
Transcriptómica

Vida – una receta para hacer proteínas
proteínaDNA RNATraducciónTranscripción
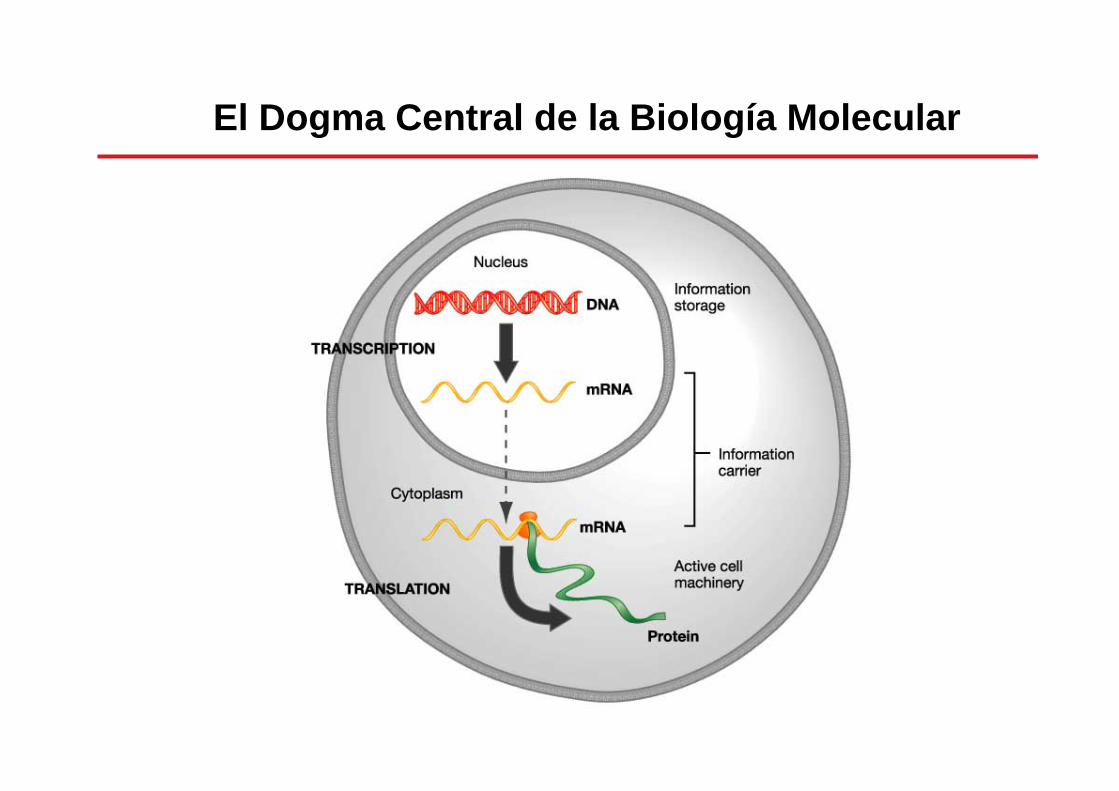
El Dogma Central de la Biología Molecular

El Dogma Central de la Biología Molecular

Genoma -> Transcriptoma ->Proteoma
¿Qué es la Información Biológica?

Tres niveles básicos de información biológica:
Genoma: la información genética común a todas las células del organismo.
Transcriptoma: la parte del genoma que se expresa en una célula en una etapa específica de su desarrollo.
Proteoma: las proteínas que interactuan para dar a la célula su carácter individual.
Del GENOMA estático, único al PROTEOMA dinámico, múltiple.
¿Qué es la Información Biológica?

La era de la genómica

La genómica se ha desarrollado como consecuencia de los avances en Biología Molecular e Informática.
La introducción y popularización de las tecnologías de alta procesividad ha cambiado drásticamente la
manera en que se abordan los problemas biológicos y se prueban las hipótesis.


• El objetivo de la genómica funcional es generar un catálogo de todos los genes y de su función.
• Para comprender el comportamiento de los sistemas biológicos y de los algoritmos genéticos que permiten el funcionamiento celular y el desarrollo de los organismos.
Genómica funcional

• La genómica funcional engloba el estudio del:
• Transcriptoma: conjunto completo de transcritos.
• Proteoma: conjunto de proteínas codificadas por un genoma.
• Interactoma: interacción de estos productos.
Genómica funcional

• Planteamiento clásico:• Dirigido por una hipótesis.• Limitado el número de genes estudiados.
• Planteamiento genómico:• No hay hipótesis de partida.• Información sobre miles de genes.
Genómica funcional

Del genotipo al fenotipo
>protein kunase
acctgttgatggcgacagggactgtatgctgatctatgctgatgcatgcatgctgactactgatgtgggggctattgacttgatgtctatc....
…codificanproteínas...
…cuya estructurainfluye en la función...
Genes en el DNA...
…además el ambiente...
…producen el fenotipo final
El paradigma pre-genómico

Secuenciacióngenoma
Espectrometría de masaspara complejos proteícos
La visión post-genómica
Microarraysde DNA
Literatura, bases de datos
¿Quién?
¿Donde, cómo y cuanto?
¿Quésabemos?
¿En qué manera?
SNPs
¿Y quién más?

Transcriptoma

Estudio de los perfiles de expresión de todos los genes presentes en el genoma.
El método más utilizado es el de microarrays de DNA,que permite el análisis simultaneo de la expresión de
miles de genes.
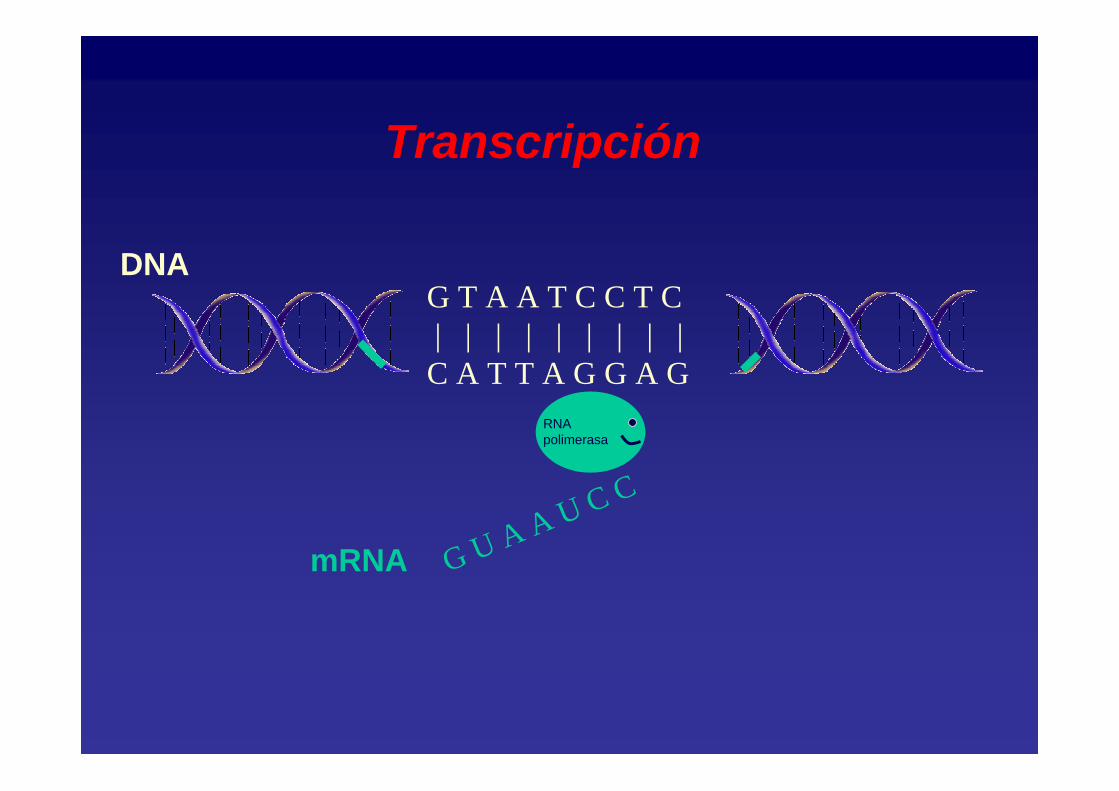
G T A A T C C T C| | | | | | | | | C A T T A G G A G
DNA
G U A A U C C
RNA polimerasa
mRNA
Transcripción

• Varios niveles de regulación: transcripción, maduración, transporte al citoplasma, degradación, traducción, post-traducción.
• Los genes no actúan de forma aislada.
• Existen redes de interacción:• Física (directa o indirecta).• Funcional.
Regulación de la expresión génica

• Pasado: técnicas tradicionales para medir la expresión génica, como Northern y RT-PCR.
• Desarrollo tecnológico:• Expressed Sequenced Tags (ESTs).• Serial analysis gene expression (SAGE).• Suppression substractive hybridization (SSH).• Microarrays de DNA.
Sistemas de detección de la expresión génica

Cambio de escala: del gen al genoma

Cambio de escala: del gen al genoma

• Independientes de conocimiento previo:• ESTs• SAGE• SSH
• Dependientes de conocimiento previo:• Microarrays de DNA
Tecnicas genómicas de alta procesividad

• Generación de colecciones de ESTs(etiquetas de secuencia expresadas).
• La complejidad de los genomas eucariotashace aconsejable no abordar inicialmente el estudio del genoma completo.
• Es preferible estudiar aquellos genes que se están expresando en un momento determinado de la vida del organismo.
ESTs (Expressed Sequenced Tags)

ESTs

• Genoteca de cDNA: colección de fragmentos de DNA clonados que representan el conjunto de genes que se están expresando en un órgano o tejido determinado, o bajo una situación particular o momento de desarrollo.
• Las genotecas de cDNA se secuencian de forma masiva para generar miles de secuencias parciales o ESTs de 200-500 bp.
ESTs
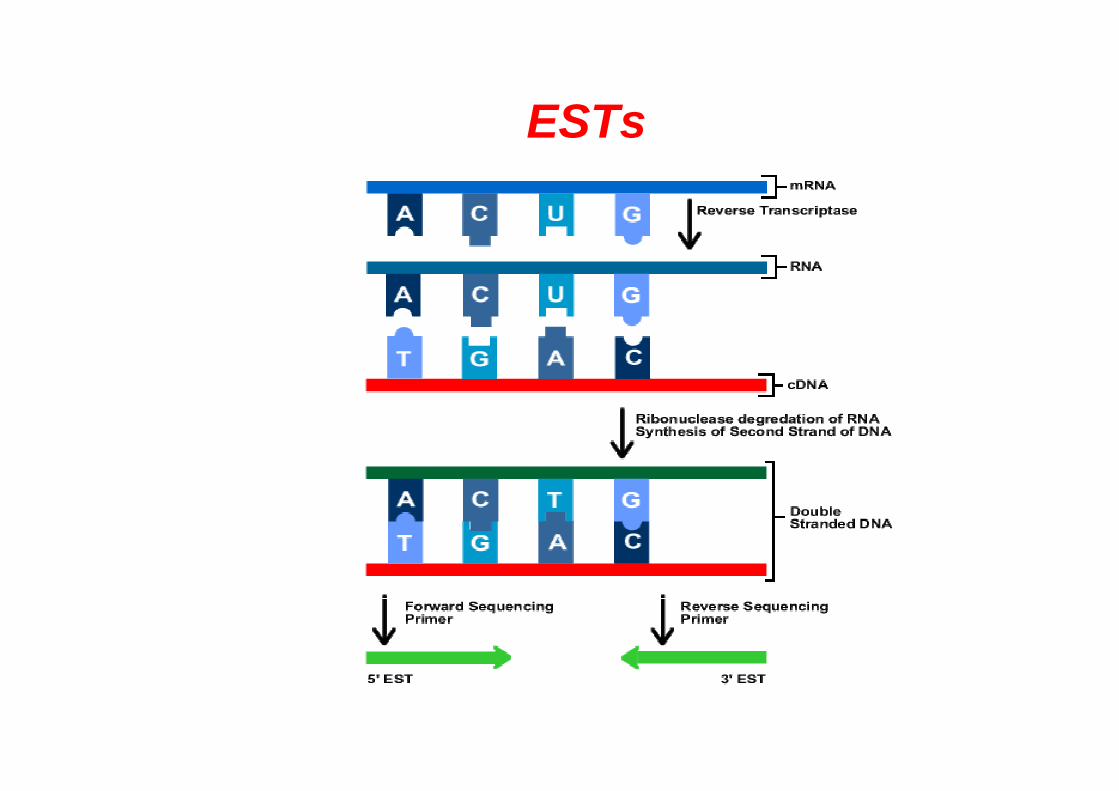
ESTs

• Las diferencias en la expresión de genes pueden ser identificadas considerando el número de veces en que aparece representada una EST particular.
• Las ESTs por su propia naturaleza, son incompletas y, hasta cierto punto, imprecisas.
• Las ESTs también suelen ser suficientes para la identificación de los genes mediante comparación con las bases de datos.
ESTs

• Versión acelerada de la secuenciación de ESTs.
• Un segmento corto procedente de un mRNA(etiqueta SAGE) es suficiente para identificar inequívocamente a un gen completo.
• La etiqueta corta tiene que estar ubicada en una posición definida dentro de la secuencia del mRNA.
Tecnología SAGE (Serial Analysis Gene Expression)

• Generación de etiquetas SAGE (Tags) de secuencias (10-14 bases).
• Ligación de las etiquetas SAGE para obtener concatémeros que pueden ser clonados y secuenciados.
• Comparación de los datos de secuencia para determinar diferencias en la expresión de los genes.
Tecnología SAGE

Tecnología SAGE
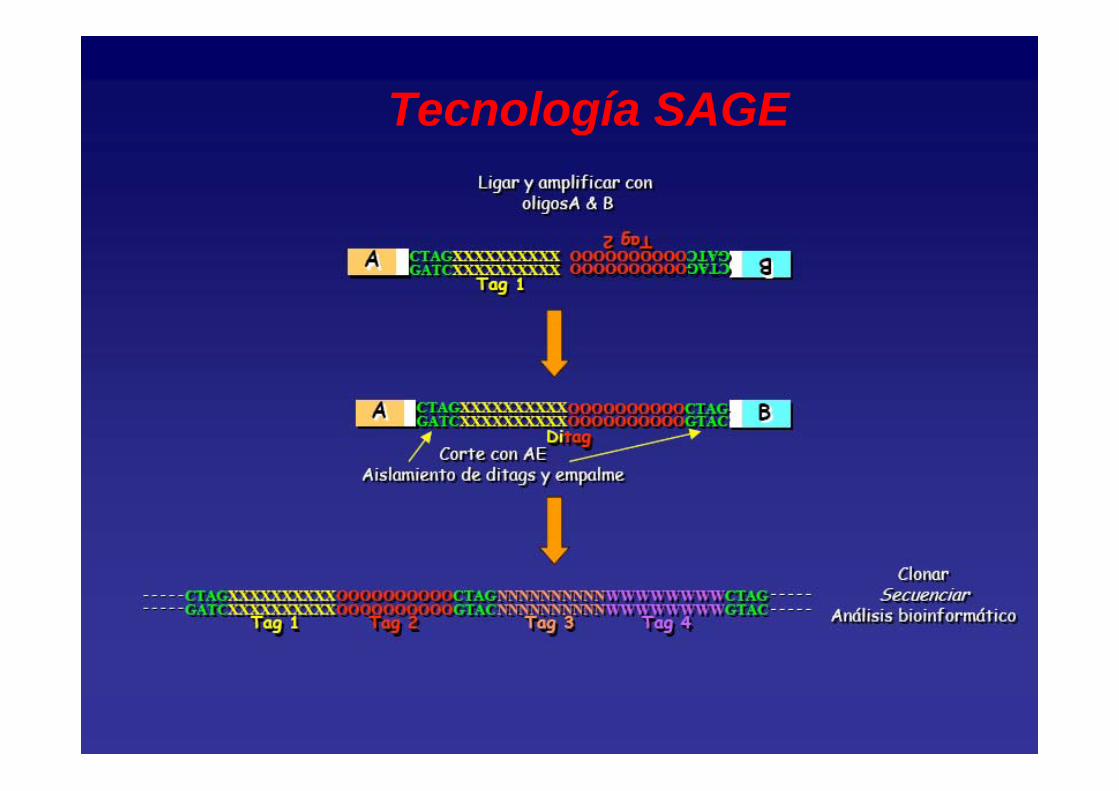
Tecnología SAGE

Tecnología SAGE

• Principalmente los ESTs brindan información de secuencia, mientras que el SAGE provee datos cuantitativos describiendo la abundancia de transcritos.
• Determina el nivel de expresión para cada gen, y contribuye al descubrimiento de nuevos genes.
• Muy buena correlación con la abundancia de RNA mensajero en la célula.
Tecnología SAGE: Ventajas

• Muy laborioso, laboratorios especializados y complejidad análisis de datos.
• Problemas técnicos:• Digestión incompleta con la enzima
que genera extremos cohesivos.
• Problemas con la secuenciación masiva.
Tecnología SAGE: Problemas

Tecnología SSH

Tecnología SSH

Microarrays de DNA

• Los microarrays de DNA surgen de la necesidad de analizar la cantidad de información procedente de los grandes proyectos de secuenciación de genomas.
• Permiten elaborar mapas finos de transcripción y proporcionan información indirecta de los niveles de proteínas.
Microarrays de DNA

• El análisis de microarrays de DNA es una nueva tecnología que permite estudiar simultáneamente la expresión de miles de genes y analizar su expresión bajo distintas condiciones experimentales.
• Los microarrays de DNA constan de miles de conjuntos ordenados de moléculas de DNA de secuencia conocida depositados en un soporte sólido (~ 2 cm2) como cristal, nylon o silicio.
• Cada combinación (gen/muestra) se localiza de forma inequívoca en un punto del microarray.
Microarrays de DNA

• Los microarrays de DNA permiten la medida simultánea de los niveles de expresión de miles de genes (sondas) en un solo experimento de hibridación con una mezcla compleja de DNA o RNA (dianas).
• Sondas: secuencias de DNA conocidas (oligonucleótidos o productos de PCR) inmovilizadas ordenadamente sobre una superficie sólida.
• Dianas: muestra problema de DNA o RNA marcada cuya abundancia será determinada por hibridación.
Microarrays de DNA
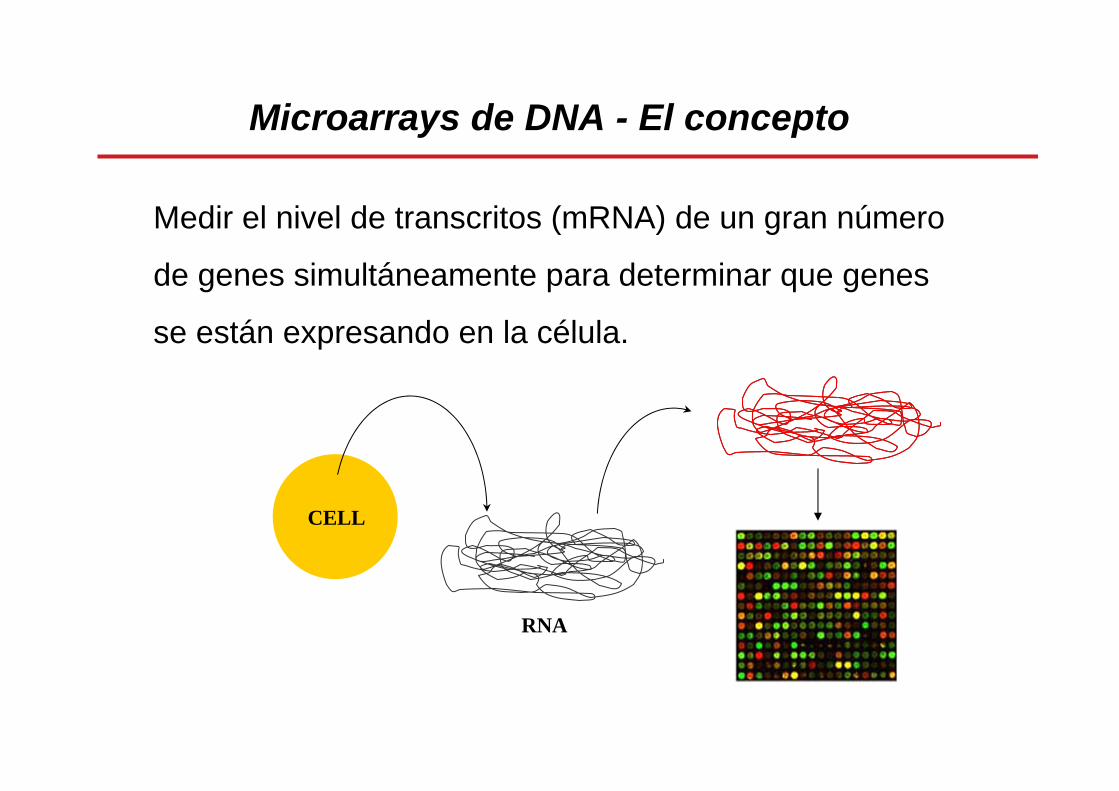
Microarrays de DNA - El concepto
Medir el nivel de transcritos (mRNA) de un gran número
de genes simultáneamente para determinar que genes
se están expresando en la célula.
CELL
RNA

• El objetivo de los experimentos con microarrays de DNA es comparar la expresión de múltiples genes(transcripción) en distintas condiciones:
• Momentos distintos del tiempo• Tejidos distintos• Tejidos sanos o enfermos (p.e. Tumores)
• Se basan en tecnologías conocidas como la hibridación y la fluorescencia.
Microarrays de DNA – El objetivo

Microarrays de DNA - Hibridación
A
A
AT
TG
GC
C
T
AT
GA
TGC
C
A
A
AT
TG
GC
C
T
AT
GA
TGC
C

Mediante hibridación, pueden detectar DNA o RNA:
Si el DNA o RNA hibridadoestá marcado fluorescentemente puede ser cuantificado mediante escaneado del chip de DNA.
Microarrays de DNA - Hibridación

• Cada sonda del microarray de DNA está diseñada para unirse a un gen de forma específica.
• Diseño de sondas específicas:• Especificidad de secuencia.• Tms homogéneas.• Sin estructuras secundarias.
• Cada sonda está dispuesta de forma ordenada sobreel microarray de DNA.
Microarrays de DNA – Sondas

Sondas de DNA específicas de gen
cDNA marcado
gen
mRNA
Microarrays de DNA – El proceso

• Los microarrays de DNA están formados por 100 - 1 millón de sondas de DNA sobre una superficie de1 cm por 1 cm (chip de DNA).
• Los resultados de microarrays de DNA se basan en el concepto de “culpable por asociación”.
• Genes que son co-regulados (patrón similar de comportamiento) es probable que estén funcionalmente relacionados formando parte del mismo proceso biológico.
Microarrays de DNA – El resultado

• Los microarrays de DNA son una revolución por su capacidad de realizar experimentos inconcebibles hasta hace poco tiempo.
• Los nuevos chips de DNA podrán contener (y por tanto estudiar) el genoma humano en 2 cm2.
• Esto genera cantidades ingentes de datos que deben ser almacenadas, procesadas y analizadas:
- Al tratarse de una nueva técnica la mayor parte de métodos, protocólos o estándares se están aún definiendo.
Microarrays de DNA – El reto

• 45 Genes de Arabidopsis y 3 genes control: total 48 señales.
El primer Microarray de DNA
• Schena et al., (1995). Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270, 467-470.

Microarrays de DNA – Experimento básico
• Un experimento básico de microarrays de DNAconsiste en:
1- Diseño y fabricación del microarray.2- Preparación de la muestra e hibridación.3- Escaneo del microarray.4- Análisis de imagen.5- Análisis de los resultados.

Preparación MuestraHibridación
Diseño Array Diseño Sonda
PREGUNTADiseño Experimental
Compra Chip/Array
RESPUESTA
Análisis Avanzado de DatosExtracción de Genes Relevantes
Análisis Estadístico
Preprocesamiento de datosNormalización
Análisis de Imagen
Microarrays de DNA

Microarrays de DNA – Experimento básico

Diseño y fabricación

Diseño y fabricación
• La primera fase del diseño del microarray de DNA consiste en la selección de los genes que se desean incorporar al experimento.
• Las secuencias necesarias pueden obtenerse, por ejemplo, de una base de datos de ESTs.
• Puede haber problemas en la identificación de las secuencias :
• Errores de secuenciación• Splicing alternativo• Contaminación

Diseño y fabricación - Sondas
• Una vez seleccionados los genes se realizan múltiples copias de cada uno mediante PCR, y los productos (sondas) se depositan en el sustrato.
• Tipos de sondas:• Genotecas de cDNA (Stanford microarrays)• Oligonucleótidos (Affymetrix)
• El soporte sólido (sustrato) del microarray suele ser cristal, y también membranas de nylon o plástico.

Diseño y fabricación – Adhesión sondas
• La adhesión de las sondas sobre el sustrato puede hacerse mediante diversas técnicas:
• Impresión mecánica (capilaridad) o microinyección (Ink-jet) → Stanford microarrays
• Fotolitografía → Affymetrix
• Existen dos tipos de tecnologías de microarrays de DNA:• Arrays de cDNAs: Stanford Microarrays.• Arrays de oligonucleótidos: Affymetrix.

Preparación de la muestra

Preparación de la muestra
• Paralelamente al diseño y fabricación del microarray que contiene los genes (sondas) cuya expresión se desea estudiar...
• ...deben prepararse las muestras problema (dianas)en las que se desea estudiar la expresión de estos genes.
• Extracción de todo el mRNA de las células en las condiciones que se desea estudiar, y posterior marcaje del mRNA.

Preparación de la muestra
1. Diseño experimental¿Pregunta?¿Réplicas?
2. Realizar experimento
4. Marcaje RNA¿Amplificación?¿Directo o indirecto?¿Tipo de marcaje?
silvestremutante
3. Precipitar RNA¿Eucariota/procariota?¿Pared celular?

Hibridación

Hibridación
• Una vez preparadas y marcadas las muestras problema (dianas) se depositan sobre las sondas en el microarray de DNA.
• Esto hará que los cDNAs o cRNAs de las muestras problema se puedan hibridar con los de cada gen contenido en las sondas del microarray de DNA.
• La hibridación tendrá lugar en un grado proporcional a la expresión del gen de cada sonda en cada muestra problema.

Hibridación
• En arrays de cDNA las muestras problema se juntan y se hibridan en un único microarray.
• En arrays de oligonucleótidos las muestras se hibridan por separado en dos microarrays idénticos.
• Tras la hibridación, el microarray se lava para eliminar el material que no se ha hibridado.
• La intensidad de la señal de hibridación resultante es proporcional a la cantidad de mRNA que corresponde a esa secuencia en la muestra original.

Hibridación
• La detección de la hibridación es un paso clave para determinar qué sondas se han unido a sus dianas complementarias procedentes de la muestra.
• Las principales técnicas de detección de la hibridación requieren del marcaje previo de las dianas:
• cDNA: Stanford Microarrays.• cRNA: Affymetrix.

Cy5 Cy3
Arrays de cDNA Arrays de oligonucleótidos
Microarrays de DNA: el paradigma de una técnica post-genómica

Microarrays de DNA - La tecnología
Stanford Microarrays
Affymetrix(GENECHIP)

Stanford Microarrays

Stanford Microarrays – Arrays de cDNA
Portas de cristal
Impresión de las sondas
Post-procesamiento
Hibridación

Stanford microarrays – Impresión robótica
Arrays de cDNA

Stanford microarrays – Impresión robótica
Mecánica (capilaridad)
Ink-jet (microinyección)



Stanford microarrays – Impresión robótica
• Tamaño del lunar: 100-300 µm (Ø)• Espaciado: 150-300 µm • Número lunares I. mecánica: 250-1000 lunares/cm2
• Número lunares Ink-jet: >2500 lunares/cm2
• Cantidad DNA: <10 pg DNA
• Tipo de substrato:• Portas recubiertos (polylisina, silano,
superaldehido, estreptavidina)• Membrana de nylon

Stanford microarrays – Perfiles de expresión génica
Clones DNA PCR Purificarproductos
Impresiónrobótica
Preparar Microarray
Muestra A AislarRNA
Marcaje con Cy5Aislar RNA y marcar
Muestra B AislarRNA Marcaje con Cy3
Mezclar, hibridar sondas y analizar datos
Hibridar al microarray Lavar Analizar datos

Stanford microarrays – Marcaje dianas (cDNA)
Fluorescencia (Cyanine 3 vs. Cyanine 5)Radioactividad (3H vs. 35S o 33P vs. 14C)
Los dos marcajes más comunes son los fluorocromosde cianina:
Cy3, absorción 554 nm, emisión 568 nmCy5, absorción 650 nm, emisión 672 nm
Pero también se utilizan fluorocromos Alexa:Alexa Fluor 546Alexa Fluor 647
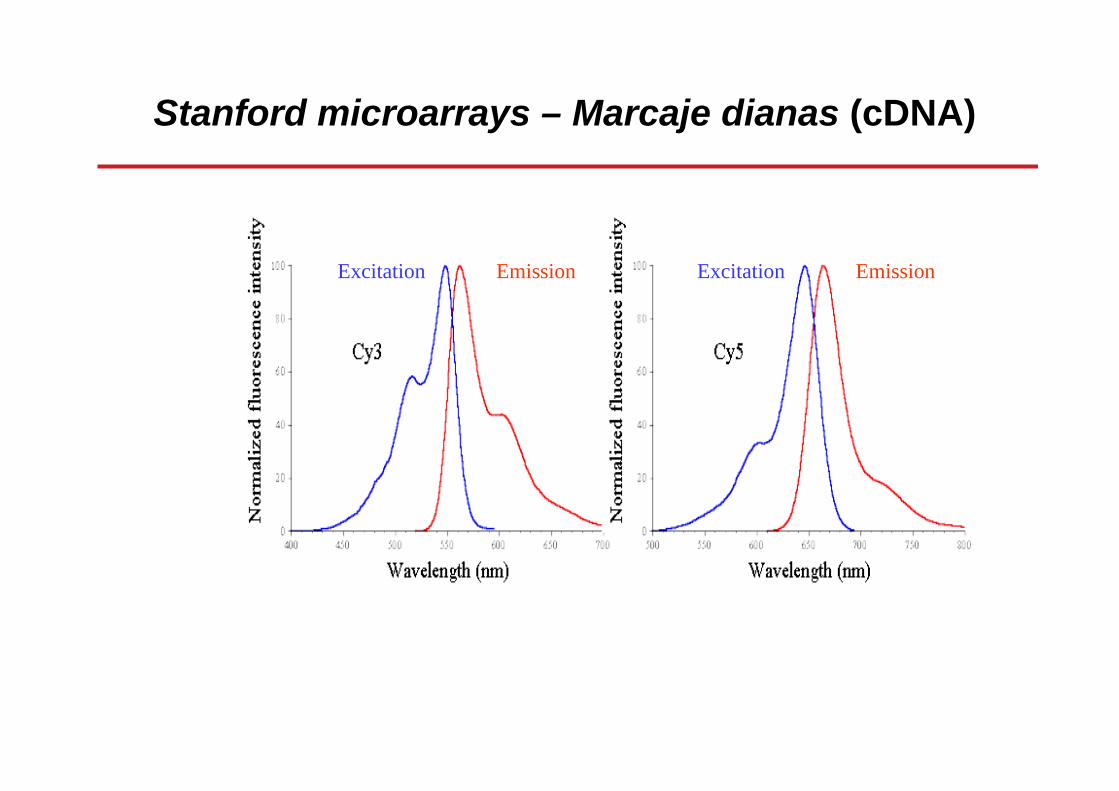
Stanford microarrays – Marcaje dianas (cDNA)
Excitation Emission Excitation Emission

Stanford microarrays – Marcaje dianas (cDNA)

Retrotranscripción: Obtener cadenas de cDNA complementarias al mRNA
G U A A U C C U CTranscriptasa
Reversa
mRNA
cDNA
C A T T A G G A GC A T T A G G A GC A T T A G G A GC A T T A G G A G
T T A G G A G
C A T T A G G A GC A T T A G G A GC A T T A G G A GC A T T A G G A G
C A T T A G G A G
Stanford microarrays: marcaje dianas (cDNA)

Cy5 Cy3
Stanford microarrays: marcaje dianas (cDNA)

Cy5 Cy3
Stanford microarrays: el proceso

mRNAmRNA
cDNAcDNA
Cy3-cDNACy5-cDNA
Muestra A Muestra BStanford microarrays
IMPRESIÓN
SONDAS

Stanford microarrays – Detección marcaje
• Las muestras hibridadas sobre el microarray se iluminan sucesivamente con luz láser de dos colores distintos para estimular la fluorescencia de uno u otro fluorocromo.
• La cantidad de mRNA unido a una muestra se puede medir por la intensidad de la fluorescencia emitida al ser iluminada por el láser del color correspondiente.

Stanford microarrays – Detección marcaje

Stanford microarrays – Detección marcaje
• Si la muestra 1 se marca con rojo y la muestra 2 con verde se obtendra en cada punto del microarray que:• Si el RNA de la muestra 1 abunda más que el de
la otra muestra se detecta como un punto rojo.• Si el RNA de la muestra 2 abunda más que el de
la otra muestra se detecta como un punto verde.• Si ambos se expresan por igual se detecta como
un punto amarillo.• Si en ninguna de las dos muestras hay mRNA se
detecta como un punto negro.

Stanford microarrays – Detección marcaje
• Las intensidades de las fluorescencias emitidas permiten determinar los niveles relativos de expresiónde los genes en ambas muestras problema.

Stanford microarrays: problemas
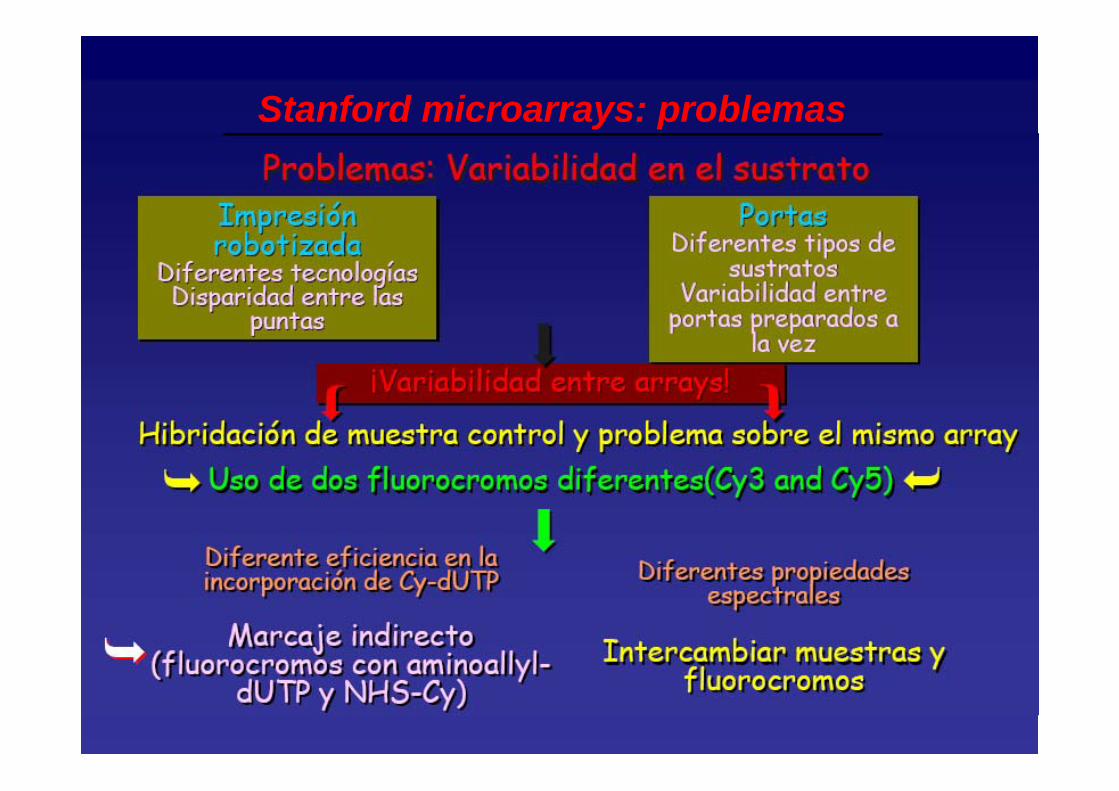
Stanford microarrays: problemas

La tecnología Affymetrix

La tecnología Affymetrix - Genechip®

La tecnología Affymetrix - Sondas
• Arrays de oligonucleótidos: síntesis in situ de oligonucleótidos de 25 bases sobre una superficie cuadrada de cristal (1.3 cm x 1.3 cm) mediante fotolitografía.
• 11-20 parejas de sondas específicas para cada gen.
• Sobrerepresentación extremos 3´de los mRNA.
• Seleccionadas para maximizar las temperaturas de hibridación y la especificidad.

La tecnología Affymetrix - Sondas
• Tamaño del lunar: ~150 µm (Ø).
• Densidad: 10.000-250.000 oligonucleótidos/cm2.
• Millones de copias de cada oligonucleótido específico (107-108 copias).
• Un array de oligonucleótidos puede contener 400.000 sondas (aproximadamente 20.000 genes).
• El array de S. cerevisae contiene 6.000 oligos, que representan todos sus genes conocidos.

La tecnología Affymetrix - Sondas
• Para cada gen existen dos sondas: una de homología perfecta (PM, Perfect Match) de 25 bases y otra con una error deliberado/mutación (MM, MisMatch) en la zona central.
• Buena calidad de datos/ baja varianza.
• La presencia de numerosos genes de control permite una casi perfecta normalización entre diferentes experimentos.
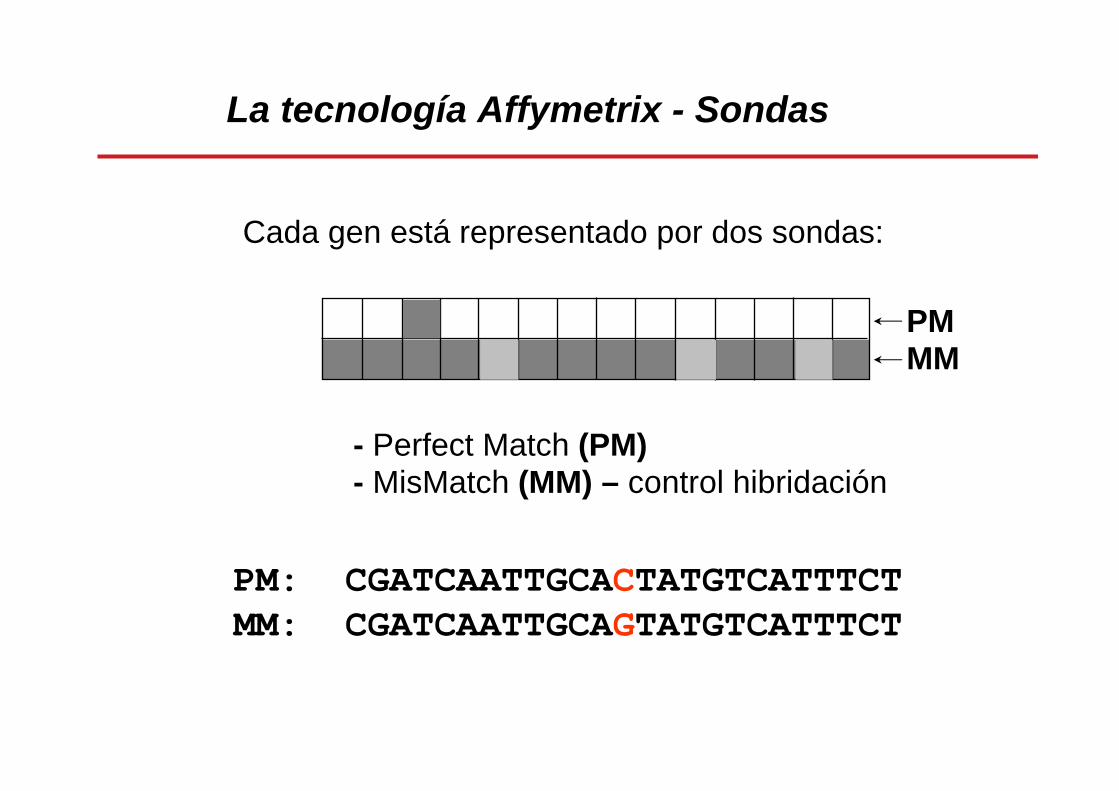
La tecnología Affymetrix - Sondas
Cada gen está representado por dos sondas:
- Perfect Match (PM)- MisMatch (MM) – control hibridación
PMMM
PM: CGATCAATTGCACTATGTCATTTCT MM: CGATCAATTGCAGTATGTCATTTCT
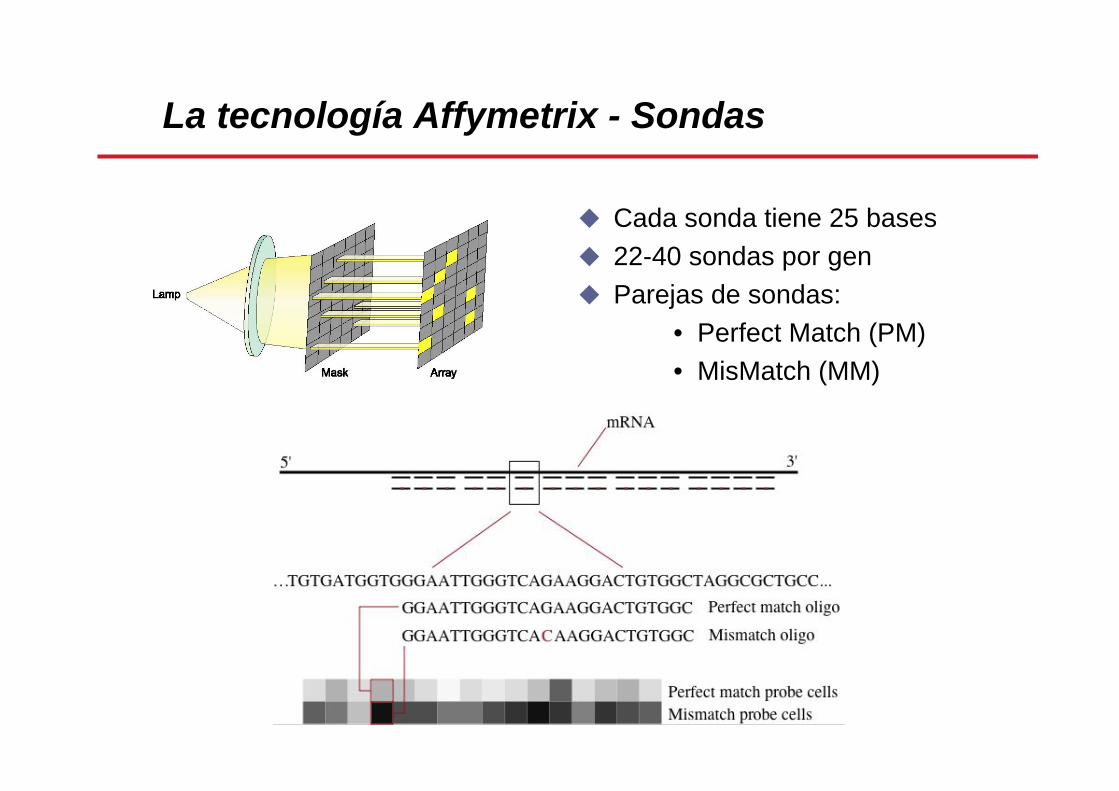
Cada sonda tiene 25 bases22-40 sondas por genParejas de sondas:
• Perfect Match (PM) • MisMatch (MM)
La tecnología Affymetrix - Sondas

Cy5 Cy3
La tecnología Affymetrix: síntesis sondas
Síntesis in situ mediante fotolitografía

TTT
T
T
TT
T
T
T
A
A
AA
A
A
A
AAA
Espaciadores unidos a la superficie de cristal con grupos protectores fotolábiles
Mask #1Mask #2
La tecnología Affymetrix – Síntesis sondas
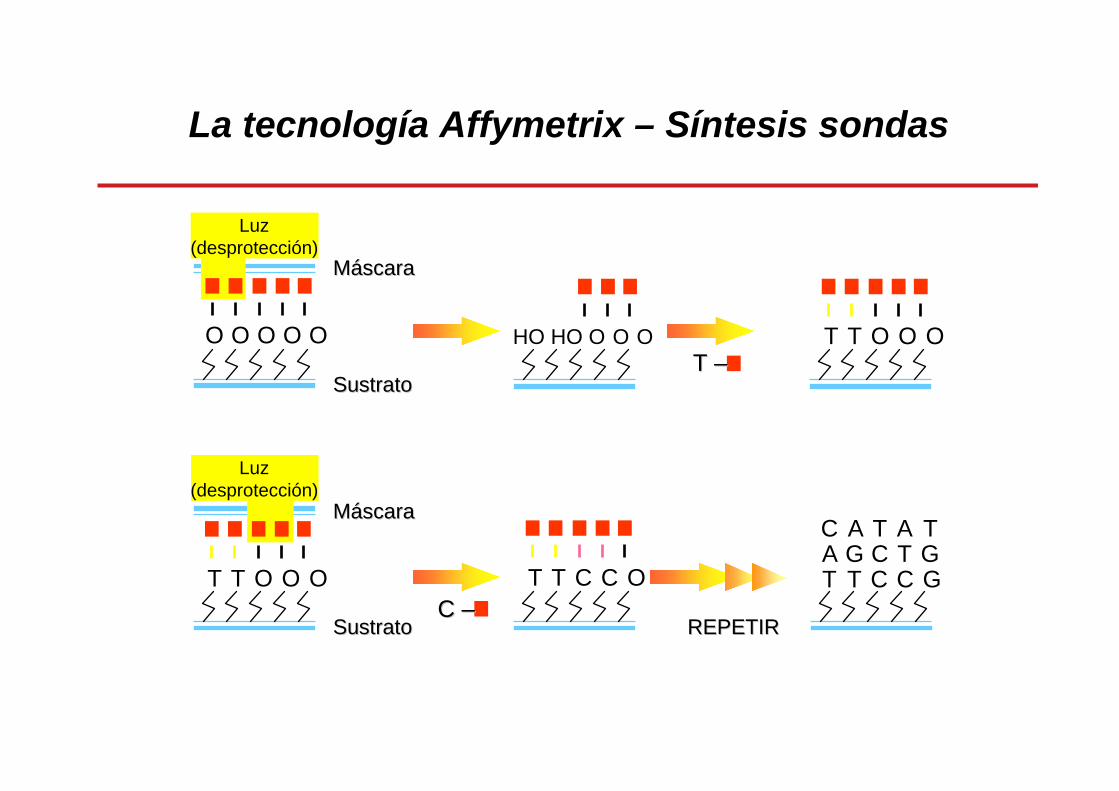
O O O O O
Luz(desprotección)
HO HO O O O T T O O O
T T C C O
Luz(desprotección)
T T O O O
C A T A TA G C T GT T C C G
MMááscarascara
SustratoSustrato
MMááscarascara
SustratoSustrato
T T ––
C C ––REPETIRREPETIR
La tecnología Affymetrix – Síntesis sondas
Cy5 Cy3
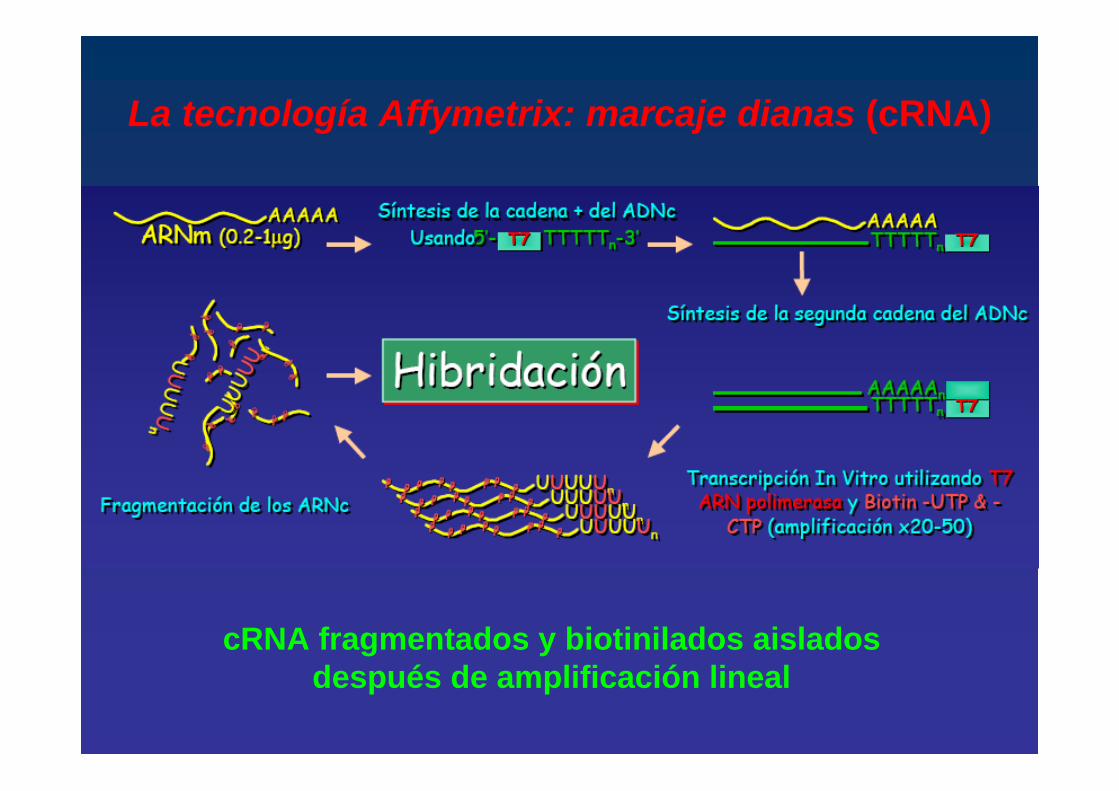
La tecnología Affymetrix: marcaje dianas (cRNA)
cRNA fragmentados y biotinilados aislados después de amplificación lineal

La tecnología Affymetrix - Equipamiento
Fluidic Station Scanner

La tecnología Affymetrix – El proceso

Cy5 Cy3
La tecnología Affymetrix: el proceso
Cy5 Cy3
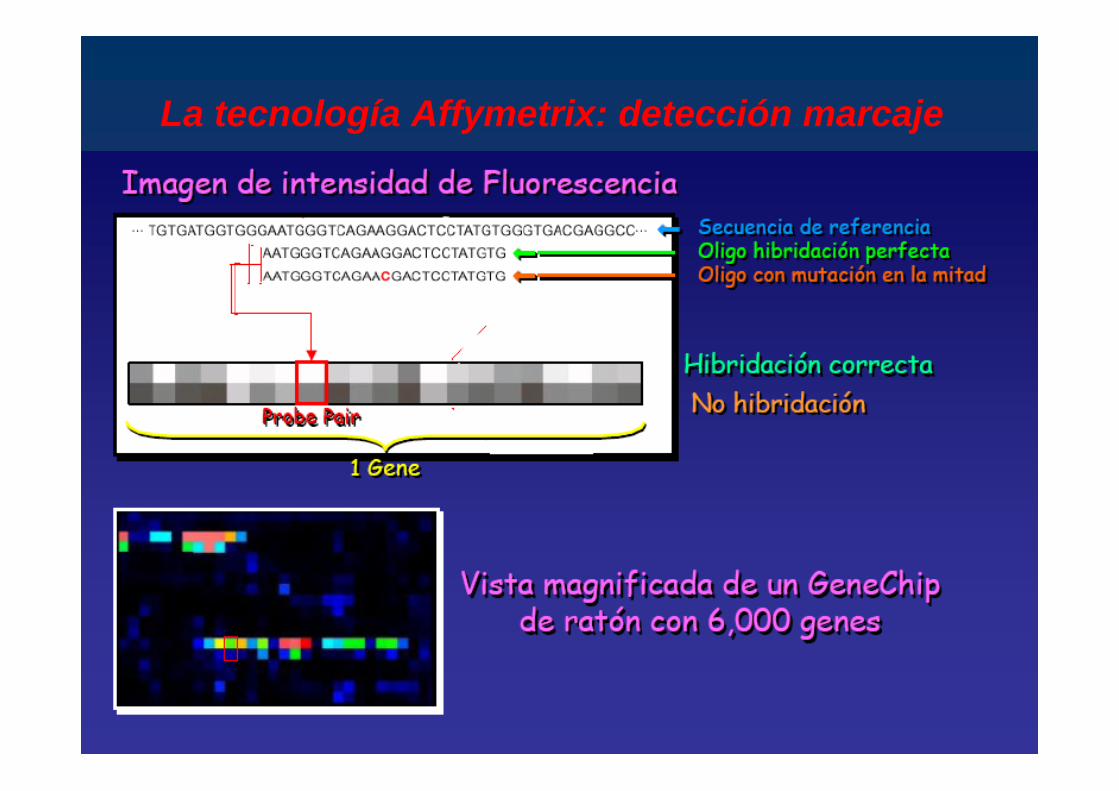
La tecnología Affymetrix: detección marcaje

La tecnología Affymetrix – El experimento

2424µµmm
Imágen de un Genechip hibridado
>200,000 differentessondas complementarias
Diana cRNA de cadenasencilla marcadoSonda Oligonucleotido
* **
**
1.28cm
GeneChipCélula de hibridación
La tecnología Affymetrix
Millones de copias de cadaoligonucleótido específico(107-108 copias)

La tecnología Affymetrix

La tecnología Affymetrix – Arrays comerciales
HumanoRatónRataArabidopsisC. elegansPerroDrosophila
E. coliP. aeruginosaPlasmodium/AnophelesVitis vinifera (uva) Xenopus laevisS. cerevisiaePez cebra

ResumenMicroarrays de DNA

Microarrays de DNA - Comparativa
Stanford microarrays: Flexible, también especies sin secuenciar Requiere menor presupuestoCalidad de datos: media-alta
Affymetrix: No flexible, sólo especies secuenciadasEquipamiento caro Calidad de datos: alta

Análisis de imágen

Preparación MuestraHibridación
Diseño Array Diseño Sonda
PREGUNTADiseño Experimental
Compra Chip/Array
RESPUESTA
Análisis Avanzado de DatosExtracción de Genes Relevantes
Análisis Estadístico
Pre-procesamiento de datosNormalización
Análisis de Imagen
Microarrays de DNA

• Esta nueva forma de experimentar requiere de nuevas herramientas de análisis y visualización de resultados.
• Cada experimento de microarrays de DNA genera una gran cantidad de datos y es preciso realizar un procesamiento apropiado de los mismos.
• Transformación de las imágenes en números.
Análisis de imágen

Análisis de imágen
Escaneado-Escáner Confocal -Escáner CCD
Formatos archivos imágen
Análisis de imágen-Localización de los puntos-Segmentación de los puntos-Evaluación calidad de los datos

Análisis de imágen – Escaneado
Escaneado del porta

Análisis de imágen – Escáner confocal
Microscopía confocal
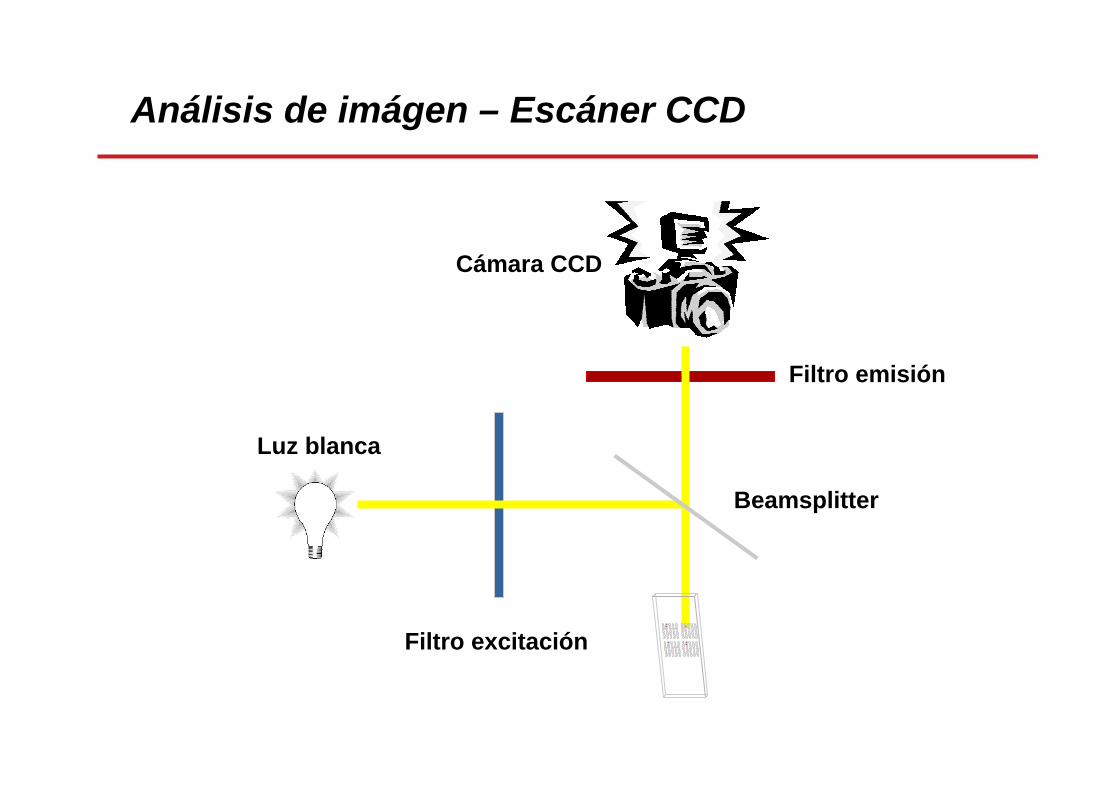
Cámara CCD
Filtro emisión
Filtro excitación
Beamsplitter
Luz blanca
Análisis de imágen – Escáner CCD

Los escáners generan un archivo gráfico y el formato de archivo de imágen más común es TIFF de 16 bits.
Un archivo TIFF de 16 bits describe cada pixel en una imagen con una intensidad entre 0 y 65535.
Normalmente dos escáners en diferentes longitudes de onda originan dos archivos monócromos que se superponen.
Canal Cy3
Canal Cy5
COMPOSICIÓN
Análisis de imágen – Formato archivos imágen

DOS COLORESMuestra 1 marcada en rojo (Cy5)Muestra 2 marcada en verde (Cy3)
Rojo: gen inducido en Muestra 1Verde: gen inducido en Muestra 2Amarillo:-niveles similares de expresiónRojo/Verde: ratio de expresión
UN COLORLa intensidad de la expresión de un gen utilizando las sondas (PM)(en algunos casos MM- control)
Los archivos gráficos generados porel escáner se analizan mediante diferentes programas informáticos.PM/MM
Análisis de imágen – Formato archivos imágen

Hígado (Cy5) / Cerebro (Cy3)
Pérfiles de expresión génica en Hígado y Cerebro
Análisis de imágen – Stanford microarrays

Affymetrix Human Genome U95A Genechip hibridado con cerebro fetal
Análisis de imágen – La tecnologia Affymetrix

La identificación y cuantificación de las señales de hibridación (puntos) puede realizarse de forma manual, automática y semiautomática.
Se dibuja una parrilla sobre la imagen para ayudar al programa en la identificación de puntos individuales.
Análisis de imágen – Localización de los puntos

ImaGene, QuantArrayHistogram method
SpotAdaptive shape
GenePix, DappleAdaptive circle
ScanAlyze, GenePix, QuantArrayFixed circle
Análisis de imágen – Segmentación de los puntos
Clasificación de cada pixel en cada imagen como señalo ruido de fondo.
Para cada punto individual obtener las medidas de:Señal, Fondo y Calidad.
Método Programa

Intensidad de los puntos: calculo de la media de los pixel en cada punto.
Corrección del ruido de fondo: local o global.
Análisis de imágen – Segmentación de los puntos

La mayor parte de las irregularidades se puedendetectar por las siguientes medidas:
Variabilidad de la intensidadDesviación del tamaño de puntoDesviación de la circularidadIntensidad de señal relativa al fondoDesviación de la posición en la parrilla
Análisis de imágen – Evaluación calidad de los datos
En base a estas medidas, se pueden descartar puntosirregulares.

Análisis de imágen – Evaluación calidad de los datos
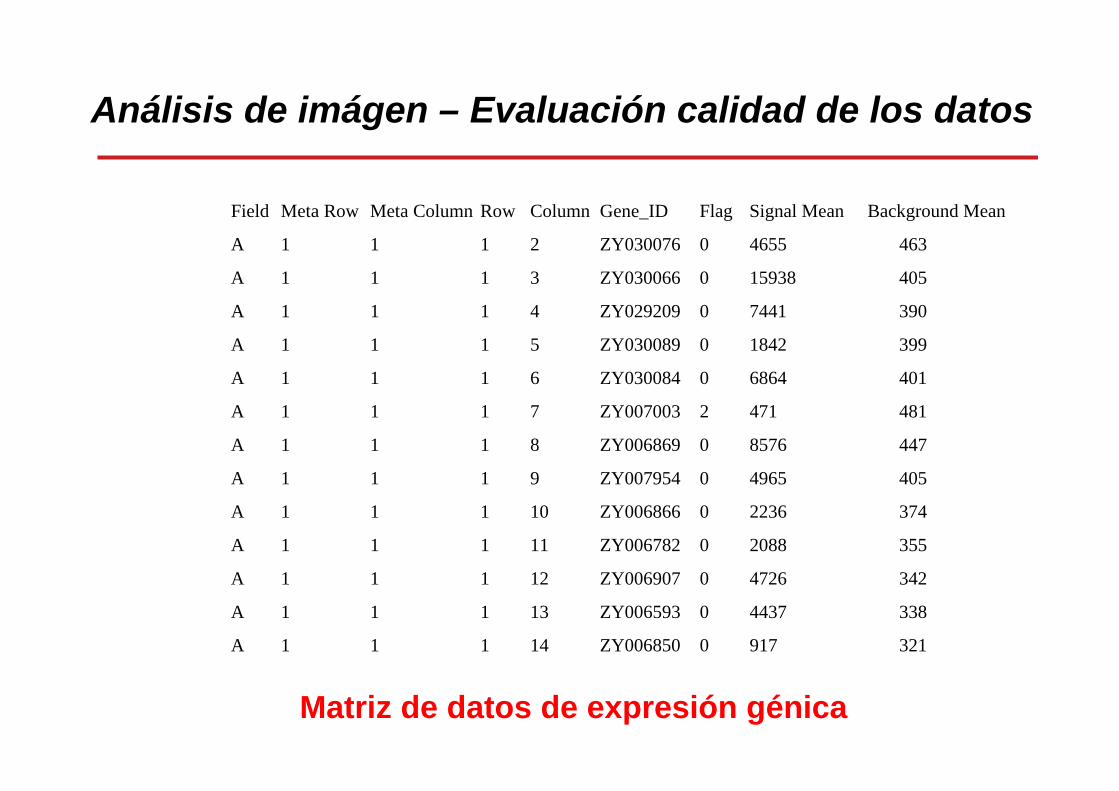
Field Meta Row Meta Column Row Column Gene_ID Flag Signal Mean Background Mean
A 1 1 1 2 ZY030076 0 4655 463
A 1 1 1 3 ZY030066 0 15938 405
A 1 1 1 4 ZY029209 0 7441 390
A 1 1 1 5 ZY030089 0 1842 399
A 1 1 1 6 ZY030084 0 6864 401
A 1 1 1 7 ZY007003 2 471 481
A 1 1 1 8 ZY006869 0 8576 447
A 1 1 1 9 ZY007954 0 4965 405
A 1 1 1 10 ZY006866 0 2236 374
A 1 1 1 11 ZY006782 0 2088 355
A 1 1 1 12 ZY006907 0 4726 342
A 1 1 1 13 ZY006593 0 4437 338
A 1 1 1 14 ZY006850 0 917 321
Análisis de imágen – Evaluación calidad de los datos
Matriz de datos de expresión génica

Normalización

Preparación MuestraHibridación
Diseño Array Diseño Sonda
PREGUNTADiseño Experimental
Compra Chip/Array
RESPUESTA
Análisis Avanzado de DatosExtracción de Genes Relevantes
Análisis Estadístico
Pre-procesamiento de datosNormalización
Análisis de Imagen
Microarrays de DNA

• En general, los niveles de expresión de genes individuales se miden por:
• log (R/G)• log (PM/MM)
• En cualquier experimento biológico es esencial conocer el grado de reproducibilidad de las medidas.
• La repetición de experimentos de microarrays es costosa.
• El factor limitante puede ser la cantidad de muestra biológica.
Normalización

• Pre-procesamiento de datos iniciales previo al análisis estadístico y análisis avanzado de los datos.
• Cada valor de intensidad proviene de una imagen independiente y es necesario hacer que estos valores sean comparables. Ajuste básico: igualar la intensidad media de las imágenes.
• Las intensidades no son únicamente concentraciones de mRNA, hay múltiples fuentes de variación que pueden afectar y desviar seriamente la interpretación de los resultados.
Normalización

Normalización – Fuentes de variación
Contaminación de tejidosDegradación Purificación RNATranscripción reversaEficiencia de amplificaciónEficiencia de marcaje(Cy3/Cy5)Soporte unión DNA
SpottingOtros temas relacionados
con la preparación del array Corrección del fondoSegmentación de la imagenEficiencia y especificidad de
hibridaciónEfectos espaciales

(a) Después de normalización por intensidad media
(b) Después de normalización por Lowess
(c) Después de normalización teniendo en cuenta efectos espaciales
Antes (izda.) y después normalización (dcha.).
(A) BoxPlots
(B) BoxPlots de subarrays
(C) MA plots (ratio versus intensidad)
A
B
C
Normalización

Análisis Estadístico

Preparación MuestraHibridación
Diseño Array Diseño Sonda
PREGUNTADiseño Experimental
Compra Chip/Array
RESPUESTA
Análisis Avanzado de DatosExtracción de Genes Relevantes
Análisis Estadístico
Pre-procesamiento de datosNormalización
Análisis de Imagen
Microarrays de DNA

• Prácticamente cualquier técnica estadística tiene cabida en los estudios de microarrays de DNA.
• La técnica de agrupamiento de datos más popular es el análisis de conglomerados:
• A partir de la matriz de datos de expresión génica.
• Busca formar “grupos naturales”(conglomerados o clusters) de genes o de condiciones experimentales que permitan responder las preguntas del estudio.
Análisis estadístico

• El análisis de conglomeradospermite visualizar aquellos genes cuyos perfiles de expresión son más similares.
• Para facilitar la visualización los números vuelven a convertirse en colores.
Análisis estadístico

• Otra forma usual de representar los datos es a través de un gráfico que muestre como varia la expresión del gen entre los distintos experimentos.
Análisis estadístico

Análisis de resultados

Preparación MuestraHibridación
Diseño Array Diseño Sonda
PREGUNTADiseño Experimental
Compra Chip/Array
RESPUESTA
Análisis Avanzado de DatosExtracción de Genes Relevantes
Análisis Estadístico
Pre-procesamiento de datosNormalización
Análisis de Imagen
Microarrays de DNA

• Una vez extraída la información de las imágenes hay que analizar e interpretar los resultados.
• ¿Cómo es posible organizar, visualizar y explorar el significado de millones de datos de expresión de miles de genes bajo cientos de condiciones distintas?.
• La forma de analizar los datos dependerá de lo que se desee averiguar.
Análisis de resultados

• Los patrones o perfiles de expresión génica se pueden estudiar desde dos puntos de vista:
• A) Comparaciones enfocadas en los genes: análisis de la expresión específica de los genes en experimentos comparativos (p.e . Tejidos diferentes).
• B) Comparaciones enfocadas en las muestras: estudio de las alteraciones del nivel de expresión en una determinada situación fisiológica o patológica con el objetivo de identificar los genes implicados.
Análisis de resultados

• A) Comparaciones enfocadas en los genes: Análisis de los valores de inducción/represión en una serie de experimentos comparativos.
• Esto permite la identificación de grupos de expresión, genes con patrones de expresión correlacionados.
• Si un número de genes se inducen/reprimen de la misma manera en varias situaciones, es probable que:
• Los genes estén regulados conjuntamente o,• Los genes estén relacionados funcionalmente
(participan en el mismo proceso biológico).
Análisis de resultados

• B) Comparaciones enfocadas en las muestras:Diferencias a nivel fenotípico son la causa de diferencias a nivel molecular que, en muchos casos, pueden detectarse midiendo los niveles de expresión génica.
• Identificación de genes implicados en procesos biológicos o condiciones experimentales de interés (p.e. Tratamiento hormonal, tumores, etc.).
• También se pueden identificar perfiles de expresión degenes con capacidad de diagnóstico.
Análisis de resultados

Análisis de resultados

• Estudios de expresión diferencial de genes• Análisis de patógenos• Identificación de enfermedades genéticas complejas• Detección de mutaciones y de Polimorfismos simples
de nucleótido (SNPs)• Farmacogenómica• Diseño y descubrimiento de fármacos• Estudios toxicológicos
Análisis de resultados - Aplicaciones

Análisis de resultados - Aplicaciones
Estudio de expresión diferencial de genesIdentificación de genes implicados en procesos biológicos de interés

Análisis de resultados - Aplicaciones
Estudio de distintos genotipos / FarmacogenómicaRespuesta a fármacos, toxicidad, predisposición desarrollo enfermedades, etc.

Recommended

















