Embed Size (px)
Citation preview

PurpurasEquipo 3

PÚRPURA: Definición Alteración en el color de la piel o mucosas por
extravasación de células sanguíneas (petequias y equimosis: lesiones purpúricas de < o > de 2 mm respectivamente.)
Se producen por fallo en los distintos mecanismos de la hemostasia 1ª: integridad de la pared vascular, o función plaquetaria.

Clasificación
1. Púrpuras trombopénicas (<40.000 plaq.) Defecto de producción
Hiperdestrucción en torrente sanguíneo

2. Púrpura no trombopénica
Alteración en función plaquetaria ↑ de P intravascular: tos, vómitos, estasis Púrpura vasculíticas (únicas palpables):
Tóxicas Infecciones Asociadas a enfermedades sistémicas

Estudio analítico diferencial
Recuento plaquetar Coagulación(Tiempo de hemorragia)

CLÍNICA DIFERENCIAL Defectos Hemostasia 1ª: afectan a vasos pequeños y
superficiales petequias y equimosis con poco sangrado.
Defectos Coagulación: grandes equimosis y sangrados internos o externos (raro petequias), con test de fragilidad capilar normal.
Vasculitis: raro sangrado externo o equimosis, a veces componente inflamatorio eritematoso.

Purpura Senil o de Bateman
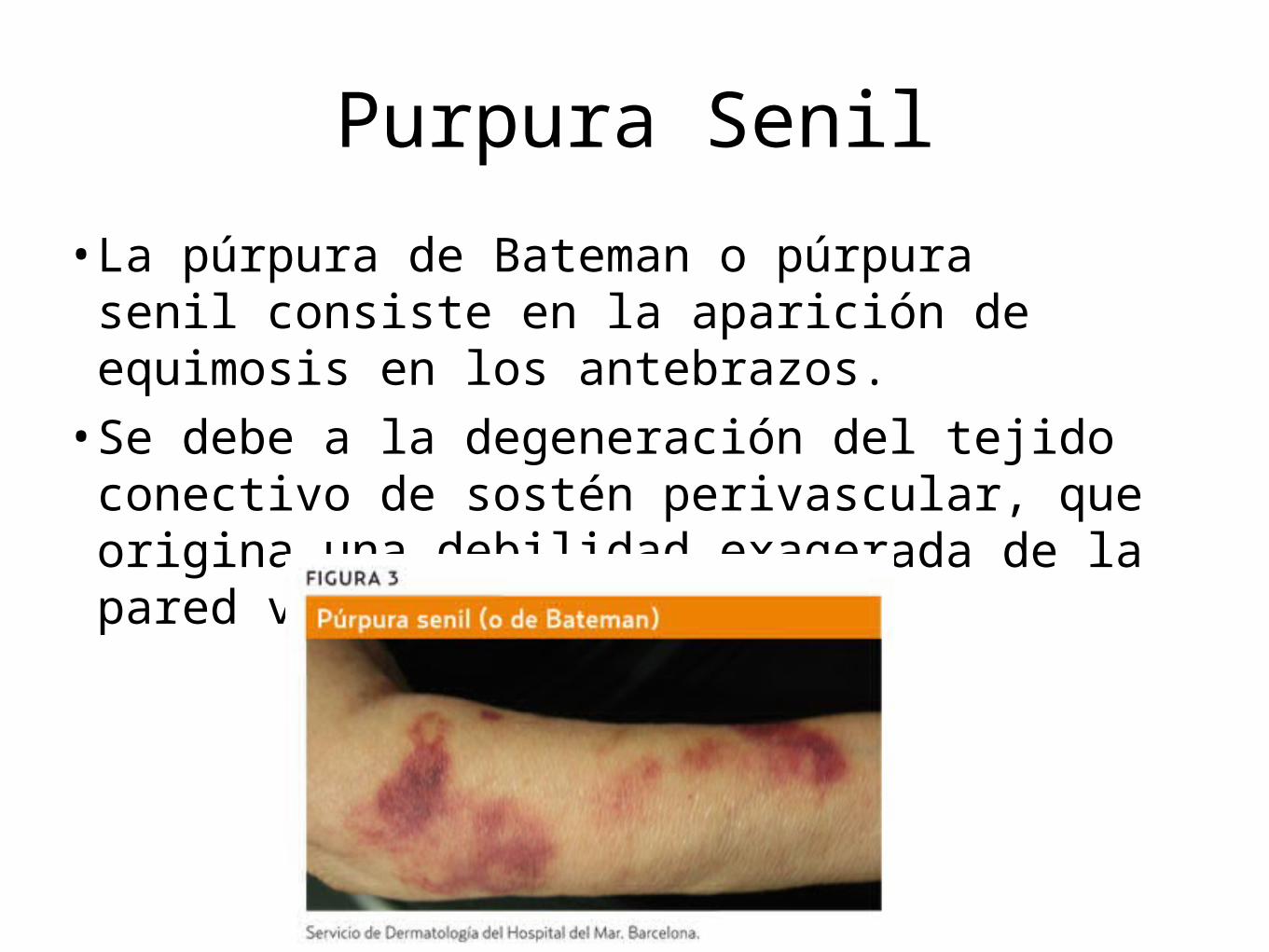
Purpura Senil
• La púrpura de Bateman o púrpura senil consiste en la aparición de equimosis en los antebrazos.
• Se debe a la degeneración del tejido conectivo de sostén perivascular, que origina una debilidad exagerada de la pared vascular.
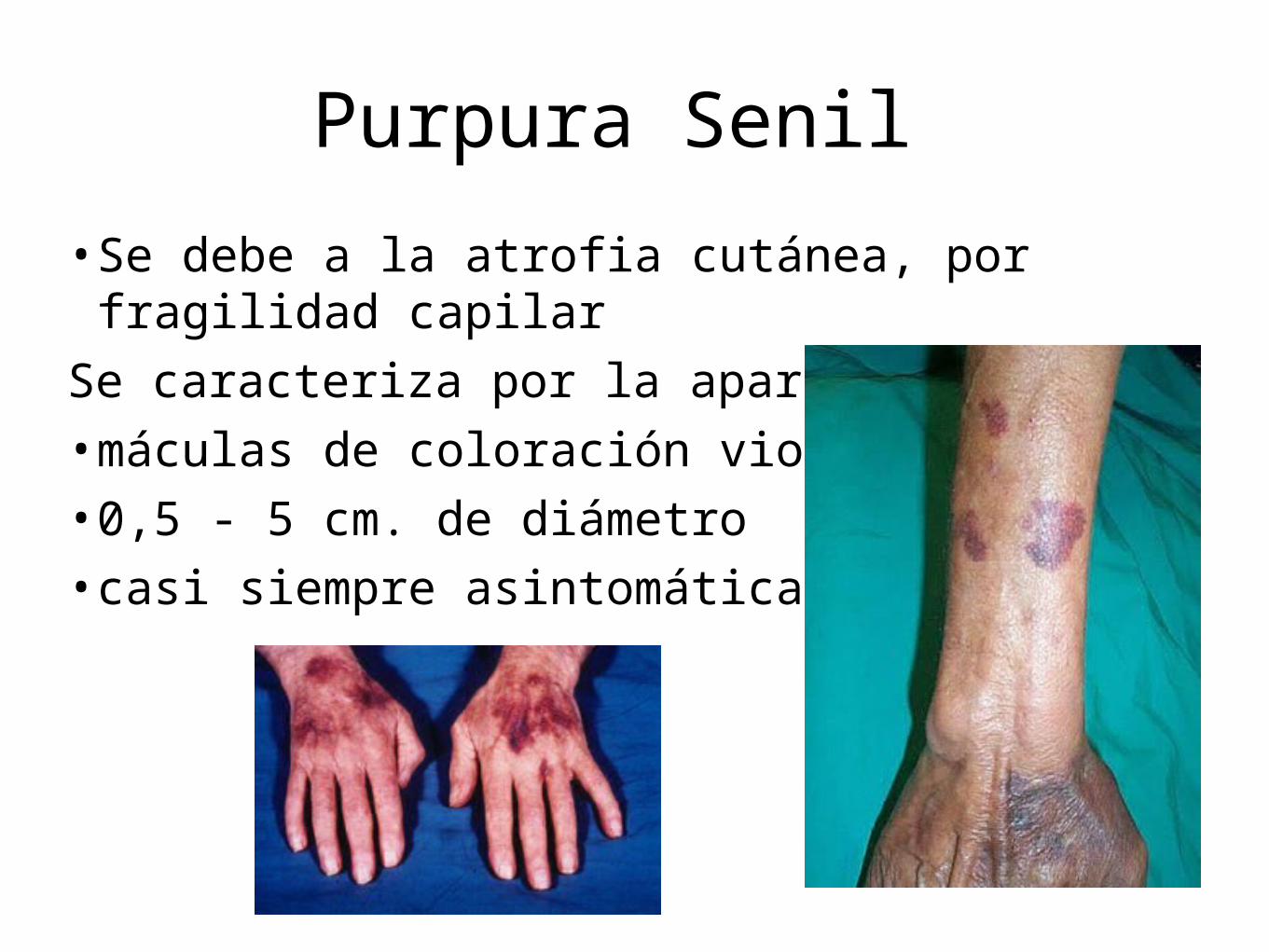
Purpura Senil
• Se debe a la atrofia cutánea, por fragilidad capilarSe caracteriza por la aparición de:• máculas de coloración violácea • 0,5 - 5 cm. de diámetro• casi siempre asintomáticas

• Ser frecuente en pacientes de edad avanzada. • Presentarse en áreas sometidas a pequeños traumatismos como
dorso de manos, antebrazos, cuello, cara y porción superior del tórax.
• Regresan espontáneamente en un periodo de varias semanas pudiendo dejar un área hiperpigmentada residual.
• No requieren de tratamiento alguno

PÚRPURA TROMBOCITOPÉNICA IDIOPÁTICA (PTI)
Destrucción plaquetaria por mecanismo autoinmune.
Incidencia de 4 a 5.3 por 100,000 personas
A cualquier edad, siendo el pico de prevalencia entre los 3 y 5 años de edad
Suele existir antec. de infección viral 1-3 semanas antes, varicela, Ebstein Barr, parvovirus e influenzae.
Ac antiplaquetarios de tipo IgG frente a glicoproteínas de membrana plaquetaria con secuestro y destrucción de las plaquetas por el sistema mononuclear fagocítico (bazo).

CLASIFICACIÓN SEGÚN EVOLUCIÓN
PTI AGUDA: Duración < 6m. Simple (80-85%): un solo brote,antecedente viral, de curso
corto y favorable, incluso sin tratamiento. Recidivante (4%): se produce una recaída tras una
normalización de más de 6 semanas sin tx.
PTI CRÓNICA: Duración > 6m. (10%). Más frecuente en niños >10a, formas de inicio insidioso,
con antecedentes viral y con recuentos entre 20,000 y 100,000/mm3.
No responde a esplenectomia Un 30% puede tener remisiones espontáneas dentro del
1º año.

13
Fisiopatologia
La PTI es un trastorno de la autoinmunidad que se caracteriza por la producción anormal de autoanticuerpos, habitualmente de la clase IgG (1 y 3) y que están dirigidos contra determinantes antigénicos sobre las glucoproteínas plaquetarias, particularmente GpIIb/IIIa.
Las plaquetas opsonizadas con anticuerpos IgG sufren una depuración acelerada por receptores Fc gama que son expresados por macrófagos, predominantemente en bazo e hígado.
13

CLÍNICA
Comienzo brusco. Plaquetas >50 000 sin sintomatologia hemorragica
Entre 30 y 50 000,petequias y equimosis al minimo traumatismo
Entre 10 y 30 000, petequias,equimosis,epistaxis,gingivorragias y/o metrorragias espontáneas
<10 000, hemorragias internas, incluyendo hemorragia en órganos vitales, por ejemplo en el sistema nervioso central.

15
De 2-6 años en primavera e invierno. Equimosis y exantema petequial generalizado y
asimétrico No adenopatías ni visceromegalias. Puede existir epistaxis, gingivorragia, hematuria,
hematemesis y melenas. Hemorragia Intracraneal en <1% (causa + frec de mortalidad)
Síndrome de Evans: PTI + anemia hemolítica autoinmune
15

EXPLORACIONES COMPLEMENTARIAS
Hemograma: trombopenia con aumento del VPM. Puede existir anemia (epistaxis severa, S. Evans), linfocitosis relativa y eosinofilia.
Estudio de coagulación y Ac antifosfolípido. Grupo sanguíneo, Rh, Coombs directo Ig, subpoblaciones linfocitarias, AC antinucleares. Serología toxoplasma, CMV, Parvovirus B19, ViH, VVZ,
VHA, B y C BQ Medulograma Sedimentación urinaria (hematuria microscópica) AC antiplaquetarios.

DIAGNÓSTICO
Se sospecha: Niño con púrpura, exploración física normal. Trombocitopenia aisladaNormalidad del resto de las series.Estudio de la coagulación normal.
Se confirma: Medulograma con celularidad normal (nº normal o ↑ de
megacariocitos). Indicado en todos los niños con clínica que no sea típica,
anomalías en el hemograma y especialmente si se inicia tratamiento con corticoides
Los Ac antiplaquetarios + confirman el diagnóstico, pero su negatividad no lo excluye.

18
Tratamiento
El tratamiento de la enfermedad va encaminado en obtener:
1- respuesta clínica 2- respuesta de laboratorio 3- respuestas completas y sostenidas o la curación de
la enfermedad. Esto se logra mediante diferentes fases de tratamiento
secuenciales que va desde el inicio con corticosteroides, seguido de la esplenectomía, hasta el empleo de inmunosupresores y anticuerpos monoclonales en caso de no respuesta 1
8

19
Depende fundamentalmente de la presentación clínica, la cuenta de plaquetas y la evolución de la enfermedad.
Se recomienda el inicio de tratamiento con corticosteroides en pacientes con cifra de plaquetas <30 x109/L y evidencia de hemorragia. Los pacientes con ausencia de síntomas y >30 x109/L plaquetas probablemente no requieran de tratamiento, excepto cuando vayan a ser sometidos un procedimiento quirúrgico o se encuentren bajo trabajo de parto
19

20
La dosis recomendada de prednisona es de 1.0 a 2.0 mg x Kg/peso/día (2 a 4 semanas) e iniciar con cifra de plaquetas <30 x109/L.
Estudios clínicos han documentado que la Inmunoglobulina endovenosa (IgG IV) produce incremento en la cifra de plaquetas en aproximadamente el 75% de los pacientes, sin embargo, la respuesta es transitoria. La IgGIV no ha demostrado efecto a largo plazo.
a)1gxKg/día(1a2días). b) 0.4 g x Kg/día por 5 días.
20

TRATAMIENTO RECOMENDACIONES GENERALES:
Ingreso si precisa transfusión o si plaquetas < 20.000/mm3.
Transfusión de plaquetas: sólo en urgencias con riesgo vital (plaquetas entre 5.000-20.000/mm3 con sangrado visceral o plaquetas < 5.000/mm3).
Siempre que la situación clínica lo permita hay que posponer el tratamiento corticoideo hasta que se realice medulograma.
Escolarización normal si recuento > 20.000/mm3, pero con restricción de la actividad física intensa y deportes de contacto.

22
El bazo es el órgano responsable de la destrucción plaquetaria y la esplenectomía permanece aún como la segunda línea de tratamiento cuando han fallado medidas terapéuticas previas.
El procedimiento no es estrictamente “curativo” debido a que el mecanismo inmunológico persiste y únicamente se remueve uno de los principales sitios de destrucción.
22

23