Embed Size (px)
Citation preview

Área Enfermedades hematológicas
Atención farmacoterapéutica alpaciente con
Alteración de la coagulaciónAutores:
Alberto Jiménez MoralesUGC de Farmacia de Granada
Pedro a. González SierraUGC Hematología y Hemoterapia
Hospital Universitario Virgen de las NievesComplejo Hospitalario Universitario Granada
Basada en el de Dipiro JT et al. Pharmacotherapy: A Pathophysiologic Approach, ninth edition, 2014
Índice
Hemofilia
Enfermedad de Von Willebrand
Déficits congénitos otros factores
Bibliografía
Atención farmacéutica en HemofiliaAtención farmacéutica en Hemofilia
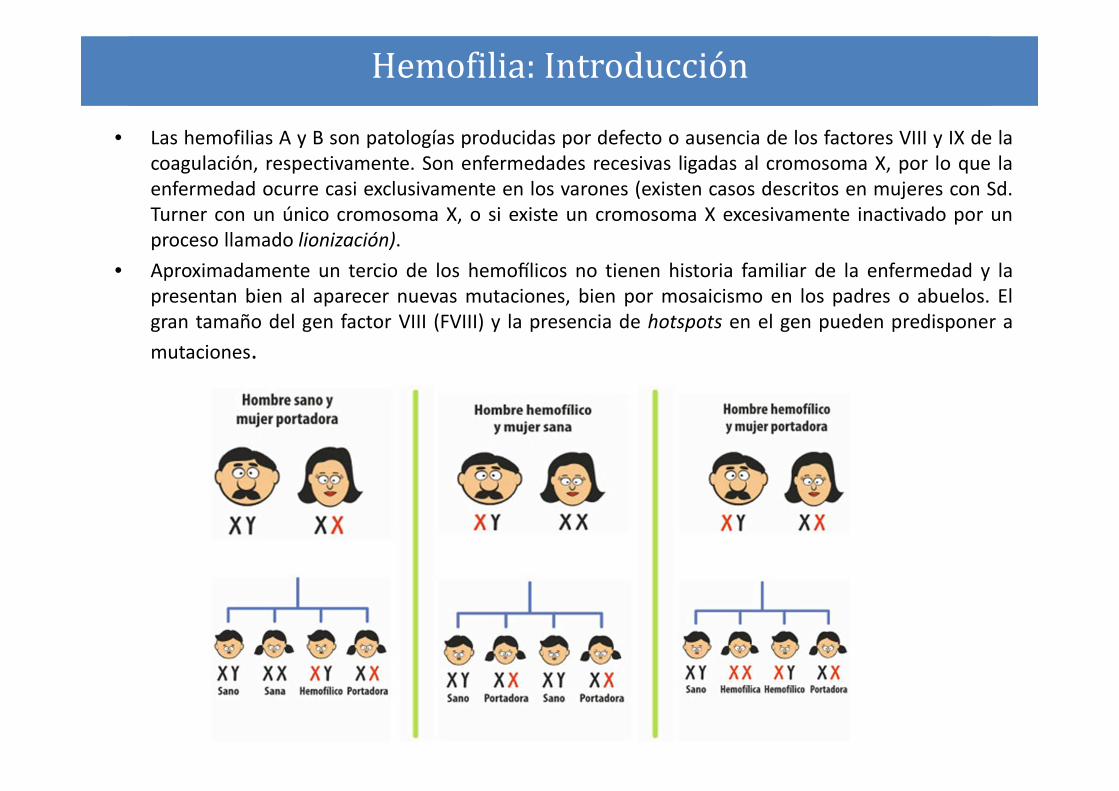
• Las hemofilias A y B son patologías producidas por defecto o ausencia de los factores VIII y IX de la
coagulación, respectivamente. Son enfermedades recesivas ligadas al cromosoma X, por lo que la
enfermedad ocurre casi exclusivamente en los varones (existen casos descritos en mujeres con Sd.
Turner con un único cromosoma X, o si existe un cromosoma X excesivamente inactivado por un
proceso llamado lionización).
• Aproximadamente un tercio de los hemo;licos no tienen historia familiar de la enfermedad y la
presentan bien al aparecer nuevas mutaciones, bien por mosaicismo en los padres o abuelos. El
gran tamaño del gen factor VIII (FVIII) y la presencia de hotspots en el gen pueden predisponer a
mutaciones.
Hemofilia: Introducción
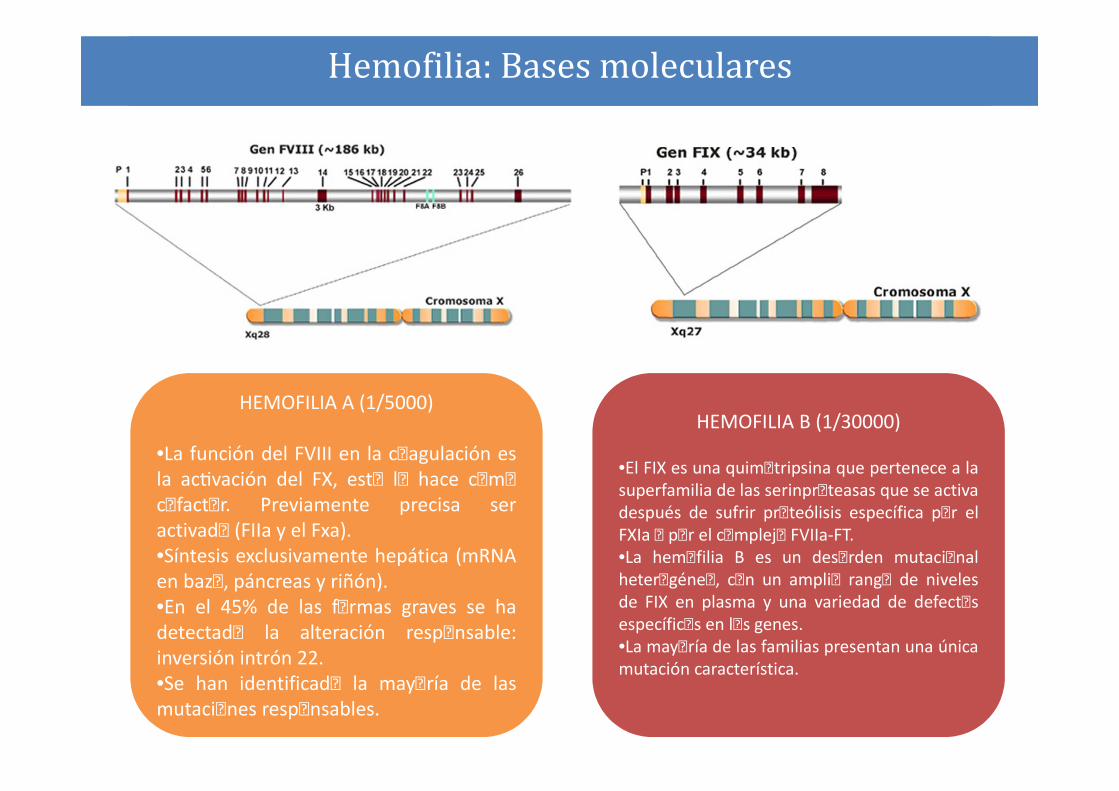
Hemofilia: Bases moleculares
HEMOFILIA A (1/5000)
•La función del FVIII en la coagulación es
la acAvación del FX, esto lo hace como
cofactor. Previamente precisa ser
activado (FIIa y el Fxa).
•Síntesis exclusivamente hepática (mRNA
en bazo, páncreas y riñón).
•En el 45% de las formas graves se ha
detectado la alteración responsable:
inversión intrón 22.
•Se han identificado la mayoría de las
mutaciones responsables.
HEMOFILIA B (1/30000)
•El FIX es una quimotripsina que pertenece a la
superfamilia de las serinproteasas que se activa
después de sufrir proteólisis específica por el
FXIa o por el complejo FVIIa-FT.
•La hemofilia B es un desorden mutacional
heterogéneo, con un amplio rango de niveles
de FIX en plasma y una variedad de defectos
específicos en los genes.
•La mayoría de las familias presentan una única
mutación característica.

• La manifestación clínica fundamental en la hemofilia es el sangrado intraarticular ymuscular. Se estima que el 80% de los episodios hemorrágicos en pacientes hemo;licosson hemartros. Durante la infancia, el tobillo es la arAculación más afectada, mientrasque el sangrado en las rodillas, codos y tobillos es más habitual en la adolescencia y vidaadulta.
• Si los episodios de hemartros de repeAción no reciben el tratamiento adecuado, seproducen cambios crónicos en la arAculación, presentándose cambios en la estructuracartilaginosa, dolor, cambios artríticos, restricción de la movilidad y deformidad articularincapacitante.
• Todo este cuadro afecta a “articulaciones diana” sometidas a sangrados frecuentes,desarrollándose, finalmente, una artropa�a crónica hemo�lica.
• Otras manifestaciones hemorrágicas menos frecuentes incluyen el sangrado digestivo,hematuria, hemorragia intracraneal, sangrado en mucosas y hemorragia prolongada trasintervenciones quirúrgicas o traumatismos.
Hemofilia: Clínica
Gravedad Nivel Factor Clínica Edad diagnóstico
Grave <1%Sangrado espontáneoHemorragia masiva tras cirugía
<1 año
Moderada 1-5%Sangrado espontáneo ocasionalHemorragia importante tras cirugía
1-2 años
Leve >5% Hemorragia importante tras cirugía >2 años

• El diagnóstico de hemofilia comienza con una revisión extensa de la historiafamiliar.
• Un TTPa prolongado sera el test de cribado inicial que nos dara la pista deldiagnóstico, aunque un test normal no descarta la enfermedad.
• El test coagulativo en una etapa y el test cromogénico son los más utilizadosen el presente para la determinación del nivel de factores.
• Debido a la variabilidad en las determinaciones, es fundamental la calibraciónfrecuente del test.
• El test de Bethesda consiste en comparar la actividad residual del FVIIIdespués de mezclarlo con plasma normal y plasma del paciente. Se trata deltest usado para cuanAficación de inhibidores frente al FVIII o FIX.
• Las técnicas de detección de mutaciones y polimorfismos del DNA, así comolas nuevas técnicas de gene arrays, son cada vez más empleadas en eldiagnóstico prenatal.
Hemofilia: Diagnóstico

Atención farmacéutica: Definición.
• La Atención farmacéutica se define como la participación activa del farmacéutico parala asistencia al paciente en la dispensación y seguimiento de un tratamientoterapéutico, cooperando así con el médico y otros profesionales sanitarios, a fin deconseguir resultados que mejoren la calidad de vida del paciente. También conlleva laparticipación del farmacéutico en actividades que proporcionen buena salud yprevengan las enfermedades.
• La Atención Farmacéutica Clínica es la práctica farmacéutica dirigida a usuarios ogrupos de usuarios, que incluye actividades de prevención de la enfermedad,educación sanitaria, farmacovigilancia, seguimiento farmacoterapéuticoindividualizado y todas aquellas otras que se relacionan con el uso racional de losmedicamentos. Dentro de la definición de atención farmacéutica clínica se incluyeel seguimiento farmacoterapéutico que es el servicio profesional que tiene comoobjetivo la detección de problemas relacionados con medicamentos (PRM), para laprevención y resolución de resultados negativos asociados a la medicación (RNM).Este servicio implica un compromiso, y debe proveerse de forma continuada,sistematizada y documentada, con el fin de alcanzar resultados concretos quemejoren la calidad de vida del paciente.
7

Atención farmacéutica: objetivos en hemofilia.
8
Eficiencia de los tratamientos de la coagulación
Minimizar los efectos adversos
La Atención farmacéutica Identificar resultados negativos de la medicación
en el paciente hemofílico
Interacciones medicamentosas
Hábitos de vida saludables

Atención farmacéutica: Efectividad del tratamiento.
Hemofilia A:
Cada unidad de FVIII por kilogramo de peso corporal que se infunda por víaintravenosa elevará el nivel plasmático de FVIII alrededor de 2 UI/dl . Dado que lavida media del factor VIII es de 8 a 12h se debe medir los niveles de factor VIII enplasma a los 15 minutos del fin de la infusión con tal de verificar la dosisadministrada.
La dosis que tenemos que administrar a nuestro paciente se calcula multiplicandoel peso en kilogramos del paciente por el aumento deseado del nivel de factor enUI/dl, multiplicado por 0,5. A la hora de administrar el factor VIII, debe infundirselentamente por vía intravenosa a un ritmo que no supere los 3 ml por minuto enadultos y 100 unidades por minuto en niños pequeños, o según lasespecificaciones de la ficha técnica del producto.
9

Atención farmacéutica: Efectividad del tratamiento.
Hemofilia B:
Cada unidad de FIX por kilogramo de peso corporal que se infunda por vía intravenosaelevará el nivel plasmático de FIX alrededor de 1 UI/dl. La vida media del factor IX es de 18 a24 horas, por ello se deberá medir el nivel de FIX del paciente alrededor de 15 minutosdespués de la infusión a fin de verificar las dosis calculadas.
El factor IX recombinante tiene una respuesta menor que los productos derivados delplasma, de manera que cada unidad de FIX por kilo de peso corporal infundida elevará laactividad del FIX en aproximadamente 0,8 UI/dl en adultos y 0,7 UI/dl en niños menores de15 años. La razón de la menor respuesta del factor IX recombinante no resulta totalmenteclara (6). Para calcular la dosis, se deberá multiplicar el peso en kilogramos del paciente por elaumento deseado del nivel de factor en UI/dl. Los concentrados de FIX deben infundirselentamente por vía intravenosa a una velocidad que no supere el volumen de 3 ml porminuto en adultos y 100 unidades por minuto en niños pequeños, o según lasrecomendaciones de la ficha técnica del producto.
10

Atención farmacéutica: Protocolos de administración de factores.
11
PROTOCOLO DEFINICIÓNTratamiento por episodios
(““““a demanda””””)
Tratamiento que se aplica cuando hay evidencia clínica de una
hemorragia.
Profilaxis continua
Profilaxis primaria
Tratamiento regular y continuo que comienza a aplicarse ante la
ausencia de una enfermedad articular osteo-cartilaginosa
documentada, determinada mediante un examen físico y/o
estudios con imágenes, y antes de que exista evidencia clínica de
una segunda hemorragia en alguna articulación grande, a partir de
los 3 años.
Profilaxis secundaria Tratamiento regular continuo* que comienza a aplicarse después
de que se han producido 2 o más hemorragias en alguna
articulación grande y antes del inicio de una enfermedad articular
documentado mediante un examen físico y estudios con imágenes.
Profilaxis terciaria Tratamiento regular continuo* que comienza a aplicarse a
continuación del inicio de la enfermedad articular que se ha
documentado mediante un examen físico y radiografías simples de
las articulaciones afectadas.
Profilaxis intermitente (““““periódica””””) Tratamiento que se aplica para prevenir hemorragias durante
períodos que no excedan 45 semanas por año.

Atención farmacéutica: Protocolos de profilaxis.
12
- El protocolo de Malmö: 25 a 40 UI/kg por dosis administrada 3 veces por semana a lospacientes con hemofilia A, y 2 veces por semana a los pacientes con hemofilia B.
- El protocolo de Utrecht: 15 a 30 UI/kg por dosis administrada 3 veces por semana a lospacientes con hemofilia A, y 2 veces por semana a los pacientes con hemofilia B.
Pese a la existencia de estos dos protocolos bien definidos se siguen muchos otrosprotocolos de profilaxis, incluso dentro de un mismo país, por lo que no se ha definido elrégimen ideal. El protocolo deberá ser lo más individualizado posible, en función de laedad, el acceso venoso, el fenotipo hemorrágico, la actividad y la disponibilidad deconcentrados de factor de coagulación. Una alternativa para el tratamiento de los niñosmás pequeños es comenzar con la profilaxis una vez por semana e ir incrementándola enfunción de las hemorragias y el acceso venoso.

Atención farmacéutica: Valoración de la efectividad de la terapia de reemplazo.
13
Se deberán evaluar los siguientes puntos y revisar y reforzar la información suministradasobre:
•El acceso venoso.•La hemostasis (registros de hemorragias).•El uso de los productos para la terapia de reemplazo y la respuesta del paciente.•El estado musculo-esquelético: disfunción y funciones detectadas mediante un examenclínico de las articulaciones y músculos, y una evaluación radiológica anual o cuando seaindicada.

Atención farmacéutica: Valoración de la efectividad de la terapia de reemplazo.
14
Existen varias puntuaciones específicas para hemofilia que permiten valorar la función ydisfunción de las articulaciones, incluyendo las actividades y la participación.
Para valorar la disfunción:
Valoración clínica: WFH Physical Examination Score (Puntuación para exámenes físicos dela FMH, conocida Como Puntaje Gilbert) y Hemophilia Joint Health Score (HJHS) (Puntajede salud articular para personas con hemofilia).
Valoración radiológica:
Puntaje de Pettersson, de imágenes de resonancia magnética (IRM).Puntaje de ultrasonido.

Atención farmacéutica: Valoración de la efectividad de la terapia de reemplazo.
15
Valoración de actividades:
Haemophilia Activities List (HAL) (Lista de Actividades para personas con hemofilia),Paediatric Haemophilia Activities List (PedHAL) (Lista pediátrica de actividades para niñoscon hemofilia), Functional Independence Score in Hemophilia (FISH) (Puntaje deindependencia funcional de personas con hemofilia).
Valorar la calidad de vida relacionada con la salud:
HaemoQol, Canadian Hemophilia Outcomes: Kids’ Life Assessment Tool [CHO-KLAT](Herramienta para la evaluación de la calidad de vida de los niños con hemofilia enCanadá).

• El tratamiento con concentrados de factor VIII/IX puede desencadenar la aparición deanticuerpos, conocidos como inhibidores, que neutralizan la función de la proteína infundida.
• La aparición de estos anticuerpos frente al FVIII/IX constituye en la actualidad la complicaciónmás importante en los pacientes con hemofilia. Ante la presencia de inhibidores, nosencontramos con una especial dificultad en el tratamiento tanto de los episodios hemorrágicoscomo de sus secuelas, ya que los recursos terapéuticos disponibles hoy en día no son siempreefectivos.
• La elección del tratamiento hemostático más adecuado para el control de un episodiohemorrágico en pacientes hemofílicos con inhibidor estará condicionada por la localización ygravedad del sangrado, el titulo del inhibidor y las características del mismo (de baja o altarespuesta).
• Definimos la Respuesta Anamnésica como un incremento de más del 50% del Wtulo del inhibi-dor tras la infusión del factor.
• Los dos agentes bypass disponibles son eficaces en el control de los episodios hemorrágicos y enla prevención de los mismos. Hoy en día, ambos productos son fundamentales, puesto queposeen diferentes perfiles terapéuticos y de seguridad y no todos los pacientes responden entodas las ocasiones a cada uno de ellos. Los fallos terapéuticos y la demora en la obtención deuna hemostasia eficaz incrementan la morbimortalidad, que puede, al menos en parte,reducirse con el uso complementario de ambos agentes.
Atención farmacéutica: Inhibidores en pacientes con hemofilia. Generalidades

Atención farmacéutica: Inhibidores en pacientes con hemofilia. Agentes By-pass
• Procede del fraccionamiento del plasma humano y está compuesto por varios factores dela coagulación vitamina K dependientes: factores II, IX y X, en su mayor parte en su formainactiva, y factor VII, éste sobre todo en su forma activa; contiene trazas de FVIII yquininas.
• En los estudios publicados se ponen en evidencia eficacias comprendidas entre un 64 y un96%
• El tratamiento se administra frecuentemente cada ocho o 24 horas a dosis de entre 50 a100 U/kg, según la gravedad de la hemorragia. La dosis total diaria nunca debería excederde 200 UI/kg debido al riesgo de trombosis relacionado con su uso.
• Respuesta anamnésica: Hasta en un 31.5% de los pacientes, seguramente debido a queestos productos contienen pequeñas cantidades de FVIII y FIX.
• Las complicaciones más temidas con la administración de los CCPA son los fenómenostrombo- embólicos y la coagulación intravascular diseminada (CID).
CONCENTRADO COMPLEJO
PROTROMBÍNICO ACTIVADO
(CCPa)(FEIBA®)
CONCENTRADO COMPLEJO
PROTROMBÍNICO ACTIVADO
(CCPa)(FEIBA®)
FACTOR VII RECOMBINANTE
ACTIVADO (rFVIIa)
(NOVOSEVEN®)
FACTOR VII RECOMBINANTE
ACTIVADO (rFVIIa)
(NOVOSEVEN®)
• Es fabricado mediante ingeniería genética y no contiene factores VIII o IX, la potencialtransmisión de virus por medio de transfusiones, así como la respuesta anamnésica delanticuerpo a los factores VIII o IX, es menos problemática que con otros agentes by-pass.
• La dosis más recomendada actualmente es de 90 a 120 μg/kg. En cuanto al intervalo deadministración, se recomienda que sea cada dos horas, dada la vida media corta delfármaco.
• Los efectos adversos más frecuentes son: fiebre, reacciones cutáneas y síntomas generales(2%). La incidencia de complicaciones tromboembólicas y la CID son extremadamente bajas.
• Por la ausencia del FVIII en el producto, no se va a producir respuesta anamnésica delinhibidor tras su administración.
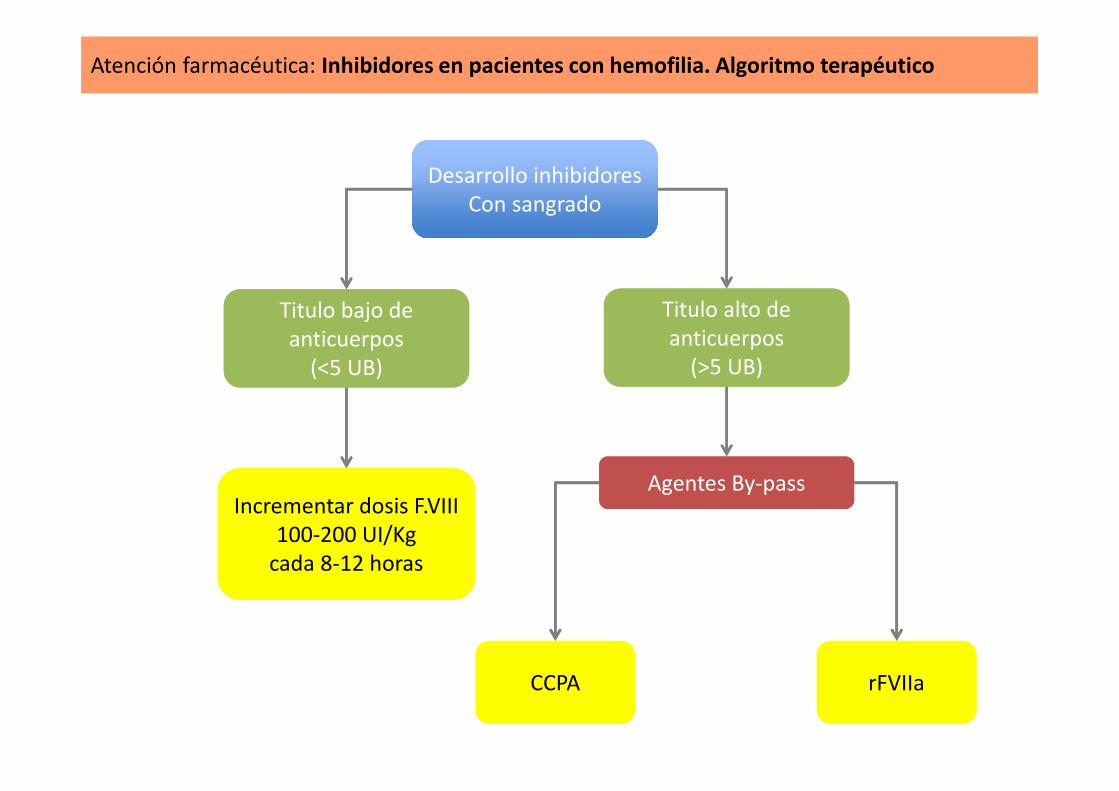
Atención farmacéutica: Inhibidores en pacientes con hemofilia. Algoritmo terapéutico
Desarrollo inhibidoresCon sangrado
Desarrollo inhibidoresCon sangrado
Titulo bajo de anticuerpos
(<5 UB)
Titulo bajo de anticuerpos
(<5 UB)
Titulo alto de anticuerpos
(>5 UB)
Titulo alto de anticuerpos
(>5 UB)
Incrementar dosis F.VIII100-200 UI/Kg
cada 8-12 horas
Incrementar dosis F.VIII100-200 UI/Kg
cada 8-12 horas
Agentes By-passAgentes By-pass
CCPA rFVIIa

Atención farmacéutica: comorbilidades en el paciente hemofílico.
19
Los pacientes hemofílicos de edad avanzada pueden padecer enfermedades relacionadascon la edad, si bien como consecuencia de su enfermedad de base son más susceptiblesde desarrollar ciertas patologías que la población normal.
Por ello desde las consultas de farmacia no sólo deberemos hacer seguimiento a losposibles eventos hemorrágicos sino también a enfermedades como procesos dolorosos,hipertensión, osteoporosis, hipercolesterolemia y diabetes mellitus.

Atención farmacéutica: comorbilidades en el paciente hemofílico. Dolor.
20
Dolor en el paciente hemofílico:
Es común que los pacientes de hemofilia sufran dolores agudos y crónicos. Para poderaplicar el tratamiento correspondiente, es fundamental hacer una evaluación correcta dela causa del dolor.
a. Dolor provocado por acceso venoso:
Por lo general, no se indica ningún medicamento. En algunos niños puede ayudar laaplicación de un spray o crema anestésica local en la zona del acceso venoso.
b. Dolor provocado por una hemorragia articular o muscular:
Si bien los concentrados de factor de coagulación deben administrarse tan pronto comosea posible para detener la hemorragia, a menudo es necesario recurrir a otrosmedicamentos para controlar el dolor. También se pueden colocar paquetes fríos oinmovilizar la zona, colocar entablillados y recurrir al uso de muletas.

Atención farmacéutica: comorbilidades en el paciente hemofílico. Dolor.
21
c. Dolor provocado por artropatía hemofílica crónica:
La artropatía hemofílica crónica se desarrolla en pacientes que no recibieron untratamiento adecuado con concentrados de factor de coagulación ante una hemorragiaarticular. El tratamiento incluye un entrenamiento funcional, adaptaciones y una correctaanalgesia. Los inhibidores COX-2 cumplen una función importante en estos casos. Debeevitarse el uso de otros anti-inflamatorios no esteroides (AINE). Si el dolor resultainhabilitante, puede recurrirse a la cirugía ortopédica .
Los pacientes que presenten dolores persistentes deben derivarse a un equipo detratamiento especializado en el dolor.

Atención farmacéutica: comorbilidades en el paciente hemofílico. Dolor
22
1º escalón Paracetamol
2º escalón
Inhibidor COX-2 (por ej., celecoxib, meloxicam,
nimesulida, entre otros;
O
Paracetamol + codeína (3 a 4 veces por día)
O
Paracetamol+ tramadol (3 a 4 veces por día)
3º escalón
Morfina: utilice un producto de liberación lenta
con un rescate de liberación rápida. Aumente
el producto de
liberación lenta si el de liberación rápida se
utiliza más de 4 veces por día
Estrategia para el manejo del dolor en el paciente hemofílico.

Atención farmacéutica: comorbilidades en el paciente hemofílico. Osteoporosis
23
La densidad mineral ósea (DMO) disminuye en las personas con hemofilia (18,19). Unnúmero elevado de artropatías, perdida de movimiento articular y atrofia muscular,conlleva a la inactividad física, lo cual está asociado con una menor DMO.Siempre que la salud de las articulaciones lo permita, se fomentará la práctica de lasactividades que impliquen ejercicios con peso (dentro de los deportes adecuados) dadoque sirven para promover el desarrollo y mantenimiento de una buena densidad ósea.También son importantes los suplementos de calcio y vitamina D y puede resultarnecesario un tratamiento con bifosfonatos. Se aconseja realizar una evaluación dentalantes de comenzar un tratamiento a largo plazo con bifosfonatos.

Atención farmacéutica: comorbilidades en el paciente hemofílico. Hipertensión.
24
La presión arterial media de los pacientes con hemofilia es más alta que la normal. Tienenel doble de probabilidades de tener hipertensión y utilizan mayor cantidad demedicamentos anti-hipertensivos que la población genera.En vista de tener un mayorriesgo de padecer hemorragias, los pacientes con hemofilia hipertensos deben recibir eltratamiento adecuado y controlarse la presión arterial en forma regular.
Ante la ausencia de otros factores de riesgo cardiovasculares, se deberá mantener lapresión sistólica en ≤140 mmHg y la diastolica en ≤90 mmHg.

Atención farmacéutica: comorbilidades en el paciente hemofílico. Hipercolesterolemia.
25
Se ha informado que los valores promedio de colesterol de los pacientes conhemofilia son inferiores que los de la población general. Es importante conocerlos valores de colesterol (colesterol total, HDL y LDL) en los pacientes de edadavanzada con hemofilia con riesgo de padecer enfermedades cardiovasculares.

Atención farmacéutica: comorbilidades en el paciente hemofílico. Diabetes.
26
La prevalencia de diabetes mellitus en pacientes con hemofilia no está biendocumentada pero se observó que es mayor en una cohorte de personas conhemofilia leve. En pacientes de edad avanzada con hemofilia, en especial entreaquellos con sobrepeso, se deberá controlar el nivel de glucosa una vez por año.
Si se indica tratamiento con insulina, pueden administrarse inyeccionessubcutáneas sin que se presenten complicaciones hemorrágicas

Atención farmacéutica: comorbilidades en el paciente hemofílico. Hábitos Saludables.
27
1. Actividad Física:
-Promover una buena condición física y un desarrollo neuromuscular normal.-Promover el fortalecimiento de los músculos, la coordinación, el estado físico en general.-Fomentar los ejercicios con peso que promuevan el desarrollo y mantenimiento de unabuena densidad ósea.-Práctica de deportes o actividades que no impliquen contacto como las caminatas, lanatación, el golf, el bádminton, la arquería, el ciclismo, el remo, la navegación y el tenis demesa.-Se recomienda evitar tanto los deportes de alto contacto y de impacto

Atención farmacéutica: comorbilidades en el paciente hemofílico. Hábitos Saludables.
28
2. Higiene Dental:
-Los pacientes deben someterse a controles dentales en forma regular.
-Los dientes deben cepillarse dos veces por día con un cepillo de dureza media pararemover la placa acumulada.
-Se deberán utilizar hilo dental o cepillos interdentales siempre que sea posible.
-Se utilizara pasta dental que contenga flúor en los lugares en los que el agua corriente nocontenga flúor natural.
-También pueden recetarse suplementos de flúor si fuera necesario.

Introducción
• La Enfermedad de von Willebrand (EvW) fue descrita por
primera vez por el médico finlandés Erik Von Willebrand
en 1926.
• Se trata de la coagulopatía congénita más frecuente, con
una incidencia en torno al 1%. Está causada por una
alteración cuantitativa o cualitativa del Factor de von
Willebrand (FvW)
• La EvW, por lo general, es congénita, transmitiéndose,
en la mayoría de los casos, con Herencia autosómica
dominante.
• Se han descrito casos adquiridos de la enfermedad,
siendo estos mucho más infrecuentes. Normalmente se
asocian a síndromes linfoproliferativos,
mieloproliferativos, otras neoplasias y trastornos
inmunológicos.
• El FvW es una glicoproteína multimérica, con múltiples
funciones. Interviene en la hemostasia primaria,
permitiendo que las plaquetas se unan de manera
estable a la superficie del vaso dañado, y también en la
hemostasia secundaria, ya que es la principal
transportadora del FVIII en el plasma.
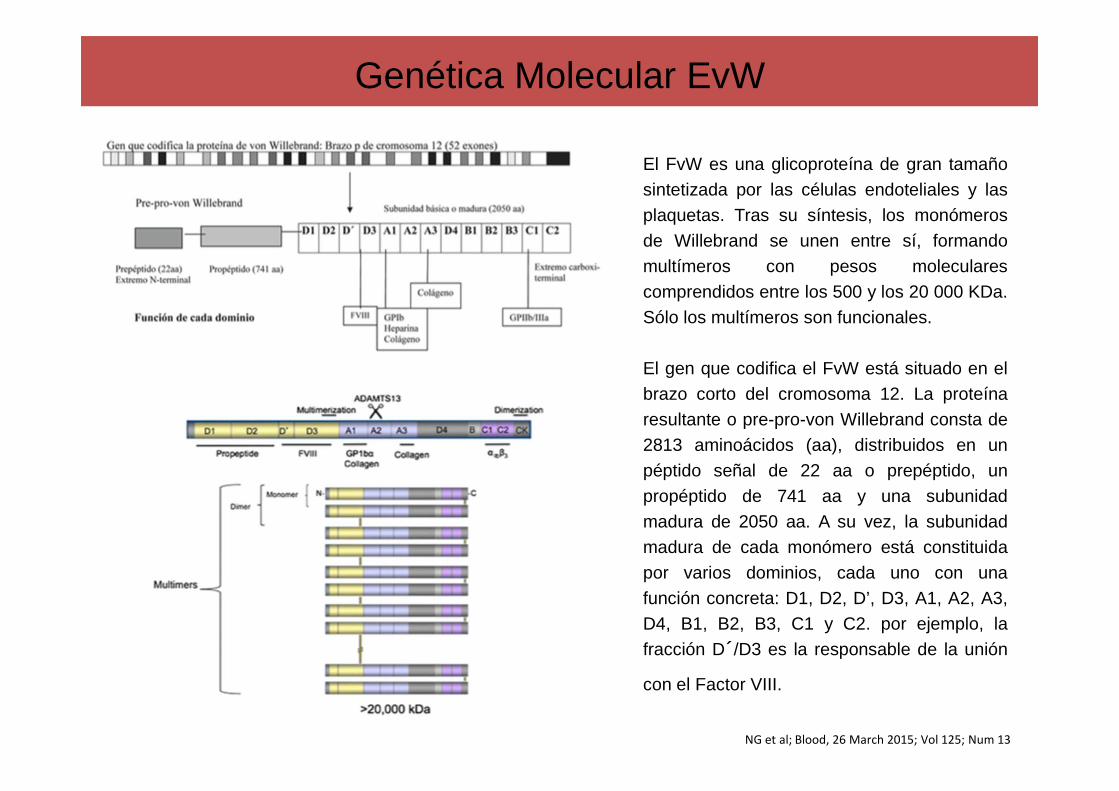
Genética Molecular EvW
El FvW es una glicoproteína de gran tamañosintetizada por las células endoteliales y lasplaquetas. Tras su síntesis, los monómerosde Willebrand se unen entre sí, formandomultímeros con pesos molecularescomprendidos entre los 500 y los 20 000 KDa.Sólo los multímeros son funcionales.
El gen que codifica el FvW está situado en elbrazo corto del cromosoma 12. La proteínaresultante o pre-pro-von Willebrand consta de2813 aminoácidos (aa), distribuidos en unpéptido señal de 22 aa o prepéptido, unpropéptido de 741 aa y una subunidadmadura de 2050 aa. A su vez, la subunidadmadura de cada monómero está constituidapor varios dominios, cada uno con unafunción concreta: D1, D2, D’, D3, A1, A2, A3,D4, B1, B2, B3, C1 y C2. por ejemplo, lafracción D´/D3 es la responsable de la unión
con el Factor VIII.
NG et al; Blood, 26 March 2015; Vol 125; Num 13

Función FvW
Tras una lesión vascular, el FvW presente en el subendotelioqueda expuesto, siendo capaz de unirse a la glicoproteínaGPIb de la membrana plaquetaria, produciéndose la unión delas plaquetas circulantes al subendotelio dañado. Así seforma una monocapa de plaquetas sobre la superficiesubendotelial.
La unión del GPIb plaquetar al FvW del subendotelio producela apertura de canales de calcio situados en latransmenbrana, con la consiguiente entrada de iones decalcio al citoplasma, produciéndose así la activaciónplaquetar.
Tras la activación, las plaquetas se degranulan, liberandograndes cantidades de FvW a la circulación sanguínea, ypresentan en su superficie de membrana proteínasadhesivas, entre las cuales merece destacar la GPIIb/IIIa, quepermite la unión más fuerte entre las plaquetas y el FvWsubendotelial y el existente en el plasma.
De esta manera las plaquetas terminan uniéndose entre sí
por medio del FvW plasmático, formando agregados.En cuanto al FVIII, éste es degradado en el plasma porfactores activados, como el FXa o la proteína C activada. ElFvW, uniéndose al FVIII, forma un complejo que lo protege dela proteolisis.
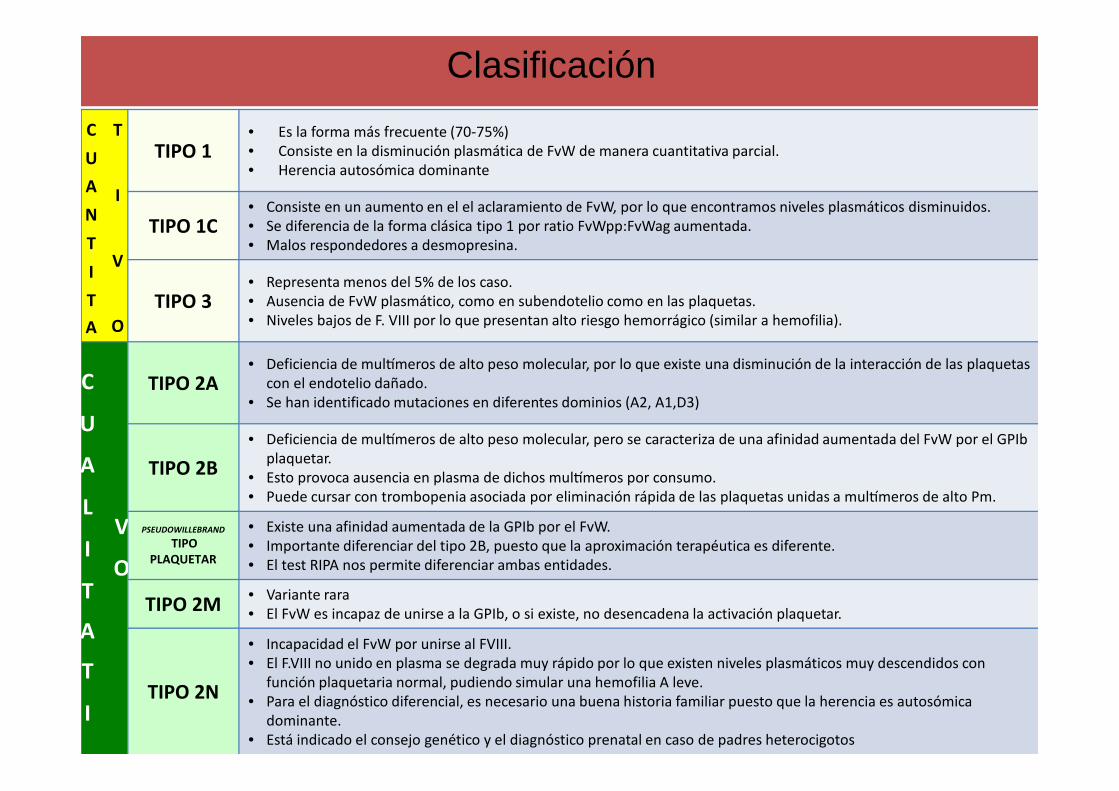
Clasificación
C
U
A
N
T
I
T
A
T
I
V
O
TIPO 1• Es la forma más frecuente (70-75%)• Consiste en la disminución plasmática de FvW de manera cuantitativa parcial.• Herencia autosómica dominante
TIPO 1C• Consiste en un aumento en el el aclaramiento de FvW, por lo que encontramos niveles plasmáticos disminuidos.• Se diferencia de la forma clásica tipo 1 por ratio FvWpp:FvWag aumentada.• Malos respondedores a desmopresina.
TIPO 3• Representa menos del 5% de los caso.• Ausencia de FvW plasmático, como en subendotelio como en las plaquetas. • Niveles bajos de F. VIII por lo que presentan alto riesgo hemorrágico (similar a hemofilia).
C
U
A
L
I
T
A
T
I
V
O
TIPO 2A• Deficiencia de mulWmeros de alto peso molecular, por lo que existe una disminución de la interacción de las plaquetas
con el endotelio dañado.• Se han identificado mutaciones en diferentes dominios (A2, A1,D3)
TIPO 2B
• Deficiencia de mulWmeros de alto peso molecular, pero se caracteriza de una afinidad aumentada del FvW por el GPIbplaquetar.
• Esto provoca ausencia en plasma de dichos mulWmeros por consumo.• Puede cursar con trombopenia asociada por eliminación rápida de las plaquetas unidas a mulWmeros de alto Pm.
PSEUDOWILLEBRAND
TIPO PLAQUETAR
• Existe una afinidad aumentada de la GPIb por el FvW.• Importante diferenciar del tipo 2B, puesto que la aproximación terapéutica es diferente.• El test RIPA nos permite diferenciar ambas entidades.
TIPO 2M• Variante rara• El FvW es incapaz de unirse a la GPIb, o si existe, no desencadena la activación plaquetar.
TIPO 2N
• Incapacidad el FvW por unirse al FVIII.• El F.VIII no unido en plasma se degrada muy rápido por lo que existen niveles plasmáticos muy descendidos con
función plaquetaria normal, pudiendo simular una hemofilia A leve.• Para el diagnóstico diferencial, es necesario una buena historia familiar puesto que la herencia es autosómica
dominante.• Está indicado el consejo genético y el diagnóstico prenatal en caso de padres heterocigotos

Clínica
• La variabilidad de las manifestaciones clínicas y la penetrancia incompleta de la EvW son algunasde sus características más destacables. La frecuencia y la intensidad de aparición de hemorragiasson distintas entre pacientes que presentan niveles similares de FvW y FVIII incluso entre losmiembros de la misma familia.
• Esta variabilidad clínica, al menos en parte, podría ser explicada por la gran variabilidad de losniveles de FvW ante distintas circunstancias. El FvW es un reactante de fase aguda y sus nivelesaumentan ante esWmulos como estrés, embarazo o cirugías o ante ciertos fármacos, como lavasopresina o los anovulatorios. Además de los niveles de FvW y su funcionalidad, otros aspectos,como los ambientales o el grupo ABO (menor nivel en los grupo 0), contribuyen a la variabilidad.
• Esta variabilidad de los niveles plasmáticos de FvW supone un problema para establecer eldiagnóstico de EvW, por lo que es obligatorio repetir la determinación de FvW en distintosperiodos de tiempo.
• La manifestación clínica más frecuente en pacientes con EvW son las hemorragias mucocutáneascomo consecuencia de la alteración de la hemostasia primaria.
• Las hemorragias más frecuentes son las metrorragias, seguidas por epistaxis, gingivorragias, hematuria y, más raramente, hematemesis y melenas.
• En el tipo 2N y tipo 3, el déficit de FVIII puede provocar hematomas de partes blandas, incluso hemartros espontáneos (al igual que en pacientes con hemofilia), en los casos con niveles de FVIII menores al 5%. Los pacientes con EvW tipo 2B pueden presentar trombopenia asociada.
• Durante el embarazo, en la EvW tipo 1 los niveles de FvW y FVIII en plasma aumentan, por lo que el riesgo hemorrágico durante el embarazo es muy bajo. No obstante, 24 o 48 horas después del parto los niveles pueden descender, con el consiguiente riesgo de hemorragias tardías.

1. Determinacion de la actividad antigenica del FVW (FvW:Ag): Es una medicion cuantitativa del FvW comoantıgeno. Es un test muy sensible y facilmente estandarizable. Se realiza mediante ELISA.Esta descendidoen la EvW tipo 1, normal o disminuido en el tipo 2 y casi indetectable en el tipo 3.
2. Determinacion de la actividad funcional de FVW (FvW:CoR): La actividad del cofactor de la ristocetina
(FvW:CoR) es el test de laboratorio mas empleado para el diagnostico. Explora la interaccion entre el FvW
y la GPIb plaquetar.
1. Cociente FvW:CoR/FvW:Ag: Muy util para diferenciar entre EvW tipo 1, en el que el cociente es igual a 1,
del tipo 2, en donde es inferior a 1. En un estudio europeo prospectivo de cohortes en el que se estudiaban
154 familias clasificadas como EvW tipo 1, se establecio que un ratio FvW:CoR/FvW:Ag menor a 0.6 era
predictivo de presencia de alteraciones cualitativas en el FvW y, por tanto, alto riesgo de tratarse de EvW
tipo 2.
1. CuantiAicacion de FVIII: Esta muy descendido en el tipo 3, pero tambien pueden verse niveles bajos en
algunas formas graves de EvW tipo 1 y en el 2N.
1. Cociente FVIII/FvW:Ag: Este cociente estara descendido tanto en la EvW tipo 2N, en la que el FvWesta
presente pero es incapaz de transportar el FVIII, como en la hemofilia A leve, en la que el paciente es
incapaz de sintetizar el FVIII. Para diferenciar estas dos entidades se realizan tecnicas de ELISA.
2. CuantificaciónPropeptido FvW:Laproporciónpropeptido yelmonómerodeFactorVonWillebrand debe
ser1:1.SucuantificaciónesútilenaquelloscasosqueestáaumentadoelaclaramientodelFvW,como
ocurreenelsubtipo1C,puestoquesedesviarálaratiodeambasproteínas.(ratioFvWpp:FvWAg)
Diagnóstico I

6. Hemograma: En pacientes con sangrados frecuentes puede existir una anemia microcítica ferropénica, lo que es especialmente frecuente en mujeres en edad fértil con menorragias. En elsubtipo 2B puede objetivarse en algunos casos trombopenia.
7. Determinación del FvW unido al colágeno: La capacidad del FvW para unirse al colágeno,determinada mediante ELISA, se ha introducido hace poco en algunos laboratorios como test descreening. Su significado es similar a la determinación de FvW:CoR, pero es un test más sensible y de más fácil realización.
6. AgluAnación plaquetaria inducida por ristocetina (RIPA): se realiza mediante agregometría. Consisteen añadir al plasma rico en plaquetas del paciente distintas concentraciones de ristocetina y medir la agregación plaquetaria. Así la mayoría de los tipos de EvW presentan una agregación descendida (senecesitan altas concentraciones de ristocetina, mayores a 1.2 mg/dl, para producir agregaciónplaquetar), excepto el tipo 2B, el cual se caracteriza por una hiperrespuesta a la ristocetina, comoconsecuencia de la hiperafinidad del FvW hacia la GPIb. Con concentraciones bajas de ristocetina,menores a 0.8 mg/dl, se consiguen importantes porcentajes de agregación.
7. Multímeros: pueden estudiarse mediante electroforesis de proteínas en gel de agarosa de baja resolución, detectándose así los déficits de mulWmeros de alto peso molecular en los tipos 2A y 2B.Haciendo estudios en geles de agarosa de alta resolución, incluso se han descrito varios subtipos 2A(IIC, IID, IIE, IIF, IIG y IIH).
8. Test de desmopresina: al diagnóstico debe realizarse para establecer la respuesta biológica individualy predecir la eficacia clínica ante futuros sangrados.
9. Test genéticos/diagnóstico prenatal: muchas mutaciones pueden ser detectadas y están disponiblespara realizar un diagnóstico definitivo y para estudios prenatales y consejo genético.
Diagnóstico II
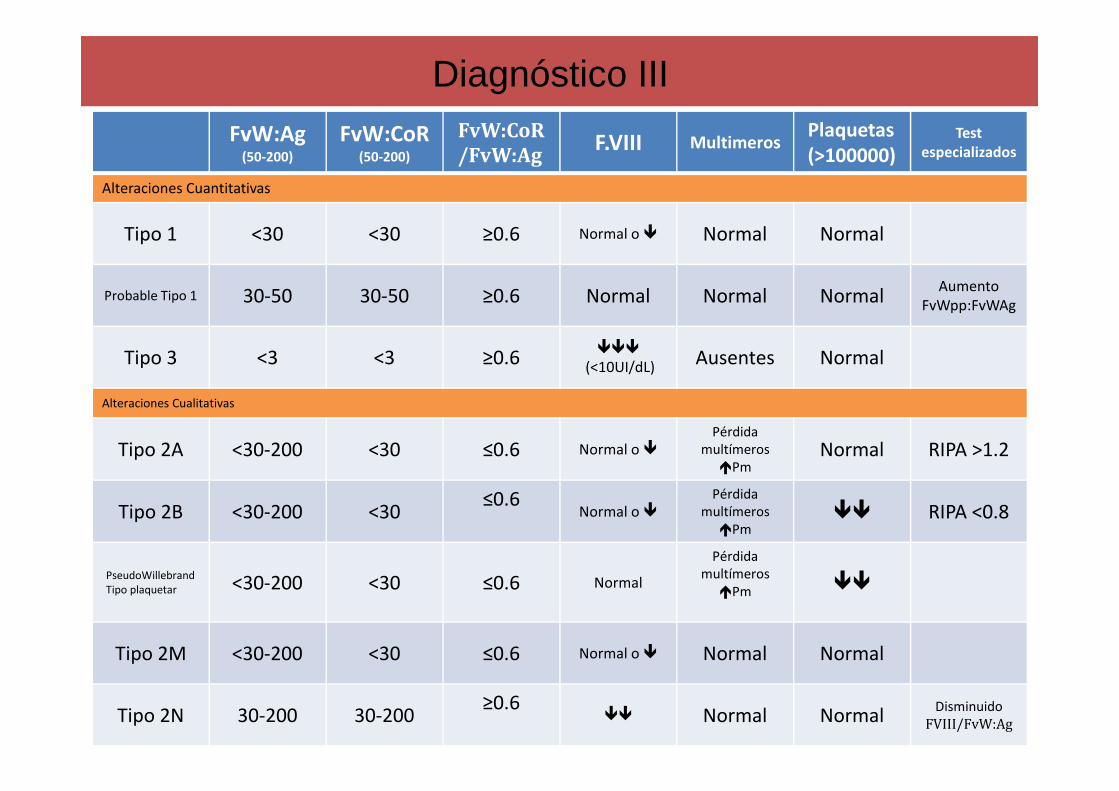
Diagnóstico III
FvW:Ag(50-200)
FvW:CoR(50-200)
FvW:CoR
/FvW:AgF.VIII Multimeros
Plaquetas(>100000)
Testespecializados
Alteraciones Cuantitativas
Tipo 1 <30 <30 ≥0.6 Normal o � Normal Normal
Probable Tipo 1 30-50 30-50 ≥0.6 Normal Normal NormalAumento
FvWpp:FvWAg
Tipo 3 <3 <3 ≥0.6���
(<10UI/dL) Ausentes Normal
Alteraciones Cualitativas
Tipo 2A <30-200 <30 ≤0.6 Normal o �Pérdida
multímeros �Pm
Normal RIPA >1.2
Tipo 2B <30-200 <30≤0.6
Normal o �Pérdida
multímeros �Pm
�� RIPA <0.8
PseudoWillebrandTipo plaquetar <30-200 <30 ≤0.6 Normal
Pérdida multímeros
�Pm ��
Tipo 2M <30-200 <30 ≤0.6 Normal o � Normal Normal
Tipo 2N 30-200 30-200≥0.6
�� Normal NormalDisminuido FVIII/FvW:Ag
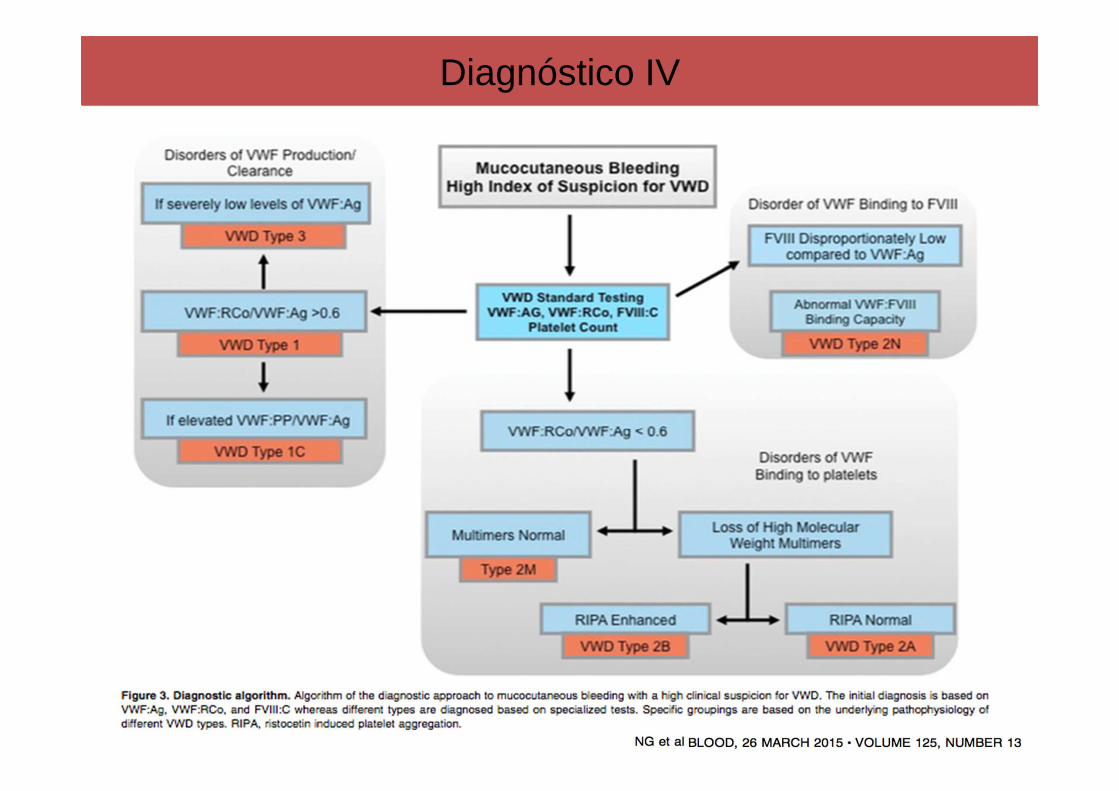
Diagnóstico IV

Tratamiento: Objetivos
• Es necesario adaptar el tratamiento al tipo específico de EvW y ala localización y la severidad del sangrado.
• Prevenir los episodios de sangrado para evitar las consecuenciasa corto y largo plazo de los mismos para que los pacientespuedan mantener una calidad de vida aceptable.
• En ocasiones, las medidas locales como la presión local, el frío oterapia tópica, pueden ser suficientes para controlar el sangradosuperficial.
• El tratamiento sistémico se empleará con el objetivo de corregirla adhesión plaquetaria y los defectos de la hemostasia,estimulando el FvW endógeno (desmopresina) o administrandoproductos que aporten FvW o F.VIII (terapia sustituiva).
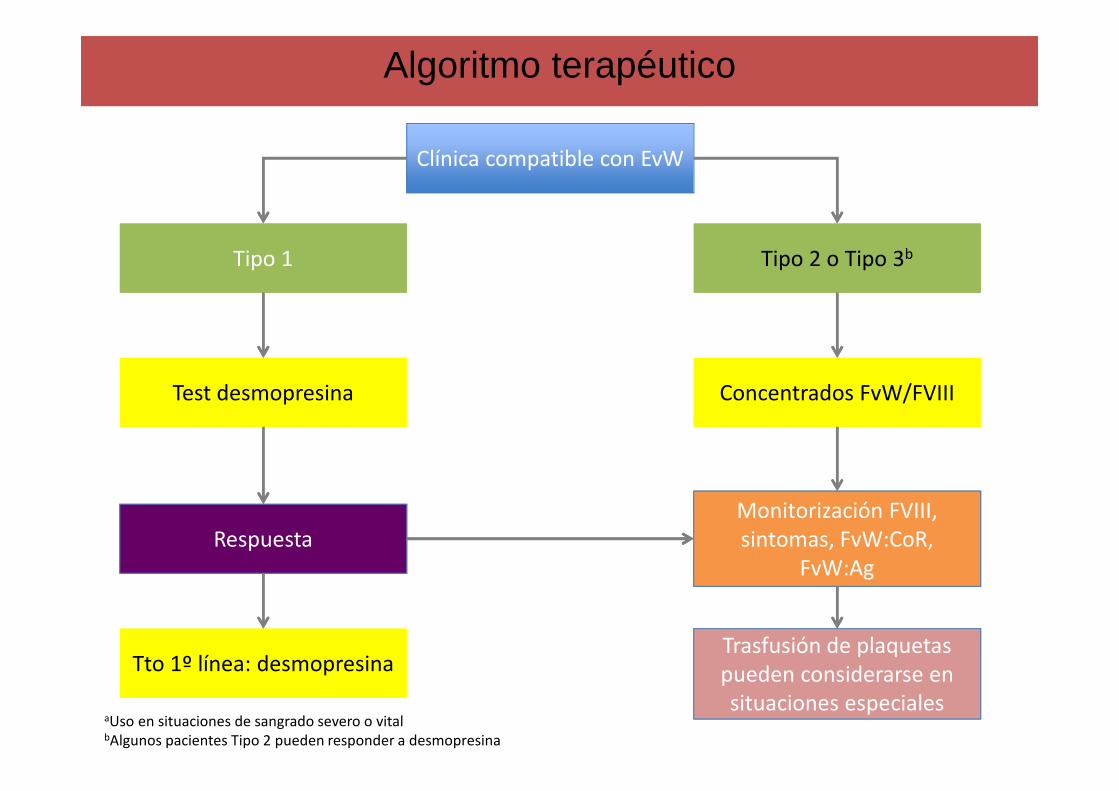
Algoritmo terapéutico
Clínica compatible con EvW
Tipo 2 o Tipo 3bTipo 2 o Tipo 3b
Respuesta
Test desmopresina Test desmopresina
Tipo 1Tipo 1
Concentrados FvW/FVIIIConcentrados FvW/FVIII
Trasfusión de plaquetas pueden considerarse en situaciones especiales
Monitorización FVIII, sintomas, FvW:CoR,
FvW:Ag
Tto 1º línea: desmopresinaTto 1º línea: desmopresina
aUso en situaciones de sangrado severo o vitalbAlgunos pacientes Tipo 2 pueden responder a desmopresina

Tratamiento: Desmopresina (Minurin®)
• El 1-deamino-8-arginina-vasopresina o DDAVP es un análogo sintético de la vasopresina, pero sin efectosvasoactivos a las dosis empleadas con frecuencia en la EvW. Es relativamente barato y seguro desde el punto devista de la transmisión de enfermedades relacionadas con la transfusión sanguínea.
• La desmopresina activa la liberación del FvW almacenado en los gránulos α de las plaquetas y los cuerpos deWeibel-Palade de las células endoteliales, incrementando así los niveles plasmáticos de FvW y FVIII, que puedenalcanzar hasta cuatro o cinco veces los valores iniciales .
• Su eficacia es variable según el subtipo de EvW: Por consiguiente, la desmopresina es el fármaco de elección en laEvW tipo 1, mientras que en el tratamiento de los pacientes con EvW tipo 2 y tipo 3 suele ser necesaria la terapiasustitutiva.
• Es recomendable evaluar la respuesta a Desmopresina al diagnóstico de la enfermedad en condiciones basales. Serealizará mediciones del FvW a la hora, 2 horas y, si es posible, a las 4 horas de la administración.
• Puede administrarse vía intravenosa (0.3 ug/Kg en SF 50-100 mL a pasar en 15-30 min) o intranasal (150 ug 1pulsación en cada orificio nasal). La vía subcutánea es algo más lenta con efecto mantenido.
• La respuesta es transitoria, con una duración de unas 4-6 horas. Puede administrarse cada 12-24 horas.
• Taquifilaxia: Es la disminución de respuesta tras administraciones repetidas. Se acentúa cuando se administra cada12 horas.
• Efectos adversos: Su infusión suele ser bien tolerada, provocando, en la mayoría de los casos, moderadoenrojecimiento facial o aumento de la frecuencia cardiaca. Sólo en muy raras ocasiones, debido a las propiedades anAdiuréticas de la desmopresina, pueden producirse cuadros de retención hídrica con insuficiencia cardiacacongestiva e hiponatremia con crisis comiciales secundarias, por lo que se recomienda especial atención ante suuso en pacientes cardiópatas y en niños.
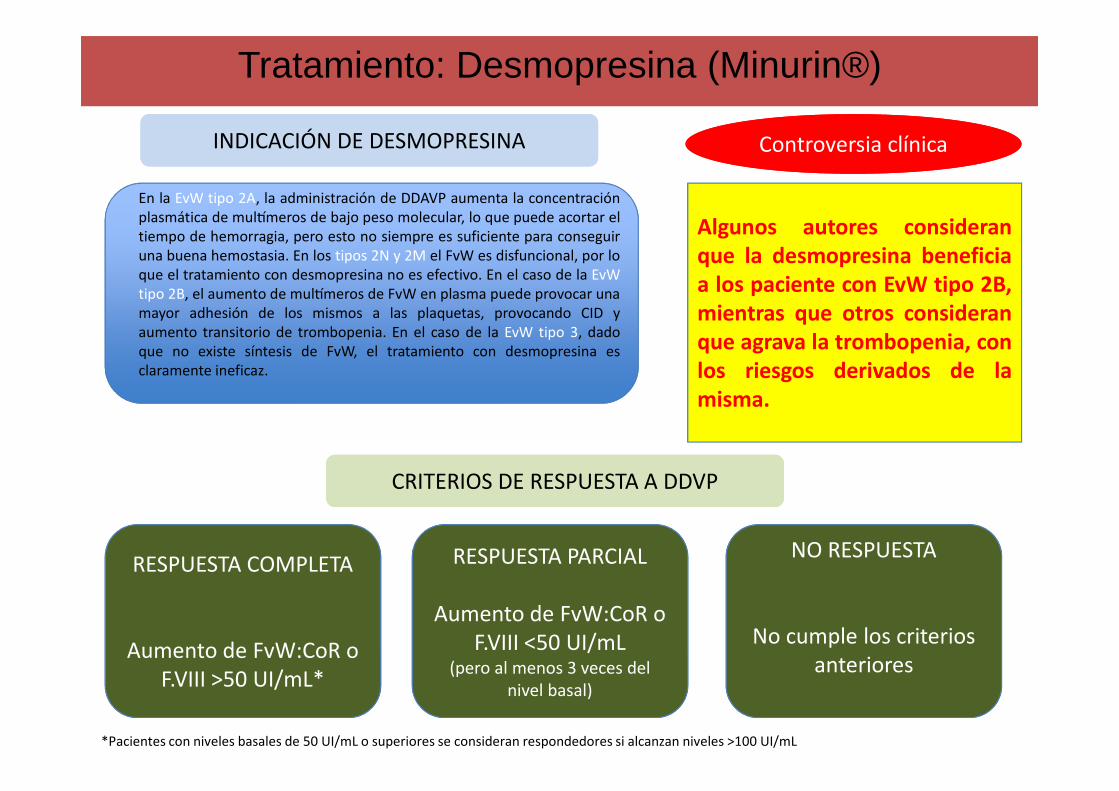
En la EvW tipo 2A, la administración de DDAVP aumenta la concentraciónplasmática de mulWmeros de bajo peso molecular, lo que puede acortar eltiempo de hemorragia, pero esto no siempre es suficiente para conseguiruna buena hemostasia. En los tipos 2N y 2M el FvW es disfuncional, por loque el tratamiento con desmopresina no es efectivo. En el caso de la EvWtipo 2B, el aumento de mulWmeros de FvW en plasma puede provocar unamayor adhesión de los mismos a las plaquetas, provocando CID yaumento transitorio de trombopenia. En el caso de la EvW tipo 3, dadoque no existe síntesis de FvW, el tratamiento con desmopresina esclaramente ineficaz.
Tratamiento: Desmopresina (Minurin®)
INDICACIÓN DE DESMOPRESINAINDICACIÓN DE DESMOPRESINA
CRITERIOS DE RESPUESTA A DDVPCRITERIOS DE RESPUESTA A DDVP
RESPUESTA COMPLETA
Aumento de FvW:CoR o F.VIII >50 UI/mL*
RESPUESTA PARCIAL
Aumento de FvW:CoR o F.VIII <50 UI/mL
(pero al menos 3 veces del nivel basal)
NO RESPUESTA
No cumple los criterios anteriores
*Pacientes con niveles basales de 50 UI/mL o superiores se consideran respondedores si alcanzan niveles >100 UI/mL
Algunos autores consideranque la desmopresina beneficiaa los paciente con EvW tipo 2B,mientras que otros consideranque agrava la trombopenia, conlos riesgos derivados de lamisma.
Controversia clínicaControversia clínica
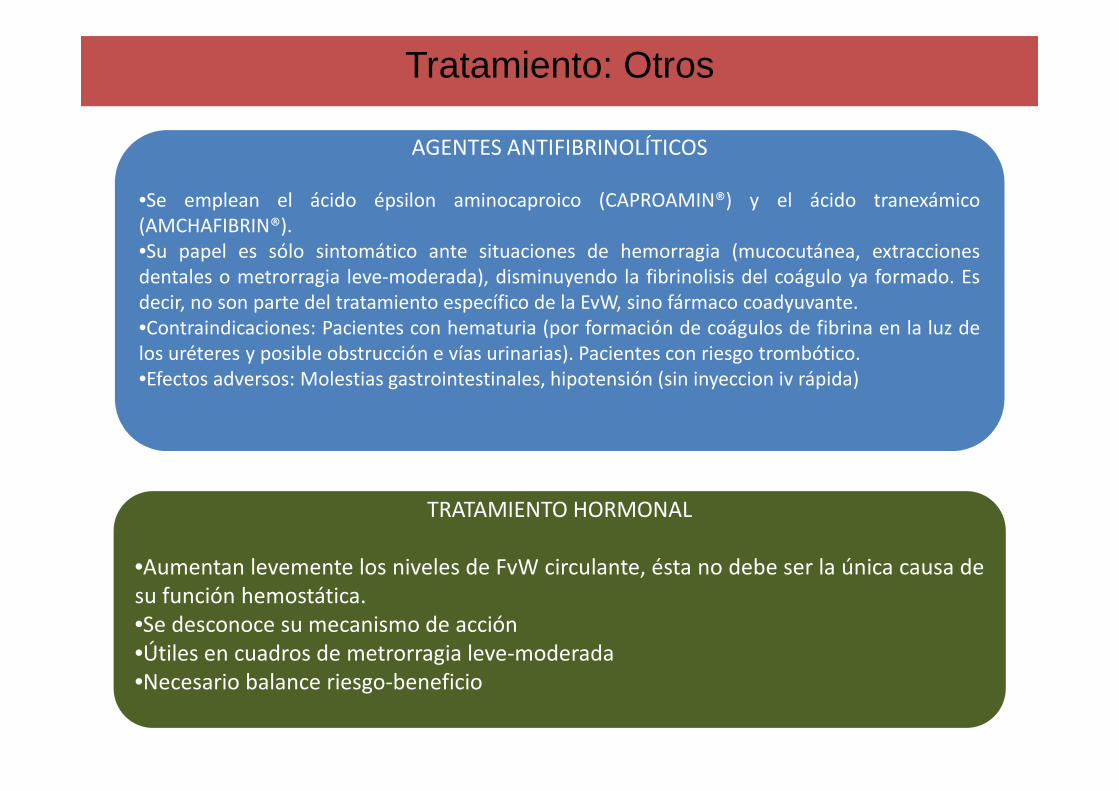
Tratamiento: Otros
AGENTES ANTIFIBRINOLÍTICOS
•Se emplean el ácido épsilon aminocaproico (CAPROAMIN®) y el ácido tranexámico(AMCHAFIBRIN®).•Su papel es sólo sintomático ante situaciones de hemorragia (mucocutánea, extraccionesdentales o metrorragia leve-moderada), disminuyendo la fibrinolisis del coágulo ya formado. Esdecir, no son parte del tratamiento específico de la EvW, sino fármaco coadyuvante.•Contraindicaciones: Pacientes con hematuria (por formación de coágulos de fibrina en la luz delos uréteres y posible obstrucción e vías urinarias). Pacientes con riesgo trombótico.•Efectos adversos: Molestias gastrointestinales, hipotensión (sin inyeccion iv rápida)
TRATAMIENTO HORMONAL
•Aumentan levemente los niveles de FvW circulante, ésta no debe ser la única causa desu función hemostática.•Se desconoce su mecanismo de acción•Útiles en cuadros de metrorragia leve-moderada•Necesario balance riesgo-beneficio
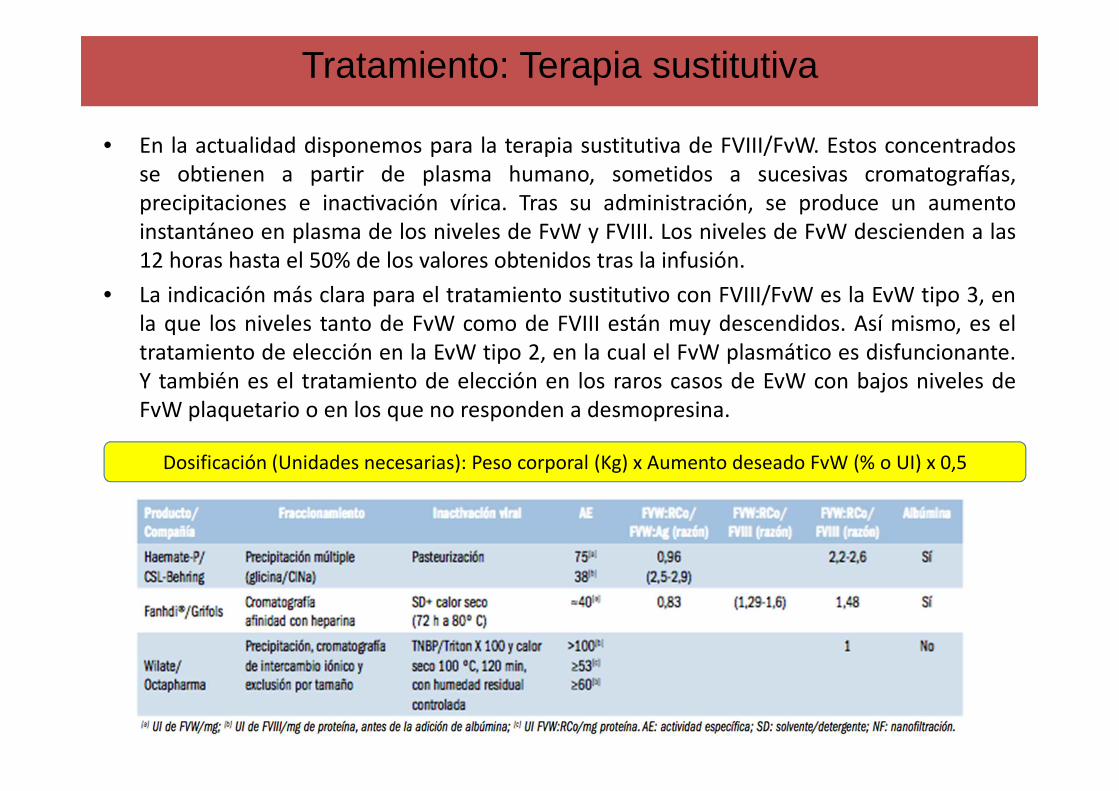
• En la actualidad disponemos para la terapia sustitutiva de FVIII/FvW. Estos concentradosse obtienen a partir de plasma humano, sometidos a sucesivas cromatogra;as,
precipitaciones e inacAvación vírica. Tras su administración, se produce un aumento
instantáneo en plasma de los niveles de FvW y FVIII. Los niveles de FvW descienden a las
12 horas hasta el 50% de los valores obtenidos tras la infusión.
• La indicación más clara para el tratamiento sustitutivo con FVIII/FvW es la EvW tipo 3, en
la que los niveles tanto de FvW como de FVIII están muy descendidos. Así mismo, es el
tratamiento de elección en la EvW tipo 2, en la cual el FvW plasmático es disfuncionante.
Y también es el tratamiento de elección en los raros casos de EvW con bajos niveles de
FvW plaquetario o en los que no responden a desmopresina.
Tratamiento: Terapia sustitutiva
Dosificación (Unidades necesarias): Peso corporal (Kg) x Aumento deseado FvW (% o UI) x 0,5
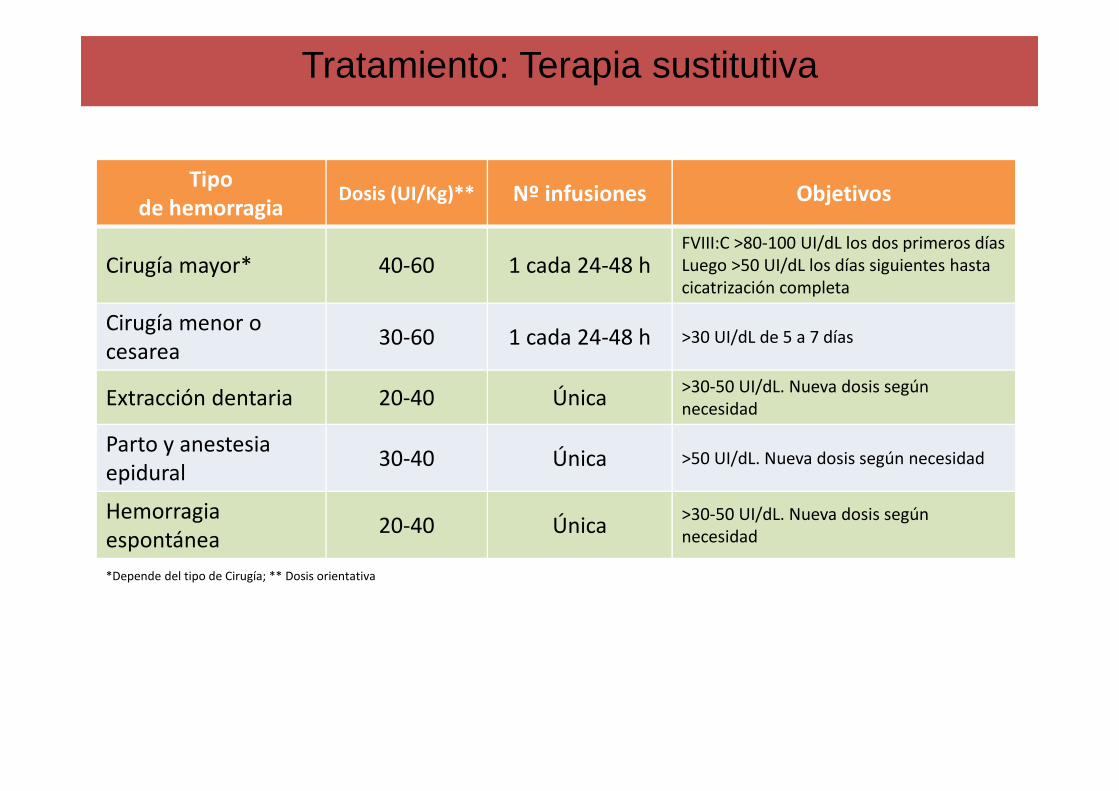
Tratamiento: Terapia sustitutiva
Tipo de hemorragia
Dosis (UI/Kg)** Nº infusiones Objetivos
Cirugía mayor* 40-60 1 cada 24-48 hFVIII:C >80-100 UI/dL los dos primeros díasLuego >50 UI/dL los días siguientes hasta cicatrización completa
Cirugía menor o cesarea
30-60 1 cada 24-48 h >30 UI/dL de 5 a 7 días
Extracción dentaria 20-40 Única>30-50 UI/dL. Nueva dosis según necesidad
Parto y anestesia epidural
30-40 Única >50 UI/dL. Nueva dosis según necesidad
Hemorragia espontánea
20-40 Única>30-50 UI/dL. Nueva dosis según necesidad
*Depende del tipo de Cirugía; ** Dosis orientativa

• En un porcentaje de pacientes tipo 3 (y algunos tipo 2), tras la administración deFVIII/FvW y corregirse los niveles plasmáticos de FvW y FVIII, no cesa la clínicahemorrágica. Se ha demostrado que esto es debido al déficit plaquetar de FvW. En estospacientes estaría indicado transfundir plaquetas.
• Durante el embarazo la mayoría de los pacientes no tienen problemas hemorrágicos,dado que los niveles de FvW y FVIII suelen aumentar, aunque hay que tener especialatención en el posparto tardío.
• En los pacientes tipo 3 sometidos a tratamiento sustitutivo, se ha descrito la formaciónde anticuerpos, sobre todo en aquellos en que la mutación causante de la enfermedadson grandes delecciones. Estos anticuerpos son de naturaleza IgG policlonales, inhibenfuertemente la funcionalidad del FvW, pero tienen nula afinidad por el FVIII. En estospacientes, el rendimiento de la terapia sustitutiva es menos eficaz del esperado. Aunqueexiste limitada evidencia, se puede emplear Factor VII recombinante (Novoseven®) conexpectativa de éxito.
• A diferencia de la hemofilia, la profilaxis secundaria prolongada en la EvW presentaescasa evidencia pero puede contemplarse en pacientes con hemorragias frecuentes eimportantes a pesar del empleo de medidas convencionales.
• Existe controversia en relación al riesgo trombótico y su profilaxis en el tratamiento de laEvW. Parece razonable monitorizar niveles de FVII.I:C
Tratamiento: Situaciones especiales

• Son entidades muy raras.
• A pesar de la escasa prevalencia de los denominados déficits raros de la coagulación, es importante sospecharlos ante una historia personal o familiarde diátesis hemorrágica o alteraciones en las pruebas de coagulación. Esnecesario realizar una exhaustiva historia clínica y un correcto estudio y diagnóstico, puesto que, en ocasiones, pueden manifestar clínica hemorrágicagrave.
• La presentación clínica es muy diversa, con hemorragias cerebrales fetalescomo en la afibrinogenemia o sangrados leves por mucosas como en el déficitde factor XII.
• Existen déficits combinados siendo los más frecuentes el tipo I (Factor V y VIII)y el tipo III (Factor II, VII; IX, X y proteína C). El resto son muy infrecuentes.
• El tratamiento de estos factores es sustitutivo del factor deficitario, y en el casoque no exista se debe realizar con Plasma Fresco Congelado (PFC). Es vital lapreparación previa a intervenciones quirúrgicas, a las dosis adecuadas paraminimizar el riesgo trombótico.
Déficits congénitos otros factores
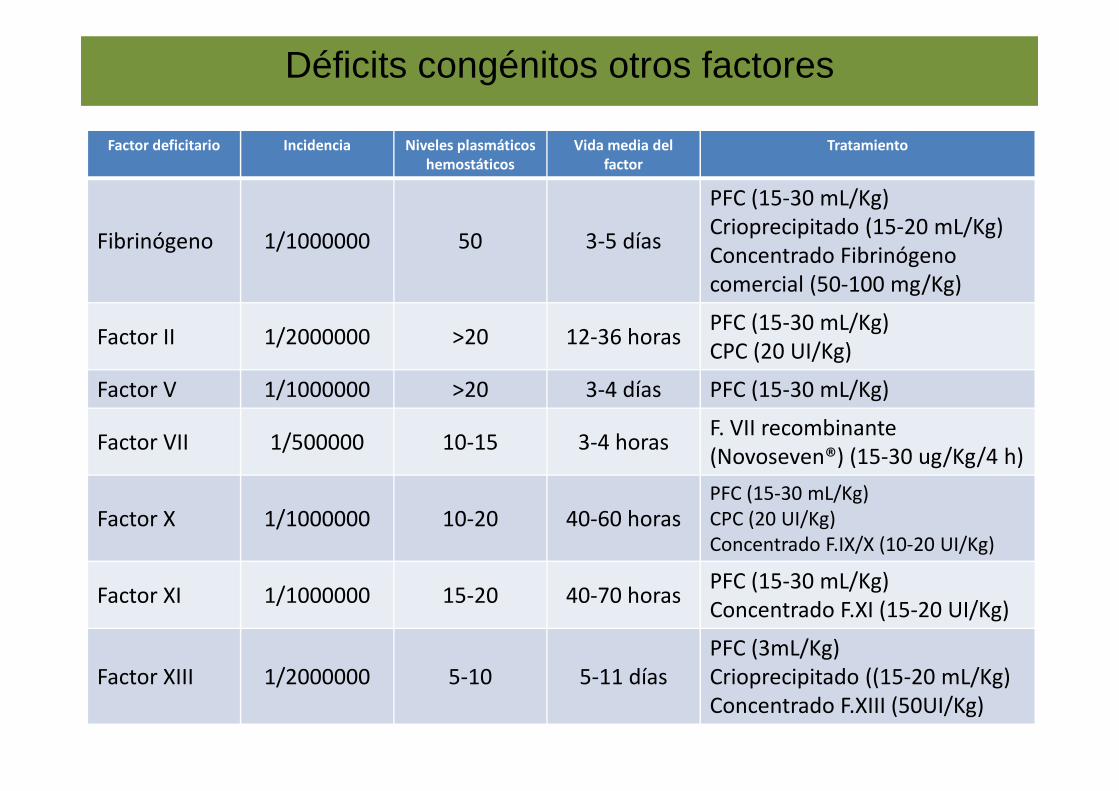
Déficits congénitos otros factores
Factor deficitario Incidencia Niveles plasmáticos hemostáticos
Vida media del factor
Tratamiento
Fibrinógeno 1/1000000 50 3-5 días
PFC (15-30 mL/Kg)Crioprecipitado (15-20 mL/Kg)Concentrado Fibrinógeno comercial (50-100 mg/Kg)
Factor II 1/2000000 >20 12-36 horasPFC (15-30 mL/Kg)CPC (20 UI/Kg)
Factor V 1/1000000 >20 3-4 días PFC (15-30 mL/Kg)
Factor VII 1/500000 10-15 3-4 horasF. VII recombinante (Novoseven®) (15-30 ug/Kg/4 h)
Factor X 1/1000000 10-20 40-60 horasPFC (15-30 mL/Kg)CPC (20 UI/Kg)Concentrado F.IX/X (10-20 UI/Kg)
Factor XI 1/1000000 15-20 40-70 horasPFC (15-30 mL/Kg)Concentrado F.XI (15-20 UI/Kg)
Factor XIII 1/2000000 5-10 5-11 díasPFC (3mL/Kg)Crioprecipitado ((15-20 mL/Kg)Concentrado F.XIII (50UI/Kg)

• Dr. Alok Srivastava et al. Guidelines for the management of hemophilia .2nd Edition
(2012).
• Miguel A. Sanz, Enric Carreras. Manual práctico de hematología clínica 2012. 4ª ed.
Molins de Rei: Ediciones Escofet Zamora SL; 2012.
• Francesco Rodeghiero, Giancarlo Castaman, and Alberto Tosetto. How I treat vonWillebrand disease. Blood, 6 Aug 2009 ; 114 (6): 1158-1165.
• David Lillicrap. Von Willebrand disease: advances in pathogenetic understanding,diagnosis, and therapy. Blood, 28 Nov 2013;122 (23): 3735-3740.
• Mike A. Laffan, Will Lester, James S. O’Donnell, et al. The diagnosis and management ofvon Willebrand disease: a United Kingdom Haemophilia Centre Doctors Organizationguideline approved by the British Committee for Standards in Haematology. Br JHaematol. 2014 Nov;167(4):453-65.
• Christopher Ng, David G. Motto, and Jorge Di Paola. Diagnostic approach to vonWillebrand disease. Blood, 26 Mar 2015; 125 (13): 2029-2037.
• Alberto Tosetto and Giancarlo Castaman. How I treat type 2 variant forms of vonWillebrand disease. Blood, 5 Feb 2015; 125 (6): 907-914.
• Batlle J, Altisent C, Aznar-Lucea JA et al. Tratamiento de la Enfermedad de VonWillebrand en España. Documento de consenso. Haematologica, Oct 2011; 96: Suplem.10.
• Curso de Coagulopatías congénitas patrocinado por Sanofi. Mod I, II y III.
Bibliografía

• Hepler CD, Strand LM. Oportunities and responsibilities in Prharmaceutical Care. AJHP
1990; 47: 533-43.
• Hepler CD, Strand LM. Oportunidades y responsabilidades en Atención Farmacéutica.Pharm Care Esp 1999; 1:43.
• Bjorkman S, Berntorp E. Pharmacokinetics of coagulation factors: clinical relevance forpatients with haemophilia. Clin Pharmacokinet 2001;40(11):815-32.
• Hemophilia of Georgia. Protocols for the treatment of hemophilia and von willebranddisease. Hemophilia of Georgia, 2012. http://www.hog.org/publications/page/ protocols-for-the-treatment-of-hemophilia-and-vonwillebrand-disease-2.
• Poon MC, Lillicrap D, Hensman C, Card R, Scully MF. Recombinant FIX recovery andinhibitor safety: A Canadian post-licensure surveillance study. Thrombosis and
Hemostasis 2002;87:431-5.
• Fischer K, Van der Bom JG, Mauser-Bunschoten EP, et al. Changes in treatment strategiesfor severe haemophilia over the last 3 decades: effects on clotting factor consumptionand arthropathy. Haemophilia 2001; 7: 446-52.
• Aronstam A, Arblaster PG, Rainsford SG, Turk P, Slattery M, Alderson MR, et al.Prophylaxis in haemophilia: a double-blind controlled trial. Br J Haematol 1976;33(1):81-Feldman BM, Pai M, Rivard GE, et al. Tailored prophylaxis in severe hemophilia A: interimresults from the first 5 years of the Canadian Hemophilia Primary Prophylaxis Study. JThromb Haemost 2006; 4(6):1228-36.
Bibliografía





























