
I
UNIVERSIDAD AUTONOMA DE NUEVO LEÓN
FACULTAD DE CIENCIAS BIOLÓGICAS
MUERTE CELULAR PROGRAMADA 4 (PDCD4) INHIBE LA PROGRESIÓN DE
CÁNCER DE MAMA INDUCIDA POR LEPTINA Y COX-2.
T E S I S
COMO REQUISITO PARCIAL PARA OBTENER EL GRADO DE DOCTOR EN CIENCIAS CON ESPECIALIDAD EN BIOTECNOLOGÍA
PRESENTA:
Q.B.P. VIANEY GONZÁLEZ VILLASANA
MAYO, 2012

I
UNIVERSIDAD AUTÓNOMA DE NUEVO LEÓN
FACULTAD DE CIENCIAS BIOLÓGICAS
MUERTE CELULAR PROGRAMADA 4 (PDCD4) INHIBE LA PROGRESIÓN DE CÁNCER DE MAMA INDUCIDA POR LEPTINA Y COX-2.
POR
Q.B.P. VIANEY GONZÁLEZ VILLASANA
COMO REQUISITO PARCIAL PARA OBTENER EL GRADO DE DOCTOR EN CIENCIAS CON ESPECIALIDAD EN BIOTECNOLOGÍA
DIRECTOR DE TESIS: Dra. YOLANDA GUTIERREZ PUENTE
DIRECTOR EXTERNO: Dra. ANA M. TARI
CO-DIRECTOR: Dr. ARTURO CHÁVEZ REYES
MAYO, 2012

II
MUERTE CELULAR PROGRAMADA 4 (PDCD4) INHIBE LA PROGRESIÓN DE
CÁNCER DE MAMA INDUCIDA POR LEPTINA Y COX-2.
COMITÉ DE TESIS
Director Interno: Dra. Yolanda Gutiérrez Puente
Director Externo: Dra. Ana María Tari
Co-Director: Dr. Arturo Chávez Reyes
Secretario: Dr. Pablo Zapata Benavides
1er. Vocal: Dr. Carlos Eduardo Hernández Luna
2do. Vocal: Dra. Diana Reséndez Pérez
3er. Vocal: Dr. Arturo Chávez Reyes
Alumno: Q.B.P. Vianey González Villasana

III
ÍNDICE
Sección
Página
DEDICATORIA...................................................................................... I
AGRADECIMIENTOS……………………………………………………… II
LUGAR DE TRABAJO…………………………………………………….. III
ABREVIATURAS Y SÍMBOLOS…………………………………………. IV
ÍNDICE DE TABLAS………………………………………………………. VIII
ÍNDICE DE FIGURAS……………………………………………………… IX
Capítulo 1.
ANTECEDENTES………………………………………………………….
1
1.1. Cáncer de mama…...………………………………………….. 1
1.2. Leptina……………….………………………………………….. 3
1.3. Ciclooxigenasa-2 (COX-2)…………………………………….. 7
1.4. Invasión celular, Metaloproteinasas e Inhibidor Tisular de
Metaloproteinasas-2 (TIMP-2)………...……………………………
13
1.5. Resistencia a tamoxifeno inducida por leptina y COX-2…... 16
1.6. JNK (Jun N-terminal quinasa)………………………………… 18
1.7. ERK (Quinasa reguladora de señales extracelulares) y
JAK2/STAT3 (Janus quinasa2/transductor de señal y activador
de la transcripción 3)…….…………………………………………..
19
1.8. Proteína de Muerte Celular Programada 4 (PDCD4)………. 20
Capítulo 2.
RESUMEN………………………………………………………………….
24
INTRODUCCIÓN................................................................................. 25
2.1. HIPÓTESIS….…………………………………………………. 27
2.2. OBJETIVO GENERAL………………………………………… 28
2.3. OBJETIVOS PARTICULARES………………………………. 28

IV
2.4. MATERIAL Y MÉTODOS…………………………………………… 29
2.4.1. Líneas celulares tumorales………………………………….. 29
2.4.2. Ensayo de invasión en Matrigel…………………………….. 29
2.4.3. Transfección de las células MCF-7/PDCD4 con siRNA
anti-TIMP-2………………..…………………………………………..
30
2.4.4. Extracción de RNA total y reacción en cadena de la
polimerasa con transcripción reversa (RT-PCR)………………….
31
2.4.5. Western Blot……..……………………………………………. 32
2.4.5.1. Obtención de las muestras ……………………………..… 32
2.4.5.2. Preparación de las muestras……………………………… 32
2.4.5.3. Electroforesis de proteínas en geles de poliacrilamida
en presencia de dodecil sulfato de sodio (SDS-PAGE)………….
32
2.4.5.4. Inmunoelectrotransferencia…..…………………………… 33
2.4.6. Ensayo de viabilidad celular (MTS)……………………....... 34
2.5. RESULTADOS……………………………………………………….. 35
2.5.1. PDCD4 inhibe la invasión inducida por leptina en las
células de cáncer de mama MCF-7………………………………..
35
2.5.2. TIMP-2 es esencial para que PDCD4 inhiba la invasión
inducida por leptina en células de cáncer de mama MCF-7…….
38
2.5.3. El silenciamiento de TIMP-2 permite la activación de
ERK1,2 y STAT3 inducida por leptina en las células MCF-
7/PDCD4……………………………………………………………..
42
2.5.4. PDCD4 inhibe la resistencia a tamoxifeno inducida por
leptina…………………………………………………………………
44
2.6. DISCUSIÓN Y CONCLUSIÓN……………………….…………….. 46
Capítulo 3.
RESUMEN…………………………………………………………………..
49
INTRODUCCIÓN.................................................................................. 50
3.1. HIPÓTESIS…………..…………………………………………. 52
3.2. OBJETIVO GENERAL……………………………………….… 53
3.3. OBJETIVOS PARTICULARES…………………………..…… 53
3.4. MATERIAL Y MÉTODOS………………………………………….. 54

V
3.4.1. Cultivo celular……………..…………………………………. 54
3.4.2. Reactivos………………………….…………………………. 54
3.4.3. Western blot……………………….. ……………………….. 55
3.4.4. Extracción nuclear de proteínas…………………………… 55
3.4.5. Ensayo de invasión en Matrigel…..……………………….. 56
3.4.6. Ensayo de viabilidad celular (MTS)……..………………… 57
3.5. RESULTADOS………………………………………………………. 58
3.5.1. La sobreexpresión de COX-2 incrementa los niveles de
JNK fosforilado……………………..………………………………..
58
3.5.2. La inhibición de JNK disminuye la invasión de las células
MCF-/COX-2…………….………………..………………………….
61
3.5.3. La inhibición de JNK no incrementa la sensibilidad de las
células MCF-7/COX-2 a tamoxifeno…..…………………………..
64
3.5.4. PDCD4 no inhibe la resistencia a tamoxifeno inducida por
PGE2………………………………………………………………….
65
3.6. DISCUSIÓN Y CONCLUSIÓN……………………….…………….
67
CONCLUSIÓN GENERAL………………………………………………. 71
APÉNDICE………………………………………………………………… 73
LITERATURA CITADA…………………………………………............. 79

I
DEDICATORIA.
A Dios: Por estar siempre conmigo, por todo lo que me ha dado y brindarme
paciencia y fuerza para no desistir y cumplir mis objetivos.
A mis Padres: María Isabel Villasana de González y Fernando González
Sánchez por confiar siempre en mi, por su paciencia e invaluables consejos, pero
sobre todo por cuidar de mi hijo Christopher como si fueran sus padres en nuestra
ausencia, que Dios los bendiga siempre.
A mi hijo Christopher: Por ser la inspiración que me dio fuerza en todo momento
para cumplir esta meta. Christopher perdóname por el tiempo que tuve que
sacrificar lejos de ti, jamás volveré a separarme de ti, te amo, eres los más
hermoso e importante en mi vida, gracias por ser mi hijo, por ser fuerte y por ser
el motor que me ayuda a funcionar y ser mejor cada día.
A Cristian: Que en los últimos años me ha apoyado e impulsado a alcanzar esta
meta, que en los momentos difíciles a permanecido a mi lado y ha sido mi soporte
para no darme por vencida, que ha compartido conmigo alegrías, penas,
sacrificios y noches en vela, pero sobre todo porque ha sido mi amor y compañero
incondicional.
A mis hermanos Diana Elizabeth, Luis Mario y Fernando: Por creer en mí, por
todo su cariño y porque han sido el motivo de mi superación.
A mi hermana Diana: Por sus incontables consejos, su compañía, cariño, apoyo
y comprensión.
A mis Abuelos: María de la Luz Sánchez Reyes, Guadalupe González Cisneros
e Ismael Villasana Sánchez por todos los buenos momentos vividos, enseñanzas
que me brindaron, porque los amo y extraño.
A mi abuelita Juana Sánchez Velásquez: Por todo su apoyo, enseñanzas,
consejos y el amor que me ha brindado.

II
AGRADECIMIENTOS.
Agradezco a tres instituciones sin las cuales este trabajo no hubiese sido
posible. Al CONACYT por el apoyo económico brindado durante la realización de
este proyecto. A la Facultad de Ciencias biológicas de la Universidad
Autónoma de Nuevo León, en la Ciudad de San Nicolás de los Garza, Nuevo
León, México y al MD Anderson Cancer Center de la Universidad de Texas
en la Ciudad de Houston TX, USA específicamente al Laboratorio de
Acarreadores de Drogas en el Departamento de Terapia Experimental: Por la
gran oportunidad que me dieron de formar parte de los mismos y el inmenso
apoyo brindado en la realización de este trabajo.
A la Dra. Ana M. Tari, Dra. Yolanda Gutiérrez Puente, Dr. Arturo Chávez
Reyes, Dr. René Nieves Alicea, DDR Gabriel Lopez-Berestein y al Maestro en
Ciencias José Luis Méndez. Porque gracias a ustedes comencé y culminé esta
meta en mi vida, por creer y confiar en mí, por sus invaluables consejos y su
asesoría científica, por estimularme a seguir creciendo profesional y
personalmente, pero sobre todo por concederme la dicha de contar con su valiosa
amistad.
Al Maestro en Ciencias Cristian Rodríguez Aguayo: Por el gran apoyo y
colaboración brindados en esta investigación, al compartirme sus conocimientos y
experiencia científica.
A Vanity McMurtry, Armando Huaringa, Dr. Pablo Vivas Mejía y Dra. Juliana
M Benito: Por su colaboración en el desarrollo de esta tesis, su disposición
incondicional en aclarar mis dudas, compartirme sus experiencias en el ámbito
científico y por su gran amistad.
I would like to express my sincere gratitude to my external Advisor Dr Ana M. Tari
for give me the opportunity of work with her, for trust in me, for her patience,
continuous support and guidance, for helping me all the time and share her
experience and knowledge, for her invaluable friendship. Thank you very much
Ana.

III
EL PRESENTE TRABAJO SE REALIZÓ EN COLABORACION CON EL LABORATORIO DE ACARREADORES DE DROGAS EN EL DEPARTAMENTO DE TERAPIA EXPERIMENTAL EN EL MD ANDERSON CANCER CENTER DE
LA UNIVERSIDAD DE TEXAS EN LA CIUDAD DE HOUSTON, TX, U.S.A.

IV
ABREVIATURAS Y SÍMBOLOS
AA Acido araquidónico
Akt/GSK3 Serina/treonina quinasa/glucógeno sintasa quinasa 3
AP-1 Proteína activadora-1
ATCC American type cell culture
Bax Proteína X asociada a Bcl-2
BCL-2 Linfoma de células B-2
bFGF Factor de crecimiento de fibroblastos básico
CDI Carcinoma ductal invasivo
CDIS Carcinoma ductal in situ
cdk2 Quinasa dependiente de ciclina 2
CO2 Dióxido de carbono
COX-1 Ciclooxigenasa-1
COX-2 Ciclooxigenasa-2
COX-3 Ciclooxigenasa-3
CSS Suero tratado con carbón vegetal
DMEM Medio Eagle modificado por Dulbecco
DMEM/F12 Medio Eagle modificado por Dulbecco
DNA Ácido desoxiribonucleico
Dra. Doctora
EDTA Ácido etilendiaminotetracético
EGFR Receptor del factor de crecimiento epidérmico
eIF4A Factor de inicio eucariótico 4A
eIF4E Factor de inicio eucariótico 4E
eIF4G Factor de inicio eucariótico 4G
ER Receptor de estrógeno
ER+ Positivo para receptor de estrógeno
ERK 1,2 Quinasa reguladora de señales extracelulares 1,2
ER Receptor de estrógeno
FBS Suero fetal bovino
g Gramos
GAPDH Gliceraldehído 3-fosfato deshidrogenasa

V
h Horas
H2Odd Agua bidestilada
HAD Hiperplasia ductal atípica
HCl Ácido clorhídrico
HEPES Ácido 4-(2-hidroxietil)-1-piperazina-etanosulfónico
HER2 Receptor del factor de crecimiento epidermal
IL-8 Interleucina-8
IC Intervalo de confianza
IMC Índice de masa corporal
JAK2/STAT3 Janus quinasa 2/ transductor de señal y activador de la
transcripción 3
JNK Jun N-terminal quinasa
kb Kilobase
KDa Kilodaltones
Kg Kilogramos
L Litro
LpB Cuerpos lipídicos
M Molar
mA Miliamperes
MAPK Proteína quinasa activada por mitógenos
MAP4K1 Proteína 4 quinasa 1 activada por mitógenos
MAPKKKs MAP quinasa quinasa quinasas
MCF-7 Michigan Cancer Foundation-7
MEC Matriz extracelular
mg Miligramos
min Minutos
miR-21 MicroRNA-21
MKK4 MAP quinasa quinasa 4
MKK7 MAP quinasa quinasa 7
mL Mililitro
mM Milimolar
mm Milímetro
MMP Metaloproteinasa de matriz

VI
mTOR Blanco de rapamicina en mamíferos
MTS (3-(4,5-dimetiltiazol-2-yl)-5-(3-carboximetoxifenil)-2-(4-sulfofenil)-
2H-tetrazolium)
Na2HPO4 Fosfato de sodio dibásico anhídrido
Na3VO4 Ortovanadato de sodio
NaCl Cloruro de sodio
NaF Fluoruro de sodio
NaH2PO4 Fosfato de sodio monobásico anhídrido
NaHCO3 Bicarbonato de sodio
NBT/BCIP 5-bromo-4-cloro-3-indoil fosfato/nitroazul de tetrazolium
ng Nanogramos
NH4Cl Cloruro de amonio
nM Nanomolar
nm Nanómetro
Ob-R Receptor de leptina
ºC Grados Celsius
PBS Buffer salino de fosfatos
PDCD4 Muerte celular programada 4
PGD2 Prostaglandina D2
PGE2 Prostaglandina E2
PGF2g Prostaglandina F2g
PGG2 Prostaglandina G2
PGH2 Prostaglandina H2
PGI2 Prostaciclina
pH Logaritmo negativo de la concentración de iones de hidrógeno
PKA Proteína quinasa A
PKC Proteína quinasa C
PMS Fenacin metosulfato
pRb Proteína del retinoblastoma
PSA Persulfato de amonio
RNA Ácido ribonucleico
RNAm RNA mensajero
rpm Revoluciones por minuto

VII
RPMI Roswell Park Memorial Institute Medium
RT-PCR Reacción en cadena de la polimerasa con transcripción reversa
s Segundos
S6K1 Proteína S6 quinasa 1
SDS Dodecil sulfato de sodio
SDS-PAGE Electroforesis en geles de poliacrilamida en presencia de dodecil
sulfato de sodio.
Ser Serina
SERM’s Moduladores selectivos del receptor de estrógeno
SHP2 Proteína tirosina fosfatasa con un dominio homólogo a Src 2
TBE Buffer tris borato EDTA
TBS Solución amortiguadora Tris-salina
TEMED N,N,N,N-tetrametil-etilendiamina
TGF-1 Factor transformador de crecimiento beta1
TIMP-2 Inhibidor tisular de metaloproteinasas-2
TNF- Factor de necrosis tumoral alfa
TPA 12-O-tetradecanoilforbol-13-acetato
TPY Treonina Prolina Tirosina
Trp Triptófano
TX Texas
TXA2 Tromboxano A2
uPAR Receptor activador del plasminógeno tipo uroquinasa
USA Por sus siglas en inglés United States of America/Estados
Unidos de America.
V Voltios
VEGF Factor de crecimiento del endotelio vascular
WT Silvestre
g Microgramo
l Microlitro
L Microlitro
m Micrómetro
M Micromolar
% Porcentaje

VIII
ÍNDICE DE TABLAS.
Tabla
Página
1. Esquema de transfección de las células MCF-7/PDCD4 con siRNA anti-TIMP-2 y control…………………………….………....
31

IX
ÍNDICE DE FIGURAS.
Figura
Página
1. Anatomía de la glándula mamaria………………………………..
2
2. Representación esquemática de las isoformas de los receptores de leptina.……………………………………………...
5
3. Localización celular y actividad enzimática de COX-2…..…….
10
4. Desarrollo del cáncer de mama ductal.………………………….
14
5. Modelo de como PDCD4 inhibe la traducción…………………..
22
6. Ensayo de invasión en Matrigel………………………………….
30
7. Sobreexpresión de PDCD4 en las células establemente transfectadas MCF-7/PDCD4………….…................................
36
8. PDCD4 inhibe la invasión inducida por leptina en células de cáncer de mama………………..………….................................
37
9. Sobreexpresión de TIMP-2 en las células MCF-7/PDCD4.….
39
10. Reducción en la expresión del RNAm de TIMP-2 en las células MCF-7/PDCD4 tratadas con siRNA anti-TIMP-2…...…
40
11. El silenciamiento de TIMP-2 incrementa la invasión de las células de cáncer de mama MCF-7/PDCD4…..........…………
41 12. El silenciamiento de TIMP-2 incrementa la activación inducida
por leptina de ERK1,2, STAT3, pero no afecta la activación de JNK en células MCF-7/PDCD4………….…….………………...
43
13. PDCD4 inhibe la resistencia a tamoxifeno inducida por leptina…..……………….………………………………………….
45
14. COX-2 no induce cambios sobre la activación de ERK1/2, p38MAPK y Akt en células MCF-7……....................................
59

X
15. COX-2 incrementa la fosforilación de JNK y los niveles de c-Jun en las células de cáncer de mama MCF-7........................
60
16. La inhibición específica de JNK, pero no la de ERK1,2 disminuye la invasión de las células MCF-7/COX-2…………..
62
17. Efecto de la inhibición de JNK en la proliferación de las células de cáncer de mama en ausencia o en presencia de Matrigel.…………….…………………………...……..…………..
63
18. La inhibición de JNK no afecto la resistencia de las células MCF-7/COX-2 a tamoxifeno……………………………………..
64
19. PDCD4 no inhibe la resistencia a tamoxifeno inducida por COX-2/PGE2……………………………………………………….
66
20. Modelo esquemático del efecto de PDCD4 sobre la invasión y resistencia a tamoxifeno inducidas por leptina y COX-2 en las células de cáncer de mama MCF-7…………………………….
72

1
Capítulo 1.
ANTECEDENTES.
1.1. Cáncer de mama.
El cáncer diagnosticado con mayor frecuencia en mujeres es el cáncer de mama.
La incidencia de esta enfermedad varía alrededor del mundo, en Estados Unidos
de América, durante el 2011, se estimaron alrededor de 39,970 muertes por
cáncer de seno (39,520 mujeres, 450 hombres). En el mismo año, se estimaron
230,480 nuevos casos de cáncer de mama invasivo en mujeres y cerca de 2,140
en hombres, mientras que los casos de cáncer de mama in situ fueron 57,650
(Society 2011). La metástasis es la principal causa de mortalidad en pacientes
con cáncer de mama. En Estados Unidos de América el rango de sobrevivencia a
5 años para mujeres diagnosticadas con cáncer de mama localizado (cáncer que
no se ha diseminado a nódulos linfáticos u otros lugares fuera de la mama) es del
98%, el cual contrasta dramáticamente con el porcentaje de sobrevivencia en
mujeres diagnosticadas con cáncer de mama y metástasis distante, el cual es de
solo 23% (Society 2011). En México el cáncer de mama es la segunda causa de
muerte en mujeres y en 2010 fallecieron 5,113 mujeres por esta causa (SALUD
2010).
El cáncer de mama es una enfermedad caracterizada por un crecimiento
descontrolado de células anormales encontradas en el tejido de la mama. Cada
mama tiene entre 15 y 20 lóbulos, los cuales a su vez tienen pequeñas secciones
denominadas lobulillos. Los lóbulos y lobulillos están conectados a través de
tubos llamados ductos (Figura 1). El tipo de cáncer de mama más común es el
ductal, originado en las células del ducto, seguido por el carcinoma lobular en
lóbulos o lobulillos. El cáncer de mama inflamatorio es el tipo de cáncer menos
frecuente y se caracteriza por enrojecimiento, edema, calor y dolor de las mamas
(Society 2011).
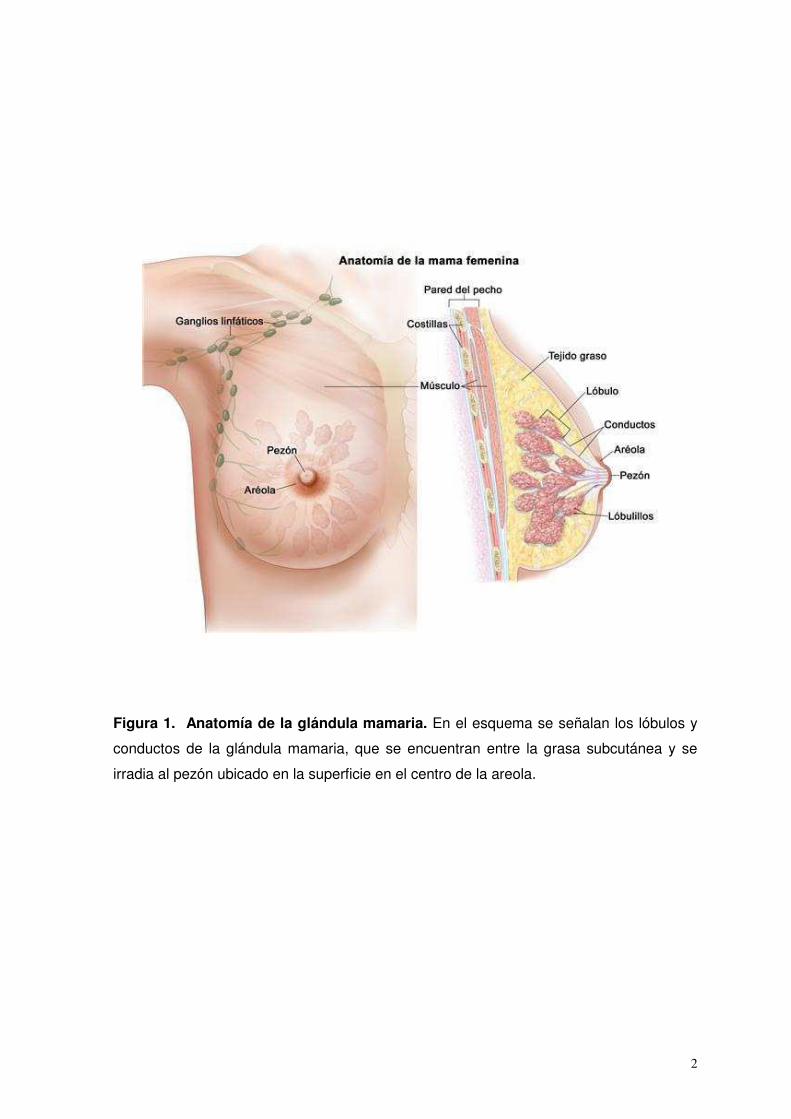
2
Figura 1. Anatomía de la glándula mamaria. En el esquema se señalan los lóbulos y
conductos de la glándula mamaria, que se encuentran entre la grasa subcutánea y se
irradia al pezón ubicado en la superficie en el centro de la areola.

3
Aunque no se conoce exactamente la causa del cáncer de mama, se sabe
que existen una gran variedad de factores de riesgo relacionados con esta
enfermedad, entre los cuales se encuentran la edad, antecedentes familiares,
menarquia precoz, menopausia tardía, densidad de la mama, uso de hormonas y
la obesidad. La obesidad se define como una acumulación anormal o excesiva de
grasa que puede ser perjudicial para la salud. Una forma simple de medir la
obesidad es el índice de masa corporal (IMC), esto es el peso de una persona en
kilogramos dividido por el cuadrado de la talla en metros. Una persona con un
IMC igual o superior a 30 es considerada obesa y con un IMC igual o superior a
25 es considerada con sobrepeso. El sobrepeso y la obesidad son factores de
riesgo para numerosas enfermedades crónicas, entre las que se incluyen la
diabetes, las enfermedades cardiovasculares y el cáncer (OMS 2011). En mujeres
post-menopáusicas una IMC >30 kg/m2 es un factor de riesgo para el desarrollo
de cáncer de mama (Protani, Coory et al. 2010). El estrógeno circulante en
mujeres post-menopáusicas es producido principalmente en el tejido adiposo, por
lo tanto, tener una mayor cantidad de tejido graso incrementa los niveles de
estrógeno y la probabilidad de desarrollar cáncer de mama (Society 2011).
Además, una elevada masa corporal se relaciona comúnmente con tumores
dependientes de hormonas y una de las hormonas relacionadas con obesidad y
cáncer de mama es leptina (Zhang, Proenca et al. 1994). Ambos factores, masa
corporal y porcentaje de grasa incrementan los niveles de leptina circulante
(McMurtry, Simeone et al. 2009).
1.2. Leptina.
La leptina es una hormona de 16 kDa fue descubierta en 1994 como un regulador
del peso corporal y balance energético que actúa en el hipotálamo y es producida
principalmente en el tejido adiposo (Zhang, Proenca et al. 1994). Aunque
inicialmente se creía que leptina se expresaba y secretaba exclusivamente por
adipocitos, ha sido identificada en tejidos como la placenta, mucosa gástrica y
colonica, testículos, ovarios y en células de epitelio mamario (Cirillo, Rachiglio et
al. 2008). En la mama, la leptina juega un papel importante en el desarrollo
normal de la glándula mamaria y la lactancia (Neville, McFadden et al. 2002). En
estudios anteriores se encontró que leptina puede modular distintos procesos en

4
órganos periféricos tales como respuesta inmune, fertilidad y hematopoyesis
(Surmacz 2007).
La regulación de la producción y secreción de leptina no está totalmente
clara, pero las hormonas sexuales parecen estar involucradas en este proceso.
Los estrógenos estimulan la liberación de leptina in vitro e in vivo, por lo que
podrían ser los responsables de encontrar una mayor cantidad de leptina en
mujeres que en hombres y de los elevados niveles de la misma durante el ciclo
menstrual (Marttunen, Andersson et al. 2000). Se ha observado que los
estrógenos regulan positivamente la expresión de leptina, mientras que la
testosterona la regula negativamente (Cirillo, Rachiglio et al. 2008).
La leptina ejecuta su actividad biológica a través de la unión a su receptor
(Ob-R), un miembro de la familia de receptores de citocinas. Se han identificado
hasta el momento seis diferentes isoformas del receptor de leptina; (ObRa-ObRf),
el dominio extracelular de cinco de las isoformas (ObRa-ObRd, ObRf) es idéntico,
y consiste de 816 aminoácidos, dos motivos de unión (Trp-Ser-X-Ser-Trp) y un
dominio de fibronectina tipo III. Las isoformas ObRa, ObRc, ObRd y ObRf están
además caracterizadas por un dominio intracelular corto formado de 32-40
residuos y un dominio transmembranal que consiste de 23 aminoácidos. Solo el
receptor ObRb contiene un dominio intracelular largo que comprende
aproximadamente 306 aminoácidos (Figura 2) (Cirillo, Rachiglio et al. 2008).
ObRb se conoce como la isoforma larga y es expresada principalmente en el
hipotálamo. Mientras que ObRa representa la isoforma corta cuya expresión es
más abundante en tejidos periféricos (Revillion, Charlier et al. 2006).

5
Figura 2. Representación esquemática de las isoformas de los receptores de
leptina. Todas las isoformas muestran el mismo dominio extracelular, solo difieren en la
longitud del dominio intracelular. La isoforma larga del receptor de leptina (obRb) contiene
dos sitios de unión a JAK2 (Box1 y Box2), un motivo de unión SHP2 (Proteína tirosina
fosfatasa con un dominio homólogo a Src 2) y un sitio de unión a STAT3. Las isoformas
cortas del receptor de leptina (obRa, obRc, obRd, obRf) presentan solo un dominio
BOX1. La isoforma soluble del receptor de leptina (obRe), la cual no se muestra en este
esquema, pierde los dominios transmembrana e intracelular (Cirillo, Rachiglio et al. 2008).
CAJA 1
CAJA 2
CAJA 1
obRb: 1165 aa
obRa, obRc,obRd, obRf: 894-965 aa
Sitio de unión a JAK2
Sitio de unión a SHP2
Sitio de unión a STAT3
Membrana celular
CAJA 1CAJA 1
CAJA 2CAJA 2
CAJA 1CAJA 1
obRb: 1165 aa
obRa, obRc,obRd, obRf: 894-965 aa
Sitio de unión a JAK2
Sitio de unión a SHP2
Sitio de unión a STAT3
Membrana celular

6
Varias líneas de evidencia sugieren que leptina y sus receptores están
involucrados en el desarrollo normal de la glándula mamaria y en el desarrollo y
progresión del cáncer de mama. Los receptores de leptina (isoformas cortas y
larga) son expresados en células normales de epitelio mamario durante el
embarazo y la lactancia (Laud, Gourdou et al. 1999). Se ha sugerido además que
leptina está involucrada en la patogénesis y progresión del cáncer de mama
dependiente de estrógenos. La síntesis de leptina en adipocitos es influenciada
por diferentes factores humorales, tales como insulina, factor de necrosis tumoral
alfa (TNF-), glucocorticoides, hormonas reproductivas y prostaglandinas.
Muchos de estos factores han sido asociados a procesos neoplásicos (Garofalo
and Surmacz 2006). En líneas celulares de cáncer de mama positivas para
receptor de estrógeno (ER+) como, T47D, MCF-7, y ZR75-1, leptina estimula la
síntesis de DNA y crecimiento celular actuando a través de múltiples cascadas de
señalización, incluyendo JAK/STAT (Janus quinasa/ transductor de señal y
activador de la transcripción), ERK1/2 (Quinasa reguladora de señales
extracelulares 1/2), y PKC- (Proteína quinasa C-). Además de las clásicas
señales de citocinas, leptina es capaz de inducir la vía de sobrevivencia Akt/GSK3
(Serina/treonina quinasa/glucógeno sintasa quinasa 3) in vitro en células de
cáncer de mama (Garofalo, Sisci et al. 2004). Leptina puede inducir la progresión
del ciclo celular incrementando los niveles de cdk2 (Quinasa dependiente de
ciclina 2) y ciclina D1, así como hiperfosforilando e inactivando al inhibidor del
ciclo celular, pRb (Proteína del retinoblastoma). En un estudio realizado por Hu y
colaboradores se observó que las células de epitelio mamario humano HBL100 y
células T4TD de cáncer de mama positiva para ER, expresaban el receptor de
leptina obRb. En estas células, leptina activó las vías STAT3, ERK y AP-1,
resultando en una mayor proliferación celular. Si embargo, solo en las células
T4TD de cáncer de mama leptina aumentó el crecimiento celular independiente
de anclaje (Hu, Juneja et al. 2002).
En cáncer de mama, leptina puede mediar angiogénesis a través de la
inducción de la expresión del VEGF (Factor de crecimiento del endotelio vascular)
y promover invasión y migración mediante la transactivación de EGFR (Receptor
del factor de crecimiento epidérmico). Además, la leptina promueve la migración e

7
invasión de células derivadas de glioma, condrosarcoma, carcinoma de colon,
células de carcinoma endometrial y hepatocelular, así como cáncer de próstata.
En las células de cáncer de próstata (DU145 y PC-3), leptina incrementa
significativamente la migración celular y la expresión inducida de VEGF, TGF-1
(Factor transformador de crecimiento beta1) y bFGF (Factor de crecimiento de
fibroblastos básico) y, por lo tanto, probablemente en general contribuye a la
progresión del cáncer de próstata (Lang and Ratke 2009). Además se ha
observado que leptina incrementa la invasión de las células de cáncer de mama a
través de la activación de JNK (Jun N-terminal quinasa), lo cual resulta en un
incremento en la actividad de MMP-2 (Metaloproteinasa de matriz-2) (McMurtry,
Simeone et al. 2009).
En las células de cáncer de mama, leptina y ambas isoformas de los
receptores se expresan a nivel de RNAm y proteína. Altos niveles de esta
hormona han sido asociados con un incremento en la incidencia de cáncer de
mama metastático. La metástasis en cáncer mamario está directamente asociada
con invasividad. Además, leptina juega un papel importante en la estimulación de
la proliferación celular y sobrevivencia. En pacientes con cáncer de mama y
obesidad, leptina podría contribuir a la resistencia a terapias antiestrógeno. Esto
es porque algunos de los efectos de los agentes antiestrógeno tales como
tamoxifeno y raloxifeno son suprimidos por altos niveles de leptina (Jansen,
Camalier et al. 2005).
1.3. Ciclooxigenasa-2 (COX-2).
Otro factor importante relacionado con invasión y resistencia a tamoxifeno en
cáncer de mama positivo para ER es la sobreexpresión de COX-2. Las
ciclooxigenasas (COXs) son también conocidas como prostaglandina H sintasas o
prostaglandina endoperóxido sintasas ya que catalizan la biosíntesis de
prostaglandina H2, el precursor de moléculas tales como prostaglandinas,
prostaciclinas y tromboxanos. Las ciclooxigenasas son enzimas bifuncionales con
dos actividades catalíticas distintas: actividad ciclooxigenasa y peroxidasa. La
actividad de ciclooxigenasa oxigena ácido araquidónico para generar

8
prostaglandina G2 (PGG2), mientras que la actividad de peroxidasa reduce este
último a prostaglandina H2 (PGH2) (Figura 3). PGH2 puede ser metabolizado por
varias prostaglandinas sintasas a prostaglandina E2 (PGE2), prostaglandina D2
(PGD2), prostaglandina F2g (PGF2g), prostaciclina (PGI2) y tromboxano A2 (TXA2)
(Taketo 1998).
La familia de las enzimas COX consiste de 3 isoenzimas: COX-1, COX-2 y
COX-3. COX-1 es expresada constitutivamente en pulmón, próstata, cerebro,
tracto gastrointestinal, riñón, hígado y bazo. En estudios recientes de biopsias y
autopsias, COX-1 fue encontrada en vasos sanguíneos, células de músculo liso y
plaquetas (Menter, Schilsky et al. 2010). La actividad de COX-1 es producir
prostaglandinas citoprotectoras, tales como prostaciclina y PGE2 (Prostaglandina
E2), las cuales parecen ser críticas para mantener la integridad de la mucosa
gástrica (Williams, Tsujii et al. 2000). COX-2 normalmente no es detectable en
muchos tejidos pero exhibe niveles de expresión basal en macrófagos, células
endoteliales, arteria coronaria, corazón, próstata, pulmón, placenta, páncreas,
cerebro y riñón (Menter, Schilsky et al. 2010). La expresión de COX-2 puede ser
inducida por varios estímulos, tales como mitógenos, citocinas, factores de
crecimiento, oncogenes, entre otros y está involucrada en reacciones
inflamatorias (Taketo 1998; Ristimaki, Sivula et al. 2002; Chandrasekharan and
Simmons 2004). COX-3 es una variante de COX-1 que retiene el intron 1 y tiene
un cambio del marco de lectura, su función biológica aún no es clara (Vesga
2004). COX-1 y COX-2 son dos proteínas, codificadas por genes con distinta
localización cromosómica: COX-1 en el cromosoma humano 9 y COX-2 en el
cromosoma 1. El gen que codifica para COX-2 es de aproximadamente 8 kb,
consiste de 10 exones y 9 intrones, mientras que el gen que codifica para COX-1
es de aproximadamente 22 kb y consiste de 11 exones y 10 intrones. COX-2
muestra una secuencia y actividad catalítica significativamente homóloga con
COX-1 (Williams, Tsujii et al. 2000; Ristimaki, Sivula et al. 2002). Las diferencias
estructurales entre los sitios catalíticos de COX-1 y COX-2 consisten en la
sustitución del aminoácido 523, el cual es una isoleucina en COX-1 por una valina
en COX-2, esta última al tener menor volumen determina una abertura en la pared
del canal del sitio activo de COX-2 haciéndolo 17% más amplio que el de COX-1
(Taketo 1998; Rizzo 2011).

9
COX-2 se localiza principalmente en el lumen del retículo endoplasmático,
en el cual se lleva a cabo la oxigenación del ácido araquidónico. La localización
nuclear de COX-2 es consistente con su papel regulador en la mitogénesis. Por
otra parte, existe evidencia de que en las células tumorales, COX-2 se localiza en
la mitocondria y en cuerpos lipídicos. La localización intracelular de COX-2 en la
mitocondria juega un papel importante en la protección contra la apoptosis
inducida por estrés oxidativo, mientras que la localización de COX-2 en los
cuerpos lipídicos influye en el crecimiento tumoral, probablemente al funcionar
como una fuente intracelular adicional para el suministro continuo de
prostaglandinas (Figura 3) (Rizzo 2011).

10
Figura 3. Localización celular y actividad enzimática de COX-2. En la figura se indica
la localización de COX-2 en el retículo endoplasmático (ER), núcleo, mitocondria (M) y
cuerpos lipídicos (LpB). Además, se muestra la oxigenación del ácido araquidónico (AA)
por COX-2 en el retículo endoplasmático. El AA es convertido a prostaglandina G2
(PGG2), a través de la actividad de ciclooxigenasa de COX-2. PGG2 es entonces reducido
por la actividad de peroxidasa de COX-2 a prostaglandina H2 (PGH2). PGH2 sirve como
substrato para prostaglandinas sintasas, las cuales son responsables de la producción de
prostaglandina E2 (PGE2), prostaglandina D2 (PGD2), prostaglandina F2g (PGF2g),
prostaciclina (PGI2) y tromboxano A2 (TXA2) (Rizzo 2011).

11
De los tres tipos de COXs, el más relevante en la terapia contra cáncer es
COX-2 ya que se ha observado que una alta expresión de esta enzima se
correlaciona con cambios malignos en una variedad de cánceres humanos,
incluyendo colorectal, mama, próstata, esófago, piel, hígado, pancreático,
cerebro, pulmón, cabeza y cuello (Singh, Berry et al. 2005). Recientemente se ha
sugerido que COX-2 podría estar involucrado directamente con el carcinoma
mamario. En el 2001, Catherine et al, encontraron que COX-2 indujo el desarrollo
de tejido tumoral en ratones transgénicos que sobreexpresaban el gen COX-2 en
glándulas mamarias, además de una reducción en los niveles de la proteína
apoptótica Bax (Proteína X asociada a Bcl-2) y un incremento en la proteína
antiapoptótica Bcl-2 (Linfoma de células B-2), sugiriendo que la disminución de
apoptosis en células del epitelio mamario contribuye a la tumorigénesis.
Estos datos indican que el aumento de la expresión de COX-2 es suficiente
para inducir tumorigénesis en glándulas mamarias. Sin embargo, el posible papel
de COX-2 en el cáncer de mama se percibió inicialmente cuando se encontraron
elevados niveles de prostaglandinas en células de tumores mamarios. En varios
tejidos tumorales de animales y humanos, incluyendo cáncer de colón humano, se
han reportado altas concentraciones de prostaglandinas (Taketo 1998; Kim,
Bossuyt et al. 2005).
Reportes recientes sugieren que COX-2 se sobreexpresa en
aproximadamente el 40% de los cánceres de mama invasivo (Singh, Berry et al.
2005) y en una alta frecuencia en carcinoma ductal pre-invasivo in situ, el cual se
asocia con un pobre pronóstico del paciente. La sobreexpresión de COX-2 está
correlacionada con parámetros que son característicos de un cáncer de mama
agresivo, tales como gran tamaño del tumor, alta proliferación celular y
sobreexpresión de HER2 (Receptor del factor de crecimiento epidermal), el cual
está presente en un 25-30% de los cánceres de mama y está asociado con una
enfermedad agresiva y un incremento de la resistencia a una variedad de terapias
contra el cáncer (Simeone, Colella et al. 2006; Howe 2007).

12
Previamente se demostró que las mujeres diagnosticadas con cáncer de
mama in situ o invasivo que tomaban aspirina al menos una vez por semana
durante 6 meses o más, tuvieron una menor asociación con el riesgo de
desarrollar cáncer de mama que las que no tomaban regularmente aspirina. La
reducción del riesgo del cáncer de seno se observó en mujeres con ER+ pero no
ER- (Terry, Gammon et al. 2004). Además, se ha observado que el ibuprofeno,
pero no el acetaminofén, reduce el riesgo del cáncer de mama y debido a que la
aspirina y el ibuprofeno pueden inhibir la actividad de COX-2, se ha especulado
que entre los numerosos mecanismos de acción de estas drogas, es la actividad
de anti-COX-2 la que reduce el riesgo de cáncer mamario (Tari, Simeone et al.
2005).
La expresión de COX-2 en pacientes con cáncer de mama es un marcador
de baja sobrevivencia libre de enfermedad y global (Ristimaki, Sivula et al. 2002;
Denkert, Winzer et al. 2004; O'Connor, Avent et al. 2004) y ha sido considerada
como un marcador de alto potencial metastásico. La expresión de COX-2 es muy
alta en tumores metastásicos y está asociada con metástasis distante (Costa,
Soares et al. 2002). COX-2 incrementa la invasión de células de cáncer de mama
in vitro (Singh, Berry et al. 2005) e in vivo (Connolly, Harmey et al. 2002).
Además se ha observado que derivados prostanoides de COX-2 como
PGE2 promueven angiogénesis, inducen invasión e incrementan metástasis.
Durante la progresión del cáncer, las prostaglandinas pueden mediar una gran
variedad de mecanismos, incluyendo proliferación celular, apoptosis, modulación
del sistema inmune y angiogénesis (Costa, Soares et al. 2002). Por otra parte, la
producción de PGE2 mediada por COX-2, eleva la actividad de aromatasa, lo cual
podría explicar el hecho de que COX-2 este involucrado en promover el
crecimiento de cáncer de mama ER+, particularmente en mujeres post-
menopáusicas (Filipovic, Giamas et al. 2011).
Anteriormente, en nuestro laboratorio se observó que COX-2 utiliza PKC
(Proteína quinasa C) para incrementar la producción de IL-8 (Interleucina-8). IL-8
a su vez activa uPA (Activador de plasminógeno de tipo urinario), resultando en
un incremento en la invasión de células de cáncer de mama (Simeone, Nieves-

13
Alicea et al. 2007). Además se reportó que PGE2 y PKC están involucrados en la
resistencia a tamoxifeno e invasión inducida por COX-2. La activación del receptor
de PGE2 y PKC incrementó la invasión de las células MCF-7 a través del Matrigel
(Tari, Simeone et al. 2005). Sin embargo, no se conoce cuales son los factores río
abajo utilizados por la vía COX-2/PGE2/PKC para inducir resistencia a tamoxifeno
e invasión en las células de cáncer de mama
Ambos leptina y COX-2 juegan un papel muy importante en invasión y
resistencia a drogas, los cuales, son procesos complejos que involucran
alteraciones genéticas y celulares.
1.4. Invasión celular, Metaloproteinasas e Inhibidor Tisular de
Metaloproteinasas-2 (TIMP-2)
La invasión de células tumorales es el proceso en el cual las células invaden
tejido adyacente penetrando a través de la membrana basal y entrando a vasos
linfáticos y sanguíneos (Chambers, Groom et al. 2002). Se ha sugerido que
existen múltiples vías para el desarrollo de cáncer de mama invasivo. Una posible
vía para la progresión de cáncer de mama ductal es que a partir de una
hiperplasia ductal atípica (HAD) pasa a un carcinoma ductal in situ (CDIS) y de
este a un carcinoma ductal invasivo (CDI) (Figura 4). CDIS puede definirse como
proliferación de células cancerosas situadas dentro de los confines del ducto
mamario. Por lo tanto, la invasión de cáncer de mama ductal requiere que las
células cancerosas penetren la membrana basal del epitelio y migren del ducto
hacia el tejido adyacente. Como consecuencia, el cáncer invasivo puede
diseminarse a tejidos circundantes, entrar al sistema vascular y colonizar sitios
distantes, lo que eventualmente conduce a la muerte de la mayoría de los
pacientes con cáncer mamario. Por lo tanto, es de gran importancia entender los
mecanismos a través de los cuales se facilita la invasión del cáncer de mama.
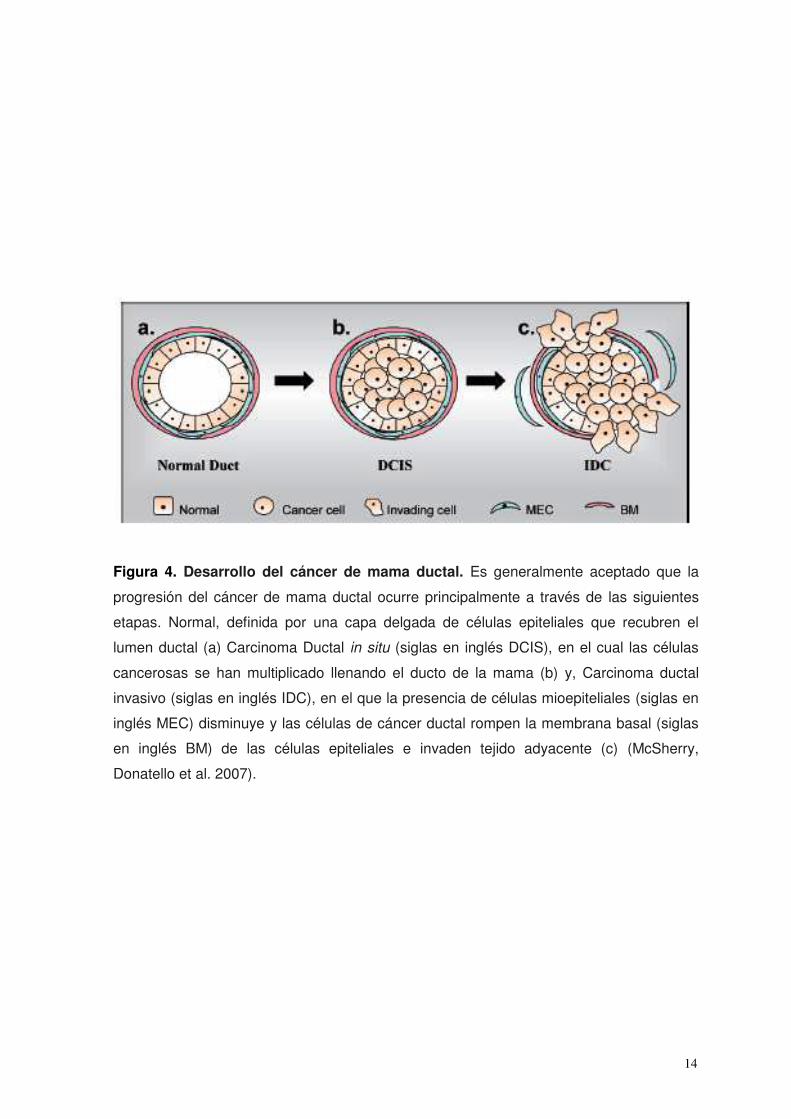
14
Figura 4. Desarrollo del cáncer de mama ductal. Es generalmente aceptado que la
progresión del cáncer de mama ductal ocurre principalmente a través de las siguientes
etapas. Normal, definida por una capa delgada de células epiteliales que recubren el
lumen ductal (a) Carcinoma Ductal in situ (siglas en inglés DCIS), en el cual las células
cancerosas se han multiplicado llenando el ducto de la mama (b) y, Carcinoma ductal
invasivo (siglas en inglés IDC), en el que la presencia de células mioepiteliales (siglas en
inglés MEC) disminuye y las células de cáncer ductal rompen la membrana basal (siglas
en inglés BM) de las células epiteliales e invaden tejido adyacente (c) (McSherry,
Donatello et al. 2007).

15
El proceso de invasión consiste en una serie de eventos complejos de los
cuales un paso inicial involucra la degradación de los componentes de la matriz
extracelular (MEC). Este proceso es catalizado por la acción de varias enzimas,
incluyendo MMPs, las cuales son una familia de proteinasas dependientes de zinc
que participan en la degradación de componentes de la matriz extracelular y que
han sido involucradas en la invasión de células tumorales, metástasis y
angiogénesis (Sacco, Cato et al. 2001; Bernardo and Fridman 2003; Ahn, Jeong
et al. 2004). Fisiológicamente, estas enzimas desempeñan un papel importante en
la remodelación de tejido normal, desarrollo embrionario, angiogénesis, ovulación,
involución de la glándula mamaria y cicatrización de heridas. Por otra parte la
expresión anormal de estas enzimas contribuye a diversos procesos patológicos,
incluyendo artritis reumatoide, crecimiento tumoral, invasión y metástasis, entre
otros (Chambers, Groom et al. 2002).
En general, se cree que el principal mecanismo por el cual las MMPs
promueven la propagación del cáncer es a través de la degradación de la MEC, la
cual se compone principalmente de membrana basal y tejido conectivo intersticial.
La membrana basal a su vez presenta como principal componente colágeno tipo
IV, el cual se cree es degradado en su mayoría por MMP-2 y MMP-9; otras
proteínas tales como laminina, proteoglicanos, entactina y osteonectina están
también presentes en la MEC. Para establecer metástasis, las células cancerosas
deben pasar a través de la membrana basal al menos tres veces. Las células de
cáncer de mama inicialmente atraviesan esta membrana cuando un carcinoma in
situ se convierte en invasivo. Posteriormente, las células malignas penetran a
través de la membrana basal durante la entrada y salida al torrente sanguíneo
(Duffy, Maguire et al. 2000).
Las MMPs presentan un dominio en común con una familia de proteínas
que juegan un papel opuesto; los inhibidores tisulares de metaloproteinasas-2
(TIMPs) los cuales inhiben a las MMPs formando uniones no covalentes
asociadas con el sitio activo de las MMPs (Ree, Florenes et al. 1997; Sacco, Cato
et al. 2001). Algunos estudios han mostrado que la sobreexpresión de TIMP-2
disminuye in vitro la invasión de células epiteliales transformadas por ras (Ahn,
Jeong et al. 2004). Además, ratones inyectados con células de cáncer de mama

16
MDA-MB-231 transfectadas con TIMP-2 presentaron un menor número de
metástasis óseas y una mayor tasa de sobrevivencia que los ratones inyectados
con células no transfectadas (Yoneda, Sasaki et al. 1997).
1.5. Resistencia a tamoxifeno inducida por leptina y COX-2.
Uno de los tratamientos empleados para el cáncer mamario dependiente de
hormonas, es el tamoxifeno, el cual ha demostrado ser también un agente
preventivo. El tamoxifeno puede prolongar considerablemente la sobrevivencia
libre de enfermedad y la remisión en más de la mitad de los pacientes con cáncer
de mama ER+ e invasivo, por lo que se cree que la reducción significativa de la
mortalidad por cáncer de mama durante la última década se debe al amplio uso
del tamoxifeno.
El tamoxifeno ha sido usado para el tratamiento sistémico de pacientes con
cáncer de mama desde hace casi tres décadas. En el carcinoma mamario el éxito
del tratamiento depende principalmente de la presencia del receptor de estrógeno
(ER). Sin embargo, cerca de la mitad de los pacientes con enfermedad avanzada
y ER+ no responden al tratamiento con tamoxifeno de manera inmediata, mientras
que en los pacientes que responden en última instancia, la enfermedad progresa
a un fenotipo de resistencia (Dorssers, van der Flier et al. 2001).
Las posibles causas de la resistencia intrínseca y adquirida han sido
atribuidas a: la farmacología del tamoxifeno, alteraciones en la estructura y
función del ER, las interacciones con el microambiente tumoral y alteraciones
genéticas en las células tumorales. Hasta el momento no ha sido identificado un
mecanismo prominente que conduzca a la resistencia a tamoxifeno (Dorssers,
van der Flier et al. 2001).
Se ha encontrado que la leptina modula tanto la síntesis de estrógeno
como la actividad del receptor de estrógeno (ER). Por ejemplo, la leptina
puede incrementar la expresión del gen de la aromatasa y la actividad de la
aromatasa en células MCF-7, lo cual podría incrementar la síntesis de estrógeno.
La leptina puede además incrementar la estabilidad del ER permitiendo

17
mantener la transcripción dependiente de ER en células de cáncer de mama en
presencia de antiestrógenos (Garofalo and Surmacz 2006). Estas observaciones
sugieren que altos niveles de leptina en pacientes obesas con cáncer mamario
podrían contribuir al crecimiento tumoral y desarrollo de resistencia a
antiestrógenos.
Garofalo et al., en el 2004 demostró que leptina es capaz de atenuar los
efectos del antiestrógeno ICI 182,780 (Fulvestrant) en células MCF-7. El
tratamiento de las células MCF-7 con fulvestrant indujo una rápida degradación
del ER en la membrana celular, lo cual redujo la expresión nuclear del receptor y
la transcripción dependiente de ER, lo que produjo a su vez, una significativa
inhibición del crecimiento. En presencia del antiestrógeno ICI 182,780, leptina
estabilizó ER, interfiriendo con las vías de degradación ubiquitina-proteosoma
del ER.
Por otra parte, la sobreexpresión de COX-2 se correlaciona con una enfermedad
agresiva e incrementa la resistencia a una gran variedad de terapias contra el
cáncer (Simeone, Li et al. 2004; Kim, Bossuyt et al. 2005; Howe 2007). En el
2002, Ristimaki et al, reportaron que las mujeres cuyos tumores de mama
invasivo eran positivos para ER pero tenían bajos niveles de COX-2,
presentaban una oportunidad del 86% de sobrevivencia libre de enfermedad a 5
años, mientras que las mujeres cuyos tumores eran positivos para ER pero que
tenían altos niveles de COX-2 presentaron una oportunidad de sobrevivencia libre
de enfermedad a 5 años del 76% (Ristimaki, Sivula et al. 2002).
Tari y colaboradores, observaron que la transfección de COX-2 en la línea
celular MCF-7, incrementó la resistencia de estas células al tamoxifeno
aproximadamente 5 veces comparado con las células MCF-7/WT. Además
reportaron que la activación del receptor de PGE2 y PKC inhibió el efecto
antiproliferativo del tamoxifeno (Tari, Simeone et al. 2005). Esta información
sugiere que los pacientes con cáncer de mama positivo para ER y que
sobreexpresan COX-2 y/o leptina podrían no estar beneficiándose de tratamientos

18
con tamoxifeno como aquellos pacientes con tumores mamarios y bajos niveles
de COX-2 y/o leptina.
Leptina estimula invasión y sobrevivencia en células de cáncer de mama
actuando a través de múltiples cascadas de señalización, tales como JNK
(McMurtry, Simeone et al. 2009), JAK2/STAT3 (Yin, Wang et al. 2004) y ERK 1,2
(Bjorbaek, Uotani et al. 1997).
1.6. JNK (Jun N-terminal quinasa)
Las Jun N-terminal quinasas son miembros de la superfamilia de las MAP
quinasas. Se han identificado 10 isoformas de JNK codificadas por tres distintos
genes denominados JNK1, JNK2 y JNK3. Una serie de MAP quinasa quinasas,
las cuales sirven como conductos para las diversas señales que activan la vía de
JNK, fosforilan y activan dos distintas MAP quinasa quinasas, MKK4 (MAP
quinasa quinasa 4) y MKK7 (MAP quinasa quinasa 7), las cuales a su vez
fosforilan el motivo TPY (Treonina, Prolina, Tirosina) de JNK permitiendo su
activación (Heasley and Han 2006). Una vez fosforilado, JNK es translocado al
núcleo donde fosforila y transactiva al factor de transcripción c-Jun. La
fosforilación de c-Jun permite la formación de la proteína AP-1 (proteína
activadora-1), la cual está involucrada en la transcripción de una gran variedad de
genes. Aunque la actividad pro-apoptótica de JNK está comenzando a ser
entendida, debe señalarse que el papel pro-apoptótico de JNK es a menudo
determinado por otros factores celulares. Esto podría incluir la activación paralela
de sobrevivencia celular ó vías antiapoptóticas. Se cree que la activación
sostenida de JNK se asocia con apoptosis, mientras que la activación aguda de
JNK esta involucrada con proliferación celular y vías de sobrevivencia
(Dhanasekaran and Reddy 2008).
Recientemente se ha demostrado que JNK desempeña un papel crucial en
obesidad y resistencia a insulina. Además, JNK2 ha sido involucrado en
proliferación celular, progresión del ciclo celular y sobrevivencia en cáncer de
próstata dependiente de andrógenos (Onuma, Bub et al. 2003). Por otra parte,
JNK ha sido implicado en la producción y activación de MMP-2 (Ispanovic and
Haas 2006) y se ha demostrado que leptina incrementa la invasión de las células

19
de cáncer de mama MCF-7 a través de la activación de la vía de JNK, la cual a su
vez incrementa la actividad de MMP-2 (McMurtry, Simeone et al. 2009).
1.7. ERK (Quinasa reguladora de señales extracelulares) y JAK2/STAT3
(Janus quinasa 2/ transductor de señal y activador de la transcripción 3)
Las acciones celulares de leptina son iniciadas a través de la unión de leptina a su
receptor específico seguido por la activación de MAPK dependiente de ras y la
fosforilación de STAT3. La p44/42 MAPK (ERK1/2) es una familia de proteínas
quinasas intracelulares, cuya actividad es regulada por la fosforilación o su sitio
de activación en residuos conservados de T (treonina) y Y (tirosina). Bajo la
estimulación de factores de crecimiento, citocinas inflamatorias o estrés físico, las
MAPKKKs (MAP quinasa quinasa quinasas) son activadas por interacción con
una familia de pequeñas GTPasas y/u otras proteínas quinasas conectando las
MAPK al receptor de superficie celular o al estímulo externo. MAPKKKs a su vez
activan a las MAPKKs, las cuales finalmente fosforilan y activan ERK. La vía de
ERK está involucrada en diferentes procesos celulares tales como proliferación
diferenciación, motilidad y muerte. ERK ha sido implicada en la producción y
activación de MMP-2 (Ispanovic and Haas 2006). Leptina activa la vía de ERK a
través de la unión a su receptor ObRa (la isoforma corta).
STAT3 pertenece a una familia de factores de transcripción citosólicos y es
activado a través de factores de crecimiento y citocinas. STAT3 es considerado
como un oncogén que promueve proliferación e inhibe apoptosis. Bajo la
estimulación de leptina, las quinasas JAKs son activadas vía transfosforilación y
fosforilan residuos de tirosina en el receptor de leptina ObRb (isoforma larga) y en
las proteínas STAT. Las proteínas STAT fosforiladas se dimerizan y translocan al
núcleo para activar la transcripción de genes. La vía de STAT3 está involucrada
en la proliferación celular inducida por leptina (Bjorbaek, El-Haschimi et al. 1999).
La invasión y resistencia a la quimioterapia juegan un papel crucial en la
progresión tumoral y son una causa significativa de morbilidad y mortalidad en
pacientes con cáncer. Por ello es extremadamente importante conocer y entender
los mecanismos que permiten la resistencia a drogas e invasión del cáncer de
mama para desarrollar nuevos y mejores tratamientos con el fin de bloquear estos

20
procesos y así prolongar la vida de los pacientes con cáncer de mama,
permitiéndoles mejorar su calidad de vida. Un nuevo y prometedor blanco
terapéutico para inhibir la progresión del cáncer de mama inducida por leptina y
COX-2 es PDCD4 (proteína de muerte celular programada 4).
1.8. Proteína de Muerte Celular Programada 4 (PDCD4)
El gen de muerte celular programada 4 fue originalmente identificado como un
gen cuya expresión es fuertemente inducida durante la apoptosis en células de
ratón (Shibahara, Asano et al. 1995). Este gen suprime la transformación de
queratinocitos de ratón in vitro inducida por 12-O-tetradecanoilforbol-13-acetato
(TPA) (Cmarik, Min et al. 1999; Yang, Jansen et al. 2001), así como la inducción y
progresión de carcinogénesis de piel en respuesta al 7,12-dimetilbenzo (a)
antraceno/TPA en modelos animales (Jansen, Camalier et al. 2005). Además, se
ha demostrado que la expresión de PDCD4 inhibe la transactivación y
transformación mediada por el factor de transcripción AP-1 (Yang, Jansen et al.
2001). Una serie de genes blanco de AP-1 han sido implicados en invasión celular
y progresión metastásica, incluyendo uPAR (Receptor activador del plasminógeno
tipo uroquinasa) y varios miembros de la familia de metaloproteinasas (Jansen,
Camalier et al. 2004).
Se ha reportado que Akt (Serina/treonina quinasa) es un modulador río
arriba de la actividad de PDCD4 que fosforila específicamente los sitios Ser67 y
Ser457 de PDCD4 in vitro e in vivo, resultando en una disminución significativa en
la capacidad de PDCD4 para interferir con la transactivación de un promotor de
respuesta a AP-1 a través de c-Jun (Wen, Shi et al. 2007). PDCD4 es regulado
también por la proteína S6 quinasa 1 (S6K1) y la ubiquitina quinasa SCFTRCP
en respuesta a la activación de la vía mTOR (Blanco de rapamicina en
mamíferos) por mitógenos. La degradación controlada de PDCD4 es esencial
para la eficiente síntesis de proteínas y, consecuentemente, para el crecimiento y
proliferación celulares (Chang, Cho et al. 2009). Por otra parte, se ha observado
que altos niveles de miR-21 (microRNA-21) regulan negativamente el RNAm de
PDCD4 al unirse a su región 3’ no traducida (3’ UTR), promoviendo así
transformación celular y progresión tumoral (Lankat-Buttgereit and Goke 2003;
Zhu, Wu et al. 2008). Además, se ha reportado que la metilación en la región CpG

21
dentro del extremo 5’ de PDCD4 bloquea la expresión de PDCD4 a nivel de
RNAm en gliomas (Xia et al., 2010).
Varias líneas de evidencia sugieren que PDCD4 juega un papel importante
en el metabolismo del RNA y/o traducción de proteínas. PDCD4 comparte
homología con factores de iniciación de la traducción eucariotas y se ha
demostrado que se une e inhibe la actividad de helicasa del factor de iniciación
eucariótico eIF4A (eIF4A es una helicasa de RNA, con dos dominios, que
desenrollan las estructuras secundarias en la región 5’ no traducida (5’UTR) y el
cap del RNAm facilitando su reconocimiento por parte de los ribosomas), lo que
subsecuentemente inhibe la traducción dependiente de cap. PDCD4 compite con
eIF4A por la unión a eIF4G, la cual es una proteína adaptadora que coordina el
ensamblaje de los factores de la traducción y la subunidad ribosomal pequeña
(Figura 5) (Lankat-Buttgereit and Goke 2003; Yang, Jansen et al. 2003; Chang,
Cho et al. 2009). PDCD4 también puede competir con eIF4G por la unión a eIF4A
y prevenir su unión al complejo de iniciación eucariota o su unión a eIF4F o a
ambos. PDCD4 parece ser el primer ejemplo de una proteína en células de
mamíferos que inhibe la traducción al atenuar la actividad de helicasa de eIF4A
(Yang, Jansen et al. 2003).
Varios tumores humanos y líneas de células tumorales muestran niveles
elevados de factores de iniciación de la traducción incluyendo eIF4A, eIF4E y
eIF4G (Jansen, Camalier et al. 2004). Por lo tanto, la inhibición de factores de
traducción mediada por PDCD4 podría regular negativamente el desarrollo del
cáncer. PDCD4 es un nuevo inhibidor de la transformación de células en cultivo y
un inhibidor en varias etapas de la carcinogénesis, lo cual sugiere que la
inhibición de la iniciación de la traducción por PDCD4 podría ser una estrategia
prometedora para la prevención del cáncer (Jansen, Camalier et al. 2004).

22
Figura 5. Modelo de como PDCD4 inhibe la traducción. (A) Modelo en el que se
muestra la unión de eIF4A y el factor eIF4G. La molécula de eIF4A se une a la región
media (4GM) y al dominio C-terminal (4GC) de eIF4G. (B) Modelo de inhibición de la
traducción por PDCD4. PDCD4 se une e inhibe la actividad de helicasa del factor eIF4A,
permitiendo la unión del factor eIF4A inactivado a la región media de eIF4G y a su vez
previniendo la asociación de eIF4A al dominio C-terminal de eIF4G, lo cual resulta en la
inhibición de la traducción. 4GN (Dominio N-teminal del factor eIF4G. (Lankat-Buttgereit
and Goke 2003; Yang, Jansen et al. 2003)

23
En células de carcinoma de colon que fueron transfectadas establemente
con PDCD4, se observó una disminución en la capacidad invasiva. Esto fue
asociado con una reducción significativa de la actividad de MAP4K1 (Proteína 4
quinasa 1 activada por mitógenos), lo que sugiere que PDCD4 bloquea la vía de
JNK al regular blancos río arriba tales como MAP4K1 (Yang, Matthews et al.
2006).
Zhang y DuBois, demostraron que la inhibición de COX-2 por el inhibidor
selectivo NS398 incrementa la expresión del RNAm de PDCD4 en células de
cáncer de colon (Zhang and DuBois 2001). Recientemente, en nuestro laboratorio
se observó que COX-2, PGE2 e IL-8 están asociados con la disminución de la
expresión de PDCD4. Mientras que la sobreexpresión de PDCD4 bloquea la
invasión inducida por PGE2 e IL-8 en células MCF-7. Además, se ha reportado
que PDCD4 inhibe la invasión de células de cáncer de mama al elevar la
expresión de TIMP-2 (Nieves-Alicea, Colburn et al. 2009).
Por otra parte, altos niveles de PDCD4 han sido correlacionados con un
aumento en la sensibilidad a antiestrógenos. Jansen et al, reportaron que la
expresión estable del antisentido de PDCD4 reduce significativamente la
sensibilidad de las células MCF-7 a geldanamicina y tamoxifeno. Mientras que la
sensibilidad a geldanamicina se incrementó significativamente en células de
cáncer renal UO-31 que sobreexpresaban PDCD4, induciendo un mayor arresto
celular y apoptosis (Jansen, Camalier et al. 2004; Jansen, Camalier et al. 2005).
Además se ha demostrado que PDCD4 puede promover significativamente la
apoptosis inducida por cisplatino a través de la vía del receptor de muerte en
células de cáncer de ovario (Xia et al., 2010).
Sin embargo, no se sabe aún si en el cáncer de mama PDCD4 puede
inhibir la invasión y resistencia a tamoxifeno inducida por leptina y COX-2.

24
Capítulo 2.
RESUMEN
La obesidad es un factor de riesgo significativo de desarrollar y morir por
cáncer de mama para las mujeres post-menopáusicas. La leptina es una
adipocina producida en altos niveles en personas obesas y sus receptores se
sobreexpresan en tumores mamarios y metástasis en nódulos linfáticos.
Anteriormente se ha demostrado que leptina estimula la invasión de células de
cáncer de mama, lo cual esta correlacionado con metástasis en cáncer mamario.
Además, leptina ha sido asociada con el desarrollo de resistencia a
antiestrógenos. Por otra parte, se ha demostrado que PDCD4 es capaz de
bloquear la invasión de células de cáncer de mama y ha sido correlacionado con
un incremento en la sensibilidad a antiestrógenos. Sin embargo, aún no se ha
determinado si PDCD4 puede bloquear la invasión y resistencia a tamoxifeno
inducida por leptina en células de cáncer de mama. En este estudio mostramos
los nuevos hallazgos de que leptina no induce invasión en las células de cáncer
de mama que sobre-expresan PDCD4 (MCF-7/PDCD4). Además TIMP-2 resultó
ser esencial para el efecto anti-invasivo de PDCD4, ya que leptina estimuló la
invasión de las células MCF-7/PDCD4 que habían sido pre-tratadas con siRNA
anti-TIMP-2. Por lo tanto, el silenciamiento de TIMP-2 permitió que leptina
incrementara la fosforilación de ERK 1,2 y JAK2/STAT3 pero no la activación de
JNK. Estos datos indican que PDCD4 utiliza a TIMP-2 para ejercer su efecto anti-
invasivo mediante la inhibición de la activación de ERK 1,2 y STAT3 inducida por
leptina. Por otra parte, mostramos que leptina incrementa la resistencia a
tamoxifeno en células MCF-7, pero más importante aún, que PDCD4 inhibe la
resistencia a tamoxifeno inducida por leptina en células de cáncer de mama.
Nuevas estrategias terapéuticas enfocadas a incrementar la expresión de PDCD4
en tumores de cáncer de mama podrían ser capaces de detener la progresión de
tumores mamarios relacionados con obesidad y de esta manera prolongar la vida
de los pacientes.

25
INTRODUCCIÓN
El cáncer de mama es el cáncer diagnosticado con mayor frecuencia en mujeres.
En Estados Unidos de América, durante el 2011, se estimaron alrededor de
39,970 muertes por cáncer de seno (39,520 mujeres, 450 hombres). En el mismo
año, se estimaron 230,480 nuevos casos de cáncer de mama invasivo en mujeres
y cerca de 2,140 en hombres, mientras que los casos de cáncer de mama in situ
fueron 57,650 (Society 2011). En México, en el 2010 fallecieron 5,113 mujeres por
esta causa (SALUD 2010). Uno de los principales factores de riesgo para las
mujeres postmenopáusicas de desarrollar cáncer de mama es la obesidad. Los
tumores de pacientes con cáncer de mama y alto índice de masa corporal son
típicamente dependientes de hormonas (Verreault, Brisson et al. 1989; Giuffrida,
Lupo et al. 1992; Huang, Newman et al. 2000). La leptina, es una hormona de 16
kDa que fue descubierta como un regulador de peso corporal y balance
energético que actúa en el hipotálamo, es una de las muchas hormonas
relacionadas con obesidad y cáncer de mama (Zhang, Proenca et al. 1994).
Ambos factores, masa corporal y el porcentaje de grasa incrementan los niveles
de leptina circulante (McMurtry, Simeone et al. 2009).
Una alta expresión de leptina y la isoforma larga de su receptor ha sido
encontrada en tumores mamarios. (Ishikawa, Kitayama et al. 2004; Caldefie-
Chezet, Damez et al. 2005; Garofalo, Koda et al. 2006; Miyoshi, Funahashi et al.
2006; Revillion, Charlier et al. 2006). En ratones deficientes de leptina, se ha
observado una disminución de tumores mamarios inducidos por oncogenes
(Cleary, Juneja et al. 2004). Además, altos niveles de leptina en tumores
mamarios se correlacionan con un mal pronóstico e incremento en la incidencia
de metástasis (Ishikawa, Kitayama et al. 2004; Garofalo, Koda et al. 2006;
Revillion, Charlier et al. 2006). La metástasis en cáncer de mama es
directamente asociada con la invasión (McMurtry, Simeone et al. 2008).
Recientemente, hemos demostrado que leptina estimula la invasión de las células
de cáncer de mama MCF-7 al activar JNK, el cual a su vez incrementa la actividad
de metaloproteinasa-2.

26
Leptina también ha sido asociada con el desarrollo de resistencia a
antiestrógenos en cáncer de mama (Garofalo, Sisci et al. 2004). Es un potente
modulador de la vía de señalización del receptor de estrógeno (Catalano, Marsico
et al. 2003; Catalano, Mauro et al. 2004; Garofalo, Sisci et al. 2004). La leptina
estimula la producción de estrógenos a través del incremento de la expresión de
la aromatasa, pero además puede activar el receptor de estrógeno de una manera
independiente de ligando. Por otra parte, esta hormona puede estabilizar el
receptor de estrógeno al interferir con su degradación mediada por el proteosoma
(Garofalo, Sisci et al. 2004). Por lo tanto, la alta expresión de leptina podría estar
asociada con un mal pronóstico en pacientes con cáncer de mama ya que
estimula invasión y resistencia a antiestrógenos. Nuevas estrategias terapéuticas
destinadas a bloquear la señalización de leptina en tumores mamarios podrían ser
capaces de impedir la progresión del cáncer de mama relacionado con obesidad
para así prolongar la vida de los pacientes.
PDCD4, originalmente identificado como un inhibidor de la transformación
celular en ratones, es un nuevo supresor de tumores. La expresión de PDCD4
está disminuida o es nula en varios tipos de tumores, incluyendo tumores de
mama invasivo ductal (Jansen, Camalier et al. 2004). Recientemente se demostró
que al incrementar la expresión de TIMP-2, la sobreexpresión de PDCD4 suprime
la invasión de las células de cáncer de mama mediada por PGE2 e IL-8 (Nieves-
Alicea, Colburn et al. 2008). Sin embargo, aún no se sabe si PDCD4 podría
suprimir la invasión y resistencia a tamoxifeno inducida por leptina en las células
de cáncer de mama
En el presente trabajo encontramos que la sobreexpresión de PDCD4
permite incrementar la transcripción de TIMP-2, el cual puede inhibir la invasión
de las células de cáncer de mama inducida por leptina al bloquear la activación de
ERK 1,2 y STAT3. Además, que la sobreexpresión de PDCD4 inhibe la
resistencia a tamoxifeno mediada por leptina en células de cáncer de mama.

27
2.1. HIPÓTESIS
PDCD4 inhibe la progresión del cáncer de mama inducida por leptina bloqueando
la invasión y resistencia a tamoxifeno.

28
2.2. OBJETIVO GENERAL
Determinar si PDCD4 inhibe la progresión inducida por leptina en las células
de cáncer de mama bloqueando la invasión y resistencia a tamoxifeno.
2.3. OBJETIVOS PARTICULARES
Determinar si PDCD4 inhibe la invasión de las células de cáncer de mama
inducida por leptina.
Analizar la activación de ERK, STAT3 y JNK inducida por leptina en células
MCF-7 que sobreexpresan PDCD4.
Examinar si TIMP-2 es esencial para el efecto anti-invasivo de PDCD4.
Determinar si PDCD4 inhibe la resistencia a tamoxifeno inducida por
leptina en células de cáncer de mama.

29
2.4. MATERIAL Y MÉTODOS
2.4.1. Líneas celulares tumorales.
En este estudio se utilizó la línea celular humana de cáncer de mama MCF-7
silvestre (MCF-7/WT), la cual fue transfectada establemente con el plásmido
pcDNA 3.1 (MCF-7/Vector) o con plásmido pcDNA 3.1 que contiene el gen de
PDCD4 (MCF-7/PDCD4). Estas células fueron adquiridas a través de la
colaboración con la Dra. Ana M Tari del MD Anderson Cancer Center en la
Ciudad de Houston, Texas, EUA. Las células fueron examinadas contra
micoplasma y mantenidas en medio DMEM/F12 (Medio Eagle modificado por
Dulbecco) (Invitrogen Corporation) suplementado con 5% de suero fetal bovino
(FBS) en 5% de CO2 y 95% de humedad a 37C. Para el mantenimiento de las
líneas celulares transfectadas, se realizaron tratamientos una vez por semana
con 500 g/mL de geneticina.
2.4.2. Ensayo de invasión en Matrigel
La invasividad de las células MCF-7/WT y MCF-7/PDCD4 tratadas con leptina se
determinó contando el número de células que invadieron a través del Matrigel, el
cual es una membrana basal artificial. Las células fueron colectadas y lavadas
con medio DMEM/F12 sin suero. Después se ajustaron a 4 x 105 células en 1 mL
de medio sin suero y se agregaron dentro de insertos especiales llamados
transwells (los cuales cuentan con un filtro con poros de 8 µm y son colocados
dentro de pozos en placas de 6 pozos (Fisher Scientific, Middleton, VA, USA)) a
los que se les recubrió con una capa de Matrigel (0.7 mg/mL: BD Bioscience,
Bedford, MA, USA). Posteriormente, al pozo donde se colocó el transwell se le
agregaron 2 mL de medio DMEM/F12 suplementado con 10% de FBS, el cual se
utilizó como agente quimiotáctico para las células de cáncer de mama (Figura 6).
Finalmente, las células fueron tratadas con 0, 10 y 100 ng/mL de leptina
empleando medio sin suero. Setenta y dos horas después, las células presentes
en la superficie superior del filtro del transwell fueron removidas con una torunda.
Mientras que las células que invadieron a través del Matrigel dentro de la parte
inferior del filtro fueron fijadas en laminillas, teñidas con el sistema de tinción
Hema-3 (Fisher Scientific), contadas y fotografiadas a una magnificación de 40X.
Se cuantificaron las células presentes en 9 campos de cada laminilla y por
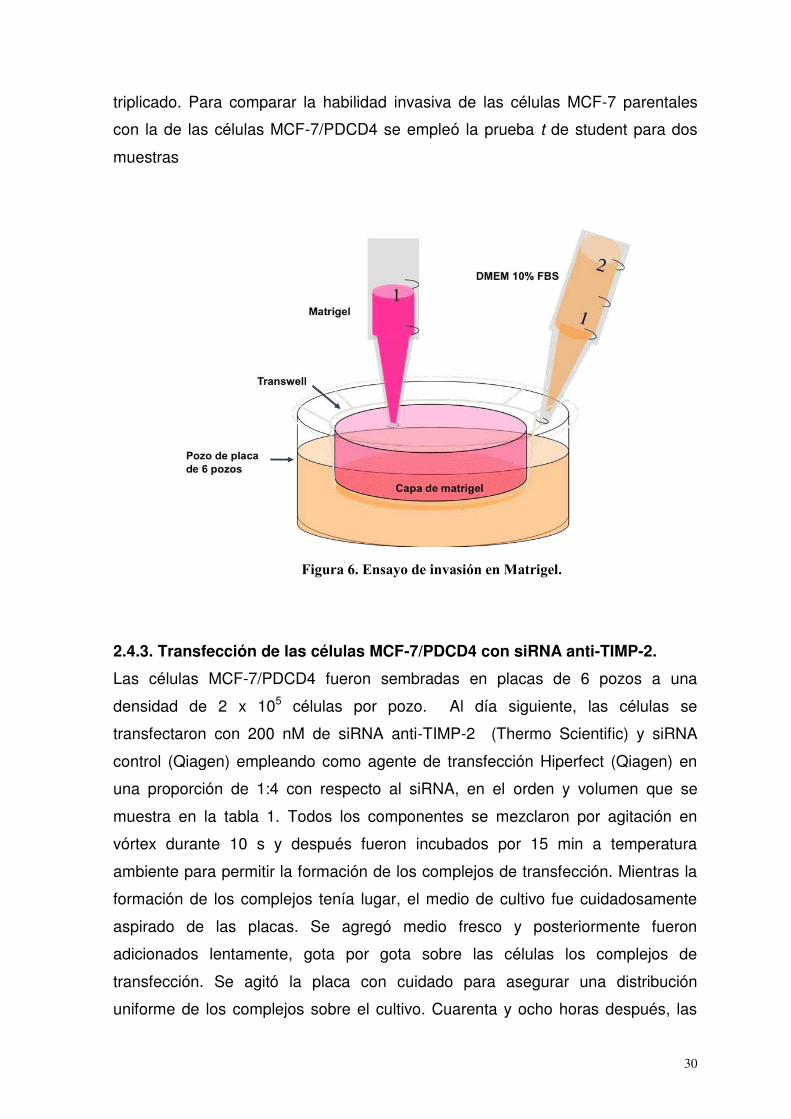
30
triplicado. Para comparar la habilidad invasiva de las células MCF-7 parentales
con la de las células MCF-7/PDCD4 se empleó la prueba t de student para dos
muestras
Figura 6. Ensayo de invasión en Matrigel.
2.4.3. Transfección de las células MCF-7/PDCD4 con siRNA anti-TIMP-2.
Las células MCF-7/PDCD4 fueron sembradas en placas de 6 pozos a una
densidad de 2 x 105 células por pozo. Al día siguiente, las células se
transfectaron con 200 nM de siRNA anti-TIMP-2 (Thermo Scientific) y siRNA
control (Qiagen) empleando como agente de transfección Hiperfect (Qiagen) en
una proporción de 1:4 con respecto al siRNA, en el orden y volumen que se
muestra en la tabla 1. Todos los componentes se mezclaron por agitación en
vórtex durante 10 s y después fueron incubados por 15 min a temperatura
ambiente para permitir la formación de los complejos de transfección. Mientras la
formación de los complejos tenía lugar, el medio de cultivo fue cuidadosamente
aspirado de las placas. Se agregó medio fresco y posteriormente fueron
adicionados lentamente, gota por gota sobre las células los complejos de
transfección. Se agitó la placa con cuidado para asegurar una distribución
uniforme de los complejos sobre el cultivo. Cuarenta y ocho horas después, las

31
células se colectaron y se sembraron a una densidad de 1 x 105 células/pozo.
Finalmente se trataron con leptina a 100 ng/mL en medio DMEM/12 5% FBS
durante 72 h más. Después de éste período de incubación las células fueron
colectadas para realizar RT-PCR y western blot. De la misma manera se
colectaron células para llevar a cabo ensayos de invasión como se describe
anteriormente.
Tabla 1. Esquema de transfección de las células MCF-7/PDCD4 con siRNA anti-TIMP-2 y control.
siRNA anti-TIMP-2 y
Control [20M]
Hiperfect Medio DMEM/F12 5% FBS Volumen Final
20 l
20 l
75 l
100 l
2.4.4. Extracción de RNA total y reacción en cadena de la polimerasa con
transcripción reversa (RT-PCR).
Para confirmar que los niveles de RNAm de TIMP-2 fueron reducidos en las
células transfectadas con siRNA anti-TIMP-2, se extrajo RNA total utilizando el kit
RNeasy (Qiagen Inc, Valencia, CA, USA) siguiendo las instrucciones del
fabricante, las células se lisaron adicionando la solución amortiguadora RLT, se
resuspendieron usando una jeringa con aguja de 0.9 mm de diámetro. Después
se agregó un volumen de etanol al 70% al homogenizado y se mezcló por
pipeteo. Se transfirieron 700 L de la muestra (incluyendo algún precipitado que
se pudo haber formado) a las columnas RNeasy, colocadas en tubos de 2 mL. Se
centrifugaron 15 s a 10000 rpm, para descartar el fluido a través de la columna.
Luego se adicionaron 700 L de la solución amortiguadora RW1 en la columna y
se centrifugó a 10000 rpm por 15 s, para descartar el fluido a través de la
columna. Posteriormente, se agregaron 500 L de solución RPE a la columna y
se centrifugó 2 min a 10000 rpm, para descartar el fluido a través de la columna.
Después la columna se colocó en tubos de 1.5 mL, se le adicionó agua libre de
RNAsas y se centrifugó a 10000 rpm durante 1 min para eluir el RNA. Para llevar
a cabo el RT-PCR se trabajó con el sistema SuperScript III one-step RT-PCR
(Invitrogen Corporation), siguiendo las instrucciones del fabricante y empleando 1
g de RNA. Las secuencias de los oligonucleótidos iniciadores (primers) utilizados

32
son las siguientes; TIMP-2: sentido, 5’-GGT CTC GCT GGA CGT TGG AG-3’ y
antisentido, 5’-GGA GCC GTC ACT TCT CTT G-3’. GAPDH: sentido, 5’-GCC
AAG GTC ATC CAT GAC AAC-3’ y antisentido, 5’-GTC CAC CAC CCT GTT GCT
GTA-3’. GAPDH se usó como control de carga. Las condiciones del PCR para
todas las reacciones que se realizaron fueron como se describe a continuación:
55oC 30 min, 94oC 2 min, 30 ciclos de 94oC durante 15 s, 57oC por 30 s, 68oC 1
min y 68oC 5 min. Los productos del PCR se analizaron en geles de agarosa al
1.5%. El corrimiento de los geles se llevo a cabo a 90 V.
2.4.5. Western Blot
2.4.5.1. Obtención de las muestras.
Después sembrar y tratar las células, éstas se tripsinizaron, colectaron y
centrifugaron a 1500 rpm, a una temperatura de 4C por 5 min. En seguida, se
realizaron dos lavados con una solución de PBS 1X e inhibidores de fosfatasas.
Posteriormente se removió el sobrenadante y el pellet celular se conservó a -80C
hasta su uso.
2.4.5.2. Preparación de las muestras.
Las células colectadas se lisaron con 50 L de buffer de lisis (1% Triton X-100,
150 mM NaCl y 25 mM de Tris, pH 7.6, 4 L/mL de NaF, 4 L/mL de Na3VO4 y 10
L/mL de inhibidor de proteasas SIGMA (St. Louis MO)) y agitación en vórtex por
30 s. Las muestras se incubaron durante 30 min a 4C (repitiendo la agitación en
vórtex cada 5 min o hasta deshacer la pastilla celular). Posteriormente se
centrifugaron a 13000 rpm por 10 min y el sobrenadante se transfirió a un tubo de
1.5 mL. Las proteínas fueron cuantificadas por el método del ácido bicinconínico,
empleando el kit Pierce BCA protein assay de Thermo Scientific (Rockford, IL).
2.4.5.3. Electroforesis de proteínas en geles de poliacrilamida en presencia
de dodecil sulfato de sodio (SDS-PAGE).
Las proteínas obtenidas fueron separadas en geles de poliacrilamida en
condiciones desnaturalizantes. En cada carril se cargaron 30 g de proteínas
totales en geles de poliacrilamida al 10%. La electroforesis se realizó en buffer de

33
corrimiento (Tris-glicina 1X) a 40 V para el gel concentrador y a 70 V para el gel
separador.
2.4.5.4. Inmunoelectrotransferencia.
Una vez terminada la electroforesis, el gel fue electrotransferido a una membrana
de nitrocelulosa (Bio-Rad) como se describe a continuación. Se preparó el
sándwich para la transferencia colocando el gel, esponjas, papel filtro y la
membrana de nitrocelulosa dentro de un recipiente con buffer de transferencia
(Tris-glicina-metanol 1X).
El sándwich se colocó dentro de la cámara (Bio-Rad), la cual se llenó con
el mismo buffer. La transferencia se llevó a cabo a 26 V constantes a 4ºC durante
24 h. Al termino de la transferencia la membrana fue teñida con Rojo Ponceau,
después se lavó con H2Odd hasta su total decoloración. Posteriormente, se
bloqueó con leche descremada al 5% en TBS-Tween 20 1X pH 7.4 por 30 min y
en agitación constante. Se realizaron 3 lavados de 10 min cada uno con TBS-
Tween 1X pH 7.4.
Después de los lavados, los anticuerpos primarios de ERK1,2 fosforilado
(T202/Y204), ERK1,2 total, STAT3 fosforilado (Y705), STAT3 total, JNK fosforilado
(T183/Y185) y JNK total se prepararon a una dilución 1:1000 con leche descremada
al 5% en TBS-Tween 1X pH 7.4 y se agregaron por separado respectivamente a
las membranas correspondientes, las cuales se dejaron en agitación constante
durante toda la noche a 4ºC. Luego se realizaron 3 lavados de 10 min con TBS-
Tween 1X pH 7.4. En seguida se agregó el anticuerpo secundario anti-Fc de
ratón o conejo (dependiendo del anticuerpo primario utilizado) a la membrana a
una dilución 1:1000 con leche descremada al 5% en TBS-Tween 1X pH 7.4 y se
dejó en agitación constante a temperatura ambiente por 2 h. Se realizaron 3
lavados de 10 min con TBS-Tween 1X pH 7.4. Posteriormente, las proteínas
fueron detectadas a través de quimioluminiscencia incubando las membranas con
el reactivo ECL (GE Healthcare, Piscataway, NJ) durante 1min. La membrana se
colocó en un film autoradiográfico durante diferentes tiempos de exposición. El
film fue revelado en la máquina de revelado radiográfico AFP IMAGING.
Finalmente, las imágenes fueron escaneadas y analizadas en el densitómetro

34
Alphalmager (Alpha Innotech Corp., San Leandro, CA). La プ-actina fue utilizada
como control de carga. Las membranas fueron incubadas durante 30 min a
temperatura ambiente con el anticuerpo primario de ß-actina adquirido de Sigma
(St. Louis, MO), el cual se preparó a una dilución de 1:10,000 con leche
descremada al 5% en TBS-Tween 1X pH 7.4. Después se realizaron 3 lavados de
10 min con TBS-Tween 1X pH 7 y se incubaron con el anticuerpo secundario anti-
Fc de ratón obtenido de Cell Signaling Technology, Inc. (3 TraskLane Danvers,
MA) (a una dilución de 1:10,000) por otros 30 min a temperatura ambiente.
2.4.6. Ensayo de viabilidad celular (MTS).
Para determinar si PDCD4 inhibe la resistencia a tamoxifeno inducida por leptina,
llevamos a cabo ensayos de viabilidad celular (MTS). Las células se sembraron
en placas de 96 pozos a una densidad de 1000 células en 100L de medio
DMEM/F12 suplementado con 5% de FBS por pozo y se incubaron durante toda
la noche. Al día siguiente, las células se trataron con diferentes concentraciones
de leptina (0, 10 y 100 ng/mL) e incubaron por 24 h, después de éste tiempo,
fueron tratadas con 0, 0.1, 1, 5 y 9 M de tamoxifeno durante 5 días.
Posteriormente se preparó la solución de trabajo, agregando 2 mL de la solución
de MTS Promega (Madison, WI) y 100 L de la solución PMS Promega (Madison,
WI). Después se agregaron 20 L de la solución MTS/PMS a cada pozo. Las
placas se leyeron en un lector de ELISA (Kinetic Microplate Reader; Molecular
Devices Corp.) a 490 nm. Los resultados se expresaron en términos de porcentaje
de inhibición del crecimiento con respecto al control (células no tratadas).

35
2.5. RESULTADOS
2.5.1. PDCD4 inhibe la invasión inducida por leptina en las células de cáncer
de mama MCF-7.
Mediante western blot confirmamos que las células MCF-7/PDCD4 expresan más
niveles de la proteína PDCD4 que las células control (MCF-7/Vector). Los niveles
de PDCD4 expresados en las células MCF-7/PDCD4 fueron 1.5 veces más altos
que en las células control (Figura 7). Posteriormente, realizamos ensayos de
invasión en Matrigel, tratando a las células MCF-7 parentales (células control
MCF-7/WT) y MCF-7/PDCD4 con 0, 10 y 100 ng/mL de leptina. En ausencia de
leptina, el número de células MCF-7/WT que invadieron a través del Matrigel fue
de 55 (15), mientras que en presencia de 10 y 100 ng/mL de leptina, el número
de células invasivas fue de 100 (18) y 140 (30), respectivamente (Figura 8A).
Estos resultados confirmaron nuestros reportes previos de que leptina estimula la
invasión de células MCF-7(McMurtry, Simeone et al. 2008). Por otra parte, leptina
no estimuló la invasión de las células MCF-7/PDCD4; en presencia de 0, 10 y 100
ng/mL de leptina, el número de células MCF-7/PDCD4 que invadieron a través del
Matrigel fue de 48 (5), 51 (4) y 47 (4) respectivamente (Figura 8B). Estos
datos, indican que altos niveles de PDCD4 bloquean la invasión inducida por
leptina en las células de cáncer de mama.
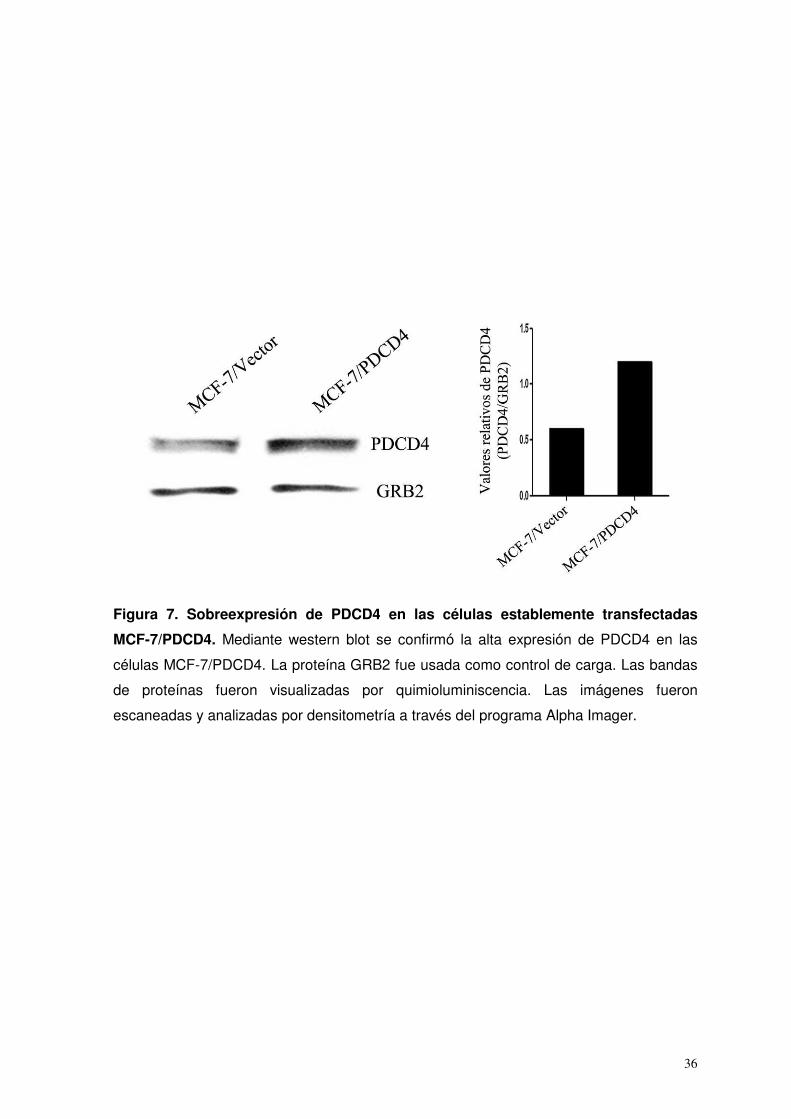
36
Figura 7. Sobreexpresión de PDCD4 en las células establemente transfectadas
MCF-7/PDCD4. Mediante western blot se confirmó la alta expresión de PDCD4 en las
células MCF-7/PDCD4. La proteína GRB2 fue usada como control de carga. Las bandas
de proteínas fueron visualizadas por quimioluminiscencia. Las imágenes fueron
escaneadas y analizadas por densitometría a través del programa Alpha Imager.

37
Figura 8. PDCD4 inhibe la invasión inducida por leptina en células de cáncer de
mama. Se realizaron ensayos de invasión en Matrigel para determinar el efecto de leptina
en la invasividad de las células (A) MCF-7/WT y (B) MCF-7/PDCD4. El número de células
que invadieron a través del Matrigel fue significativamente más alto en las células
tratadas con leptina que en las células no tratadas. Las columnas indican el número
promedio de las células invasivas ± desviación estándar, (*,P0.05).
A
B

38
2.5.2. TIMP-2 es esencial para que PDCD4 inhiba la invasión inducida por
leptina en células de cáncer de mama MCF-7.
Debido a que los resultados anteriores muestran que la sobreexpresión de
PDCD4 inhibe la invasión de células de cáncer de mama inducida por leptina y a
que en estudios anteriores se observó que la sobreexpresión de PDCD4 bloquea
la invasión de células de cáncer de mama inducida por PGE2 e IL-8 a través del
incremento de la expresión de TIMP-2, (Nieves-Alicea, Colburn et al. 2008)
decidimos determinar si TIMP-2 podría ser esencial para el efecto anti-invasivo de
PDCD4. Primeramente realizamos RT-PCR para determinar los niveles basales
de RNAm de TIMP-2 en las células MCF-7/WT, MCF-7/Vector y MCF-7/PDCD4.
Los niveles de RNAm de TIMP-2 fueron aproximadamente 2.1 y 1.5 más altos en
las células MCF-7/PDCD4 que en las células MCF-7/WT y MCF-7/Vector,
respectivamente (Figura 9). Con el fin de determinar si TIMP-2 es esencial para
que PDCD4 pueda bloquear la invasión de las células de cáncer de mama
inducida por leptina, tratamos las células MCF-7/PDCD4 con siRNA anti-TIMP-2 y
control (Figura 10), y después realizamos ensayos de invasión en Matrigel
tratando a las células con leptina (100 ng/mL). En presencia de leptina, el número
de células MCF-7/PDCD4 no tratadas y tratadas con siRNA control que invadieron
a través del Matrigel fue de 60 (3) y 62 (5) respectivamente, mientras que bajo
las mismas condiciones, el número de células MCF-7/PDCD4 tratadas con siRNA
anti-TIMP-2 que invadieron a través del Matrigel fue de 220 (20) (Figura 11).
Estos resultados sugieren que en presencia de leptina y cuando la expresión de
TIMP-2 es disminuida, la capacidad invasiva de las células MCF-7/PDCD4
aumenta significativamente, indicando que TIMP-2 es esencial para que PDCD4
pueda bloquear la invasión de las células de cáncer de mama inducida por
leptina.

39
Figura 9. Sobreexpresión de TIMP-2 en las células MCF-7/PDCD4. Se realizó RT-PCR
para determinar los niveles de mRNA de TIMP-2 en las células de cáncer de mama.
GAPDH fue usado como control de carga. Las bandas de DNA fueron visualizadas,
fotografiadas y analizadas por densitometría a través del Alpha Imager.
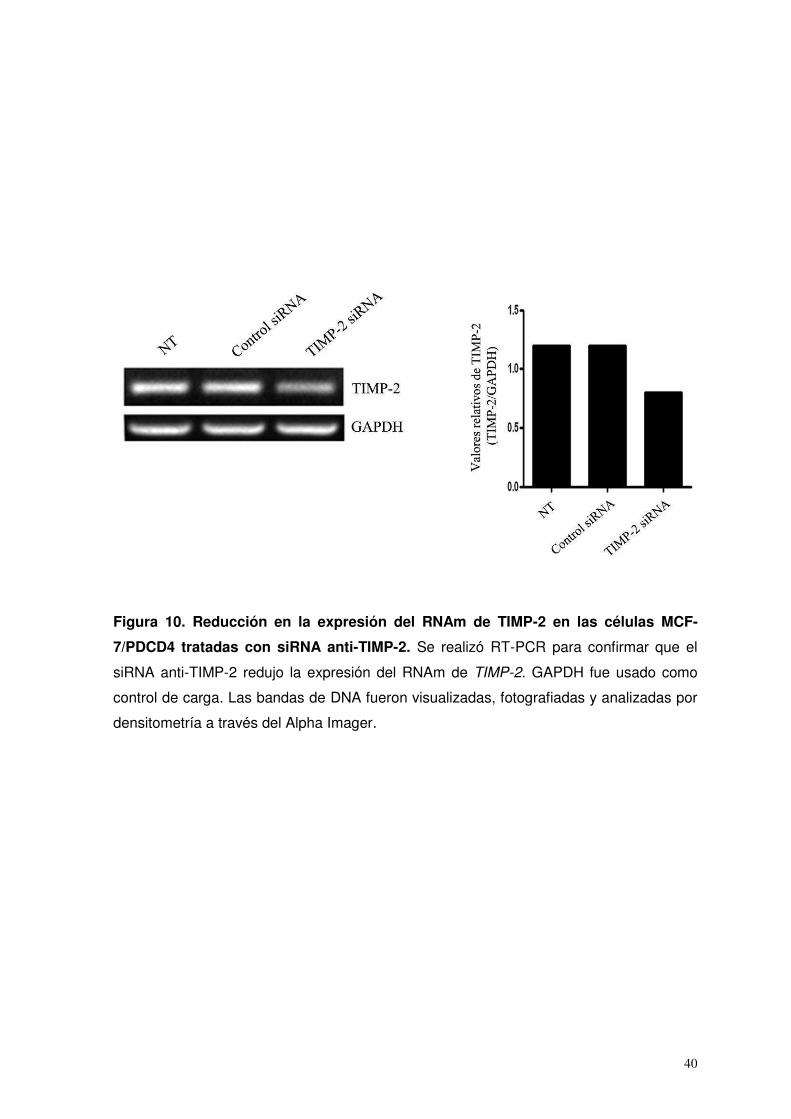
40
Figura 10. Reducción en la expresión del RNAm de TIMP-2 en las células MCF-
7/PDCD4 tratadas con siRNA anti-TIMP-2. Se realizó RT-PCR para confirmar que el
siRNA anti-TIMP-2 redujo la expresión del RNAm de TIMP-2. GAPDH fue usado como
control de carga. Las bandas de DNA fueron visualizadas, fotografiadas y analizadas por
densitometría a través del Alpha Imager.

41
Figura 11. El silenciamiento de TIMP-2 incrementa la invasión de las células de
cáncer de mama MCF-7/PDCD4. El número de células que invadieron a través del
Matrigel fue significativamente más alto en las células tratadas con siRNA anti-TIMP-2
que en las células transfectadas con el siRNA control. Las columnas indican el número
promedio de las células invasivas ± desviación estándar, (**,P0.01).

42
2.5.3. El silenciamiento de TIMP-2 permite la activación de ERK1,2 y STAT3
inducida por leptina en las células MCF-7/PDCD4.
Nuestros resultados anteriores sugieren que TIMP-2 es esencial para que PDCD4
pueda bloquear la invasión de las células de cáncer de mama inducida por leptina
y como ya se ha demostrado que leptina activa ERK1,2, STAT3 y JNK en cáncer
de mama,(Catalano, Marsico et al. 2003; Catalano, Mauro et al. 2004; Yin, Wang
et al. 2004; Chen, Chang et al. 2006; Gonzalez, Cherfils et al. 2006; Ray, Nkhata
et al. 2007; Saxena, Sharma et al. 2007; Jiang, Yu et al. 2008; McMurtry, Simeone
et al. 2008) nuestro siguiente objetivo fue determinar si el mecanismo por el cual
PDCD4 y TIMP-2 bloquean la invasión inducida por leptina en las células de
cáncer de mama es a través de la activación de ERK1,2, STAT3 y/o JNK. Por
tanto, pre-tratamos las células MCF-7/PDCD4 con siRNA anti-TIMP-2 y siRNA
control, antes de tratarlas con leptina. En seguida, realizamos western blot para
determinar los niveles de fosforilación de ERK1,2, STAT3 y JNK. En presencia de
leptina, las células MCF-7/PDCD4 tratadas con siRNA anti-TIMP-2 aumentaron
tres veces los niveles de ERK1,2 fosforilado comparado con las células tratadas
con el siRNA control (Figura 12A). Además en presencia de leptina, las células
MCF-7/PDCD4 tratadas con siRNA anti-TIMP-2 aumentaron 2.5 veces más la
fosforilación de STAT3 comparado con las células tratadas con el siRNA control
(Figura 12B). Sin embargo, bajo las mismas condiciones, no se observó diferencia
en los niveles de JNK fosforilado entre las células MCF-/PDCD4 tratadas con
siRNA anti-TIMP-2 y control (Figura 12C). Estos resultados muestran que el
silenciamiento de TIMP-2 permite que leptina estimule la activación de ERK1,2 y
STAT3 en las células MCF-7/PDCD4. De manera interesante el silenciamiento de
TIMP-2 permitió un incremento en la fosforilación de ERK 1,2 pero una
disminución en los niveles de ERK 1,2 total, mientras que el silenciamiento de
TIMP-2 incremento los niveles de fosforilación de STAT3 sin afectar los niveles
de STAT3 total. En este caso, se sugiere que podría existir un mecanismo de
retroalimentación que regula los niveles de ERK fosforilado a partir de los niveles
de ERK total.

43
Figura 12. El silenciamiento de TIMP-2 incrementa la activación inducida por leptina
de ERK1,2, STAT3, pero no afecta la activación de JNK en células MCF-7/PDCD4.
Mediante western blot se determinaron los niveles de (A) ERK1,2 fosforilado y ERK1,2
total, (B) los niveles de STAT3 fosforilado y STAT3 total y (C) los niveles de JNK
fosforilado y JNK total. -actina fue usada como control de carga. Las bandas de

44
proteínas fueron visualizadas por quimioluminiscencia. Las imágenes fueron escaneadas
y analizadas por densitometría a través del programa Alpha Imager.
2.5.4. PDCD4 inhibe la resistencia a tamoxifeno inducida por leptina.
La leptina puede incrementar la expresión del gen de la aromatasa y su actividad
en células MCF-7, lo cual podría aumentar la síntesis de estrógeno. Leptina
puede además incrementar la estabilidad del receptor de estrógeno alfa (ER)
permitiendo mantener la transcripción dependiente de ER en células de cáncer
de mama en presencia de antiestrógenos (Garofalo and Surmacz., 2006). Estas
observaciones sugieren que altos niveles de leptina en pacientes obesos con
cáncer mamario podrían contribuir al crecimiento tumoral y desarrollo de
resistencia a antiestrógenos. Por otra parte, altos niveles de PDCD4 han sido
correlacionados con un incremento en la sensibilidad a antiestrógenos. Para
determinar si PDCD4 puede inhibir la resistencia a tamoxifeno inducida por leptina
en células MCF-7, realizamos ensayos de viabilidad celular, pre-tratando a la
células MCF-7/Vector y MCF-7/PDCD4 con 0, 10 y 100 ng/mL de leptina durante
24 h. En seguida las células fueron tratadas con 0, 0.1, 1, 5 y 9 M de tamoxifeno
durante 5 días. Nuestros resultados muestran que las células MCF-7/PDCD4 son
más sensibles al tamoxifeno que las células MCF/Vector (Figura 13A). En
presencia de 10 ng/mL de leptina, las células MCF-7/Vector llegaron a ser más
resistentes a tamoxifeno, mientras que la leptina no estimuló la resistencia a
tamoxifeno en las células MCF-7/PDCD4 (Figura 13B). Aunque en presencia de
100 ng/mL de leptina la resistencia a tamoxifeno en las células MCF-7/Vector no
se incrementó, la sensibilidad a tamoxifeno en las células MCF-7/PDCD4 se
mantuvo (figura 13C).

45
Figura 13. PDCD4 inhibe la resistencia a tamoxifeno inducida por leptina. Las células MCF-7/Vector y MCF-7/PDCD4 se pre-trataron con 0 (A), 10 (B) y 100 (C) ng/mL de leptina durante 24 h. Después de este tiempo de incubación, fueron tratadas con 0, 0.1, 1, 5 y 9 M de tamoxifeno por 5 días. La viabilidad celular se realizó mediante MTS
Leptina [0 ng/ml]
Leptina [10 ng/ml]
Leptina [100 ng/ml]

46
2.6. DISCUSIÓN Y CONCLUSIÓN.
La obesidad es un factor de riesgo de mortalidad para las mujeres diagnosticadas
con cáncer de mama; las mujeres con un índice de masa corporal de al menos 40
tienen 2.1 veces más riesgo de morir por cáncer de mama (Calle, Rodriguez et al.
2003). La leptina es un enlace vital entre obesidad y cáncer de mama, ya que
altos niveles de leptina han sido asociados con un mal pronóstico e incremento en
la incidencia de metástasis (Ishikawa, Kitayama et al. 2004; Garofalo, Koda et al.
2006; Revillion, Charlier et al. 2006). Nuevas estrategias terapéuticas son
necesarias para impedir la progresión inducida por leptina en cáncer mamario.
En este estudio, mostramos por primera vez que PDCD4 inhibe la invasión
de células de cáncer de mama inducida por leptina y que tal y como en reportes
anteriores (Nieves-Alicea, Colburn et al. 2008) TIMP-2 es crítico para el efecto
anti-invasivo de PDCD4. La leptina estimuló la invasión de células MCF-7/PDCD4
cuando la expresión de TIMP-2 disminuyó. Interesantemente, se ha observado
que la expresión de PDCD4 está solo ligeramente disminuida en muestras de
carcinoma ductal in situ en comparación con células de epitelio normal, pero esta
marcadamente disminuida en muestras de carcinoma ductal invasivo (Wen, Shi et
al. 2007). Por lo tanto, PDCD4 podría actuar como un supresor de la invasión en
células de cáncer de mama.
En este proyecto estudiamos también el mecanismo a través del cual
PDCD4 y TIMP-2 inhiben la invasión inducida por leptina en células de cáncer de
mama. Se sabe que leptina estimula la fosforilación de ERK 1,2, STAT3 y JNK en
células de cáncer de mama (Catalano, Marsico et al. 2003; Catalano, Mauro et al.
2004; Yin, Wang et al. 2004; Chen, Chang et al. 2006; Gonzalez, Cherfils et al.
2006; Ray, Nkhata et al. 2007; Saxena, Sharma et al. 2007; Jiang, Yu et al. 2008;
McMurtry, Simeone et al. 2008). Nosotros encontramos que el silenciamiento de
TIMP-2 permite que leptina aumente la activación de ERK 1,2 y STAT3 en células
MCF-7/PDCD4, sugiriendo que PDCD4 utiliza TIMP-2 para contrarrestar la
activación mediada por leptina de ERK 1,2 y STAT3 para bloquear la invasión. En
efecto ERK 1,2 y STAT3 son esenciales para la invasión estimulada por leptina,
ya que se ha observado que inhibidores farmacológicos de JAK2/STAT3 y ERK

47
1,2 bloquean la invasión inducida por leptina en células de carcinoma
hepatocelular y endometrial (Sharma, Saxena et al. 2006; Saxena, Sharma et al.
2007).
Sin embargo, el silenciamiento de TIMP-2 no cambió los niveles de
fosforilación de JNK mediada por leptina, indicando que TIMP-2 no es utilizado
por PDCD4 en la regulación de la actividad de JNK. Se ha mostrado que PDCD4
inhibe la transcripción de MAP4K1 (Yang, Matthews et al. 2006), una quinasa río
arriba de JNK, inhibiendo de este modo la activación de c-Jun y la transcripción
dependiente de AP-1 (Yang, Matthews et al. 2006). Alternativamente, PDCD4
podría inhibir la transcripción dependiente de AP-1 al interferir con el
reclutamiento del co activador p300 a través de c-Jun, y suprimir la fosforilación
de c-Jun a través de JNK (Bitomsky, Bohm et al. 2004). Por lo que en nuestro
caso PDCD4 podría estar regulando a JNK a través de la inhibición de MAP4K1 o
del reclutamiento del co activador p300 o mediante otras proteínas pero no a
través de TIMP-2.
Por otra parte, mostramos que leptina incrementa la resistencia a
tamoxifeno en células MCF-7, ya que en presencia de leptina (10ng/mL) y
tamoxifeno (9M) se obtuvo un 13% de inhibición de la proliferación en células
MCF-7/Vector, comparado con el 40% de inhibición observado en las células
tratadas solo con tamoxifeno (9M). Además, observamos que en las células
MCF-7/PDCD4, en presencia de leptina (10ng/mL) y tamoxifeno (9M) el
porcentaje de inhibición del crecimiento fue de 57%, indicando que PDCD4 inhibe
la resistencia a tamoxifeno inducida por leptina en células de cáncer de mama. En
base a estos resultados se sugiere que PDCD4 podría no solo actuar como un
supresor de la invasión si no también como un supresor de cáncer mamario en
pacientes con cáncer de mama obesos.
La leptina puede incrementar la expresión del gen de la aromatasa y su
actividad en células MCF-7, lo cual podría aumentar la síntesis de estrógeno.
Leptina puede además incrementar la estabilidad del receptor de estrógeno alfa
(ER) permitiendo mantener la transcripción dependiente de ER en células de

48
cáncer de mama en presencia de antiestrógenos (Garofalo and Surmacz., 2006).
Estas observaciones sugieren que altos niveles de leptina en pacientes obesos
con cáncer mamario podrían contribuir al crecimiento tumoral y desarrollo de
resistencia a antiestrógenos
En conclusión, nuestros resultados demuestran que la sobreexpresión de
PDCD4 permite incrementar la transcripción de TIMP-2, el cual puede inhibir la
invasión de células de cáncer de mama inducida por leptina al bloquear la
activación de ERK 1,2 y STAT3. Además, la sobreexpresión de PDCD4 inhibe la
resistencia a tamoxifeno mediada por leptina en células de cáncer de mama
(Figura 20). Nuevas estrategias terapéuticas dirigidas a incrementar la expresión
de PDCD4 en tumores de mama podrían ser capaces de impedir la progresión del
cáncer de mama relacionado con obesidad para prolongar y mejorar la calidad de
vida de los pacientes y probablemente llegar a curarlos.

49
Capítulo 3.
RESUMEN
La expresión elevada de ciclooxigenasa-2 (COX-2) en tumores de mama ha sido
asociada con una menor tasa de sobrevivencia en pacientes con tumores
positivos para ER. Nosotros proponemos que COX-2 reduce la tasa de
sobrevivencia en pacientes con cáncer de mama positivo para ER debido a que
disminuye la sensibilidad a tamoxifeno e incrementa la actividad invasiva de las
células de cáncer de mama. Anteriormente, en nuestro laboratorio se demostró
que la transfección del gen COX-2 en las células de cáncer de mama MCF-7
(células positivas para ER y sensibles a tamoxifeno) disminuyó la sensibilidad a
tamoxifeno aproximadamente 5 veces e incrementó su actividad invasiva
alrededor de 3 veces más comparado con las células MCF-7 no transfectadas.
También, se demostró que COX-2 utiliza PGE2 para activar PKC e inducir estos
efectos tumorigénicos. Sin embargo, no se conocen hasta el momento cuales
factores río abajo son utilizados por la vía COX-2/PGE2/PKC para inducir
resistencia a tamoxifeno e invasión de las células de cáncer de mama. Por otra
parte, se ha reportado que la sobreexpresión de COX-2, PGE2 y IL-8 está
asociada con una baja expresión de PDCD4, mientras que la sobreexpresión de
PDCD4 bloquea la invasión inducida por PGE2 y IL-8. En este estudio,
reportamos que COX-2 incrementa la fosforilación de JNK, pero no la de ERK 1,2,
p38 ó AKT. La inhibición de JNK pero no la de ERK 1,2, redujo la invasión
mediada por COX-2. Aunque, la inhibición de JNK no alteró la resistencia a
tamoxifeno mediada por COX-2 en las células que sobreexpresan COX-2. La
sobreexpresión de PDCD4 no afectó la resistencia a tamoxifeno inducida por
COX-2. Nosotros concluimos que JNK es esencial para que COX-2 induzca
invasión, pero no resistencia a tamoxifeno. También concluimos que aún y
cuando PDCD4 bloquea la invasión inducida por COX-2 en células de cáncer de
mama, PDCD4 no bloquea la resistencia a tamoxifeno inducida por COX-2 en
células de cáncer de mama positivas para ER.

50
INTRODUCCIÓN
Diferentes estudios han demostrado altos niveles de la proteína COX-2 en
tumores sólidos (Eberhart, Coffey et al. 1994; Soslow, Dannenberg et al. 2000;
Kirschenbaum, Liu et al. 2001). En cáncer de mama, la expresión de COX-2 es un
marcador de sobrevivencia global y libre de enfermedad (Ristimaki, Sivula et al.
2002; Denkert, Winzer et al. 2003; O'Connor, Avent et al. 2004). Ristimaki y
colaboradores reportaron que la expresión elevada de COX-2 estaba asociada
con una baja tasa de sobrevivencia en pacientes con tumores mamarios positivos
para ER. Esta asociación fue particularmente significativa para los tumores de
mama positivos para ER que sobreexpresaban COX-2 y que no tenían expresión
de p53 ó amplificación de HER2/neu. Además, observaron que las mujeres cuyos
tumores de mama invasivo eran positivos para ER pero tenían bajos niveles de
COX-2, presentaban una oportunidad del 86% (IC de 95%, 84-89) de
sobrevivencia libre de enfermedad distante a 5 años, mientras que las mujeres
cuyos tumores eran positivos para ER pero que tenían altos niveles de COX-2
presentaron una oportunidad de sobrevivencia libre de enfermedad distante a 5
años del 76% (IC de 95%, 72-81) (Ristimaki, Sivula et al. 2002).
Los pacientes con cáncer de mama que tienen tumores positivos para ER
son comúnmente tratados con moduladores selectivos del receptor de estrógeno
(SERM’s). En nuestro laboratorio, se demostró previamente que la transfección de
COX-2 dentro de la línea celular MCF-7, la cual es sensible a tamoxifeno y
positiva para ER redujo la sensibilidad a tamoxifeno 5 veces más que las células
no transfectadas (Tari, Simeone et al. 2005). Estos resultados sugieren que los
pacientes con cáncer de mama positivo para ER que sobreexpresan COX-2
podrían no estar beneficiándose del tratamiento con tamoxifeno, como aquellos
pacientes con tumores positivos para ER que presentan bajos niveles de COX-2.
Niveles elevados de COX-2 han sido asociados con un alto potencial metastásico
en tumores mamarios. La expresión de COX-2 es alta en tumores metastásicos
(Badawi and Badr 2003) y esta asociada con metástasis a nódulos linfáticos y
distante (Costa, Soares et al. 2002; Denkert, Winzer et al. 2004; Ranger, Thomas
et al. 2004). Además, se ha observado que COX-2 incrementa la invasión de las

51
células de cáncer de mama in vitro (Rozic, Chakraborty et al. 2001; Prosperi,
Mallery et al. 2004; Singh, Berry et al. 2005). Nosotros y otros investigadores
(Prosperi et al) demostramos que las células MCF-7 transfectadas con COX-2
(MCF-7/COX-2) son 3 veces más invasivas que las células MCF-7 parentales
(Simeone, Nieves-Alicea et al. 2007). Por otra parte, demostramos que PGE2 e IL-
8 disminuyeron la expresión de PDCD4, pero que la sobreexpresión de PDCD4
inhibió la invasión inducida por COX-2 a través del incremento en los niveles de
TIMP-2 (Nieves-Alicea, Colburn et al. 2009). Nosotros proponemos que la
reducción en la sensibilidad a tamoxifeno y el incremento de la actividad invasiva
inducida por COX-2 podrían contribuir a la reducción del rango de sobrevivencia
en pacientes con tumores de mama positivos para ER que sobreexpresan COX-
2.
Se sabe que COX-2 activa la proteína quinasa C (PKC) (Fiebich,
Schleicher et al. 2001; Timoshenko, Xu et al. 2003; Gerlo, Verdood et al. 2004;
Chang, Ai et al. 2005). En nuestro laboratorio, se mostró que la activación de PKC
incrementó la invasión de las células MCF-7 a través del Matrigel (Simeone,
Nieves-Alicea et al. 2007) y redujo los efectos anti-proliferativos de tamoxifeno en
las células MCF-7 (Tari, Simeone et al. 2005). Sin embargo, no se conoce cuales
son los factores río abajo utilizados por COX-2 y PKC para incrementar la
invasividad y reducir la sensibilidad a tamoxifeno de las células de cáncer de
mama. Altos niveles de COX-2 han sido asociados con la activación de la familia
de la proteína quinasa activada por mitógenos (MAPK) y la quinasa Akt (Guan,
Buckman et al. 1998; Kim and Kim 2004; Ispanovic and Haas 2006; Wu, Luo et al.
2006; Park, Kim et al. 2007; Looby, Abdel-Latif et al. 2009). Pero no se sabe si
estas quinasas regulan la resistencia a tamoxifeno y la actividad invasiva inducida
por COX-2 en las células de cáncer de mama. Además, hemos establecido que
PDCD4 inhibe la invasión inducida por COX-2 de las células MCF-7, pero no se
sabe aún si PDCD4 afecta la resistencia a tamoxifeno inducida por COX-2. En
este estudio reportamos que COX-2 incrementa la actividad de JNK para mediar
invasión, pero no la resistencia a tamoxifeno en las células de cáncer de mama
MCF-7. También reportamos que PDCD4 no bloquea la resistencia a tamoxifeno
inducida por COX-2 en células de cáncer de mama positivas para ER-, a pesar
de que PDCD4 bloquea la invasión inducida por COX-2 en las células de cáncer
de mama MFC-7.

52
3.1. HIPÓTESIS
JNK es esencial para que COX-2 induzca invasión y resistencia a tamoxifeno,
mientras que PDCD4 inhibe la progresión del cáncer de mama al bloquear la
resistencia a tamoxifeno inducida por COX-2.

53
3.2. OBJETIVO GENERAL
Determinar los factores río abajo utilizados por COX-2 y PKC para incrementar
la invasividad y la resistencia a tamoxifeno de las células de cáncer de mama,
así como analizar si PDCD4 inhibe la resistencia a tamoxifeno inducida por
COX-2 en las células de cáncer de mama.
3.3. OBJETIVOS PARTICULARES
Determinar si COX-2 incrementa la activación de ERK 1,2, p38MAPK, Akt y
JNK en las células de cáncer de mama MCF-7.
Dilucidar cual (es) de estas quinasas son esenciales para que COX-2
induzca invasión y resistencia a tamoxifeno en las células de cáncer de
mama MCF-7.
Establecer si PDCD4 inhibe la resistencia a tamoxifeno inducida por COX-2
en las células de cáncer de mama MCF-7.

54
3.4. MATERIAL Y MÉTODOS.
3.4.1. Cultivo celular.
La línea celular de cáncer de mama MCF-7 parental (MCF-7/WT) y la línea celular
MCF-7/COX-2, la cual fue generada a través de la transfección estable del
plásmido que codifica el gen COX-2 en las células MCF-7 fueron adquiridas a
través de la colaboración con la Dra. Ana M Tari del MD Anderson Cancer Center
en la Ciudad de Houston, Texas, EUA. Las células fueron examinadas contra
micoplasma y mantenidas en medio DMEM/F12 (medio de cultivo Eagle
modificado por Dulbecco) (Invitrogen Corporation) suplementado con 5% de suero
fetal bovino (FBS) y 500 g/mL de geneticina (G418) en 5% de CO2 y 95% de
humedad a 37C. En el caso de las células transfectadas, se trataron con, para
mantener solo las células transfectadas. Para confirmar la sobreexpresión de
COX-2 en las células MCF-7/COX-2, se realizó western blot utilizando el
anticuerpo monoclonal anti-COX-2 (Cayman Chemical, Ann Arbor, MI) a una
dilución de 1:1000, y el anticuerpo secundario (anti-ratón) obtenido de Cell
Signaling Technology, Inc. (3 TraskLane Danvers, MA) a una dilución de 1:1000.
3.4.2. Reactivos.
Citrato de tamoxifeno, Gö6976 (inhibidor de proteína quinasa C), SP600125
(inhibidor de JNK) y PD98059 (inhibidor de ERK) se compraron en EMD
Chemicals (San Diego, CA). Las soluciones de trabajo (10 mM) de tamoxifeno,
Gö6976, SP600125 y PD98059 fueron preparadas en DMSO y almacenadas a
-20oC. Todos los reactivos se diluyeron en medio de cultivo a la concentración
final indicada. El Matrigel se obtuvo de BD Biosciences (Bedford, MA). Los
anticuerpos específicos para ERK fosforilado (T202/Y204), p38MAPK fosforilado
(T183/Y185), JNK fosforilado (T183/Y185) y Akt fosforilado (S473), ERK total, JNK total,
p38MAPK total, Akt total y c-Jun se obtuvieron de Cell Signaling Technology
(Danvers, MA). Los anticuerpos específicos para -actina e Histona H3 se
compraron en Sigma-Aldrich Chemical Co. (St. Louis, MO). Los anticuerpos
secundarios anti-ratón y anti-conejo conjugados con peroxidasa de rábano se
compraron de Amersham Life Sciences (Cell Signaling Technology)

55
3.4.3. Western blot
Las células MCF-7/WT y MCF-7/COX-2 fueron sembradas a una concentración
de 4 X 105 células/pozo en una placa de 6 pozos. Dos días después, las células
se colectaron y se aislaron proteínas totales para posteriormente realizar el
western blot como se ha descrito previamente (sección 2.4.5.). La concentración
de proteínas se determinó por el método del ácido bicinconínico empleando el kit
Pierce BCA protein assay (Thermo Scientific). Las muestras se corrieron en geles
de poliacrilamida al 12%, y se transfirieron a membranas de nitrocelulosa (Bio-
Rad). Posteriormente las membranas se bloquearon con leche descremada al 5%
en TBS-Tween 20 1X pH 7.4 por 30 min y en agitación constante. Se realizaron 3
lavados de 10 min cada uno con TBS-Tween 20 1X pH 7.4. Después de los
lavados, cada anticuerpo primario con excepción de -actina se agregó por
separado respectivamente a las membranas correspondientes, a una dilución de
1:1000 durante toda la noche a 4C. Se realizaron lavados nuevamente,
enseguida se añadieron los anticuerpos secundarios correspondientes.
Posteriormente, las proteínas fueron detectadas a través de quimioluminiscencia
(Kirkegaard & Perry Laboratories, Gaithersburg, MD) y analizadas con el
densitómetro Alphalmager (Alpha Innotech Corp., San Leandro, CA). ß-actina fue
utilizada como control de carga, como ya se índico anteriormente (sección 2.4.5).
Para determinar el efecto de la inhibición de PKC en la fosforilación de
JNK, las células MCF-7/COX-2 se sembraron a una densidad de 4 X 105
células/pozo en una placa de 6 pozos. Dos días después, las células se trataron
con el inhibidor Gö6976 a diferentes concentraciones (0, 25, 50 nM) durante 8 h.
En seguida, se colectaron las células tratadas y no tratadas, se aislaron las
proteínas para después realizar western blot como ya se ha descrito (sección
2.4.6.).
3.4.4. Extracción de proteínas nucleares.
Para determinar los niveles de c-Jun en las células MCF-7/WT y MCF-7/COX-2,
se sembraron 4 X 105 células por pozo en placas de 6 pozos. Después se
obtuvieron y lisaron las pastillas celulares con 250 L de buffer de lisis (10 mM
HEPES, 10 mM KCl, 0.5% Nonidet P-40, pH 7.9) en hielo por 15 min. Los núcleos

56
se sedimentaron por centrifugación a 13,000 rpm por 1 min y las proteínas
nucleares fueron extraídas con 50 L de buffer de extracción nuclear (20 mM
HEPES, 400 mM NaCl, pH 7.9). La concentración de proteína nuclear se
determinó con el kit Bio-Rad DC protein assay (Bio-Rad), siguiendo las
instrucciones del fabricante. El western blot se llevó a cabo como se describió
anteriormente, en este caso los geles de poliacrilamida al 12 % se cargaron con
50 g de proteína nuclear. Después de la transferencia se agregó el anticuerpo
primario específico para c-Jun a una dilución de 1:500, seguido por el anticuerpo
secundario anti-conejo a una dilución 1:1000. La Histona H3 fue utilizada como
control de carga. Las membranas se incubaron con el anticuerpo anti-histona H3
(dilución 1:5000) durante 30 min a temperatura ambiente, después de los lavados
se agregó el anticuerpo secundario anti-ratón (dilución 1:5000) por 30 min a
temperatura ambiente.
3.4.5. Ensayo de invasión en Matrigel
El efecto de la inhibición de JNK en la invasión de las células de cáncer de mama
MCF-7/COX-2 fue determinado in vitro a través de ensayos de invasión en
Matrigel. Transwells para placas de 6 pozos, con un filtro de policarbonato y poros
de 8 たm (Fisher Scientific, Middleton, VA), se cubrieron con Matrigel (0.7 mg/mL)
e incubaron a temperatura ambiente por 40 min. Las células MCF-7/COX-2 se
sembraron a una concentración de 4 x 105 células/pozo en 500 L de medio
DMEM al 5% FBS, luego de 24 h se pre-trataron con SP600125 y PD98059 (0, 5
y 10 M) 2 h antes de agregar el Matrigel en los transwells. Posteriormente se
realizó el ensayo de invasión en Matrigel como se indicó previamente (sección
2.4.2). Después de las 72 h de incubación, las células que invadieron a través del
Matrigel fueron fijadas en laminillas, teñidas, contadas y fotografiadas. Se
cuantificaron las células presentes en 9 campos de cada laminilla y por triplicado.
La invasividad de las células MCF-7/COX-2 se expresó como el número de
células que invadieron la parte inferior del filtro.

57
3.4.6. Ensayo de viabilidad celular (MTS)
Para determinar el efecto de la inhibición de JNK en la proliferación de las células
de cáncer de mama MCF-7/COX-2, éstas células se sembraron en una placa de
96 pozos a una densidad de 1000 células por pozo en presencia y ausencia de
Matrigel en 0.1 mL de DMEM/F12 suplementado con 5% FBS. Al día siguiente,
las células fueron tratadas con varias concentraciones de SP600125 por 3 días.
Después de esta incubación, se determinó la proliferación celular mediante MTS
(Madison, WI) previamente descrito (sección 2.4.6.).
Para determinar la sensibilidad de las células MCF-7/COX-2 a tamoxifeno,
las células se sembraron en placas de 96 pozos a una densidad de 1000 células
en 100 µL de medio DMEM/F12 suplementado con 5% de FBS por pozo y se
incubaron durante toda la noche. Al día siguiente, se reemplazó el medio por
DMEM/F12 suplementado con 5% de CSS (por sus siglas en inglés charcoal
stripped serum). Después de 24 h, las células se pre-trataron con SP600125 (0, 5
y 10 M) y luego de 4 h fueron tratadas con diferentes concentraciones de
tamoxifeno (0, 0.1, 1, 5 y 9 M) por 5 días. Al final de esta incubación, se
determinó la proliferación celular a través del ensayo MTS. Los resultados se
expresaron en términos de porcentaje de inhibición del crecimiento con respecto
al control (células no tratadas).

58
3.5. RESULTADOS.
3.5.1. La sobreexpresión de COX-2 incrementa los niveles de JNK
fosforilado.
Altos niveles de COX-2 han sido asociados con la activación de la familia de la
proteína quinasa activada por mitógenos (MAPK) y la quinasa Akt (Guan,
Buckman et al. 1998; Kim and Kim 2004; Ispanovic and Haas 2006; Looby, Abdel-
Latif et al. 2009). En este estudio, nosotros tratamos de determinar si MAPK y/o
Akt son utilizados por COX-2 para inducir resistencia a tamoxifeno e invasión en
las células de cáncer de mama. Para ello, primeramente determinamos si la
sobreexpresión de COX-2 altera los niveles de fosforilación de proteínas de la
familia MAPK ó la de Akt. Los niveles de fosforilación de ERK, p38MAPK y Akt
fueron muy similares entre las células MCF-7/COX-2 y MCF-7/WT (Figura 14).
Sin embargo, observamos un aumento en la fosforilación de JNK en las células
MCF-7/COX-2 comparado con las células MCF-7/WT (Figura 15A). Esto
corresponde con los altos niveles de c-Jun encontrados en las células MCF-
7/COX-2 en comparación con las células MCF-7/WT (Figura 15B). Por otra parte,
se sabe que COX-2 activa a PKC (Fiebich, Schleicher et al. 2001; Timoshenko,
Xu et al. 2003; Gerlo, Verdood et al. 2004; Chang, Ai et al. 2005). Para determinar
si JNK esta río abajo de PKC en la vía mediada por COX-2/PGE2/PKC para
inducir resistencia a tamoxifeno e invasión celular, tratamos a las células MCF-
7/COX-2 con el inhibidor de PKC Gö6976 (0, 25 y 50 nM) durante 8 h.
Posteriormente, se analizaron los niveles de JNK fosforilado mediante western
blot. La inhibición de PKC resultó en la disminución de JNK fosforilado en las
células MCF-7/COX-2 (Figura 15C), sugiriendo que PKC regula la activación de
JNK inducida por COX-2.

59
Figura 14. COX-2 no induce cambios sobre la activación de ERK1/2, p38MAPK y Akt
en células MCF-7. En las células MCF-7/COX-2 y MCF-7/WT se determinaron los
niveles de fosforilación de ERK (A), p38MAPK (B) y Akt (C).

60
Figura 15. COX-2 incrementa la fosforilación de JNK y los niveles de c-Jun en las
células de cáncer de mama MCF-7. Mediante western blot se determinaron los niveles
de fosforilación de JNK (A) y los niveles nucleares de c-Jun (B) en las células MCF-
7/COX-2 y en las células MCF-7/WT. La inhibición de PKC disminuye la fosforilación de
JNK en las células MCF-7/COX-2 (C).

61
3.5.2. La inhibición de JNK disminuye la invasión de las células MCF-7/COX2
Las MAPKs se encuentran implicadas en el control de procesos angiogénicos
(migración, proliferación) a través de ERK y JNK los cuales están específicamente
implicados en la producción y activación de MMP-2 (Ispanovic and Haas 2006).
Para determinar si JNK es esencial para que COX-2 induzca invasión en las
células de cáncer de mama, las células MCF-7/COX-2 fueron tratadas con
SP600125 (inhibidor químico contra JNK) y debido a que no pudimos observar
cambios sobre la activación de ERK1/2 inducidos por COX-2, decidimos usar
como control negativo en los ensayos de invasión en Matrigel, las células tratadas
con el inhibidor de PD98059 (inhibidor químico contra ERK). La inhibición de JNK
redujo la invasión de las células MCF-7/COX-2 (Figura 16A). A concentraciones
de 5 y 10 M, el inhibidor SP600125 disminuyó la actividad invasiva de las células
MCF-7/COX-2 aproximadamente un 75% y 80% respectivamente. Sin embargo y
como se esperaba, bajo las mismas condiciones, el inhibidor PD98059 no afectó
la actividad invasiva de las células MCF-7/COX-2 (Figura 16B). Estos resultados
indican que JNK regula la invasión inducida por COX-2 en las células de cáncer
de mama.
Sin embargo, debido a que JNK ha sido asociado con proliferación celular
(Xu, Heidenreich et al. 1996; Du, Lyle et al. 2004), se determinó el efecto de la
inhibición de JNK en la proliferación de las células de cáncer de mama en
ausencia o en presencia de Matrigel (17) para simular las condiciones usadas en
el ensayo de invasión. Las dosis de 5 y 10 M del inhibidor de JNK, indujeron una
inhibición de la proliferación en las células MCF-7/COX-2 del 23.4% y 21.8%
respectivamente en presencia de Matrigel. Mientras que en ausencia de Matrigel
se observó una inhibición de la proliferación del 20.2% y 16.3% en las dosis de 5
y 10 M del inhibidor de JNK respectivamente. Estos resultados sugieren, que
JNK no influye en la proliferación celular. Por lo tanto, la disminución observada
en los ensayos de invasión en Matrigel es debida a la inhibición del proceso de
invasión celular regulada por JNK y no por causa de la inhibición de la
proliferación celular.
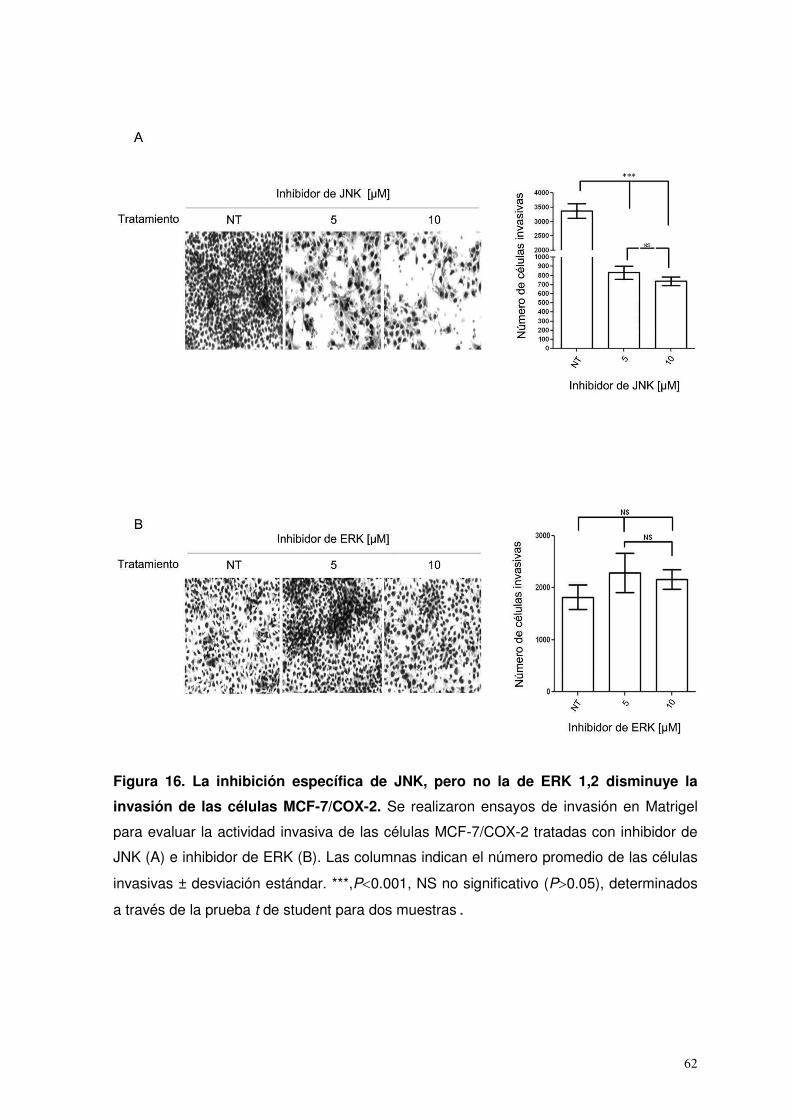
62
Figura 16. La inhibición específica de JNK, pero no la de ERK 1,2 disminuye la
invasión de las células MCF-7/COX-2. Se realizaron ensayos de invasión en Matrigel
para evaluar la actividad invasiva de las células MCF-7/COX-2 tratadas con inhibidor de
JNK (A) e inhibidor de ERK (B). Las columnas indican el número promedio de las células
invasivas ± desviación estándar. ***,P0.001, NS no significativo (P0.05), determinados
a través de la prueba t de student para dos muestras.

63
Figura 17. Efecto de la inhibición de JNK en la proliferación de las células de
cáncer de mama en ausencia o en presencia de Matrigel. La inhibición de JNK no
afectó la proliferación de las células MCF-7/COX-2 cultivadas en medio con y sin
Matrigel.

64
3.5.3. La inhibición de JNK no incrementa la sensibilidad de las células MCF-
7/COX-2 a tamoxifeno.
Previamente en nuestro laboratorio se reportó que las células MCF-7/COX-2 son
altamente resistentes a tamoxifeno (Tari, Simeone et al. 2005). Por lo que en este
estudio determinamos si JNK es además esencial para la resistencia a tamoxifeno
inducida por COX-2. A una concentración de 5 M, tamoxifeno inhibió el
crecimiento de las células MCF-7/COX2 solo un 20% (Figura 18). En presencia de
5 y 10 M de SP600125 y 5 M de tamoxifeno, la inhibición del crecimiento de las
células MCF-7/COX-2 fue del 30% y 40% respectivamente (Figura 18) (P0.05).
Estos resultados indican que JNK no regula la resistencia a tamoxifeno inducida
por COX-2.
Figura 18. La inhibición de JNK no afecto la resistencia de las células MCF-7/COX-2
a tamoxifeno. Se llevaron a cabo ensayos de viabilidad celular MTS para evaluar el
efecto de SP600125 en la sensibilidad a tamoxifeno de las células MCF-7/COX-2.

65
3.5.4. PDCD4 no inhibe la resistencia a tamoxifeno inducida por PGE2
La Prostaglandina E2 (PGE2) es una de las prostaglandinas más secretadas en
tumores y se cree que es el principal producto tumorigénico derivado de COX-2
(Chen and Smyth 2011). Previamente, en nuestro laboratorio se demostró que
COX-2 disminuye el efecto del tamoxifeno a través de PGE2 en las células de
cáncer de mama (Tari, Simeone et al. 2005). Por otra parte, se ha correlacionado
al incremento en los niveles de PDCD4 con un incremento en la sensibilidad de
las células MCF-7 a antiestrógenos tales como tamoxifeno (Jansen, Camalier et
al. 2004). Para determinar si PDCD4 puede inhibir la resistencia a tamoxifeno
inducida por COX-2 en las células MCF-7, llevamos a cabo ensayos de MTS. Las
células MCF-7/Vector y las MCF-7/PDCD4 se sembraron y trataron con 0, 1 y 10
M de PGE2 por 24 h, después se trataron con diferentes concentraciones de
tamoxifeno (0, 0.1, 1, 5 y 9 M) durante 5 días. Las células MCF-7/PDCD4 son
más sensibles al tamoxifeno que las células MCF-7/Vector (Figura 19A). En
presencia de 1 y 10 M de PGE2 (Figura 19B y 19C) las células MCF-7/Vector y
MCF-7/PDCD4 llegaron a ser más resistentes al tamoxifeno, sugiriendo que la
sobreexpresión de PDCD4 no afecta la resistencia a tamoxifeno inducida por
COX-2/PGE2.

66
Figura 19. PDCD4 no inhibe la resistencia a tamoxifeno inducida por COX-2/PGE2.
Las células MCF-7/Vector y MCF-7/PDCD4 se pre-trataron con 0(A), 1(B) y 10 (C) M de
PGE2 por 24 h. Posteriormente, se trataron con 0,0.1, 1,5 y 9 M de tamoxifeno durante 5
días. Finalmente, se realizaron los ensayos de viabilidad celular MTS.
PGE2 0 M
PGE2 1 M
PGE2 10 M

67
3.6. DISCUSIÓN Y CONCLUSIÓN.
El cáncer de mama es el tipo de cáncer más frecuentemente diagnosticado en
mujeres y la metástasis su principal causa de mortalidad. La naturaleza agresiva
de las células cancerosas es determinada por vías de señalización complejas que
regulan funciones clave, incluyendo proliferación celular, sobrevivencia,
resistencia a drogas, migración e invasión. La expresión de COX-2 en pacientes
con cáncer de mama es un marcador de una baja sobrevivencia global y libre de
enfermedad (Ristimaki, Sivula et al. 2002; Denkert, Winzer et al. 2004; O'Connor,
Avent et al. 2004) y ha sido considerado como un marcador de alto potencial
metastásico. COX-2 se sobreexpresa en aproximadamente el 40% de los tumores
de mama (Soslow, Dannenberg et al. 2000; Ristimaki, Sivula et al. 2002; Denkert,
Winzer et al. 2004; Singh, Berry et al. 2005). La relevancia de la expresión de
COX-2 en cáncer también se desprende de los estudios clínicos que indican que
la presencia de COX-2 se correlaciona, en ciertos tumores con un fenotipo más
agresivo (incluyendo una desregulación del crecimiento y proliferación celular, un
incremento en la habilidad para escapar de la apoptosis, neovascularización
sostenida, incremento de invasión, metástasis y la habilidad para evadir el
sistema inmune) y, sobre todo, con un bajo pronóstico clínico (Rizzo 2011).
Estudios recientes han relacionado la expresión de COX-2 con la
activación de la vía de las MAPK y Akt (Guan, Buckman et al. 1998; Kim and Kim
2004; Ispanovic and Haas 2006; Wu, Luo et al. 2006; Park, Kim et al. 2007;
Looby, Abdel-Latif et al. 2009). La familia de las MAPK incluye tres subfamilias:
ERK, p38 y JNK. Recientemente, se ha demostrado que la activación de ERK,
p38 y JNK promueve migración celular en cáncer de mama (You, Mi et al. 2009).
En este estudio, nosotros observamos que COX-2 incrementó la activación de
JNK; esta activación se confirmó con los niveles nucleares de c-Jun encontrados
en las células MCF-7/COX-2. Por otro lado, se sabe que COX-2 activa las vías de
PKA y PKC (Fiebich, Schleicher et al. 2001; Timoshenko, Xu et al. 2003; Gerlo,
Verdood et al. 2004). Previamente en nuestro laboratorio se reportó que la
activación de PKC, mas no la de PKA, estaba involucrada en la invasión inducida
por COX-2 en las células de cáncer de mama MCF-7 (Simeone, Nieves-Alicea et
al. 2007). Aquí, nosotros encontramos que la activación de JNK en las células

68
MCF-7/COX-2 es regulada por PKC, sugiriendo que en la vía que hemos
propuesto (COX-2/PGE2/PKC), JNK se encuentra río abajo de PKC. Este
resultado es consistente con reportes previos como el de Lopez-Bergami et al,
que mostraron que PKC contribuye al incremento de la activación de JNK en las
células SW1 (células de melanoma de ratón) y HEK 293T (células embrionarias
humanas de riñón 293), además revelaron que la inhibición de PKC redujo la
fosforilación de JNK, disminuyendo la respuesta pro-apoptótica después de
radiación ultravioleta (Lopez-Bergami and Ronai 2008). En el 2002 Chen et al,
demostraron que la activación de JNK es mediada a través de la vía de PKC, lo
cual se asocia con la inducción de apoptosis por TPA (Chen, Wu et al. 2002). En
estos reportes los autores correlacionan la activación de JNK a través de PKC con
la actividad apoptótica de JNK. En nuestro trabajo, correlacionamos la activación
de JNK a través de PKC con la actividad invasiva de JNK.
Las MAPK están implicadas en el control de procesos angiogénicos,
específicamente a través de la activación de ERK1,2 y JNK, los cuales están
involucrados en la producción y activación de MMP-2 (Ispanovic and Haas 2006).
En el 2009, You et al, describió una vía para el mantenimiento de la migración de
las células de cáncer de mama MDA-MB-231, en la cual metabolitos del ácido
araquidónico (AA) son capaces de activar PKC que sirve como un activador río
arriba de ERK 1,2, que a su vez activa la vía de señalización de -catenina, ciclina
D1 y survivina lo cual contribuye a la proliferación celular y migración (You, Mi et
al. 2009). En contraste con este estudio, nuestros resultados indican que ERK1,2
no juega un papel en la invasión inducida por COX-2 en las células de cáncer de
mama MCF-7 y que es solo a través de la activación de JNK que COX-2 regula la
invasión en éstas células. Por lo tanto, sugerimos que JNK es el factor río abajo
que es utilizado por la vía COX-2/PGE2/PKC para inducir invasión en las células
de cáncer de mama.
El tamoxifeno es el fármaco más ampliamente utilizado en el tratamiento
del cáncer de mama (Dorssers, van der Flier et al. 2001). Desafortunadamente,
cerca de la mitad de los pacientes con cáncer de mama positivo para ER no
responden al tamoxifeno y muchos de los pacientes que responden adquieren
resistencia al tamoxifeno, permitiendo así la progresión de la enfermedad. Son

69
muchos los mecanismos que podrían contribuir al desarrollo de este fenotipo de
resistencia, estos incluyen: pérdida del ER en el tumor, farmacología del
tamoxifeno, alteraciones en la estructura y función del ER o la activación de vías
tales como JNK y COX-2. Anteriormente, se ha encontrado un incremento
sustancial en la actividad de JNK en tumores de cáncer de mama resistentes a
tamoxifeno. En 1999, Johnston et al, encontraron que la regulación positiva tanto
de la unión de AP-1 al DNA y de la actividad de JNK ocurre en tumores de mama
positivos para ER, que han adquirido resistencia a tamoxifeno (Johnston, Lu et al.
1999). En el 2000, Shiff et al, demostraron que el incremento en la actividad de
JNK y fosforilación de c-Jun están involucrados con el fenotipo de resistencia a
tamoxifeno (Schiff, Reddy et al. 2000). Sin embargo, en el presente trabajo,
observamos que JNK no regula la resistencia a tamoxifeno inducida por COX-2,
sugiriendo que COX-2 regula la activación de JNK para inducir invasión pero no
para incrementar la resistencia a tamoxifeno. Por lo tanto, la resistencia a
tamoxifeno a través de la sobreexpresión de COX-2 pudiera ser causado a través
de la activación de las vías de Akt (Kirkegaard, Witton et al. 2005) o ERK1,2 (Li,
Wang et al. 2012), las cuales se han sido asociadas también a la resistencia a
tamoxifeno en células de cáncer mama.
Se sabe que la sobreexpresión de COX-2, incrementa la producción de
PGs, tales como PGE2 (Chen and Smyth 2011). Anteriormente, nuestro grupo
demostró que COX-2 inhibe el efecto del tamoxifeno en las células de cáncer de
mama (Tari, Simeone et al. 2005). Además se observó que el efecto de COX-2
sobre la resistencia a tamoxifeno estaba mediado por PGE2. Por otra parte, se ha
reportado que altos niveles de PDCD4 están correlacionados y específicamente
contribuyen con la actividad anti-tumoral del tamoxifeno en las células MCF-7
(Jansen, Camalier et al. 2004). Consistente con este reporte, nosotros
encontramos que la sobreexpresión de PDCD4 incrementa la sensibilidad al
tamoxifeno en las células de cáncer de mama MCF-7. Sin embargo, en presencia
de PGE2, la sobreexpresión de PDCD4 no mantiene o incrementa la sensibilidad
al tamoxifeno en las células MCF-7.

70
En conclusión, en este estudio, encontramos que COX-2 incrementa la
actividad de JNK para regular la invasión, pero no la resistencia a tamoxifeno en
las células de cáncer de mama MCF-7. Además aunque PDCD4 inhibe la invasión
inducida por COX-2, PDCD4 no inhibe la resistencia a tamoxifeno inducida por
COX-2 en las células de cáncer de mama, positivas para ER MCF-7 (Figura 20).

71
CONCLUSIÓN GENERAL.
Se encontró que leptina incrementa la resistencia a tamoxifeno en las células
MCF-7, y que la sobreexpresión de PDCD4 incrementa la transcripción de TIMP-
2, permitiendo la inhibición de la invasión inducida por leptina, al bloquear la
activación de ERK 1,2 y STAT3. También se reportó que la sobreexpresión de
PDCD4 inhibe la resistencia a tamoxifeno inducida por leptina en las células MCF-
7. Además en este trabajo, se encontró que PKC induce la activación de JNK 1/2
con la subsecuente activación de c-Jun para regular la invasión de las células
MCF-7. También se observó, que aunque PDCD4 inhibe la invasión inducida por
COX-2, PDCD4 no inhibe la resistencia a tamoxifeno inducida por COX-2 en las
células de cáncer de mama, positivas para ER MCF-7 (como se muestra en el
siguiente modelo esquemático).

72
Figura 20. Modelo esquemático del efecto de PDCD4 sobre la invasión y resistencia
a tamoxifeno inducidas por leptina y COX-2 en las células de cáncer de mama
MCF-7.
Induce
TIMP-2TIMP-2
IncrementaLeptinaLeptina
Incrementa
STAT3STAT3ERK
1,2
ERK
1,2
InvasiónInvasión
Induce
COX-2
Resistencia a
tamoxifeno
Resistencia a
tamoxifeno
Incrementa
PKC
JNK1/2
activa
Invasión
Induce
Incrementa
PGE2PGE2
Resistencia a
tamoxifeno
Resistencia a
tamoxifeno
Induce
PKCPKC
PGE2
activa activa
c-Jun
activa
Induce
Induce
TIMP-2TIMP-2
IncrementaLeptinaLeptina
Incrementa
STAT3STAT3ERK
1,2
ERK
1,2
InvasiónInvasión
Induce
COX-2
Resistencia a
tamoxifeno
Resistencia a
tamoxifeno
Incrementa
PKC
JNK1/2
activa
Invasión
Induce
Incrementa
PGE2PGE2
Resistencia a
tamoxifeno
Resistencia a
tamoxifeno
Induce
PKCPKC
PGE2
activa activa
c-Jun
activa
Induce
TIMP-2TIMP-2
IncrementaLeptinaLeptina
Incrementa
STAT3STAT3ERK
1,2
ERK
1,2
InvasiónInvasión
Induce
COX-2COX-2
Resistencia a
tamoxifeno
Resistencia a
tamoxifeno
Incrementa
PKCPKC
JNK1/2JNK1/2
activa
InvasiónInvasión
Induce
Incrementa
PGE2PGE2
Resistencia a
tamoxifeno
Resistencia a
tamoxifeno
Induce
PKCPKC
PGE2PGE2
activa activa
c-Junc-Jun
activa
Induce
Inhibicion
No Inhibicion
Inhibicion
No Inhibicion

73
APÉNDICE
Soluciones utilizadas para el ensayo de invasión.
Matrigel 0.7mg/mL
Medio sin suero (DMEM/F12) 3 mL
Matrigel 200 µL
Leptina 1mg/mL
Leptina 1mg
H2Odd 1mL
Soluciones usadas para RT-PCR
Preparación de la muestra
Buffer de muestra 6X 3.3L
Muestra 20 L
Preparación del marcador de peso molecular de DNA
Buffer de muestra 6X 1 L
Marcador de peso molecular de DNA 2L
Buffer de corrida ó TBE 5X (solución stock)
Tris-Base 54 g
Ácido Bórico 27.5 g
EDTA 0.5M pH8.0 20 mL
Buffer de corrida ó TBE 1X
Buffer de corrida TBE 5X 100 mL
H2Odd 400 mL
Gel de agarosa al 2%
Agarosa 0.8 g

74
TBE 1X 40 mL
Bromuro de etidio* 2 L
* 5l de bromuro de etidio por cada 100 mL de agarosa
Soluciones para la preparación de las muestras para Western blot.
PBS 10X (solución stock)
NaCl 80 g
KCl 2 g
KH2PO4 2 g
Ajustar el pH a 7.4 y aforar a 1 L con H2Odd
PBS 1X con inhibidores de fosfatasas
PBS 10X 5 mL
H2Odd 45 mL
NaF 1M 50 L
Na3VO4 1M 50 L
Buffer de lisis
NaCl 2.19 g
Tris 25 mM 0.76 g
Triton X-100 1%
H2Odd 200 mL
Ajustar el pH a 7.6 y aforar a 250 mL con H2Odd
Buffer de lisis con inhibidores de proteasas y fosfatasas
Buffer de lisis 1 mL
NaF 1M 4L
Na3VO4 1M 4L
Inhibidor de proteasas* 10L
* Agregar 10 l de inhibidor de proteasas/mL

75
Soluciones para la preparación de geles de poliacrilamida SDS-PAGE
Acrilamida/bisacrilamida 30%
Acrilamida 30 g
Bisacrilamida 1g
Aforar a 100 mL con H2Odd
Buffer Tris-HCl 1 M pH 6.8
Tris-HCl 60.5 g
H2Odd 800 mL
Ajustar el pH a 6.8 y Aforar a 1 L con H2Odd
Buffer Tris-Base 1.5 M pH 8.8
Tris-Base 182 g
H2Odd 800 mL
Ajustar el pH a 8.8 y Aforar a 1 L con H2Odd
PSA 10%
PSA 1 g
H2Odd 10 mL
SDS 10%
SDS 10 g
Aforar a 100 mL con H2Odd
Solución para gel concentrador de poliacrilamida
H2Odd 1.4 mL
Acrilamida/Bisacrilamida 30% 0.33 mL
Tris 0.5 M pH 6.8 0.25 mL
SDS 10% 0.02 mL
PSA 10% 0.02 mL
TEMED 0.002 mL

76
Solución para gel separador al 10% de poliacrilamida.
H2Odd 4 mL
Acrilamida/Bisacrilamida 30% 3.3 mL
Tris 1.5 M pH 8.8 2.5 mL
SDS 10% 0.1 mL
PSA 10% 0.1 mL
TEMED 0.004 mL
Soluciones para el corrimiento electroforético de proteínas en geles de
poliacrilamida.
Buffer de corrimiento (Tris-Glicina) 10X
Tris-Base 30 g
Glicina 144 g
H2Odd 1 L
Buffer de corrimiento (Tris-Glicina) 1X
Buffer de Corrida 10X 50 mL
H2Odd 450 mL
SDS 10% 5 mL
Buffer de muestra 1X
SDS 30% 1.2 mL
ß-mercaptoetanol 0.9 mL
Glicerol 1.8 mL
Azul de Bromofenol 2.5% 0.18 mL
Tris-HCl 0.5 M pH 6.8 1.1 mL
H2Odd 0.82 mL
Soluciones para la inmunoelectrotransferencia
Buffer de Transferencia (Tris-glicina) 10X
Tris-Base 30 g
Glicina 144 g
H2Odd 1 L

77
Buffer de transferencia (Tris-glicina) 1X
Buffer de transferencia 10X 100 mL
Metanol 200 mL
H2Odd 700 mL
Buffer TBS 10X (solución stock)
Tris-HCl 31.5 g
NaCl 80 g
Ajustar el pH a 7.6 y aforar a 1 L con H2Odd
Buffer TBS-Tween 20 1X (TBS-T)
Buffer TBS 10X 100 mL
Tween-20 1 mL
H2Odd 900 mL
Leche descremada al 5%
Leche descremada 5 g
Buffer TBS-T 1X 100 mL
Rojo de Ponceau-S (solución stock)
Rojo de Ponceau-S 2 g
Ácido tricloroacético 30 g
Ácido sulfosalicílico 30 g
Aforar a 100 mL con H2Odd
Rojo de Ponceau-S (solución de trabajo)
Rojo de Ponceau-S stock 10 mL
H2Odd 90 mL
Soluciones utilizadas para el ensayo de MTS.
MTS (Solución stock)
MTS 42 mg

78
D-PBS 1X sin calcio y magnesio 21 mL
Ajustar el pH a 6.5, cubrirlo con papel aluminio, filtrarlo y conservarlo a -20 C
PMS (Solución stock)
PMS 0.92 mg
D-PBS 1X sin calcio y magnesio 1 mL
Cubrirlo con papel aluminio, filtrarlo y conservarlo a -20 C
MTS/PMS (Solución de trabajo)
MTS 2 mL
PMS 100 L
Soluciones utilizadas para la extracción nuclear de proteínas
Buffer de lisis
HEPES 10 mM
KCl 10 mM
Nonidet P-40 0.5%
Ajustar el pH a 7.9
Buffer de extracción nuclear
HEPES 20 mM
NaCl 400 mM
Ajustar el pH a 7.9

79
LITERATURA CITADA
Ahn, S. M., S. J. Jeong, et al. (2004). "Retroviral delivery of TIMP-2 inhibits H-ras-induced migration and invasion in MCF10A human breast epithelial cells." Cancer Lett 207(1): 49-57.
Badawi, A. F. and M. Z. Badr (2003). "Expression of cyclooxygenase-2 and peroxisome proliferator-activated receptor-gamma and levels of prostaglandin E-2 and 15-deoxy-Delta(12.14)-prostaglandin J(2) in human breast cancer and metastasis." International Journal of Cancer 103(1): 84-90.
Bernardo, M. M. and R. Fridman (2003). "TIMP-2 (tissue inhibitor of metalloproteinase-2) regulates MMP-2 (matrix metalloproteinase-2) activity in the extracellular environment after pro-MMP-2 activation by MT1 (membrane type 1)-MMP." Biochem J 374(Pt 3): 739-745.
Bitomsky, N., M. Bohm, et al. (2004). "Transformation suppressor protein Pdcd4 interferes with JNK-mediated phosphorylation of c-Jun and recruitment of the coactivator p300 by c-Jun." Oncogene 23(45): 7484-7493.
Bjorbaek, C., K. El-Haschimi, et al. (1999). "The role of SOCS-3 in leptin signaling and leptin resistance." J Biol Chem 274(42): 30059-30065.
Bjorbaek, C., S. Uotani, et al. (1997). "Divergent signaling capacities of the long and short isoforms of the leptin receptor." J Biol Chem 272(51): 32686-32695.
Caldefie-Chezet, F., M. Damez, et al. (2005). "Leptin: a proliferative factor for breast cancer? Study on human ductal carcinoma." Biochem Biophys Res Commun 334(3): 737-741.
Calle, E. E., C. Rodriguez, et al. (2003). "Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults." N Engl J Med 348(17): 1625-1638.
Catalano, S., S. Marsico, et al. (2003). "Leptin enhances, via AP-1, expression of aromatase in the MCF-7 cell line." J Biol Chem 278(31): 28668-28676.
Catalano, S., L. Mauro, et al. (2004). "Leptin induces, via ERK1/ERK2 signal, functional activation of estrogen receptor alpha in MCF-7 cells." J Biol Chem 279(19): 19908-19915.
Chambers, A. F., A. C. Groom, et al. (2002). "Dissemination and growth of cancer cells in metastatic sites." Nat Rev Cancer 2(8): 563-572.
Chandrasekharan, N. V. and D. L. Simmons (2004). "The cyclooxygenases." Genome Biology 5(9).
Chang, J. H., Y. H. Cho, et al. (2009). "Crystal structure of the eIF4A-PDCD4 complex." Proc Natl Acad Sci U S A 106(9): 3148-3153.
Chang, S. H., Y. Ai, et al. (2005). "The prostaglandin E2 receptor EP2 is required for cyclooxygenase 2-mediated mammary hyperplasia." Cancer Res 65(11): 4496-4499.
Chen, C., Y. C. Chang, et al. (2006). "Leptin-induced growth of human ZR-75-1 breast cancer cells is associated with up-regulation of cyclin D1 and c-Myc and down-regulation of tumor suppressor p53 and p21WAF1/CIP1." Breast Cancer Res Treat 98(2): 121-132.

80
Chen, E. P. and E. M. Smyth (2011). "COX-2 and PGE2-dependent immunomodulation in breast cancer." Prostaglandins Other Lipid Mediat 96(1-4): 14-20.
Chen, Y., Q. Wu, et al. (2002). "Activation of JNK by TPA promotes apoptosis via PKC pathway in gastric cancer cells." World J Gastroenterol 8(6): 1014-1018.
Cirillo, D., A. M. Rachiglio, et al. (2008). "Leptin signaling in breast cancer: an overview." J Cell Biochem 105(4): 956-964.
Cleary, M. P., S. C. Juneja, et al. (2004). "Leptin receptor-deficient MMTV-TGF-alpha/Lepr(db)Lepr(db) female mice do not develop oncogene-induced mammary tumors." Exp Biol Med (Maywood) 229(2): 182-193.
Cmarik, J. L., H. Min, et al. (1999). "Differentially expressed protein Pdcd4 inhibits tumor promoter-induced neoplastic transformation." Proc Natl Acad Sci U S A 96(24): 14037-14042.
Connolly, E. M., J. H. Harmey, et al. (2002). "Cyclo-oxygenase inhibition reduces tumour growth and metastasis in an orthotopic model of breast cancer." Br J Cancer 87(2): 231-237.
Costa, C., R. Soares, et al. (2002). "Cyclo-oxygenase 2 expression is associated with angiogenesis and lymph node metastasis in human breast cancer." J Clin Pathol 55(6): 429-434.
Denkert, C., K. J. Winzer, et al. (2004). "Prognostic impact of cyclooxygenase-2 in breast cancer." Clin Breast Cancer 4(6): 428-433.
Denkert, C., K. J. Winzer, et al. (2003). "Elevated expression of cyclooxygenase-2 is a negative prognostic factor for disease free survival and overall survival in patients with breast carcinoma." Cancer 97(12): 2978-2987.
Dhanasekaran, D. N. and E. P. Reddy (2008). "JNK signaling in apoptosis." Oncogene 27(48): 6245-6251.
Dorssers, L. C. J., S. van der Flier, et al. (2001). "Tamoxifen resistance in breast cancer - Elucidating mechanisms." Drugs 61(12): 1721-1733.
Duffy, M. J., T. M. Maguire, et al. (2000). "Metalloproteinases: role in breast carcinogenesis, invasion and metastasis." Breast Cancer Res 2(4): 252-257.
Eberhart, C. E., R. J. Coffey, et al. (1994). "Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas." Gastroenterology 107(4): 1183-1188.
Fiebich, B. L., S. Schleicher, et al. (2001). "Mechanisms of prostaglandin E2-induced interleukin-6 release in astrocytes: possible involvement of EP4-like receptors, p38 mitogen-activated protein kinase and protein kinase C." J Neurochem 79(5): 950-958.
Filipovic, A., G. Giamas, et al. (2011). "The potential role of cyclooxygenase-2 (COX-2) during early breast cancer therapy." Ann Oncol 22(8): 1700-1702.
Garofalo, C., M. Koda, et al. (2006). "Increased expression of leptin and the leptin receptor as a marker of breast cancer progression: possible role of obesity-related stimuli." Clin Cancer Res 12(5): 1447-1453.
Garofalo, C., D. Sisci, et al. (2004). "Leptin interferes with the effects of the antiestrogen ICI 182,780 in MCF-7 breast cancer cells." Clin Cancer Res 10(19): 6466-6475.
Garofalo, C. and E. Surmacz (2006). "Leptin and cancer." J Cell Physiol 207(1): 12-22.

81
Gerlo, S., P. Verdood, et al. (2004). "Mechanism of prostaglandin (PG)E2-induced prolactin expression in human T cells: cooperation of two PGE2 receptor subtypes, E-prostanoid (EP) 3 and EP4, via calcium- and cyclic adenosine 5'-monophosphate-mediated signaling pathways." J Immunol 173(10): 5952-5962.
Giuffrida, D., L. Lupo, et al. (1992). "Relation between steroid receptor status and body weight in breast cancer patients." Eur J Cancer 28(1): 112-115.
Gonzalez, R. R., S. Cherfils, et al. (2006). "Leptin signaling promotes the growth of mammary tumors and increases the expression of vascular endothelial growth factor (VEGF) and its receptor type two (VEGF-R2)." J Biol Chem 281(36): 26320-26328.
Guan, Z., S. Y. Buckman, et al. (1998). "Interleukin-1beta-induced cyclooxygenase-2 expression requires activation of both c-Jun NH2-terminal kinase and p38 MAPK signal pathways in rat renal mesangial cells." J Biol Chem 273(44): 28670-28676.
Heasley, L. E. and S. Y. Han (2006). "JNK regulation of oncogenesis." Mol Cells 21(2): 167-173.
Howe, L. R. (2007). "Inflammation and breast cancer. Cyclooxygenase/prostaglandin signaling and breast cancer." Breast Cancer Res 9(4): 210.
Hu, X., S. C. Juneja, et al. (2002). "Leptin--a growth factor in normal and malignant breast cells and for normal mammary gland development." J Natl Cancer Inst 94(22): 1704-1711.
Huang, W. Y., B. Newman, et al. (2000). "Hormone-related factors and risk of breast cancer in relation to estrogen receptor and progesterone receptor status." Am J Epidemiol 151(7): 703-714.
Ishikawa, M., J. Kitayama, et al. (2004). "Enhanced expression of leptin and leptin receptor (OB-R) in human breast cancer." Clin Cancer Res 10(13): 4325-4331.
Ispanovic, E. and T. L. Haas (2006). "JNK and PI3K differentially regulate MMP-2 and MT1-MMP mRNA and protein in response to actin cytoskeleton reorganization in endothelial cells." Am J Physiol Cell Physiol 291(4): C579-588.
Jansen, A. P., C. E. Camalier, et al. (2005). "Epidermal expression of the translation inhibitor programmed cell death 4 suppresses tumorigenesis." Cancer Res 65(14): 6034-6041.
Jansen, A. P., C. E. Camalier, et al. (2004). "Characterization of programmed cell death 4 in multiple human cancers reveals a novel enhancer of drug sensitivity." Mol Cancer Ther 3(2): 103-110.
Jiang, H., J. Yu, et al. (2008). "Upregulation of survivin by leptin/STAT3 signaling in MCF-7 cells." Biochem Biophys Res Commun 368(1): 1-5.
Johnston, S. R., B. Lu, et al. (1999). "Increased activator protein-1 DNA binding and c-Jun NH2-terminal kinase activity in human breast tumors with acquired tamoxifen resistance." Clin Cancer Res 5(2): 251-256.
Kim, H. J. and T. Y. Kim (2004). "IGF-II-mediated COX-2 gene expression in human keratinocytes through extracellular signal-regulated kinase pathway." Journal of Investigative Dermatology 123(3): 547-555.
Kim, J. H., V. Bossuyt, et al. (2005). "Cyclooxygenase-2 expression in postmastectomy chest wall relapse." Clin Cancer Res 11(14): 5199-5205.

82
Kirkegaard, T., C. J. Witton, et al. (2005). "AKT activation predicts outcome in breast cancer patients treated with tamoxifen." J Pathol 207(2): 139-146.
Kirschenbaum, A., X. Liu, et al. (2001). "The role of cyclooxygenase-2 in prostate cancer." Urology 58(2 Suppl 1): 127-131.
Lang, K. and J. Ratke (2009). "Leptin and Adiponectin: new players in the field of tumor cell and leukocyte migration." Cell Commun Signal 7: 27.
Lankat-Buttgereit, B. and R. Goke (2003). "Programmed cell death protein 4 (pdcd4): a novel target for antineoplastic therapy?" Biol Cell 95(8): 515-519.
Laud, K., I. Gourdou, et al. (1999). "Detection and regulation of leptin receptor mRNA in ovine mammary epithelial cells during pregnancy and lactation." FEBS Lett 463(1-2): 194-198.
Li, Z., N. Wang, et al. (2012). "Role of PKC-ERK signaling in tamoxifen-induced apoptosis and tamoxifen resistance in human breast cancer cells." Oncol Rep 27(6): 1879-1886.
Looby, E., M. M. Abdel-Latif, et al. (2009). "Deoxycholate induces COX-2 expression via Erk1/2-, p38-MAPK and AP-1-dependent mechanisms in esophageal cancer cells." BMC Cancer 9: 190.
Lopez-Bergami, P. and Z. Ronai (2008). "Requirements for PKC-augmented JNK activation by MKK4/7." Int J Biochem Cell Biol 40(5): 1055-1064.
Marttunen, M. B., S. Andersson, et al. (2000). "Antiestrogenic tamoxifen and toremifene increase serum leptin levels in postmenopausal breast cancer patients." Maturitas 35(2): 175-179.
McMurtry, V., A. M. Simeone, et al. (2008). "Leptin utilizes Jun N-terminal kinases to stimulate the invasion of MCF-7 breast cancer cells." Clin Exp Metastasis.
McMurtry, V., A. M. Simeone, et al. (2009). "Leptin utilizes Jun N-terminal kinases to stimulate the invasion of MCF-7 breast cancer cells." Clin Exp Metastasis 26(3): 197-204.
McSherry, E. A., S. Donatello, et al. (2007). "Molecular basis of invasion in breast cancer." Cell Mol Life Sci 64(24): 3201-3218.
Menter, D. G., R. L. Schilsky, et al. (2010). "Cyclooxygenase-2 and cancer treatment: understanding the risk should be worth the reward." Clin Cancer Res 16(5): 1384-1390.
Miyoshi, Y., T. Funahashi, et al. (2006). "High expression of leptin receptor mRNA in breast cancer tissue predicts poor prognosis for patients with high, but not low, serum leptin levels." Int J Cancer 118(6): 1414-1419.
Neville, M. C., T. B. McFadden, et al. (2002). "Hormonal regulation of mammary differentiation and milk secretion." J Mammary Gland Biol Neoplasia 7(1): 49-66.
Nieves-Alicea, R., N. H. Colburn, et al. (2008). "Programmed Cell Death 4 inhibits breast cancer cell invasion by increasing Tissue Inhibitor of Metalloproteinases-2 expression." Breast Cancer Res Treat.
Nieves-Alicea, R., N. H. Colburn, et al. (2009). "Programmed cell death 4 inhibits breast cancer cell invasion by increasing tissue inhibitor of metalloproteinases-2 expression." Breast Cancer Res Treat 114(2): 203-209.
O'Connor, J. K., J. Avent, et al. (2004). "Cyclooxygenase-2 expression correlates with diminished survival in invasive breast cancer treated with mastectomy and radiotherapy." Int J Radiat Oncol Biol Phys 58(4): 1034-1040.
OMS (2011) "Obesidad y sobrepeso." Centro de prensa 311.

83
Onuma, M., J. D. Bub, et al. (2003). "Prostate cancer cell-adipocyte interaction - Leptin mediates androgen-independent prostate cancer cell proliferation through c-Jun NH2-terminal kinase." Journal of Biological Chemistry 278(43): 42660-42667.
Park, S. A., E. H. Kim, et al. (2007). "KG-135 inhibits COX-2 expression by blocking the activation of JNK and AP-1 in phorbol ester-stimulated human breast epithelial cells." Signal Transduction Pathways, Pt C 1095: 545-553.
Prosperi, J. R., S. R. Mallery, et al. (2004). "Invasive and angiogenic phenotype of MCF-7 human breast tumor cells expressing human cyclooxygenase-2." Prostaglandins Other Lipid Mediat 73(3-4): 249-264.
Protani, M., M. Coory, et al. (2010). "Effect of obesity on survival of women with breast cancer: systematic review and meta-analysis." Breast Cancer Res Treat 123(3): 627-635.
Ranger, G. S., V. Thomas, et al. (2004). "Elevated cyclooxygenase-2 expression correlates with distant metastases in breast cancer." Anticancer Res 24(4): 2349-2351.
Ray, A., K. J. Nkhata, et al. (2007). "Effects of leptin on human breast cancer cell lines in relationship to estrogen receptor and HER2 status." Int J Oncol 30(6): 1499-1509.
Ree, A. H., V. A. Florenes, et al. (1997). "High levels of messenger RNAs for tissue inhibitors of metalloproteinases (TIMP-1 and TIMP-2) in primary breast carcinomas are associated with development of distant metastases." Clinical Cancer Research 3(9): 1623-1628.
Revillion, F., M. Charlier, et al. (2006). "Messenger RNA expression of leptin and leptin receptors and their prognostic value in 322 human primary breast cancers." Clin Cancer Res 12(7 Pt 1): 2088-2094.
Ristimaki, A., A. Sivula, et al. (2002). "Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer." Cancer Res 62(3): 632-635.
Rizzo, M. T. (2011). "Cyclooxygenase-2 in oncogenesis." Clin Chim Acta 412(9-10): 671-687.
Rozic, J. G., C. Chakraborty, et al. (2001). "Cyclooxygenase inhibitors retard murine mammary tumor progression by reducing tumor cell migration, invasiveness and angiogenesis." Int J Cancer 93(4): 497-506.
Sacco, M. G., E. M. Cato, et al. (2001). "Systemic gene therapy with anti-angiogenic factors inhibits spontaneous breast tumor growth and metastasis in MMTVneu transgenic mice." Gene Ther 8(1): 67-70.
SALUD, S. D. (2010). SE REFUERZAN ACCIONES PARA LA LUCHA CONTRA EL CÁNCER DE MAMA. MÉXICO.
Saxena, N. K., D. Sharma, et al. (2007). "Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells." Cancer Res 67(6): 2497-2507.
Schiff, R., P. Reddy, et al. (2000). "Oxidative stress and AP-1 activity in tamoxifen-resistant breast tumors in vivo." J Natl Cancer Inst 92(23): 1926-1934.
Sharma, D., N. K. Saxena, et al. (2006). "Leptin promotes the proliferative response and invasiveness in human endometrial cancer cells by activating multiple signal-transduction pathways." Endocr Relat Cancer 13(2): 629-640.

84
Shibahara, K., M. Asano, et al. (1995). "Isolation of a novel mouse gene MA-3 that is induced upon programmed cell death." Gene 166(2): 297-301.
Simeone, A. M., S. Colella, et al. (2006). "N-(4-Hydroxyphenyl)retinamide and nitric oxide pro-drugs exhibit apoptotic and anti-invasive effects against bone metastatic breast cancer cells." Carcinogenesis 27(3): 568-577.
Simeone, A. M., Y. J. Li, et al. (2004). "Cyclooxygenase-2 is essential for HER2/neu to suppress N- (4-hydroxyphenyl)retinamide apoptotic effects in breast cancer cells." Cancer Res 64(4): 1224-1228.
Simeone, A. M., R. Nieves-Alicea, et al. (2007). "Cyclooxygenase-2 uses the protein kinase C/interleukin-8/urokinase-type plasminogen activator pathway to increase the invasiveness of breast cancer cells." International Journal of Oncology 30(4): 785-792.
Singh, B., J. A. Berry, et al. (2005). "COX-2 overexpression increases motility and invasion of breast cancer cells." Int J Oncol 26(5): 1393-1399.
Society, A. C. (2011). "Breast Cancer Facts & Figures 2011-2012." Atlanta: American Cancer Society, Inc. Soslow, R. A., A. J. Dannenberg, et al. (2000). "COX-2 is expressed in human
pulmonary, colonic, and mammary tumors." Cancer 89(12): 2637-2645. Surmacz, E. (2007). "Obesity hormone leptin: a new target in breast cancer?"
Breast Cancer Res 9(1): 301. Taketo, M. M. (1998). "Cyclooxygenase-2 inhibitors in tumorigenesis (part I)." J
Natl Cancer Inst 90(20): 1529-1536. Tari, A. M., A. M. Simeone, et al. (2005). "Cyclooxygenase-2 protein reduces
tamoxifen and N-(4-hydroxyphenyl)retinamide inhibitory effects in breast cancer cells." Lab Invest 85(11): 1357-1367.
Terry, M. B., M. D. Gammon, et al. (2004). "Association of frequency and duration of aspirin use and hormone receptor status with breast cancer risk." Jama-Journal of the American Medical Association 291(20): 2433-2440.
Timoshenko, A. V., G. Xu, et al. (2003). "Role of prostaglandin E2 receptors in migration of murine and human breast cancer cells." Exp Cell Res 289(2): 265-274.
Verreault, R., J. Brisson, et al. (1989). "Body weight and prognostic indicators in breast cancer. Modifying effect of estrogen receptors." Am J Epidemiol 129(2): 260-268.
Vesga, O. E. P. (2004). "Ciclooxigenasa 3: La nueva iso-enzima en la familia." MedUNAB 7: 181-184.
Wen, Y. H., X. Shi, et al. (2007). "Alterations in the expression of PDCD4 in ductal carcinoma of the breast." Oncol Rep 18(6): 1387-1393.
Williams, C. S., M. Tsujii, et al. (2000). "Host cyclooxygenase-2 modulates carcinoma growth." J Clin Invest 105(11): 1589-1594.
Wu, G., J. Luo, et al. (2006). "Involvement of COX-2 in VEGF-induced angiogenesis via P38 and JNK pathways in vascular endothelial cells." Cardiovasc Res 69(2): 512-519.
Yang, H. S., A. P. Jansen, et al. (2003). "The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation." Molecular and Cellular Biology 23(1): 26-37.
Yang, H. S., A. P. Jansen, et al. (2001). "A novel transformation suppressor, Pdcd4, inhibits AP-1 transactivation but not NF-kappa B or ODC transactivation." Oncogene 20(6): 669-676.

85
Yang, H. S., A. P. Jansen, et al. (2001). "A novel transformation suppressor, Pdcd4, inhibits AP-1 transactivation but not NF-kappaB or ODC transactivation." Oncogene 20(6): 669-676.
Yang, H. S., C. P. Matthews, et al. (2006). "Tumorigenesis suppressor Pdcd4 down-regulates mitogen-activated protein kinase kinase kinase kinase 1 expression to suppress colon carcinoma cell invasion." Mol Cell Biol 26(4): 1297-1306.
Yang, H. S., C. P. Matthews, et al. (2006). "Tumorigenesis suppressor Pdcd4 down-regulates mitogen-activated protein kinase kinase kinase kinase 1 expression to suppress colon carcinoma cell invasion." Molecular and Cellular Biology 26(4): 1297-1306.
Yin, N., D. Wang, et al. (2004). "Molecular mechanisms involved in the growth stimulation of breast cancer cells by leptin." Cancer Res 64(16): 5870-5875.
Yoneda, T., A. Sasaki, et al. (1997). "Inhibition of osteolytic bone metastasis of breast cancer by combined treatment with the bisphosphonate ibandronate and tissue inhibitor of the matrix metalloproteinase-2." J Clin Invest 99(10): 2509-2517.
You, J. C., D. Mi, et al. (2009). "A Positive Feedback between Activated Extracellularly Regulated Kinase and Cyclooxygenase/Lipoxygenase Maintains Proliferation and Migration of Breast Cancer Cells." Endocrinology 150(4): 1607-1617.
Zhang, Y., R. Proenca, et al. (1994). "Positional cloning of the mouse obese gene and its human homologue." Nature 372(6505): 425-432.
Zhang, Z. and R. N. DuBois (2001). "Detection of differentially expressed genes in human colon carcinoma cells treated with a selective COX-2 inhibitor." Oncogene 20(33): 4450-4456.
Zhu, S., H. Wu, et al. (2008). "MicroRNA-21 targets tumor suppressor genes in invasion and metastasis." Cell Res 18(3): 350-359.
Recommended

















