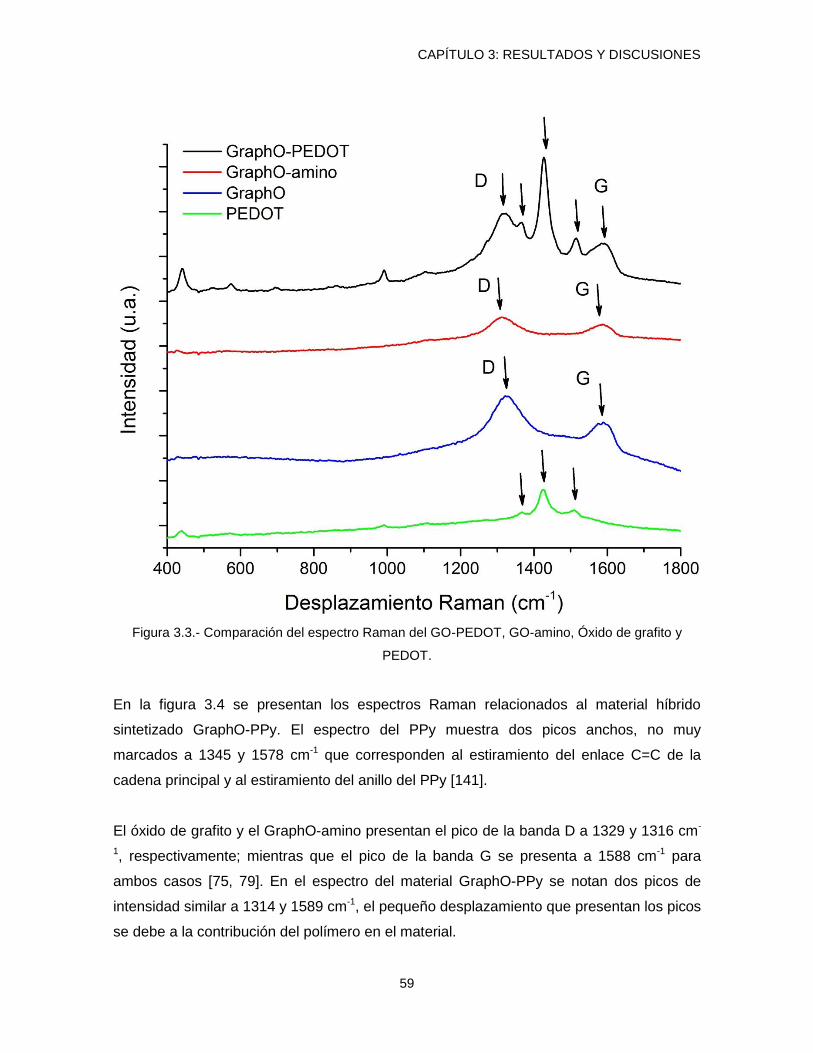
CAPÍTULO 3: RESULTADOS Y DISCUSIONES
59
Figura 3.3.- Comparación del espectro Raman del GO-PEDOT, GO-amino, Óxido de grafito y
PEDOT.
En la figura 3.4 se presentan los espectros Raman relacionados al material híbrido
sintetizado GraphO-PPy. El espectro del PPy muestra dos picos anchos, no muy
marcados a 1345 y 1578 cm-1 que corresponden al estiramiento del enlace C=C de la
cadena principal y al estiramiento del anillo del PPy [141].
El óxido de grafito y el GraphO-amino presentan el pico de la banda D a 1329 y 1316 cm-
1, respectivamente; mientras que el pico de la banda G se presenta a 1588 cm-1 para
ambos casos [75, 79]. En el espectro del material GraphO-PPy se notan dos picos de
intensidad similar a 1314 y 1589 cm-1, el pequeño desplazamiento que presentan los picos
se debe a la contribución del polímero en el material.

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
60
Figura 3.4.- Comparación del espectro Raman del GO-PPy, GO-amino, Óxido de grafito y PPy.
Respecto a la relación de intensidades en las señales D y G, se puede observar que en
los materiales grafíticos, el pico D es más intenso que el pico G, lo que indica un alto nivel
de desorden en el material. En la tabla 3.1 se presenta la relación ID/IG con la cual se
puede notar la formación de defectos y desorden, posiblemente debido a una oxidación
excesiva del material, durante la obtención del óxido de grafito. También se puede notar
que la relación de las intensidades de las bandas para los materiales híbridos disminuye
en comparación con el óxido de grafito, lo que indica que los carbonos del grafito en éstos
materiales contienen menos defectos. Además, los materiales híbridos muestran un
desplazamiento de la banda D hacia números de onda más bajos, lo que confirma una
construcción más ordenada debido a la fuerte interacción electrónica [142].

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
61
Tabla 3.1.- Bandas D, G y la relación ID/IG.
3.3.- Difracción de Rayos X
En la figura 3.5 se muestra el patrón de difracción del PEDOT, el cual presenta diferentes
picos de difracción, en los ángulos 2θ = 6.7°, 12.5°, 19.2°; el pico de difracción a 26.1° se
atribuye a la distancia interplanar del apilamiento de los anillos; para el PEDOT en la
literatura se presentan los picos a 2θ = 6.4°, 12.7°, 19.3 y 2θ = 25.9° [143].
En la figura 3.5, se presenta también en el extremo derecho superior, el patrón de
difracción del grafito en hojuela que se empleó para la síntesis, el cual presenta un pico
intenso y agudo a los 2θ = 26.8°, correspondiente al plano (002), lo que sugiere el alto
grado de ordenamiento de las hojas, la distancia interplanar calculada es de 3.32 Å,
calculada mediante la Ley de Bragg. También se puede observar que óxido de grafito,
presenta un pico marcado de difracción en 2θ = 15.2°, que corresponde al plano (001)
[144, 145]; se presenta otro pico de difracción con menor intensidad a 2θ = 25.43° el cual
corresponde al grafito, en el plano (002). La presencia de éste pico indica que se realizó
una oxidación incompleta del materia, la extensión del pico sugiere un desordenamiento
de las hojas del grafito [146]. La distancia interplanar del óxido de grafito es de 7.25 Å,
este incremento se puede deber a la presencia de los grupos funcionales y otros defectos
estructurales [144, 145, 147].
En la señal obtenida por la difracción del GraphO-PEDOT aparece un pico a los 2θ =
12.2°, lo que da una distancia interplanar de 7.25 Å, que corresponde al óxido, y dos picos

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
62
sobrepuestos, uno por la presencia de residuos de grafito a los 2θ = 26.56° y el otro por la
presencia del PEDOT en el material. También se puede apreciar tanto en el óxido de
grafito como en el GraphO-PEDOT la presencia de un pico a los 2θ = 41.9° y 42.1°,
respectivamente, que es la fase interplanar (100); indicando de ésta forma que ambos
materiales tiene una red constante (2.1 Å) similar a la del grafito, por lo que la estructura
del grafito no pudo ser completamente deformada a través del proceso de oxidación [147].
Figura 3.5.- Patrón de difracción de rayos X del PEDOT, óxido de grafito, GraphO-PEDOT y del
grafito.
En la figura 3.6 se muestra el patrón de difracción del PPy, donde se puede observar una
señal ancha y mayormente amorfa, con una separación intermolecular, aproximadamente

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
63
a 2θ = 22.7°. En la literatura éste pico es reportado a 2θ = 25° [148-152]. El GraphO-PPy
presenta tres picos de difracción a 2θ = 12.1°, 26.39°, 42.32°; éste material presenta una
distancia interplanar de 7.32 Å; en el segundo pico se observa la presencia del PPy por el
hombro que aparece a valores de 2θ menores, se debe al traslape de los picos del óxido
de grafito y del PPy. En cuanto a las señales que presentó el óxido de grafito, éstos ya
fueron descritos anteriormente.
Figura 3.6.- Patrones de difracción de rayos X del PPy, óxido de grafito, GraphO-PPy y grafito.
En la tabla 3.2 se presenta un resumen de los parámetros de la difracción de rayos X de
los diversos materiales sintetizados. En esta tabla, Lc se refiere al tamaño medio de los
cristales en el material; se muestra además el número medio de hojas apiladas a lo largo
del eje-C. Se observa que existe un notable incremento de la distancia entre el grafito y

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
64
los materiales sintetizados, siendo el GraphO-PPy el que presenta una mayor distancia,
además de que también tiene menor cantidad de hojas apiladas.
Tabla 3.2.- Parámetros XRD de los materiales sintetizados.
3.4.- Análisis Termogravimétrico
En la figura 3.7 se muestran las curvas del análisis de TGA del grafito, óxido de grafito,
PEDOT, PPy, GraphO-amino, GraphO-PEDOT y GraphO-PPy; realizadas en atmósfera
de nitrógeno. El PPy presenta dos pasos de pérdida de masa, la primera hasta
aproximadamente 150°C corresponde a la evaporación del agua o algunas impurezas
volátiles presentes en el material, la segunda pérdida de masa se atribuye a la
descomposición térmica de las cadenas del PPy; la temperatura de descomposición es de
229 °C [151, 153, 154], quedando una masa residual del 32 % a los 900 °C. Para el caso
del PEDOT, se mostró estable hasta los 200°C con una pérdida aproximada del 5 % del
peso debido a la evaporación de agua, a partir de los 300°C comienza la descomposición
del polímero; la temperatura de descomposición se presenta a los 360 °C [155], quedando
a los 900 °C una masa residual cercana a los 28 %.
Como se puede observar, el grafito no presenta ninguna caída, mostrándose estable
hasta los 900 °C. Para el caso del óxido de grafito, se presentan tres pérdidas de masa; la
primera pérdida de masa que va hasta los 150 °C se debe a la evaporación del agua; la
segunda pérdida entre los 150 °C hasta 300 °C, es debido a la pirolisis de los grupos
funcionales lábiles del oxígeno (anhídrido carboxílico o grupos lactona), produciendo CO,

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
65
CO2 y vapor; el tercer paso superior a los 300 °C correspondiente a funcionalidades más
estables del oxígeno como el fenol, el carbonilo o la quinona [156, 157]. La masa residual
del óxido de grafito es 60 %, hasta los 800 °C.
Para el GraphO-amino, GraphO-PEDOT y GraphO-PPy se presenta una primera pérdida
de masa hasta los 150 °C que se debe a la evaporación de agua; la segunda caída que
va desde los 150 °C hasta los 260 °C, se debe a la pirólisis de los grupos funcionales del
oxígeno presentes en el óxido de grafito; que ya se mencionaron anteriormente; mientras
que en los materiales híbridos, se observa una caída adicional por la degradación del
polímero. En éstos tres últimos materiales se nota una mayor estabilidad térmica
comparada con el óxido de grafito; esto se puede atribuir a la formación de nuevos
enlaces químicos, entre el grupo amino, los polímeros y el óxido de grafito [158]. El GO-
amino presenta un 66 % de masa residual tras el análisis.
De los dos materiales híbridos sintetizados, el GraphO-PPy tuvo una menor pérdida de
masa durante el análisis, conservando una masa residual de 64 % una vez que alcanzó la
temperatura máxima de la prueba de 900 °C; mientras que el GraphO-PEDOT tuvo un 61
% de material residual.
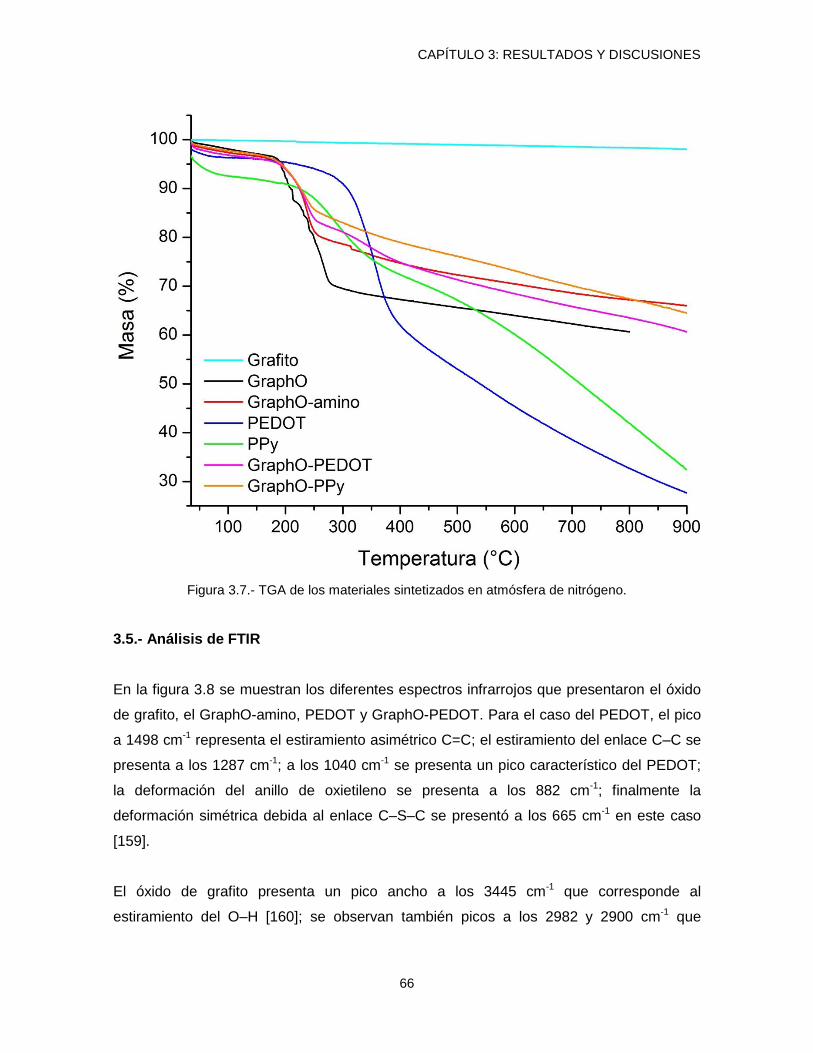
CAPÍTULO 3: RESULTADOS Y DISCUSIONES
66
Figura 3.7.- TGA de los materiales sintetizados en atmósfera de nitrógeno.
3.5.- Análisis de FTIR
En la figura 3.8 se muestran los diferentes espectros infrarrojos que presentaron el óxido
de grafito, el GraphO-amino, PEDOT y GraphO-PEDOT. Para el caso del PEDOT, el pico
a 1498 cm-1 representa el estiramiento asimétrico C=C; el estiramiento del enlace C–C se
presenta a los 1287 cm-1; a los 1040 cm-1 se presenta un pico característico del PEDOT;
la deformación del anillo de oxietileno se presenta a los 882 cm-1; finalmente la
deformación simétrica debida al enlace C–S–C se presentó a los 665 cm-1 en este caso
[159].
El óxido de grafito presenta un pico ancho a los 3445 cm-1 que corresponde al
estiramiento del O–H [160]; se observan también picos a los 2982 y 2900 cm-1 que

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
67
corresponden al estiramiento simétrico y asimétrico del enlace –CH [161], el pico a los
1618 cm-1 se atribuye a la vibración del esqueleto de dominios grafíticos no oxidados
[162], el grupo C–O–C se encuentra a los 1389 cm-1 [160], mientras que el estiramiento
del C–O se hace notar a los 1061 cm-1 [160].
Para el caso del GraphO-amino el pico ancho a 3370 cm-1 se debe al estiramiento del O–
H; los picos del estiramiento simétrico y asimétrico del –CH se pueden observar a los
2982 y 2912 cm-1 [161]; la vibración del esqueleto de dominios grafíticos no oxidados se
presenta a los 1629 cm-1, éste pico es menos intenso que en el caso del óxido de grafito
[162]; otro pico presente es por el enlace C–O–C a los 1391 cm-1, que corresponde a los
grupos epoxi, éste pico es menos intenso que en el caso del óxido de grafito, esto podría
deberse a los enlaces que se formaron entre el grupo epoxi y el grupo amino del
componente usado para la hibridación, lo que hizo que la cantidad de grupos epoxi
disminuyera significativamente [160]; el estiramiento del C–O se encuentra a los 1066 cm-
1 [160]; tomando en cuenta toda la estructura del componente amino que se empleó para
la hibridación, la otra terminación de éste es un anillo de pirrol, que es la parte en donde
se realiza el crecimiento de las cadenas poliméricas, por lo que se presentan dos picos a
894 y 764 cm-1 que son característicos del pirrol [163].
El material GraphO-PEDOT presenta un ensanchamiento y en algunos casos
desplazamiento de los picos con respecto al GraphO-amino. La señal ancha de O–H se
encuentra a los 3295 cm-1 siendo menos intenso que en caso del GraphO-amino; a los
2920 cm-1 se aprecia un sólo pico del enlace –CH, esto se puede explicar por el
ensanchamiento de los picos de estiramiento simétrico y asimétrico por la presencia del
polímero, lo que hace que se aprecie un sólo pico; el pico correspondiente a la vibración
del esqueleto de dominios grafíticos no oxidados aparece menos intenso que el GraphO-
amino a los 1633 cm-1; a 1364 cm-1 se encuentra un pico ancho que hace referencia al
enlace C–O–C, el ensanchamiento de éste pico se relaciona con la presencia de otros
picos del polímero en ésta zona; el estiramiento del enlace C–O se encuentra a los 1074
cm-1, lo que se puede apreciar es un ensanchamiento del pico además de la aparición de
un hombro hacia números de onda más altos relacionados a la presencia de picos
correspondientes al PEDOT. Finalmente, la deformación simétrica del enlace C–S–C, del
anillo tiofeno aparece a los 696 cm-1; así como también está la presencia del pico a 759

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
68
cm-1, que como ya se mencionó anteriormente característico del pirrol, debido al
componente amino usado para la hibridación.
Figura 3.8.- Espectros de FTIR correspondientes al óxido de grafito, GraphO-amino, GraphO-
PEDOT, PEDOT; obtenidos por ATR.
En la figura 3.9 se muestran los espectros infrarrojos del óxido de grafito, el GraphO-
amino, el PPy y del GraphO-PPy. En el PPy a números de onda de 2982 y 2895 cm-1 se
presenta la vibración de tensión asimétrica y simétrica del CH2 [151, 164]. La banda de
estiramiento del anillo C–N se observa a 1466 cm-1 [163]. El pico a 1560 cm-1 se asocia a
la vibración de estiramiento del C–C en el anillo del PPy [165]. La vibración de las bandas
de =C–H en el plano se encuentran entre 1186 cm-1 [163]. Los picos a 1052 cm-1 se

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
69
deben a la vibración de estiramiento correspondiente a C=C [166]. Los picos presentes a
los 951, 894 y 778 cm-1 son característicos del PPy [163].
Figura 3.9.- Espectros FTIR del óxido de grafito, GO-amino, PPy y GO-PPy; obtenidos por ATR.
Los espectros del óxido de grafito y del GraphO-amino ya fueron analizados
anteriormente por lo que en éste caso sólo se hará referencia al espectro del GraphO-
PPy. En el material híbrido GraphO-PPy el pico del enlace H–O se encuentra a los 3388
cm-1, el cual es menos intenso que el del GraphO-amino; a 2988 y 2900 cm-1 se aprecian
los dos picos de estiramiento simétrico y asimétrico del enlace C–H, respectivamente; el
pico de la vibración del esqueleto de dominios grafíticos no oxidados aparece a los 1635

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
70
cm-1, pero a comparación del GraphO-amino es más ancho y menos definido; a los 1557
cm-1 se hace presente el estiramiento del enlace C–C del anillo de PPy; a 1393 y 1056
cm-1 aparecen los picos de los enlaces C–O–C y el estiramiento del C–O,
respectivamente; mientras que los picos a 1220, 926 y 767 son característicos del PPy
[163].
Una vez que se analizaron los espectros infrarrojos de cada uno de los materiales
sintetizados, se puede comprobar que se llevó a cabo el anclaje del compuesto con
terminación amino para obtener el GraphO-amino; además que en los materiales híbridos
sí existe una diferencia entre los picos de los materiales individuales, lo que confirma la
existencia de nuevos enlaces entre estos materiales.
3.6.- Caracterización electroquímica
3.6.1.- Voltamperometría cíclica
En la figura 3.10 se pueden observar los resultados obtenidos por voltamperometría
cíclica referente al óxido de grafito, PEDOT, PPy, GraphO-PEDOT y GraphO-PPy
En la figura 3.10a se puede observar la voltamperometría cíclica del óxido de grafito a
diferentes velocidades de barrido, donde se puede apreciar un pico de oxidación a los
0.46 VSCE y otro de reducción a 0.33 VSCE, los cuales se pueden deber a los grupos
funcionales presentes en el material que le dan una contribución pseudocapacitiva. En el
trabajo de Y.-H. Lee, K.-H. Chang, et al., se reportaron picos de oxidación y de reducción,
el primero se presentó a los 0.65 V Vs RHE (0.41 VSCE) mientras que el de reducción se
observó a los 0.60 V vs RHE (0.36 VSCE) [167]; el corrimiento presente en la literatura con
los que se presentaron en lo experimental del trabajo, puede deberse a los grupos
funcionales y el porcentaje de cada uno de ellos en el óxido de grafito; además de que los
potenciales fueron medidos empleando diferentes electrodos de referencia lo que
desplaza los valores del potencial.
En la figura 3.10b se muestran las voltamperometrías cíclicas del PEDOT, donde se
observa que con forme se va aumentando la velocidad de barrido las curvas van

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
71
presentando una mayor inclinación por la resistencia del material, ya que a mayor
velocidad de barrido aumenta la corriente porque es necesario alcanzar los mismos
valores de potencial a menor tiempo; lo que hace que los picos de oxidación-reducción de
las cadenas del PEDOT no sean apreciables en la caracterización. Se empleó una
ventana de potencial de -0.3 a 1 V vs SCE. En la literatura se han reportado picos de
oxidación-reducción de las cadenas poliméricas a 0.47 y 0.35 V vs Ag/AgCl (0.4 y 0.3
VSCE); el hecho de que no se observen los picos en el material se debe a problemas de
difusión entre las especies involucradas en las reacciones redox, el seno del material y los
iones en el electrolito [168].
En la figura 3.10c se observa el material híbrido que presenta dos picos pronunciados
principalmente por los grupos funcionales presentes en el óxido de grafito, uno de
oxidación a 0.44 V vs SCE y otro de reducción a 0.33 V vs SCE; también se puede
observar un aumento de la densidad de corriente con respecto al óxido de grafito, lo que
se puede atribuir a la contribución de la pseudocapacitancia del polímero en el material
híbrido, sin embargo, la contribución de la doble capa es mayor que la de las reacciones
redox por lo que no se aprecian los picos de oxidación-reducción del PEDOT. La ventana
de potencial empleada fue de -0.3 a 1 V vs SCE [169].

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
72
Figura 3.10.- a) Voltamperometrías cíclicas a diferentes velocidades de barrido para a) óxido de
grafito, b) PEDOT y c) GraphO-PEDOT; en 2M H2SO4.
En la figura 3.11 se muestran las voltamperometrías cíclicas a diferentes velocidades de
barrido del óxido de grafito, del PPy y del GraphO-PPY; utilizando una ventana de
potencial de -0.3 a 1 V vs SCE.
En la figura 3.11a, se muestra la voltamperometría del óxido de grafito, en donde se
observa un pico de oxidación a 0.32 V vs SCE y otro pico de reducción a los 0.06 V vs
SCE, los que se atribuyen a los grupos funcionales del óxido de grafito [167]. Para el caso

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
73
del PPy (figura 3.11b), los picos de oxidación–reducción no son notorios, debido a la
resistencia que presenta el material, la cual va aumentando con forme aumenta la
velocidad de barrido; mostrando una forma alargada y estrecha de la voltamperometría;
ya se presenta resistencia por difusión por la fricción entre la migración de los complejos
de carga y las moléculas del electrolito [169]. En la literatura se han repostado picos redox
a -0.2 V vs SCE para el pico anódico y para el pico catódico a -0.25 V vs SCE [170].
Figura 3.11.- a) Voltamperometrías cíclicas a diferentes velocidades de barrido para a) óxido de
grafito, b) PPy y c) GraphO-PPy; en 2M H2SO4.

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
74
En la figura 3.11c se pueden identificar dos picos en material híbrido GraphO-PPy, uno de
oxidación a los 0.49 V y el de reducción a los 0.39 V, que se deben principalmente por los
grupos funcionales del óxido de grafeno del material híbrido. La densidad de corriente del
material aumentó a comparación del óxido de grafito, aunque también aumentó la
resistencia, la cual se hace más notable a velocidades de barrido mayores.
A partir de las curvas de voltamperometría cíclica, se puede obtener el comportamiento
del material, considerando que la capacitancia está definida como la medida de la
capacidad para almacenar energía eléctrica a una diferencia de potencial en un electrodo
(ver tabla 3.3) [3]. Basándose en la ecuación 1.1 se realizaron los cálculos para obtener el
área bajo la curva y así calcular la capacitancia especifica.
Teniendo los valores de Q de descarga de todos los ciclos, que están dados en mA/h, se
toman los valores máximos de cada uno de los ciclos para posteriormente sacar un
promedio de éstos; una vez que se obtiene el dato, éste se multiplica por 3600 y el
resultado se divide entre 1000, obteniendo como resultado faradios. Luego los faradios se
dividen entre el peso del material activo que se tiene en los electrodos, que en éste caso
son 6 mg; de esta forma se obtiene la capacitancia específica del material.
Tabla 3.3.- Valores de capacitancia calculados por voltamperometría cíclica.
En la figura 3.12a se presenta la capacitancia del óxido de grafito, PEDOT y en la figura
3.12b se tienen las del óxido de grafito, PPy y del GraphO-PPy; ambas con una velocidad
de barrido de 10 mV/s. En la figura 3.12a la curva del material híbrido presenta mayor

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
75
capacitancia que la del óxido de grafito, además de que los picos de óxido-reducción son
más notables en el GraphO-PEDOT; sin embargo el PEDOT presenta una mayor
capacitancia que el óxido de grafito y el GraphO-PEDOT, aunque también presenta una
inclinación que se puede deber a la resistencia en la difusión de iones en el polímero.
En la figura 3.12b se puede observar que el material híbrido presenta mayor capacitancia
que la del óxido de grafito, sin embargo, el GraphO-PPy al igual que PPy presentan una
inclinación que se debe a la resistencia que presenta el material, la cual puede ser
atribuida a la difusión de iones al interior del material.
Figura 3.12.- Capacitancia específica del óxido de grafito, PEDOT y GO-PEDOT a), y del óxido de
grafito, PPy y del GO-PPy; velocidad de barrio 10 mV/s.
3.6.2.- Ciclos galvanostáticos de carga-descarga
En la figura 3.13 se presentan las curvas de carga-descarga de los materiales sintetizados
a una corriente constante de 2 mA. El óxido de grafito muestra un comportamiento de
carga-descarga triangular, lo que se atribuye a procesos de doble capa electroquímica
puros que se llevan a cabo en el material [171], sin embargo, principalmente en la fase de
descarga es donde se evalúa el comportamiento capacitivo. En PEDOT y PPy, el
comportamiento de la curva de descarga es no-lineal, debido a los procesos

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
76
pseudocapacitivos que se llevan a cabo en las cadenas de los polímeros; los cuales, al
ser más lentos, aumentan el tiempo de carga-descarga que presenta el material [146].
En los dos materiales híbridos se puede notar que existen dos etapas de potencial en las
curvas; para el caso del GraphO-PEDOT de 0 a 0.47 V vs SCE y de 0.47 a 1 V vs SCE,
en el GraphO-PPy de 0 a 0.37 V vs SCE y de 0.37 a 1 V vs SCE. La primera etapa, donde
se lleva a cabo una descarga más lenta no lineal, se atribuye a la presencia tanto de la
capacitancia de la doble capa electroquímica, así como por las reacciones farádicas, por
la contribución de los polímeros en el material híbrido; la segunda etapa, que tiene un
comportamiento de descarga lineal y más corta es puramente por la capacitancia de la
doble capa electroquímica [146, 171]. La descarga de los materiales híbridos consta de
tres partes: una componente resistiva de caída de potencial repentina, IR, por causa de la
resistencia interna del material; otro elemento es la capacitancia correspondiente la
diferencia de potencial debido a la separación de los iones de la doble capa
electroquímica, en la región interfacial del electrodo, y por último se encuentra la
componente farádica con un tiempo mayor de descarga por las reacciones de adsorción
de iones [172, 173].

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
77
Figura 3.13.- Ciclos galvanostáticos de los materiales sintetizados a 2 mA, en 2M H2SO4.
La caída IR en los polímeros se puede explicar por el grado de aglomeración que tienen,
lo cual se observa en las micrografías SEM, que en el caso del PPy presenta una mayor
aglomeración que el PEDOT, lo cual se hace manifiesta como una caída de potencial en
la carga-descarga del material [173]. Estando los polímeros aglomerados, se presenta
una resistencia al momento de la difusión, porque a las cargas se le dificulta el acceso a
los poros del material.
En la tabla 3.4 se muestra la resistencia interna (ERS) del óxido de grafito, de los
polímeros y de los materiales híbridos; se puede observar que el PPy es el que presenta
la mayor resistencia y GraphO es el que tiene la menor resistencia. La resistencia GraphO
aumentó al agregarle el polímero, siendo el que contiene PPy el que tiene la mayor
resistencia de ambos materiales híbridos. En todos los casos, la eficiencia es cercana al

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
78
90 %, sin embargo los materiales híbridos tienen los valores de eficiencia más grandes
con respecto a los demás materiales caracterizados, presentando valores similares;
mientras que la eficiencia del PEDOT es la más baja de todos los materiales, que se
puede debe al proceso de dopado-desdopado que ocurre durante el almacenamiento de
carga.
Tabla 3.4.- Valores de ERS y eficiencia obtenidos por las curvas galvanostáticas.
En la tabla 3.5 se compara la capacitancia de los materiales obtenidas por diferentes
técnicas electroquímicas, ciclos galvanostáticos a 2 mA y por voltamperometría cíclica a 5
mV/s de velocidad de barrido; los valores que se reportan son similares en la mayoría de
los casos, siendo los dos polímeros los que muestran mayor discrepancia. Las
variaciones presentes son en mayor medida en los parámetros que cada una de las
técnicas toma en cuenta para calcular la capacitancia específica; además para éste caso,
sólo se está evaluando la capacitancia del material en sí y aplicado ya como un electrodo
en un supercondensador. Los materiales híbridos muestran un aumento de la
capacitancia con respecto GraphO, con lo que sí se observa una mejoría en el
almacenamiento de energía de los materiales híbridos.

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
79
Tabla 3.5.- Valores de capacitancia obtenidos por ciclos galvanostáticos y voltamperometría cíclica.
3.6.3.- Espectroscopia de impedancia electroquímica
En la figura 3.14 se presenta el diagrama Nyquist para el óxido de grafito, PPy, PEDOT,
GraphO-PEDOT y GraphO-PPy; en un intervalo de frecuencia de 100 KHz a 10 mHz, con
una amplitud de onda de 0.5 mA, usando la técnica de espectroscopia de impedancia
electroquímica galvánica (GEIS) a corriente controlada. El recuadro insertado en la gráfica
representa la región de alta frecuencia, en donde se pueden observar los valores de
resistencia del electrolito (intersección de la línea de impedancia con el eje real), con
valores que van de 1.19 Ω hasta 3.09 Ω; en el PPy es de 3.09 Ω, para el PEDOT es de
1.19 Ω, en el óxido de grafito es de 1.5 Ω, el GraphO-PEDOT 1.55 Ω y para el GraphO-
PPy 1.77 Ω.

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
80
Figura 3.14.- Diagramas Nyquist de los materiales sintetizados, en 2M H2SO4.
Empleando los valores obtenidos experimentalmente, se realizaron los circuitos
equivalentes de los materiales que se muestran en la figura 3.14. Se realizó el ajuste
empleando el software ZView 3.14c. Se realizaron dos circuitos equivalentes,
dependiendo del comportamiento que presentaban los materiales; la figura 3.14a
representa el circuito equivalente de los dos polímeros; en la figura 3.14b se presenta el
circuito equivalente de los materiales híbridos y del óxido de grafito.
El primer componente del circuito equivalente de la figura 3.15a engloba la resistencia de
la solución, la resistencia intrínseca del soporte y la resistencia por el contacto entre el
material y el colector de corriente, R1; en la figura 3.15a y b, se tiene una CPE1 que se
debe a la contribución de la doble capa electroquímica entre la interfase
electrodo/electrolito; en el circuito 3.15 la CPE1 se encuentra conectada en paralelo a la

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
81
resistencia de transferencia de carga R2 a través del material, el último componente es
una CPE2, atribuida a la capacitancia de las reacciones farádicas en los electrodos [174].
Figura 3.15.- Circuitos equivalentes: a) PEDOT y PPy; b) GraphO, GraphO-PEDOT y GraphO-PPy.
En la tabla 3.6 se muestran los ajustes de los datos experimentales en los circuitos
equivalentes del óxido de grafito, PPy, PEDOT, GraphO-PEDOT y GraphO-PPy; se
presentan los valores de la resistencia (R1 y R2), los elementos de fase constante (CPE1 y
CPE2), el porcentaje de error y la Chi cuadrada (χ2). La R1 es similar en todos los casos,
va de 1.2 hasta 2.8 Ω, siendo el PPy el que presenta la mayor resistencia. Las
resistencias por transferencia de carga son elevadas, en el caso del óxido de grafito es
tan grande del orden de 1020 Ω que pude ser despreciable. La CPE1 de los materiales es
del orden de 10-2 a 10-4 F, lo que son valores bajos, mostrando una capacitancia no ideal.
Los valores obtenidos de resistencias por impedancia se pueden comparar con la ERS, ya
que ésta es la suma de todas las resistencia obtenidas a través del sistema; los
materiales en cuanto a la resistencia se encuentran de la siguiente manera, comenzando
con el material con una menor resistencia, GraphO < GraphO-PEDOT < GraphO-PPy <
PEDOT < PPy; tanto la ERS obtenidas por ciclos galvanostáticos, así como los valores de
resistencia por impedancia concuerdan entre sí (ver tabla 3.7); los valores de resistencia
tanto del Graph y GraphO-PEDOT son similares tanto en los ciclos galvanostáticos y en la
impedancia.
Los altos valores de resistencia de los polímeros se debe posiblemente a la baja
conductividad de los polímeros obtenidos, aunado a la aglomeración de las partículas, lo
que hace que a los iones se les dificulte desplazarse dentro del material y les sea más
difícil introducirse a los poros [175], tal y como se observo en las micrografías SEM. Este
mismo comportamiento se observa con los valores calculados de ERS en la tabla 9, en
donde el PPy es el que presenta la mayor resistencia de los materiales sintetizados

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
82
Tabla 3.6.- Valores obtenidos de ajustes a partir de los circuitos equivalentes.
Tabla 3.7.- Comparación de las ERS de los materiales sintetizados obtenidas por impedancia y por
ciclos galvanostáticos.
Con las micrografías SEM del GraphO-PPy y del GraphO-PEDOT se puede ver que las
hojas de óxido de grafeno se encuentran cubiertas de polímero, sin embargo, en el
GraphO-PPy es más notoria ésta capa de polímero sobre la hoja, lo que podría dificultar
el transporte de carga entre la interface electrodo/electrolíto, al presentar una mayor

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
83
inclinación en la línea vertical del GraphO-PPy, respecto a la del GraphO-PEDOT, en el
diagrama de Nyquist (figura 3.14).
Finalmente, observando la resistencia total que presentan los materiales, y comparándola
con el comportamiento que se obtuvo mediante las técnicas de voltamperometría cíclica y
carga-descarga, se puede decir que el material más resistivo sintetizado es el PPy,
mientras que el óxido de grafito es el menos resistivo de ellos.

CAPÍTULO 3: RESULTADOS Y DISCUSIONES
84

CONCLUSIONES
85
CONCLUSIONES
Se comprobó la obtención de los materiales híbridos GraphO-PEDOT y GraphO-PPy,
mediante los espectros FTIR, debido a la presencia de picos característicos tanto de los
polímeros: PPy y PEDOT; así como del GraphO-amino. La presencia del compuesto
amino en las hojas del óxido de grafito, también se comprobó debido a la presencia de los
picos del anillo aromático del PPy.
Por medio de la microscopía electrónica de barrido, se observó que los materiales
híbridos obtenidos presentaron una morfología de nanopartículas aglomeradas, las cuales
principalmente se depositaron sobre la superficie de las hojas de GraphO-amino, siendo
las de menor tamaño las de GraphO-PPy con 26 nm de diámetro. La obtención de
nanopartículas, y no de nanotubos como se esperaba, puede deberse a que durante la
polimerización no se formaron las micelas, para darle así la forma tubular. En el caso del
GraphO-PEDOT, se observó únicamente una mayor irregularidad en la superficie de
GraphO-amino.
Por medio de la espectroscopia Raman se determinó el grado de desorden mediante la
relación ID/IG; siendo el óxido de grafito el que presentó mayor desorden, mientras que el
GraphO-PPy mostró mayor orden es su estructura; lo que hace referencia a la existencia
de otras interacciones electrónicas en los materiales híbridos.
Mediante la difracción de rayos X se demostró la intercalación de grupos funcionales entre
las capas de óxido de grafito. GraphO-PPy presentó menos láminas (10) en los
aglomerados, mientras que el compuesto GraphO-PEDOT presentó 12 láminas de óxido
de grafeno. De acuerdo a la nomenclatura que se ha establecido, se puede decir que se
obtuvieron materiales híbridos a base de óxido de grafeno multicapas.
Haciendo una comparación del TGA entre los dos materiales híbridos, se observa que
GraphO-PPy presentó una mayor estabilidad térmica, conservando una masa residual de
64.43 % a 900 °C; mientras que GraphO-PEDOT conservó una masa residual de 60.62 %
a 900 °C.

CONCLUSIONES
86
Respecto al comportamiento electroquímico de los materiales obtenidos, se observó por
medio de voltamperometría cíclica, que el óxido de grafito tiene una capacitancia
promedio de 13.5 F/g a una velocidad de barrido de 5 mV/s, la cual fue mayor en los
materiales híbridos, siendo para GraphO-PEDOT de 19 F/g, y de 18 F/g para GraphO-
PPy, siendo el primero quien presenta una mejor capacitancia.
Por medio de los ciclos de carga–descarga a corriente constante de 2 mA/cm2, se observó
que el óxido de grafito fue el que presentó menor resistencia interna, mientras que el PPy
fue el del valor más alto de resistencia. Además, también se observaron dos etapas de
descarga, debido a la presencia de reacciones farádicas y de doble capa electroquímicas;
con lo que también podemos corroborar la contribución del polímero al material híbrido.
La espectroscopia de impedancia electroquímica de los materiales híbridos no mostró una
presencia notoria de los procesos de transferencia de carga, por la contribución de los
polímeros, lo que podría deberse a que los polímeros se están comportando más como
semiconductores que como un conductor como tal. Además, se observó que en el óxido
de grafito no se presentó una capacitancia pura, debido a que la presencia de los grupos
funcionales contribuye a otros procesos a bajas frecuencias, provocando así una
inclinación en la línea capacitiva.
En base a lo anteriormente mencionado, se puede deducir que el material híbrido
GraphO-PPy presentó mejores propiedades físico-químicas que GraphO-PEDOT;
teniendo una mejor estabilidad térmica, mayor distancia interplanar, menor número de
hojas en apilamiento y un menor desorden en su estructura. Sin embargo, en el
comportamiento electroquímico GraphO-PEDOT tuvo mejores características frente al
GraphO-PPy; presentando una menor resistencia total y una mayor capacitancia en
comparación al GraphO-PPy.
Finalmente, también se comprobó que los materiales híbridos sí tuvieron una mayor
capacitancia y mayor tiempo de ciclos de carga-descarga en comparación al óxido de
grafito, aunque la resistencia equivalente aumento en los materiales híbridos por la
presencia de los polímeros.

REFERENCIAS
87
REFERENCIAS
1. P.F. Ribeiro; B.K. Johnson; et al., Energy storage systems for advanced power
applications. Proceedings of the IEEE. 89, 1744-1756, 2002.
2. D.A. Scherson; Y. Kiya; et al., Batteries and electrochemical capacitors. Phys.
Today. 61, 43-47, 2008.
3. A.J. Bard; G. Inzelt; et al., Electrochemical dictionary. Springer, 2008.
4. J. Garche, Encyclopedia of electrochemical power sources. Elservier, 2009.
5. H.D. Abruna; Y. Kiya; et al., Batteries and electrochemical capacitors. Physics
Today. 61, 43-47, 2008.
6. J. Chmiola; G. Yushin; et al., Anomalous increase in carbon capacitance at pore
sizes less than 1 nanometer. Science Magazine. 313, 1760-1763, 2006.
7. S.-L. Kuo y N.-L. Wu, Electrochemical capacitor of MnFe2O4 with NaCl electrolyte.
Electrochem. Solid St. 8, 495-499, 2005.
8. M.M. Bruno; N.G. Cotella; et al., Aplicación de materiales nanoestructurados en
supercapacitores electroquímicos, Jornadas SAM/CONAMET/Simposio Materia,
Argentina, 2003.
9. O. Barbieri; M. Hahn; et al., Capacitance limits of hign surface area activated
carbons for double layer capacitors. Carbon. 43, 1303-1310, 2005.
10. M.D. Stoller; S. Park; et al., Graphene-based ultracapacitors. Nano Lett. 8, 3498-
3502, 2008.
11. P. Simon y Y. Gogotsi, Materials for electrochemical capacitors. Nature Materials.
7, 845-854, 2008.
12. H.-K. Jeong; Y.P. Lee; et al., Evidence of Graphitic AB Stacking Order of Graphite
Oxides. Journal of the American Chemical Society. 130, 1362-1366, 2008.
13. C. Porter; G. Yushin; et al., Electrochemical performance of carbon onions,
nanodiamonds, carbon black and multiwalled nanotubes in electrical doble layer
capacitors. Carbon. 45, 2511-2518, 2007.
14. F. Béguin y E. Frąckowiak, Carbons for electrochemical energy storage and
conversion systems. CRC Press Taylor & Francis Group, 2010.
15. Y. Zhang; H. Feng; et al., Progress of electrochemical capacitor electrode
materials: A review. Int. J. Hydrogen Energ. 34, 4889-4899, 2009.

REFERENCIAS
88
16. D.A.C. Brownson; D.K. Kampouris; et al., An overview of graphene in energy
production and storage applications. J. Power Sources. 196, 4873-4885, 2011.
17. S. Cosnier y A. Karyakin, Electropolymerization: Concepts, materials and
applications. Wiley-VCH Verlag & Co. KGaA., Germany, 2010.
18. R. Kötz y M. Carlen, Principles and applications of electrochemical capacitors.
Electrochim. Acta. 45, 2483-2498, 2000.
19. M.S. Halper y J.C. Ellenborgen, Supercapacitors: A brief overview, 2006, MITRE
Nanosystems Group.
20. M. Jayalakshmi y K. Balasubramanian, Simple capacitors to supercapacitors - An
overview. Int. J. Electrochem. Sc. 3, 1196-1217, 2008.
21. S.R. Prabhu Gnanakan; M. Rajasekhar; et al., Synthesis of polythiophene
nanoparticles by surfactant-assisted dilute polymerization methid for high
performance redox supercapacitors. Int. J. Electrochem. Sc. 4, 1289-1301, 2009.
22. A.K. Shukla; S. Sampath; et al., Electrochemical supercapacitors: Energy storage
beyond batteries. Struct. Chem. 79, 1656-1661, 2000.
23. X.-F. Wang; D.-B. Ruan; et al., Hybrid electrochemical supercapacitors based on
polyaniline and activated carbon electrodes. Acta Phys. Chim. Sin. 21, 261-266,
2005.
24. Y.I. Yoon y J.M. Ko, CoNi oxide/carbon-nanofiber composite electrodes for
supercapacitors. Int. J. Electrochem. Sc. 3, 1340-1347, 2008.
25. F. Lufrano y P. Staiti, Mesoporous carbon materials as electrodes for
electrochemical supercapacitors. Int. J. Electrochem. Sc. 5, 903-916, 2010.
26. E. Frackowiak; J. Machnikowski; et al., Novel carbonaceous materials for
application in the electrochemical supercapacitors, in New carbon based materials
for electrochemical energy storage systems, I.V. Barsuko, et al., Editors. 2006,
Springer. p. 5-20.
27. A.K. Shukla, Electrochemical power sources 2. Fuel cells and supercapacitors.
Resonanace. 6, 72-81, 2001.
28. J. Wang, Analytical electrochemistry. Wiley - VCH, 2000.
29. O. Haas y E.J. Cairns, Electrochemical energy storage. Annu. Rep. Prog. Chem.,
Sect. C. 95, 163-197, 1999.
30. B.E. Conwat; V. Birss; et al., The role and utilization of pseudocapacitance for
energy storage by supercapacitors. J. Power Sources. 66, 1-14, 1997.

REFERENCIAS
89
31. W. Schmickler, Electronic effects in the electric double layer. Chemical Reviews.
96, 3177-3200, 1996.
32. E. Frackowiak y F. Béguin, Carbon materials for the electrochemical storage of
energy in capacitors. Carbon. 39, 937-950, 2001.
33. C.M.A. Brett y A.M.O. Brett, Electrochemistry: Principles, methods, and
applications. Oxford University Press, 1993.
34. B.E. Conwat, Electrochemical supercapacitors: Scientific fundamentals and
technological applications. Kluwer Academic \ Plenum Publishers, 1999.
35. S. Srinivasan, Fuel cells: From fundamentals to applications. Springer, 2006.
36. B.E. Conway; V. Birss; et al., The role and utilization of pseudocapacitance for
energy storage by supercapacitors. J. Power Sources. 66, 1-14, 1997.
37. B.E. Conway, Electrochemical supercapacitors: Scientific fundamentals and
technological applications. Kluwer Academic \ Plenum Publishers, 1999.
38. J. Liu; J. An; et al., Synthesis of graphene-polypyrrole nanotube composite and its
applications in supercapacitor electrode. J. Electrochem. Soc. 159, A828-A833,
2012.
39. X.W. Huang; Z.W. Xie; et al., Electric double layer capacitors using activated
carbon prepares form pyrolytic treatmen of sugar as their electrodes. Synth. Met.
135, 235-236, 2003.
40. E. Gomibuchi; T. Ichikawa; et al., Electrode properties of a double layer capacitor
of nano-structured graphite produced by ball milling under a hydrogen atmosphere.
Carbon. 44, 983-988, 2006.
41. B. Fang y L. Binder, Enhanced surface hydrophobisation for improved performance
of carbon aerogel electrochemical capacitor. Electrochim. Acta. 52, 6916-6921,
2007.
42. W. Xing; S.Z. Qiao; et al., Superior electric double layer capacitors using ordered
mesoporous carbons. Carbon. 44, 216-224, 2006.
43. Y. Zhao; M.-B. Zheng; et al., Easy synthesis of ordered meso/macroporous carbon.
Mater Lett. 62, 548-551, 2008.
44. B. Xu; F. Wu; et al., Activated carbon fiber cloths as electrodes for high
performance electric double layer capacitors. Electrochim Acta. 52, 4595-4598,
2007.

REFERENCIAS
90
45. T. Katakabe; T. Kaneko; et al., Electric double-layer capacitors ising bucky gels
consisting of an ionic liquid and carbon nanotubes. J. Electrochem. Soc. 152,
A1913-1916, 2005.
46. Y. Honda; T. Haramoto; et al., Aligned MWCNT sheet electrodes prepared by
transfer methodology providing high-power capacitor performance. Electrochem.
Solid St. 10, A106-110, 2007.
47. S.R. Sivakkumar; J.M. Ko; et al., Performance evaluation of CNT/polypyrrole/MnO2
composite electrodes for electrochemica capacitors. Electrochim Acta. 52, 7377-
7385, 2007.
48. I.-H. Kim y K.-B. Kim, Electrochemical characterization of hydrous ruthenium oxide
thin-film electrodes for electrochemica capacitors applications. J. Electrochem.
Soc. 153, A383-389, 2006.
49. W. Sugimoto; K. Yokoshima; et al., Fabrication of thin-film, flexible, and transparent
electrodes composed of ruthenic acid nanosheets by electrophoretic deposition
and application to electrochemical capacitors. J. Electrochem. Soc. 153, A255-260,
2006.
50. W. Sugimoto; H. Iwata; et al., Electrochemical capacitor behavior of layered
ruthenic acid hydrate. J. Electrochem. Soc. 151, A1181-1187, 2004.
51. N.A. Choudhury; A.K. Shukla; et al., Cross-linked polymer hydrogel electrolytes for
electrochemical capacitors. J. Electrochem. Soc. 153, A614-620, 2006.
52. G. Bo; Z. Xiaogang; et al., Amorphous Ru1- y CryO2 liaded on TiO2 nanotubes for
electrochemica capacitors. Electrochim Acta. 52, 1028-1032, 2006.
53. X.-H. Yang; Y.-G. Wang; et al., Interfacial synthesis of porous MnO2 and its
application in electrochemical capacitor. Electrochimica Acta. 53, 752-757, 2007.
54. T. Brousse; M. Toupin; et al., A hybrid activated carbonmanganese dioxide
capacitor using a mild aqueous electrolyte. J. Electrochem. Soc. 151, A614-622,
2004.
55. S.-W. Hwang y S.-H. Hyun, Synthesis and characterization of tin oxide/carbon
aerogel composite electrodes for electrochemical supercapacitors. Journal of
Power Sources. 172, 451-459, 2007.
56. D.-D. Zhao; S.-J. Bao; et al., Preparation of hexagonal nanoporous nickel
hydroxide film and its application for electrochemical capacitor. Electrochemistry
Communications. 9, 869-874, 2007.

REFERENCIAS
91
57. G.-h. Yuan; Z.-h. Jiang; et al., Electrochemical behavior of activated-carbon
capacitor material loaded with nickel oxide. Carbon. 43, 2913-2917, 2005.
58. Z. Fan; J. Chen; et al., Preparation and capacitive properties of cobalt–nickel
oxides/carbon nanotube composites. Electrochimica Acta. 52, 2959-2965, 2007.
59. H. Liu; P. He; et al., A novel nickel-based mixed rareearth oxide/activated carbon
supercapacitor using room temperature ionic liquid electrolyte. Electrochim Acta.
51, 1925-1931, 2006.
60. C. Chen; D. Zhao; et al., Influence of addition of tantalum oxide on electrochemical
capacitor performance of molybdenum nitride. Mater. Chem. Phys. 97, 156.161,
2006.
61. W. Liu; Y. Soneda; et al., Low-temperature preparation and electrochemical
capacitance of WC/carbon composites with high specific surface area. Carbon. 45,
2759-2767, 2007.
62. S.-L. Kuo y N.-L. Wu, Electrochemical capacitor of MnFe2O4 with organic Li-ion
electrolyte. Electrochem. Solid St. 10, A171-175, 2007.
63. D. Choi y P.N. Kumta, Nanocrystalline TiN derived by a two-step halide approach
for electrochemical capacitors. J. Electrochem. Soc. 153, A2298-2303, 2006.
64. Z.J. Lao; K. Konstantinov; et al., Synthesis of vanadium pentoxide powders with
enhanced surface-area for electrochemical capacitors. Journal of Power Sources.
162, 1451-1454, 2006.
65. A. Balducci; W.A. Henderson; et al., Cycling stability of hybrid activated
carbon//poly(3-methylthiophene) supercapacitor with N-butyl-N-methylpyrrolidinium
bis(trifluoromethenesulfonyl)imide ionic liquid as electrolyte. Electrochim Acta. 50,
2233-2237, 2005.
66. E.C. Rios; A.V. Rosario; et al., Poly(3-methylthiophene)/MnO2 composite
electrodes as electrochemical capacitors. J. Power Sources. 163, 1137-1142,
2007.
67. B. Muthulakshmi; D. Kalpana; et al., Electrochemical deposition of polypyrrole for
symmetric supercapacitors. Journal of Power Sources. 158, 1533-1537, 2006.
68. K.R. Prasad y N. Miura, Polyaniline–MnO2 composite electrode for high energy
density electrochemical capacitor. Electrochem. Solid St. 7, A425-428, 2004.
69. J.-H. Kim; Y.-S. Lee; et al., Polypyrrole/carbon composite electrode for high-power
electrochemical capacitors. Electrochimica Acta. 52, 1727-1732, 2006.

REFERENCIAS
92
70. H. Wan; Y. Liang; et al., Advanced asymetrical supercapacitors based on graphene
hybrid materials. Nano Res. 4, 729-736, 2011.
71. F. Alvi; M.K. Ram; et al., Graphene-polyethylenedioxythiophene conductiong
polymer nanocomposite based supercapacitor. Electrochim. Acta. 56, 9406-9412,
2011.
72. A.C. Neto; F. Guinea; et al., Drawing conclusions from graphene. Phys. World. 19,
33-37, 2006.
73. C. Rodríguez González y O. Vasilievna Kharissova, Propiedades y aplicaciones
del grafeno. Ingenierias. 11, 17-23, 2008.
74. X. Dong; P. Wang; et al., Growth of large-sized graphene thin-films by liquid
precursor-based chemical vapor deposition under atmospheric pressure. Carbon.
49, 3672-3678, 2011.
75. C.N.R. Rao; K. Biswas; et al., Graphene, the new nanocarbon. J. Mater. Chem. 19,
2457-2469, 2009.
76. A.K. Geim y K.S. Novoselov, The rise of graphene. Nat. Mater. 6, 183-191, 2007.
77. M.J. Allen; V.C. Tung; et al., Honeycomb carbon: A review of graphene. Chem.
Rev. 110, 132-145, 2010.
78. C.-Y. Su; A.-Y. Lu; et al., High-quality thin graphene films from fast electrochemical
exfoliation. ACS Nano. 5, 2332-2339, 2011.
79. Y. Zhu; S. Murali; et al., Graphene and graphene oxide: Synthesis, properties, and
applications. Adv. Mater. 22, 3906-3924, 2010.
80. A. Bianco; H.-M. Cheng; et al., All in the graphene family – A recommended
nomenclature for two-dimensional carbon materials. Carbon. 65, 1-6, 2013.
81. Y.H. Hu; H. Wang; et al., Thinnest two-dimensional nanomaterial-graphene for
solar energy. ChemSusChem. 3, 782-796, 2010.
82. T. Kuilla; S. Bhadra; et al., Recent advances in graphene based polymer
composites. Prog. Polym. Sci. 35, 1350-1375, 2010.
83. W. Choi; I. Lahiri; et al., Synthesis of graphene and its applications: Areview. Crit.
Rev. Solid State. 35, 52-71, 2010.
84. B.C. Brodie, On the atomic weight of graphite. Phil. Trans. R. Sco. Lond. 149, 249-
259, 1859.
85. F. Ali; N. Agarwal; et al., Chemical route to the formation of graphene. Curr. Sci.
India. 97, 683-685, 2009.

REFERENCIAS
93
86. D.R. Dreyer; S. Park; et al., The chemistry of graphene oxide. Chem. Soc. Rev. 39,
228-240, 2010.
87. L.-X. Dong y Q. Chen, Properties, synthesis, and characterization of graphene.
Front. Mater. Sci. China. 4, 45-51, 2010.
88. A. Elschener; S. Kirchmeyer; et al., PEDOT: Principles and applications of an
intrinsically conductive polymer. CRC Press Taylor & Francis Group, 2011.
89. H.R. Kricheldorf; O. Nuyken; et al., Handbook of polymer synthesis. Marcel Dekker,
New York, 2005.
90. T. Fernández Otero, Polímeros conductores: síntesis, propiedades y aplicaciones
electroquímicas. Revista Iberoamericana de Polímeros. 4, 2003.
91. M. Codina Cabeza y R. Salvatella Anzo, Estudio experimental de generación y
propiedades físicas de polímeros conductores, Maestro en Química Industrial,
Universitat Politècnica de Catalunya, Barcelona, 2007.
92. Committee on Polymer Science and Engineering, Polymer Science and
Engineering:The Shifting Research Frontiers. The National Academies Press,
1994.
93. W.E.J. Jones; J. Chiguma; et al., Electrically and thermally coducting
nanocomposites for electronic applications. Materials. 3, 1478-1496, 2010.
94. Y. Xu; J. Wang; et al., Capacitance properties of poly(3,4-
ethylenedioxythiophene)/polypyrrole composites. Journal of Power Sources. 159,
370-373, 2006.
95. R. Liu; S.I. Cho; et al., Poly(3,4-ethylenedioxythiophene) nanotubes as electrode
materials for a high-powered supercapacitor. Nanotechnology. 19, 2008.
96. R. Ramya; R. Sivasubramanian; et al., Conducting polymers-based
electrochemical supercapacitors—Progress and prospects. Electrochimica Acta.
101, 109-129, 2013.
97. A. Rudge; J. Davey; et al., Conducting polymers as active materials in
electrochemical capacitors. J. Power Sources. 47, 89-107, 1994.
98. F. López Carrasquero, Fundamentos de polímeros. Universidad de Los Andes,
Venezuela, 2004.
99. P. J. Flory, Principles of polymer chemistry. Cornell University Press, New York,
1953.
100. A. L. Gupta, Polymer chemistry. Pragati publications, 2010.

REFERENCIAS
94
101. G. Odian, Principles of polymerization. John Wiley & Sons, Inc., New York, 2004.
102. A. Kumar y R.K. Gupta, Fundamentals of polymer engineering. Marcel Dekker,
inc., 2003.
103. R. Ansari, Polypyrrole conducting electroactive polymers: synthesis and stability
studies. E-J Chem. 3, 186-201, 2006.
104. T.A. Skotheim y J.R. Reynolds, Handbook of Conductiong Polymers - Conjugated
polymers: Theory, synthesis, properties, and characterization. CRC Press Taylor &
Francis Group, 2007.
105. D. Braun; H. Cherdron; et al., Polymer synthesis: Theory and practice. Springer,
2005.
106. R. Ansari, In -Situ cyclic voltammetry and cyclic resistometry analyses of
conducting electroactive polymer membranes. Int. J. ChemTech Res. 1, 1398-
1402, 2009.
107. J. Arias Pardilla, Síntesis y caracterización de polímeros conductores basados en
anilinas sustituidas y su aplicación en electrocatálisis, Doctor en Ciencias de los
Materiales, Universidad de Alicante, Alicante, 2007.
108. G.G. Wallace; G.M. Spinks; et al., Conductive electroactive polymers. CRC Press,
2009.
109. N. Alonso-Vante, Electroquímica y electrocatálisis: Materiales - Aspectos
fundamenteles y aplicaciones. e-libro.net, 2002.
110. L. Groenendaal; F. Jonas; et al., Poly(3,4-ethylenedioxythiophene) and its
derivatives: past, present, and future. Adv. Mater. 12, 481-494, 2000.
111. D. Nilsson, An organic electrochemical transistor for priented sensor and logic,
Linköping, Sweden, 2005, Linköpings Universitet.
112. R. Liu; J. Duay; et al., Synthesis and characterization of RuO2/poly(3,4-
ethylenedioxythiophene) composite nanotubes for supercapacitors. Phys. Chem.
Chem. Phys. 12, 4309-4316, 2010.
113. R.K. Hiremath; M.K. Rabinal; et al., Simple stup to measure electrical properties of
polymeric films. Rev. Sci. Instrum. 77, 126106-126108, 2006.
114. Y. Han; L. Hao; et al., Preparation and electrochemical performances of graphite
oxide/polypyrrole composites. Synthetic Metals. 160, 2336-2340, 2010.

REFERENCIAS
95
115. S. Biswas y L.T. Drzal, Multilayered nanoarchitecture of graphene nanosheets and
polypyrrole nanowires for high permance supercapacitor electrodes. Chem. Mater.
22, 5667-5671, 2010.
116. D.A. Skoog; F.J. Holler; et al., Principles of instrumental analysis. McGraw-Hill,
2001.
117. S. Amelinckx; D.v. Dyck; et al., Electron microscopy: Principles and fundamentals.
VCH, 1997.
118. Z. Ni; Y. Wang; et al., Raman spectroscopy and imaging of graphene. Nano Res.
1, 273-291, 2008.
119. Z. Luo; C. Cong; et al., The origin of sub-bands in the Raman D-band of graphene.
Carbon. 50, 4252-4258, 2012.
120. J. Cazes y G.W. Ewing, Ewing's Analytical intrumentation handbook. Mercel
Dekker, 2005.
121. C. Kittel, Introduction to solid state physics. John Wiley & Sons, Inc., 2005.
122. Y. Waseda; E. Matsubara; et al., X-Ray diffraction crystallography: Introduction,
examples and solved problems. Springer, Berlin, 2011.
123. B.D. Cullity, Elements of X-Ray Diffraction. Addison-Wesley publishing company,
Inc., 1956.
124. R. Jabari Seresht; M. Jahanshahi; et al., Synthesize and characterization of
graphene nanosheets with high surface area and nano-porous structure. Applied
Surface Science. 276, 672-681, 2013.
125. V.S. Ramachandran; R.M. Paroli; et al., Handbook of thermal analysis of
construction materials. William Andrew, 2002.
126. T. Hatakeyama y F.X. Quinn, Thermal analysis: Fundamentals and applications to
polymer science. John Wiley & sons, 1999.
127. T. Theophanides, Infrared spectroscopy: Materials science, engineering and
technology. InTech, Croatia, 2012.
128. Q. Hao; H. Wang; et al., Morphology-controlled fabrication of sulfonated
graphene/polyniline nanocomposites by liquid/liquid interfacial polymerization and
investigation of their electrochemical properties. Nano Res. 4, 323-333, 2011.
129. D.E. Pacheco Catalán, Síntesis de polímeros electroconductores para la aplicación
en dispositivos energéticos no-convencionales, Doctor en materiales poliméricos,
Centro de investigación científica de yucatán, A. C., Mérida, Yucatán, 2009.

REFERENCIAS
96
130. Y. Liu; H. Wang; et al., Graphene/polypyrrole intercalating nanocomposites as
supercapacitors electrode. Electrochimica Acta. 112, 44-52, 2013.
131. J. Jang y H. Yoon, Facile fabrication of polypyrrole nanotubes using reverse
microemulsion polymerization. Chem. Commun.720-721, 2003.
132. M. Hernández; M.d.M. Bernal; et al., Overall performance of natural
rubber/graphene nanocomposites. Composites Science and Technology. 73, 40-
46, 2012.
133. E. Morales y J.L. Acosta, Supercondensadores electroquímicos basados en
polímeros conductores micro/nanoestructurados. Red Nacional de Pilas de
Combustible, Baterias Avanzadas e Hidrogeno457-461, 2006.
134. H. Yang; C. Shan; et al., Covalent functionalization of polydisperse chemically-
converted graphene sheets with amine-terminated ionic liquid. Chem. Commun. 0,
3880-3882, 2009.
135. N. Sakmeche; J.-J. Aaron; et al., Usefulness of aqueous anionic micellar media for
electrodeposition of poly-(3,4-ethylenedioxythiophene) films on iron, mild steel and
aluminium. Electrochimica Acta. 45, 1921-1931, 2000.
136. M. Souque; D. de Caro; et al., Tetrathiafulvalenium–bromide systems as
nanosticks and nanoparticles stabilized by PEDOT. Synthetic Metals. 161, 1001-
1004, 2011.
137. K.-H. Kim; J.-Y. Kim; et al., Synthesis of manganese dioxide/poly(3,4-
ethylenedioxythiophene) core/sheath nanowires by galvanic displacement reaction.
Journal of Electroceramics. 29, 149-154, 2012.
138. A. A. Farah; S. A. Rutledge; et al., Conductivity enhancement of poly(3,4-
ethylenedioxythiophene)-poly(styrenesulfonate) films post-spincasting. J. Appl.
Phys. 112, 113709-113717, 2012.
139. Y. Zhang; D. Li; et al., High quality graphene sheets from graphene oxide by hot-
pressing. Carbon. 54, 143-148, 2013.
140. L. Zhou; H. Gu; et al., Study on the synthesis and surface enhanced Raman
spectroscopy of graphene-based nanocomposites decorated with noble metal
nanoparticles. Colloids and Surfaces A: Physicochemical and Engineering Aspects.
430, 103-109, 2013.

REFERENCIAS
97
141. Q. Cheng; Y. He; et al., Surfactant-assisted polypyrrole/titanate composite
nanofibers: Morphology, structure and electrical properties. Synthetic Metals. 158,
953-957, 2008.
142. Y. Liu; R. Deng; et al., Carboxyl-functionalized graphene oxide-polyaniline
composite as a promising supercapacitor material. Journal of Materials Chemistry.
22, 13619-13624, 2012.
143. M.G. Han y S.H. Foulger, Facile Synthesis of Poly(3,4-ethylenedioxythiophene)
Nanofibers from an Aqueous Surfactant Solution. Small. 2, 1164-1169, 2006.
144. K. Awasthi; R. Kumar; et al., Synthesis of nano-carbon (nanotubes, nanofibers,
graphene) materials. Bull. Mater. Sci. 34, 607-614, 2011.
145. P. Karthika; N. Rajalakshmi; et al., Functionalized exfoliated graphene oxide as
supercapacitor electrodes. Soft. Nanoscience Letters. 2, 59-66, 2012.
146. Z. Gao; W. Yang; et al., Electrochemical synthesis of layer-by-layer reduced
graphene oxide sheets/polyaniline nanofibers composite and its electrochemical
performance. Electrochimica Acta. 91, 185-194, 2013.
147. H.K. Jeong; M.H. Jin; et al., Tailoring the characteristics of graphite oxides by
different oxidation times. J. Phys. D: Appl. Phys. 42, 1-6, 2009.
148. C. Zhu; J. Zhai; et al., Graphene oxide/polypyrrole nanocomposites: one-step
electrochemical doping, coating and synergistic effect for energy storage. Journal
of Materials Chemistry. 22, 6300-6306, 2012.
149. L. Mao; H.S.O. Chan; et al., Cetyltrimethylammonium bromide intercalated
graphene/polypyrrole nanowire composites for high performance supercapacitor
electrode. RSC Advances. 2, 10610-10617, 2012.
150. S. Konwer; R. Boruah; et al., Studies on Conducting Polypyrrole/Graphene Oxide
Composites as Supercapacitor Electrode. Journal of Electronic Materials. 40, 2248-
2255, 2011.
151. C. Bora y S.K. Dolui, Fabrication of polypyrrole/graphene oxide nanocomposites by
liquid/liquid interfacial polymerization and evaluation of their optical, electrical and
electrochemical properties. Polymer. 53, 923-932, 2012.
152. D. Zhang; X. Zhang; et al., Enhanced capacitance and rate capability of
graphene/polypyrrole composite as electrode material for supercapacitors. Journal
of Power Sources. 196, 5990-5996, 2011.

REFERENCIAS
98
153. B. Zhang; Y. Xu; et al., A Facile Synthesis of Polypyrrole/Carbon Nanotube
Composites with Ultrathin, Uniform and Thickness-Tunable Polypyrrole Shells.
Nanoscale Research Letters. 6, 1-9, 2011.
154. S. Sahoo; G. Karthikeyan; et al., Electrochemical characterization of in situ
polypyrrole coated graphene nanocomposites. Synthetic Metals. 161, 1713-1719,
2011.
155. T.L. Kelly; K. Yano; et al., Supercapacitive Properties of PEDOT and Carbon
Colloidal Microspheres. ACS Applied Materials & Interfaces. 1, 2536-2543, 2009.
156. S. Stankovich; D. Dikin; et al., Synthesis of graphene-based nanosheets via
chemical reduction of exfoliated graphite oxide. Carbon. 45, 1558-1565, 2007.
157. K. Haubner; J. Morawski; et al., The route to functional graphene oxide.
Chemphyschem. 11, 2131-1239, 2010.
158. Y. Liu; J. Zhou; et al., Synthesis, characterization and optical limiting property of
covalently oligothiophene-functionalized graphene material. Carbon. 47, 3113-
3121, 2009.
159. C.-Y. Chu; J.-T. Tsai; et al., Synthesis of PEDOT-modified graphene composite
materials as flexible electrodes for energy storage and conversion applications.
International Journal of Hydrogen Energy. 37, 13880-13886, 2012.
160. H. Feng; B. Wang; et al., Polypyrrole/hexadecylpyridinium chloride-modified
graphite oxide composites: Fabrication, characterization, and application in
supercapacitors. Journal of Power Sources. 246, 621-628, 2014.
161. A. Kaniyoor; R. Imran Jafri; et al., Nanostructured Pt decorated graphene and multi
walled carbon nanotube based room temperature hydrogen gas sensor.
Nanoscale. 1, 382-386, 2009.
162. S. Stankovich; R.D. Piner; et al., Synthesis and exfoliation of isocyanate-treated
graphene oxide nanoplatelets. Carbon. 44, 3342-3347, 2006.
163. H.-Y. Woo; W.-G. Jung; et al., Synthesis and dispersion of polypyrrole
nanoparticles in polyvinylpyrrolidone emulsion. Synthetic Met. 160, 588-591, 2010.
164. L.M. Yee; H.N.M.E. Mahmud; et al., Polypyrrole-polyethylene glycol conducting
polymer composite films: Preparation and characterization. Synthetic Met. 157,
386-389, 2007.
165. S. Bose; T. Kuila; et al., In-situ synthesis and characterization of electrically
conductive polypyrrole/graphene nanocomposites. Polymer. 51, 5921-5928, 2010.

REFERENCIAS
99
166. W.-J. Zou; S.-S. Mo; et al., Preparation of mesoporous carbon/polypyrrole
composite materials and their supercapacitive properties. J. Electrochem. Sci Eng.
1, 67-73, 2011.
167. Y.-H. Lee; K.-H. Chang; et al., Differentiate the pseudocapacitance and double-
layer capacitance contributions for nitrogen-doped reduced graphene oxide in
acidic and alkaline electrolytes. Journal of Power Sources. 227, 300-308, 2013.
168. J. Jang; J. Bae; et al., Selective Fabrication of Poly(3,4-ethylenedioxythiophene)
Nanocapsules and Mesocellular Foams Using Surfactant-Mediated Interfacial
Polymerization. Advanced Materials. 18, 354-358, 2006.
169. C. Lei; P. Wilson; et al., Effect of poly(3,4-ethylenedioxythiophene) (PEDOT) in
carbon-based composite electrodes for electrochemical supercapacitors. Journal of
Power Sources. 196, 7823-7827, 2011.
170. C. Shi, Polypyrrole and composite electrodes for electrochemical supercapacitors,
Master, McMaster University, Ontario, 2010.
171. Q. Wu; Y. Xu; et al., Supercapacitors Based on Flexible Graphene/Polyaniline
Nanofiber Composite Films. ACS Nano. 4, 1963-1970, 2010.
172. S. Hassan; M. Suzuki; et al., Synthesis of MnO2–chitosan nanocomposite by one-
step electrodeposition for electrochemical energy storage application. Journal of
Power Sources. 246, 68-73, 2014.
173. J. Yan; T. Wei; et al., Preparation of graphene nanosheet/carbon
nanotube/polyaniline composite as electrode material for supercapacitors. J. Power
Sources. 195, 3041-3045, 2010.
174. M.C. Turhan; A. Sezai Sarac; et al., Electrochemical impedance characterization
and potential dependence of poly[3,4-(2,2-dibutylpropylenedioxy)thiophene]
nanostructures on single carbon fiber microelectrode. Synthetic Metals. 162, 511-
515, 2012.
175. Q. Cheng; J. Tang; et al., Graphene and nanostructured MnO2 composite
electrodes for supercapacitors. Carbon. 49, 2917-2925, 2011.
Recommended















