
1
EVALUACIÓN DE LA ASOCIACIÓN MELOXICAM-ATORVASTATINA SOBRE LA
RESPUESTA NEURONAL Y GLIAL EN UN MODELO MURINO DE ISQUEMIA
CEREBRAL POR EMBOLISMO ARTERIAL
LINA MARIA DE LOS REYES
Trabajo de grado como requisito parcial para optar por el título de
Magister en Ciencias Biológicas
Director
ÁNGEL ENRIQUE CÉSPEDES RUBIO
Doctor en Ciencias Básicas Biomédicas
UNIVERSIDAD DEL TOLIMA
FACULTAD DE CIENCIAS
MAESTRÍA EN CIENCIAS BIOLÓGICAS
IBAGUÉ - TOLIMA
2014

2

3
A Dios por darme lo necesario para alcanzar los objetivos planteados.
A mi esposo y a mi hijo por su apoyo incondicional
A mi madre y hermano por acompañarme en esta travesía.

4
AGRADECIMIENTOS
El autor expresa sus agradecimientos:
Al Dr. Ángel Enrique Céspedes por su valiosa dirección y pertinente orientación.
A la Dra. Liliana Francis Turner por su preciado apoyo y oportuna colaboración.
A los integrantes de los grupos de investigación en Enfermedades Neurodegenerativas
y Modelos Experimentales para las Ciencias Zoohumanas por su compañerismo y
capacidad para compartir sus conocimientos.
A los laboratorios de Biotecnología Animal y Toxicología de la Universidad del Tolima por
su apoyo logístico.
Al Comité Central de Investigaciones de la Universidad del Tolima por la financiación de
este proyecto.

5
CONTENIDO
Pág.
INTRODUCCIÓN 14
1. OBJETIVOS 18
1.1 OBJETIVO GENERAL 18
1.2 OBJETIVOS ESPECÍFICOS 18
2. MARCO REFERENCIAL 19
2.1 ENFERMEDADES CEREBROVASCULARES 19
2.1.1 Isquemia Cerebral 19
2.1.2 Fisiopatología de la Isquemia Cerebral 20
2.2 CÉLULAS GLIALES 32
2.2.1 Astrocitos 32
2.2.3 Microglía 37
2.2.3 Células progenitoras NG2 45
2.3 MODELOS DE ISQUEMIA CEREBRAL 45
2.3.1 Modelos focales de isquemia cerebral 48
2.3.2 Modelos globales de isquemia cerebral 51
2.4 TERAPÉUTICA DE LA ISQUEMIA CEREBRAL 52
2.4.1 Estatinas 55
2.4.2 Inhibidores de la ciclooxigenasa 61
3. MATERIALES Y METÓDOS 70
3.1 REACTIVO BIOLÓGICO 66
3.2 MODELO DE ISQUEMIA CEREBRAL POR EMBOLIZACIÓN ARTERIAL 66

6
3.3 DISEÑO EXPERIMENTAL 69
3.4 EVALUACIÓN NEUROLÓGICA 70
3.5 PERFUSIÓN Y PROCESAMIENTO DE LOS CEREBROS 70
3.6 PROCESAMIENTO HISTOLÓGICO 71
3.6.1 Supervivencia celular e integridad de la mielina 73
3.6.2 Neurodegeneración 74
3.6.3 Inmunohistoquímica 74
3.7 OBTENCIÓN Y PROCESAMIENTO DE IMÁGENES 76
3.7.1 Astrocitos 76
3.7.2 Microglía 77
3.7.3 Ciclooxigenasa 2 77
3.8 ANÁLISIS ESTADÍSTICO 77
4. RESULTADOS 79
4.1 EMBOLIZACIÓN ARTERIAL 79
4.2 EVALUACIÓN NEUROLÓGICA 80
4.3 SUPERVIVENCIA CELULAR 81
4.4 NEURODEGENERACIÓN 83
4.5 INMUNORREACTIVIDAD DE CÉLULAS GLIALES 85
4.5.1 Células precursoras NG2 85
4.5.2 Reactividad de la microglía 86
4.5.3 Reactividad de los astrocitos 93
4.5.6 Reactividad de la enzima ciclooxigenasa 2 101
5. DISCUSIÓN 105
5.1 EMBOLIZACIÓN ARTERIAL 105
5.2 EVALUACIÓN NEUROLÓGICA 106
5.3 SUPERVIVENCIA CELULAR 106

7
5.4 NEURODEGENERACIÓN 107
5.5 INMUNORREACTIVIDAD DE CÉLULAS GLIALES 108
5.5.1 Células precursoras NG2 108
5.5.2 Reactividad microglial 109
5.5.4 Reactividad de la ciclooxigenasa 2 113
6. CONCLUSIONES 117
RECOMENDACIONES 119
REFERENCIAS 120

8
LISTA DE FIGURAS
Pág.
Figura 1. Distribución de las zonas de lesión en la isquemia cerebral. 21
Figura 2. Metabolismo del glutamato neuronal soportado por los astrocitos. 34
Figura 3. Transformación fenotípica de la microglía ramificada a fagocítica. 39
Figura 4. Inducción de isquemia cerebral experimental mediante embolización arterial.
68
Figura 5. Administración de los tratamientos, pruebas comportamentales y
procesamiento de los cerebros después de la inducción experimental de la isquemia. 71
Figura 6. Zonas evaluadas mediante técnicas histoquímicas e inmunohistoquímicas en
un modelo de infarto cerebral. 72
Figura 7. Registro del score neurológico de los grupos isquémicos frente al simulado
placebo. 80
Figura 8. Densidad de la población neuronal y astrocitaria en la corteza
somatosensorial primaria de los grupos simulados e isquémicos. 81
Figura 9. Patrón de la citoarquitectura en la isquemia cerebral y frente a los
tratamientos con meloxicam, atorvastina y su asociación en la corteza somatosensorial
primaria y la CA1 del hipocampo. 82
Figura 10. Cambios en la arquitectura celular en zonas focales de la isquemia cerebral
por embolismo arterial. 83
Figura 11. Distribución de neuronas en degeneración en la isquemia cerebral embólica.
84
Figura 12. Células Fluoro Jade B positivas en la corteza piriforme en la isquemia
cerebral por embolismo arterial. 84
Figura 13. Esquematización de la distribución de células precursoras NG2 en un corte
coronal (Bregma -2,8) por efecto de la isquemia cerebral embólica. 85
Figura 14. Inmunoreactividad de células NG2 en animales simulados e isquémicos
placebo en la zona paraventricular. 86
Figura 15. Esquematización de la distribución de células microgliales en la isquemia
cerebral por embolismo arterial. Bregma -2,8. 87

9
Figura 16. Distribución de células microgliales en el foco isquémico a las 120 horas, en
isquemia cerebral embólica. 88
Figura 17. Cambios morfológicos microgliales en zonas perifocales y focales en los
animales isquémicos. 89
Figura 18. Inmunorreactividad de células microgliales en la isquemia cerebral embólica
y frente a los tratamientos con meloxicam, atorvastatina y su asociación en la fimbria
del hipocampo. 90
Figura 19. Evaluación de la reactividad microglial en la isquemia cerebral por
embolismo arterial y frente a los tratamientos con meloxicam, atorvastatina y su
asociación, en la fimbria del hipocampo. 91
Figura 20. Densidad y longitud microglial de los grupos simulados tratados con
meloxicam, atorvastatina y su asociación en la fimbria del hipocampo. 92
Figura 21. Reactividad astrocitaria en isquemia cerebral embólica y frente a los
tratamientos con meloxicam, atorvastatina y su asociación, en la zona hipocampal CA1.
94
Figura 22. Densidad global de los astrocitos en la isquemia cerebral embólica y frente
a los tratamientos con meloxicam, atorvastina y su asociación en la CA1 hipocampal. 95
Figura 23. Reactividad astrocitaria en la isquemia cerebral y frente a los tratamientos
con meloxicam, atorvastatina y su asociación, en la corteza somatosensorial. 96
Figura 24. Densidad global de los astrocitos en la isquemia cerebral embólica y frente
a los tratamientos con meloxicam, atorvastatina y su asociación, en la corteza
somatosensorial. 97
Figura 25. Reactividad de los astrocitos en la isquemia cerebral embólica y frente a los
tratamientos con meloxicam, atorvastatina y su asociación, en el cuerpo estriado. 98
Figura 26. Densidad global de los astrocitos en isquemia cerebral embólica y frente a
los tratamientos con meloxicam, atorvastatina y su asociación, en el cuerpo estriado. 98
Figura 27. Evaluación de la densidad y longitud celular de los astrocitos, en los grupos
simulados tratados con meloxicam, atorvastatina y su asociación, en la CA1
hipocampal. 99

10
Figura 28. Evaluación de la densidad y longitud celular de los astrocitos en los grupos
simulados tratados con meloxicam, atorvastatina y su asociación, en la corteza
somatosensorial. 100
Figura 29. Esquematización de la distribución de la reactividad de COX 2 en la
isquemia cerebral por embolismo arterial. 101
Figura 30. Inmunomarcaje de COX 2 en la isquemia cerebral y frente a los tratamientos
con meloxicam, atorvastatina y su asociación, en la corteza somatosensorial primaria y
el giro dentado del hipocampo. 102
Figura 31. Evaluación de la reactividad de COX-2 en la isquemia cerebral embólica y
frente a los tratamientos con meloxicam, atorvastatina y su asociación, en la corteza
somatosensorial y en el giro dentado del hipocampo. 103

11
RESUMEN
El accidente cerebrovascular es la segunda causa de muerte y la primera de
discapacidad en el mundo, siendo más del 85% de origen isquémico.
Se estudió el efecto de la atorvastatina y el meloxicam en forma única y asociada sobre
la respuesta neuronal, astrocitaria y microglial en un modelo de infarto cerebral por
embolia arterial. Se emplearon 32 ratas Wistar hembras sometidas a embolia arterial
carotidea y posterior tratamiento con meloxicam, atorvastatina y su asociación a 6, 24,
48 y 72 horas; se evaluó la reactividad de las proteínas COX-2, GFAP y OX-42 como
marcadores de la actividad de la enzima ciclooxigenasa 2, los astrocitos y la microglía
respectivamente, empleando la técnica de inmunohistoquímica convencional. La
neurodegeneración, la supervivencia celular y la integridad de la mielina fueron
evaluadas utilizando las tinciones Flourojade y Luxol Fast Blue (método Kluver Barrera
modificado), mediante análisis densitométrico y morfológico.
Los datos obtenidos fueron evaluados empleando análisis de varianza y pruebas no
paramétricas de comparación múltiple. La isquemia cerebral por embolia arterial
incrementó significativamente (p<0,001) la reactividad astrocitaria y microglial, en tanto,
la atorvastatina, el meloxicam y su asociación la redujeron.
La isquemia produjo acortamiento de las proyecciones astrocitarias, engrosamiento
celular, ruptura de las expansiones protoplásmicas (clasmatodendrosis) y cambios
morfológicos microgliales propios de diversas etapas de actividad. En zonas perifocales,
se incrementó la inmunoreactividad de COX-2 y se redujo en el foco isquémico; en tanto,
el meloxicam y la atorvastatina redujeron significativamente la inmunoreactividad
perifocal, restableciendo el marcaje de COX-2 en el foco isquémico.
En conclusión, la asociación meloxicam – atorvastatina atenúa la respuesta astrocitaria
y microglial del proceso inflamatorio luego de la isquemia cerebral por embolia arterial,

12
reduciendo la neurodegeneración y restableciendo el equilibrio morfológico y funcional
del encéfalo.
Palabras Clave: Astrocitos, Atorvastatina, Ciclooxigenasa, Isquemia Cerebral,
Meloxicam, Microglía.

13
ABSTRACT
Stroke is the second leading cause of death and the first of disability in the world, with
more than 85% of the cases having ischemic origin.
To evaluate in an embolism model of stroke the effect of atorvastatin and meloxicam on
neurons, astrocytes and microglia. This evaluation was done administering each
medication individually and in association. Wistar rats were subjected to carotid arterial
embolism and treatment with meloxicam and atorvastatin to 6, 24, 48 and 72 hours. Using
immunohistochemistry, we evaluated the immunoreactivity of COX-2 protein, GFAP and
OX-42 in neurons, astrocytes and microglia by densitometric and morphological studies.
Neurodegeneration, cells survival and myelin integrity were analyzed with Fluoro Jade
and Luxol Fast Blue (Kluver`s Barrera method modified). Data were evaluated by analysis
of variance and non-parametric multiple comparison.
Cerebral ischemia by arterial embolism increased significantly the reactivity of microglia
and astrocytes (p <0.001), whereas it was reduced by single or associated treatments
with atorvastatin and meloxicam. Ischemia produced astrocytic shortening, cellular
thickening, protoplasmic rupture expansions (clasmatodendrosis) and microglial
morphological changes characteristic of various stages of activity. In perifocal areas, the
immunoreactivity of COX-2 was increased in the ischemic focus it was reduced, while
meloxicam and atorvastatin significantly reduced (p <0.001) the perifocal
immunoreactivity, restoring the marking of cyclooxygenase in the ischemic focus.
These results suggest that meloxicam–atorvastatin association attenuates astrocytic and
microglial response in the inflammatory process after cerebral ischemia by arterial
embolism, reducing neurodegeneration and restoring morphological and functional
balance of nervous tissue.
Keywords: astrocytes, microglia, atorvastatin, brain ischemia, cyclooxygenase,
meloxicam.

14
INTRODUCCIÓN
El accidente cerebrovascular (ACV) es la tercera causa de muerte en las Américas
(Organización Panamericana de la Salud, 2013) y la primera de discapacidad en los
países industrializados (Arango Dávila, Escobar Betancourt, Cardona Gómez, &
Pimienta Jiménez, 2004; Céspedes Rubio, Wandosell, & Cardona Gómez, 2010; Denes,
Thornton, Rothwell, & Allan, 2010), siendo el 85% de los casos de tipo isquémico (Casals
et al., 2011; Lapuente, Rengifo, Ávila Rodriguez, & Céspedes Rubio, 2013; Shen, Wu,
Zhu, & Sun, 2013; Woodruff et al., 2011); aproximadamente, el 75% de las isquemias
cerebrales son de origen embólico (Woodruff et al., 2011).
La isquemia cerebral, es ocasionada por la oclusión de una arteria principal,
interrumpiéndose el flujo de sangre al cerebro e instaurándose un fallo energético por
disminución en el aporte de oxígeno y glucosa, con alteración en la producción de ATP
y de los gradientes iónicos, así mismo con liberación de elevadas cantidades de
glutamato (Arango Dávila et al., 2004; Culmsee & Krieglstein, 2005; Hossmann, 2012;
Rojas, Zurru, Romano, Patrucco, & Cristiano, 2007; F. Silva, Quintero, & Zarruc, 2009).
El glutamato liberado en exceso al espacio extracelular, conlleva a la muerte neuronal
por excitotoxicidad debido a la sobreestimulación de los receptores N-metil-D-aspartato
(NMDA) y α-amino-3-hidroxi-5-metil-4-isoxazolpropiónico (AMPA), conduciendo al
aumento en el influjo de calcio intracelular que junto con un proceso de acidosis
metabólica en el tejido isquémico, contribuyen al daño y a la activación de proteínas y
enzimas que participan en los procesos de muerte celular. La necrosis que ocurre
principalmente en el foco isquémico y la apoptosis en las zonas de penumbra (Arango-
Dávila et al., 2004; Weinstein & Möller, 2011; Z.-G. Zhang, Sun, Zhang, & Yang, 2013)
En el proceso inflamatorio del cerebro, diversos factores solubles pueden mediar,
influenciar y regular la actividad glial y neuronal, activando diversas células como los
astrocitos y la microglía, células que migran hacia el sitio del daño y liberan citoquinas

15
proinflamatorias (Franke, Verkhratsky, Burnstock, & Illes, 2012; Weinstein & Möller,
2011).
Por otro lado, estas células se encargan de proteger al cerebro mediante variadas
acciones entre las que se encuentran, la captación de iones y neurotransmisores del
espacio extracelular que ayudan a limitar el efecto excitotóxico a las células vecinas,
como es el caso de los astrocitos (W. Kang & Hébert, 2011; Ouyang, Voloboueva, Xu, &
Giffard, 2007; Weinstein & Möller, 2011).
De otra parte y frente al daño tisular, la microglía activa su programa inmunoefector
modificando su morfología, proliferando e incrementando la expresión de antígenos de
superficie inmunomoduladores, como la integrina CD11b (OX-42), además, de
encargarse de la fagocitosis de restos celulares en caso de presentarse degeneración
neuronal (Gehrmann, Matsumoto, & Kreutzberg, 1995; Weinstein & Möller, 2011).
Adicionalmente, otras células gliales, las precursoras NG2 se pueden diferenciar a
células productoras de mielina (oligodendrocitos) en respuesta a la isquemia,
(Richardson, Kaylene, Tripathi, & McKenzie, 2011; Sakry, Karram, & Trotter, 2011;
Wigley, Hamilton, Nishiyama, Kirchhoff, & Butt, 2007), jugando un papel crucial en el
proceso de remielinización (Anderova et al., 2011; Honsa, Pivonkova, Dzamba, Filipova,
& Anderova, 2012; Lecca, Ceruti, Fumagalli, & Abbracchio, 2012; Maldonado, Vélez Fort,
& Angulo, 2011)
Junto con la activación de las células gliales, durante la isquemia cerebral se incrementa
la liberación de factores mediadores de la inflamación, entre estos las prostaglandinas,
que son producidas por la actividad enzimática de la ciclooxigenasa (COX), a partir del
ácido araquidónico (Hara, Kong, Sharp, & Weinstein, 1998).
Existen dos isoformas principales de COX, la COX 1 y la COX 2; en el cerebro, se
expresa constitutivamente la COX 2 y representa la principal isoforma bajo condiciones
fisiológicas (Stefanovic, Bosetti, & Silva, 2006; Yang & Chen, 2008), sin embargo la

16
expresión y actividad de COX 2 como enzima inducible, es marcadamente elevada frente
a una variedad de estímulos pro-inflamatorios y de activación de los receptores NMDA
(C. Chen, Magee, & Bazan, 2002; Yang & Chen, 2008).
Aunque se ha logrado avanzar sustancialmente en el conocimiento de la fisiopatología
de la isquemia cerebral, faltan aún por aclarar muchos de los eventos celulares y
subcelulares que se suscitan en las neuronas, en la astroglía y la microglía. De otra parte,
existen pocas alternativas terapéuticas aprobadas para el tratamiento de la isquemia
cerebral, con excepción del factor activador de plasminógeno tisular recombinante (rt-
PA) y ciertos fármacos trombolíticos de variada eficacia (Fernández Gómez et al., 2008).
Por las anteriores razones, se hace necesario buscar nuevas alternativas de tratamiento
para la isquemia cerebral, como pudiese ser la asociación farmacológica de un inhibidor
preferencial de la COX 2 (meloxicam) y un fármaco inhibidor de una enzima pivote en la
síntesis del colesterol (atorvastatina), que ha demostrado favorecer la recuperación
tisular y reducir el daño neuronal por diversos mecanismos (Céspedes-Rubio et al.,
2010).
Teniendo como referencia estos antecedentes, se planteó como objetivo evaluar en un
modelo de infarto cerebral por embolismo arterial, el efecto de la atorvastatina y el
meloxicam en forma individual y asociada sobre la respuesta celular de astrocitos,
microglía, células progenitoras NG2 e inflamación, a través de los biomarcadores GFAP,
OX-42, NG2 y COX 2, determinando los patrones de inmunoreactividad de estas
proteínas en el tejido isquémico, así como la neurodegeneración, la integridad de la
mielina y la muerte neuronal mediante las tinciones vitales Flourojade B®, Luxol Fast Blue
y cresil violeta.
En este documento se presenta una reseña de la fisiopatología de la isquemia cerebral,
las células gliales, los modelos experimentales de isquemia cerebral y las alternativas
terapéuticas, así como la descripción de las metodologías empleadas, los resultados

17
obtenidos, la discusión, las conclusiones y las recomendaciones para futuras
investigaciones.

18
1. OBJETIVOS
1.1 OBJETIVO GENERAL
Evaluar en un modelo murino de infarto cerebral por embolismo arterial, los cambios
morfológicos en poblaciones neuronales y gliales y de la respuesta inmune de diversos
territorios encefálicos, así como los efectos de la asociación meloxicam – atorvastatina.
1.2 OBJETIVOS ESPECÍFICOS
Determinar los cambios morfológicos ocasionados por la isquemia cerebral inducida por
embolismo arterial en neuronas y células gliales de corteza, hipocampo, cuerpo estriado,
cuerpo calloso, sustancia nigra, amígdala y núcleos talámicos, mediante marcadores de
supervivencia, inflamación, neurodegeneración, mielinización y muerte neuronal (Luxol
Fast Blue, cresil violeta, Fluorojade y COX-2) por técnicas histoquímicas,
inmunohistoquímicas y de fluorescencia.
Evaluar la inmunoreactividad de los astrocitos (anti-GFAP), células NG2 glía (anti-NG2)
y microglía (anti-OX-42) por efecto de la isquemia cerebral embolizante y frente a la
asociación meloxicam – atorvastatina, a través de técnicas histoquímicas e
inmunohistoquímicas.

19
2. MARCO REFERENCIAL
2.1 ENFERMEDADES CEREBROVASCULARES
La enfermedad cerebrovascular es definida por la Organización Mundial de la Salud
como el desarrollo de signos neurológicos focales o globales que comprometen la
función cerebral, con síntomas de 24 horas o más, o que llevan a la muerte sin otra causa
que el origen vascular (Arauz, A. & Ruíz Franco, A. 2012)
La Guía de Práctica Clínica para el Manejo de Pacientes con Ictus en Atención Primaria,
publicada en el 2009, clasifica los infartos cerebrales en dos grandes tipos según la
naturaleza de la lesión encefálica: Isquémica la cual se presenta aproximadamente en el
85% de los casos y hemorrágica en 15% restante (Unidad de evaluación de tecnologías
sanitarias de la agencia Laín Entralgo de la comunidad de Madrid, 2009).
2.1.1 Isquemia Cerebral. Se debe a la falta de aporte sanguíneo a una zona del
parénquima cerebral, caracterizada por un déficit encefálico focal como consecuencia de
una alteración circulatoria, generalmente por la oclusión de un vaso principal. Esta
alteración puede ser cuantitativa o cualitativa; la primera, es ocasionada por la reducción
en la cantidad de sangre que llega al cerebro, como consecuencia de trombosis, embolia
o por bajo gasto cardiaco; en la segunda, la calidad de la sangre se encuentra alterada
por anemia, trombocitemia o policitemia.
Existen diferentes clasificaciones de la isquemia cerebral dependiendo de la naturaleza
de la lesión, etiología, tamaño, morfología, topografía, forma de instauración y evolución
posterior. La más empleada es la clasificación del estudio Trial of Org 10172 in Acute
Stroke Treatment (TOAST) que define 5 grupos (Arauz & Ruíz Franco, 2012; Rojas et
al., 2007), como se describe a continuación.

20
2.1.1.1 Ateroesclerosis de grandes vasos: Definido por síntomas neurológicos debido a
ateroesclerosis intra o extracraneal. La ateroesclerosis extracraneal afecta
principalmente la bifurcación carotidea, la porción proximal de la carótida interna y el
origen de las arterias vertebrales. En este subtipo la isquemia cerebral, es el resultado
de la oclusión trombótica (aterotrombosis) o tromboembólica (embolismo arteria-arteria)
de los vasos.
2.1.1.2 Cardioembolismo: Se debe a la oclusión de una arteria cerebral por un émbolo
originado a partir del corazón.
2.1.1.3 Enfermedad de pequeño vaso cerebral: El infarto lacunar es una isquemia
cerebral menor de 15 mm de diámetro, localizado en el territorio irrigado por una arteriola.
Principalmente ocurren en las arterias lenticuloestriadas o talamoperforantes.
2.1.1.4 Otras causas. Las más frecuentes son vasculopatías no ateroescleróticas como:
disección arterial cervico-cerebral, fibrodisplasia muscular, enfermedad de Takayasu,
vasculitis del Sistema Nervioso Central y enfermedad de Moya-Moya.
2.1.1.5 Etiología no determinada: Incluye las isquemias cerebrales con más de una
etiología posible; aquellas en las cuales no es posible se puede determinar la causa, a
pesar de una evaluación completa y las que presentan una evaluación incompleta.
A partir de la interrupción del flujo sanguíneo, hacia una zona del parénquima cerebral,
se desencadenan una serie de eventos bioquímicos y moleculares que determinan la
vida o muerte de las células cerebrales (Arauz & Ruíz Franco, 2012; Rojas et al., 2007;
F. A. Silva et al., 2006)
2.1.2 Fisiopatología de la Isquemia Cerebral. La isquemia cerebral es un evento
dinámico, que provoca el desarrollo de procesos excitotóxicos, inflamatorios y
microvasculares los cuales pueden conducir a la necrosis del tejido en el foco de la lesión;
sin embargo, una parte del tejido cerebral puede ser recuperado si la reperfusión ocurre

21
antes de que el daño isquémico se torne irreversible (Jordán, Ikuta, García García,
Calleja, & Segura, 2007). En la periferia del área necrótica se puede diferenciar una
segunda zona, conocida como penumbra, en donde la disminución del flujo sanguíneo
es menos grave debido a la irrigación sanguínea colateral (Arango Dávila et al., 2004);
estas áreas, se pueden visualizar en la figura 1.
Figura 1. Distribución de las zonas de lesión en la isquemia cerebral.
a. Foco isquémico. b. Penumbra.
Fuente: Grupo END
Inicialmente las neuronas de las zonas de penumbra aunque han sido afectadas pueden
mantener su potencial de membrana, sin embargo más tardíamente tienden a un proceso
de degeneración y muerte apoptótica. El preciso momento y las vías celulares implicadas
en la muerte neuronal todavía no se han dilucidado completamente; las evidencias
sugieren que están involucradas la disminución de ATP, la excitotoxicidad por glutamato,
la acumulación de especies reactivas de oxigeno (ROS), la activación de las vías
apoptóticas y la activación de los procesos inflamatorios (Culmsee & Krieglstein, 2005;
Di Filippo et al., 2008)
Por otro lado, los niveles disminuidos de ATP traen como consecuencia el mal
funcionamiento de las bombas iónicas, conduciendo a la despolarización de las
membranas y a la activación de los receptores de glutamato N-metil-D-Aspartato (NMDA)
y los canales de calcio dependientes del voltaje, causando la hiperexcitación de la zona
aledaña al infarto, aumentando el gasto energético debido al intento de las células por

22
repolarizar sus membranas (Arango-Dávila et al., 2004; Nakka, Gusain, Mehta, &
Raghubir, 2008)
La sobrecarga intracelular de calcio activa quinasas y proteasas ejecutoras como las
caspasas que finalmente van a promover la muerte celular; a su vez, falla el metabolismo
energético de la mitocondria, promoviendo el desequilibrio iónico, lo que produce
acidosis, resultando en la activación de los canales iónicos sensibles al pH ácido,
perturbando la homeostasis de los iones sodio y calcio (Di Filippo et al., 2008). Además,
los niveles de ROS entre los que se incluyen el anión superóxido (O2-), el peróxido de
hidrogeno (H2O2), el radical hidroxilo (OH-), así como las especies reactivas de nitrógeno,
el monóxido de nitrógeno (NO-) y el peroxinitrito (ONOO-), se elevan especialmente
durante la fase de reperfusión (Culmsee & Krieglstein, 2005; Nakka et al., 2008).
La producción de estas especies reactivas de oxígeno y nitrógeno, se encuentran
mediadas por diferentes mecanismos fisiopatológicos entre los que se destacan: 1) la
activación del metabolismo del ácido araquidónico y la enzima óxido nítrico sintasa
neuronal (nNOS), 2) el aporte de los neutrófilos que se infiltran desde el tejido sanguíneo
y 3) la síntesis y activación de la óxido nítrico sintasa inducible (iNOS) y la ciclooxigenasa
2 (COX 2) (Arango Dávila et al., 2004).
De otra parte, el daño isquémico a nivel celular induce diferentes subrutinas de muerte
celular entre la apoptosis y la necrosis; la muerte neuronal isquémica, podría estar
regulada por los miembros de la familia Bcl-2, que promueven o previenen la formación
de un poro de transición en la membrana externa de la mitocondria (Bax, Bad, Bid y Bcl-
2 y Bcl-x), activación de caspasas y factores de transcripción como el factor nuclear
kappa B (NF-kB) o el factor supresor de tumores p53 (Culmsee & Krieglstein, 2005).
Otros eventos celulares que están involucrados en el daño posisquémico incluyen la
activación de la población glial y la invasión de leucocitos que contribuyen a incrementar
los niveles de citoquinas, producción excesiva de NO y otras respuestas inflamatorias.
Igualmente, se ha encontrado la participación del daño vascular en los mecanismos

23
patológicos de la isquemia, sugiriendo una comunicación entre células endoteliales
afectadas y las neuronas del tejido isquémico (Culmsee & Krieglstein, 2005)
Conjuntamente a los eventos antes mencionados, la isquemia cerebral activa una serie
de respuestas moleculares que pueden ser una consecuencia de la reacción inmediata
de las neuronas al daño, de la determinación del destino de la neurona afectada y de la
coordinación de mecanismos de reparación de las neuronas y tejidos. Entre estos
mecanismos se encuentran la activación de genes de expresión rápida (IEG), inducción
de proteínas de choque térmico (HSP), de genes relacionados con citoquinas
proinflamatorias y moléculas de adhesión celular, elevación de la síntesis de enzimas
como la iNOS y COX 2, transcripción de genes relacionados con muerte celular
programada y genes relacionados con factores de crecimiento (Arango-Dávila et al.,
2004).
A continuación se ampliaran algunos de los mecanismos involucrados en la muerte
celular como resultado de una isquemia cerebral.
2.1.2.1 Excitotoxicidad y Acidosis. El daño isquémico conduce a la disminución de las
reservas de energía intracelular, produciendo alteración en el funcionamiento de la célula
mediante la interrupción de los procesos dependientes de ATP, predominantemente la
bomba de sodio/potasio ATPasa (Na+/K+ ATPasa), lo que a su vez induce la alteración
del gradiente iónico a lo largo de la membrana. Esto causa un incremento de K+
extracelular y una entrada de Na+, Cl- y Ca++ en las células. El incremento en la
concentración de K+ extracelular induce despolarización e inversión de los
transportadores de aminoácidos (Jordán et al., 2007; Nakka et al., 2008).
En estas condiciones, los receptores ionotrópicos de glutamato y los canales iónicos
dependientes de voltaje son activados llevando a la elevación del Ca++ citosólico. La
liberación masiva de aminoácidos excitatorios, particularmente el glutamato, podría
deberse a la inversión de los transportadores de glutamato y a la exocitosis dependiente
de Ca++ (Jordán et al., 2007; Nakka et al., 2008).

24
Igualmente, la acumulación citotóxica del calcio ha sido establecida como un paso clave
en la muerte celular neuronal isquémica, lo cual podría establecer un vínculo entre los
procesos necróticos y apoptóticos. La sobrecarga de calcio, desencadena la muerte de
la célula mediante la activación de proteasas, lipasas y DNAsas, cambiando el balance
de la muerte neuronal apoptótica a necrótica debido al agotamiento de las reservas
energéticas o a la amplificación de otras vías (Culmsee & Krieglstein, 2005; Mehta,
Manhas, & Raghubir, 2007; Trump & Berezesky, 1995).
De otra parte, se ha encontrado que la sobrecarga de calcio intracelular es mediada
principalmente a través de la estimulación de los receptores de glutamato NMDA, ácido
α-amino-3-hidroxi-5-metil-4-isoxazolpropiónico (AMPA)/kainato permeables al calcio,
receptores metabotrópicos y canales de calcio dependientes de voltaje. Además, los
receptores ionotrópicos de glutamato promueven una excesiva entrada de Na+
provocando tumefacción y edema celular (Culmsee & Krieglstein, 2005; Harukuni &
Bhardwaj, 2006; Nakka et al., 2008)
La acumulación fatal de calcio en las células apoptóticas también puede ser el resultado
de la disociación de las bombas iónicas. Existe evidencia de la disociación de la bomba
de calcio de la membrana plasmática (PMCA) y el intercambiador Na+/Ca++ en neuronas
apoptóticas, los cuales se encargan de rectificar y remover grandes cantidades de calcio
acumuladas en el citosol (Culmsee & Krieglstein, 2005; Nakka et al., 2008).
La acidosis es otra característica de la isquemia cerebral, en donde el pH ácido juega un
papel importante en el proceso patológico; sin embargo, el mecanismo por el cual la
acidosis conduce al daño isquémico no está totalmente claro. Se ha demostrado la
activación de canales de calcio dependientes de pH ácido (ASIC), en particular la
subunidad ASIC1a, a la cual se le ha atribuido ser responsable de la acidosis,
independiente del receptor del glutamato en el daño isquémico (Culmsee & Krieglstein,
2005; Nakka et al., 2008).

25
Por otro lado, en la isquemia cerebral, las caspasas activadas también podrían
eventualmente disociar estas bombas iónicas, lo que podría resultar en la alteración de
la homeostasis del calcio cambiando finalmente de señalización apoptótica a necrótica
(Culmsee & Krieglstein, 2005; Jordán et al., 2007; Nakka et al., 2008; Trump &
Berezesky, 1995).
2.1.2.2 Estrés oxidativo. La isquemia cerebral y la reperfusión son los principales eventos
responsables del estrés oxidativo debido a la generación de radicales libres. Los
radicales libres pueden causar daño en las membranas a través de la peroxidación de
los fosfolípidos. También, pueden dañar componentes celulares fundamentales como
son los ácidos nucleicos, llevando a la muerte celular por la vía apoptótica o necrótica
(Heo, Han, & Lee, 2005; Nakka et al., 2008)
Entre las ROS que son particularmente responsables del estrés oxidativo se encuentran
el óxido nítrico (NO) y el anión superóxido (O2-); estos dos radicales libres, reaccionan
con otros para formar el peroxinitrito (ONOO-), el cual posee un gran poder oxidante. La
sobreproducción de NO inducida por la isquemia, es en parte, causada por el incremento
del Ca++ intracelular mediado por glutamato, resultando en una sobre-regulación de la
óxido nítrico sintasa inducible (iNOS) dependiente de calmodulina (Mehta et al., 2007;
Nakka et al., 2008)
La isquemia cerebral ocasiona aumento de la actividad de NOS-1/nNOS (dependiente
de Ca++) en las neuronas y posiblemente en la glía; además, se cree que es un suceso
secundario a la alteración de la recaptación de glutamato en las sinapsis y la activación
de los receptores NMDA, resultando en la elevación del Ca++ intracelular, mientras que
el incremento de la actividad de la NOS-3 en las células endoteliales y la NOS-2 en el
tejido isquémico probablemente se derive de los neutrófilos y macrófagos infiltrados, así
como de los astrocitos y la microglía (Nakka et al., 2008)
Por otro lado, el NO juega un doble papel en la patología isquémica, posee un efecto
benéfico actuando como un potente vasodilatador y un efecto citotóxico debido a que

26
inhibe enzimas importantes en el metabolismo oxidativo como son el complejo I y II de la
cadena transportadora de electrones mitocondrial (Harukuni & Bhardwaj, 2006)
De otra parte, las ROS pueden ocasionar la peroxidación de los lípidos de la membrana,
generando aldehídos tóxicos como el 4-hidroxinonenal (marcador de daño oxidativo) que
afecta la función de las ATPasas iónicas y los transportadores de glucosa y glutamato,
amplificando de esta manera la alteración de la homeostasis del calcio. Además del daño
a los lípidos y a las proteínas de membrana, las ROS pueden ocasionar daños en el
ADN, lo que activa señales apoptóticas a través del factor supresor de tumores p53 y la
poli (ADP-ribosa) polimerasa (PARP) (Culmsee & Krieglstein, 2005; Mehta et al., 2007;
Nakka et al., 2008).
El daño oxidativo del ADN consiste en lesiones químicas altamente específicas como
bases modificadas (8-hidroxil-2’-deoxiguanosina) por radicales hidroxilo que rompen la
hebra de ADN. Una característica prominente del daño cerebral isquémico, que ha sido
detectado en la zona del infarto en desarrollo minutos después del insulto y antes de la
fragmentación internucleosomal, es representativo de las etapas tardías de la muerte
celular programada (Culmsee & Krieglstein, 2005; Harukuni & Bhardwaj, 2006).
Las rupturas en el ADN activan la PARP-1 que construye polímeros de adenosin difosfato
ribosa usando la nicotinamida adenina dinucleótido (NAD) como sustrato. Entre las
dianas de la PARP-1 se encuentran proteínas asociadas al ADN y enzimas como las
histonas, las topoisomerasas y la ligasa 2. Adicional a la disminución de NAD+ y ATP,
debido a la masiva utilización de NAD+ por la PARP-1, el mal funcionamiento de la
enzimas poli ADP ribosiladas podría estar involucrado en la señalización apoptótica
mediada por la PARP-1. Por otro lado, se ha identificado que la translocación del factor
inductor de la apoptosis (AIF) de la mitocondria al núcleo es un mecanismo corriente
abajo de la señalización apoptótica mediada por PARP-1 (Culmsee & Krieglstein, 2005;
Harukuni & Bhardwaj, 2006).

27
Se ha encontrado, además, sobrerregulación del factor de transcripción p53 en el tejido
cerebral isquémico, el cual podría activar la muerte celular por apoptosis mediante la
transcripción de genes proapoptóticos de la familia Bcl2 como Bax o proteínas de la
familia BH3 como PUMA y NOXA, que generan daño en la membrana mitocondrial y
posterior activación de las caspasas. La activación de PARP-1 y el factor p53 son eventos
tempranos después de una isquemia cerebral y aparentemente juegan un papel
fundamental en las fases iniciales de la muerte celular programada inducida por la
isquemia (Culmsee & Krieglstein, 2005; Nakka et al., 2008).
Otro mecanismo de producción de ROS durante la isquemia es la activación de la
fosfolipasa A2 (PLA2), que libera el ácido araquidónico a partir de los fosfolípidos. El
metabolismo oxidativo del ácido araquidónico ha sido considerado una fuente importante
de generación de ROS. Adicionalmente, las ROS pueden estimular vías de señalización
específicas como la de las quinasas activadas por mitogénos (MAPK) que
posteriormente contribuirán al daño celular (Mehta et al., 2007)
2.1.2.3 Señalización de estrés. La isquemia cerebral activa intrincadas vías de
señalización que son cruciales en la supervivencia o en el daño celular. Una de estas
cascadas involucra la vía de las MAPK que participa en la transducción de la señal desde
el ambiente extracelular hacia el núcleo y otras dianas intracelulares, mediante
fosforilaciones secuenciales. La familia de las MAPK tiene tres miembros principales: la
quinasa regulada por señales extracelulares (ERK), la p38 y la quinasa con dominio N-
terminal c-jun o proteínas quinasas activadas por estrés (JNK o SAPK). Estas proteínas
se activan por diferentes estímulos, pero sus vías efectoras confluyen en una misma
cascada de señalización (Mehta et al., 2007).
ERK es activada en respuesta a factores de crecimiento, estrés oxidativo, incremento del
Ca++ o estimulación del receptor NMDA y señales mediadas desde la sinapsis hacia el
núcleo. Se ha propuesto que la activación de ERK, media en la protección parcialmente,
mediante la activación de neurotrofinas (NT), particularmente el factor neurotrófico
derivado del cerebro (BDNF). Esta protección, esta probablemente relacionada a la

28
sobre-regulación de receptores tirosina quinasa (RTK) y contra-regulación de las
proteínas Bax/Bcl2 (Marosi & Mattson, 2014; Mehta et al., 2007; Nakka et al., 2008; Rami,
Bechmann, & Sthehle, 2008).
Las MAPK activadas por estrés, p38 y JNK funcionan principalmente como mediadores
mediante la fosforilación de enzimas intracelulares, factores de transcripción, proteínas
citosólicas involucradas en supervivencia celular, producción de citoquinas
proinflamatorias y apoptosis. Además, la activación de p38 a través del NO producido
por la nNOS seguido de la interacción del receptor de glutamato estimulado, ha sido
involucrada con la muerte celular neuronal. Existe evidencia de que la vía p38/JNK media
señales de muerte a través de proteínas pro-apoptóticas relacionadas con la mitocondria
como son Bax, Bak, DP5 y Bim (Maulik, Ashraf, Mishra, & Delivoria-papadopoulos, 2008;
Mehta et al., 2007; Nakka et al., 2008).
2.1.2.4 Daño mitocondrial. El daño mitocondrial ha sido considerado como el punto de
no retorno en la cascada de muerte celular iniciada en las neuronas después de la
isquemia. Luego de un insulto isquémico, la disfunción de la mitocondria es una de las
principales causas del agotamiento del ATP y posterior alteración de la homeostasis del
Ca++. Adicionalmente, el daño de la membrana de la mitocondria permite la liberación de
proteínas proapoptóticas como la citocromo c, Smac/DIABLO o HtrA2/Omi que activan
la vía dependiente de las caspasas (Arango Dávila et al., 2004; Culmsee & Krieglstein,
2005; Nakka et al., 2008).
Después de la isquemia, la estimulación del receptor de muerte podría activar la
caspasa-1 o la caspasa-8 (caspasas iniciadoras), las cuales activan la cascada de
muerte y finalmente la activación de caspasas ejecutoras como la caspasa-3 y caspasa-
9. Aunque, la vía de las caspasas no es la única involucrada en los procesos de muerte
celular posisquemia (Yuan & Yankner, 2000). Existen vías independientes que se activan
corriente abajo del daño mitocondrial, las cuales pueden ser activadas por la liberación
del AIF o la endonucleasa G, que inducen daño al ADN celular (Arango Dávila et al.,
2004; Culmsee & Krieglstein, 2005; Nakka et al., 2008).

29
La liberación mitocondrial de la AIF ocurre horas antes de la liberación del citocromo c y
la activación de la caspasa-3, aunque no se conoce exactamente como la AIF ejerce su
función apoptogénica, la activación de la PARP-1 ha sido identificada como clave en la
apoptosis mediada por la AIF; así mismo, se ha propuesto la participación potencial de
las calpaínas y las proteínas proapoptoticas Bax, BimEL y tBid para la liberación
mitocondrial de AIF en neuronas apoptóticas, mostrando múltiples rutas para la liberación
y translocación de la AIF (Arango-Dávila et al., 2004; Culmsee & Krieglstein, 2005; Nakka
et al., 2008).
2.1.2.5 Daño vascular. La disfunción de los mecanismos regulatorios del endotelio son
eventos claves en la patología de la isquemia cerebral, particularmente, en la
transformación hemorrágica posterior a la isquemia. La unidad neurovascular formada
por el endotelio microvascular, los astrocitos, las neuronas y la matriz extracelular es
esencial para el funcionamiento apropiado del cerebro. La interacción compleja y
dinámica entre estos componentes en la interfase sangre-vasculatura-parénquima
después de la isquemia cerebral, determina la extensión del daño neuronal (Culmsee &
Krieglstein, 2005; G.J. Del Zoppo, 2010; Mehta et al., 2007); además, el metabolismo de
las células nerviosas es influenciado por esta unidad a través de dos barreras anatómicas
y funcionales, la barrera hematoencefálica (BHE), formada por las uniones estrechas de
las células endoteliales, la lámina basal que inhiben la transmigración de las células
sanguíneas circulantes y actúan como filtro para el paso de solutos desde la sangre hacia
el parénquima cerebral. La isquemia cerebral involucra la ruptura de la BHE, alterando
las uniones estrechas e induciendo a la degradación de la lámina basal mediante la
excesiva activación enzimática de la matriz de metaloproteinasas (MMP) (Culmsee &
Krieglstein, 2005; G.J. Del Zoppo, 2010; Lakhan, Kirchgessner, & Hofer, 2009; Mehta et
al., 2007).
En la isquemia cerebral la pérdida de las proteínas de la matriz extracelular como la
laminina, el colágeno IV, la fibronectina y el perlecano ha sido atribuido a la rápida
generación de proteasas de matriz en respuesta a la isquemia. Se ha demostrado la
rápida aparición de miembros de cuatro familias de proteasas en el territorio isquémico

30
luego de la oclusión de la arteria cerebral media (en primates no humanos), en los
microvasos y cerca de las neuronas: 1)Las MMP entre las que se encuentran la MMP-9
y la MMP-2, 2) La L-catepsina, 3) La heparinasa y 4) La uroquinasa (u-PA); involucradas
con la degradación de la lámina basal, resultando en la pérdida del contacto entre las
células endoteliales, los astrocitos y la neuronas (Culmsee & Krieglstein, 2005; G.J. Del
Zoppo, 2010; Lakhan et al., 2009; Mehta et al., 2007).
Por otro lado, los cambios en la integridad de la BHE inducen la acumulación, la adhesión
y la transmigración de leucocitos, mientras que las proteínas integrales de membrana
(por ejemplo, integrinas) modifican estructuralmente la lámina basal y la matriz
extracelular, iniciando la activación de cascadas inflamatorias que posteriormente
conllevan al daño cerebral (G.J. Del Zoppo, 2010; Mehta et al., 2007).
2.1.2.6 Inflamación. La isquemia cerebral inicia una plétora de reacciones inflamatorias
que pueden progresar durante días luego del insulto (Mehta et al., 2007). Entre los
principales mediadores de la inflamación se encuentran incluidas las citoquinas,
moléculas de adhesión, quimioquinas y leucocitos. Luego de la interrupción del flujo de
sangre hacia una zona del cerebro, las células inician una reacción inflamatoria que
estimula la infiltración de leucocitos, formación de edema, necrosis e infarto del tejido
(Culmsee & Krieglstein, 2005; Harukuni & Bhardwaj, 2006; Nakka et al., 2008).
Por otro lado, tres clases de moléculas de adhesión celular, las selectinas, las integrinas
y la superfamilia de las inmunoglubulinas intervienen en la interacción leucocito-célula
endotelial. Las selectinas se encuentran involucradas en la afinidad leucocito-endotelio,
promoviendo la marginación y el rodamiento de los leucocitos, conduciendo a la
acumulación de neutrófilos en el microvaso y posterior daño cerebral (Denes et al., 2010;
Nakka et al., 2008).
Para la migración de los leucocitos polimorfonucleares (PMN) es necesaria la expresión
de la integrina β2 CD11b/CD18 (Mac-1) y CD11a/CD18 (LFA-1) en la superficie del
leucocito. En cambio, en las células endoteliales activadas se presenta la expresión de

31
moléculas de adhesión que pertenecen a la familia de las inmunoglobulinas ICAM-1 e
ICAM-2, moléculas de adhesión endotelio-plaqueta 1 (CD3), molécula de adhesión
intercelular adresina-1 (CD146) y la molécula de adhesión intercelular vascular-1
(CD106), que actúan como ligandos para las integrinas permitiendo de esta manera el
proceso de transmigración de los leucocitos hacia el parénquima cerebral (Denes et al.,
2010; Nakka et al., 2008).
A pesar de lo anterior, para la expresión de las moléculas de adhesión celular se requiere
la activación de mecanismos intracelulares, en los cuales las citoquinas juegan un papel
importante. Citoquinas como la interleuquina-1β (IL-1), la IL-6, el factor de necrosis
tumoral α (TNF- α) o IL-10, factor de crecimiento transformante β (TGF β) y quimioquinas
como la proteína quimio-atrayente de monocitos-1 (MCP-1), proteína inflamatoria de
macrófagos-1 (MIP-1), quimioquina derivada de queratinocitos (CXC) y fractalquina
(CX3CL1), entre otros, son producidas por una variedad de tipos celulares activados
entre los que se encuentran las células endoteliales, las plaquetas, los leucocitos y los
fibroblastos (Denes et al., 2010; Lakhan et al., 2009; Nakka et al., 2008).
Por otro lado, se ha evidenciado el incremento de la expresión de IL-1 después de una
isquemia permanente o transitoria en microglía, astrocitos y neuronas. También se ha
demostrado que la IL-1 produce sobre-regulación de la selectina-E, ICAM-1, ICAM-2 y
CD106 en la superficie de las células endoteliales cerebrales, (Lakhan et al., 2009; Nakka
et al., 2008). Además, se ha comprobado que las enzimas iNOS y COX-2 se
desempeñan predominantemente como intermediarios inflamatorios y se ha indicado
que los productos de la reacción de COX-2 estimula la expresión de iNOS y la generación
citotóxica de NO (Culmsee & Krieglstein, 2005; Lucas, Rothwell, & Gibson, 2006).
Finalmente, la isquemia cerebral produce múltiples respuestas en la región afectada
entre las que se incluyen la activación de las células gliales, las cuales se activan no solo
en el sector focal y en la penumbra sino también en zonas alejadas (Arango-Dávila et al.,
2004).

32
2.2 CÉLULAS GLIALES
Las células gliales son una población celular altamente heterogénea generalmente
clasificada como astrocitos, oligodendrocitos, microglía y células NG2 glía o sinantocitos
(Franke et al., 2012).
2.2.1 Astrocitos. Los astrocitos son el tipo de células no neuronales más abundantes del
SNC y son fundamentales para el apropiado funcionamiento del SNC (Y. Chen &
Swanson, 2003; Li et al., 2008; López-Bayghen & Ortega, 2010). Los astrocitos son
clásicamente divididos en tres grandes grupos de acuerdo a su morfología y organización
espacial: 1) Astrocitos radiales, que se encuentran rodeando los ventrículos y se
caracterizan morfológicamente por poseer procesos largos no ramificados, 2) Astrocitos
protoplásmicos, distribuidos en la sustancia gris, que muestran procesos cortos
altamente ramificados y 3) Astrocitos fibrosos, localizados en la sustancia blanca, de
forma estrellada con procesos largos poco ramificados (Y. Chen & Swanson, 2003;
Franke et al., 2012; Sofroniew & Vinters, 2010).
En la corteza cerebral el cuerpo de los astrocitos ocupa dominios estrictamente
delineados dentro del neuropilo, formando múltiples contactos con estructuras
neuronales, mientras que los pies vasculares envuelven los vasos sanguíneos del SNC
(Bushong, Martone, & Ellisman, 2004; Franke et al., 2012). En el hipocampo de los
roedores, los procesos finamente ramificados que emanan de un solo astrocito contactan
probablemente varios cientos de dendritas de múltiples neuronas y se ha calculado que
posiblemente envuelven entre veinte mil a cien mil sinapsis, mientras que en la sustancia
blanca los procesos gliales terminan en los nódulos de Ranvier (Franke et al., 2012).
Las células astrogliales cumplen variadas funciones en el SNC, entre las que se
encuentran: 1) controlar el desarrollo del sistema nervioso guiando la migración de los
axones en desarrollo y de ciertos neuroblastos, 2) regular los procesos de sinaptogénesis
(Franke et al., 2012; López-Bayghen & Ortega, 2010; Sofroniew & Vinters, 2010), 3)
mantener la homeostasis del medio extracelular mediante la captación de transmisores

33
liberados durante la actividad sináptica como es el caso del glutamato, la alanina, la
glicina y el ácido gama amino butírico (GABA); 4) la amortiguación de iones como el K+
y el H+, 5) el transporte de agua, 6) la liberación de factores neurotróficos, 7) el transporte
de metabolitos y desechos metabólicos, 8) la formación de la barrera hematoencefálica
y 9) la regulación del flujo de sangre en los microvasos (Abbott, Rönnbäck, & Hansson,
2006; Y. Chen & Swanson, 2003; López-Bayghen & Ortega, 2010; Pascual, González-
Llanos, Cerdán, & Carceller, F. Roda, 2000; Sofroniew & Vinters, 2010; Takano,
Oberheim, Cotrina, & Nedergaard, 2009).
Los astrocitos pueden cumplir con esta variedad de funciones gracias a que contienen
una extensa dotación enzimática, elevada cantidad de mitocondrias y gran variedad de
canales iónicos y transportadores de diversos metabolitos (López Bayghen & Ortega,
2010; Pascual et al., 2000).
Durante la actividad sináptica los neurotransmisores, principalmente el glutamato, es
captado por los astrocitos y convertido en su precursor por acción enzimática, como se
explica a continuación. El glutamato (Glu) liberado durante la neurotransmisión es
tomado por los astrocitos vecinos a través de los transportadores de aminoácidos
excitatorios (TAAE). El Glu tomado por el astrocito es convertido a glutamina (Gln) por la
glutamina sintetasa, la cual es abundante en los astrocitos y ausente en las neuronas.
La Gln es liberada por los astrocitos vía el sistema de transportadores N (SN1) y tomada
por las neuronas por medio del transportador de aminoácidos acoplado al sodio (SAT),
en las neuronas la Gln es desaminada a Glu por medio de la glutaminasa mitocondrial.
El Glu neuronal también puede ser producido a partir del α-cetoglutarato (α-KG), el cual
proviene en parte de un segundo sistema de transporte astrocito-neurona. En ese caso,
los astrocitos toman aminoácidos ramificados como la leucina (Leu) y transfieren el grupo
amino a α-KG por la transaminasa de aminoácidos de cadena ramificada (BCAA)
produciendo α- cetoisocaproato (KIC) y Glu, a su vez, KIC puede ser transferido a las
neuronas para la formación de nuevas moléculas de α-KG, como se muestra en la figura
2 (Y. Chen & Swanson, 2003; Franke et al., 2012; Pascual et al., 2000; Sofroniew &
Vinters, 2010)

34
Figura 2. Metabolismo del glutamato neuronal soportado por los astrocitos.
Fuente: Modificado de Chen y Swanson, 2003
Por otro lado, los astrocitos modulan su comportamiento y el de otras células del SNC
mediante la liberación de moléculas transmisoras como el ATP, la prostaglandina E2
(PGE2), el TNF-α, glutamato, d-serina y factores de crecimiento como el factor de
crecimiento transformante beta (TGF-β), el factor neurotrófico derivado de la glía (GDNF),
el factor de crecimiento fibroblástico beta (FGF-β), que juegan un papel en la señalización
parácrina entre astrocitos y neuronas, células endoteliales y microglía (Abbott et al.,
2006; Franke et al., 2012; Takano et al., 2009). Así mismo, los astrocitos son
consideradas células más resistentes a la hipoxia y a la disminución de glucosa que las
neuronas debido a que son células capaces de almacenar glicógeno y utilizar la glucosa
en condiciones anaeróbicas. Aunque, la glicólisis anaerobia permite la supervivencia del
astrocito, la acumulación de ácido durante la hipoxia puede tener efectos deletéreos en
su metabolismo a mediano y largo plazo (Y. Chen & Swanson, 2003; Hulse, Winterfield,
Kunkler, & Kraig, 2001).
De otra parte, el astrocito puede regular el pH extracelular mediante la utilización de la
enzima anhidrasa carbónica y el intercambio de iones empleando contratransportadores
H+/Na+ permitiendo intercambiar el exceso de H+ intracelular por Na+ extracelular,
ocasionando entrada de agua y el aumento del volumen celular; este exceso de agua es

35
posteriormente regulado mediante la apertura de canales iónicos y acuaporinas (Y. Chen
& Swanson, 2003; Pascual et al., 2000).
En cuanto a las técnicas inmuno-histoquímicas, estas han permitido detectar marcadores
específicos a nivel celular que se utilizan como herramientas esenciales en la
identificación y caracterización celular. La expresión de la proteína acídica glial fibrilar
(GFAP) ha llegado a ser considerada un marcador sensible y confiable que revela
principalmente el cuerpo estrellado de la mayoría de los astrocitos reactivos (Bushong et
al., 2004; Sofroniew & Vinters, 2010). GFAP, es una proteína estructural que pertenece
a los filamentos intermedios del citoesqueleto y es requerida por los astrocitos para su
normal funcionamiento y para el proceso conocido como astrogliosis y formación de la
cicatriz glial en un estado patológico (Y. Chen & Swanson, 2003; Sofroniew & Vinters,
2010).
En consecuencia, el desempeño de los astrocitos puede influenciar de manera crítica la
supervivencia neuronal, el crecimiento de las neuritas y otros procesos que contribuyen
a la recuperación del cerebro después de la isquemia cerebral (Y. Chen & Swanson,
2003; Takano et al., 2009).
2.2.1.2 Fisiopatología de los astrocitos en la isquemia cerebral. La gliosis reactiva es una
de las respuestas de los astrocitos a la isquemia cerebral, siendo la sobrerregulación de
la expresión de la proteína GFAP, una de las características principales (Franke et al.,
2012; Li et al., 2008; Stoll, Jander, & Schroeter, 1998). Además, algunos autores han
descrito que el incremento de los filamentos intermedios, predominantemente GFAP y
bajo ciertas circunstancias vimentina, indican el paso de un estado quiescente a uno
reactivo (Li et al., 2008; Stoll et al., 1998). Junto con el aumento de GFAP se ha
observado que los astrocitos experimentan hipertrofia e hiperplasia considerándose
entre las propiedades más llamativas de la respuesta de estas células a la isquemia
cerebral (Stoll et al., 1998).

36
La astrogliosis por su parte, se ha definido como un espectro de cambios moleculares,
celulares y funcionales de los astrocitos en respuesta a todas las formas de daño del
SNC; los cambios sufridos por los astrocitos reactivos varían dependiendo de la
gravedad del insulto generando un continuo de alteraciones progresivas en la expresión
molecular, la hipertrofia celular y en casos severos proliferación y formación de cicatriz
glial (Sofroniew & Vinters, 2010).
Estos cambios son regulados de una manera específica por moléculas de señalización
intra e intercelular, que pueden alterar potencialmente la actividad del astrocito hacia la
pérdida o ganancia de funciones, con un impacto benéfico o perjudicial en las células
neuronales y no neuronales que los rodean (Sofroniew & Vinters, 2010). Además se han
descrito variados tipos de astrogliosis, entre los que se encuentran: 1) La astrogliosis
anisomórfica, la cual ha sido observada como una encapsulación del tejido dañado,
caracterizada por la desaparición de la organización de los dominios astrogliales y la
formación de un plexo denso o pared; 2) La astrogliosis isomórfica, representada por la
hipertrofia astroglial con la preservación de la organización de los dominios gliales que a
menudo es reversible y benéfica para la regeneración después del insulto (Franke et al.,
2012).
De otra parte, experimentos in vivo e in vitro han corroborado la importancia de la
señalización purinérgica en la respuesta reactiva de los astrocitos a una injuria debido a
que se ha demostrado que los purinoreceptores regulan la remodelación morfológica, la
proliferación y la sobrerregulación de la síntesis de GFAP, como también los procesos
de quimiotaxia y quimioquinesis en los astrocitos activados (Franke et al., 2012).
Dentro de las primeras horas después de la injuria, las células neuronales y gliales
experimentan la muerte celular en el foco, desencadenando el reclutamiento de células
inmunoefectoras como la microglía y los monocitos periféricos. Por su lado, los astrocitos
migran desde el parénquima adyacente no dañado hacia la periferia del foco para iniciar
el proceso de reparación mediante la secreción de neurotrofinas y la provisión de un
sustrato que soporte el crecimiento axonal (Franke et al., 2012; Stoll et al., 1998).

37
Además, los astrocitos exhiben variabilidad local y regional que es regulada por un gran
número de moléculas de señalización intracelular y extracelular. Entre los ligandos
intercelulares que podrían iniciar la astrogliosis o regular aspectos específicos de ella, se
encuentran los factores de crecimiento y las citoquinas como las IL-1, IL-6 e IL-10, el
factor neurotrófico ciliar (CNTF), el TNF-α, el interferón γ (INF γ), el TGFβ, el factor de
crecimiento fibroblástico 2 (FGF2), mediadores de la inmunidad innata,
neurotransmisores, ROS y NO, entre otros (Franke et al., 2012; Sofroniew & Vinters,
2010).
Estos mediadores pueden ser liberados por todas las células del sistema nervioso,
incluidas neuronas, microglía, oligodendrocitos, pericitos, células endoteliales y otros
astrocitos en respuesta a todas las formas de insulto del SNC (Sofroniew & Vinters,
2010). También se ha sugerido que los astrocitos bajo condiciones patológicas, pueden
cambiar su fenotipo de una célula de soporte metabólico a una célula inmunocompetente
capaz de inducir inflamación mediante la producción de una variedad de factores
proinflamatorios (G J Del Zoppo, 2009; Franke et al., 2012; R. Y. Kim et al., 2014).
2.2.3 Microglía. Las células microgliales comprenden alrededor del 20% de toda la
población glial en el cerebro. Estas células de origen mesodérmico, del linaje
monocito/macrófago, quedaron confinadas en el SNC durante el desarrollo embrionario
y se especializaron en las típicas células ramificadas conocidas como microglía residente
(Gehrmann et al., 1995; Gomes-Leal, 2012; Harry & Kraft, 2012). La microglía es
considerada la célula inmune residente del cerebro, que se encuentra involucrada en
procesos regulatorios críticos para el desarrollo, el mantenimiento del ambiente neuronal
y la reparación. Estas células son morfológicamente distintas de las neuronas y los
astrocitos y se han descrito varias clasificaciones. Según su forma se han catalogado
como formas ramificadas, formas intermedias y las formas ameboideas; en cuanto a su
función se han descrito como quiescentes/vigilantes, reactivas/activadas y
ameboideas/fagocíticas (Harry & Kraft, 2012; Kitamura, Yanagisawa, Takata, &
Taniguchi, 2009).

38
La microglía es una célula activa que constantemente está inspeccionando el
parénquima cerebral a su alrededor mediante movimientos dinámicos de sus procesos,
mientras que su soma se mantiene estático, lo que les permite revisar su microambiente.
La extensión de esta vigilancia depende de la distribución de la microglía, el nivel de
ramificación de cada célula y la tasa de remodelación de sus procesos, facultándolas
para responder rápidamente a los cambios ambientales, entre los que se incluyen
alteraciones en la actividad cerebral o perturbaciones en el parénquima cerebral. La
microglía a menudo provee la primera línea de defensa contra los microorganismos y a
través de una íntima comunicación con las neuronas, puede ser la primera en detectar
cambios críticos en la actividad neuronal (Harry & Kraft, 2012; Kitamura et al., 2009).
En el cerebro en desarrollo, la microglía se encarga de fagocitar el excedente celular que
experimenta apoptosis, pero también están activamente involucradas en la
determinación del destino celular (eliminación/supervivencia) de las neuronas, además
son promotoras de la migración, el crecimiento axonal y la diferenciación terminal de
diferentes tipos de neuronas a través de la liberación de componentes de matriz
extracelular, factores solubles y el contacto directo célula-célula (Vilhardt, 2005)
En el cerebro adulto, bajo condiciones fisiológicas la función fagocítica de la microglía se
mantiene regulada asegurándose que las células microgliales permanezcan en un
estado vigilante no activado, las neuronas son unas de las células encargadas de este
control por medio del contacto directo o la liberación de neurotransmisores, péptidos y/o
factores de crecimiento (Gomes, 2012; Vilhardt, 2005)
Perturbaciones en la homeostasis del SNC maduro, inician una rápida transformación
fenotípica de la microglía ramificada, hacia un fenotipo activado, que posteriormente
podría evolucionar a fagocítico, como se muestra en la figura 3. La definición de la
activación microglial inicialmente se basó en el criterio morfológico y a menudo era
descrito como un proceso estereotípico y gradual, sin embargo, en la actualidad la
activación de la microglía se considera un proceso adaptativo y variable (Biber,
Neumann, Inoue, & Boddeke, 2007).

39
Figura 3. Transformación fenotípica de la microglía ramificada a fagocítica.
La flecha azul indica el incremento en la activación. 1. La microglía posee procesos
ramificados con un soma pequeño. 2-5. Incremento del tamaño del soma y retracción de
los procesos. 6. Célula con fenotipo ameboideo y transformación a fagocítica.
Fuente: Modificado de Jonas et al, 2012.
La microglía al detectar ligeros cambios en el microambiente, extiende sus procesos
hacia el lugar de daño y elimina los detritos celulares con sus procesos mediante
actividad fagocítica, protegiendo su microambiente. Cuando se presentan señales
patológicas severas de tipo endógeno (disfunción neuronal o muerte, agregación
anormal de proteínas, e interacción entre células inmunes) o exógeno (infecciones), la
sobrerregulación de múltiples funciones efectoras y la expresión de diversos mediadores
moleculares permite a la microglía responder específica y apropiadamente hacia el
insulto para la reparación del tejido, soporte neurotrófico, inducción de inflamación o
activación de linfocitos (Vilhardt, 2005).
La adquisición gradual de estas funciones, se produce paralelamente a la transformación
morfológica y a cambios discretos en la expresión génica. La microglía activada,
morfológicamente engrosa y acorta sus procesos en respuesta a señales tales como
neurotransmisores, citoquinas, quimioquinas, factores de crecimiento y componentes de
los tejidos dañados. Así mismo, la microglía activa/reactiva produce citoquinas
proinflamatorias como prostaglandinas, factor de necrosis tumoral α (TNF-α),

40
interleuquina 1-β (IL-1 β), e IL-6, como también, radicales libres (ROS e intermediaros
nitrogenados), que se piensan contribuyen a la muerte neuronal (Kitamura et al., 2009;
Vilhardt, 2005).
Por otro lado, la microglía reactiva igualmente libera factores neurotróficos y de
crecimiento, como el factor neurotrófico derivado de la glía (GDNF, por sus siglas en
inglés), factor de crecimiento tipo insulina (IGF, por sus siglas en inglés), factor de
crecimiento de hepatocitos (HGF, por sus siglas en inglés), neurotrofinas (NTs, por sus
siglas en inglés) y el factor de crecimiento derivado de plaquetas (PDGF, por sus siglas
en inglés) y elimina las células muertas o en proceso de muerte por fagocitosis, ayudando
probablemente a procesos neuroprotectores (Kettenmann, Kirchhoff, & Verkhratsky,
2013; Kitamura et al., 2009; Vilhardt, 2005). Junto con los cambios fenotípicos, la
activación de la microglía también involucra proliferación y reclutamiento a la zona de
daño (Gehrmann et al., 1995).
Hasta hace poco tiempo, las neuronas se consideraban víctimas de la microglía reactiva
y con baja influencia sobre su actividad, pero hallazgos como los que se describen a
continuación han objetado esta apreciación: 1) las neuronas intercambian información
con las células gliales, 2) el daño neuronal inicia el proceso temprano de activación de la
microglía, 3) se han encontrado variados efectos de las neuronas o los sobrenadantes
neuronales en la función de la microglía en cultivos in vitro, 4) las neuronas son células
altamente activas y vulnerables que requieren soporte microglial y 5) el daño de un nervio
periférico activa específicamente la microglía del SNC en el correspondiente sitio de
inervación del nervio lesionado, indicando una comunicación directa entre las neuronas
dañadas y la microglía (Biber et al., 2007).
Adicionalmente, se han descrito dos tipos de señal que controlan el comportamiento
microglial en respuesta a un daño; la primera, correspondiente a la atracción de la
microglía al sitio de daño y la segunda, que permite el reconocimiento de la microglía por
su diana y la activación de la fagocitosis. También se han detallado otros estímulos
clasificados como señales “on”, que incluyen factores que aparecen o se incrementan en

41
el contexto patológico y señales “off”, que comprenden factores que desaparecen o
disminuyen durante el proceso patológico, estas señales “off” se encuentran
constitutivamente en el microambiente de las neuronas sanas y la pérdida de estas
señales genera cambios en la función microglial, por el contrario, las señales “on” son
producidas a demanda para iniciar un definido programa de activación microglial (pro o
antiinflamatorio) (Biber et al., 2007; Kettenmann et al., 2013).
A continuación se ampliara acerca de las moléculas que sirven como señales para
controlar la actividad microglial.
2.2.2.1 Señalización “Off”. Existe evidencia que la función de la microglía en diferentes
enfermedades del cerebro está controlada por la señales “off”, debido a que la remoción
experimental de estas señales han producido sobre-activación de la microglía (Biber et
al., 2007).
Se ha encontrado que miembros de la superfamilia de las inmunoglobulinas son
reguladores de las funciones de las células mieloides. Las moléculas mejor
caracterizadas de esta superfamilia en el SNC son CD200 y CD47, las cuales se
expresan constitutivamente en la membrana externa de las neuronas. El receptor para
CD200 en el cerebro se ha encontrado principalmente en la microglía (conocido como
OX-42 en la rata) y experimentalmente ha sido demostrado que el bloqueo del receptor
CD200r, en encefalomielitis autoinmune experimental aumenta los signos clínicos de la
enfermedad e intensifica la actividad de la microglía, así mismo la actividad de este
receptor ha sido relacionada con el control de la liberación del TNF- α (Biber et al., 2007;
Harry & Kraft, 2012; Vilhardt, 2005).
Otra molécula expresada constitutivamente por las neuronas sanas es el ligando de
quiomioquinas CX3C -1 (CX3CL1), también conocido como fractalkina o neurotactina,
mientras que el correspondiente receptor se encuentra en la microglía, los hallazgos
experimentales tanto in vivo como in vitro sugieren que la señalización CX3CL1-CX3CR1
en la comunicación neurona-microglía juega un papel importante, ya que la exposición

42
del cocultivo neurona-microglía al ligando CX3CL1 reduce la muerte neuronal
inflamatoria. Además la deficiencia del receptor CX3R1 in vivo ha sido asociado con
elevada actividad microglial acompañada de la muerte neuronal, indicando que la
actividad neurotóxica de la microglía es inhibida por la señalización CX3CL1-CX3CR1
(Biber et al., 2007; Harry & Kraft, 2012).
Así mismo, la microglía posee receptores para los neurotransmisores glutamato, GABA,
noradrenalina, purinas y dopamina, que pueden cambiar las propiedades
electrofisiológicas de su membrana. En cultivos celulares, se ha observado que el
tratamiento con neurotransmisores, exceptuando el glutamato, inhibe la liberación de
factores proinflamatorios (por ejemplo, ON, IL-1β, TNF-α), inducida por lipopolisacáridos,
aunque, esta inhibición al parecer, es dependiente del tipo de neurotransmisor, por lo
cual se plantea que la microglía vigila la actividad de la neurona basada en los niveles
del neurotransmisor local y esta señal local inhibe la liberación de factores
proinflamatorios (Biber et al., 2007; Gomes Leal, 2012; Harry & Kraft, 2012).
2.2.2.2 Señalización “On”. Las señales “on” se presentan en neuronas que están en
riesgo de daño o dañadas y pueden iniciar funciones neuroprotectoras o neurotóxicas en
la microglía, una de estas señales “on” son las purinas. Purinas como el ATP y el UTP,
se fugan o se liberan desde las neuronas ya sea por daño o sobreactivación y
posteriormente van a estimular sus correspondientes receptores (P2X4, P2X7, P2Y2,
P2Y6 y P2Y12), en la microglía (Biber et al., 2007).
Así mismo, se ha demostrado que la estimulación del receptor purinérgico P2Y12, afecta
la extensión de los procesos de la microglía y es esencial para dirigirla hacia el sitio de
injuria (quimiotaxia), la expresión de este receptor se ha encontrado principalmente en la
microglía ramificada y es regulado durante la activación de la microglía, indicando que
este receptor detecta la liberación de purinas en etapas tempranas del daño. Sin
embargo, el receptor P2Y6 no se ha encontrado relacionado a cambios morfológicos en
la microglía, pero, si con la iniciación de la actividad fagocítica (Biber et al., 2007; Gomes-
Leal, 2012; Lecca et al., 2012).

43
Por otro lado, se ha detectado microglía activada alrededor de las neuronas a las pocas
horas de una injuria, sugiriendo que las neuronas emiten señales atrayentes
(quimioquinas). Entre las quimioquinas que se han caracterizado, se encuentran
CX3CL1, CCL21 y CXCL10, las cuales han sido capaces de inducir la migración de la
microglía en condiciones in vitro; además CX3CL1 se ha encontrado relacionado con la
remoción de espinas dendríticas durante el desarrollo del sistema nervioso (Biber et al.,
2007; Harry & Kraft, 2012; Kettenmann et al., 2013).
El glutamato es otra señal “on” y ha sido demostrado que la microglía expresa una
variedad de receptores para este neurotransmisor, como los tipo AMPA (GluR1- GluR4),
kainato, y miembros de los tres grupos de receptores metabotrópicos (mGluRs). También
se conoce que la activación de varios receptores inicia la liberación microglial de TNF-α
que junto con el ligando Fas, derivado de la microglía, conducen a neurotoxicidad. Sin
embargo, experimentalmente se ha demostrado que la activación del receptor GluR2 se
encuentra asociado a neuroprotección mediante la producción microglial del factor
neurotrófico derivado del cerebro (BDNF), y que los receptores mGlu del grupo III
reducen la producción de neurotoxinas microgliales, cambiando su actividad hacia un
fenotipo más neurotrófico, indicando que el glutamato podría servir como un mecanismo
auto-limitante de la neurotoxicidad de la microglía (Biber et al., 2007; Polazzi & Monti,
2010).
De otra parte, las metaloproteinasas de matriz (MMP), son enzimas proteolíticas que
degradan macromoléculas extracelulares y están involucradas en la remodelación del
tejido, la migración celular, la reparación, la angiogénesis y en varias condiciones
neuropatológicas, existe evidencia de la liberación de MMP-3 de líneas celulares
neuronales en proceso de apoptosis y en cultivos de neuronas mesencefálicas
apoptoticas, adicionalmente, se ha demostrado que el dominio catalítico de la MMP-3
recombinante media la liberación de TNF- α, IL-6 e IL-1β en sobrenadantes de cultivos
microgliales, por lo que MMP-3 podría ser considerada una señal neuroglial apoptotica
directa en el proceso de neurodegeneración (Biber et al., 2007).

44
Otra vía de señalización involucrada en la activación microglial, es la integrada por el
receptor TREM2 y la molécula de señalización DAP12, este receptor se encuentra en un
subgrupo de la microglía y ha sido implicado en la restricción del fenotipo pro-inflamatorio
de los macrófagos y la estimulación de las moléculas asociadas con la presentación de
antígenos a los linfocitos T, además, se ha demostrado experimentalmente que la
estimulación de TREM2 induce la fosforilación de DAP12 e incrementa la actividad
fagocítica de la microglía in vitro, así mismo en modelos knockdown de este receptor se
ha visto inhibida la fagocitosis de neuronas apoptóticas y el incremento de la
transcripción de genes de los mediadores proinflamatorios TNF-α y óxido nítrico
sintetasa-2. Entonces, la posible deficiencia de TREM2 resulta en una remoción
disminuida de neuronas apoptóticas y genera la producción de citoquinas
proinflamatorias, indicando que la señalización TREM2-DAP12 es responsable por la
homeostasis del SNC mediante el apagado de la actividad inflamatoria microglial (Biber
et al., 2007; Harry & Kraft, 2012)
Finalmente, la señalización on y off ha sido definida de acuerdo a su mecanismo de
acción, no por su resultado, teóricamente, la actividad fagocítica de la microglía puede
ser iniciada por la ausencia de señales “off” o la presencia de señales “on”. La microglía
fagocítica puede tener efectos benéficos por la remoción de células muertas, estructuras
no funcionales y desechos celulares tóxicos. Pero, también hay evidencias que sugieren
que la sobreactivación microglial puede ser extremadamente perjudicial luego de un
desorden neural agudo como el infarto cerebral, encontrándose que la inhibición de la
activación microglial reduce la muerte neuronal en la zona hipocampal y la corteza en
modelos de isquemia cerebral, mostrando un papel dual de la microglía en enfermedades
del SNC (Biber et al., 2007; Gomes-Leal, 2012).

45
2.2.3 Células progenitoras NG2. El condroitin sulfato proteoglicano NG2 es un tipo de
proteína transmembranal expresada por un rango de células dentro y fuera del SNC de
los mamíferos. Las células que expresan el proteoglicano NG2 representan entre el 5%
y 10% de las células gliales y se encuentran uniformemente distribuidas tanto en la
sustancia gris como en la blanca del SNC en desarrollo y adulto, algunas de estas células
proliferan aún en el cerebro adulto, implicando un reemplazo continuo de esta población
(Sakry et al., 2011; Trotter, Karram, & Nishiyama, 2010)
Las células NG2 se han descrito clásicamente como células precursoras de
oligodendrocitos (OPC, por sus siglas en ingles), desde que ellas usualmente
coexpresan marcadores de OPC como el receptor α del factor de crecimiento derivado
de plaquetas (PDGFR- α) y O4, dando origen a la mayoría de los oligodendrocitos en el
encéfalo, constituyendo un reservorio de células, que ayudarían a reemplazar la mielina
dañada. En la última década, estas células han generado un gran interés debido a que
ellas muestran una combinación de características inesperadas en las OPCs, entre las
se incluyen: compleja morfología estrellada, tendencia a asociarse íntimamente con
cuerpos neuronales y dendritas y habilidad latente para producir astrocitos y neuronas
en un contexto patológico (Gallo, Mangin, Kukley, & Dietrich, 2008; Maldonado et al.,
2011; Richardson et al., 2011).
En patologías agudas del SNC, como la isquemia global y la desmielinización, los
polidendrocitos sobreexpresan el proteoglicano NG2 e incrementan significantemente su
tasa de proliferación y aparentemente los cambios fisiopatológicos en el sitio de la lesión
podrían habilitar a los polidendrocitos para desplegar una gran plasticidad de linajes
celulares y de esta manera contribuir a la reparación y a la regeneración (Honsa et al.,
2012; S. H. Kang, Furukaya, Yang, Rothstein, & Bergles, 2010)
2.3 MODELOS DE ISQUEMIA CEREBRAL
Los modelos animales son herramientas que han ayudado a mejorar el entendimiento de
los mecanismos subyacentes al daño producido por la isquemia cerebral, así como al

46
desarrollo de aproximaciones terapéuticas (Bacigaluppi, Comi, & Hermann, 2010a;
Casals et al., 2011; Liu & McCullough, 2011; Turner, Jickling, & Sharp, 2011)
Múltiples modelos han sido específicamente diseñados para abordar factores de riesgo
concretos como ateroesclerosis, hipercolesterolemia, hipertensión arterial, obesidad,
envejecimiento, para determinar procesos de reparación, así como, probar estrategias
neuroprotectoras y de recanalización (Bacigaluppi, Comi, & Hermann, 2010b). En la
actualidad, se encuentran disponibles una variedad de modelos animales para el estudio
de la isquemia cerebral, en diferentes especies, entre los que se incluyen, los roedores,
los caninos, los conejos, los gatos, los cerdos, las ovejas, los jerbos e inclusive los
primates no humanos (Bacigaluppi et al., 2010a; Howells et al., 2010; Liu & McCullough,
2011).
Recientemente, los modelos de isquemia cerebral que utilizan animales pequeños han
logrado ocupar un lugar privilegiado, a nivel preclínico, debido a que las variables
fisiológicas pueden ser monitoreadas sin dificultades y se puede alcanzar un número de
individuos estadísticamente relevante sin costos excesivos (Bacigaluppi et al., 2010b).
Entre los animales pequeños, el uso de los roedores se ha logrado difundir por las
razones que se exponen a continuación: 1) La anatomía circulatoria craneal es muy
similar a la humana, 2) los roedores genéticamente modificados han sido particularmente
útiles para el estudio de componentes de la fisiopatología de la isquemia y de esta
manera diseñar nuevas intervenciones preventivas, neuroprotectoras o terapéuticas, 3)
numerosos test de comportamiento neurosensoriales y motores han sido estandarizados
en roedores, facilitando la medición de recuperación funcional luego de una isquemia
experimental y 4) la facilidad de tener mayor cantidad de reactivo biológico bajo
condiciones de bienestar animal (Liu & McCullough, 2011; Woodruff et al., 2011). La rata
es el animal más comúnmente empleado para los modelos de isquemia cerebral, ya que
su tamaño permite un fácil monitoreo de las condiciones fisiológicas y fácil manipulación
de las estructuras vasculares (Bacigaluppi et al., 2010b)

47
En general, los modelos de isquemia cerebral se han clasificado en dos categorías
principales, los modelos de isquemia global, los modelos de isquemia focal y existe una
tercera categoría en la que se encuentra el modelo de isquemia hipoxia (Liu &
McCullough, 2011; Woodruff et al., 2011).
En la isquemia cerebral focal el flujo de sangre a la zona afectada casi siempre es mayor
que en el global, por lo que el insulto deberá ser más prolongado para ocasionar daño,
indicando que las condiciones metabólicas son diferentes con respecto a la global
(Woodruff et al., 2011); además, debido a la duración y heterogeneidad del insulto, los
procesos que se desencadenan son mucho más complejos que en una isquemia global,
haciendo que el modelo focal se asemeje más a lo que sucede en el accidente vascular
encefálico en los humanos (Woodruff et al., 2011).
Debido a que la isquemia cerebral en humanos usualmente resulta de una oclusión
trombótica o embólica de una arteria cerebral principal, generalmente la arteria cerebral
media (ACM), los modelos de isquemia cerebral focal simulan la isquemia en humanos
a través de esta arteria (Bacigaluppi et al., 2010b; DiNapoli, Rosen, Nagamine, & Crocco,
2006; Liu & McCullough, 2011)
A continuación se explicaran algunos de los modelos que se emplean para reproducir la
isquemia cerebral.

48
2.3.1 Modelos focales de isquemia cerebral. La isquemia cerebral aguda ocasionada por
la oclusión de una arterial cerebral puede ser reproducida empleando diferentes técnicas.
Estas técnicas son llamadas según el método que se emplea para la inducción
experimental de la isquemia. En algunos casos se realiza una oclusión mecánica distal
o proximal de la ACM, en otros, se ocasiona una oclusión embólica mediante la inyección
de coágulos preformados o de trombina a través de la ACM, o la inducción de
fototrombosis después de la inyección intravenosa de rosa de bengala. Además de los
modelos de infarto cerebral arterial, también se han desarrollado modelos para reproducir
la trombosis venosa cerebral, una forma rara de infarto que resulta de la trombosis de
los senos venosos cerebrales (Bacigaluppi et al., 2010b)
2.3.1.1 Modelo de oclusión mecánica de la ACM. Existen principalmente dos sitios de
oclusión en este modelo, la oclusión proximal y la oclusión distal. El modelo de oclusión
proximal de la ACM (pMCAO, por sus siglas en inglés), es usualmente inducido a través
de la oclusión mecánica directa de la ACM, mediante la inserción de una sutura de nylon
no recubierta o recubierta de silicona o poli-L-lisina en la Arteria Carótida Interna (ACI),
que subsecuentemente es avanzada hacia el círculo de Willis para ocluir la ACM en su
origen. La severidad del daño isquémico puede ser modelada dejando la sutura
transitoriamente, usualmente entre 30 a 120 minutos antes de removerla para permitir la
reperfusión. En el caso de la oclusión permanente, se deja la sutura y no se permite la
reperfusión (Bacigaluppi et al., 2010b; Céspedes, Arango, & Cardona, 2013; Henninger
et al., 2009; Popp, Jaenisch, Witte, & Frahm, 2009; Woodruff et al., 2011).
El menor tiempo de oclusión de la ACM, ocasiona muerte neuronal selectiva en el cuerpo
estriado, expresión de proteínas de choque térmico e inducción de la vía de señalización
apoptótica en el tejido subyacente (corteza). Tiempos de oclusión más prolongados
producen infartos cerebrales que involucran tanto el estriado como la corteza que pueden
estar asociados con la muerte del animal, cuando hay formación de edema (Bacigaluppi
et al., 2010b)

49
El modelo de oclusión vascular distal de la ACM (dMCAO, por sus siglas en inglés), se
realiza mediante craneotomía para exponer y manipular directamente las ramas de la
ACM. Usualmente la ACM es ocluida distal al origen de las ramas lenticuloestriadas, así
como en el modelo proximal, la ACM puede ser ocluida transitoriamente por medio de un
clip o permanentemente mediante electrocoagulación. La dMCAO también puede ser
combinada con la oclusión transitoria o permanente de la arteria carótida común (ACC)
ipsilateral y/o contralateral (Bacigaluppi et al., 2010b; Howells et al., 2010; Woodruff et
al., 2011).
En este modelo el daño isquémico puede involucrar la corteza frontal, parietal y temporal,
también podría verse afectada la sustancia blanca por debajo de la corteza y una
pequeña porción del cuerpo estriado lateral. Además, este modelo ha sido considerado
particularmente apropiado para estudios de neuroplasticidad y neuroregeneración,
debido a que puede reproducir el daño en la corteza sin que se vean dañadas estructuras
cerebrales profundas (Bacigaluppi et al., 2010b).
2.3.1.2 Modelos de isquemia cerebral tromboembólica. Debido a que la isquemia en
humanos es frecuentemente causada por tromboembolismo cerebral, se han generado
diferentes modelos que simulan cercanamente la oclusión embólica de las arterias
cerebrales. Las isquemias embólicas puede ser inducidas en animales mediante la
inyecciones de macroesferas sintéticas de gran tamaño (300-400 µm de diámetro), o
pequeñas microesferas (menos de 50 µm) en la ACI. En el primer caso, se producen
grandes infartos similares a los provocados por el modelo oclusión transitoria de la ACM,
en el segundo caso, se ocasionan pequeños infartos multifocales (Bacigaluppi et al.,
2010b; Liu & McCullough, 2011).
Otros modelos embólicos, en lugar de emplear microesferas, utilizan coágulos autólogos
preformados que son inyectados directamente en la ACC, en la ACI o en el origen de la
ACM (Bacigaluppi et al., 2010b; DiNapoli et al., 2006; Henninger et al., 2009; Howells et
al., 2010; Kudo, Aoyama, Ichimori, & Fukunaga, 1982; C. Wang, Yang, & Shuaib, 2001).
Otras variantes de estos modelos, son la generación in situ de coágulos autólogos

50
mediante diferentes técnicas como son, la inyección directa de trombina purificada en la
ACM, o mediante la inserción de un catéter en la arteria carótida externa (ACE) o en la
ACC, el cual se encuentra conectado a una jeringa que contiene trombina bovina, esta
jeringa se carga con sangre y el catéter es retenido por unos minutos (3-15 minutos) para
permitir la formación del coágulo, una vez se ha formado el coágulo se avanza el catéter
hasta el origen de la ACM y se inyecta, luego de unos minutos se retira el catéter y se
liga la arteria en la que se insertó el catéter (Bacigaluppi et al., 2010b; Casals et al., 2011;
Henninger, Sicard, Schmidt, Bardutzky, & Fisher, 2006; Howells et al., 2010; Woodruff et
al., 2011).
De otra parte, los modelos embólicos se han empleado principalmente para el estudio de
terapias trombolíticas, ya que producen infartos relativamente variables que hacen más
difícil probar intervenciones terapéuticas neuroprotectoras (Bacigaluppi et al., 2010b).
2.3.1.4 Modelo de endotelina y fototrombosis. Debido a que los modelos más empleados
(dMCAO, pMCAO), son técnicamente exigentes para el investigador, se han desarrollado
modelos más simples para inducir una lesión isquémica focal como una alternativa. Una
de estas alternativas consiste en la inyección de sustancias vasoconstrictoras como la
endotelina 1 (ET-1, por sus siglas en inglés), que ocasionan la disminución aguda del
flujo sanguíneo en el área inyectada. En la materia gris la ET-1 produce pequeñas
lesiones con muerte neuronal y astrocitaria y respuesta microglial retardada. En la
sustancia blanca, provoca perturbación axonal y alteración en los oligodendrocitos
seguido de desmielinización e incremento en la reactividad astrocitaria (Bacigaluppi et
al., 2010b; Casals et al., 2011; Howells et al., 2010).
Otra técnica para inducir un daño isquémico localizado es la fototrombosis, la cual
consiste en la inyección intravenosa de un colorante fotosensible (rosa de bengala o
flouresceina) y la posterior iluminación transcranial, permitiendo la coagulación de la
sangre que se encuentra en la zona irradiada (Bacigaluppi et al., 2010b; Casals et al.,
2011; De Ryck, Van Reempts, Borgers, Wauquier, & Janssen, 1989; Howells et al., 2010)

51
2.3.2 Modelos globales de isquemia cerebral. La isquemia cerebral global se caracteriza
por la reducción crítica del flujo sanguíneo cerebral en la totalidad del encéfalo,
induciendo daño neuronal selectivo en la región hipocampal CA1. La severidad del infarto
es dependiente de la duración de la isquemia, adicionalmente otras áreas del cerebro
podrían estar involucradas en este proceso de daño celular por la no restauración del
flujo sanguíneo. Los modelos de isquemia cerebral global generalmente son empleados
para el estudio del daño cerebral ocurrido después de una resucitación (Bacigaluppi et
al., 2010b)
La isquemia cerebral global puede ser inducida mediante diferentes técnicas.
2.3.2.1 Modelo de oclusión de 4 vasos (4-VO). Este modelo consiste en la oclusión
reversible de la ACC, combinada con la interrupción permanente de las arterias
vertebrales mediante electrocauterización, resultando en una isquemia bilateral del
encéfalo y el prosencéfalo. Este modelo presenta diversas ventajas, entre las que se
encuentran, daño neuronal isquémico altamente predecible, una baja incidencia de
ataques convulsivos y la no necesidad de anestesia. El modelo de 4-VO ha sido
ampliamente utilizado para estudiar la efectividad de potenciales agentes terapéuticos
(Bacigaluppi et al., 2010b; Céspedes et al., 2013; Woodruff et al., 2011).
2.3.2.2 Modelo de oclusión de 2 vasos (2-VO). Este modelo es inducido bajo anestesia
general y requiere la administración de un relajante muscular, por otro lado, ha sido bien
documentado que solamente la oclusión bilateral de la ACC es insuficiente para disminuir
el flujo sanguíneo cerebral por debajo del umbral isquémico, se debe reducir el flujo
sanguíneo cerebral mediante hipotensión al mismo tiempo que se ocluyen las arterias
carótidas. La ligación bilateral de la ACC junto con la reducción del flujo sanguíneo a 50
mm Hg provocan igual o más daño que el producido por el modelo de 4-VO (Bacigaluppi
et al., 2010b; Liu & McCullough, 2011; Woodruff et al., 2011).

52
2.3.2.3 Modelo de isquemia global en jerbos. Este modelo se induce a través de la
ligación bilateral temporal de la ACC sin hipotensión, debido a que los jerbos no
presentan arterias comunicantes posteriores, lo que genera una hipoperfusión
prolongada y una reducción del 25% del flujo sanguíneo cerebral. Esta reducción del flujo
sanguíneo produce pequeños focos necróticos en la corteza y en los ganglios basales
acompañados por gliosis reactiva y proliferación de pequeños vasos. (Bacigaluppi et al.,
2010b; Howells et al., 2010; Woodruff et al., 2011).
2.4 TERAPEUTICA DE LA ISQUEMIA CEREBRAL
A pesar de los avances en el entendimiento de la fisiopatología de la isquemia cerebral
las opciones terapéuticas para el tratamiento del infarto cerebral isquémico agudo son
muy limitadas. Solamente ha sido aprobado un medicamento para uso clínico, el
activador de plasminógeno tisular recombinante (rt-PA), utilizado como terapia
trombolítica dentro de las 3 horas desde el inicio del ataque, mejorando la recuperación
funcional y disminuyendo los déficits neurológicos de los pacientes, sin embargo esta
mejora en la recuperación es alcanzada a expensas de un incremento en la incidencia
de hemorragia intracraneal, la cual ocurre aproximadamente en el 6% de los pacientes
(Jordán et al., 2007; Lakhan et al., 2009; Woodruff et al., 2011).
Debido a que la mayoría de los pacientes no llegan a los centros clínicos dentro de las 3
horas siguientes al inicio del ataque isquémico o no reciben el tratamiento oportuno con
rt-PA, el tratamiento exitoso del infarto cerebral agudo, se mantiene como uno de los
principales retos para la medicina clínica (Lakhan et al., 2009). Además, se ha
demostrado que el rt-PA puede exacerbar la muerte neuronal excitotóxica, sugiriendo un
efecto adverso de este medicamento que podría en parte, contrarrestar su actividad
trombolítica (Woodruff et al., 2011)
Actualmente, el esfuerzo clínico para el tratamiento del infarto cerebral isquémico se ha
encaminado principalmente hacia dos objetivos, reabrir las arterias ocluidas con el
consecuente restablecimiento de la perfusión cerebral y obtener un efecto neuroprotector

53
que garantice la viabilidad celular hasta que los mecanismos fisiológicos naturales o
derivados del tratamiento farmacológico detengan los efectos de la isquemia (Fernández
Gómez et al., 2008).
El aumento del conocimiento concerniente a los eventos fisiopatológicos moleculares ha
llevado a considerar cada etapa de la cascada isquémica como una posible diana
terapéutica para la protección de las neuronas. La neuroprotección es un término
empleado para describir el supuesto efecto de diferentes intervenciones para prevenir,
proteger y limitar el daño del tejido nervioso; en isquemia, este concepto involucra la
inhibición de una cascada de eventos moleculares patológicos que ocurren durante la
isquemia y que conducen al influjo de calcio, la producción de radicales libres y la muerte
celular (Jordán et al., 2007).
Diferentes fármacos han sido empleados experimentalmente para inhibir la muerte
celular necrótica o apoptótica, ocasionadas por el infarto cerebral isquémico, las cuales
son mediadas, en parte, por la entrada excesiva de calcio resultante de la activación de
los receptores de glutamato, canales de calcio dependiente de voltaje y del compromiso
de las bombas de Ca++, generando una prolongada elevación de la concentración de
Ca++ intracelular; fármacos como nimodipine y flunarazine que bloquean canales de Ca++,
han sido probados efectivamente en modelos animales, pero al ser empleados en
ensayos clínicos no se han encontrado diferencias significativas en los resultados frente
a los grupos placebo (Canova, Roatta, Micieli, & Bosone, 2012; Jordán et al., 2007;
Woodruff et al., 2011).
Otras aproximaciones farmacológicas para el tratamiento de la isquemia cerebral, han
intentado disminuir la excitotoxicidad mediante el desarrollo de componentes que
interfieren con la activación de los receptores de glutamato, como los antagonistas de
los receptores NMDA, no competitivos dizocilpine, dextromethorphan, aptiganel,
cerezine y competitivos selfotel, elipodril que han demostrado eficacia en modelos
animales pero sin efectos significativos en humanos. También se han ensayado
antagonistas de receptores no NMDA, como zonampanel, SPD-502 sin resultados

54
alentadores (Ginsberg, 2008; Lai, Zhang, & Wang, 2014; Qu et al., 2012; Waszkielewicz
et al., 2013; Woodruff et al., 2011; Xu & Pan, 2013)
Además de la excitotoxicidad por glutamato, la producción de radicales libres se
incrementa en el foco y en la zona de penumbra y se piensa que son la principal causa
del daño observado en estas regiones; existen muchas sustancias, que pueden bloquear
la producción de radicales libres o inhibir su activación que han demostrado su eficacia
en modelos experimentales de isquemia cerebral, como edaravone, tetramethylpirazine,
alpha-phenyl-N-tert-butyl-nitrone, FR210575, NXY-59 y EGb-761, pero en humanos no
se han logrado reproducir estos resultados (H. Chen et al., 2011; Woodruff et al., 2011;
Xu & Pan, 2013).
Existe evidencia suficiente que sugiere la participación de las caspasas en el proceso de
muerte neuronal apoptótica. Diferentes grupos han estudiado los efectos de la inhibición
de las capasas sobre la neurodegeneración inducida por la isquemia empleando
inhibidores como z-VAD en forma de fluorometilcetona (FMK), ácido
diclorobenzoiloxopentanoico (dcb), z-DEVD-FMK y XBIR3 los cuales han sido
neuroprotectores en modelos de isquemia cerebral ofreciendo protección neuro-
funcional (Akpan et al., 2011; Fricker, Vilalta, Tolkovsky, & Brown, 2013; Ma, Qiu, Hirt,
Dalkara, & Moskowitz, 2001; Woodruff et al., 2011)
Para disminuir los efectos de la inflamación se han desarrollado diferentes estrategias
como son la inhibición de la adhesión de los leucocitos al endotelio activado a través de
anticuerpos anti-selectinas y anticuerpos anti-ICAM-1, los cuales han disminuido los
volúmenes de infarto en modelos experimentales de isquemia cerebral focal transitoria;
sin embargo, en un ensayo clínico en el cual se empleó el anticuerpo murino anti-ICAM-
1 (enlimomab), se encontró empeoramiento del déficit neurológico y mortalidad
(Ginsberg, 2008; Woodruff et al., 2011)
Se han ensayado otros fármacos, como agentes anti-integrinas CD11b/CD18 (UK-
279276), sin efectos neuroprotectores y con efectos secundarios como leucopenia e

55
inmunosupresión (Jin, Yang, & Li, 2010), inhibidores de la producción de óxido nítrico
como Lubeluzole, mostrando incremento significativo de desórdenes de conducción
cardiaca (De Keyser et al., 1997; Lai et al., 2014; Xu & Pan, 2013), agonistas de
neurotransmisores como la serotonina (repinotan) y activadores de canales de potasio
como BMS-204352, sin efectos protectores (Ginsberg, 2008; Xu & Pan, 2013).
Durante décadas los investigadores han intentado abordar el tratamiento de la isquemia
cerebral centrándose en dianas individuales de la cascada isquémica sin resultados
exitosos, sugiriendo que durante la cascada isquémica se activan una variedad de
procesos que corren en paralelo, por lo que la atención debería centrarse en agentes
neuroprotectores de múltiples dianas terapéuticas (Xu & Pan, 2013)
Entre estos agentes de múltiples dianas terapéuticas se encuentran el magnesio, la
progesterona, la eritropoyetina y sus derivados, la albumina y las estatinas (Xu & Pan,
2013)
2.4.1 Estatinas. Las estatinas son un diverso grupo de fármacos que actúan como
potentes inhibidores de la biosíntesis del colesterol, que impiden la acción de la enzima
3-hidroxi-3-metilglutaril coenzima A reductasa (HMGCoA reductasa), la cual se encarga
de la conversión de la hidroxi metil glutaril coenzima A en ácido mevalónico, un precursor
del colesterol (Pella, Rybar, & Mechirova, 2005).
Las estatinas fueron originalmente identificadas como metabolitos secundarios de
hongos y en la actualidad se encuentran disponibles comercialmente una gran variedad
de éstas, tanto naturales, producidas por el hongo Aspergillus terreus (lovastatina,
simvastatina, pravastatina), como sintéticas (atorvastatina, fluvastatina y rosuvastatina)
(Liao & Laufs, 2005). Aunque las estatinas comparten el mismo mecanismo de acción,
su farmacocinética es diferente, todas las estatinas son bien absorbidas en el intestino
cuando se administran oralmente, su primer paso por el hígado reduce su
biodisponibilidad en un 5 – 30% y una vez en el plasma se unen a la albumina lo cual
determina la variabilidad de la vida media entre las estatinas. Atorvastatina y

56
rosuvastatina son las estatinas que poseen una vida media más larga entre 15-30 horas
para la primera y 20,8 horas para la segunda, mientras que fluvastatina, pravastatina,
lovastatina y simvastatina tienen una vida media entre 0,5 a 3 horas (Butterfield, Barone,
& Mancuso, 2011).
Debido a sus diferentes estructuras químicas, las estatinas exhiben grados variables de
afinidad por los lípidos (lipofilicidad). Los profármacos simvastatina y lovastatina tienen
los índices de lipofilicidad más altos, atorvastatina, fluvastatina y pitavastatina presentan
un índice intermedio, en cambio, pravastatina posee el índice más bajo, lo cual puede
influir en su capacidad para atravesar la barrera hematoencefálica (Butterfield et al.,
2011; Wood, Eckert, Igbavboa, & Müller, 2010)
Más de dos tercios del colesterol del organismo, se sintetiza en el hígado, por lo tanto, la
inhibición de la biosíntesis de colesterol hepático es la principal fuente para reducir los
niveles de colesterol sérico (Liao & Laufs, 2005). Las estatinas son uno de los agentes
más efectivos para tal fin, debido a que reducen el colesterol ligado a lipoproteínas de
baja densidad (c-LDL). Por consiguiente, se ha determinado éste como el mecanismo
principal en la reducción de la incidencia del ictus y de eventos cardiovasculares (Castilla
Guerra, Fernandez Moreno, López Chozas, & Jiménez Hernandez, 2007; Tsai et al.,
2011).
Luego del descubrimiento de las estatinas en 1976 y su frecuente uso para el control de
la hipercolesterolemia y la prevención de la enfermedad cardiovascular, se le han
atribuido una serie de beneficios adicionales o efectos pleiotrópicos, no dependientes de
la reducción de c-LDL, sino de la inhibición de las subsecuentes asociaciones en la ruta
del mevalonato (Liao, 2005), que no sólo es precursor del colesterol sino también de
muchos componentes isoprenoides no esteroidales que incluyen geranil pirofosfato
(GPP), farnesil pirofosfato (FPP), geranil geranil pirofosfato (GGPP) y el escualeno,
vitales para diferentes funciones que incluyen desde la síntesis del colesterol hasta el
crecimiento y diferenciación celular (Pella et al., 2005; Zhou & Liao, 2010).

57
Al disminuir, mediante el uso de las estatinas, las concentraciones de intermediarios de
la ruta del mevalonato como el FPP y el GGPP, claves para la isoprenilación o
modificación postraduccional de proteínas transmembrana tales como, Ras, Rho y Rac,
se logra inhibir gran cantidad de funciones de dichas macromoléculas logrando reducir
eventos de inflamación, trombogenicidad y vasoconstricción importantes en la patología
vascular (Liao, 2005; Schönbeck & Libby, 2004; C. Y. Wang, Liu, & Liao, 2008; Wood et
al., 2010). Entre los efectos pleiotrópicos de las estatinas se incluyen la estabilización de
la placa aterosclerótica, la disminución del estrés oxidativo, el efecto antiinflamatorio, y
antitrombótico, la mejora de la reactividad vasomotora y función endotelial, y algunas
acciones inmunomoduladoras (Castilla Guerra, Fernandez-Moreno, et al., 2007; Zeiser
et al., 2008).
Las estatinas mejoran la función endotelial mediante la estimulación y sobrerregulación
de la óxido nitroso sintetasa (eNOS) de los vasos, enzima que ejerce funciones
antiateroscleróticas paracrinas en el endotelio, debido a que aumenta la síntesis,
liberación y actividad del NO endotelial; parando el proceso aterogénico, estimulando la
relajación vascular e inhibiendo la agregación plaquetaria, la proliferación de la
musculatura lisa vascular y las interacciones entre los leucocitos y el endotelio, efecto
diferente al ocasionado por el NO como radical libre producido por la óxido nítrico
sintetasa inducible, como se había descrito anteriormente (Liao, 2005; Sawada & Liao,
2009; Takemoto & Liao, 2001).
Esta clase de fármacos aumentan la expresión del activador tisular de plasminógeno y
disminuyen la expresión de la endotelina 1, un potente vasoconstrictor y mitógeno que
se genera en el mismo endotelio (Minoru et al., 2005; Takemoto & Liao, 2001). También,
se ha demostrado aumento significativo de la respuesta vasodilatadora del endotelio
vascular, favoreciendo el flujo sanguíneo, mediante la producción de prostaciclina,
explicando su actividad vasodilatadora, antiagregante y antiinflamatoria (Liao, 2005;
Ramos-Esquivel & León Céspedes, 2007).

58
De igual forma, las estatinas inducen angiogénesis al promover la proliferación,
migración y supervivencia de células progenitoras endoteliales circulantes y acelerando
la formación de la estructura vascular mediante la activación de la vía fosfatidilinositol 3-
Kinasa y la eNOS; de esta forma, aumentan la neovascularización inducida por la
isquemia y mejoran la función tisular posisquemia (Liao, 2005; Schönbeck & Libby,
2004).
Las estatinas contribuyen a la estabilidad de la placa ateromatosa, mediante la reducción
del tamaño de la placa y la modificación de las propiedades fisicoquímicas del núcleo
lipídico (Pella et al., 2005), ya que disminuyen la acumulación de macrófagos en las
lesiones ateroscleróticas, aumentan el contenido de colágeno de la placa y disminuyen
su componente inflamatorio y protrombótico, mediante la reducción de diversas enzimas
y citocinas, ejemplo de ello, es la disminución de los niveles de interferón ɣ (que inhibe
la formación de colágeno y aumenta la inestabilidad de la placa), de metaloproteinasas
(enzimas que debilitan la cápsula fibrosa) y de moléculas de adhesión proinflamatorias
como la VCAM-1 (molécula de adhesión vascular 1) o la ICAM-1(molécula de adhesión
intercelular 1), entre otras (Castilla-Guerra, Fernandez-Moreno, et al., 2007; Liao, 2005).
Además, la estatinas inhiben la expresión de fibrinógeno y la formación de trombina,
reducen la agregación plaquetaria, la expresión de COX 2, tromboxano A2 (TxA2) o TxB2
y mejoran la síntesis de prostaciclina, contribuyendo a disminuir la activación plaquetaria
(Schönbeck & Libby, 2004; Takemoto & Liao, 2001).
Las estatinas poseen efectos antiinflamatorios mediante la disminución en la expresión
de moléculas de adhesión, las cuales participan en la primera fase de la aterogénesis y
de los procesos inflamatorios. Así mismo, impiden las reacciones químicas intracelulares
que llevan a la expresión y modulación de sustancias y promotores inflamatorios (como
NF-κB, IL-1, IL-6), que son dependientes de proteínas GTPasas y requieren isoprenilarse
para su activación (Castilla-Guerra, Fernandez-Moreno, et al., 2007; Kandadai, Meunier,
Lindsell, Shaw, & Elkind, 2012; Ramos-Esquivel & León Céspedes, 2007).

59
Por otro lado, se le atribuye a las estatinas propiedades antioxidantes, puesto que al
reducir la isoprenilación de proteínas Rac 1, impiden que estas se trasladen del citosol
hacia la membrana celular en el proceso de activación de la NADPH oxidasa, logrando
así la reducción de radicales libres (Kandadai et al., 2012; Ramos Esquivel & León
Céspedes, 2007).
Otro efecto observado con las estatinas es la modulación del crecimiento celular, debido
a que las proteínas Ras y Rho están implicadas en la regulación del ciclo celular, ya que,
regulan señales de traducción que se encargan de la transcripción de genes involucrados
en la proliferación y diferenciación celular, así como en la apoptosis (Ramos Esquivel &
León-Céspedes, 2007; C. Y. Wang et al., 2008). Ras puede promover la progresión del
ciclo celular a través de la activación de la proteína quinasa activada por mitógeno,
mientras que Rho provoca la proliferación celular a través de la desestabilización de la
proteína p27 Kip1. La inhibición de Rho por las estatinas es el mecanismo predominante
por el que se inhibe la proliferación vascular de células del musculo liso (SMC) inducida
por PDGF (Liao, 2005; Takemoto & Liao, 2001).
Además, se ha sugerido que la actividad de Rac 1 se encuentra involucrada en procesos
neurodegenerativos luego de la isquemia cerebral y también en su recuperación a largo
plazo, puesto que Rac 1 no solo se encuentra implicada en la actividad sináptica, sino
también, en procesos de muerte celular mediante la regulación de la activación de
quinasas c-jun N-terminal, promoviendo la supervivencia neuronal (Gutiérrez Vargas et
al., 2010), del mismo modo, se ha evidenciado que la neuroprotección ejercida por el
tratamiento con Atorvastatina en modelo de excitotoxicidad por glutamato es dependiente
de la activación de Rac 1 (Posada Duque, Velasquez Carvajal, Eckert, & Cardona
Gomez, 2013)
Un efecto pleiotrópico no relacionado con la inhibición de la síntesis del mevalonato, es
la unión de las estatinas con un nuevo sitio alostérico dentro de la integrina β2 antígeno-
1 asociado a la función linfocitaria (LFA-1), el cual juega un papel importante en el
transporte de leucocitos y la activación de las células T, bloqueando así la adhesión de

60
linfocitos al endotelio en un grado suficiente para suprimir la respuesta inflamatoria
(Schönbeck & Libby, 2004). Por esta vía las estatinas favorecen la liberación de las
interleuquinas 4, 5 y 10, el factor de crecimiento transformante beta (TGF-β),
favoreciendo la respuesta TH2, aboliendo la respuesta inmune TH1, que libera IL-2, IL-
12, o interferón ɣ (IFN-ɣ), lo cual reduce significativamente la infiltración inflamatoria y a
la vez promueve la diferenciación del linfocito TH0 a TH2, sugiriendo una acción protectora
de las estatinas en enfermedades autoinmunes mediadas por linfocitos TH1(Ramos
Esquivel & León-Céspedes, 2007; Zeiser et al., 2008). Actualmente, se conoce que las
estatinas suprimen efectivamente la expresión del complejo mayor de
histocompatibilidad tipo II en células específicas, por un efecto dependiente de la
inhibición de la enzima 3-hidroxi-3-metilglutaril CoA reductasa, quizás mediante un
mecanismo de expresión génica a nivel transcripcional, reprimiendo de este modo la
activación de células T (Kandadai et al., 2012; Liao, 2005).
Aunque el papel de las estatinas en la prevención del infarto cerebral ha sido durante
mucho tiempo tema de controversia debido a que inicialmente no se hallaba una
correlación entre los niveles séricos de colesterol y el infarto cerebral en estudios
prospectivos, en la actualidad está plenamente establecido que las estatinas disminuyen
su incidencia y este efecto se debe en gran parte al grado de reducción del colesterol
LDL asociado a lipoproteínas de baja densidad (Castilla Guerra, Fernández Moreno,
López Chozas, & Jiménez Hernández, 2007).
Inicialmente, la prevención primaria y secundaria del infarto cerebral se debe
fundamentalmente a la disminución en los niveles de colesterol sérico. Sin embargo, se
han demostrado efectos neuroprotectivos de las estatinas en los accidentes
cerebrovasculares isquémicos, debido a su capacidad para regular la expresión y
actividad de eNOS en los vasos sanguíneos cerebrales, aumentando la producción de
NO y por ende el flujo sanguíneo. Además, éste aumento del NO tiene impacto en la
severidad del daño y viabilidad del tejido isquémico al atenuar la expresión de la P-
selectina y la adhesión leucocitaria (Liao, 2005; Takemoto & Liao, 2001).

61
Además, las estatinas contribuyen a la neuroprotección en isquemia a través de su
capacidad antiaterosclerotica y estabilización de la placa ateromatosa, su acción
antiinflamatoria y la movilización de células progenitorias endoteliales, reduciendo el
tamaño del infarto y contribuyendo a la disminución en la incidencia de accidentes
cerebrovasculares.(Liao, 2005).
Las estatinas han demostrado su efectividad para el tratamiento del infarto cerebral
isquémico en modelos animales, puesto que su administración ha logrado disminuir el
tamaño de la lesión, aumentar el flujo sanguíneo cerebral, reducir el volumen del infarto
y mejorar la permeabilidad microvascular (Ding et al., 2006; Juha Yrjänheikki et al., 2005;
L. Zhang et al., 2009). También, se ha encontrado que posee efecto neuroprotector
reduciendo la pérdida neuronal y la neurodegeneración al parecer mediante la
aceleración de los mecanismos de supervivencia celular junto con la regulación de
moléculas de adhesión como p-120 catenina y αN-catenina (Céspedes-Rubio et al.,
2010). Así mismo, la atorvastatina ha aumentado la ventana terapéutica de otros
medicamentos como el activador de plasminógeno tisular recombinante (rtPA) sin
incrementar la incidencia de una transformación hemorrágica (Ding et al., 2006; L. Zhang
et al., 2009).
2.4.2 Inhibidores de la ciclooxigenasa. La neuroinflamación ha sido implicada en la
patogenia o en la progresión de diferentes desórdenes neurológicos y
neurodegenerativos crónicos o agudos, la prostaglandina sintasa H o ciclooxigenasa
juega un papel central en la cascada inflamatoria mediante la conversión del ácido
araquidónico en prostanoides bioactivos. Existen dos isoformas de ciclooxigenasas COX
1 y COX 2 las cuales son codificadas por diferentes genes y se ha descrito una variante
de la COX 1, denominada COX 3 (Aïd & Bosetti, 2011; Camara Lemarroy, Gonzalez
Moreno, Guzman-de la Garza, & Fernandez-Garza, 2012; Yagami, 2006).
La producción de los prostanoides inicia con la inducción de la cascada del ácido
araquidónico que involucra la liberación de ácidos grasos poliinsaturados desde los
fosfolípidos de membrana por la enzima fosfolipasa A (PLA2) (Bosetti, 2007),

62
posteriormente, las COX catalizan la conversión de ácido araquidónico (AA) a
prostaglandinas (PG) y tromboxanos (TXB), los cuales actúan como ligandos autocrinos
y paracrinos en muchas respuestas fisiológicas y patológicas, la COX 1 se conoce como
la isoforma constitutiva y la COX 2 como la isoforma inducible, ambas utilizan los mismos
sustratos y catalizan las mismas reacciones, deoxigenación del ácido araquidónico para
producir la prostaglandina G2 (PGG2) y una reacción peroxidasa que convierte la PGG2
en prostaglandina H (PGH2), la cual es transformada a PGE2, PGF2α, PGD2, PGI2 y TBX2
a través de procesos enzimáticos específicos (Aïd & Bosetti, 2011; Bailey, 1989).
Estos prostanoides interactúan con receptores específicos de membrana de la
superfamilia de receptores acoplados a proteínas G, jugando un papel importante en
muchas respuestas celulares y en procesos patológicos como modulación de la reacción
inflamatoria y su resolución, erosión del cartílago, citoprotección y ulceración
gastrointestinal, angiogénesis y cáncer, hemostasia y trombosis, hemodinámica renal y
progresión de enfermedades renales, ateroprotección y progresión de ateroesclerosis
(Capone et al., 2007).
En el sistema nervioso central el AA y sus productos finales se encuentran implicados en
diversos procesos fisiológicos como la señalización sináptica, la activación neuronal, la
liberación de neurotransmisores, la nocicepción, la expresión de genes, el flujo
sanguíneo cerebral, el ciclo sueño/vigilia y el apetito. Además, las alteraciones en el
metabolismo del AA han sido implicadas en diferentes desórdenes neurológicos,
neurodegenerativos y siquiátricos, entre los que se incluyen epilepsia, isquemia,
demencia asociada al síndrome de inmunodeficiencia adquirida, esclerosis lateral
amiotrófica, Alzheimer, Parkinson y esquizofrenia (Bosetti, 2007)
Aunque las isoformas COX comparten un 60% de homología en su secuencia de
aminoácidos y ambas son proteínas integrales de membrana del retículo endoplasmático
y el núcleo, difieren en sus mecanismos regulatorios, localización celular y función (Aïd
& Bosetti, 2011).

63
La COX 2 fue identificada inicialmente como un elemento clave en la respuesta
inflamatoria aguda debido a que su expresión es rápidamente inducida por varios
inflamógenos y posee diferentes elementos transcripcionales regulatorios, entre los que
se incluyen, el factor nuclear kappa beta (NF-kB), Sp1, una caja TATA, un potenciador
de proteínas de unión beta CAAT, un elemento de respuesta a AMP cíclico, los cuales
interactúan con factores de transcripción generados por múltiples vías de señalización;
en cambio, COX 1 no posee caja TATA ni CAAT, contiene un alto contenido GC y varios
elementos Sp1. La COX 2 contiene una única secuencia de 27 aminoácidos cerca de su
extremo carboxilo terminal, que corresponde a un elemento involucrado en la
inestabilidad de la proteína (Aïd & Bosetti, 2011).
Debido a que COX 2 es típicamente inducida por estímulos inflamatorios en la mayoría
de los tejidos se presumía que era la isoforma responsable de la propagación de la
respuesta inflamatoria y tradicionalmente considerada como la mejor diana para los
fármacos antiinflamatorios (Capone et al., 2007). Algunos fármacos antiinflamatorios
como la aspirina y los fármacos antiinflamatorios no esteroidales (NSAIDs) e inhibidores
selectivos de COX 2 (coxibs) generan acciones antipiréticas, analgésicas y
antiinflamatorias a través de la inhibición competitiva del sitio activo de COX. Los coxibs
(celecoxib, etirocoxib, rofecoxib y valdecoxib), fueron desarrollados con el objetivo de
reducir la incidencia de efectos gastrointestinales adversos asociados con la
administración de NSAIDs, como consecuencia de la inhibición de los prostanoides
derivados de COX 1, sin embargo, en los coxibs se han encontrado otros efectos no
deseados como son el aumento de la incidencia de infarto del miocardio y las
enfermedades cerebrovasculares (Capone et al., 2007).
Además de los inhibidores selectivos de COX 2, se han producido antiinflamatorios como
el meloxicam que tienen una actividad inhibitoria preferencial contra la isoforma inducible
COX 2 sobre la isoforma constitutiva COX 1 (Goverdhan, Sravanthi, & Mamatha, 2012)
COX 1 y COX 2 se expresan constitutivamente en el cerebro. En condiciones fisiológicas
COX 1 es expresada principalmente en la microglía y en las células perivasculares y

64
COX 2 se encuentra en las dendritas posinápticas y en la terminales excitatorias,
particularmente en la corteza, el hipocampo y la amígdala, con asociaciones neuronales
y vasculares, por lo que COX 2 ha sido involucrada en la actividad sináptica, en la
potenciación a largo plazo, en la consolidación de la memoria y en el acoplamiento
neurovascular durante la hiperemia. Además, COX 2 y no COX 1 puede oxigenar
endocanabinoides lo cual representa una importante vía metabólica para la regulación
de la transmisión sináptica excitatoria (Aïd & Bosetti, 2011; Yagami, 2006)
En modelos animales de neuroinflamación se ha demostrado que la inhibición selectiva
de COX 2 incrementa el daño neuronal, la activación glial, la expresión de citoquinas
cerebrales, la permeabilidad de la barrera hematoencefálica y la infiltración de leucocitos,
por lo que se sugiere que los productos derivados de COX 2 pueden mediar un efecto
protector en la progresión y/o en la resolución de la inflamación luego de la activación de
la inmunidad innata del cerebro, puesto que al bloquear selectivamente a COX 2 se
estarían inhibiendo mediadores claves que ayudarían al tejido a retornar a un estado no
inflamado y posterior homeostasis (Aïd & Bosetti, 2011).
El meloxicam, como inhibidor preferencial de COX 2, ha sido estudiado en diferentes
modelos entre los que se encuentra el estrés oxidativo, el cual es uno de los procesos
patogénicos comunes en enfermedades neurodegenerativas como Alzheimer,
Parkinson, Esclerosis Lateral Amiotrófica, Huntington y desórdenes neurológicos y
siquiátricos relacionados, encontrándose reducción de la pérdida neuronal y disminución
del estrés oxidativo mediante la supresión de la sobrecarga de iones metálicos,
especialmente el aluminio (Yu, Jiang, Su, Yu, & Yang, 2014). Además, en un modelo de
daño cognitivo, el meloxicam asociado a un inhibidor de la monoamino oxidasa
(seleginine), lograron mejorar la actividad antiamnésica a través de la inhibición de la
peroxidación lipídica, aumento de las enzimas antioxidantes y disminución de la actividad
de la acetilcolinestarasa en el cerebro (Goverdhan et al., 2012).
Por otro lado, en un modelo de isquemia cerebral mediante oclusión de la ACM, en el
que se empleó meloxicam como analgésico, se encontró reducción significativa del

65
volumen de infarto, lo que fue asociado a la disminución de la producción de
prostanglandinas proinflamatorias potencialmente perjudiciales para el tejido cerebral
(Jacobsen et al., 2013)
En un modelo de isquemia/reperfusión, el meloxicam atenuó los efectos de la isquemia
sobre la subregulación de la expresión de los receptores NMDA y el aumento de
marcadores de inflamación como GFAP y CD11b, sugiriendo que meloxicam disminuye
los marcadores de inflamación y bloquea la regulación hacia abajo de los genes del
receptor NMDA (Montori et al., 2010), así mismo, se ha demostrado que regula el
transporte vesicular del glutamato que se ve alterado por la inflamación como respuesta
a la isquemia (Llorente et al., 2013). También, en un modelo de hipoxia-reoxigenación
en cortes de hipocampo, el meloxicam ejerció un efecto citoprotector mediante la
inhibición de la lactato deshidrogenasa y el incremento de la interleuquina 10 (López
Villodres et al., 2012).
Finalmente, no se ha logrado aprobar ninguna terapia diferente al tratamiento con
trombolíticos como el rt-PA, el cuál logra un único objetivo que es la restauración del flujo
de sangre hacia la zona afectada sin ofrecer otras ventajas y cuya ventana terapéutica
es muy limitada, lo cual hace necesario plantear estrategias terapéuticas que permitan
incidir desde diferentes frentes la cascada fisiopatológica desencadenada por el infarto
cerebral, como podrían ser los tratamientos combinando atorvastatina como fármaco de
efectos pleiotrópicos dependientes de la inhibición de las subsecuentes asociaciones en
la ruta del mevalonato y un inhibidor preferencial de COX 2 como el meloxicam, el cual
puede ayudar a modular la respuesta mediada por las prostaglandinas y los prostanoides
durante la fase aguda inflamatoria de la lesión cerebral.

66
3. MATERIALES Y MÉTODOS
3.1 REACTIVO BIOLÓGICO
Se emplearon 39 ratas hembras de la línea Wistar (no consanguínea), con un peso de
211 ± 15 gramos, mantenidas en el Bioterio de Experimentación de la Universidad del
Tolima (BEAUT), bajo las siguientes condiciones ambientales: 22 ± 2°C de temperatura,
70 ± 5% de humedad relativa, ciclo de luz / oscuridad de 12 horas, agua ad libitum y una
ración diaria de alimento comercial (15 g/animal).
Los procedimientos experimentales se efectuaron siguiendo las normas internacionales
de protección y manejo de animales de laboratorio (Acadamies, 2011), atendiendo la ley
84 de 1989 de protección animal en Colombia y la resolución 8430/1993, contando con
la aprobación del comité local de ética de la Universidad del Tolima.
3.2 MODELO DE ISQUEMIA CEREBRAL POR EMBOLIZACIÓN ARTERIAL
Para la inducción de la isquemia cerebral por embolismo arterial se siguió el protocolo
estandarizado por el grupo en Enfermedades Neurodegenerativas de la Universidad del
Tolima, END (Hernández, Trujillo, & Céspedes Rubio, 2010). Se retiró el alimento 12
horas antes de los procedimientos quirúrgicos, para facilitar la acción de los anestésicos.
Los procedimientos quirúrgicos se realizaron en el quirófano del Laboratorio de
Biotecnología Animal de la Universidad del Tolima (LABIOAUT). Los animales fueron
anestesiados mediante la asociación Xilacina 2% (5 mg/Kg) y Ketamina 5% (90 mg/Kg)
vía intraperitoneal y atropina sulfato 1:1000 (100 µg/Kg) vía subcutánea como vagolítico
y protector del miocardio.
Seguidamente, se monitoreó la temperatura rectal con termómetro digital, se rasuró la
zona del cuello, se lavó y se desinfectó con etanol al 70%. Se ubicó el animal en posición
decúbito supino y se inmovilizó adecuadamente (Figura 4A).

67
La disección de la arteria carótida común (ACC), se realizó mediante una incisión
mediana longitudinal anterior en sentido cráneo-caudal (cervicotomía) de
aproximadamente 3 cm de longitud hasta la horquilla esternal, abarcando la piel, el tejido
celular subcutáneo y la fascia del músculo cutáneo. En la línea media, se separaron los
músculos esternohioideos mediante divulsión roma, con un retractor manual se replegó
lateralmente el músculo esternomastoideo derecho, se ubicó otro retractor entre el
músculo digástrico y el esternomastoideo, mientras se separó el músculo omohoideo
hasta que se visualizó la ACC (Figura 4B) y el nervio vago surcando la cara externa de
la arteria carótida interna (ACI), seguidamente se aíslo la ACI y se separó del nervio
vago.
El protocolo de embolización se efectuó siguiendo la metodología propuesta por Kudo y
colaboradores (Kudo et al., 1982), con algunas modificaciones realizadas por el grupo
de Enfermedades Neurodegenerativas (END), como se describe a continuación.
Se expuso la ACC derecha a nivel de la bifurcación en arteria carótida interna (ACI) y
arteria carótida externa (ACE), se colocó temporalmente una pinza microvascular de
aneurisma justo arriba del origen de la ACI y se dispuso una banda de látex alrededor de
la ACC 15 mm debajo de su bifurcación (Figura 4C).
A través de una microincisión en la ACC, se introdujo un catéter de poliestireno N°24,
avanzándolo cranealmente a través de la ACI hacia la bifurcación de la arteria cerebral
media (ACM) y la arteria cerebral anterior (ACA) a cuyo nivel se inyectó el coágulo
preformado resuspendido en un volumen de 400 µL de solución salina isotónica (NaCl).
La preparación del coágulo se realizó in situ siguiendo el método propuesto por Wang y
colaboradores (C. X. Wang, Yang, Yang, & Shuaib, 2001; C. Wang et al., 2001), con las
siguientes innovaciones: se cargaron 50 µl de sangre de la ACC mediante ligera presión
negativa con jeringuilla de 1 cc acoplada al catéter, conteniendo 50 µl de alfa trombina
bovina (α-tb 13 NIH/U -HTI Lab, BCT 1020); luego de 3 minutos, se formó el coágulo, se
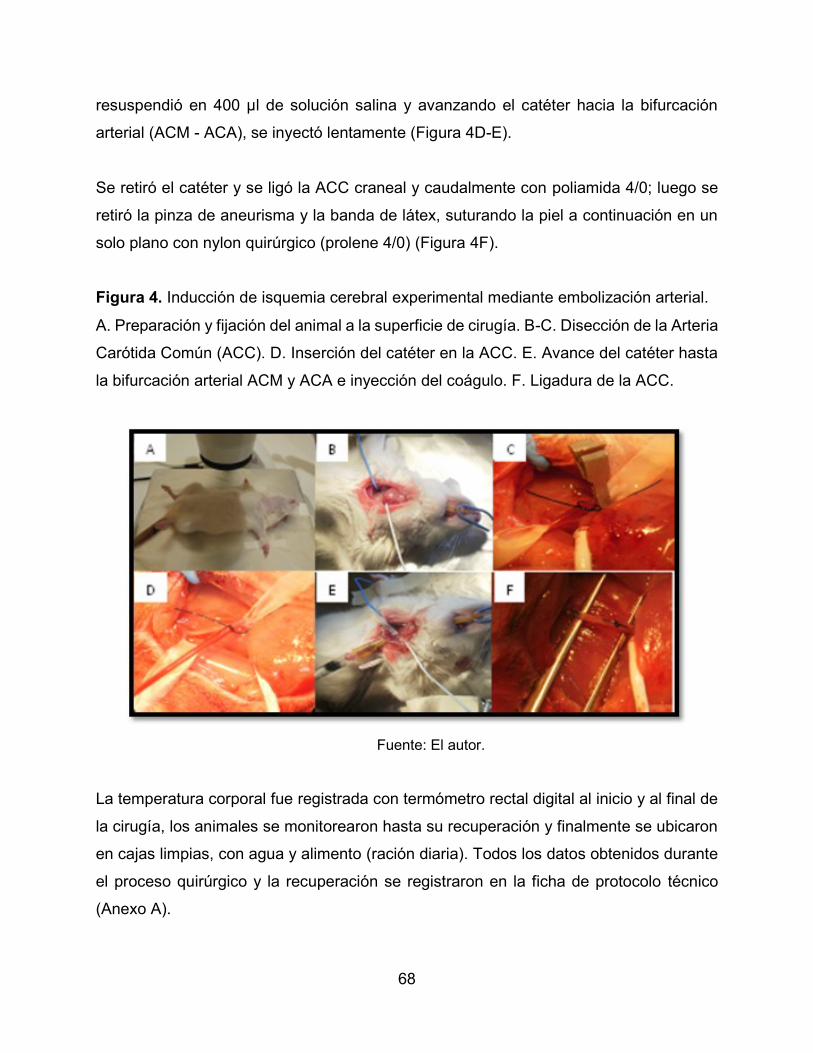
68
resuspendió en 400 µl de solución salina y avanzando el catéter hacia la bifurcación
arterial (ACM - ACA), se inyectó lentamente (Figura 4D-E).
Se retiró el catéter y se ligó la ACC craneal y caudalmente con poliamida 4/0; luego se
retiró la pinza de aneurisma y la banda de látex, suturando la piel a continuación en un
solo plano con nylon quirúrgico (prolene 4/0) (Figura 4F).
Figura 4. Inducción de isquemia cerebral experimental mediante embolización arterial.
A. Preparación y fijación del animal a la superficie de cirugía. B-C. Disección de la Arteria
Carótida Común (ACC). D. Inserción del catéter en la ACC. E. Avance del catéter hasta
la bifurcación arterial ACM y ACA e inyección del coágulo. F. Ligadura de la ACC.
Fuente: El autor.
La temperatura corporal fue registrada con termómetro rectal digital al inicio y al final de
la cirugía, los animales se monitorearon hasta su recuperación y finalmente se ubicaron
en cajas limpias, con agua y alimento (ración diaria). Todos los datos obtenidos durante
el proceso quirúrgico y la recuperación se registraron en la ficha de protocolo técnico
(Anexo A).
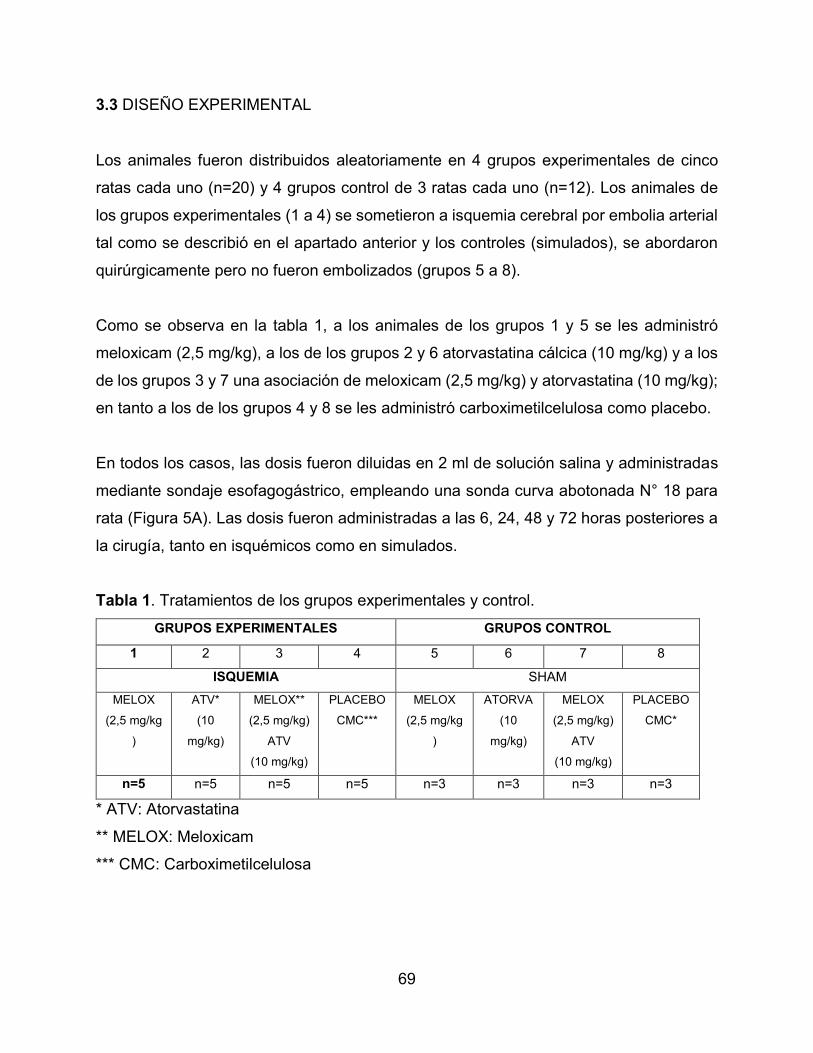
69
3.3 DISEÑO EXPERIMENTAL
Los animales fueron distribuidos aleatoriamente en 4 grupos experimentales de cinco
ratas cada uno (n=20) y 4 grupos control de 3 ratas cada uno (n=12). Los animales de
los grupos experimentales (1 a 4) se sometieron a isquemia cerebral por embolia arterial
tal como se describió en el apartado anterior y los controles (simulados), se abordaron
quirúrgicamente pero no fueron embolizados (grupos 5 a 8).
Como se observa en la tabla 1, a los animales de los grupos 1 y 5 se les administró
meloxicam (2,5 mg/kg), a los de los grupos 2 y 6 atorvastatina cálcica (10 mg/kg) y a los
de los grupos 3 y 7 una asociación de meloxicam (2,5 mg/kg) y atorvastatina (10 mg/kg);
en tanto a los de los grupos 4 y 8 se les administró carboximetilcelulosa como placebo.
En todos los casos, las dosis fueron diluidas en 2 ml de solución salina y administradas
mediante sondaje esofagogástrico, empleando una sonda curva abotonada N° 18 para
rata (Figura 5A). Las dosis fueron administradas a las 6, 24, 48 y 72 horas posteriores a
la cirugía, tanto en isquémicos como en simulados.
Tabla 1. Tratamientos de los grupos experimentales y control.
GRUPOS EXPERIMENTALES GRUPOS CONTROL
1 2 3 4 5 6 7 8
ISQUEMIA SHAM
MELOX
(2,5 mg/kg
)
ATV*
(10
mg/kg)
MELOX**
(2,5 mg/kg)
ATV
(10 mg/kg)
PLACEBO
CMC***
MELOX
(2,5 mg/kg
)
ATORVA
(10
mg/kg)
MELOX
(2,5 mg/kg)
ATV
(10 mg/kg)
PLACEBO
CMC*
n=5 n=5 n=5 n=5 n=3 n=3 n=3 n=3
* ATV: Atorvastatina
** MELOX: Meloxicam
*** CMC: Carboximetilcelulosa

70
3.4 EVALUACIÓN NEUROLÓGICA
Los animales isquémicos y simulados fueron evaluados neurológicamente a las 24, 48,
72 y 96 horas post-cirugía (Figura 5B), mediante los test neurológicos descritos por otros
autores, con algunas modificaciones como se describe a continuación (Bederson et al.,
1986; Juha Yrjänheikki et al., 2005).
El test asigna valores entre 0 y 6 dependiendo del grado de afectación neurológica. El
valor 0 indica que el animal no presenta movimientos espontáneos, 1 muestra
movimientos circulares espontáneamente, 2 se mueve circularmente si es tirado de la
cola, 3 movimientos circulares si es levantado y tirado de la cola, 4 resistencia reducida
a la presión lateral, 5 flexión consistente de uno o ambos miembros anteriores, 6
extensión normal de ambos miembros anteriores cuando es izado de la superficie (Anexo
B). Además, se realizó una evaluación neurológica complementaria en donde se tuvieron
en cuenta diferentes parámetros como el reflejo de agarre, el reflejo de corrección,
reacción de posición por pérdida de apoyo, localización visual, reflejo corneal, reflejo
palpebral, estimulación auditiva intensa y prueba de equilibrio en barra horizontal y
plataforma inclinada, modificado a partir del examen neurológico descrito por Belayev y
colaboradores (Belayev, Busto, Zhao, Fernandez, & Ginsberg, 1999).
3.5 PERFUSIÓN Y PROCESAMIENTO DE LOS CEREBROS
48 horas después de la administración de la última dosis de tratamiento, los animales
fueron sacrificados bajo anestesia general con una mezcla de Xilazina (10 mg/kg) y
Pentobarbital sódico (60 mg/kg) i.p., e inmediatamente fueron perfundidos con 200 ml de
solución salina isotónica (0,9% NaCl) por vía intracardíaca y avance aórtico, seguido de
200 ml de paraformaldehído (PFA) 4% en buffer fosfato 0,1 M pH 7,4. Los cerebros
fueron extraídos y postfijados 48 horas en PFA 4% a 4°C (Figura 5C) y luego tratados
con gradientes de sacarosa (7%, 25% y 30%) para reducir el edema intersticial y
posteriormente se sumergieron en una solución criopreservante (etilenglicol 30%, glicerol

71
30%, buffer fosfato 0,4 M 25% y agua desionizada 15%) a -20°C hasta el momento de
ser cortados.
Luego de cinco lavados consecutivos con PB 0,1M en agitación (shaker) y a temperatura
ambiente, los cerebros crioconservados se cortaron coronalmente, entre los bregma -2,0
a -4,5 (Paxinos & Watson, 1998), con un Vibratomo (Vibratome® 1500) a 50 µm de
espesor, en buffer fosfato (PB 0,1M) a 4°C (Figura 5D). Los cortes obtenidos se
almacenaron en cajas multipozo y se mantuvieron en criopreservante a -20°C hasta el
montaje de las técnicas histoquímicas e inmunohistoquímicas.
Figura 5. Administración de los tratamientos, pruebas comportamentales y
procesamiento de los cerebros después de la inducción experimental de la isquemia.
A. Administración de medicamento mediante gavaje. B. Evaluación neurológica, prueba
de equilibrio en barra transversal. C. Cerebro perfundido. D. Vibratome® 1500 empleado
para obtener los cortes coronales.
Fuente: Grupo en Enfermedades Neurodegenerativas
3.6 PROCESAMIENTO HISTOLÓGICO
El procesamiento histológico se realizó en las instalaciones del Laboratorio de
Toxicología de la Universidad del Tolima.

72
Para la evaluación de los marcadores histoquímicos e inmunohistoquímicos se
emplearon cortes comprendidos entre los bregmas -2,8 a -3,8; para el montaje de cada
una de las pruebas, se emplearon 2 cortes por unidad experimental. Para todos los
marcadores, se evaluaron los siguientes territorios encefálicos: la corteza
somatosensorial, la corteza motora y la corteza piriforme, las zonas CA1, CA2, CA3,
fimbria y giro dentado del hipocampo, la zona paraventricular, la capsula interna, el
putamen caudado, el tracto óptico y los núcleos talámicos (Figura 6).
Figura 6. Zonas evaluadas mediante técnicas histoquímicas e inmunohistoquímicas en
un modelo de infarto cerebral.
1. Corteza somatosensorial primaria ( ). 2. Corteza piriforme ( ). 3. CA1 hipocampal
( ). 4. CA2 hipocampal ( ). 5. CA3 hipocampal ( ). 6. Giro dentado del hipocampo
( ). 7. Fimbria del hipocampo ( ). 8. Zona paraventricular ( ). 9. Capsula interna
( ). 10. Putamen caudado ( ). 11. Tracto óptico ( ). 12 Núcleos talámicos ( ).
Fuente: Modificado de Paxinos 1998.
Para todos los marcadores, se realizó una revisión visual de las zonas anteriormente
señaladas, pero se escogieron para análisis por densitometría las zonas que revelaban

73
variaciones constantes en la reactividad en los individuos del grupo isquémico placebo
frente al grupo simulado placebo.
3.6.1 Supervivencia celular e integridad de la mielina. La supervivencia neuronal y
astrocitaria se evaluó mediante el método Kluver Barrera. El método Kluver Barrera es
una tinción diferencial que permite visualizar la mielina mediante el colorante luxol fast
blue y los somas celulares utilizando el colorante cresil violeta acetato.
Los cortes se depositaron en cajas multipozo con PB 0,1 M. Se lavaron 3 veces con una
solución de PB 0,1 M: Tritón X-100 0,05% para retirar el criopreservante. Se montaron
en láminas gelatinizadas (gelatina porcina 1% Cas # 9000-70-8) y se dejaron secar 24
horas a temperatura ambiente.
Se siguió el protocolo publicado por Klatt, E.C. en WebPath: Internet Pathology
Laboratory, con algunas adaptaciones a las condiciones del laboratorio. Los cortes se
sometieron a deshidratación en alcohol al 95% por 5 minutos, luego se transfirieron a
solución de Luxol fast blue 1% (L0294 Sigma) y se dejaron durante toda la noche en
estufa a 50°C (12 horas) (Klatt, 2010).
Al siguiente día, los cortes se sometieron a diferenciación mediante inmersiones en
etanol 95% (30 segundos), PB 0,1 M (30 segundos), carbonato de litio 0,05%, Sigma
255823, (60 segundos), etanol 70% (30 segundos) y PB 0,1 M (30 segundos); los cortes
se revisaron al microscopio y si era necesario se repetía la diferenciación. Seguidamente,
los cortes se sometieron a tinción con Cresil Violeta 0,25% (C5042 Sigma), pH 4,5
durante 10 minutos, se retiró el exceso de colorante lavándolos con PB. 0,1 M y se
deshidrataron en gradientes ascendente de alcoholes al 95% (3 minutos), etanol absoluto
(3 minutos) y xileno (5 minutos); finalmente, los cortes se cubrieron con laminillas y
sellante resinoso (Consult mount, 9990440 Thermo Scientific). Los lavados y la tinción
con cresil violeta se realizaron a una temperatura de 37°C.

74
3.6.2 Neurodegeneración. Se analizó mediante la tinción con Flourojade B®, el cual es
un fluorocromo aniónico capaz de teñir selectivamente somas y neuritas de neuronas en
degeneración (Liu, Schafer, & McCullough, 2009).
Los cortes previamente lavados se montaron en láminas gelatinizadas y secaron durante
24 horas a temperatura ambiente. Se fijaron en xileno (5 minutos, 2 veces),
posteriormente se adicionó una solución de hidróxido de sodio 1% disuelto en etanol
80% (1 minuto), se hidrataron en etanol 70% (2 minutos), se lavaron con PB. 0,1 M, se
agregó permanganato de potasio 0,06% (17 minutos a 37°C, 131527 Panreac), se
lavaron con PB. 0,1 M, se añadió Fluorojade B® 0,0004% (30 minutos a 37°C, AG310
Millipore), se lavaron con PB 0,1 M, se secaron a temperatura ambiente protegidos de la
luz, se cubrieron con laminillas y medio de montaje (Fluoromount, F4680 Sigma) y se
sellaron con esmalte transparente.
Para la evaluación de la neurodegeneración se empleó microscopio un de
epifluorescencia Zeiss Axio Lab. A1 FL-LED, en el filtro de GFP a una longitud de onda
de 470 nm (emisión) y magnificación de 20X.
3.6.3 Inmunohistoquímica. Los cortes previamente lavados se recolectaron en cajas
multipozo con PB 0,1 M.La inmunohistiquímica convencional se realizó empleando la
técnica avidina-biotina peroxidasa. Los cortes se trataron con una solución de inhibición
de la peroxidasa endógena durante 20 minutos (Metanol: PB 0,1M 1:1 y H2O2 1%), se
lavaron tres veces con PB 0,1M y se sometieron a solución de bloqueo (BSA 1%, Triton
X-100 3% y PB 0,1M) durante 90 minutos a temperatura ambiente.
Posteriormente, se incubaron con el anticuerpo primario diluido en solución de
incubación (Triton X-100 0,3%, BSA 0,3%, PB 0,1M), a 4°C toda la noche; luego, se
sumergieron en los anticuerpos secundarios biotinilados correspondientes por 120
minutos a TA, previo lavado con PB 0,1M. Seguidamente, se adicionó el complejo
avidina-biotina-peroxidasa (Kit ABC peroxidase, Pierce #32020) 1:250 A y 1:250 B
durante 120 minutos a temperatura ambiente, protegidos de la luz y en agitación
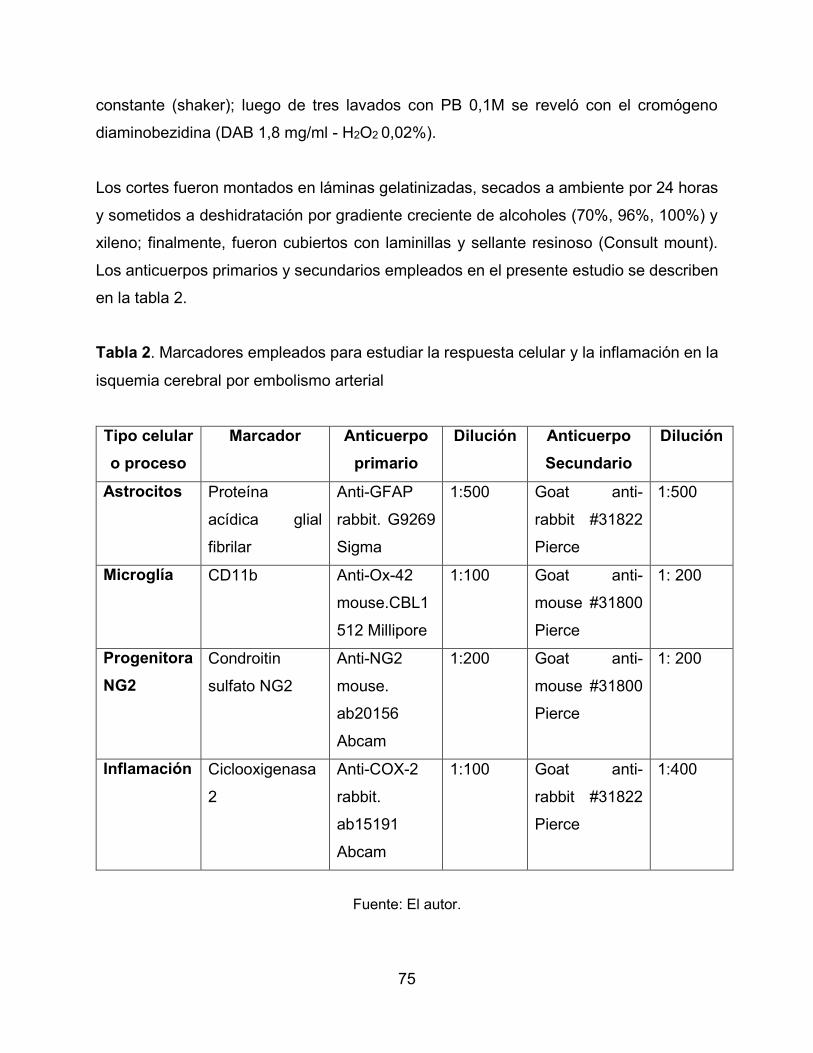
75
constante (shaker); luego de tres lavados con PB 0,1M se reveló con el cromógeno
diaminobezidina (DAB 1,8 mg/ml - H2O2 0,02%).
Los cortes fueron montados en láminas gelatinizadas, secados a ambiente por 24 horas
y sometidos a deshidratación por gradiente creciente de alcoholes (70%, 96%, 100%) y
xileno; finalmente, fueron cubiertos con laminillas y sellante resinoso (Consult mount).
Los anticuerpos primarios y secundarios empleados en el presente estudio se describen
en la tabla 2.
Tabla 2. Marcadores empleados para estudiar la respuesta celular y la inflamación en la
isquemia cerebral por embolismo arterial
Tipo celular
o proceso
Marcador Anticuerpo
primario
Dilución Anticuerpo
Secundario
Dilución
Astrocitos Proteína
acídica glial
fibrilar
Anti-GFAP
rabbit. G9269
Sigma
1:500 Goat anti-
rabbit #31822
Pierce
1:500
Microglía CD11b Anti-Ox-42
mouse.CBL1
512 Millipore
1:100 Goat anti-
mouse #31800
Pierce
1: 200
Progenitora
NG2
Condroitin
sulfato NG2
Anti-NG2
mouse.
ab20156
Abcam
1:200 Goat anti-
mouse #31800
Pierce
1: 200
Inflamación Ciclooxigenasa
2
Anti-COX-2
rabbit.
ab15191
Abcam
1:100 Goat anti-
rabbit #31822
Pierce
1:400
Fuente: El autor.

76
3.7 OBTENCIÓN Y PROCESAMIENTO DE IMÁGENES
Para la evaluación de la supervivencia celular y la integridad de la mielina, la reactividad
de las células gliales (astrocitos y microglía) y de la enzima COX 2, se empleó un
microscopio óptico (Motic BA210) con cámara digital (Moticam 2000). A diferentes
magnificaciones, 10X para supervivencia celular e integridad de la mielina, astrocitos y
microglía y 40X para astrocitos, microglía y COX 2.
Las fotografías fueron homogenizadas mediante el programa Adobe Photoshop CS4 y
posteriormente, se procesaron las imágenes digitalizadas con el software Fiji-Image J
1.46.
La respuesta de las células gliales, neuronales y la COX 2 se estudió mediante
densitometría global y/o individual dependiendo del marcador, para esto se emplearon la
imágenes digitales en formato de 8 bits (escala de grises).
3.7.1 Astrocitos. La reactividad de los astrocitos se analizó mediante densitometría global
e individual; para la densitometría global se utilizaron imágenes digitalizadas a una
magnificación de 10X, se sustrajo el fondo y se estableció el umbral por defecto en
blancos y negros, según el protocolo para mediciones densitométricas globales del grupo
END.
Para la medición de las densidades individuales y la longitud, se emplearon fotografías
digitalizadas a una magnificación de 40X, empleando el protocolo para mediciones
densitométricas individuales del grupo END; además, se estableció una escala no
globalizada en donde se determinó la distancia en pixeles (800) y se fijó la distancia
conocida en micras correspondiente al objetivo de 40X (200 µm).
Posteriormente, se midió la longitud de los astrocitos o el espacio que ocupan
espacialmente en la misma imagen, empleando la herramienta a mano alzada,

77
circunscribiendo el astrocito desde la proyección más larga de un cuadrante pasando por
el soma hasta la proyección más larga de otro cuadrante.
El número de células a medir por unidad experimental se obtuvo mediante la fórmula
para estimar el tamaño muestral (Fernández, 2001), como se describe a continuación:
𝑛 =𝑍 ∝2× 𝑃 × 𝑄
𝑑2
En donde Zα2 = 1,962 (Intervalo de confianza 95%)
P= 0,05 (Proporción esperada 5%)
Q= 1 – P (1 – 0,05 = 0,95)
d= Precisión (8%= 0,0064)
Obteniéndose un tamaño muestral n= 28, 5 células por tratamiento.
3.7.2 Microglía. Se cuantificó la reactividad de la proteína OX-42 en imágenes
digitalizadas a una magnificación de 10X, para visualizar mayor cantidad de microglía
por campo, empleando densitometría individual. Se midieron 10 células por unidad
experimental.
3.7.3 Ciclooxigenasa 2. Se midió la inmunoreactividad de la proteína COX 2 en imágenes
digitalizadas a una magnificación de 40X, mediante densitometría individual.
Los datos se obtuvieron en unidades densitométricas relativas; para el análisis de los
resultados se utilizó el valor correspondiente a la densidad interior aleatoria.
3.8 ANÁLISIS ESTADÍSTICO
Los resultados obtenidos (densitometría global, longitud y densidad celular), fueron
consignados en matrices de cálculo en donde se relacionaron cada uno de los individuos,
contra cada uno de los tratamientos. Posteriormente se calcularon parámetros
descriptivos como la media y la desviación estándar. Los datos organizados fueron

78
evaluados para los supuestos de normalidad y bondad de ajuste con ayuda del software
estadístico Infostat versión libre 2011 (Di Rienzo et al., 2011), mediante la prueba de
Shapiro Willks y Kolmogorov Smirnov.
Al obtener como resultado, la no distribución normal de los datos, se aplicó análisis de
varianza (ANOVA) no paramétrico mediante la prueba de Kruskal Wallis. Para conocer
en donde se encontraban las diferencias significativas establecidas en los ANOVA se
utilizó el test de Dunns, que realiza comparaciones múltiples entre las medias de los
tratamientos. Adicionalmente, se realizó análisis no pareado (t-Student), para evaluar el
efecto intrínseco de los medicamentos en los tratamientos controles, empleando el
programa Graph Pad (Prism v 5,0), y utilizando un nivel de confianza p < 0,05 como
significancia estadística.

79
4. RESULTADOS
4.1 EMBOLIZACIÓN ARTERIAL
Durante los procedimientos quirúrgicos se registró la temperatura inicial post-anestesia
y final post-operatoria obteniéndose una temperatura rectal inicial y final promedio de
35,6 ± 1,3 °C y 32,3 ±0,6 °C respectivamente. Desde el inicio de la cirugía hasta la
inyección del coágulo se emplearon 14,1 ±3,1 minutos y el tiempo total de la cirugía fue
de 22,5±3,8 minutos. La tasa de supervivencia fue del 74,1% y la tasa de mortalidad
intraoperatoria y posoperatoria fue de 7,4% y 18,5% respectivamente, como se
encuentra resumido en la tabla 3.
Tabla 3. Resumen de los datos registrados durante la cirugía y el posoperatorio.
Parámetros Valores
Temperatura inicial 35,6 ± 1,3 °C
Temperatura final 32,3 ± 0,6 °C
Tiempo de embolización 14,9 ± 3,1 minutos
Tiempo de cirugía 22,5 ± 3,8 minutos
Supervivencia 74,1%
Fallecidos en cirugía 7,4%
Fallecidos post-operatorio 18,5%
Fallecidos total 25,9%
Por otro lado, la concentración óptima para producir el coágulo in vivo para el modelo de
embolización arterial fue de 13 NIH/U, puesto que la concentración propuesta
inicialmente (40 NIH/U), generó elevada mortalidad intraoperatoria y posoperatoria.

80
4.2 EVALUACIÓN NEUROLÓGICA
Los resultados obtenidos muestran que todos los individuos isquémicos tratados con
meloxicam (IM), atorvastatina (IA) y la asociación meloxicam-atorvastatina (IAM), así
como los isquémicos placebo (IP), presentaron déficits neurológicos similares a las 24
horas posisquemia, obteniendo valores entre 3 y 4 del score, mostrando movimientos
circulares si eran levantados de la cola o resistencia reducida a la presión lateral,
evidenciando una afectación motora después la isquemia.
Por otro lado, se observó recuperación gradual de todos los individuos (tratados y no
tratados) después de 96 horas, presentando valores entre 5 y 6 del test, evidenciando
flexión consistente de uno de los miembros anteriores en el 30% de los individuos y
extensión normal de ambos miembros anteriores en otros (70%), como se muestra en la
figura 7, sin diferencias significativas entre los grupos isquémicos IM, IA, IAM frente al
grupo IP.
Figura 7. Registro del score neurológico de los grupos isquémicos frente al simulado
placebo.
SP: Simulado placebo. IP: Isquemia placebo. IM: Isquemia meloxicam. IA: Isquemia
atorvastatina. IAM: Isquemia asociación atorvastatina+meloxicam. Se asignaron valores
arbitrarios (AU), de acuerdo a la respuesta neurológica en cada test.
Fuente: El autor

81
Adicionalmente, se evaluaron ciertos reflejos que permiten detectar otros déficits que
podrían ser imperceptibles con el test de Bederson modificado, encontrándose que en el
80% de los animales, se afectó el reflejo palpebral, también la sensibilidad auditiva que
se alteró en el 35% de los animales, seguido por la corrección de posición por perdida
de apoyo (30%), en menor grado se perturbaron el equilibrio en barra horizontal (25%),
el equilibrio en plataforma inclinada (15%), y por último el reflejo de corrección que se vio
afectado en la evaluación neurológica a las 24 horas en el 15% de los casos, el cual se
recuperó a las 48 horas de la isquemia.
4.3 SUPERVIVENCIA CELULAR
En el modelo de isquemia cerebral por embolización arterial, a las 120 horas poscirugía,
no se observó disminución significativa de la población neuronal y astrocitaria ni de la
mielina en zonas perifocales y/o de penumbra, como la corteza somatosensorial primaria
y la CA1 del hipocampo (Figura 8).
Figura 8. Densidad de la población neuronal y astrocitaria en la corteza somatosensorial
primaria de los grupos simulados e isquémicos.
SP: Simulado placebo. IP: Isquemia placebo. SM: Simulado meloxicam. IM: Isquemia
meloxicam. SA: Simulado atorvastatina. IA: Isquemia atorvastatina. SAM: Simulado
asociación meloxicam+atorvastatina. IAM: Isquemia asociación
meloxicam+atorvastatina. Los valores densitométricos son presentados en unidades
relativas (U.R). Los datos son expresados como la media ± error estándar.
Fuente: El autor

82
Además, se visualizó una citoarquitectura homogénea y armónica en los componentes
celulares de las capas 3 – 5 de la corteza somatosensorial y en la región CA1 del
hipocampo (Figura 9).
Figura 9. Patrón de la citoarquitectura en la isquemia cerebral y frente a los tratamientos
con meloxicam, atorvastina y su asociación en la corteza somatosensorial primaria y la
CA1 del hipocampo.
A-H: Corteza Somatosensorial. I-P: CA1 del hipocampo. P: placebo, M: meloxicam, A:
atorvastatina, AM: Asociación meloxicam-atorvastatina. Imágenes magnificadas a 10X.
Barra de escala= 100 µm.
Fuente: El autor.
Contrario a lo observado en las zonas perifocales y de penumbra, como se muestra en
la figura 10, en el foco isquémico se advirtió pérdida de la arquitectura y la integridad
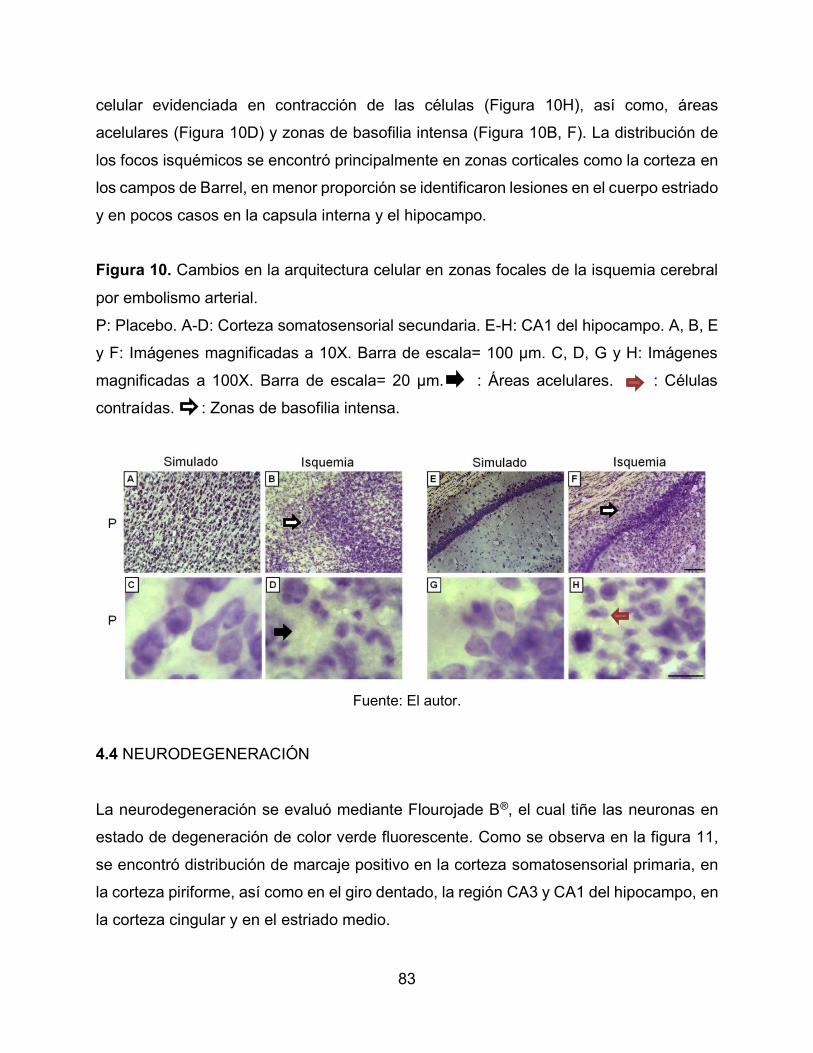
83
celular evidenciada en contracción de las células (Figura 10H), así como, áreas
acelulares (Figura 10D) y zonas de basofilia intensa (Figura 10B, F). La distribución de
los focos isquémicos se encontró principalmente en zonas corticales como la corteza en
los campos de Barrel, en menor proporción se identificaron lesiones en el cuerpo estriado
y en pocos casos en la capsula interna y el hipocampo.
Figura 10. Cambios en la arquitectura celular en zonas focales de la isquemia cerebral
por embolismo arterial.
P: Placebo. A-D: Corteza somatosensorial secundaria. E-H: CA1 del hipocampo. A, B, E
y F: Imágenes magnificadas a 10X. Barra de escala= 100 µm. C, D, G y H: Imágenes
magnificadas a 100X. Barra de escala= 20 µm. : Áreas acelulares. : Células
contraídas. : Zonas de basofilia intensa.
Fuente: El autor.
4.4 NEURODEGENERACIÓN
La neurodegeneración se evaluó mediante Flourojade B®, el cual tiñe las neuronas en
estado de degeneración de color verde fluorescente. Como se observa en la figura 11,
se encontró distribución de marcaje positivo en la corteza somatosensorial primaria, en
la corteza piriforme, así como en el giro dentado, la región CA3 y CA1 del hipocampo, en
la corteza cingular y en el estriado medio.
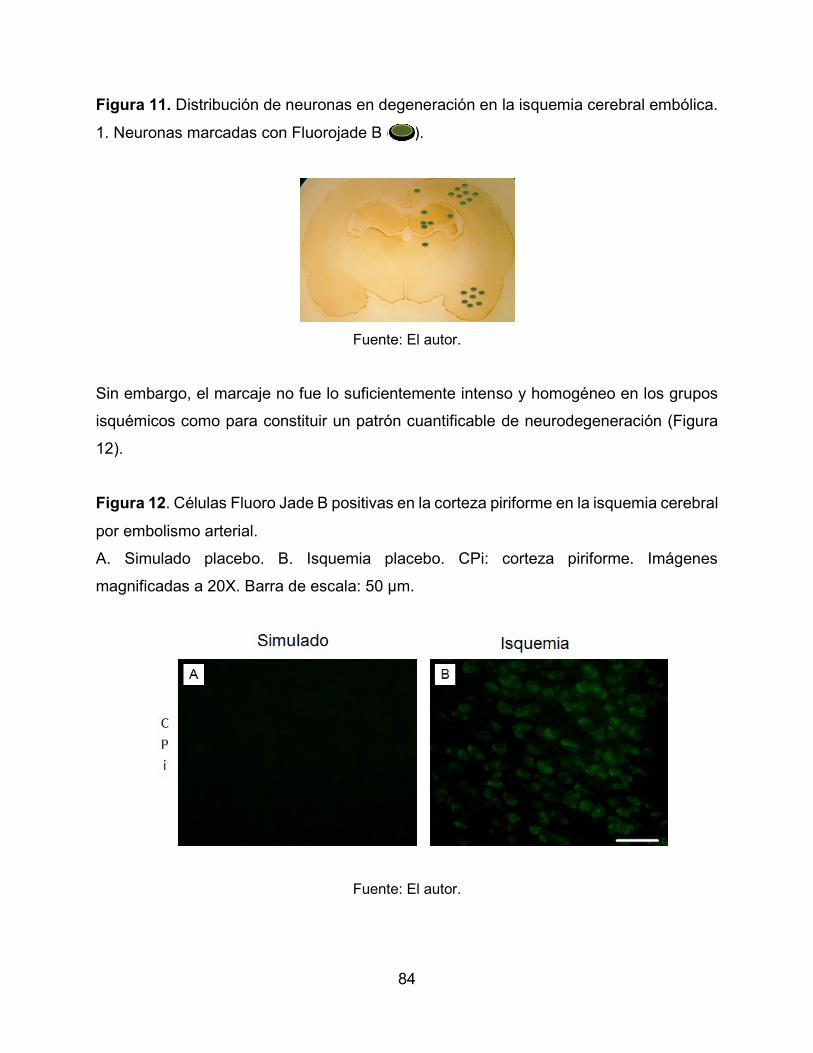
84
Figura 11. Distribución de neuronas en degeneración en la isquemia cerebral embólica.
1. Neuronas marcadas con Fluorojade B ( ).
Fuente: El autor.
Sin embargo, el marcaje no fue lo suficientemente intenso y homogéneo en los grupos
isquémicos como para constituir un patrón cuantificable de neurodegeneración (Figura
12).
Figura 12. Células Fluoro Jade B positivas en la corteza piriforme en la isquemia cerebral
por embolismo arterial.
A. Simulado placebo. B. Isquemia placebo. CPi: corteza piriforme. Imágenes
magnificadas a 20X. Barra de escala: 50 µm.
Fuente: El autor.

85
4.5 INMUNORREACTIVIDAD DE CÉLULAS GLIALES
4.5.1 Células precursoras NG2. Se encontró inmunomarcaje positivo para células
precursoras NG2 o polidendrocitos en tres de cinco casos; este marcaje, se evidenció en
individuos en los que más del 50% del corte coronal en el hemisferio ipsilateral fue
afectado por la isquemia.
La distribución de células positivas para NG2 se advirtió en el cuerpo calloso de la zona
paraventricular (ZPV), como se puede ver en la Figura 13.
Figura 13. Esquematización de la distribución de células precursoras NG2 en un corte
coronal (Bregma -2,8) por efecto de la isquemia cerebral embólica.
1. Célula precursora NG2 ( ).
Fuente: El autor
Adicionalmente, se observó que estas células presentaron una morfología estrellada con
un soma pequeño y varios procesos largos y delgados (Figura 14D). De otra parte, en
los animales simulados no se localizó marcaje positivo para NG2.

86
Figura 14. Inmunoreactividad de células NG2 en animales simulados e isquémicos
placebo en la zona paraventricular.
A-B: Imágenes magnificadas a 10X. Barra de escala: 100
µm. C-D: Imágenes magnificadas a 40X. Barra de escala: 50 µm. La flecha negra señala
una célula NG2.
Fuente: El autor.
4.5.2 Reactividad de la microglía. En los individuos simulados se observó que el marcaje
con Anti-OX42 se distribuyó principalmente en la sustancia blanca (cuerpo calloso, zona
paraventricular, capsula interna y en la fimbria del hipocampo), las células marcadas
demostraron una morfología típica de una microglía residente, cuerpo celular pequeño y
varios procesos largos y delgados, morfología la cual se clasificó como microglía
ramificada (Figura 15A-D).
En los animales isquémicos, se percibieron cambios morfológicos consistentes con
diferentes estados de activación microglial, dependiendo de la cercanía al foco isquémico
como se puede ver en la figura 15E-H. En las zonas perifocales, las células microgliales
presentaron un soma de mayor tamaño con retracción de los procesos o pérdida de los
mismos en el lado contrario a la lesión y un proceso dirigido hacia la zona de la lesión
(Figuras 154E-H y 16C-D)

87
Figura 15. Esquematización de la distribución de células microgliales en la isquemia
cerebral por embolismo arterial. Bregma -2,8.
1. Microglía ramificada ( ). 2. Microglía ramificada reactiva ( ). 3. Microglía
ameboide ( ). P: placebo. M: meloxicam. A: Atorvastatina. AM: Asociación
atorvastatina-meloxicam.
Fuente: El autor.
La morfología ameboide se evidenció en la corteza somatosensorial, en el hipocampo,
en el putamen caudado y en el estriado medial. En la sustancia blanca de los animales
isquémicos, se encontró principalmente microglía ramificada reactiva, en la cual, se ha
aumentado el tamaño del soma y se han retraído sus procesos (Figura 15E-H).

88
A las 120 horas posisquemia, en el foco isquémico se agruparon células de morfología
ameboide con un cuerpo celular redondeado y sin procesos como se puede notar en las
figuras 15E-H y 16 y 17E-F.
Figura 16. Distribución de células microgliales en el foco isquémico a las 120 horas, en
isquemia cerebral embólica.
Reactividad de OX-42 en el foco isquémico de la corteza somatosensoria, zona CA2 del
hipocampo (B), núcleos talámicos (C) y putamen caudado (D) a 10X (barra de escala:
100 µm). En el recuadro de la imagen A se muestra una célula con morfología ameboidea
a 100X (barra de escala 20 µm).
Fuente: El autor.
En zonas de penumbra, también se observó marcaje de OX-42, pero no circunscrita a
una forma celular específica que se denominó microglía activada ver Figura 17A.
A B
C D
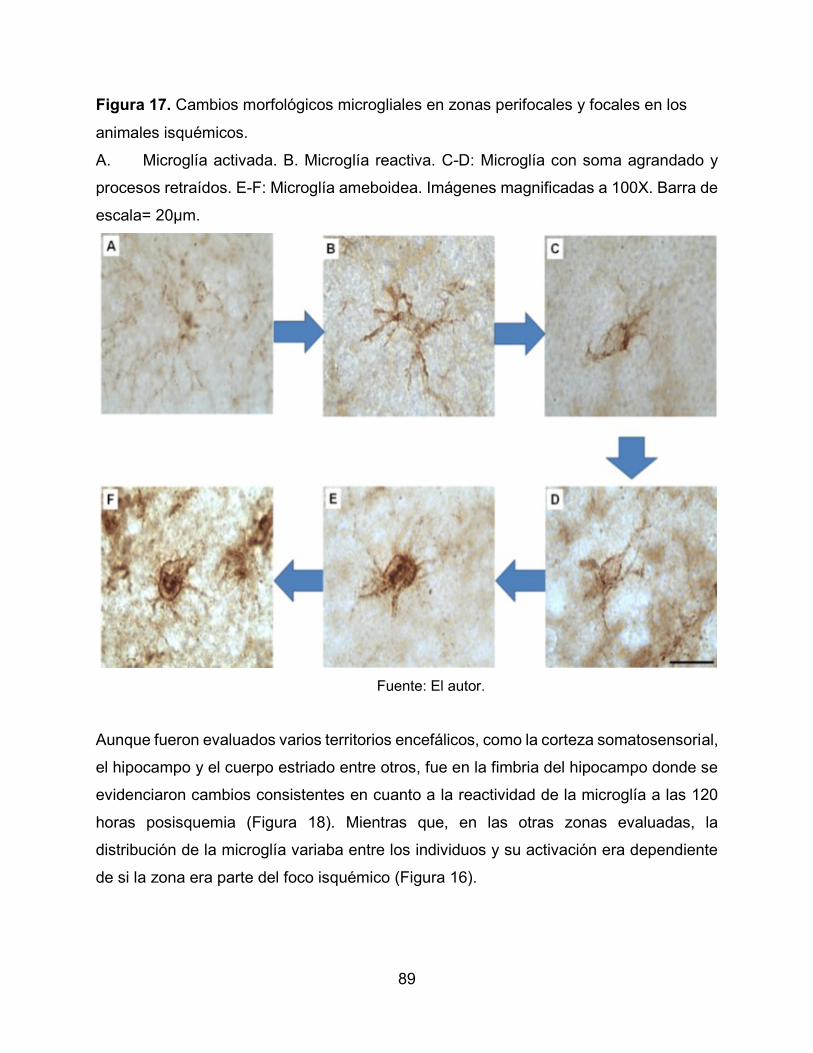
89
Figura 17. Cambios morfológicos microgliales en zonas perifocales y focales en los
animales isquémicos.
A. Microglía activada. B. Microglía reactiva. C-D: Microglía con soma agrandado y
procesos retraídos. E-F: Microglía ameboidea. Imágenes magnificadas a 100X. Barra de
escala= 20µm.
Fuente: El autor.
Aunque fueron evaluados varios territorios encefálicos, como la corteza somatosensorial,
el hipocampo y el cuerpo estriado entre otros, fue en la fimbria del hipocampo donde se
evidenciaron cambios consistentes en cuanto a la reactividad de la microglía a las 120
horas posisquemia (Figura 18). Mientras que, en las otras zonas evaluadas, la
distribución de la microglía variaba entre los individuos y su activación era dependiente
de si la zona era parte del foco isquémico (Figura 16).

90
Figura 18. Inmunorreactividad de células microgliales en la isquemia cerebral embólica
y frente a los tratamientos con meloxicam, atorvastatina y su asociación en la fimbria del
hipocampo.
P: placebo, M: meloxicam, A: atorvastatina, AM: Asociación meloxicam-atorvastatina. A-
H: Imágenes magnificadas a 10X. Barra de escala: 100 µm. I-P: Imágenes magnificadas
a 100X. Barra de escala: 20 µm.
Fuente: El autor.
En la fimbria del hipocampo, se observó una acentuada reactividad microglial en los
grupos, isquemia placebo (IP), isquemia meloxicam (IM), isquemia asociación meloxicam
- atorvastatina (IAM), con relación a los controles ver figura 17E, F, H, M, N, P. Así mismo,
se hallaron diferencias altamente significativas en la reactividad microglial entre isquemia

91
placebo (IP) y simulado placebo (SP) (****P <0,0001); simulado meloxicam (SM) e IM
(¥¥¥¥P <0,0001), simulado asociación meloxicam-atorvastatina (SAM) e isquemia
asociación atorvastatina-meloxicam (IAM) (++++P <0,0001) y entre IP e isquemia
atorvastatina (IA) (€€€€P <0,0001) como se nota en la figura 19.
Figura 19. Evaluación de la reactividad microglial en la isquemia cerebral por embolismo
arterial y frente a los tratamientos con meloxicam, atorvastatina y su asociación, en la
fimbria del hipocampo.
SP: Simulado placebo. IP: Isquemia placebo. SM: Simulado meloxicam. IM: Isquemia
meloxicam. SA: Simulado atorvastatina. IA: Isquemia atorvastatina. SAM: Simulado
asociación meloxicam+atorvastatina. IAM: Isquemia asociación
meloxicam+atorvastatina. A. Las gráficas de barras corresponden a unidades relativas
de densidad. *P <0,05, 1****P <0,0001, ¥¥¥¥P <0,0001, ++++P <0,0001, €€€€P <0,0001.
Los datos son expresados como la media ± error estándar. B. Reactividad de OX-42 de
la célula microglial en la isquemia placebo (1) y en la isquemia tratada con atorvastatina
(2) a 100X (barra de escala= 20 µm).
Fuente: El autor
Contrario a lo observado en los grupos isquémicos tratados con meloxicam y la
asociación meloxicam-atorvastatina, el tratamiento con atorvastatina disminuyo el
inmunomarcaje de OX-42 a tal punto que la reactividad de la microglía en respuesta a la
isquemia mostró un comportamiento similar al SP, en cuanto a la densidad celular como
se visualiza en las figuras 18G, O y 19. Además, se encontró aumento importante de la
A B 1
2

92
densidad microglial en los grupos simulado meloxicam (SM) y simulado asociación
meloxicam-atorvastatina (SAM) frente al SP (*P <0,05).
En el grupo IP, se constató que la microglía a las 120 horas posisquemia, aumenta su
densidad celular y disminuye su longitud al compararla con el SP (53,8±2,2 µm versus
62,4±1,7 µm). Así mismo, en los grupos isquémicos tratados con meloxicam y la
asociación meloxicam atorvastatina se evidenció incremento de la densidad y la longitud
celular (77,90±1,7 µm y 72,96±1,1, respectivamente) al confrontarla con el grupo IP,
como se puede ver en las Figuras 18A, E; I, M y 19.
Adicionalmente, en los grupos simulados se notaron cambios en la densidad y la longitud
de la microglía frente a los tratamientos, por lo que se analizaron estos parámetros
mediante un test no pareado (t-Student), encontrándose aumento significativo en la
densidad celular en los grupos SM, SAM (****P <0,0001) y SA (**P=0,0012; P<0,01),
93640 U.R., 96700 U.R. y 73610 U.R. respectivamente, versus 56870 U.R del SP (figura
20A); también, se halló aumento significativo en la longitud microglial del grupo SA
(*P=0,029) con un promedio de 68,8±1,7 µm frente a 62,4± 2,2 µm del SP (Figura 20B).
Figura 20. Densidad y longitud microglial de los grupos simulados tratados con
meloxicam, atorvastatina y su asociación en la fimbria del hipocampo.
SP: Simulado placebo. SM: Simulado meloxicam. SA: Simulado atorvastatina. SAM:
Simulado asociación meloxicam+atorvastatina. Las gráficas de barras corresponden a
unidades relativas de densidad. *P <0,05. Los datos son expresados como la media ±
error estándar.
Fuente: El autor.

93
4.5.3 Reactividad de los astrocitos. Se evaluó el efecto de la isquemia y de los
tratamientos a las 120 horas post-cirugía, sobre la reactividad de los astrocitos en
diversos territorios encefálicos como: la zona hipocampal CA1, la corteza
somatosensorial y el putamen caudado.
En la CA1 hipocampal, se observaron cambios morfológicos en los astrocitos
representados en aumento de la densidad celular y disminución de la longitud celular,
43±2,7 µm en el IP frente a 51±2,8 µm del SP. Contrario a los IP, los astrocitos de los
grupos IM, IA e IAM, aumentaron su longitud (55,1±1,9 µm, 61,7±2,2 µm y 57,4±1,3 µm,
respectivamente), como se visualiza en las figura 21N-P.
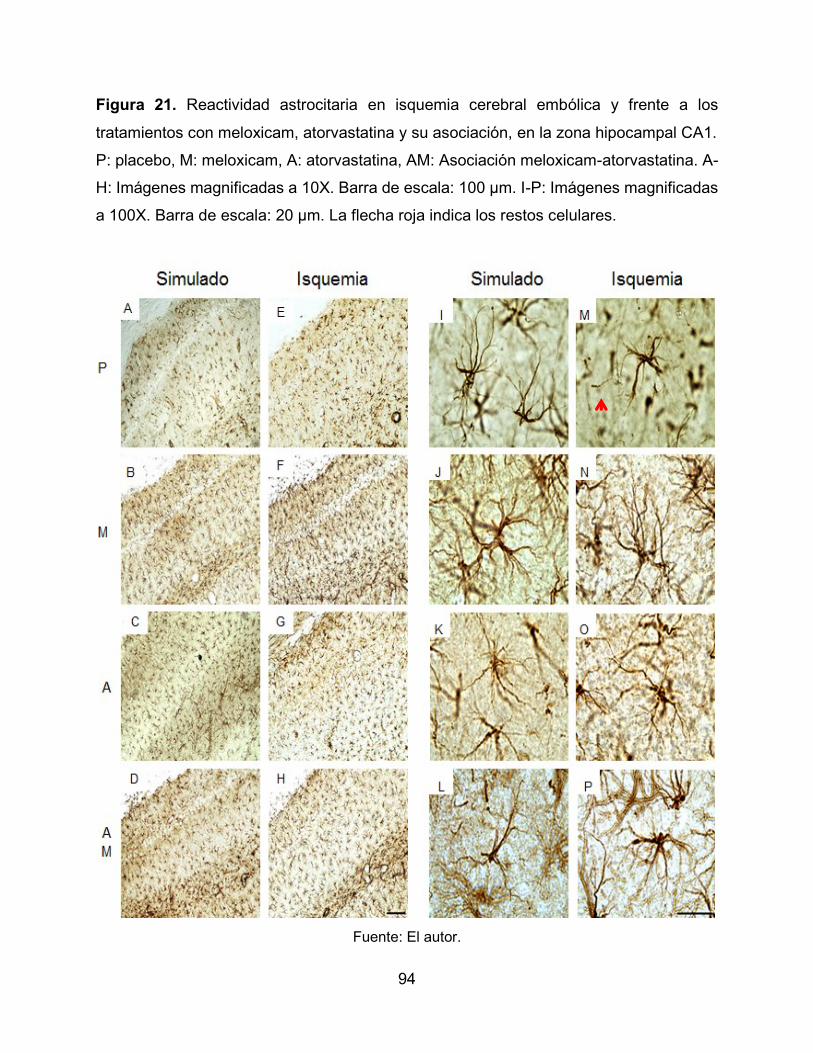
94
Figura 21. Reactividad astrocitaria en isquemia cerebral embólica y frente a los
tratamientos con meloxicam, atorvastatina y su asociación, en la zona hipocampal CA1.
P: placebo, M: meloxicam, A: atorvastatina, AM: Asociación meloxicam-atorvastatina. A-
H: Imágenes magnificadas a 10X. Barra de escala: 100 µm. I-P: Imágenes magnificadas
a 100X. Barra de escala: 20 µm. La flecha roja indica los restos celulares.
Fuente: El autor.
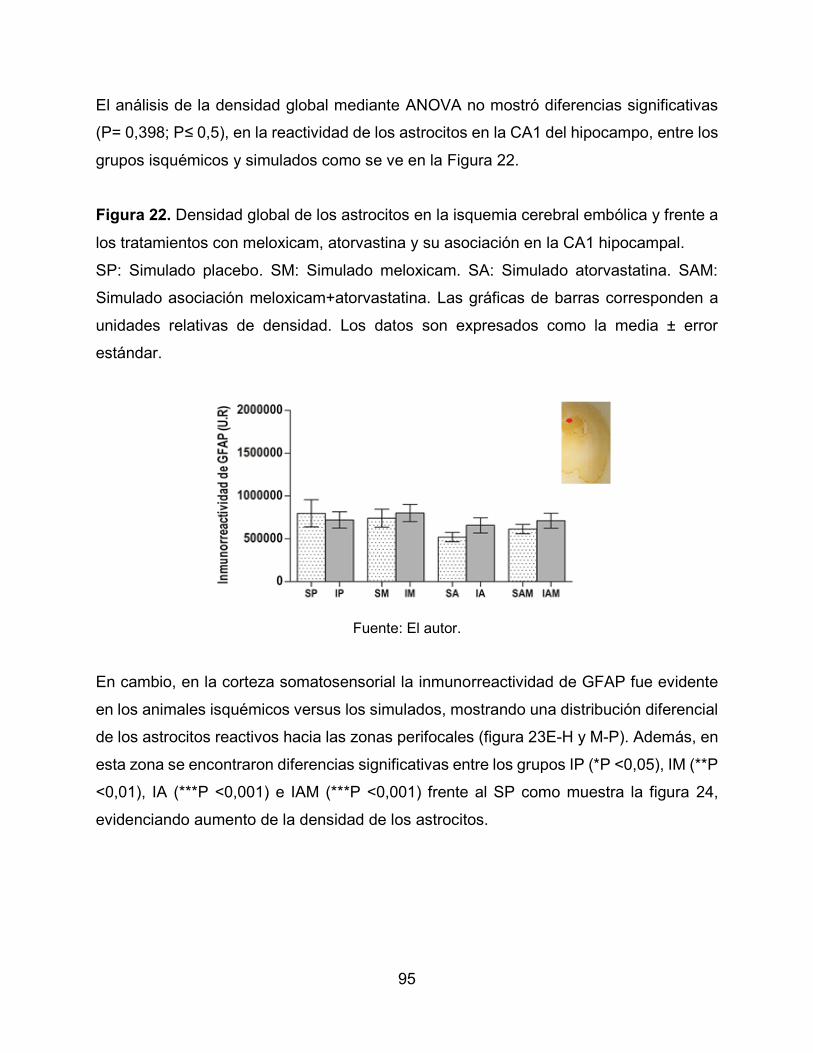
95
El análisis de la densidad global mediante ANOVA no mostró diferencias significativas
(P= 0,398; P≤ 0,5), en la reactividad de los astrocitos en la CA1 del hipocampo, entre los
grupos isquémicos y simulados como se ve en la Figura 22.
Figura 22. Densidad global de los astrocitos en la isquemia cerebral embólica y frente a
los tratamientos con meloxicam, atorvastina y su asociación en la CA1 hipocampal.
SP: Simulado placebo. SM: Simulado meloxicam. SA: Simulado atorvastatina. SAM:
Simulado asociación meloxicam+atorvastatina. Las gráficas de barras corresponden a
unidades relativas de densidad. Los datos son expresados como la media ± error
estándar.
Fuente: El autor.
En cambio, en la corteza somatosensorial la inmunorreactividad de GFAP fue evidente
en los animales isquémicos versus los simulados, mostrando una distribución diferencial
de los astrocitos reactivos hacia las zonas perifocales (figura 23E-H y M-P). Además, en
esta zona se encontraron diferencias significativas entre los grupos IP (*P <0,05), IM (**P
<0,01), IA (***P <0,001) e IAM (***P <0,001) frente al SP como muestra la figura 24,
evidenciando aumento de la densidad de los astrocitos.
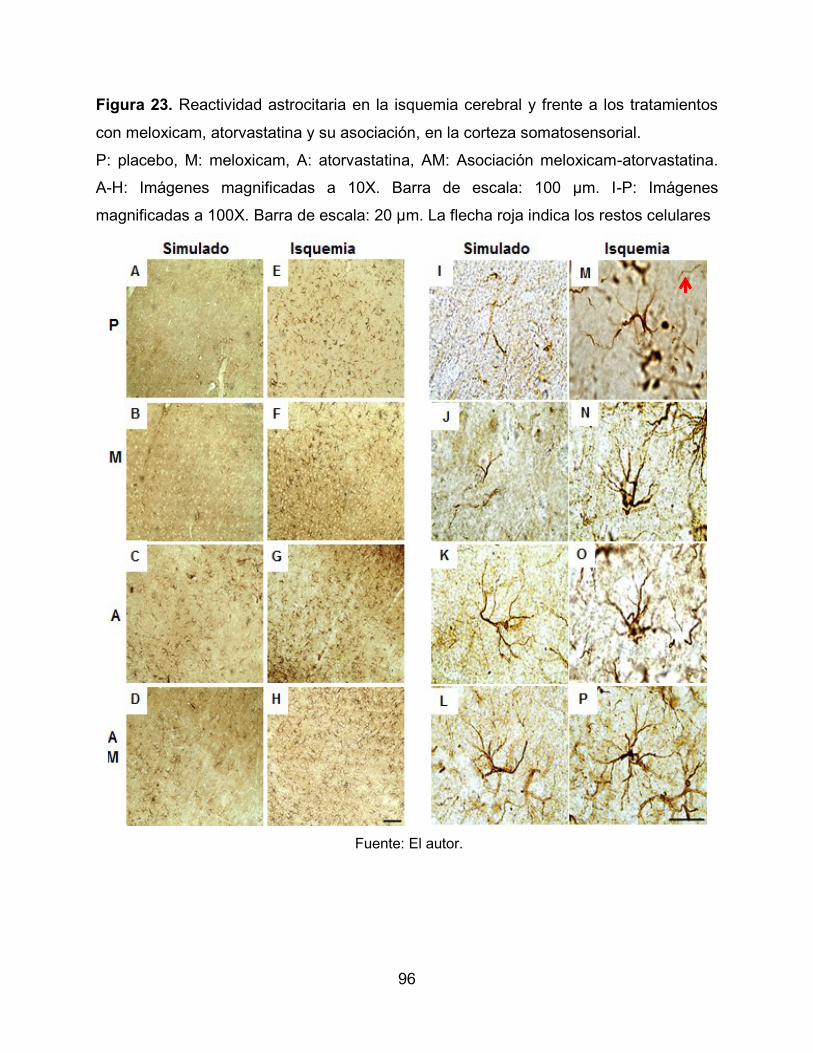
96
Figura 23. Reactividad astrocitaria en la isquemia cerebral y frente a los tratamientos
con meloxicam, atorvastatina y su asociación, en la corteza somatosensorial.
P: placebo, M: meloxicam, A: atorvastatina, AM: Asociación meloxicam-atorvastatina.
A-H: Imágenes magnificadas a 10X. Barra de escala: 100 µm. I-P: Imágenes
magnificadas a 100X. Barra de escala: 20 µm. La flecha roja indica los restos celulares
Fuente: El autor.
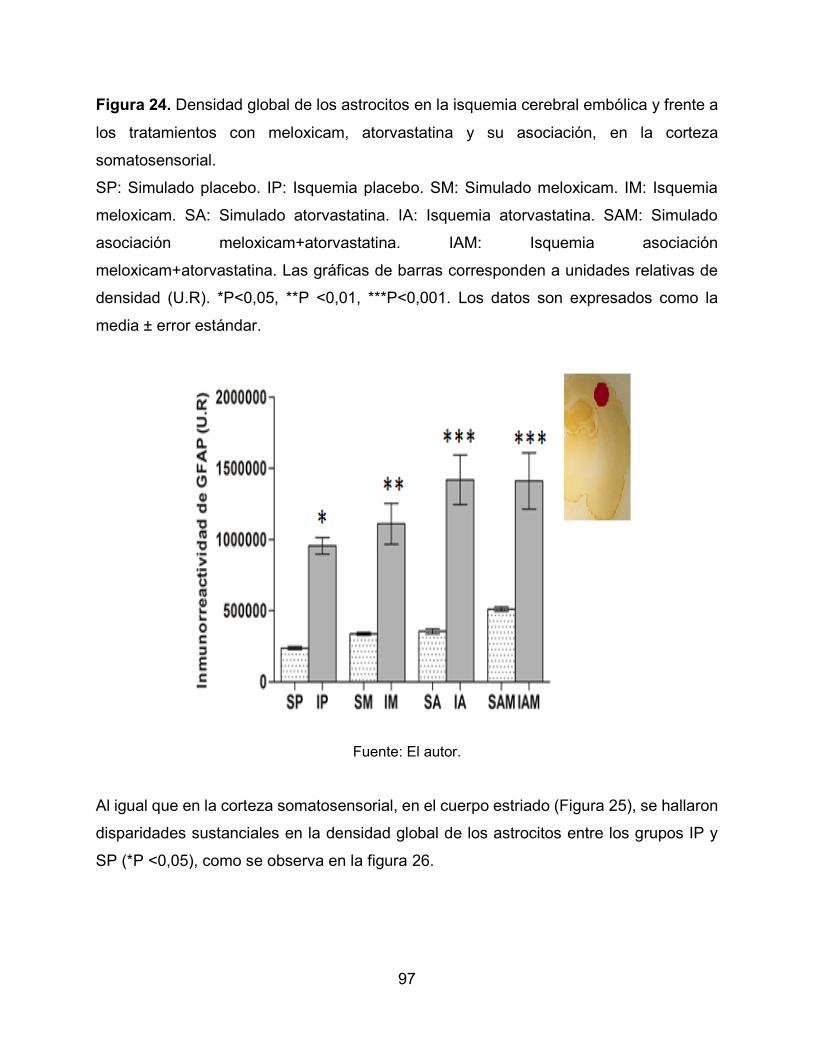
97
Figura 24. Densidad global de los astrocitos en la isquemia cerebral embólica y frente a
los tratamientos con meloxicam, atorvastatina y su asociación, en la corteza
somatosensorial.
SP: Simulado placebo. IP: Isquemia placebo. SM: Simulado meloxicam. IM: Isquemia
meloxicam. SA: Simulado atorvastatina. IA: Isquemia atorvastatina. SAM: Simulado
asociación meloxicam+atorvastatina. IAM: Isquemia asociación
meloxicam+atorvastatina. Las gráficas de barras corresponden a unidades relativas de
densidad (U.R). *P<0,05, **P <0,01, ***P<0,001. Los datos son expresados como la
media ± error estándar.
Fuente: El autor.
Al igual que en la corteza somatosensorial, en el cuerpo estriado (Figura 25), se hallaron
disparidades sustanciales en la densidad global de los astrocitos entre los grupos IP y
SP (*P <0,05), como se observa en la figura 26.
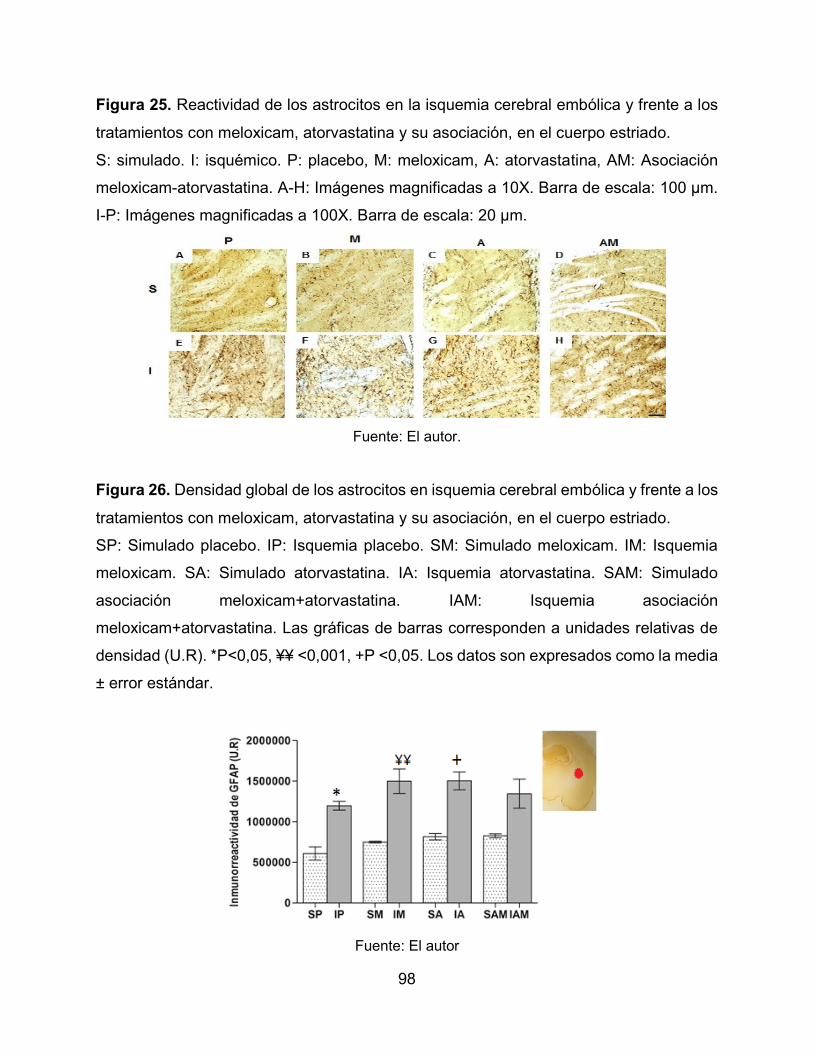
98
Figura 25. Reactividad de los astrocitos en la isquemia cerebral embólica y frente a los
tratamientos con meloxicam, atorvastatina y su asociación, en el cuerpo estriado.
S: simulado. I: isquémico. P: placebo, M: meloxicam, A: atorvastatina, AM: Asociación
meloxicam-atorvastatina. A-H: Imágenes magnificadas a 10X. Barra de escala: 100 µm.
I-P: Imágenes magnificadas a 100X. Barra de escala: 20 µm.
Fuente: El autor.
Figura 26. Densidad global de los astrocitos en isquemia cerebral embólica y frente a los
tratamientos con meloxicam, atorvastatina y su asociación, en el cuerpo estriado.
SP: Simulado placebo. IP: Isquemia placebo. SM: Simulado meloxicam. IM: Isquemia
meloxicam. SA: Simulado atorvastatina. IA: Isquemia atorvastatina. SAM: Simulado
asociación meloxicam+atorvastatina. IAM: Isquemia asociación
meloxicam+atorvastatina. Las gráficas de barras corresponden a unidades relativas de
densidad (U.R). *P<0,05, ¥¥ <0,001, +P <0,05. Los datos son expresados como la media
± error estándar.
Fuente: El autor
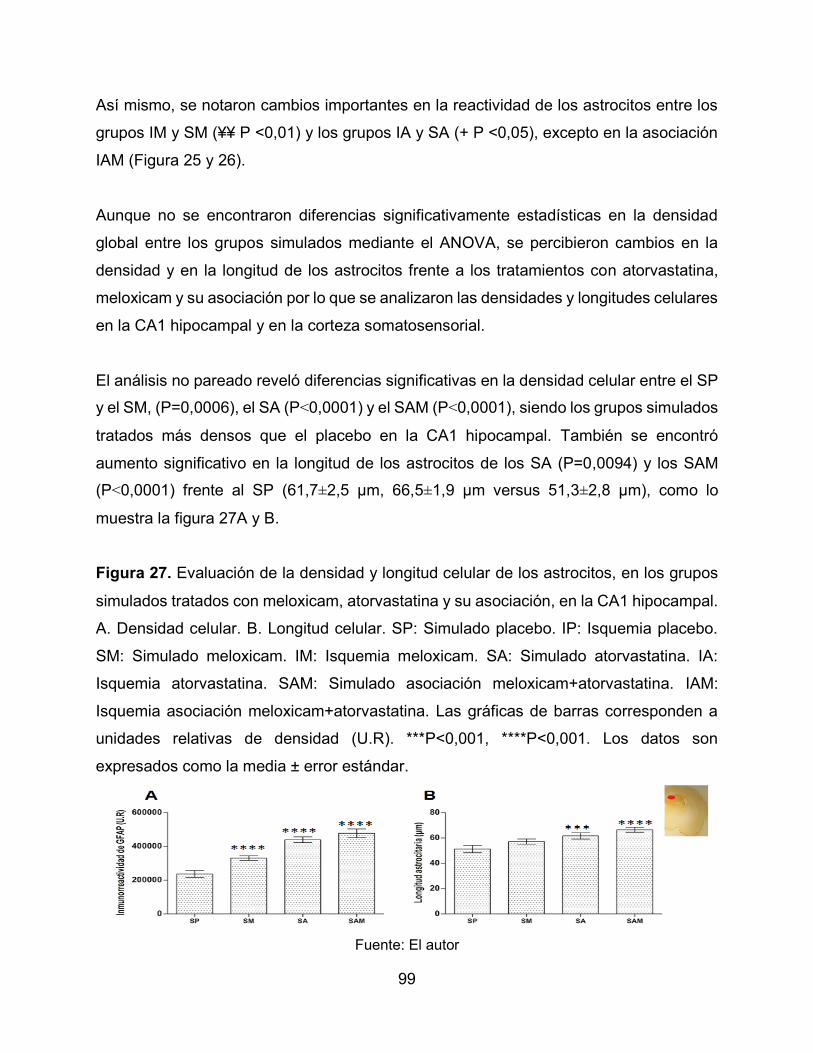
99
Así mismo, se notaron cambios importantes en la reactividad de los astrocitos entre los
grupos IM y SM (¥¥ P <0,01) y los grupos IA y SA (+ P <0,05), excepto en la asociación
IAM (Figura 25 y 26).
Aunque no se encontraron diferencias significativamente estadísticas en la densidad
global entre los grupos simulados mediante el ANOVA, se percibieron cambios en la
densidad y en la longitud de los astrocitos frente a los tratamientos con atorvastatina,
meloxicam y su asociación por lo que se analizaron las densidades y longitudes celulares
en la CA1 hipocampal y en la corteza somatosensorial.
El análisis no pareado reveló diferencias significativas en la densidad celular entre el SP
y el SM, (P=0,0006), el SA (P<0,0001) y el SAM (P<0,0001), siendo los grupos simulados
tratados más densos que el placebo en la CA1 hipocampal. También se encontró
aumento significativo en la longitud de los astrocitos de los SA (P=0,0094) y los SAM
(P<0,0001) frente al SP (61,7±2,5 µm, 66,5±1,9 µm versus 51,3±2,8 µm), como lo
muestra la figura 27A y B.
Figura 27. Evaluación de la densidad y longitud celular de los astrocitos, en los grupos
simulados tratados con meloxicam, atorvastatina y su asociación, en la CA1 hipocampal.
A. Densidad celular. B. Longitud celular. SP: Simulado placebo. IP: Isquemia placebo.
SM: Simulado meloxicam. IM: Isquemia meloxicam. SA: Simulado atorvastatina. IA:
Isquemia atorvastatina. SAM: Simulado asociación meloxicam+atorvastatina. IAM:
Isquemia asociación meloxicam+atorvastatina. Las gráficas de barras corresponden a
unidades relativas de densidad (U.R). ***P<0,001, ****P<0,001. Los datos son
expresados como la media ± error estándar.
Fuente: El autor

100
En la corteza somatosensorial, se evidenció incremento significativo de la densidad de
las células astrogliales en los grupos SA (P<0,0001) y SAM (P<0,0001) al compararlas
con el SP. Igualmente, los astrocitos extendieron sus longitudes considerablemente en
los grupos SA (P<0,0001) y SAM (P<0,0001) con 53,5±1,6 µm en el SA, 45,3±3,6 µm el
SAM frente a 29,5±4,2 µm del SP (figura 28B).
Figura 28. Evaluación de la densidad y longitud celular de los astrocitos en los grupos
simulados tratados con meloxicam, atorvastatina y su asociación, en la corteza
somatosensorial.
A. Densidad celular. B. Longitud celular. SP: Simulado placebo. IP: Isquemia placebo.
SM: Simulado meloxicam. IM: Isquemia meloxicam. SA: Simulado atorvastatina. IA:
Isquemia atorvastatina. SAM: Simulado asociación meloxicam+atorvastatina. IAM:
Isquemia asociación meloxicam+atorvastatina. Las gráficas de barras corresponden a
unidades relativas de densidad (U.R). ***P<0,001, ****P<0,001. Los datos son
expresados como la media ± error estándar.
Fuente: El Autor

101
4.5.6 Reactividad de la enzima ciclooxigenasa 2. Se encontró distribución de la
inmunoreactividad de la enzima COX 2 en todos los sectores y capas corticales, así como
en el hipocampo (giro dentado y CA3), de los grupos simulados (Figura 29A).
Figura 29. Esquematización de la distribución de la reactividad de COX 2 en la isquemia
cerebral por embolismo arterial.
A. Simulado placebo. B. Isquemia placebo. 1. Marcaje de células positivas para COX-2
( ). 2. Marcaje de células positivas para COX-2 en zonas perifocales y de penumbra
( ).
Fuente: El autor
En los grupos isquémicos, se observó disminución de la reactividad de COX-2 en zonas
muy definidas del foco e incremento en zonas perifocales como la corteza
somatosensorial primaria y de penumbra como el giro dentado del hipocampo, en el
grupo IP frente al SP como lo muestra las Figura 29B y 30E, L.
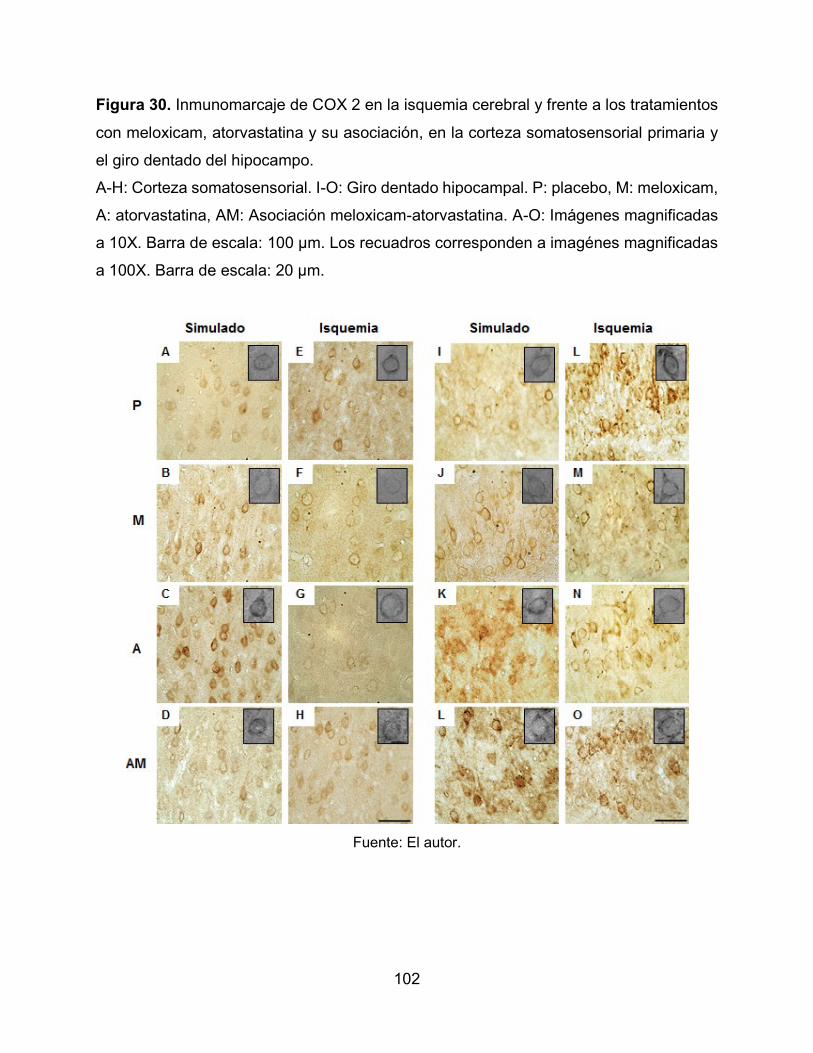
102
Figura 30. Inmunomarcaje de COX 2 en la isquemia cerebral y frente a los tratamientos
con meloxicam, atorvastatina y su asociación, en la corteza somatosensorial primaria y
el giro dentado del hipocampo.
A-H: Corteza somatosensorial. I-O: Giro dentado hipocampal. P: placebo, M: meloxicam,
A: atorvastatina, AM: Asociación meloxicam-atorvastatina. A-O: Imágenes magnificadas
a 10X. Barra de escala: 100 µm. Los recuadros corresponden a imagénes magnificadas
a 100X. Barra de escala: 20 µm.
Fuente: El autor.

103
Además, en la corteza somatosensorial primaria del grupo IP, se halló hiperreactividad
de COX 2 con diferencias altamente significativas en relación al grupo SP (***P <0,001),
como se visualiza en las figuras 30A, 30E y 31A.
En los grupos tratados, se evidenció una marcada reducción en la inmunoreactividad de
COX 2 que difieren considerablemente del grupo IP; frente al grupo IM (¥¥P <0,01), IA
(€€€P <0,001), IAM (++P <0,01). Ver figuras 30E-H y 31A. Además, se observaron
diferencias significativas entre los grupos SA e IA (***P <0,001), notándose una mayor
reactividad de COX 2 en el SA con respecto a IA, (Figuras 30C, 30G y 31A).
Figura 31. Evaluación de la reactividad de COX-2 en la isquemia cerebral embólica y
frente a los tratamientos con meloxicam, atorvastatina y su asociación, en la corteza
somatosensorial y en el giro dentado del hipocampo.
SP: Simulado placebo. IP: Isquemia placebo. SM: Simulado meloxicam. IM: Isquemia
meloxicam. SA: Simulado atorvastatina. IA: Isquemia atorvastatina. SAM: Simulado
asociación meloxicam+atorvastatina. IAM: Isquemia asociación
meloxicam+atorvastatina. Las gráficas de barras corresponden a unidades relativas de
densidad (U.R) de datos densitométricos celulares. *P<0,05, †P<0,05, ¥¥P<0,01,
++P<0,01, €€€ P<0,01, ***P<0,001, ****P<0,001. Los datos son expresados como la
media ± error estándar.
Fuente: El autor.

104
En el giro dentado del hipocampo, se notó incremento sustancial de la reactividad entre
los SP e IP (*P<0,05) como se ve en las figuras 30I, 30L y 31B. Aunque no se advirtieron
diferencias en los grupos tratados frente al placebo, si se puede observar en las figuras
30M-O y 31B, una tendencia a la disminución de la reactividad de COX-2 en los grupos
isquémicos tratados con meloxicam, atorvastatina y la asociación meloxicam-
atorvastatina.
Finalmente, se percibió elevación significativa (*p<0,05).de la reactividad de COX 2 en
el giro dentado del hipocampo, en el grupo SAM (Figura 31B).

105
5. DISCUSIÓN
5.1 EMBOLIZACIÓN ARTERIAL
La embolización arterial tuvo una duración promedio de 22,5 ± 3,8 minutos, otros autores
han reportado tiempos similares (15 y 25 minutos) en modelo embólico con coágulo
preformado (Lan, Chopp, Zhang, Jiang, & Ewing, 1997; C. Wang et al., 2001), la poca
fluctuación en el tiempo de cirugía puede sugerir que se ha logrado estandarizar la
técnica quirúrgica, haciendo que este modelo sea reproducible y poco variable, puesto
que la mayoría de las lesiones se encontraron en las regiones de irrigación de la ACM,
principalmente en corteza y estriado. Así mismo Kudo y colaboradores (1982),
encontraron alta reproducibilidad y predictibilidad en las zonas de lesión, en modelo
embólico con coágulo preformado, atribuido a la consistencia en el tamaño y a las
características físicas del émbolo, lo que podría sugerir que en este estudio se produjeron
coágulos estables en cuanto a sus características físicas y tamaño producido con alfa
trombina bovina (α-tb).
Por otro lado, el modelo de isquemia cerebral por embolismo arterial con coágulo
producido con α-tb in situ, generó una mortalidad del 26%, resultados similares a los
mostrados por otros autores, quienes con modelo embólico con coágulo preformado
obtuvieron entre 33% a 37,5% de mortalidad, empleando microcoágulos resuspendidos
en solución salina (DiNapoli et al., 2006; Henninger et al., 2006). Aunque, Shehadag y
colaboradores en 2011, lograron disminuir la mortalidad al 11%, mediante la inyección
de un único coágulo de aproximadamente 0,8 µl de volumen, resuspendido en 3 µl de
solución salina.
La mortalidad obtenida con este modelo podría deberse a la ubicación del coágulo o que
durante el proceso de lisis los fragmentos que probablemente se liberen, migren y
lesionen áreas claves para la supervivencia del animal (Henninger et al., 2009).

106
5.2 EVALUACIÓN NEUROLÓGICA
Los resultados de la evaluación neurológica mostraron que los animales isquémicos, a
las 24 horas presentaban déficits neurológicos caracterizados por movimientos circulares
si eran levantados de la cola, coincidente con lo encontrado por otros investigadores, en
el mismo periodo de evaluación (DiNapoli et al., 2006; Henninger et al., 2006).
Igualmente, Wang, Yang y Shuaib (2001) mostraron que los animales sometidos a
isquemia cerebral embólica con coágulo preformado, a las 48 horas presentaron
resistencia reducida a la presión lateral y flexión consistente de una de la extremidades
anteriores, déficits que también fueron registrados en este trabajo, los cuales muy
probablemente son secuelas de las lesiones producidas en la corteza frontal y el estriado
(Bederson et al., 1986).
Por otro lado, otros investigadores encontraron déficits neurológicos evidentes a las 24
horas posisquemia y tendencia a la recuperación a las 48 y 72 horas, en el modelo de
oclusión temporal de la ACM, en el modelo global de oclusión de 4 vasos y en el modelo
embólico, similar a lo observado en la presente investigación (Alonso de Leciñana,
Gutiérrez, Roda, Carceller, & Díez-Tejedor, 2006; Céspedes et al., 2013).
5.3 SUPERVIVENCIA CELULAR
Como se describió en el capítulo de resultados, este modelo de embolización arterial
produjo lesiones en las zonas cerebrales irrigadas por la ACM, principalmente en la
corteza y el estriado, concordante con las zonas de daño demostradas en el modelo
embólico (Alonso de Leciñana et al., 2006; Chen Xu, Yi, Qiu, & Shuaib, 2000; DiNapoli
et al., 2006; Henninger et al., 2009; Kudo et al., 1982; C. Wang et al., 2001), en el modelo
de oclusión temporal de la ACM (Liu et al., 2009; Popp et al., 2009) y en el modelo de
oclusión permanente de la ACM (Belayev et al., 1999).
Adicionalmente, Alonso de Leciñana y colaboradores (2006) y Casal y colaboradores
(2011) además del daño en la corteza cerebral, describieron focos de lesión de tamaño

107
variable en los ganglios basales y en el núcleo caudado, zonas que también se hallaron
afectadas por la isquemia en este trabajo aunque no en todos los casos. Atribuyendo la
variabilidad de las lesiones, a la diferencia entre individuos en la eficacia de la circulación
colateral leptomeníngea hacia el área del cerebro dependiente de la arteria ocluida
(ACM), lo cual podría generar un flujo sanguíneo residual y de esta manera un volumen
cambiante de tejido viable después de la oclusión (Alonso de Leciñana et al., 2006)
Como resultado de la isquemia también se encontraron pequeños focos de lesión en
regiones del hipocampo como son CA2, CA3 y giro dentado, en unos pocos individuos
(3 animales); estos resultados, son contrarios a lo obtenido por DiNapoli y colaboradores
(2006), quienes solo evidenciaron lesiones en la corteza y el estriado, pero concordante
con los obtenidos por Kudo, Aoyama, Ichimori y Fukunaga (1982). Estas lesiones
probablemente resultan de la movilización del coágulo o de sus fragmentos, los cuales
posteriormente podrían ocluir arterias como la arteria cerebral posterior (ACP) (DiNapoli
et al., 2006).
5.4 NEURODEGENERACIÓN
La distribución de células positivas para Fluoro - jade B a las 120 horas posisquemia en
este modelo embólico, corresponde con territorios focales y perifocales como la corteza
somatosensoria primaria y de penumbra como las zonas hipocampales CA1, CA3 y giro
dentado, corteza piriforme, cingular y estriado medio, lo cual es concordante con lo
encontrado en otros estudios con modelo de oclusión temporal de la ACM, modelo
embólico e inducción de la isquemia mediante termocoagulación (Butler, Kassed,
Sanberg, Willing, & Pennypacker, 2002; Duckworth et al., 2005; Giraldi Guimarães,
Rezende Lima, Bruno, & Mendez Otero, 2009; Liu et al., 2009).
De otra parte, en este estudio no se encontró un marcaje suficientemente intenso y
homogéneo en los grupos isquémicos como para constituir un patrón cuantificable de
neurodegeneración, lo que podría ser explicado por lo descrito por otros autores quienes
evaluaron temporalmente procesos de neurodegeneración empleando Fluoro-jade (FJ),

108
en periodos de tiempo que comprendían desde las 6 horas hasta los 28 días,
encontrando un pico máximo de la tinción en modelos murinos entre los días 3 y 4
posinjuria y que luego de este pico las células FJ positivas empiezan a declinar hasta
que la tinción es similar a los controles en el día 7, demostrando que Fluorojade podría
no ser apropiado para evaluar el daño ocasionado por la isquemia después de las 72
horas (Butler et al., 2002; Giraldi-Guimarães et al., 2009; Liu et al., 2009).
Lo anterior podría indicar que probablemente a las 120 horas posisquemia las células en
degeneración han empezado a experimentar cambios bioquímicos dependientes del pH
del medio, que podrían ocasionar una pérdida de afinidad por el colorante, evidenciada
en la falta de intensidad y homogeneidad del FJ.
5.5 INMUNOREACTIVIDAD DE CÉLULAS GLIALES
La isquemia cerebral por embolismo a las 120 horas ocasionó cambios en los patrones
de reactividad con respecto a la distribución y densidad de los marcadores NG2, OX-42,
GFAP y COX 2, al compararlos con los grupos control.
5.5.1 Células precursoras NG2. No se evidenció reactividad de células precursoras NG2
en los grupos simulados y se encontró reactividad de NG2 en unos pocos individuos
isquémicos en el cuerpo calloso de la zona paraventricular, estos resultados son
contrarios a lo mostrado por otros autores quienes hallaron aumento de células NG2
positivas en los individuos isquémicos en la zona subventricular y en la materia blanca
alrededor del infarto (cuerpo calloso y estriado), a diferentes intervalos de tiempo, desde
las 24 horas hasta 7 días posinsulto (Morris, Chopp, Zhang, Lu, & Zhang, 2010; Sun et
al., 2013; L. Zhang et al., 2010).
En este estudio, la falta de inmunoreactividad tanto en individuos control como
isquémicos podría estar relacionado a que el proteoglicano NG2 en los mamíferos, posee
un gran dominio extracelular que contiene múltiples sitios de escisión para proteasas,
cerca de su dominio transmembranal, lo que podría resultar en la deposición del

109
ectodominio en la matriz extracelular haciendo que el reconocimiento de la proteína
mediante anticuerpos sea particularmente difícil (Trotter et al., 2010).
La reactividad positiva para NG2 encontrada en individuos isquémicos placebo, podría
deberse a una respuesta compensatoria puesto que en estos se advirtió una gran
proporción de tejido afectado por la isquemia, estimulando lo suficiente a las células
progenitoras como para permitir el marcaje con este anticuerpo.
5.5.2 Reactividad microglial. El aumento de la reactividad de la proteína OX-42 ha sido
documentada por otros autores en isquemia cerebral experimental (D. H. Kim et al., 2010;
Madinier et al., 2009; Vinet et al., 2012; J. Wang, Yang, Liu, Zhao, & Chen, 2013). El
incremento en la reactividad de OX-42 ha sido directamente relacionada al proceso de
activación microglial (Madinier et al., 2009), por lo cual se podría suponer que la microglía
detectada en este estudio se encuentra en un estado activado.
En el estudio realizado por Kim y colaboradores (2010), también fue evidente la
activación microglial en el hipocampo (CA1, CA3 y giro dentado) a tiempos similares; sin
embargo, en la presente investigación no se encontró reactividad de OX-42 en estas
zonas (Figura 14), sólo en los casos en donde el foco isquémico se extendió a ese
territorio, lo cual pudo obedecer a migración de las células de microglía hacia el área
lesionada (D. H. Kim et al., 2010).
Otros autores, han descrito que después de 24 horas de la lesión, la mayoría de las
células de microglía se encuentran en zonas limítrofes y en el centro de la lesión,
exhibiendo diferentes estados de activación, representados en cambios morfológicos
como son retracción de los procesos, aumento de tamaño del soma celular y morfología
redondeada tipo ameboide, tal como se presentó en este estudio (Madinier et al., 2009;
J. Wang et al., 2013).
Con relación a las formas ameboideas, se ha demostrado en otros trabajos que la
proteína ED-1 se expresa en las células microgliales en proceso de fagocitosis, y esta a

110
su vez está colocalizada con la proteína OX-42 a partir del estado 5A de activación en el
cual las células muestran formas redondeadas con un único proceso prominente y
alargado hacia el sitio lesionado, como se ve en la figura 15D (Jonas et al., 2012); sin
embargo en la presente investigación no se abordó la expresión o la modulación de la
proteína ED-1. Estados morfológicos similares fueron observados en este estudio, en
zonas perifocales y dentro del foco isquémico cortical, por lo que se podría inferir que se
trata de células con actividad fagocítica dentro del proceso de limpieza de restos
celulares. La transformación microglial de una forma ramificada a una forma ameboide,
ha sido relacionada a la vez con un cambio de actividad para iniciar el proceso de
fagocitosis (Kettenmann et al., 2013).
En la fimbria del hipocampo, el aumento significativo de la inmunorreactividad de OX-42
permitió establecer una morfología primordialmente ramificada, lo cual ha sido
relacionado en otros estudios a un fenotipo neuroprotector (J. Wang et al., 2013) y como
parte de los procesos de neuroplasticidad (Kettenmann et al., 2013; Madinier et al., 2009)
y remodelación sináptica, orquestando un balance entre la sinaptogénesis y la muerte
neuronal en territorios lesionados (Bessis, Béchade, Bernard, & Roumier, 2007; Sozmen,
Hinman, & Carmichael, 2012; Thored et al., 2009). Esto puede indicar que la microglía
ramificada es probablemente la responsable del mantenimiento de las sinapsis entre
redes neuronales que conectan al hipocampo con otras zonas cerebrales. La ausencia
de formas ameboideas y la presencia de formas ramificadas de la microglía
probablemente están a favor de la integridad de los cordones axonales que cruzan hacia
diversos territorios estableciendo la red sináptica.
Además, se encontró una reducción significativa de la reactividad de la microglía en la
fimbria del hipocampo en los individuos isquémicos tratados con atorvastatina,
modulando probablemente el comportamiento de la microglía, lo que coincide con otros
autores que han encontrado que la administración de atorvastatina posisquemia reduce
la respuesta inflamatoria, mediante la reducción de la cantidad de células microgliales y
la inhibición del factor de necrosis tumoral-α (TNF- α) (Saito et al., 2014).

111
En este trabajo, se encontró aumento de la densidad de la célula microglial en individuos
sanos tratados con meloxicam, atorvastatina y su asociación y aumento de la longitud de
la célula en el tratamiento con atorvastatina. Cambios en la densidad y la morfología de
la microglía en individuos sanos ha sido documentada por otros autores (Steger et al.,
2014), quienes encontraron que la ingestión crónica de cafeína produjo disminución de
la densidad global de la microglía en la corteza motora primaria y somatosensorial
primaria, así como a nivel subcortical (tálamo, hipotálamo y estriado), además
observaron retracción de los procesos celulares y el aumento del tamaño del soma,
posiblemente como un indicador de actividad microglial.
Teniendo en cuenta lo anterior, no se puede decir que la microglía de individuos sanos
tratados con meloxicam, atorvastatina y su asociación, se encuentra en un estado
activado y para establecer un verdadero efecto de estos tratamientos sobre esta
población celular en individuos sanos se requieren estudios más detallados.
5.5.3 Reactividad de los astrocitos. El aumento significativo en la reactividad de la
proteína GFAP en la corteza somatosensorial y el cuerpo estriado, coincide con lo
descrito por Stoll, Jander y Schroeter en 1998 quienes manifiestan que la
sobrerregulación de esta proteína es una de las características más relevantes de la
respuesta celular astrocitaria frente a la isquemia cerebral, junto con la hipertrofia e
hiperplasia de los astrocitos. De otra parte, la sobreexpresión de GFAP en astrocitos
reactivos (hipertróficos), ha sido relacionada con procesos de neuroprotección, en donde
se ha demostrado que ayuda en la regulación del glutamato extracelular (Li et al., 2008);
Franke y colaboradores (2012), describen un tipo de astrogliosis (isomórfica), que es
representada por la hipertrofia astroglial con la preservación de la organización de los
dominios gliales, que a menudo es reversible y benéfica para la regeneración posinsulto.
Junto con el aumento de tamaño en el cuerpo celular y el engrosamiento de las
proyecciones astrocitarias en el grupo IP, se observaron astrocitos fragmentados con
reactividad positiva para GFAP, lo que puede interpretarse como daño celular glial

112
(clasmatodendrosis), proceso que ha sido asociado por algunos autores, a pérdida en la
integridad celular consecuente a acidosis astrocitaria (Hulse et al., 2001).
En un estudio realizado por Ouyang, Voloboueva, Xu y Giffard (2007), se demostró que
ante la disminución de oxígeno y glucosa, los astrocitos de la zona hipocampal CA1,
reducían su capacidad para captar glutamato extracelular, debido a la pérdida de función
del receptor GLT-1 como consecuencia de la alta producción de especies reactivas de
oxígeno en la mitocondria, llevando a un descenso en la reactividad de GFAP y la
consecuente pérdida en la función del astrocito, lo cual a la vez, fue relacionado con
muerte neuronal. Igualmente, otros investigadores han demostrado que existe una
correlación entre la pronunciada proliferación de los astrocitos y la fragmentación del
DNA en las neuronas, sugiriendo una conexión intrínseca entre astrogliosis y muerte
neuronal retardada (W. Wang et al., 2008).
Teniendo en cuenta que el presente estudio se realizó a las 120 horas posisquemia, se
observó en el grupo IP una población de astrocitos heterogénea (funcionales y no
funcionales), probablemente los astrocitos inmunorreactivos corresponden a células
hipertróficas en proceso de recaptación de glutamato extracelular, lo cual estaría a favor
de la supervivencia neuronal.
De otra parte, se ha demostrado que el proceso excitotóxico inducido por la isquemia
además de disminuir la capacidad de los transportadores de glutamato también reduce
la actividad de la enzima glutamina sintetasa y que el tratamiento con atorvastatina
previene la disminución en la captación del glutamato y la reducción en la actividad de la
glutamina sintetasa, favoreciendo la regulación de la concentración de glutamato
extracelular, mediante la modulación del potencial redox de las células (Vandresen-Filho
et al., 2013), puesto que la glutamina sintetasa es muy sensible al estrés oxidativo y su
actividad está fuertemente relacionada con la suficiencia de los astrocitos para capturar
el glutamato (Jian et al., 2011; Vandresen Filho et al., 2013).

113
En los grupos IM, IA e IAM la inmunoreactividad de GFAP fue significativamente mayor
(P<0,01) respecto al control, aunque con morfología astrocitaria homogénea, lo cual
podría indicar que el meloxicam en forma individual o asociada a la atorvastatina
posiblemente favorece la supervivencia de los astrocitos que han reaccionado a la lesión
pero mantienen su función. Mostrando que, posiblemente los tratamientos modulan
positivamente la respuesta de los astrocitos frente al daño, manteniendo su integridad
celular y conservando su capacidad como reguladores de la homeostasis.
5.5.4 Reactividad de la ciclooxigenasa 2. Simultáneamente al incremento en la
reactividad de GFAP y OX-42 por efecto de la isquemia cerebral, se aumentó
significativamente la reactividad de la enzima COX 2, principalmente en la corteza
somatosensorial primaria y en el giro dentado del hipocampo, lo cual concuerda con lo
reportado por otros autores, quienes encontraron una inducción dramática de la enzima
COX 2 en zonas perifocales (Hirst et al., 1999; Nakayama et al., 1998; Stark & Bazan,
2011; Yokota et al., 2004).
El aumento en la reactividad de COX-2 ha sido asociado a la activación de la isoforma
inducible de la enzima, vinculada a la elevación de prostaglandinas proinflamatorias
como la PGE2, que median procesos de daño neuronal, plasticidad sináptica aberrante
y respuestas inmunoinflamatorias (Hirst et al., 1999; Nakayama et al., 1998; Stark &
Bazan, 2011; Yokota et al., 2004; J Yrjänheikki et al., 1999). Se ha demostrado que la
activación de la isoforma inducible de la COX 2 se hace a través de la estimulación de
los receptores NMDA, principalmente extrasinápticos (Stark & Bazan, 2011).
En el presente estudio, el aumento en la reactividad de COX 2 a las 120 horas
posisquemia, probablemente corresponde a un estímulo sostenido de activación de la
isoforma inducible de COX 2 concordante con un proceso inflamatorio persistente.
De otra parte, la actividad de COX 2 ha sido asociada a la proliferación de células
progenitoras neurales (CPN), bajo condiciones fisiológicas y en zonas neurogénicas
como la zona subgranular del giro dentado del hipocampo y en algunas circunstancias

114
las CPN pueden ser desviadas de su vía normal hacia sitios de lesión como respuesta a
un daño (Goncalves et al., 2010). Sasaki y colaboradores en 2003, comprobaron que la
isquemia cerebral incrementa transitoriamente la neurogénesis en el giro dentado del
hipocampo y se encuentra ligada a la actividad de COX 2 en células neuronales y en
astrocitos actuando posiblemente como moduladores de la proliferación de las CPN
(Sasaki et al., 2003).
En el presente estudio, se encontró en los animales isquémicos no tratados, un aumento
significativo en la inmunorreactividad de COX 2 en el giro dentado (GD); sin embargo en
los grupos isquémicos tratados con meloxicam y atorvastatina, también se encontró un
incremento en la reactividad de COX 2 en esta zona respecto a los controles pero sin
diferencias entre ellos. Es posible que la persistente reactividad de COX 2 en GD en los
grupos tratados, se corresponda con la capacidad del hipocampo para iniciar procesos
neurogénicos a partir de un estímulo lesivo, indicando, probablemente, que las células
del giro dentado del hipocampo podrían estar favoreciendo procesos de reparación
tisular.
Contrario a lo encontrado en el hipocampo, se observó una reducción significativa en la
reactividad de COX-2 de IM (P<0,01), IA (P<0,001) e IAM (P<0,01), en la corteza
somatosensorial de los grupos isquémicos tratados, concordando con lo descrito por
otros autores, cuando se han empleado antiinflamatorios no esteroidales como
inhibidores selectivos y no selectivos de la enzima COX 2 (Hara et al., 1998; Nakayama
et al., 1998). Gómez-Hernández y colaboradores en el 2006, encontraron disminución en
la expresión de la COX 2, descenso de la enzima responsable de la síntesis de
prostaglandina E2 (mPGE2) y reducción en la expresión de los receptores EP-1, EP-3 y
EP-4 de la prostaglandina E2 en placas ateroescleróticas, luego del tratamiento con
atorvastatina (Gómez-Hernandez et al., 2006). Así mismo, Piermartiri y colaboradores en
2010 en un modelo in vitro de la enfermedad de Alzheimer, hallaron disminución en la
expresión de COX 2 y protección en las células neuronales frente a la peroxidación
lipídica, después del tratamiento con atorvastatina (Piermartiri et al., 2010).

115
Algunos autores han descrito que la disminución en la reactividad de COX 2 durante un
proceso patológico podría favorecer la supervivencia de las células neuronales y la
plasticidad sináptica (Gómez-Hernandez et al., 2006; Hara et al., 1998; Nakayama et al.,
1998; Piermartiri et al., 2010; Sasaki et al., 2003; Strauss, 2008) y que la actividad
prolongada de la COX 2 podría afectar el metabolismo del ácido araquidónico a nivel
neuronal, desviando la síntesis de metabolitos benéficos hacia la producción de radicales
libres (Strauss, 2008).
Aunque existe evidencia de que el incremento de la síntesis de productos derivados de
COX 2 tiene efectos perjudiciales sobre el cerebro, como el aumento de estrés oxidativo,
cambios inapropiados en el flujo sanguíneo cerebral, disrupción de la barrera
hematoencefálica, trastornos de la función neuronal e inducción de proteínas dañinas y
que la inhibición selectiva de COX 2 protege las neuronas (Candelario Jalil, González
Falcón, García Cabrera, León, & Fiebich, 2004), contradictoriamente, también se ha
demostrado que algunos prostanoides derivados de COX-2 protegen a las células frente
a un daño (Horiguchi, Snipes, Kis, Shimizu, & Busija, 2006).
Estudios en modelos de excitotoxicidad e hipoxia in vitro han evidenciado que ciertos
receptores de prostaglandinas pueden mediar efectos tóxicos, pero un número de ellos
parecen ejercer efectos paradójicamente protectores (Andreasson, 2010). Entre los
receptores de prostaglandinas que se han estudiado se encuentran los DP1 y DP2,
receptores de la prostaglandina D2, constatándose que los efectos neuroprotectores se
encuentran relacionados a la activación del receptor DP1 y a su vez a la señalización
dependiente de AMP cíclico (AMPc), contrariamente, el receptor DP2 promueve la
pérdida neuronal (Liang, Wu, Hand, & Andreasson, 2005).
Así mismo, los receptores EP1, EP2, EP3 y EP4 de la prostaglandina E2 han sido
evaluados, encontrándose que tienen cascadas de señalización muy distintas y
potencialmente antagónicas, demostrándose que la activación de los receptores EP2 y
EP4 favorecen la supervivencia neuronal mediante el incremento de la producción de
AMPc, mientras que, el receptor EP1 aumenta el daño neuronal incrementando los

116
niveles de calcio intracelular y el EP3 produce efectos neurotóxicos a través de la
disminución el AMPc intracelular o la activación de la proteína Rho quinasa (Ikeda-
Matsuo et al., 2010).
Teniendo en cuenta lo anterior, la inhibición de la COX 2 en la isquemia cerebral en fase
aguda, podría potencialmente inhibir la activación de cascadas de señalización que
favorezcan la supervivencia neuronal. En contraste, la sobreestimulación de COX 2
generaría la producción de metabolitos potencialmente dañinos, por lo que el empleo de
un inhibidor preferencial de COX-2 como meloxicam y un fármaco promisorio como
atorvastatina probablemente permitiría mantener cierta cantidad de enzimas activas y de
esta manera probablemente beneficiar procesos neuroprotectores.
Finalmente, esta investigación evidencia que el tratamiento posisquemia con
atorvastatina consiguió disminuir significativamente la reactividad microglial,
adicionalmente, atorvastatina y meloxicam, en forma individual o asociada, lograron una
reducción importante de la inmunorreactividad de la forma inducible de la COX 2, y
aunque se observó un incremento en la reactividad de los astrocitos (GFAP positivo),
éstos mantuvieron las características morfológicas que tipifican células gliales en
diversas fases de actividad en pro del equilibrio y la plasticidad celular, con lo cual se
reduce el riesgo de muerte celular a 120 horas posisquemia y les confiere un efecto
neuroprotector al reducir la citotoxicidad en el tejido afectado y al favorecer la modulación
en la actividad glial.

117
6. CONCLUSIONES
La isquemia cerebral por embolismo arterial mediante alfa trombina bovina, produce
lesiones en los territorios correspondientes a los irrigados por la ACM y en raras
ocasiones genera lesiones en territorios de la ACP.
Los tratamientos con meloxicam y atorvastatina en forma individual o asociada no
lograron mejorar significativamente los déficits neurológicos a las 120 horas
posisquemia.
El modelo embólico con alfa trombina bovina a las 120 horas, produjo pérdida de la
arquitectura celular en el foco isquémico y mantuvo los patrones citoarquitectónicos en
las zonas perifocales y de penumbra.
La tinción con Fluoro Jade B a las 120 horas posisquemia, no consiguió mostrar procesos
de neurodegeneración consistentes en los grupos isquémicos placebo ni en los tratados
con meloxicam, atorvastatina en forma individual o asociada.
La isquemia cerebral por embolismo arterial a las 120 horas, aumenta la reactividad de
los astrocitos y la microglía e induce cambios morfológicos propios de la activación de
estas células en zonas perifocales y de penumbra.
La isquemia cerebral embólica a las 120 horas, en el grupo isquémico placebo produce
una población de astrocitos heterogénea, astrocitos hipertróficos, en proceso de
captación de glutamato y otros que probablemente no logran superar el insulto y
experimentan pérdida de su integridad y funcionalidad, en cambio los tratamientos con
meloxicam y atorvastatina en forma individual y asociada permiten a los astrocitos
mantener su integridad y posiblemente su funcionalidad como reguladores del ambiente
extracelular, modulando positivamente su actividad frente a la isquemia cerebral.

118
El tratamiento con atorvastatina, a las 120 horas posisquemia, reduce la reactividad de
OX-42 modulando la actividad microglial, en tanto el tratamiento con meloxicam y la
asociación de este con atorvastatina no logró reducir la hiperreactividad posisquemia.
La isquemia cerebral embólica a las 120 horas, incrementa significativamente la
reactividad de la COX 2 en zonas perifocales y de penumbra y los tratamientos con
meloxicam y atorvastatina en forma individual y asociada reducen el estímulo de la COX
2 disminuyendo la respuesta inflamatoria.
Finalmente, el tratamiento posisquemia individual con atorvastatina logró reducir
significativamente la inmunorreactividad microglial, en tanto la asociación con meloxicam
no alcanzo este propósito. Aunque fue evidenciado un incremento en la reactividad de
los astrocitos (GFAP positivo), éstos mantuvieron las características morfológicas que
tipifican células gliales en diversas fases de actividad en pro del equilibrio y la plasticidad
celular, con lo cual se reduce el riesgo de muerte celular a 120 horas posisquemia y les
confiere un efecto neuroprotector al reducir la citotoxicidad en el tejido afectado y al
favorecer la modulación en la actividad glial. Posiblemente la atorvastatina en forma
individual o asociada al meloxicam inhibe la expresión de la ciclooxigenasa 2 en el tejido
isquémico.

119
RECOMENDACIONES
Teniendo en cuenta los resultados obtenidos en este trabajo se recomienda:
Evaluar el posible efecto intrínseco de los tratamientos con meloxicam, atorvastina y su
asociación sobre los astrocitos, la microglía y la COX 2, empleado triple marcaje, en
cultivos mixtos de corteza e hipocampo.
Estudiar los niveles de prostangladina E2, en animales isquémicos y simulados tratados
con meloxicam, atorvastatina y su asociación mediante microdiálisis para establecer
correlaciones entre los tratamientos, la producción de esta prostaglandina y procesos de
neuroprotección.
Examinar la modulación de los receptores neurotóxicos EP1 y EP3, de la prostaglandina
E2, por la isquemia cerebral embólica y los tratamientos con atorvastatina y meloxicam
y su asociación.
Analizar el efecto de los tratamientos con meloxicam, atorvastatina y su asociación sobre
procesos neurogénicos, debido a que es conocido que la neurogénesis es un proceso
relacionado a la actividad de la enzima COX 2, empleando Ki-67 como marcador de
proliferación celular.
Estimar el efecto de la isquemia cerebral embólica y los tratamientos con meloxicam,
atorvastatina y su asociación sobre las poblaciones neuronales empleando el anticuerpo
anti-NeuN para cuantificar pérdida neuronal.

120
REFERENCIAS
Abbott, N. J., Rönnbäck, L., & Hansson, E. (2006). Astrocyte-endothelial interactions at
the blood-brain barrier. Nature Reviews. Neuroscience, 7, 41–53. doi:10.1038/nrn1824
Acadamies, N. R. C. of the N. (2011). Guide for the care and use of laboratory animals.
Aïd, S., & Bosetti, F. (2011). Targeting cyclooxygenases-1 and -2 in neuroinflammation:
therapeutic implications. Biochimic, 93(1), 46–51.
doi:10.1016/j.biochi.2010.09.009.Targeting
Akpan, N., Serrano-saiz, E., Zacharia, B. E., Otten, M. L., Ducruet, A., Snipas, S. J., Troy,
C. M. (2011). Intranasal delivery of caspase-9 inhibitor reduces caspase-6-dependent
axon/neuron loss and improves neurological function after stroke. J Neurosci, 31(24),
8894–904. doi:10.1523/JNEUROSCI.0698-11.2011.Intranasal
Alonso de Leciñana, M., Gutiérrez, M., Roda, J. M., Carceller, F., & Díez-Tejedor, E.
(2006). Effect of combined therapy with thrombolysis and citicoline in a rat model of
embolic stroke. Journal of the Neurological Sciences, 247, 121–9.
doi:10.1016/j.jns.2006.03.022
Anderova, M., Vorisek, I., Pivonkova, H., Benesova, J., Vargova, L., Cicanic, M., Sykova,
E. (2011). Cell death/proliferation and alterations in glial morphology contribute to
changes in diffusivity in the rat hippocampus after hypoxia-ischemia. Journal of Cerebral
Blood Flow and Metabolism : Official Journal of the International Society of Cerebral Blood
Flow and Metabolism, 31, 894–907. doi:10.1038/jcbfm.2010.168
Andreasson, K. (2010). Prostaglandin signalling in cerebral ischaemia. British Journal of
Pharmacology, 160, 844–6. doi:10.1111/j.1476-5381.2010.00715.x
Arango-Dávila, C., Escobar-Betancourt, M., Cardona-Gómez, G. P., & Pimienta-Jiménez,
H. (2004). Fisiopatología de la isquemia cerebral focal: aspectos básicos y proyección a

121
la clínica. Revista de Neurología, 39(2), 156–165. Retrieved from
http://cat.inist.fr/?aModele=afficheN&cpsidt=16066866
Arauz, A., & Ruíz-Franco, A. (2012). Enfermedad vascular cerebral. Revista de La
Facultad de Medicina de La UNAM, 55(3), 11–21. Retrieved from
http://new.medigraphic.com/cgi-bin/resumenMain.cgi?IDARTICULO=14130
Bacigaluppi, M., Comi, G., & Hermann, D. (2010a). Animal models of ischemic stroke.
Part one: modeling risk factors. The Open Neurology Journal, 4, 26–33.
doi:10.2174/1874205X01004020026
Bacigaluppi, M., Comi, G., & Hermann, D. (2010b). Animal models of ischemic stroke.
Part two: modeling cerebral ischemia. The Open Neurology Journal, 4, 34–8.
doi:10.2174/1874205X01004020034
Bailey, J. M. (1989). THE NEW YORK ACADEMY BIOCHEMISTRY AND
PHARMACOLOGY OF CYCLOOXYGENASE INHIBITORS *. Bulletin of the New York
Academy of Medicine, 65(1), 5–15.
Bederson, J. B., Pitts, L. H., Tsuji, M., Nishimura, M. C., Davis, R. L., & Bartkowski, H.
(1986). Rat middle cerebral artery occlusion: evaluation of the model and development of
a neurologic examination. Stroke, 17(3), 472–476. doi:10.1161/01.STR.17.3.472
Belayev, L., Busto, R., Zhao, W., Fernandez, G., & Ginsberg, M. D. (1999). Middle
cerebral artery occlusion in the mouse by intraluminal suture coated with poly-L-lysine:
neurological and histological validation. Brain Research, 833(2), 181–90. Retrieved from
http://www.ncbi.nlm.nih.gov/pubmed/10375693
Bessis, A., Béchade, C., Bernard, D., & Roumier, A. (2007). Microglial Control of Neuronal
Death and Synaptic Properties. Glia, 55, 233–38. doi:10.1002/glia

122
Biber, K., Neumann, H., Inoue, K., & Boddeke, H. (2007). Neuronal “On” and “Off” signals
control microglia. Trends in Neurosciences, 30(11), 596–602.
doi:10.1016/j.tins.2007.08.007
Bosetti, F. (2007). Arachidonic acid metabolism in brain physiology and pathology:
lessons from genetically altered mouse models. J Neurochem, 102(3), 577–86.
Bushong, E. a, Martone, M. E., & Ellisman, M. H. (2004). Maturation of astrocyte
morphology and the establishment of astrocyte domains during postnatal hippocampal
development. International Journal of Developmental Neuroscience : The Official Journal
of the International Society for Developmental Neuroscience, 22(2), 73–86.
doi:10.1016/j.ijdevneu.2003.12.008
Butler, T. L., Kassed, C. A., Sanberg, P. R., Willing, A. E., & Pennypacker, K. R. (2002).
Neurodegeneration in the rat hippocampus and striatum after middle cerebral artery
occlusion. Brain Research, 929, 252–60. Retrieved from
http://www.ncbi.nlm.nih.gov/pubmed/11864631
Butterfield, A. D., Barone, E., & Mancuso, C. (2011). Cholesterol-independent
neuroprotective and neurotoxic activities of statins: prespectives for statin use in
Alzheimer disease and other age-related neurodegenerative disorders. Pharmacol Res,
64(3), 180–6. doi:10.1016/j.phrs.2011.04.007.CHOLESTEROL-INDEPENDENT
Camara-Lemarroy, C. R., Gonzalez-Moreno, E. I., Guzman-de la Garza, F. J., &
Fernandez-Garza, N. E. (2012). Arachidonic acid derivatives and their role in peripheral
nerve degeneration and regeneration. The Scientific World Journal, 168953.
doi:10.1100/2012/168953
Candelario-Jalil, E., González-Falcón, A., García-Cabrera, M., León, O. S., & Fiebich, B.
L. (2004). Wide therapeutic time window for nimesulide neuroprotection in a model of

123
transient focal cerebral ischemia in the rat. Brain Research, 1007, 98–108.
doi:10.1016/j.brainres.2004.01.078
Canova, D., Roatta, S., Micieli, G., & Bosone, D. (2012). Cerebral oxygenation and
haemodynamic effects induced by nimodipine in healthy subjects. Functional Neurology,
27(3), 169–76. Retrieved from
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3812769&tool=pmcentrez&re
ndertype=abstract
Capone, M. L., Tacconelli, S., Di Francesco, L., Sacchetti, A., Sciulli, M. G., & Patrignani,
P. (2007). Pharmacodynamic of cyclooxygenase inhibitors in humans. Prostaglandins &
Other Lipid Mediators, 82, 85–94. doi:10.1016/j.prostaglandins.2006.05.019
Casals, J. B., Pieri, N. C. G., Feitosa, M. L. T., Ercolin, A. C. M., Roballo, K. C. S., Barreto,
R. S. N., … Ambrósio, C. E. (2011). The use of animal models for stroke research: a
review. Comparative Medicine, 61(4), 305–13. Retrieved from
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3155396&tool=pmcentrez&re
ndertype=abstract
Castilla-Guerra, L., Fernandez-Moreno, M. C., López-Chozas, J. M., & Jiménez-
Hernandez, M. D. (2007). Estatinas y enfermedad cerebrovascular: nuevas perspectivas
en la prevención del ictus. Revista de Neurología, 44(2), 95–100.
Castilla-Guerra, L., Fernández-Moreno, M., López-Chozas, J., & Jiménez-Hernández, M.
(2007). Estatinas y enfermedad cerebrovascular : nuevas perspectivas en la prevención
del ictus. Revista de Neurología, 44(2), 95–100.
Céspedes, Á. E., Arango, C. A., & Cardona, G. P. (2013). Análisis comparativo de
marcadores de lesión en modelos de isquemia cerebral focal y global en ratas.
Biomédica, 33, 292–305. doi:http://dx.doi.org/10.775/biomedica.v33i2.830

124
Céspedes-Rubio, A., Wandosell, F., & Cardona-Gómez, G. P. (2010). p120 Catenin/N-
catenin are molecular targets in the neuroprotection and neuronal plasticity mediated by
atorvastatin after focal cerebral ischemia. Journal of Neuroscience Research, 88, 3621–
3634. doi:10.1002/jnr.22511
Chen, C., Magee, J. C., & Bazan, N. (2002). Cyclooxygenase-2 Regulates Prostaglandin
E 2 Signaling in Hippocampal Long-Term Synaptic Plasticity Cyclooxygenase-2
Regulates Prostaglandin E 2 Signaling in Hippocampal Long-Term Synaptic Plasticity. J
Neurphysiol, 87, 2851–2857.
Chen, H., Yoshioka, H., Kim, G. S., Jung, J. E., Okami, N., Sakata, H., … Chan, P. H.
(2011). Oxidative stress in ischemic brain damage: mechanisms of cell death and
potential molecular targets for neuroprotection. Antioxidants & Redox Signaling, 14(8),
1505–17. doi:10.1089/ars.2010.3576
Chen Xu, W., Yi, Y., Qiu, L., & Shuaib, A. (2000). Neuroprotective activity of tiagabine in
a focal embolic model of cerebral ischemia. Brain Research, 874, 75–7. Retrieved from
http://www.ncbi.nlm.nih.gov/pubmed/10936225
Chen, Y., & Swanson, R. A. (2003). Astrocytes and Brain Injury. Journal of Cerebral Blood
Flow & Metabolism, 23, 137–149. doi:10.1097/01.WCB.0000044631.80210.3C
Culmsee, C., & Krieglstein, J. (2005). Mechanisms of neuronal degeneration after
ischemic stroke – Emerging targets for novel therapeutic strategies. Drug Discovery
Today: Disease Mechanisms, 2(4), 463–470. doi:10.1016/j.ddmec.2005.11.006
De Keyser, J., Van de Velde, V., Schellens, R. L., Hantson, L., Tritsmans, L., Gheuens,
J., … Van Gorp, J. (1997). Safety and pharmacokinetics of the neuroprotective drug
lubeluzole in patients with ischemic stroke. Clinical Therapeutics, 19(6), 1340–51.
Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9444444

125
De Ryck, M., Van Reempts, J., Borgers, M., Wauquier, A., & Janssen, P. a. (1989).
Photochemical stroke model: flunarizine prevents sensorimotor deficits after neocortical
infarcts in rats. Stroke, 20, 1383–1390. doi:10.1161/01.STR.20.10.1383
Del Zoppo, G. J. (2009). INFLAMMATION AND THE NEUROVASCULAR UNIT IN THE
SETTING OF FOCAL CEREBRAL ISCHEMIA. Neuroscie, 158, 972–82.
doi:10.1016/j.neuroscience.2008.08.028
Del Zoppo, G. J. (2010). The neurovascular unit in the setting of stroke. Journal of Internal
Medicine, 267(2), 156–171. doi:10.1111/j.1365-2796.2009.02199.x.The
Denes, A., Thornton, P., Rothwell, N. J., & Allan, S. M. (2010). Inflammation and brain
injury: acute cerebral ischaemia, peripheral and central inflammation. Brain, Behavior,
and Immunity, 24(5), 708–23. doi:10.1016/j.bbi.2009.09.010
Di Filippo, M., Tozzi, A., Costa, C., Belcastro, V., Tantucci, M., Picconi, B., & Calabresi,
P. (2008). Plasticity and repair in the post-ischemic brain. Neuropharmacology, 55, 353–
362. doi:10.1016/j.neuropharm.2008.01.012
Di Rienzo, J. A., Balzarini, M., Casanoves, F., Gonzalez, L., Tablada, M., & Robledo, C.
W. (2011). InfoStat. Universidad Nacional de Córdoba.
DiNapoli, V. a, Rosen, C. L., Nagamine, T., & Crocco, T. (2006). Selective MCA occlusion:
a precise embolic stroke model. Journal of Neuroscience Methods, 154, 233–8.
doi:10.1016/j.jneumeth.2005.12.026
Ding, G., Jiang, Q., Li, L., Zhang, L., Zhang, Z. G., Panda, S., … Chopp, M. (2006). MRI
of combination treatment of embolic stroke in rat with rtPA and atorvastatin. Journal of
the Neurological Sciences, 246(1-2), 139–47. doi:10.1016/j.jns.2006.02.020

126
Duckworth, E. A. M., Butler, T. L., De Mesquita, D., Collier, S. N., Collier, L., &
Pennypacker, K. R. (2005). Temporary focal ischemia in the mouse: technical aspects
and patterns of Fluoro-Jade evident neurodegeneration. Brain Research, 1042, 29–36.
doi:10.1016/j.brainres.2005.02.021
Fernández, P. (2001). Determinación del tamaño muestral. Retrieved from
http://www.fisterra.com/mbe/investiga/9muestras/tamano_muestral2.pdf
Fernández-Gómez, F. J., Hernández, F., Argandoña, L., Galindo, M. F., Segura, T., &
Jordán, J. (2008). Farmacología de la neuroprotección en el ictus isquémico agudo. Rev
Neurol, 47(5), 253–260.
Franke, H., Verkhratsky, A., Burnstock, G., & Illes, P. (2012). Pathophysiology of
astroglial purinergic signalling. Purinergic Signalling, 8(3), 629–57. doi:10.1007/s11302-
012-9300-0
Fricker, M., Vilalta, A., Tolkovsky, A. M., & Brown, G. C. (2013). Caspase inhibitors protect
neurons by enabling selective necroptosis of inflamed microglia. The Journal of Biological
Chemistry, 288(13), 9145–52. doi:10.1074/jbc.M112.427880
Gallo, V., Mangin, J.-M., Kukley, M., & Dietrich, D. (2008). Synapses on NG2-expressing
progenitors in the brain: multiple functions? The Journal of Physiology, 586(16), 3767–
81. doi:10.1113/jphysiol.2008.158436
Gehrmann, J., Matsumoto, Y., & Kreutzberg, G. W. (1995). Microglia : intrinsic
immuneffector cell of the brain. Brain Research Reviews, 20, 269–287.
Ginsberg, M. D. (2008). Neuroprotection for ischemic stroke: past, present and future.
Neuropharmacology, 55(3), 363–89.
doi:10.1016/j.neuropharm.2007.12.007.NEUROPROTECTION

127
Giraldi-Guimarães, A., Rezende-Lima, M., Bruno, F. P., & Mendez-Otero, R. (2009).
Treatment with bone marrow mononuclear cells induces functional recovery and
decreases neurodegeneration after sensorimotor cortical ischemia in rats. Brain
Research, 1266, 108–20. doi:10.1016/j.brainres.2009.01.062
Gomes-Leal, W. (2012). Microglial physiopathology: how to explain the dual role of
microglia after acute neural disorders? Brain and Behavior, 2(3), 345–56.
doi:10.1002/brb3.51
Gómez-Hernandez, A., Sánchez-Gálan, E., Martín-Ventura, J. L., Vidal, C., Blanco-Colio,
L. M., Ortego, M., … Egido, J. (2006). Atorvastatin Reduces the Expression of
Prostaglandin E 2 Receptors in Human Carotid Atherosclerotic Plaques and Monocytic
Cells. J Cardiovasc Pharmacol, 47(1), 60–9.
Goncalves, M. B., Williams, E.-J., Yip, P., Yáñez-Muñoz, R. J., Williams, G., & Doherty,
P. (2010). The COX-2 inhibitors, meloxicam and nimesulide, suppress neurogenesis in
the adult mouse brain. British Journal of Pharmacology, 159(5), 1118–25.
doi:10.1111/j.1476-5381.2009.00618.x
Goverdhan, P., Sravanthi, A., & Mamatha, T. (2012). Neuroprotective effects of
meloxicam and selegiline in scopolamine-induced cognitive impairment and oxidative
stress. International Journal of Alzheimer’s Disease, 2012, 974013.
doi:10.1155/2012/974013
Gutiérrez-Vargas, J., Castro-Alvarez, J. F., Velásquez-Carvajal, D., Montañez-
Velásquez, M. N., Céspedes-Rubio, Á., & Cardona-Gómez, G. P. (2010). Rac1 activity
changes are associated with neuronal pathology and spatial memory long-term recovery
after global cerebral ischemia. Neurochemistry International, 57, 762–73.
doi:10.1016/j.neuint.2010.08.014

128
Hara, K., Kong, D. L., Sharp, F. R., & Weinstein, P. R. (1998). Effect of selective inhibition
of cyclooxygenase 2 on temporary focal cerebral ischemia in rats. Neuroscience Letters,
256, 53–6. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9832215
Harry, J. G., & Kraft, A. D. (2012). Microglia in the developing brain: a potential target with
life time effects. Neurotoxicology, 33(2), 191–206.
doi:10.1016/j.neuro.2012.01.012.Microglia
Harukuni, I., & Bhardwaj, A. (2006). Mechanisms of brain injury after global cerebral
ischemia. Neurologic Clinics, 24(1), 1–21. doi:10.1016/j.ncl.2005.10.004
Henninger, N., Bouley, J., Bråtane, B. T., Bastan, B., Shea, M., & Fisher, M. (2009). Laser
Doppler flowmetry predicts occlusion but not tPA-mediated reperfusion success after rat
embolic stroke. Experimental Neurology, 215, 290–7.
doi:10.1016/j.expneurol.2008.10.013
Henninger, N., Sicard, K. M., Schmidt, K. F., Bardutzky, J., & Fisher, M. (2006).
Comparison of ischemic lesion evolution in embolic versus mechanical middle cerebral
artery occlusion in Sprague Dawley rats using diffusion and perfusion imaging. Stroke; a
Journal of Cerebral Circulation, 37, 1283–7. doi:10.1161/01.STR.0000217223.72193.98
Heo, J. H., Han, S. W., & Lee, S. K. (2005). Free radicals as triggers of brain edema
formation after stroke. Free Radical Biology & Medicine, 39(1), 51–70.
doi:10.1016/j.freeradbiomed.2005.03.035
Hernández, M., Trujillo, L., & Céspedes Rubio, A. (2010). Protocolo quírurgico para la
oclusión unilateral de la arteria cerebral media por coágulo endógeno.
Hirst, W. D., Young, K. A., Newton, R., Allport, V. C., Marriott, D. R., & Wilkin, G. P. (1999).
Expression of COX-2 by normal and reactive astrocytes in the adult rat central nervous
system. Molecular and Cellular Neuroscience, 13, 57–68.

129
Honsa, P., Pivonkova, H., Dzamba, D., Filipova, M., & Anderova, M. (2012).
Polydendrocytes display large lineage plasticity following focal cerebral ischemia. PloS
One, 7(5), e36816. doi:10.1371/journal.pone.0036816
Horiguchi, T., Snipes, J. A., Kis, B., Shimizu, K., & Busija, D. W. (2006). Cyclooxygenase-
2 mediates the development of cortical spreading depression-induced tolerance to
transient focal cerebral ischemia in rats. Neuroscience, 140, 723–30.
doi:10.1016/j.neuroscience.2006.02.025
Hossmann, K.-A. (2012). The two pathophysiologies of focal brain ischemia: implications
for translational stroke research. Journal of Cerebral Blood Flow and Metabolism, 32(7),
1310–6. doi:10.1038/jcbfm.2011.186
Howells, D. W., Porritt, M. J., Rewell, S. S. J., O’Collins, V., Sena, E. S., van der Worp,
H. B., … Macleod, M. R. (2010). Different strokes for different folks: the rich diversity of
animal models of focal cerebral ischemia. Journal of Cerebral Blood Flow and
Metabolism : Official Journal of the International Society of Cerebral Blood Flow and
Metabolism, 30, 1412–31. doi:10.1038/jcbfm.2010.66
Hulse, R., Winterfield, J., Kunkler, P., & Kraig, R. (2001). Astrocytic clasmatodendrosis in
hipocampal organ culture. Glia, 33(2), 169–79.
Ikeda-Matsuo, Y., Tanji, H., Ota, A., Hirayama, Y., Uematsu, S., Akira, S., & Sasaki, Y.
(2010). Microsomal prostaglandin E synthase-1 contributes to ischaemic excitotoxicity
through prostaglandin E2 EP3 receptors. British Journal of Pharmacology, 160, 847–59.
doi:10.1111/j.1476-5381.2010.00711.x
Jacobsen, K. R., Fauerby, N., Raida, Z., Kalliokoski, O., Hau, J., Johansen, F. F., &
Abelson, K. S. (2013). Effects of buprenorphine and meloxicam analgesia on induced
cerebral ischemia in C57BL/6 male mice. Comparative Medicine, 63(2), 105–13.
Retrieved from

130
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3625051&tool=pmcentrez&re
ndertype=abstract
Jian, Z., Yan-Xia, W., Fang-Fang, D., He-Zou, L., Zheng-Wen, M., Pei-Hua, L., & Xiao-
Ming, X. (2011). Glutamine synthetase downregulation reduces astrocyte protection
against glutamate excitotoxicity to neurons. Neurochem Int, 56(4), 577–584.
doi:10.1016/j.neuint.2009.12.021.Glutamine
Jin, R., Yang, G., & Li, G. (2010, May). Inflammatory mechanisms in ischemic stroke: role
of inflammatory cells. Journal of Leukocyte Biology. doi:10.1189/jlb.1109766
Jonas, R. a, Yuan, T.-F., Liang, Y.-X., Jonas, J. B., Tay, D. K. C., & Ellis-Behnke, R. G.
(2012). The spider effect: morphological and orienting classification of microglia in
response to stimuli in vivo. PloS One, 7(2), e30763. doi:10.1371/journal.pone.0030763
Jordán, J., Ikuta, I., García-García, J., Calleja, S., & Segura, T. (2007). Stroke
pathophysiology : management challenges and new treatment advances. Journal of
Physiology and Biochemistry, 63(3), 261–277.
Kandadai, M., Meunier, J., Lindsell, C., Shaw, G., & Elkind, M. (2012). Short-term high-
dose effect of Lovastatin on thrombolisis by rt-PA in a human whole-blood in vitro clot
model. Curr Neurovasc Res, 9(3), 207–13.
Kang, S. H., Furukaya, M., Yang, J. K., Rothstein, J. D., & Bergles, D. E. (2010). NG2+
CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life
and following neurodegeneration. Neuron, 68(4), 668–681.
doi:10.1016/j.neuron.2010.09.009.
Kang, W., & Hébert, J. M. (2011). Signaling pathways in reactive astrocytes, a genetic
perspective. Mol Neurobiol, 43(3), 147–154. doi:10.1007/s12035-011-8163-7.

131
Kettenmann, H., Kirchhoff, F., & Verkhratsky, A. (2013). Microglia: new roles for the
synaptic stripper. Neuron, 77(1), 10–8. doi:10.1016/j.neuron.2012.12.023
Kim, D. H., Kim, J. M., Park, S. J., Lee, S., Yoon, B. H., & Ryu, J. H. (2010). Early-
activated microglia play a role in transient forebrain ischemia-induced neural precursor
proliferation in the dentate gyrus of mice. Neuroscience Letters, 475, 74–9.
doi:10.1016/j.neulet.2010.03.046
Kim, R. Y., Hoffman, A. S., Itoh, N., Ao, Y., Spence, R., Sofroniew, M. V, & Voskuhl, R.
R. (2014). Astrocyte CCL2 sustains immune cell infiltration in chronic experimental
autoimmune encephalomyelitis. Journal of Neuroimmunology, 274, 53–61.
doi:10.1016/j.jneuroim.2014.06.009
Kitamura, Y., Yanagisawa, D., Takata, K., & Taniguchi, T. (2009). Neuroprotective
function in brain microglia. Current Anaesthesia & Critical Care, 20(1), 142–147.
doi:10.1016/j.cacc.2008.12.007
Klatt, E. C. (2010). Luxol fast blue - modified kluver’s - myelin sheath. WebPath: Internet
laboratory Pathology. Retrieved from http://www-
medlib.med.utah.edu/WebPath/webpath.html
Kudo, M., Aoyama, a., Ichimori, S., & Fukunaga, N. (1982). An animal model of cerebral
infarction. Homologous blood clot emboli in rats. Stroke, 13(4), 505–508.
doi:10.1161/01.STR.13.4.505
Lai, T. W., Zhang, S., & Wang, Y. T. (2014). Excitotoxicity and stroke: identifying novel
targets for neuroprotection. Progress in Neurobiology, 115, 157–88.
doi:10.1016/j.pneurobio.2013.11.006

132
Lakhan, S. E., Kirchgessner, A., & Hofer, M. (2009). Inflammatory mechanisms in
ischemic stroke: therapeutic approaches. Journal of Translational Medicine, 7, 97.
doi:10.1186/1479-5876-7-97
Lan, R., Chopp, M., Zhang, Z. G., Jiang, Q., & Ewing, J. R. (1997). A rat model of focal
embolic cerebral ischemia. Brain Research, 766, 83–92. doi:10.1016/S0006-
8993(97)00580-5
Lapuente, C., Rengifo, C., Ávila Rodriguez, M., & Céspedes Rubio, A. (2013). Regulatory
effect of Dimethyl Sulfoxide (DMSO) on astrocytic reactivity in a murine model of cerebral
infarction by arterial embolization. Colombia Médica, 44(Lapuente C), 32–37. Retrieved
from http://colombiamedica.univalle.edu.co/index.php/comedica/article/view/1154
Lecca, D., Ceruti, S., Fumagalli, M., & Abbracchio, M. P. (2012). Purinergic trophic
signalling in glial cells: functional effects and modulation of cell proliferation,
differentiation, and death. Purinergic Signalling, 8, 539–57. doi:10.1007/s11302-012-
9310-y
Li, L., Lundkvist, A., Andersson, D., Wilhelmsson, U., Nagai, N., Pardo, A. C., … Pekny,
M. (2008). Protective role of reactive astrocytes in brain ischemia. Journal of Cerebral
Blood Flow and Metabolism : Official Journal of the International Society of Cerebral Blood
Flow and Metabolism, 28(3), 468–81. doi:10.1038/sj.jcbfm.9600546
Liang, X., Wu, L., Hand, T., & Andreasson, K. (2005). Prostaglandin D2 mediates
neuronal protection via the DP1 receptor. Journal of Neurochemistry, 92, 477–86.
doi:10.1111/j.1471-4159.2004.02870.x
Liao, J. K. (2005). Effects of statins on 3-Hydroxy-3-Methylglutaryl Coenzyme A
Reductase Inhibition Beyond Low-Density Lipoprotein Cholesterol. Am J Cardiol, 96(5),
24–33. doi:10.1016/j.amjcard.2005.06.009.Effects

133
Liao, J. K., & Laufs, U. (2005). Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol,
45, 89–118. doi:10.1146/annurev.pharmtox.45.120403.095748.PLEIOTROPIC
Liu, F., & McCullough, L. (2011). Middle cerebral artery occlusion model in rodents:
methods and potential pitfalls. Journal of Biomedicine & Biotechnology, 464701.
doi:10.1155/2011/464701
Liu, F., Schafer, D., & McCullough, L. (2009). TTC, flouro-jade B and NeuN staining
confirm evolving phases of infarction induced by middle cerebral artery oclusion. J
Neurosci Methods, 179(1), 1–8. doi:10.1016/j.jneumeth.2008.12.028.TTC
Llorente, I. L., Pérez-Rodríguez, D., Burgin, T. C., Gonzalo-Orden, J. M., Martínez-
Villayandre, B., & Fernández-López, A. (2013). Age and meloxicam modify the response
of the glutamate vesicular transporters (VGLUTs) after transient global cerebral ischemia
in the rat brain. Brain Research Bulletin, 94(2013), 90–7.
doi:10.1016/j.brainresbull.2013.02.006
López-Bayghen, E., & Ortega, A. (2010). Células gliales y actividad sináptica : control
traduccional del acople metabólico. Revista de Neurología, 50(10), 607–615.
López-Villodres, J. A., De La Cruz, J. P., Muñoz-Marin, J., Guerrero, A., Reyes, J. J., &
González-Correa, J. (2012). Cytoprotective effect of nonsteroidal antiinflammatory drugs
in rat brain slices subjected to reoxygenation after oxygen-glucose deprivation. European
Journal of Pharmaceutical Sciences : Official Journal of the European Federation for
Pharmaceutical Sciences, 45, 624–31. doi:10.1016/j.ejps.2012.01.001
Lucas, S.-M., Rothwell, N. J., & Gibson, R. M. (2006). The role of inflammation in CNS
injury and disease. British Journal of Pharmacology, 147 Suppl , S232–40.
doi:10.1038/sj.bjp.0706400

134
Ma, J., Qiu, J., Hirt, L., Dalkara, T., & Moskowitz, M. A. (2001). Synergistic protective
effect of caspase inhibitors and bFGF against brain injury induced by transient focal
ischaemia. British Journal of Pharmacology, 133, 345–50. doi:10.1038/sj.bjp.0704075
Madinier, A., Bertrand, N., Mossiat, C., Prigent-Tessier, A., Beley, A., Marie, C., &
Garnier, P. (2009). Microglial involvement in neuroplastic changes following focal brain
ischemia in rats. PloS One, 4(12), e8101. doi:10.1371/journal.pone.0008101
Maldonado, P. P., Vélez-Fort, M., & Angulo, M. C. (2011). Is neuronal communication
with NG2 cells synaptic or extrasynaptic? Journal of Anatomy, 219, 8–17.
doi:10.1111/j.1469-7580.2011.01350.x
Marosi, K., & Mattson, M. P. (2014). BDNF mediates adaptive brain and body responses
to energetic challenges. Trends in Endocrinology and Metabolism: TEM, 25(2), 89–98.
doi:10.1016/j.tem.2013.10.006
Maulik, D., Ashraf, Q. M., Mishra, O. P., & Delivoria-papadopoulos, M. (2008). Activation
of p38 mitogen-activated protein kinase ( p38 MAPK ), extracellular signal-regulated
kinase ( ERK ) and c-jun N-terminal kinase ( JNK ) during hypoxia in cerebral cortical
nuclei of guinea pig fetus at term : Role of nitric oxide. Neuroscience Letters, 439, 94–99.
doi:10.1016/j.neulet.2008.02.037
Mehta, S. L., Manhas, N., & Raghubir, R. (2007). Molecular targets in cerebral ischemia
for developing novel therapeutics. Brain Research Reviews, 54(1), 34–66.
doi:10.1016/j.brainresrev.2006.11.003
Minoru, A., Thomas, S., Yoshimura, S. I., Toshihisa, S., Mori, T., Qiu, J., … Moskowitz,
M. A. (2005). Protective effects of statins after embolic focal cerebral ischemia in
endothelial nitric oxide synthase knockout mice. J Cereb Blood Flow Metab, 25(6), 722–
9. doi:10.1038/sj.jcbfm.9600070.Protective

135
Montori, S., Dos-Anjos, S., Martínez-Villayandre, B., Regueiro-Purriños, M. M., Gonzalo-
Orden, J. M., Ruano, D., & Fernández-López, A. (2010). Age and meloxicam attenuate
the ischemia/reperfusion-induced down-regulation in the NMDA receptor genes.
Neurochemistry International, 56, 878–85. doi:10.1016/j.neuint.2010.03.013
Morris, D. C., Chopp, M., Zhang, L., Lu, M., & Zhang, Z. G. (2010). Thymosin B4 improves
functional neurological outcome in a rat model of embolic stroke. Neuroscience, 169(2),
674–82. doi:10.1016/j.neuroscience.2010.05.017.Thymosin
Nakayama, M., Uchimura, K., Zhu, R. L., Nagayama, T., Rose, M. E., Stetler, R. a, …
Graham, S. H. (1998). Cyclooxygenase-2 inhibition prevents delayed death of CA1
hippocampal neurons following global ischemia. Proceedings of the National Academy of
Sciences of the United States of America, 95, 10954–9. Retrieved from
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=28002&tool=pmcentrez&rend
ertype=abstract
Nakka, V. P., Gusain, A., Mehta, S. L., & Raghubir, R. (2008). Molecular mechanisms of
apoptosis in cerebral ischemia: multiple neuroprotective opportunities. Molecular
Neurobiology, 37, 7–38. doi:10.1007/s12035-007-8013-9
Organización Panamericana de la Salud, P. (2013). Causas principales de mortalidad en
las Américas. Observatorio Regional de Salud. Retrieved from
http://ais.paho.org/phip/viz/mort_causasprincipales_lt_oms.asp
Ouyang, Y.-B., Voloboueva, L. A., Xu, L.-J., & Giffard, R. G. (2007). Selective dysfunction
of hippocampal CA1 astrocytes contribute to delayed neuronal damage after transient
forebrain ischemia. J Neurosci, 27(16), 4253–4260. doi:10.1523/JNEUROSCI.0211-
07.2007.
Pascual, J. M., González-Llanos, F., Cerdán, S., & Carceller, F. Roda, J. M. (2000).
Fisiopatología de las células gliales en la isquemia cerebral. Neurocirugía, 11, 247 – 259.

136
Paxinos, G., & Watson, C. (1998). Atlas the Rat Brain in Stereotaxic Coordinates (Fourth
Edi.). Estados Unidos: Academic Press.
Pella, D., Rybar, R., & Mechirova, V. (2005). Pleiotropic Effects of Statins. Acta Cardiol
Sin, 21, 190–8.
Piermartiri, T. C. B., Figueiredo, C. P., Rial, D., Duarte, F. S., Bezerra, S. C., Mancini, G.,
… Tasca, C. I. (2010). Atorvastatin prevents hippocampal cell death, neuroinflammation
and oxidative stress following amyloid-β(1-40) administration in mice: evidence for
dissociation between cognitive deficits and neuronal damage. Experimental Neurology,
226, 274–84. doi:10.1016/j.expneurol.2010.08.030
Polazzi, E., & Monti, B. (2010). Microglia and neuroprotection: from in vitro studies to
therapeutic applications. Progress in Neurobiology, 92(1), 293–315.
doi:10.1016/j.pneurobio.2010.06.009
Popp, A., Jaenisch, N., Witte, O. W., & Frahm, C. (2009). Identification of ischemic regions
in a rat model of stroke. PloS One, 4(3), e4764. doi:10.1371/journal.pone.0004764
Posada-Duque, R. A., Velasquez-Carvajal, D., Eckert, G. P., & Cardona-Gomez, G. P.
(2013). Atorvastatin requires geranylgeranyl transferase-I and Rac1 activation to exert
neuronal protection and induce plasticity. Neurochemistry International, 62, 433–45.
doi:10.1016/j.neuint.2013.01.026
Qu, Z.-W., Miao, W.-Y., Hu, S.-Q., Li, C., Zhuo, X.-L., Zong, Y.-Y., … Zhang, G.-Y. (2012).
N-methyl-D-aspartate receptor-dependent denitrosylation of neuronal nitric oxide
synthase increase the enzyme activity. PloS One, 7(12), e52788.
doi:10.1371/journal.pone.0052788

137
Rami, A., Bechmann, I., & Sthehle, J. (2008). Exploiting endogenous anti-apoptotic
proteins for novel therapeutic strategies in cerebral ischemia. Progress in Neurobiology,
85, 273–96. doi:10.1016/j.pneurobio.2008.04.003
Ramos-Esquivel, A., & León-Céspedes, C. (2007). Revisión Efectos no hipolipemiantes
de las estatinas. Acta Médica Costarricense, 49(4), 182–9.
Richardson, W., Kaylene, M. Y., Tripathi, R. B., & McKenzie, I. (2011). NG2-glia as
multipotent neural stem cells - fact or fantasy? Neuron, 70(4), 661–673.
doi:10.1016/j.neuron.2011.05.013.
Rojas, J., Zurru, M., Romano, M., Patrucco, L., & Cristiano, E. (2007). Accidente
cerebrovascular isquémico en mayores de 80 años. Medicina (Buenos …, (67), 701–704.
Retrieved from http://www.scielo.org.ar/scielo.php?pid=S0025-
76802007000600005&script=sci_arttext&tlng=en
Saito, T., Nito, C., Ueda, M., Inaba, T., Kamiya, F., Muraga, K., … Katayama, Y. (2014).
Continuous oral administration of atorvastatin ameliorates brain damage after transient
focal ischemia in rats. Life Sciences, 94, 106–14. doi:10.1016/j.lfs.2013.11.018
Sakry, D., Karram, K., & Trotter, J. (2011). Synapses between NG2 glia and neurons.
Journal of Anatomy, 219, 2–7. doi:10.1111/j.1469-7580.2011.01359.x
Sasaki, T., Kitagawa, K., Sugiura, S., Omura-Matsuoka, E., Tanaka, S., Yagita, Y., …
Hori, M. (2003). Implication of cyclooxygenase-2 on enhanced proliferation of neural
progenitor cells in the adult mouse hippocampus after ischemia. Journal of Neuroscience
Research, 72(4), 461–71. doi:10.1002/jnr.10595
Sawada, N., & Liao, J. (2009). Targeting eNOS and beyond: emerging heterogeneity of
the role of endothelial Rho proteins in stroke protection. Expert Rev Neurother, 9(8),
1171–86. doi:10.1586/ern.09.70.Targeting

138
Schönbeck, U., & Libby, P. (2004). Inflammation, immunity, and HMG-CoA reductase
inhibitors: statins as antiinflammatory agents? Circulation, 109(21 Suppl 1), II18–26.
doi:10.1161/01.CIR.0000129505.34151.23
Shen, H., Wu, X., Zhu, Y., & Sun, H. (2013). Intravenous administration of achyranthes
bidentata polypeptides supports recovery from experimental ischemic stroke in vivo. PloS
One, 8(2), e57055. doi:10.1371/journal.pone.0057055
Silva, F. A., Zarruk, J. G., Quintero, C., Arenas, W., Rueda-Clausen, C., Silva, S. Y., &
Estupiñan, A. (2006). Enfermedad cerebrovascular en Colombia. Revista Colombiana de
Cardiología, 13(2), 85–89.
Silva, F., Quintero, C., & Zarruc, J. (2009). Enfermedad cerebrovascular. In Guía
Neurologica 8 (pp. 22–29).
Sofroniew, M. V, & Vinters, H. V. (2010). Astrocytes: biology and pathology. Acta
Neuropathologica, 119, 7–35. doi:10.1007/s00401-009-0619-8
Sozmen, E. G., Hinman, J. D., & Carmichael, S. T. (2012). Models that matter: white
matter stroke models. Neurotherapeutics : The Journal of the American Society for
Experimental NeuroTherapeutics, 9, 349–58. doi:10.1007/s13311-012-0106-0
Stark, D. T., & Bazan, N. G. (2011). Synaptic and extrasynaptic NMDA receptors
differentially modulate neuronal COX-2 function, lipid peroxidation, and neuroprotection.
J Neurosci, 31(39), 13710–21. doi:10.1523/JNEUROSCI.3544-11.2011.Synaptic
Stefanovic, B., Bosetti, F., & Silva, A. C. (2006). Modulatory role of cyclooxygenase-2 in
cerebrovascular coupling. NeuroImage, 32(1), 23–32.
doi:10.1016/j.neuroimage.2006.03.014

139
Steger, R., Kamal, A., Lutchman, S., Intrabartolo, L., Sohail, R., & Brumberg, J. C. (2014).
Chronic caffeine ingestion causes microglia activation, but not proliferation in the healthy
brain. Brain Research Bulletin, 106, 39–46. doi:10.1016/j.brainresbull.2014.05.004
Stoll, G., Jander, S., & Schroeter, M. (1998). Inflammation and glial responses in ischemic
brain lesions. Progress in Neurobiology, 56(2), 149–71. Retrieved from
http://www.ncbi.nlm.nih.gov/pubmed/9760699
Strauss, K. I. (2008). Antiinflammatory and neuroprotective actions of COX2 inhibitors in
the injured brain. Brain, Behavior, and Immunity, 22, 285–98.
doi:10.1016/j.bbi.2007.09.011
Sun, J., Fang, Y., Ren, H., Chen, T., Guo, J., Yan, J., … Liao, H. (2013). WIN55,212-2
protects oligodendrocyte precursor cells in stroke penumbra following permanent focal
cerebral ischemia in rats. Acta Pharmacologica Sinica, 34(1), 119–28.
doi:10.1038/aps.2012.141
Takano, T., Oberheim, N., Cotrina, M. L., & Nedergaard, M. (2009). Astrocytes and
ischemic injury. Stroke; a Journal of Cerebral Circulation, 40(3 Suppl), S8–12.
doi:10.1161/STROKEAHA.108.533166
Takemoto, M., & Liao, J. K. (2001). Pleiotropic Effects of 3-Hydroxy-3-Methylglutaryl
Coenzyme A Reductase Inhibitors. Arteriosclerosis, Thrombosis, and Vascular Biology,
21, 1712–19. doi:10.1161/hq1101.098486
Thored, P., Heldmann, U., Gomes-Leal, W., Gisler, R., Darsalia, V., Taneera, J., …
Lindvall, O. (2009). Long-term accumulation of microglia with proneurogenic phenotype
concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia,
57, 835–49. doi:10.1002/glia.20810

140
Trotter, J., Karram, K., & Nishiyama, A. (2010). NG2 cells: properties, progeny and origin.
Brain Research Reviews, 63(1-2), 72–82. doi:10.1016/j.brainresrev.2009.12.006.NG2
Trump, B. F., & Berezesky, I. . (1995). Calcium-mediated cell injury and cell death. FASEB
Journal: Official Publication of the Federation of Americam Societies for Experimental
Biology, 9, 219–228.
Tsai, N.-W., Lin, T.-K., Chang, W.-N., Jan, C.-R., Huang, C.-R., Chen, S.-D., … Lu, C.-H.
(2011). Statin pre-treatment is associated with lower platelet activity and favorable
outcome in patients with acute non-cardio-embolic ischemic stroke. Critical Care, 15(4),
R163. doi:10.1186/cc10303
Turner, R. J., Jickling, G. C., & Sharp, F. R. (2011). Are Underlying Assumptions of
Current Animal Models of Human Stroke Correct: from STAIRs to High Hurdles?
Translational Stroke Research, 2, 138–43. doi:10.1007/s12975-011-0067-3
Unidad de evaluación de tecnologías sanitarias de la agencia Laín Entralgo de la
comunidad de Madrid. (2009). Guía de práctica clínica para el manejo de pacientes con
ictus en atención primaria.
Vandresen-Filho, S., Martins, W. C., Bertoldo, D. B., Mancini, G., Herculano, B. a, de
Bem, A. F., & Tasca, C. I. (2013). Atorvastatin prevents cell damage via modulation of
oxidative stress, glutamate uptake and glutamine synthetase activity in hippocampal
slices subjected to oxygen/glucose deprivation. Neurochemistry International, 62, 948–
55. doi:10.1016/j.neuint.2013.03.002
Vilhardt, F. (2005). Microglia: phagocyte and glia cell. The International Journal of
Biochemistry & Cell Biology, 37(1), 17–21. doi:10.1016/j.biocel.2004.06.010

141
Vinet, J., Weering, H. R. J. V, Heinrich, A., Kälin, R. E., Wegner, A., Brouwer, N., … Biber,
K. (2012). Neuroprotective function for ramified microglia in hippocampal excitotoxicity.
Journal of Neuroinflammation, 9(1), 27. doi:10.1186/1742-2094-9-27
Wang, C. X., Yang, Y., Yang, T., & Shuaib, A. (2001). A focal embolic model of cerebral
ischemia in rats: introduction and evaluation. Brain Research Protocols, 7(2), 115–20.
Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11356377
Wang, C. Y., Liu, P. Y., & Liao, J. (2008). Pleiotropic effects of statin therapy. Trends Mol
Med, 14(1), 37–44. doi:10.1016/j.molmed.2007.11.004.Pleiotropic
Wang, C., Yang, T., & Shuaib, A. (2001). An improved version of embolic model of brain
ischemic injury in the rat. Journal of Neuroscience Methods, 109(2), 147–51. Retrieved
from http://www.ncbi.nlm.nih.gov/pubmed/11513949
Wang, J., Yang, Z., Liu, C., Zhao, Y., & Chen, Y. (2013). Activated microglia provide a
neuroprotective role by balancing glial cell-line derived neurotrophic factor and tumor
necrosis factor-α secretion after subacute cerebral ischemia. International Journal of
Molecular Medicine, 31, 172–8. doi:10.3892/ijmm.2012.1179
Wang, W., Redecker, C., Yu, Z.-Y., Xie, M.-J., Tian, D.-S., Zhang, L., … Witte, O. W.
(2008). Rat focal cerebral ischemia induced astrocyte proliferation and delayed neuronal
death are attenuated by cyclin-dependent kinase inhibition. Journal of Clinical
Neuroscience, 15, 278–85. doi:10.1016/j.jocn.2007.02.004
Waszkielewicz, A. M., Gunia, A., Szkaradek, N., Słoczyńska, K., Krupińska, S., & Marona,
H. (2013). Ion channels as drug targets in central nervous system disorders. Current
Medicinal Chemistry, 20, 1241–85. Retrieved from
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3706965&tool=pmcentrez&re
ndertype=abstract

142
Weinstein, J. R., & Möller, T. (2011). Microglia in ischemic brain injury. Future Neurol,
5(2), 227–246. doi:10.2217/fnl.10.1.
Wigley, R., Hamilton, N., Nishiyama, A., Kirchhoff, F., & Butt, A. M. (2007). Morphological
and physiological interactions of NG2-glia with astrocytes and neurons. Journal of
Anatomy, 210, 661–70. doi:10.1111/j.1469-7580.2007.00729.x
Wood, W. G., Eckert, G. P., Igbavboa, U., & Müller, W. (2010). Statins and
neuroprotection. a prescription to move the field forward. Ann N Y Acad Sci, 1199, 69–
76. doi:10.1111/j.1749-6632.2009.05359.x.STATINS
Woodruff, T. M., Thundyil, J., Tang, S.-C., Sobey, C. G., Taylor, S. M., & Arumugam, T.
V. (2011). Pathophysiology, treatment, and animal and cellular models of human ischemic
stroke. Molecular Neurodegeneration, 6, 11. doi:10.1186/1750-1326-6-11
Xu, S.-Y., & Pan, S.-Y. (2013). The failure of animal models of neuroprotection in acute
ischemic stroke to translate to clinical efficacy. Medical Science Monitor Basic Research,
19, 37–45. Retrieved from
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3638705&tool=pmcentrez&re
ndertype=abstract
Yagami, T. (2006). Cerebral Arachidonate Cascade in Dementia : Alzheimer ’ s Disease
and Vascular Dementia. Current Neuropharmacology, 4, 87–100.
Yang, H., & Chen, C. (2008). Cyclooxygenase-2 in synaptic signaling. Curr Pharm Des,
14(14), 1443–1451.
Yokota, C., Kaji, T., Kuge, Y., Inoue, H., Tamaki, N., & Minematsu, K. (2004). Temporal
and topographic profiles of cyclooxygenase-2 expression during 24 h of focal brain
ishemia in rats. Neuroscience Letters, 357, 219–22. doi:10.1016/j.neulet.2003.12.109

143
Yrjänheikki, J., Koistinaho, J., Kettunen, M., Kauppinen, R. A., Appel, K., Hüll, M., &
Fiebich, B. L. (2005). Long-term protective effect of atorvastatin in permanent focal
cerebral ischemia. Brain Research, 1052(2), 174–9. doi:10.1016/j.brainres.2005.06.004
Yrjänheikki, J., Tikka, T., Keinänen, R., Goldsteins, G., Chan, P. H., & Koistinaho, J.
(1999). A tetracycline derivative, minocycline, reduces inflammation and protects against
focal cerebral ischemia with a wide therapeutic window. Proceedings of the National
Academy of Sciences of the United States of America, 96(23), 13496–500. Retrieved
from
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=23976&tool=pmcentrez&rend
ertype=abstract
Yu, L., Jiang, R., Su, Q., Yu, H., & Yang, J. (2014). Hippocampal neuronal metal ion
imbalance related oxidative stress in a rat model of chronic aluminum exposure and
neuroprotection of meloxicam. Behavioral and Brain Functions : BBF, 10(1), 6.
doi:10.1186/1744-9081-10-6
Yuan, J., & Yankner, B. a. (2000). Apoptosis in the nervous system. Nature, 407(6805),
802–9. doi:10.1038/35037739
Zeiser, R., Maas, K., Youssef, S., Dürr, C., Steinman, L., & Negrin, R. S. (2008).
Regulation of different inflammatory diseases by impacting the mevalonate pathway.
Immunology, 127, 18–25. doi:10.1111/j.1365-2567.2008.03011.x
Zhang, L., Chopp, M., Jia, L., Cui, Y., Lu, M., & Zhang, Z. G. (2009). Atorvastatin extendes
the therapeutic window for tPA to 6 h after the onset of embolic stroke in rats. J Cereb
Blood Flow Metab, 29(11), 1816–1824. doi:10.1038/jcbfm.2009.105.Atorvastatin
Zhang, L., Chopp, M., Zhang, R. L., Wang, L., Zhang, J., Wang, Y., … Zhang, Z. G.
(2010). Erythropoietin amplifies stroke-induced oligodendrogenesis in the rat. PloS One,
5(6), e11016. doi:10.1371/journal.pone.0011016

144
Zhang, Z.-G., Sun, X., Zhang, Q.-Z., & Yang, H. (2013). Neuroprotective effects of ultra-
low-molecular-weight heparin on cerebral ischemia/reperfusion injury in rats: involvement
of apoptosis, inflammatory reaction and energy metabolism. International Journal of
Molecular Sciences, 14(1), 1932–9. doi:10.3390/ijms14011932
Zhou, Q., & Liao, J. (2010). Pleiotropic effects of statins: basic research and clinical
perspectives. Circ J, 74(5), 818–26.

145
ANEXOS

146
Anexo A. Formato protocolo técnico.

147
Anexo B. Formato evaluación Neurológica.
Recommended

















