
Gaona de Gaona María Dolores
Hernández López Rubí
Juárez Pérez Rosario
Morales Isaias
Palacios Gonzales Beatriz
Pantle Castillo Joshelin
Varela Trujillo Claudia

Enfermedad hereditaria
Globulos rojos en forma semilunar
Caracterizado por una anomalía de la hemoglobina
Tienden a aglutinarse y viven solo de 10 a 20 días
Mutación genética
Un bebe nace con AD
si hereda dos genes Hb
S
Es autosómica recesiva



Sustitución de ácido glutámico por valina
Cadena de β-globina en el cromosoma 11
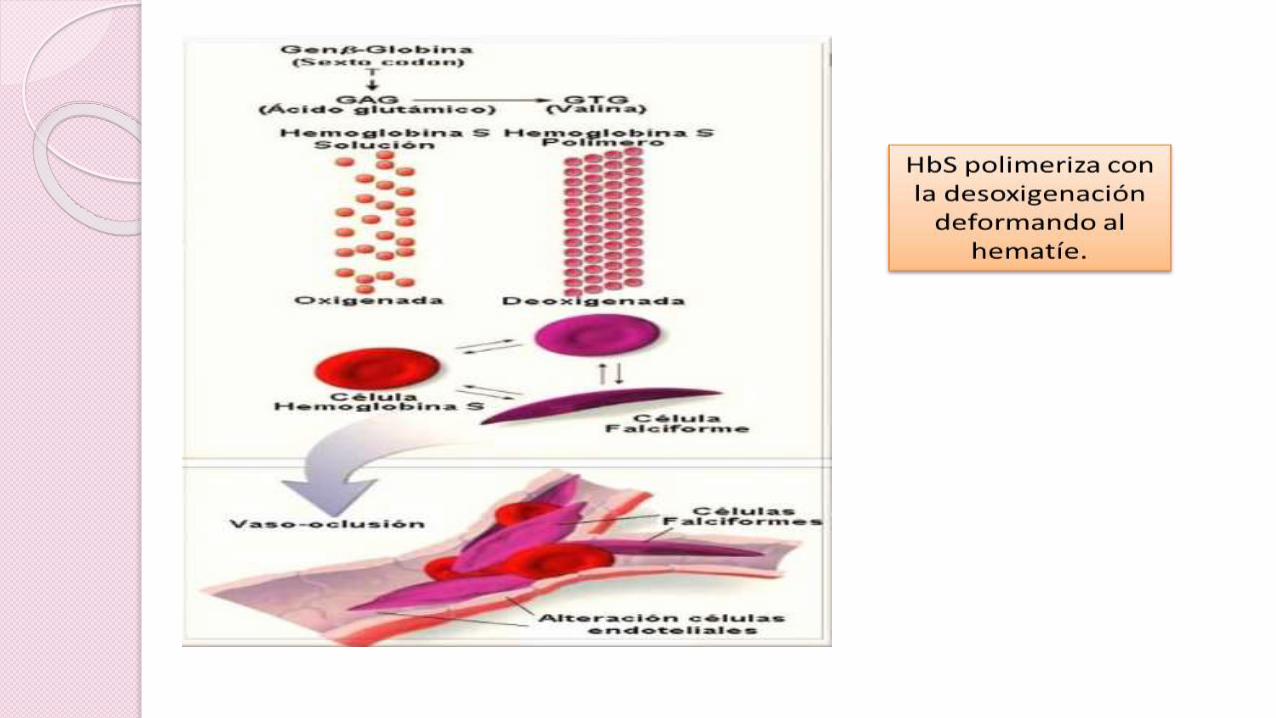


Forma homocigota o anemia
falciforme (HbSS)
Afecta a uno solo de los alelos que codifican
la cadena β.
Afecta a los 2 alelos del gen correspondiente
a la cadena β.
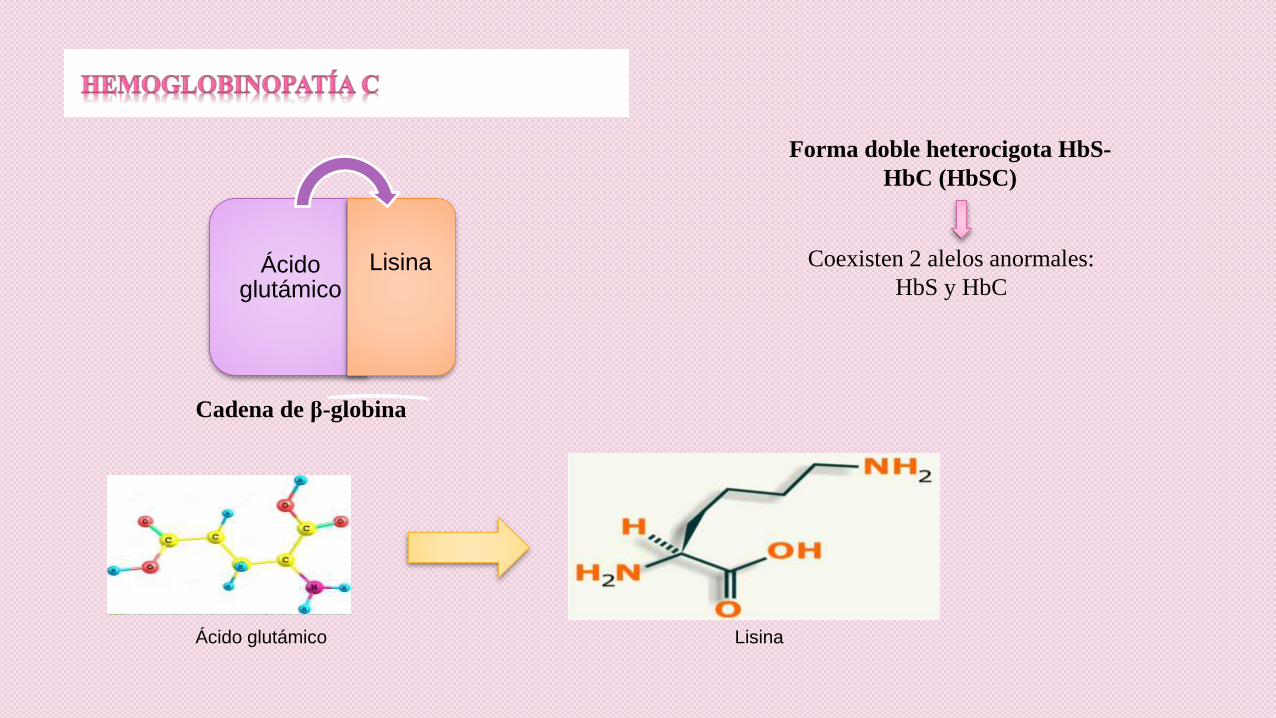
Ácido glutámico
Lisina
Cadena de β-globina
Ácido glutámico Lisina
Forma doble heterocigota HbS-
HbC (HbSC)
Coexisten 2 alelos anormales:
HbS y HbC

Glicina Ácido aspartico
Cadena β-globina (posicición 16)
Glicina
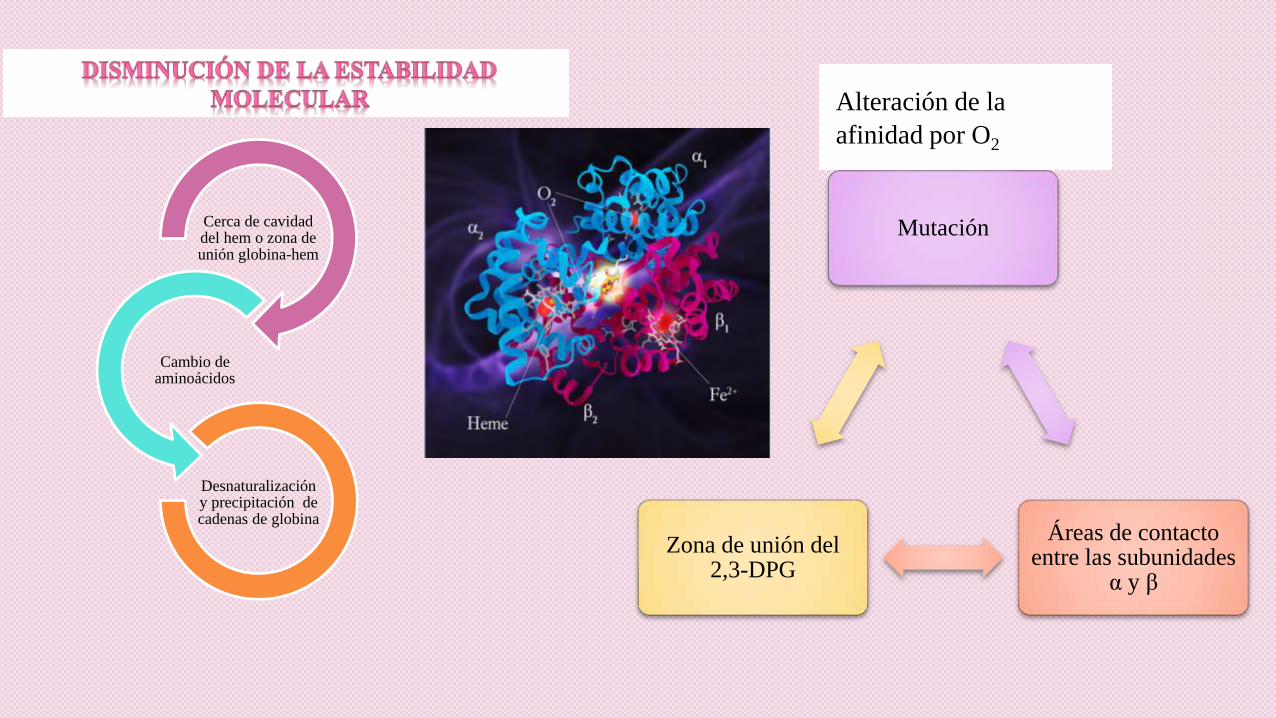
Alteración de la
afinidad por O2
Cerca de cavidad del hem o zona de unión globina-hem
Cambio de aminoácidos
Desnaturalización y precipitación de cadenas de globina
Mutación
Áreas de contacto entre las subunidades
α y β
Zona de unión del 2,3-DPG


• Leve fatiga
• Falta de aire
• Dolor de pecho y cabeza
• Piel pálida
• Retraso en el crecimiento
• Frío en manos y pies
• Ictericia
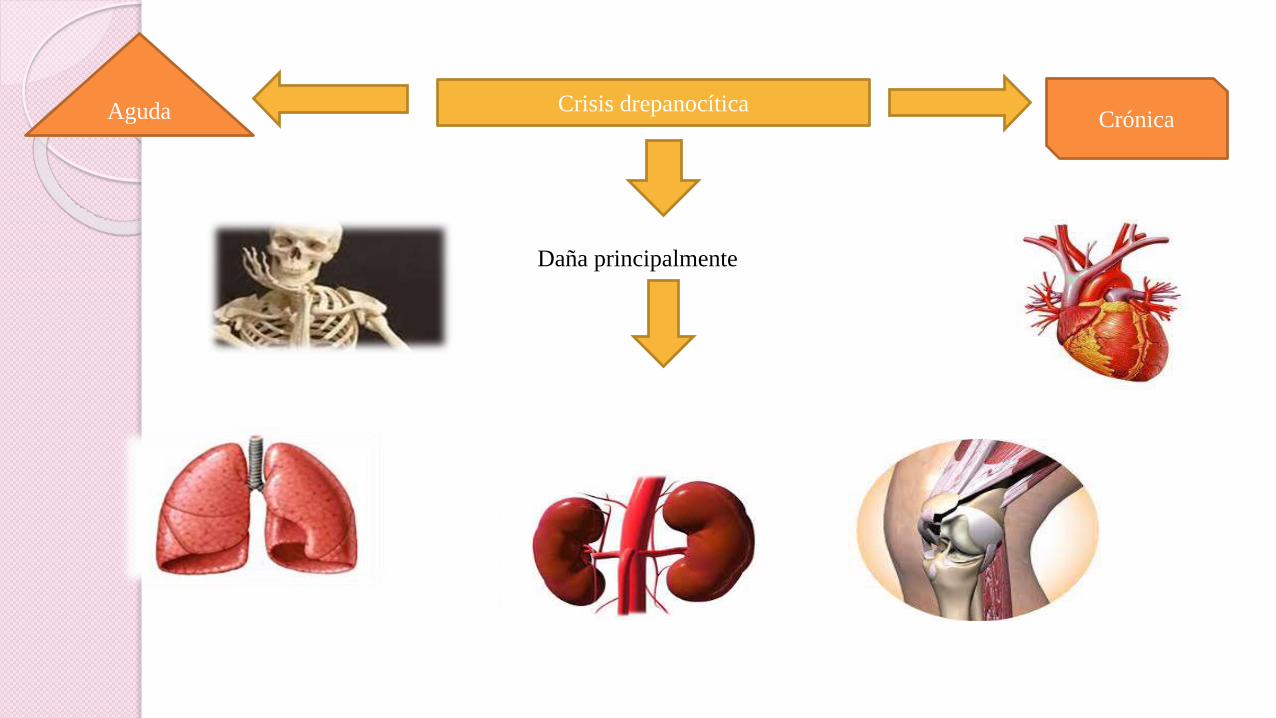
Crisis drepanocítica
Daña principalmente
Aguda Crónica

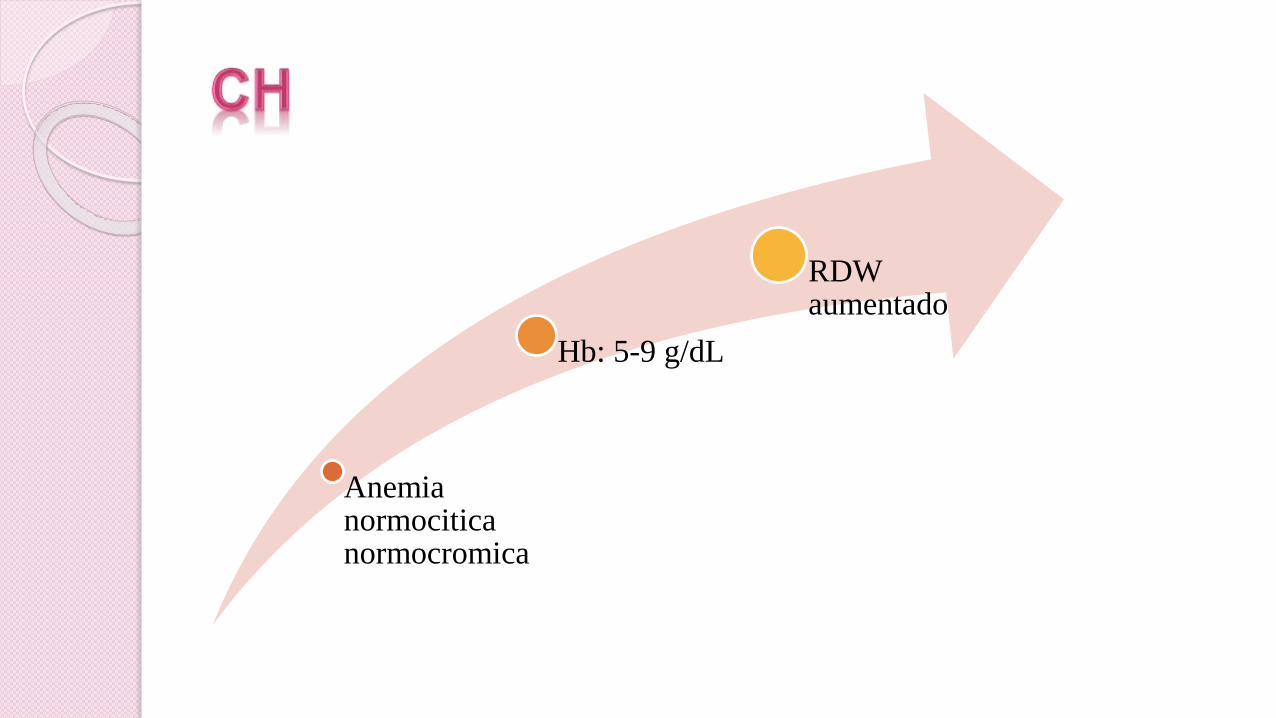
Anemia normociticanormocromica
Hb: 5-9 g/dL
RDW aumentado

Anisocitosis, poiquilocitosis e hipocromia.
• Dianocitos.
A veces se advierten drepanocitosen extensiones teñidas.
• Queratocitos, esquistocitos.
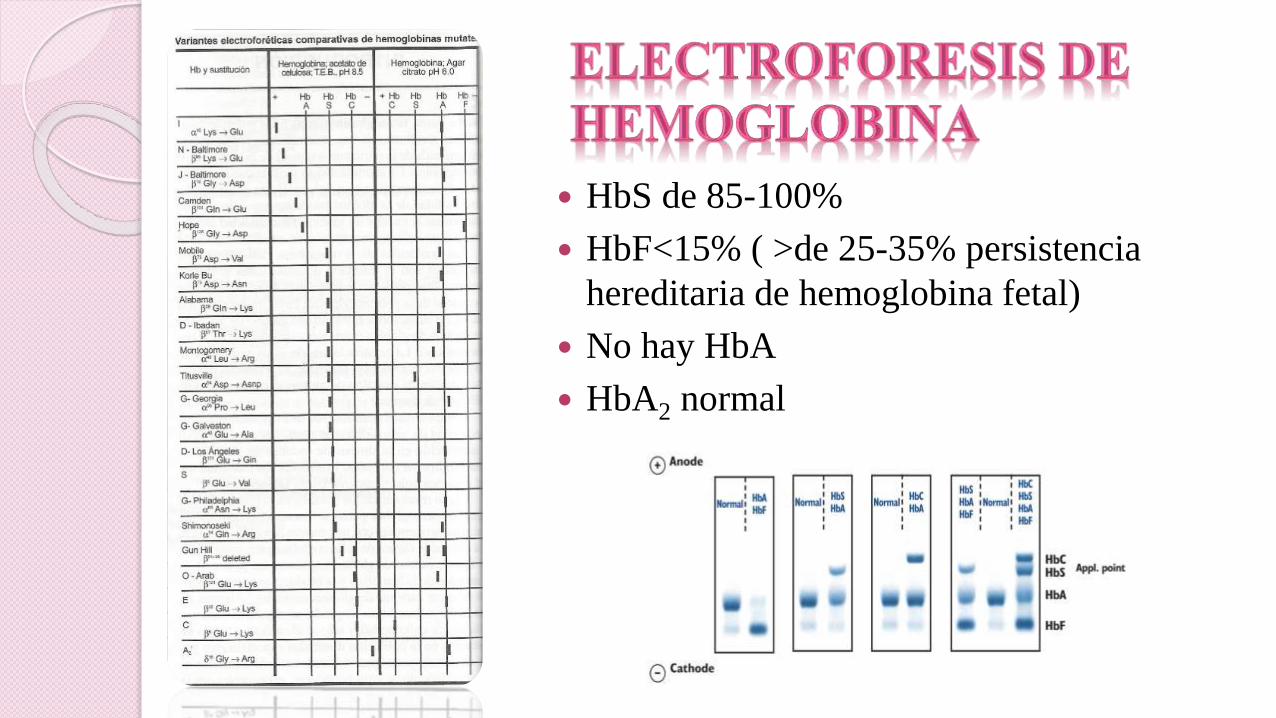
HbS de 85-100%
HbF<15% ( >de 25-35% persistencia
hereditaria de hemoglobina fetal)
No hay HbA
HbA2 normal

Confirma HbS HgS Causa células
falciformes.
Sangre
(lisada) +
ditionito de
sodio(agente
reductor)
Turbidez
HgS polimerizada
(insoluble)

Gota de sangre
+
Pirosulfito de sodio o
metabisulfito

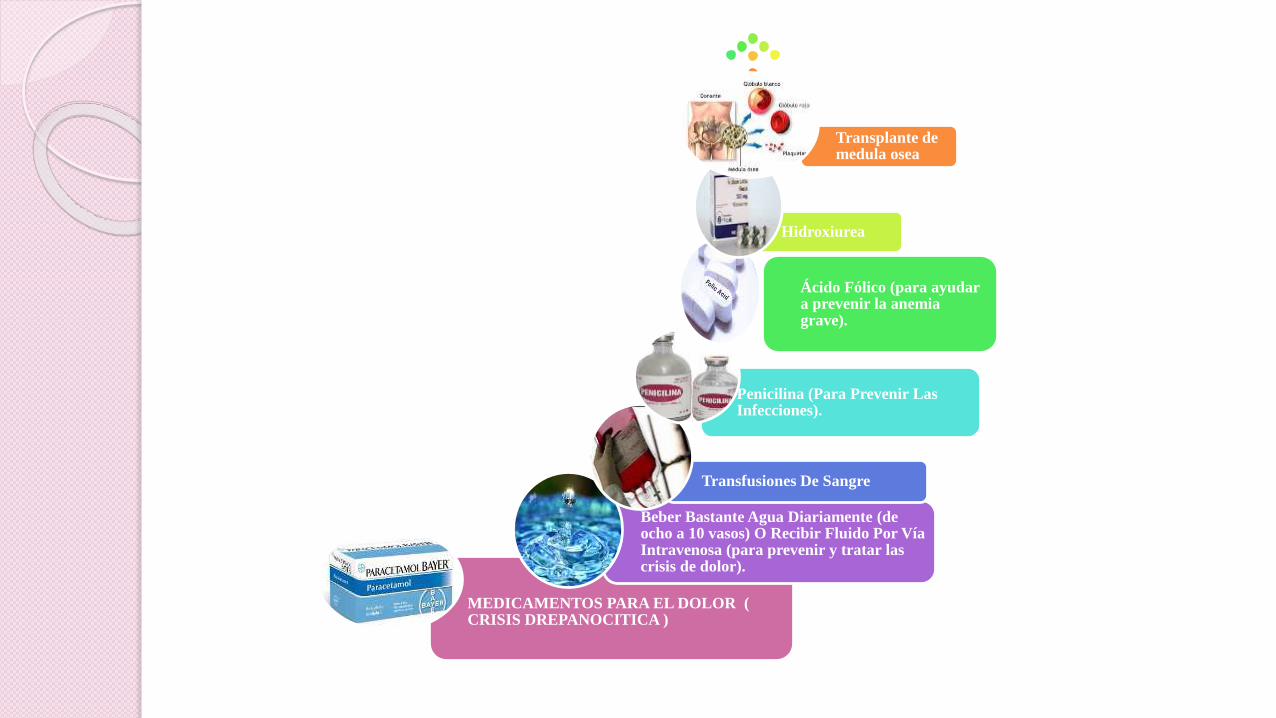
Ibuprofeno y aspirina
MEDICAMENTOS PARA EL DOLOR ( CRISIS DREPANOCITICA )
Beber Bastante Agua Diariamente (de ocho a 10 vasos) O Recibir Fluido Por Vía Intravenosa (para prevenir y tratar las crisis de dolor).
Transfusiones De Sangre
Penicilina (Para Prevenir Las Infecciones).
Ácido Fólico (para ayudar a prevenir la anemia grave).
Hidroxiurea
Transplante de medula osea
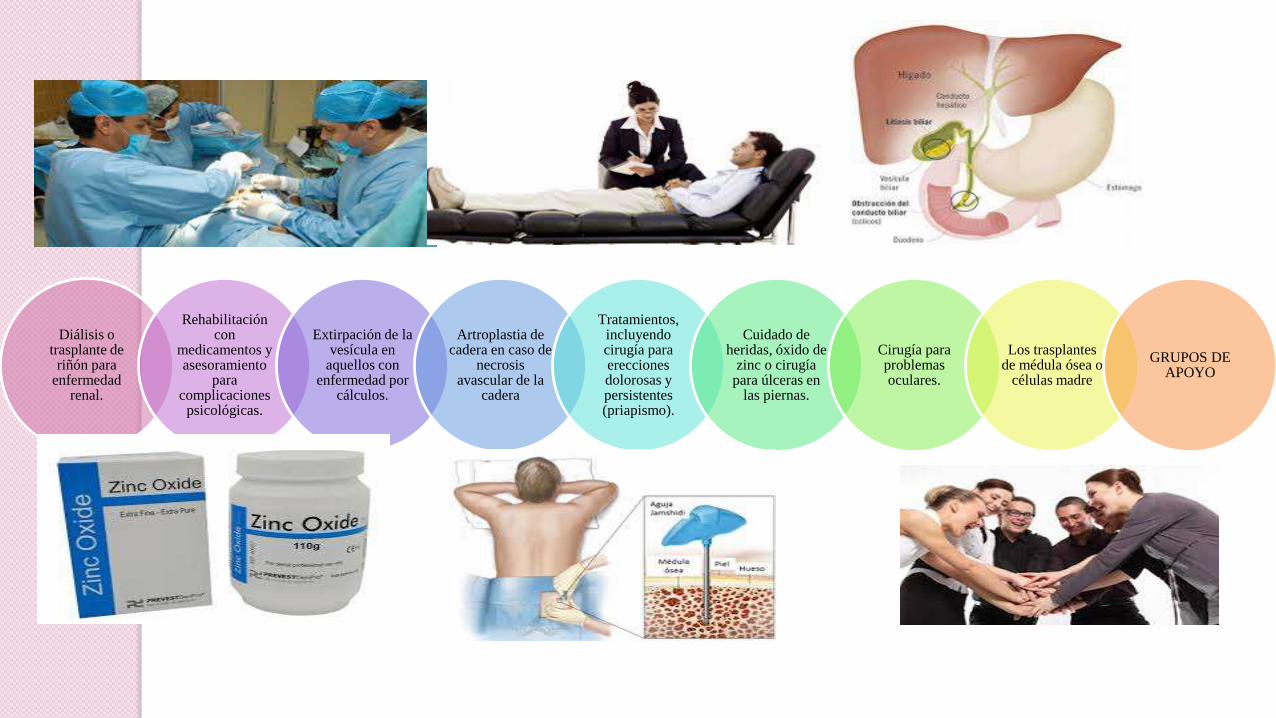
Diálisis o trasplante de
riñón para enfermedad
renal.
Rehabilitación con
medicamentos y asesoramiento
para complicaciones
psicológicas.
Extirpación de la vesícula en
aquellos con enfermedad por
cálculos.
Artroplastia de cadera en caso de
necrosis avascular de la
cadera
Tratamientos, incluyendo cirugía para erecciones dolorosas y persistentes (priapismo).
Cuidado de heridas, óxido de
zinc o cirugía para úlceras en
las piernas.
Cirugía para problemas oculares.
Los trasplantes de médula ósea o
células madre
GRUPOS DE APOYO

La anemia drepanocítica se puede presentar sólo cuando dos personas portadoras del rasgo
drepanocítico tienen un hijo juntos
Es posible diagnosticar esta anemia durante el embarazo.
Si usted tiene anemia drepanocítica, puede prevenir el cambio en la forma de los glóbulos
rojos:
• Tomando mucho líquido.
• Recibiendo suficiente oxígeno.
• Tratando rápidamente las infecciones.
• Procure que le hagan un examen físico o chequeos médicos cada 3 a 6 meses para constatar
que esté obteniendo la nutrición y actividad suficientes y que esté recibiendo las vacunas
apropiadas. También se recomiendan exámenes regulares de los ojos.

• Es importante mantener buenos niveles de oxígeno y prevenir la deshidratación. Las siguientes medidas pueden
ayudar a prevenir las crisis drepanocíticas:
• Evitar la actividad extenuante, el estrés, el tabaquismo, las grandes alturas, los vuelos en aviones no presurizados
y otros eventos que reduzcan el nivel de oxígeno.
• Tener siempre muchos líquidos a la mano.
• Evitar la exposición excesiva al sol.
• Contemple la posibilidad de que el niño lleve puesto un brazalete de alerta médica. Comparta la información
anterior con profesores y otros cuidadores cuando sea necesario

Dolor Abdominal Agudo debido a Infarto Esplénico en un paciente con
Enfermedad Heterocigota de Células Falciformes expuesto a la altura

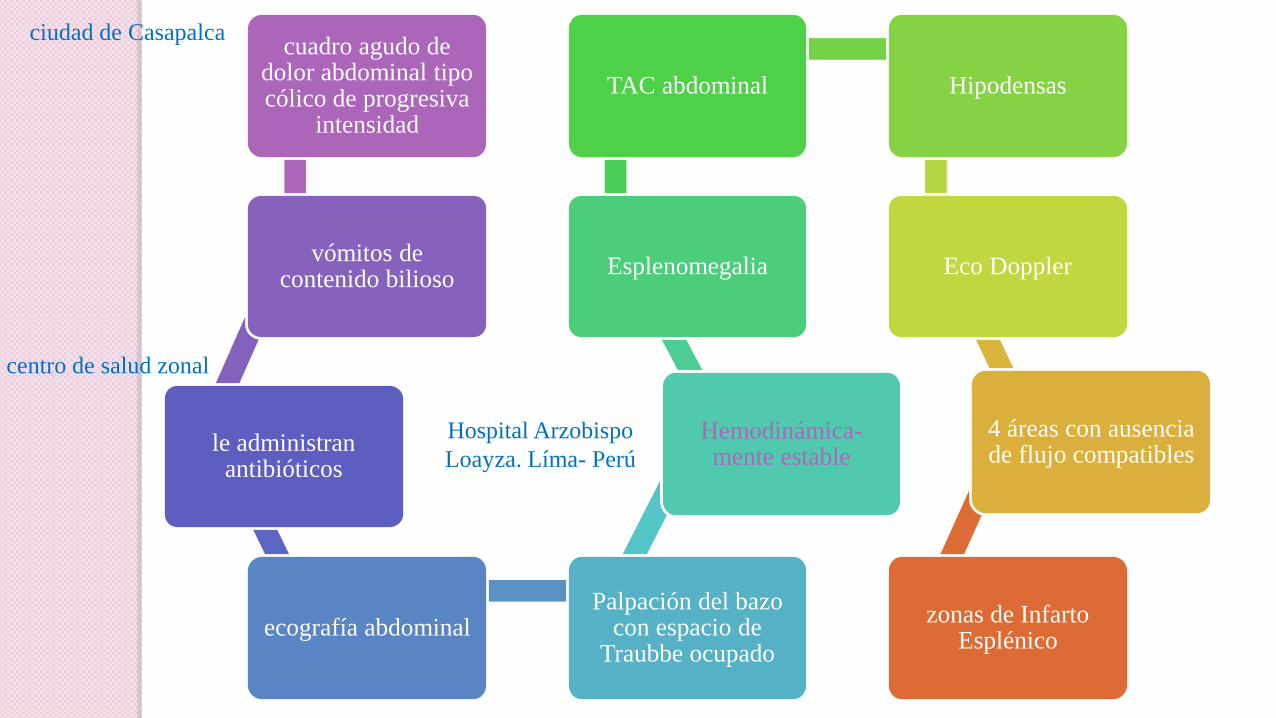
cuadro agudo de dolor abdominal tipo cólico de progresiva
intensidad
vómitos de contenido bilioso
le administran antibióticos
ecografía abdominal Palpación del bazo
con espacio de Traubbe ocupado
Hemodinámica-mente estable
Esplenomegalia
TAC abdominal Hipodensas
Eco Doppler
4 áreas con ausencia de flujo compatibles
zonas de Infarto Esplénico
ciudad de Casapalca
centro de salud zonal
Hospital Arzobispo
Loayza. Líma- Perú


Con el diagnóstico planteado de Infarto Esplénico se realizan los siguientes exámenes
Leucocitos: 18,300 ( B: 0%, A: 3, E: 1%, S: 82%, L: 10%)
Hb: 14gr/dl
Hto:41%
Frotis de Sangre Periférica con Hipocromía y Anisocitosis
plaquetas normales
Amilasa: 77 U/I
Sedimento Urinario con 5-10 leucocitos por campo,cilindros granulosos, filamentos mucoides
Glucosa: 82mg/dl
Urea:31mg/dl
Perfi l Hepático dentro de límites normales

Se solicitó entonces Electroforesis de Hemoglobina:
Determinación de Hb A1 : 59% (VN: 94-99%)
Hb A2: 2.3% (VN: 1,0-3,5%)
Hb Fetal: 1.7%
Hb S: 37.0% (VN:0%)
conclusión: Heterocigote de Hemoglobina AS.
Se le solicita un hemograma de control a los 5 días con disminución del número de
leucocitos a 10,600(A:0%)
Hb:12.4 gr/dl
Sedimento Urinario con presencia de algunos hematíes
El paciente experimenta una notable mejoría clínica con tratamiento sintomático conservador, y sale
de alta a los 8 días de su ingreso en el pabellón

No reveló presencia de patología cardiaca ni enfermedad
autoinmune asociada e estado de hipercoagulabilidad.
Antecedente epidemiológico de su procedencia
Dolor abdominal
Posibilidad diagnóstica de una hemoglobinopatía.
El estudio de electroforesis de hemoglobina:
confirmó la presencia de una depranocitosis o
falciformismo (Sickle cell)
Variedad heterocigota de células falciformes (Sickle cell
Trait).

El caso descrito corresponde a un síndrome doloroso
abdominal por infarto esplénico asociado a enfermedad
heterocigota de células falciformes, condición que deberá
tenerse en cuenta en los cuadros de dolor abdominal agudo
asociado a la exposición de grandes alturas.
El tratamiento fue conservador consistente en hidratación,
analgesia y adecuada oxigenación, La evolución fue
favorable estando en condiciones de alta a los 8 días de su
hospitalización del Servicio de Medicina

https://umm.edu/health/medical/spanishency/.../anemia-drepanocitica
http://www.cirbiomedicas.uady.mx/sgc/documentos/procedimiento3/M-CIRB-AADC-02.pdf
Fuente del contenido:
◦ Fuente del contenido:Centro Nacional de Defectos Congénitos y Discapacidades del Desarrollo de los CDC, Centros para el Control y
la Prevención de Enfermedades
◦ Versión en español aprobada por CDC Multilingual Services – Order # 213266
Powars D, Chan LS, Schroeder Wa. The variable expression of sickle cell disease is genetically determined. Semin Hematol
1990;27:360_76.
Stuart M, Nagel R. Sickle-cell disease. Lancet 2004;364:1343-59.
Walters MC, Storb R, Patience M, Leisenring W, Taylor T, Sanders JE, et al. Impact of bone marrow transplantation for symptomatic sickle
cell disease: An interim report. Multicenter investigation of bone marrow transplantation for sickle cell disease. Blood 2000;95:1918-24.
Charache S, Terrin ML, Moore RD, Dover GT, Barton FB, Eckert SU, et al. Effect of hydroxyurea on the frequency of painful crisis in
sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Eng J Med 1995;332:1317-22.
Recommended

















