Embed Size (px)
Citation preview

8/4/2019 Productos Esteriles
http://slidepdf.com/reader/full/productos-esteriles 1/14
FARMACIA II
1
INTRODUCCIÓN
Para la fabricación de productos estériles es necesario cumplir con requisitosespeciales, con vistas a minimizar los riesgos de contaminación microbiológica,partículas y por endotoxinas, lo que depende en gran medida de la habilidad,conocimientos, entrenamiento y actitud del personal involucrado. La seguridad enla esterilidad u otro parámetro de calidad no será establecida sobre ningúnproceso terminal o prueba al producto terminadoLa presente regulación constituye un complemento a la Regulación No. 16-2000
“Directrices sobre Buenas Prácticas de Fabricación de Productos Farmacéuticos” y es de estrictocumplimiento para efectuar la fabricación de productos estériles.
DEFINICIONESÁrea crítica: Es un área limpia clasificada diseñada para prevenir lacontaminación microbiana, en la cual existe un alto nivel de control microbiológico,ya que en ella tienen lugar pasos del proceso donde no se permite lacontaminación microbiana. Estas áreas usualmente se clasifican como A. Elambiente que rodea a la misma garantizará por diseño la clasificación.Aéreas controladas: Son áreas donde el nivel de control del ambiente aplicado al
y/o los pasos de fabricación minimizará el riesgo de contaminación microbianaCondiciones de reposo: Es la condición donde la instalación cuenta con el equipamiento
de producción instalado y operando pero no con el personal de producción presente.
Esterilidad: Ausencia de microorganismos vivos.Esterilización: Destrucción y/o remoción de toda forma de vida por medio deagentes físicos o químicos.Desinfección: Proceso donde se emplea un agente antimicrobiano con el objetivode destruir microorganismos patógenos en objetos inanimados.Higienización: Proceso que se realiza en las áreas limpias clasificadas, con vistasa reducir la contaminación microbiológica a un nivel permisible según losresultados históricos del control o a un nivel de referencia según los establecidos
en la presente regulación.Indicadores biológicos: Son preparaciones estandarizadas de microorganismosseleccionados para comprobar la efectividad de los procesos de esterilización.Indicadores químicos: Se utilizan para comprobar que se alcanza la temperaturade esterilización en el punto en que son colocados. Existen diferentes tipos deestos indicadores; por fusión (ámpulas deÁcido Benzoico) y otros que cambian de color por la reacción química que tienelugar, pero algunas veces el cambio de color ocurre antes de que el tiempo de
8/4/2019 Productos Esteriles
http://slidepdf.com/reader/full/productos-esteriles 2/14
FARMACIA II
2
esterilización sea completado. Por esta razón no es una prueba concluyente, osea, no indican la efectividad del proceso.Producto estéril: Producto que requiere esterilidad, la cual es diseñada ygarantizada a través de la validación. El mismo puede obtenerse a partir de unprocesamiento aséptico o mediante un proceso con esterilización en el envase
final. Esta calidad corresponde tanto a Ingredientes Activos Farmacéuticos comopara producto terminado.Sistema abierto: Es un sistema que no está diseñado para garantizar lascondiciones de sistema cerrado.Superficie crítica: Superficie que entra en contacto con el producto estéril, porejemplo, envases primarios (ampolletas, bulbos), cierres (tapones), bandejas deliofilización, bombas de llenado, inyectores, etc.
REQUISITOS GENERALES
La fabricación de productos estériles se realizará en áreas limpias clasificadas; el
acceso se hará a través de esclusas de aire independientes, tanto para el personalcomo para las materias primas y materiales.Se garantizará por diseño el acceso controlado a las áreas de fabricación.Las áreas limpias clasificadas se mantendrán con un grado de limpieza talque garanticen las operaciones que son llevadas a cabo y se lessuministrará solamente aire que haya pasado a través de filtros de altaeficiencia.Las operaciones de preparación de materiales, formulación del producto,llenado y esterilización se realizarán en locales separados dentro del árealimpia clasificada, ubicadas secuencialmente y de forma tal que segarantice un flujo unidireccional de materiales, productos y personal.
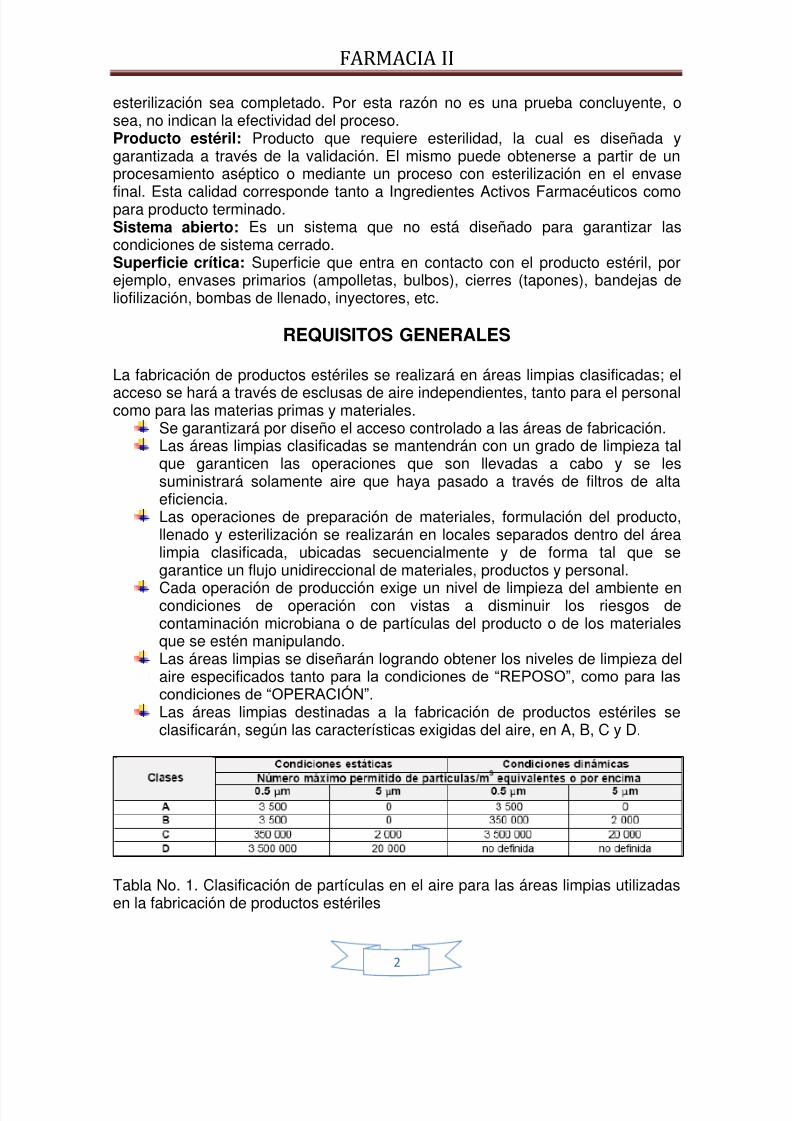
Cada operación de producción exige un nivel de limpieza del ambiente encondiciones de operación con vistas a disminuir los riesgos decontaminación microbiana o de partículas del producto o de los materialesque se estén manipulando.Las áreas limpias se diseñarán logrando obtener los niveles de limpieza delaire especificados tanto para la condiciones de “REPOSO”, como para lascondiciones de “OPERACIÓN”. Las áreas limpias destinadas a la fabricación de productos estériles seclasificarán, según las características exigidas del aire, en A, B, C y D.
Tabla No. 1. Clasificación de partículas en el aire para las áreas limpias utilizadasen la fabricación de productos estériles

8/4/2019 Productos Esteriles
http://slidepdf.com/reader/full/productos-esteriles 3/14
FARMACIA II
3
Para alcanzar las clases B, C y D; el número de cambios de aire seestablecerá en dependencia de las dimensiones del local, el número depersonal que labora en el mismo y el tipo de operación que se lleva a cabo.El sistema de acondicionamiento y ventilación de aire estará provisto de
filtros de alta eficiencia, como por ejemplo, filtros HEPA.Los requisitos y límites de partículas/ m3 permitidos para la clase D enoperación dependerán de la naturaleza de las operaciones que se realicen.El número máximo de partículas para condiciones estáticas será alcanzadodespués de un período de recuperación máximo de 15 a 20 minutos alconcluir las operaciones.Las condiciones de partículas para la clase A en condiciones de operaciónserán mantenidas en la zona que rodea inmediatamente al producto,siempre que el producto o el contenedor abierto estén expuestos alambiente.
TECNOLOGÍA DE CONFORMAR / LLENAR/ SELLAREsta tecnología trae acoplado un sistema de aire efectivo clase A; se instalará almenos en un ambiente de clasificación C.Se proveerá al personal de ropa protectora de las clases A y/o B.El ambiente cumplirá con los límites de viables y no viables para las condicionesde reposo y el límite de viables para las condiciones de operación.La utilización de este tipo de tecnología requiere el cumplimiento de aspectos talescomo: calificación del diseño e instalación del equipamiento, validación de losprocedimientos de limpieza y esterilización en el lugar, y del ambiente del árealimpia que rodea al equipo, entrenamiento del operario requeridos para garantizar
la clase.FABRICACIÓN DE PRODUCTOS ESTÉRILES
Productos esterilizados en su envase final
La preparación de materiales se realizará, almenos, en un ambiente clase D.La operación de formulación en sistemacerrado se realizará al menos en un ambientede clasificaciónD, con el objetivo de minimizar el riesgo de
contaminación microbiana y particuladaadecuado para la filtración y la esterilización.Cuando exista riesgo para el producto de contaminación microbiana, porejemplo, debido a que el producto sustente activamente el crecimientomicrobiano, o sea, almacenado por períodos prolongados antes de laesterilización, la formulación se realizará como mínimo en un ambienteclase C.

8/4/2019 Productos Esteriles
http://slidepdf.com/reader/full/productos-esteriles 4/14
FARMACIA II
4
El llenado de productos parenterales con esterilización terminal se realizarácomo mínimo en un ambiente de clasificación A rodeado de al menos unambiente C.La preparación y llenado de colirios, cremas, suspensiones y emulsiones serealizará generalmente en un ambiente de clasificación C antes de la
esterilización terminal.Productos fabricados asépticamente
Los materiales después de limpios y esterilizados, se manipularán en unambiente que garantice esta condición es decir como mínimo en unambiente de clase C, siempre que los mismos estén protegidos para evitarsu recontaminación.La formulación que va a ser esterilizada por filtración se efectuará en unambiente de clasificación C.La formulación con materias de partida estériles, que no puedan sersometidas al menos a una filtración esterilizante se realizará en unambiente clasificación A rodeado de un ambiente de clasificación BEl llenado se realizará en un ambiente A rodeado de un ambiente B.La transferencia de envases parcialmente cerrados, como ocurre en elproceso de liofilización, se realizará en un ambiente de clasificación Arodeado de un ambiente B, antes de completar el sellado, o en bandejasselladas de transferencia en un ambiente clasificación B.La preparación y llenado de colirios, cremas, suspensiones, y emulsionesse realizará en un ambiente de clasificación A con un entorno declasificación B, cuando el producto esté expuesto al ambiente y no seaposteriormente filtrado.
Las materias primas utilizadas en la preparación de los productos que sonsometidos a una filtración esterilizante, contendrán una carga bacteriana talque puedan ser removida mediante esta operación.
PERSONAL
En las áreas limpias sólo estará presente el númeromínimo de personal, especialmente durante elprocesamiento aséptico. Las inspecciones y controlesse realizarán desde el exterior de las áreas limpias.
Se establecerán programas de capacitación paratodo el personal empleado, incluyendo el de limpiezay mantenimiento, en disciplinas relacionadas con lasBuenas Prácticas de Fabricación para losProductos Estériles, la higiene y aspectos básicos demicrobiología. Se comprobará sistemáticamente queel personal conozca, entienda y desempeñesatisfactoriamente los procedimientos de operación.

8/4/2019 Productos Esteriles
http://slidepdf.com/reader/full/productos-esteriles 5/14
FARMACIA II
5
En caso que sea necesario el ingreso a dichas áreas de personas que no hayanrecibido esta formación, serán supervisados cuidadosamente.El personal involucrado en la fabricación de los productos estériles recibiráinstrucciones de la obligación de informar sobre cualquier situación que puedacausar la liberación de una cantidad o tipo anormal de contaminantes. Se
establecerán chequeos médicos periódicos para detectar tales situaciones. Unapersona competente designada será la responsable de decidir acerca de lasmedidas que se adoptarán con respecto al personal que pudiera causarsituaciones anormales de peligro microbiológico.El acceso a las esclusas de personal se realizará vistiendo ropas protectoras deuso dentro de las áreas de producción. El cambio de ropas y el aseo personal serealizarán según un procedimiento escrito de eficacia comprobada, diseñado paraminimizar la contaminación del área limpia o el arrastre de contaminantes hacialas mismas.Las personas que accedan a las áreas limpias cumplirán las siguientes medidasque serán supervisadas por el personal designado:
El acceso a las áreas limpias clasificadas se realizará de manera controlada,limitándose al personal autorizado en cada caso.No se permitirá la entrada de personas que puedan causar la liberación de unacantidad o tipo anormal de contaminantes.No se llevaran relojes de pulsera ni joyas, manteniéndose las uñas cortas y sinesmalte, así como no se utilizarán cosméticos.Se seguirá el procedimiento establecido para el lavado y desinfección de lasmanos y antebrazos antes de proceder a vestir la ropa estéril de las áreas limpiasclasificadas.Luego de colocarse los guantes sólo se manipularán los objetos y materialesrequeridos para el proceso que se está efectuando, en caso contrario se
procederá al cambio de guantes.La salida eventual de personal de los cuartos limpios sólo se autorizará en loscasos absolutamente imprescindibles.
INSTALACIONES Y LOCALES
Las instalaciones se diseñarán de forma tal que se evite el ingresoinnecesario de personal de supervisión y control. El diseño de las áreasclase B permitirá que todas las operaciones puedan ser observadas desdeel exterior.En las áreas limpias todas las superficies expuestas serán lisas,
impermeables y sin ranuras ni fisuras con el fin de minimizar la liberación oacumulación de partículas o microorganismos y permitir la aplicaciónrepetidas de agentes de limpieza e higienizantes.Para reducir la acumulación de polvo y garantizar una limpieza ehigienización efectiva, la relación de dimensiones de áreas y equipos sediseñará convenientemente, evitando que no existan lugares de difícilacceso. Además se habilitarán de un número mínimo de repisas, estantes yarmarios.

8/4/2019 Productos Esteriles
http://slidepdf.com/reader/full/productos-esteriles 6/14
FARMACIA II
6
Las puertas se diseñarán evitando lugares difíciles de limpiar; por estarazón no es conveniente el uso de puertas corredizas. Las mismas abrirándesde el área de mayor grado de exigencia de limpieza hacia la de menorgrado.En caso de existir falsos techos es necesario que sean integrales y sellados
herméticamente para prevenir la contaminación proveniente del espaciolibre exterior.En la instalación de tuberías y conductos no quedarán huecos ni espaciosque dificulten la limpieza efectiva y en su diseño el trazado garantizará quesu recorrido sea fundamentalmente por el exterior de las áreas limpias.Siempre que sea posible, se evitará la instalación de sumideros y drenajes,o bien excluirlos de las áreas donde se realicen operaciones asépticas.Donde haya necesidad de instalarlos, se diseñarán, ubicarán y mantendránde tal manera que se reduzca al mínimo el riesgo de contaminaciónmicrobiana; contarán con trampas con cierres de agua u otro métodoalternativo para evitar el reflujo.
EQUIPAMIENTO
Para la fabricación de productos estériles se escogerán equipos que puedan sereficientemente limpiados y esterilizados por medio de vapor, calor seco u otrosmétodos aprobado por la Agencia
Nacional de Control de Medicamentos.Los equipos, accesorios y sistemas de apoyo se diseñarán e instalarán deforma que las operaciones, el mantenimiento y las reparaciones puedanrealizarse desde el exterior del área limpia. Si fuese necesario esterilizar,
esta operación se efectuará, siempre que sea posible, luego del montajecompleto del equipo.Cuando las operaciones de mantenimiento se realicen dentro del árealimpia, ésta se limpiará, desinfectará e higienizará antes de recomenzar elproceso si no se han mantenido los niveles exigidos de limpieza y/o asepsiadurante el trabajo.No se introducirán en las áreas limpias materiales o herramientas quepuedan desprender partículas o gérmenes.Todos los equipos y sistemas serán sometidos a un plan de mantenimiento,calibración y verificación planificado. Se registrará la autorización de usootorgada después del mantenimiento de los mismos.
El diseño y operación de los sistemas de apoyo crítico garantizarán lafabricación consistente de un producto terminado de calidad estéril.Las instalaciones de tratamiento, purificación, almacenamiento ydistribución de agua de uso farmacéutico, se diseñarán, construirán ymantendrán de tal forma que se asegure la producción consistente de aguade la calidad requerida según su uso. En su funcionamiento dichas plantasno excederán la capacidad para la que fueron diseñadas y en la

8/4/2019 Productos Esteriles
http://slidepdf.com/reader/full/productos-esteriles 7/14
FARMACIA II
7
producción, almacenamiento y distribución se impedirá el crecimientomicrobiano.
LIMPIEZA E HIGIENIZACIÓN
Las áreas limpias clasificadas se limpiarán e higienizarán de conformidadcon un programa escrito aprobado por el departamento de Aseguramientode la Calidad. El uso y/o rotación de los higienizantes estará basado en losresultados del programa de vigilancia ambiental, donde se determine laaparición de cepas de microorganismos resistentes.La luz ultravioleta no se usará en sustitución de la higienización o de ladesinfección química.Los higienizantes y agentes de limpieza se controlarán para detectar suposible contaminación microbiana, las diluciones se mantendrán enrecipientes limpios e identificados y no se almacenarán a menos que seanesterilizadas. Los higienizantes y agentes de limpieza utilizados en las
áreas de clasificación A y B se esterilizarán antes de su uso. Si unrecipiente está parcialmente vacío, no se rellenará.Los materiales y/o utensilios utilizados para efectuar la limpieza ehigienización se esterilizarán antes de su uso para evitar unarecontaminación en la propia higienización.Los agentes de limpieza e higienizantes serán evaluados para su seleccióny aplicación utilizando métodos adecuados de control.Se asegurará que el personal designado para ejecutar el y/o losprocedimientos de limpieza este apropiadamente calificado, de forma talque conozca, comprenda y ejecute correctamente los procedimientos y seobtenga consistentemente, el grado de limpieza establecido.
Se seguirán procedimientos escritos de eficacia comprobada para efectuarel proceso de limpieza de áreas y superficies críticas.La supervisión sistemática de los resultados del proceso de limpieza serealizará por personal calificado.
PROCESAMIENTO
Durante todas las etapas del proceso se tomarán las precauciones para reducir almínimo la contaminación, incluso durante las etapas anteriores a la esterilización.No se fabricarán productos que contengan organismos microbiológicos vivos enáreas usadas para el procesamiento de otros productos farmacéuticos, ni tampocose efectuará el llenado de recipientes con dichos productos, sin embargo, lasvacunas de organismos muertos o de los extractos bacterianos pueden serllenados, luego de su inactivación en las mismas áreas que otros productosfarmacéuticos estériles previa validación de los procedimientos de inactivación,limpieza y esterilización de equipos y de toda superficie donde la contaminaciónpueda migrar.

8/4/2019 Productos Esteriles
http://slidepdf.com/reader/full/productos-esteriles 8/14
FARMACIA II
8
La validación del proceso de llenado aséptico incluirá la simulación del procesousando medios nutritivos. Las pruebas de simulación del proceso reunirán lassiguientes características:Se simularán lo más fielmente posible las operaciones reales, teniendo en cuentafactores tales como la complejidad de las operaciones, el número de empleados
que están trabajando y el tiempo de duración e incluirán todas las etapas críticasdel procesamiento.Ha de ser posible que en el (los) medio(s) seleccionado(s) se pueda cultivar unamplio espectro de microorganismos, incluyendo aquellos que se esperaríaencontrar en el ambiente donde se efectúe el llenado.Incluirán un número suficiente de unidades de producción para que se tenga unalto grado de seguridad de que, de existir, podrían ser detectados aún los nivelesbajos de contaminación.El número de recipientes usados para el llenado del medio será suficiente demodo que permita una evaluación válida o sea un mínimo de 3 000 unidades deproducción en cada llenado de caldo (valor guía).
Se procurará llegar al nivel cero de crecimiento, se considerará inaceptablecualquier cifra superior a0.1% de unidades contaminadas con un 95% nivel de confianza. Todacontaminación será investigada.12.3.6 La simulación del proceso se repetirá a intervalos definidos y luego decualquier modificación significativa del equipamiento, del personal y del proceso.
ESTERILIZACIÓNAspectos generalesEl método de esterilización adoptado seconformará de acuerdo a lo establecido en el
Registro del medicamento y se prestará unaatención especial cuando el método empleado nosea especificado enla farmacopea; o bien cuandose emplee en un producto que no sea una simplesolución acuosa o aceitosa.Antes de que se adopte un proceso deesterilización, se demostrará su idoneidad para elproducto y su eficacia para lograr las condicionesdeseadas de esterilización en todas las partes decualquier tipo de carga que se someta a dicho proceso, mediante medicionesfísicas o por el uso de indicadores biológicos, cuando sea apropiado.La validez del proceso se verificará a intervalos periódicos, al menos anualmente osiempre que se hayan introducido modificaciones importantes en el equipamiento.Se mantendrán los registros de los resultados.Para lograr una esterilización efectiva, todo el material se someterá al tratamientonecesario y el proceso se diseñará para garantizar que se alcance este objetivo.Se prestará atención especial a la distribución que se realice de las cargas.Se prestará atención a la preparación del material a esterilizar, todos losrecipientes y paquetes serán cerrados y cubiertos de manera que el aire que entresea convenientemente filtrado. No se empleará nunca celofán ni otras envolturas

8/4/2019 Productos Esteriles
http://slidepdf.com/reader/full/productos-esteriles 9/14
FARMACIA II
9
impermeables al vapor para cubrir materiales que van hacer esterilizados por calorhúmedo. Se garantizará que no quede aire aprisionado entre los materiales que seesterilizarán por calor húmedo, por que el aire incluido no puede ser reemplazadopor el vapor.
ESTERILIZACIÓN POR CALORCada ciclo de esterilización por calor se registrará en un gráfico de temperaturacontra tiempo con una escala de tamaño adecuado. La temperatura se registrarámediante una sonda colocada en el punto más frío de la carga o de la cámaracargada. Habiéndose determinado este punto durante la validación,preferiblemente la temperatura será verificada comparándola con otra temperaturatomada mediante otra sonda independiente colocada en la misma posición. Lamencionada tabla de temperatura contra tiempo o bien una fotocopia de la mismaformará parte del registro del lote. Pueden emplearse también indicadoresquímicos y biológicos pero estos no reemplazarán a los controles efectuados por
medios físicos.Se dejará transcurrir suficiente tiempo para que toda la carga alcance latemperatura requerida antes de que comience la medición del tiempo o período deesterilización, para cada tipo de carga se determinará y establecerá dicho tiempo.Luego de la etapa de temperatura alta de un ciclo de esterilización por calor sertomarán las precauciones necesarias para evitar la contaminación de la cargaesterilizada durante la etapa de enfriamiento. Todo líquido o gas de enfriamientoen contacto con el producto será esterilizado a menos que pueda ser demostradoque cualquier recipiente no hermético no será aprobado para su uso.
ESTERILIZACIÓN POR CALOR HÚMEDO
El proceso se controlará mediante los parámetros temperatura y presión. Lainstrumentación de control será independiente de la instrumentación para lavigilancia y registro.Donde se utilicen sistemas automatizados para la vigilancia y control, se realizaránperiódicamente estudios de validación que aseguren los requerimientos críticosdel proceso.Si se trata de esterilizadores que tienen un drenaje en el fondo de la cámara seránecesario registrar la temperatura en esta posición, durante todo el período deesterilización. Cuando forme parte del ciclo una fase de vacío, entonces seefectuarán controles regulares para verificar si la cámara pierde hermeticidad.
Los productos a ser esterilizados, siempre que no se trate de recipientesherméticamente cerrados, se envolverán en un material que permita la eliminacióndel aire y la penetración del vapor, pero que impida a su vez la recontaminacióndespués de la esterilización, todas las partes de la cámara estarán en contactocon el agua o vapor saturado a la temperatura requerida y por el tiempo requerido.

8/4/2019 Productos Esteriles
http://slidepdf.com/reader/full/productos-esteriles 10/14
FARMACIA II
10
ESTERILIZACIÓN POR CALOR SECO
Cuando se emplea el proceso de esterilización por calor seco, el aire circularádentro de la cámara manteniéndose una presión positiva, para impedir la entradade aire no estéril. El aire suministrado ha de ser pasado por un filtro que retenga
microorganismos. Si el proceso de esterilización con calor seco tiene también porobjeto la eliminación de pirógenos, como parte de la comprobación han deefectuarse pruebas de impugnación empleando endotoxinas.Para este método de esterilización terminal las condiciones de operación serán almenos 180C por 2 horas.Para la despirogenización de los materiales se utilizarán condiciones de operaciónde al menos 220C por 30 minutos, en este caso se demostrará una reducción de3 log de endotoxinas resistentes al calor.
ESTERILIZACIÓN POR RADIACIÓN
Se permitirá la esterilización por radiación principalmente para materiales yproductos sensibles al calor.Debido a que muchos productos farmacéuticos y materiales de envase sonsensibles a la radiación, se permitirá emplear este método cuando laausencia de efectos nocivos sobre el producto sea demostradaadecuadamente.No se aceptará la radiación ultravioleta como método de esterilizaciónterminal.La dosis de radiación será medida durante el procesamiento. Con este finse utilizarán indicadores dosimétricos independientes de la taza deradiación, que indiquen cuantitativamente la dosis recibida por el producto
mismo. Los dosímetros se incluirán en la carga en un número y distanciasuficiente como para garantizar que siempre haya un dosímetro dentro dela cámara.Cuando se trate de dosímetros plásticos se emplearán dentro del tiempolímite fijado después de su calibración. Se verificará las absorbancias de losdosímetros transcurrido un corto período de tiempo después de suexposición a la radiación.Se demostrará satisfactoriamente que la cantidad o dosis de radiaciónionizante absorbida no tenga efectos perjudiciales para el productoterminado y por ende para la salud humana.En los procedimientos de validación se asegurará que se tengan en cuenta
debidamente los efectos de las variaciones en la densidad de los envases.Los materiales han de manipularse de tal forma que se evite la confusión omezcla entre los que han sido irradiados y los que no. Cada recipientecontará con un sensor de radiación que indique que ha sido sometido altratamiento con radiación.Los discos de color sensibles a la radiación podrán utilizarse paradiferenciar los envases que han sido sometidos a la radiación de los que nohan pasado este proceso; estos discos no son indicadores de una

8/4/2019 Productos Esteriles
http://slidepdf.com/reader/full/productos-esteriles 11/14
FARMACIA II
11
esterilización completa. La información obtenida se incluirá en el protocolodel lote.
ESTERILIZACIÓN CON OXIDO DE ETILENO
Debido a su gran toxicidad y el ser explosivo en algunas proporciones con el airese establecerán requisitos de seguridad para la manipulación del oxido de etileno.La concentración máxima que se permitirá por escape a la atmósfera será entre10 y 25 ppm respectivamente.Es esencial el contacto directo entre el gas y las células microbianas; se tomaránprecauciones para evitar la presencia de organismos que puedan estar envueltosen materiales tales como cristales o proteína seca. La naturaleza y cantidad de losmateriales de envasado pueden influir significativamente en el proceso.Antes de la exposición al se establecerá un equilibrio entre los materiales, lahumedad y la temperatura requeridas para el proceso. El tiempo empleado en estaoperación se considerará en relación con la necesidad de reducir al mínimo
posible el tiempo transcurrido antes de la esterilización.Cada ciclo de esterilización será controlado mediante indicadores biológicos,utilizando un número adecuado de prueba en toda la carga. La informaciónobtenida por este medio formará parte del registro del lote..
ESTERILIZACIÓN POR FILTRACIÓN
Siempre que sea posible se practicará la esterilización en el envase final. Lacombinación de ambos procesos filtración y esterilización en el envase final,brinda una mayor seguridad. Ciertas soluciones y líquidos que no pueden seresterilizados en el envase final, pueden ser filtradas a través de un filtro estéril con
poros de tamaño nominal de 0.22 micras o menos, o de uno que tengacaracterísticas equivalentes de retención de microorganismos y cargados enrecipientes previamente esterilizados.Mediante tales filtros podrán eliminarse bacterias y hongos, pero no todos los virusy micoplasmas. Se tendrá en cuenta la posibilidad de complementar el proceso defiltración con algún grado de tratamiento térmico.Debido a los riegos potenciales que podría significar el empleo del método defiltración, a diferencia de otros métodos de esterilización, será aconsejableemplear un filtro de doble capa de filtración o efectuarse una segunda filtracióncon otro filtro retenedor de microorganismos inmediatamente antes del llenado. Lafiltración final estéril se llevará a cabo lo más cerca posible al punto de llenado.
No se emplearán filtros que desprendan fibras. El uso de filtros que contienenasbeto se descartará totalmente.Los filtros se seleccionarán de acuerdo a su compatibilidad con el producto y a lascaracterísticas del proceso como: Volumen del producto filtrado, rango de flujo,presión diferencial, temperatura y características químicas del producto.

8/4/2019 Productos Esteriles
http://slidepdf.com/reader/full/productos-esteriles 12/14
FARMACIA II
12
PRODUCTO TERMINADO
El sistema de cierre de los envases primarios serávalidado, los envases cerrados mediante fusión, porejemplo, ámpulas de vidrio o plásticas estarán sujetas
a un ensayo de integridad del 100 %. Se comprobarála integridad de otros envases empleandoprocedimientos adecuados.Los envases primarios cerrados herméticamente alvacío han de verificarse mediante el control demuestras de los mismos para establecer si el vacío seha mantenido después de transcurrido un tiempo predeterminado.Los recipientes llenos de productos parenterales se inspeccionaránindividualmente. Si la inspección es visual se efectuará bajo condicionesadecuadas y controladas de iluminación. Los inspectores se someterán acontroles regulares de la vista, con anteojos puestos si los usan normalmente, y
durante las inspecciones han de tener descansos frecuentes. Si se utilizan otrosmétodos de inspección estos se comprobarán y el equipamiento empleado serácontrolado a intervalos regulares. Los métodos serán validados.
CONTROL DE LA CALIDAD
En la prueba de esterilidad se incluirán no sólo muestras representativas de todoel lote, si no también muestras tomadas de las partes del lote más expuestas alriesgo de contaminación, como por ejemplo:En el caso de productos que han sido llenados asépticamente, entre las muestrasse incluirán las provenientes de recipientes llenados al inicio y al final del lote yluego de alguna interrupción importante del trabajo.b) Si se trata de productos que son esterilizados en sus recipientes finales, seobtendrán muestras de la parte que potencialmente sea la más fría de la carga.La prueba de esterilidad a la que se somete el producto final será consideradasólo como la última de una serie de medidas de control mediante las cuáles seasegura la esterilidad.Los lotes que no pasan la prueba inicial de esterilidad, no pueden ser aprobadossobre la base de una segunda prueba a menos que se demuestre que lascondiciones ambientales y los materiales usados durante la prueba no cumplen lasespecificaciones establecidas y esta pueda ser invalidada.Se procederá a liberar un producto cuando además de pasar la prueba deesterilidad cumpla con las condiciones establecidas durante la fabricación para elaseguramiento de la esterilidadEn los casos donde la liberación paramétrica haya sido autorizada, se prestaráuna atención especial a la validación y al monitoreo del proceso de producciónentero.Cuando se trata de productos inyectables se considerará el control del agua y delos productos intermedios y final para verificar si no contienen endotoxinas,empleando un método bien establecido de la farmacopea que haya sidocomprobado para cada tipo de producto. Para las soluciones de infusión de gran

8/4/2019 Productos Esteriles
http://slidepdf.com/reader/full/productos-esteriles 13/14
FARMACIA II
13
volumen, el control del agua o de los productos intermedios se efectuará en todoslos casos. Además de las pruebas exigidas para obtener la autorización decomercialización del producto final. Cuando una muestra no pasa la prueba, seinvestigarán las causas y se adoptarán las medidas correctivas necesarias.
VALIDACIÓNTodos los sistemas ingenieros de apoyo serán validados especialmente loscríticos; por ejemplo:
Sistema de calefacción, ventilación y acondicionamiento de aire de áreaslimpias clasificadas.Aire comprimido y gases en contacto con el productoVapor puroAgua purificadaAgua para inyecciónVacío central
Todos los equipos importantes que desempeñan procesos de apoyo crucial ocríticos serán validados; por ejemplo:
Esterilización por calor húmedo (autoclave)Esterilización por calor seco y despirogenización (horno y túnel)LiofilizadorCentrifuga de flujo continuo.
Todas las etapas del proceso de producción serán validadas, haciendo énfasis enlas etapas criticas del mismo con el objetivo de asegurar la esterilidad del lote, porejemplo:
Lavado, limpieza, esterilización y/o despirogenización de envases primariosy componentes.FormulaciónHomogeneidad de la mezclaCarga bacteriana antes de la filtraciónCarga bacteriana después de la filtraciónLlenado AsépticoIntegridad del selladoInspecciónMateria partículada en el producto terminado
Las validaciones de los procesos de esterilización se efectuarán sobre todo losprocedimientos que tengan un efecto en la esterilidad del producto.La validación de los procedimientos de limpieza especialmente en las áreas yequipamientos comunes a varios productos es indispensable principalmente enaquellos que por su potencia o toxicidad constituyen un riesgo.

8/4/2019 Productos Esteriles
http://slidepdf.com/reader/full/productos-esteriles 14/14
FARMACIA II
14
EMPAQUETADO
Antes de ser usados, los productos sanitarios estériles son almacenados hasta elmomento de su uso. Para prevenir la recontaminación durante el almacenamiento,los materiales deben ser empaquetados; esto implica, que el contenido de un
equipo debe ser esterilizado con su envoltura. Por lo tanto, el embalaje deberápermitir que el agente esterilizante penetre en el paquete y haga contacto contodos y cada uno de los instrumentos que forman el contenido. Posteriormente,después de la esterilización, éste deberá impedir que los microorganismos puedanpenetrar en el paquete y alcanzar su contenido; el envoltorio deberá actuar comouna barrera antimicrobiana. El embalaje deberá garantizar le esterilidad hasta elmomento de uso del producto. Envolturas dañadas o poco resistentes hacen quetodo el trabajo de limpieza, empaquetado y esterilización haya sido en vano.
TRANSPORTE HASTA EL USUARIO
Cuando los productos estériles sean requeridos para una nueva intervenciónquirúrgica, el departamento que los necesite puede pedirlos al almacén deproductos estériles, el cual registrará los productos pedidos y los transportará alusuario haciendo uso de un sistema de contenedores o mediante carros cerrados.Cuando los productos sanitarios estériles tengan que ser transportados fuera de laInstitución, tendrán que tomarse medidas adicionales para asegurar la integridadde los productos. Al mismo tiempo, es absolutamente necesario un protocoloadecuado en el que se estipulen las condiciones de entrega al usuario.















![Manual de Procedimientos Prep No Esteriles 2006[1]](https://img.pdfslide.es/doc/110x75/5571fb71497959916994e283/manual-de-procedimientos-prep-no-esteriles-20061.jpg)


![Procesos Industriales de Formas Farmacéuticas Sólidas[No Esteriles]](https://img.pdfslide.es/doc/110x75/5695cf7e1a28ab9b028e5517/procesos-industriales-de-formas-farmaceuticas-solidasno-esteriles.jpg)










