Embed Size (px)
Citation preview


Trastorno cromosómico hereditario, afecta a lamayoría de los bebés durante sus primerosmeses de vida.
Afecta al SNC
Frecuente en judíos ashkenazi (1/4000)
Déficit de beta-hexosaminidasa A y B

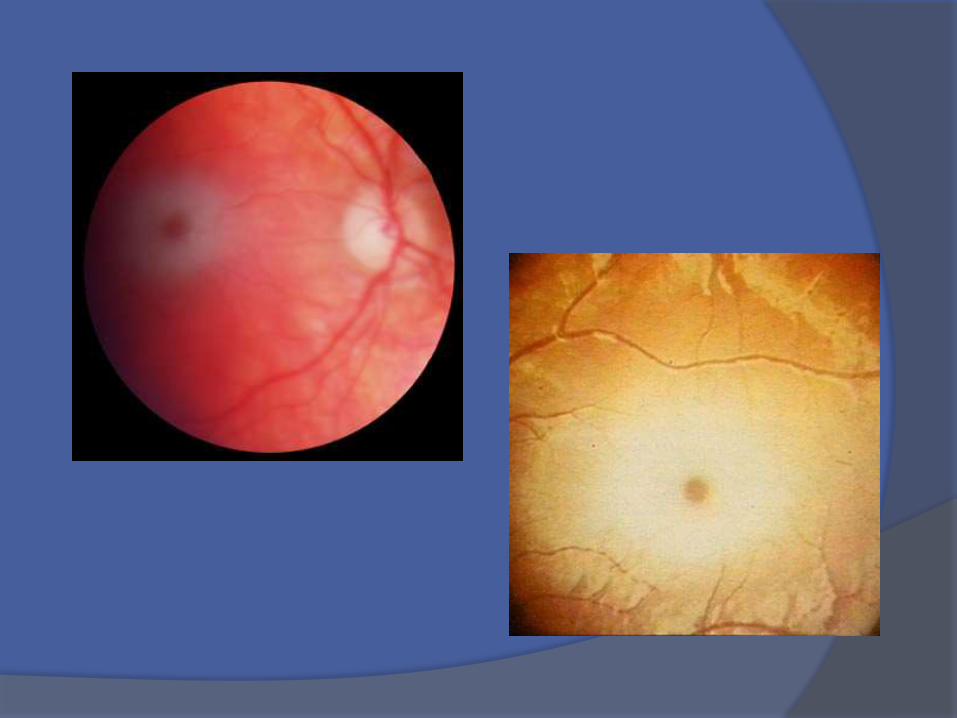
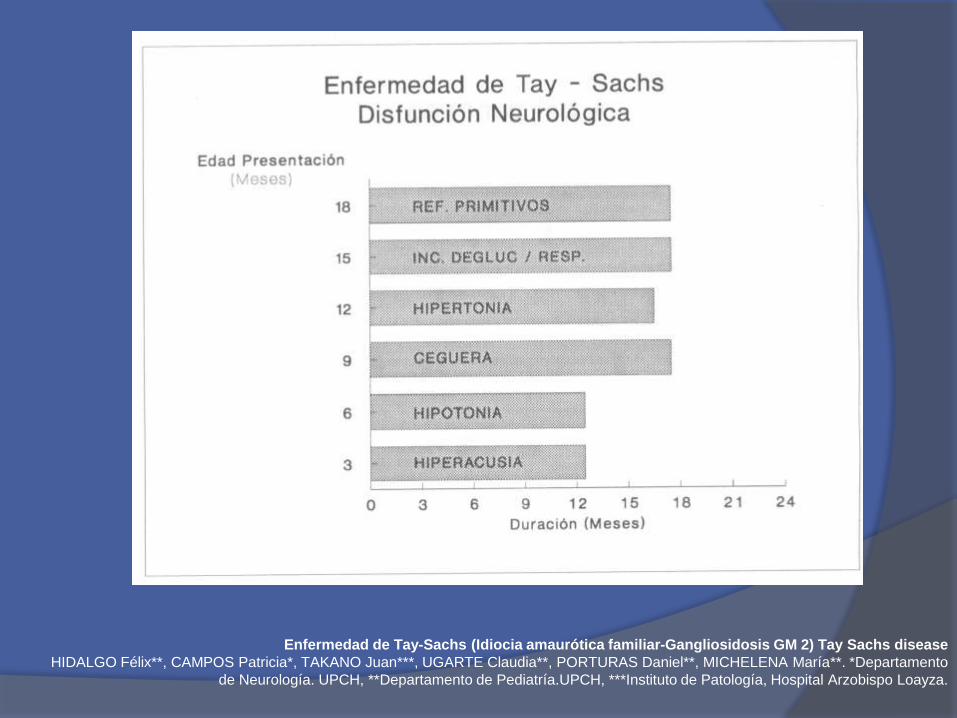
A los 5 meses hiperacusia + reducción delcontacto ocular, enfoque visual y ceguera
Retraso psicomotor con hipotonía grave al finaldel primer año de vida y convulsiones
Aumento tamaño craneal sin hidrocefalia
No alteraciones óseas
Muerte entre los 2-4 años
Forma tardía ó juvenil: ataxia y disartria
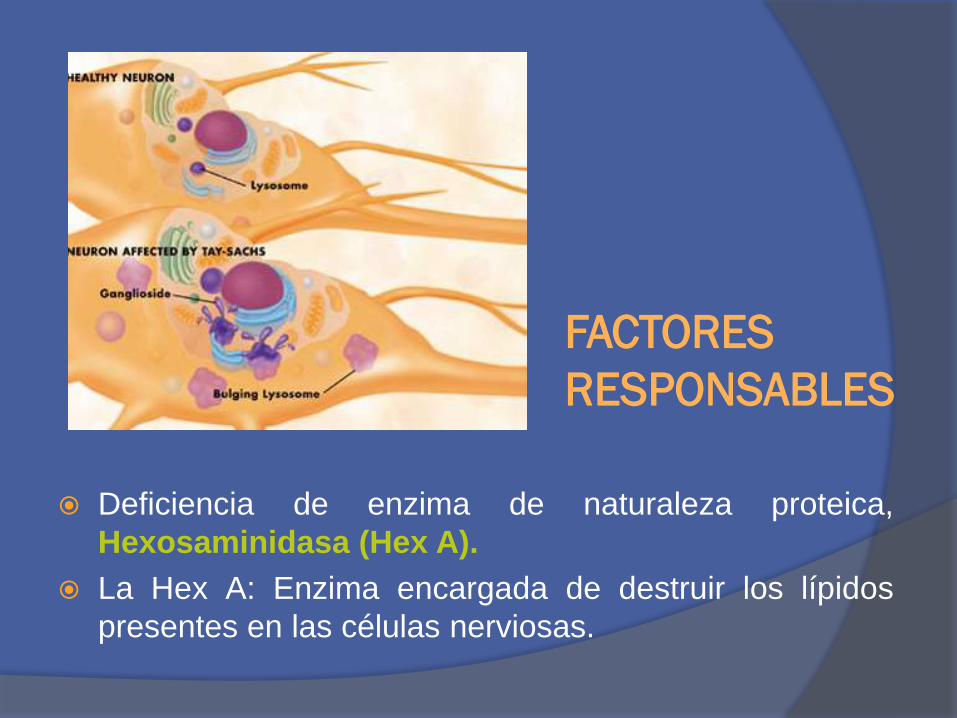
FACTORES
RESPONSABLES
Deficiencia de enzima de naturaleza proteica,
Hexosaminidasa (Hex A).
La Hex A: Enzima encargada de destruir los lípidos
presentes en las células nerviosas.
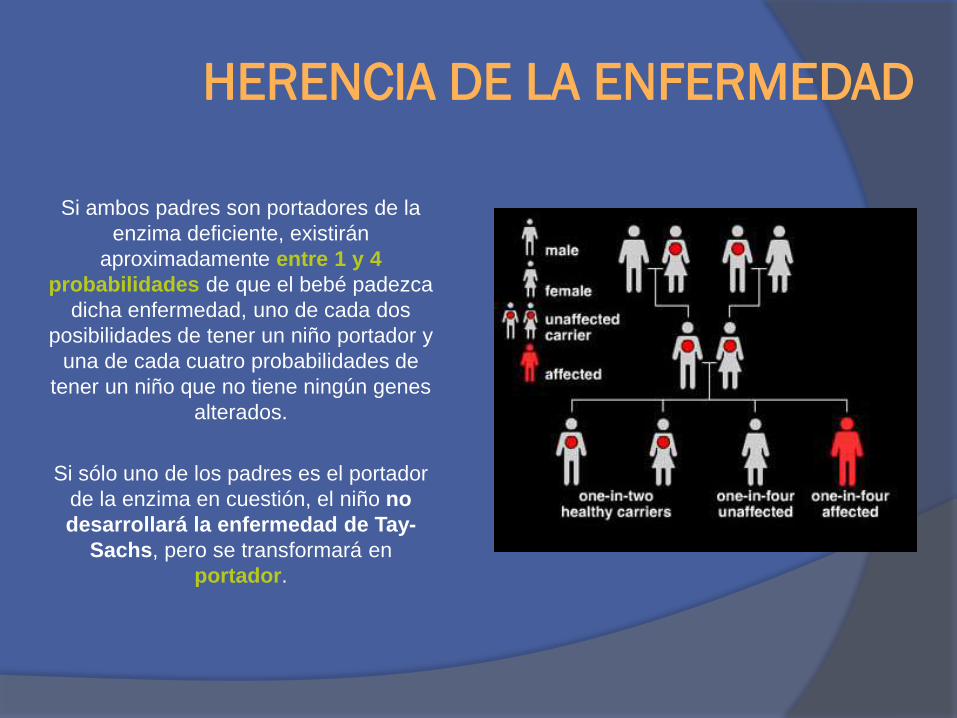
HERENCIA DE LA ENFERMEDAD
Si ambos padres son portadores de la
enzima deficiente, existirán
aproximadamente entre 1 y 4
probabilidades de que el bebé padezca
dicha enfermedad, uno de cada dos
posibilidades de tener un niño portador y
una de cada cuatro probabilidades de
tener un niño que no tiene ningún genes
alterados.
Si sólo uno de los padres es el portador
de la enzima en cuestión, el niño no
desarrollará la enfermedad de Tay-
Sachs, pero se transformará en
portador.

Los portadores de la enfermedad de
Tay-Sachs sólo cuentan con la mitad de
los niveles de HexA.
Dosaje sérico o PCR.
Amniocentesis
Muestreo de Vellosidades Coriónicas
DIAGNÓSTICO LABORATORIAL

SÍNTOMASSaludables hasta que cumplen los cuatro meses de edad.
Los síntomas más comunes incluyen:
› Declinación del contacto visual.
› Desarrollo de reflejos exagerados.
› Desarrollo físico y mental muy lento.
› Pérdida gradual de la movilidad.
› Atrofia muscular
› Parálisis.
› Ceguera gradual.
› Sordera gradual.
› Convulsiones.
› Demencia.

Enfermedad de Tay-Sachs (Idiocia amaurótica familiar-Gangliosidosis GM 2) Tay Sachs disease
HIDALGO Félix**, CAMPOS Patricia*, TAKANO Juan***, UGARTE Claudia**, PORTURAS Daniel**, MICHELENA María**. *Departamento
de Neurología. UPCH, **Departamento de Pediatría.UPCH, ***Instituto de Patología, Hospital Arzobispo Loayza.

Enfermedad de Tay-Sachs (Idiocia amaurótica familiar-Gangliosidosis GM 2) Tay Sachs disease
HIDALGO Félix**, CAMPOS Patricia*, TAKANO Juan***, UGARTE Claudia**, PORTURAS Daniel**, MICHELENA María**. *Departamento
de Neurología. UPCH, **Departamento de Pediatría.UPCH, ***Instituto de Patología, Hospital Arzobispo Loayza.

Enfermedad de Tay-Sachs (Idiocia amaurótica familiar-Gangliosidosis GM 2) Tay Sachs disease
HIDALGO Félix**, CAMPOS Patricia*, TAKANO Juan***, UGARTE Claudia**, PORTURAS Daniel**, MICHELENA María**. *Departamento
de Neurología. UPCH, **Departamento de Pediatría.UPCH, ***Instituto de Patología, Hospital Arzobispo Loayza.
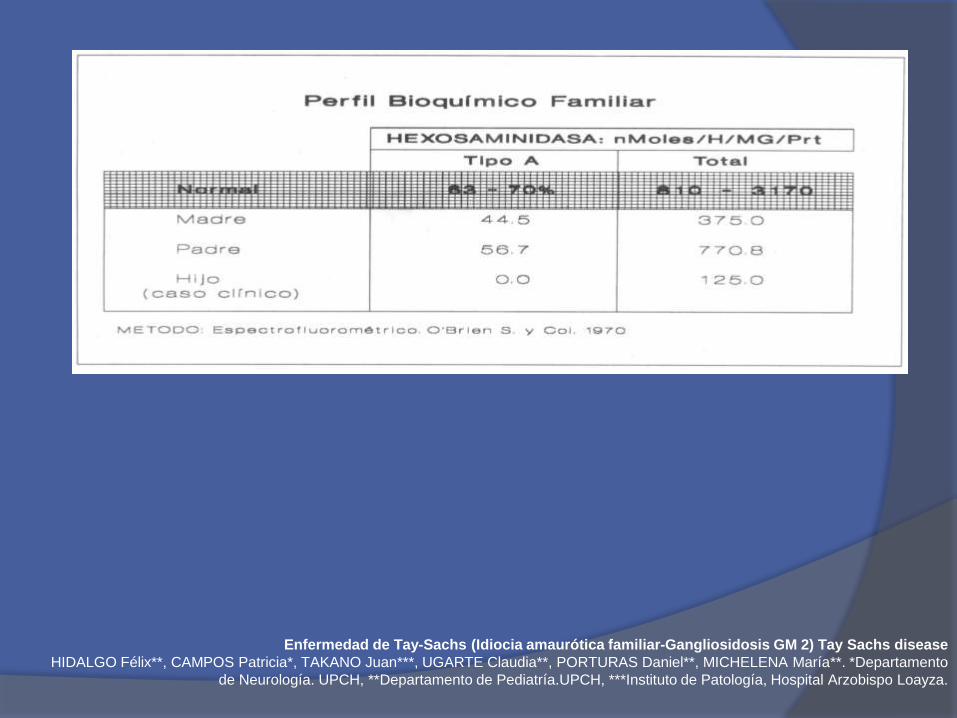
Enfermedad de Tay-Sachs (Idiocia amaurótica familiar-Gangliosidosis GM 2) Tay Sachs disease
HIDALGO Félix**, CAMPOS Patricia*, TAKANO Juan***, UGARTE Claudia**, PORTURAS Daniel**, MICHELENA María**. *Departamento
de Neurología. UPCH, **Departamento de Pediatría.UPCH, ***Instituto de Patología, Hospital Arzobispo Loayza.
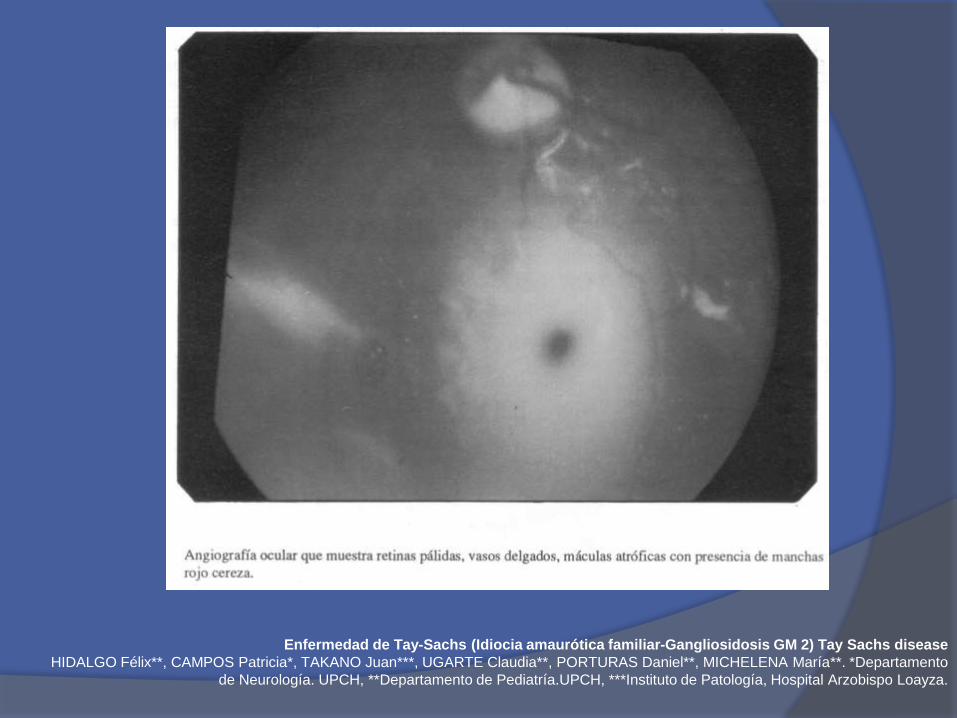
Enfermedad de Tay-Sachs (Idiocia amaurótica familiar-Gangliosidosis GM 2) Tay Sachs disease
HIDALGO Félix**, CAMPOS Patricia*, TAKANO Juan***, UGARTE Claudia**, PORTURAS Daniel**, MICHELENA María**. *Departamento
de Neurología. UPCH, **Departamento de Pediatría.UPCH, ***Instituto de Patología, Hospital Arzobispo Loayza.
Enfermedad de Tay-Sachs (Idiocia amaurótica familiar-Gangliosidosis GM 2) Tay Sachs disease
HIDALGO Félix**, CAMPOS Patricia*, TAKANO Juan***, UGARTE Claudia**, PORTURAS Daniel**, MICHELENA María**. *Departamento
de Neurología. UPCH, **Departamento de Pediatría.UPCH, ***Instituto de Patología, Hospital Arzobispo Loayza.


Gaucher
Descrita 1882 por PhillipCharles Gaucher.
Desorden multisistémico que resulta de mutaciones autosómicas recesivas en el gen codificador de la enzima glucocerebrosidasa (GBA)
Incidencia 1:40,000 – 60,000 (población gral)
Autosómica recesiva
β –glucocerebrosidasa: Hidrólisis intracelular de la glucosilceramida
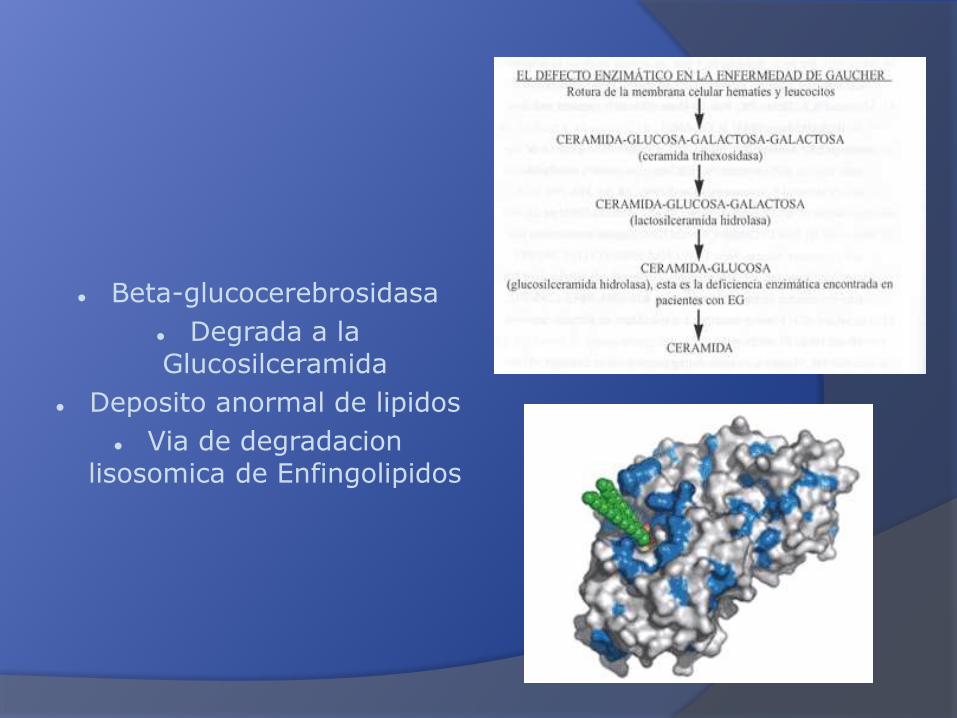
Beta-glucocerebrosidasa
Degrada a la Glucosilceramida
Deposito anormal de lipidos
Via de degradacionlisosomica de Enfingolipidos
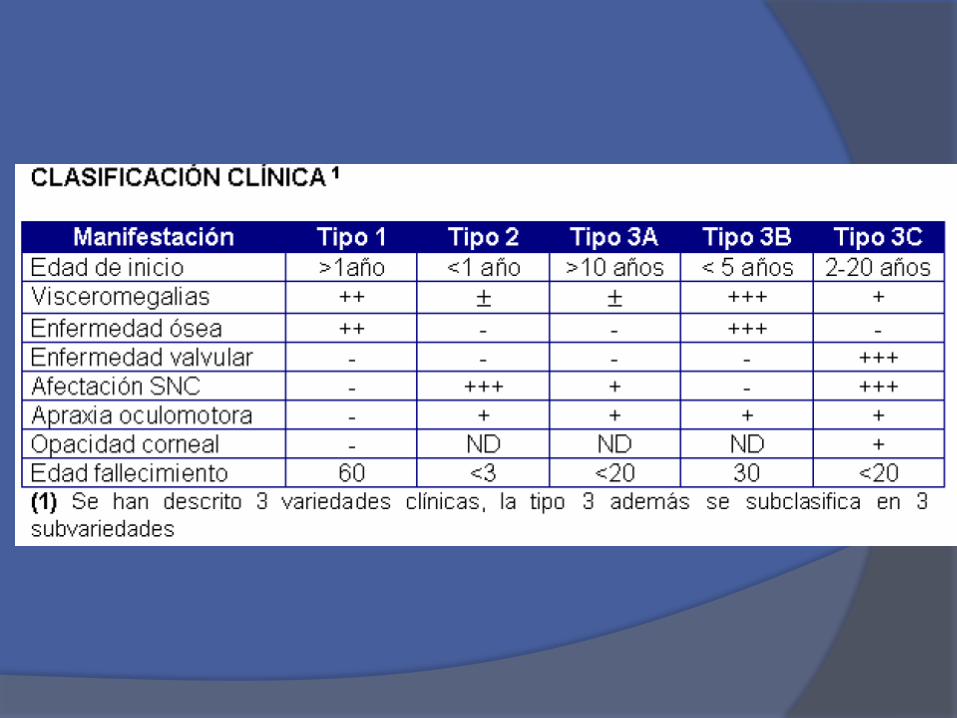
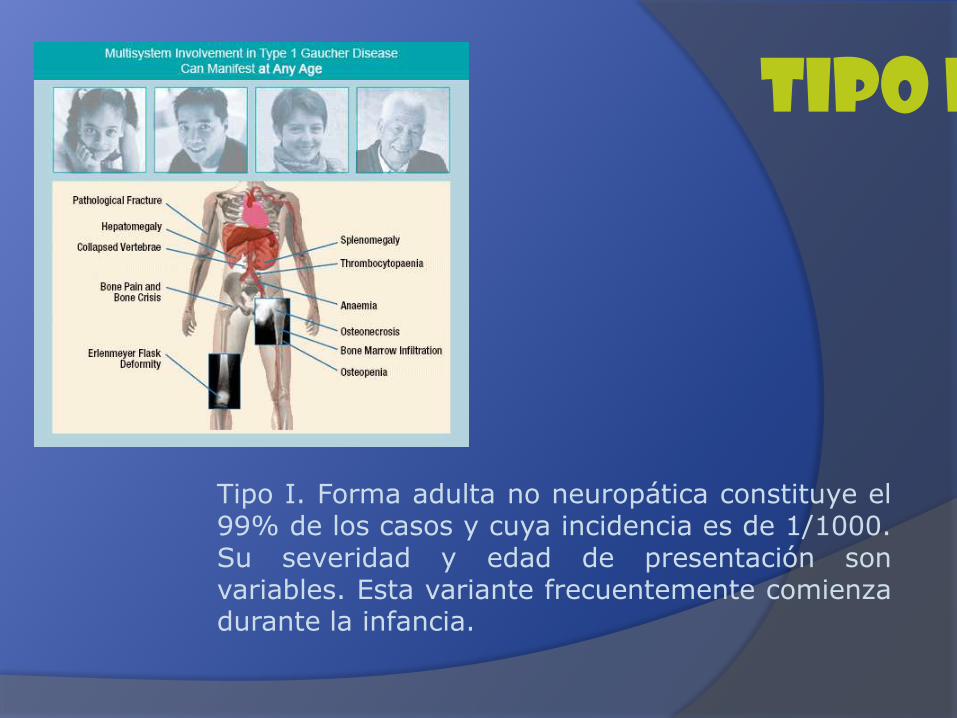
Tipo I. Forma adulta no neuropática constituye el99% de los casos y cuya incidencia es de 1/1000.Su severidad y edad de presentación sonvariables. Esta variante frecuentemente comienzadurante la infancia.
Tipo I

Forma infantil o
aguda neuropática,
se presenta en edad
pediátrica en
pacientes entre 1 a
12 meses, la
mortalidad antes de
los 2 años es alta.
Tipo II
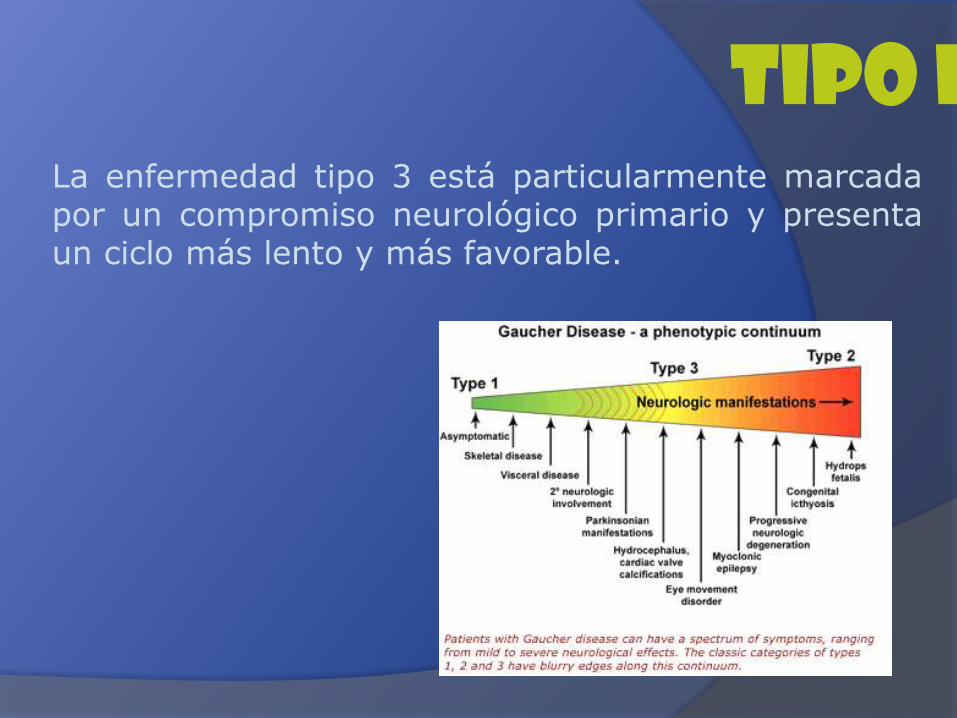
Tipo IIILa enfermedad tipo 3 está particularmente marcadapor un compromiso neurológico primario y presentaun ciclo más lento y más favorable.
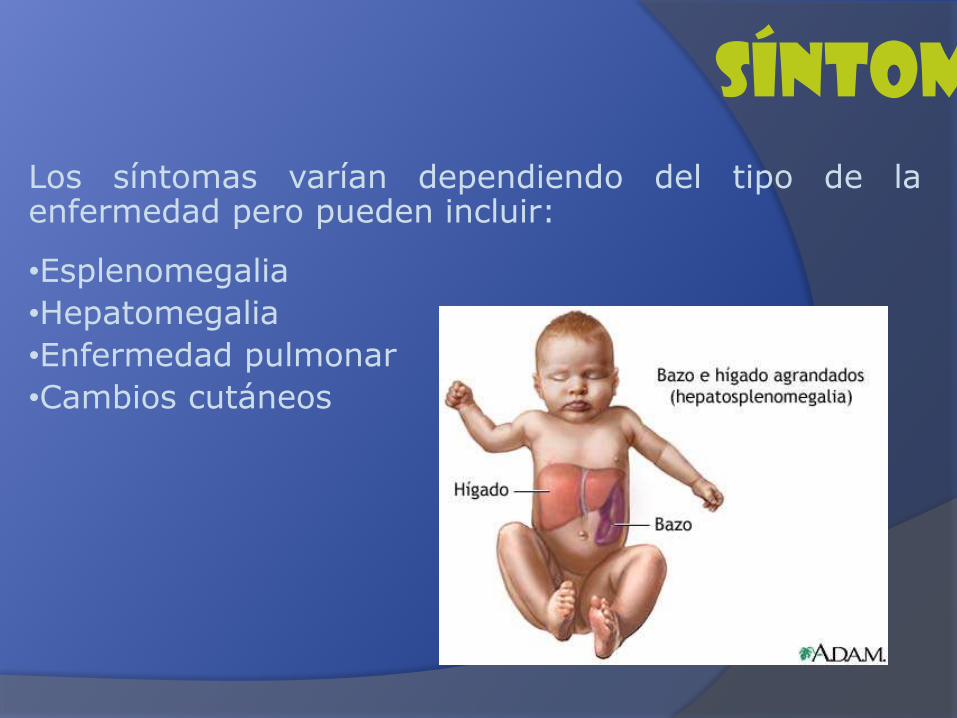
Los síntomas varían dependiendo del tipo de laenfermedad pero pueden incluir:
•Esplenomegalia
•Hepatomegalia
•Enfermedad pulmonar
•Cambios cutáneos
Síntomas

•Deterioro cognitivo
•Dolor y fracturas óseas
•Tendencia a la formación de hematomas
•Fatiga
•Convulsiones
•Edema grave al nacer
•Problemas con las válvulascardíacas
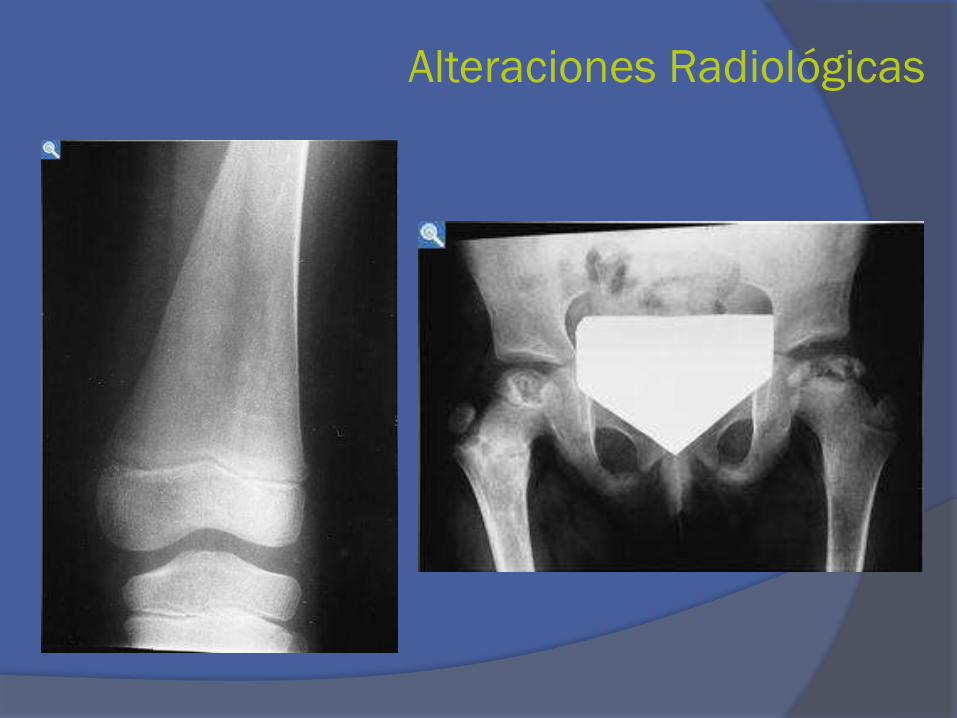
Alteraciones Radiológicas
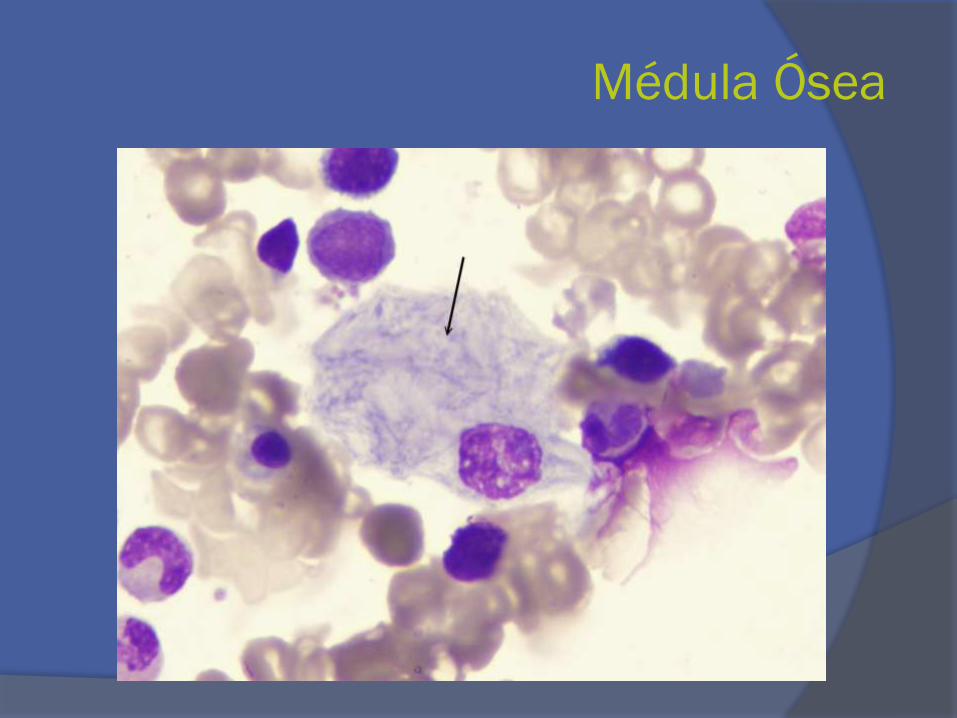
Médula Ósea

- Clínico: esplenomegalia y anemia leve no explicada
- Aspiración de médula ósea
- Genética molecular : Cromosoma1
Diagnóstico

Complicaciones
•Convulsiones
•Anemia
•Trombocitopenia
•Infartos Óseos

Pronóstico
• Depende del subtipo de la enfermedad.• La forma infantil de esta enfermedad puede
conducir a la muerte temprana; de hecho, lamayoría de los niños afectados muere antes delos 5 años de edad.
• Con la disponibilidad de la enzima sintética, lamayoría de los pacientes con la forma crónicaadulta de la enfermedad pueden esperar unaexpectativa de vida normal o casi normal.

Tratamiento Esplenectomía en desuso.
Hay disponibilidad de la terapia de
reemplazo enzimático, reemplazo
del sustrato y, en algunos casos, se
puede requerir un trasplante de
médula ósea.


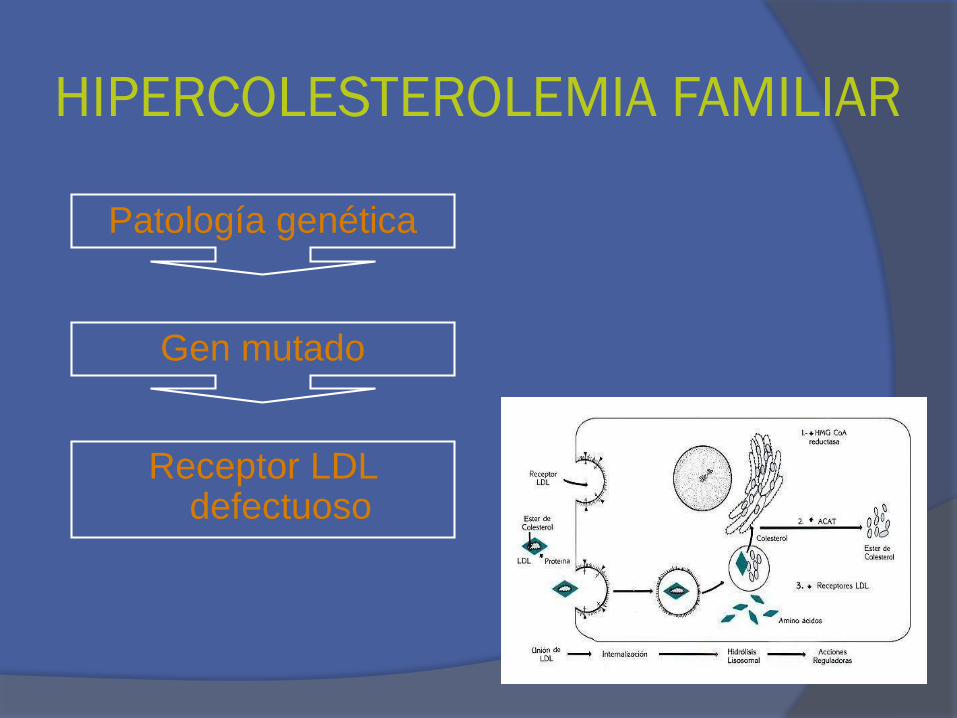
HIPERCOLESTEROLEMIA FAMILIAR
Receptor LDL defectuoso
Patología genética
Gen mutado

GRADOS
Hipercolesterolemia poligénica grave:
Hipercolesterolemia familiar combinada:
Niveles de colesterol > 220mg/dl
Niveles de colesterol y triglicéridos > 350mg/dl
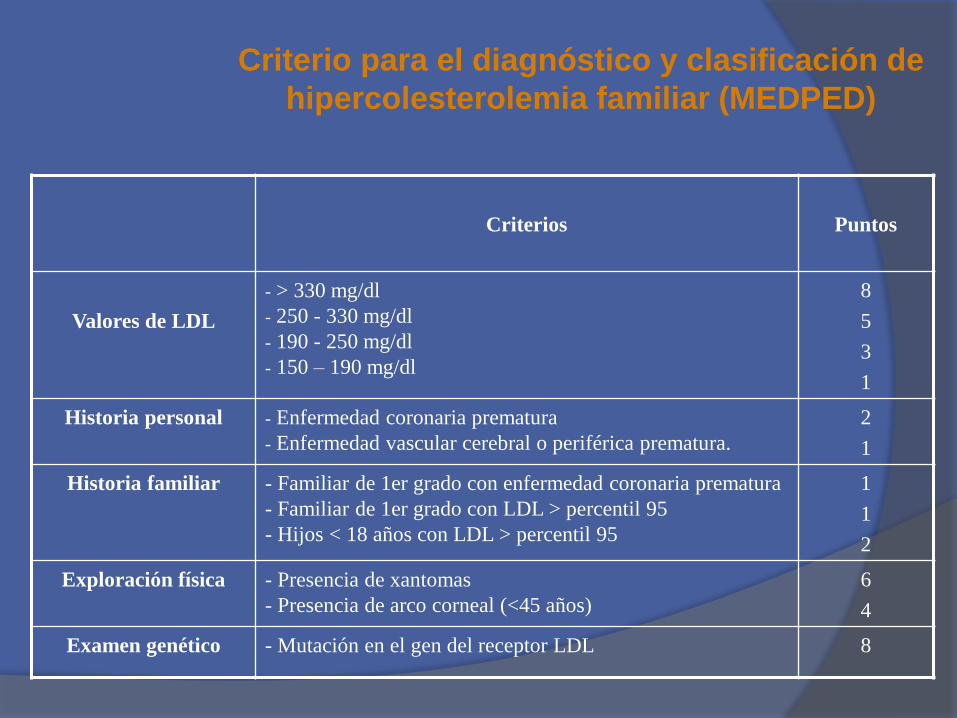
Criterios Puntos
Valores de LDL
- > 330 mg/dl
- 250 - 330 mg/dl
- 190 - 250 mg/dl
- 150 – 190 mg/dl
8
5
3
1
Historia personal - Enfermedad coronaria prematura
- Enfermedad vascular cerebral o periférica prematura.
2
1
Historia familiar - Familiar de 1er grado con enfermedad coronaria prematura
- Familiar de 1er grado con LDL > percentil 95
- Hijos < 18 años con LDL > percentil 95
1
1
2
Exploración física - Presencia de xantomas
- Presencia de arco corneal (<45 años)
6
4
Examen genético - Mutación en el gen del receptor LDL 8
Criterio para el diagnóstico y clasificación de
hipercolesterolemia familiar (MEDPED)

Puntos Probabilidad
mutación
8 puntos Heterocigoto seguro
6-7 puntos Heterocigoto probable
3-5 puntos Heterocigoto posible

Hipercolesterolemia. Abordaje terape´utico
A. Mora´is Lo´peza, R.A. Lama Morea, J. Dalmau Serrab y Comite´ de Nutricio´n de la AEP,
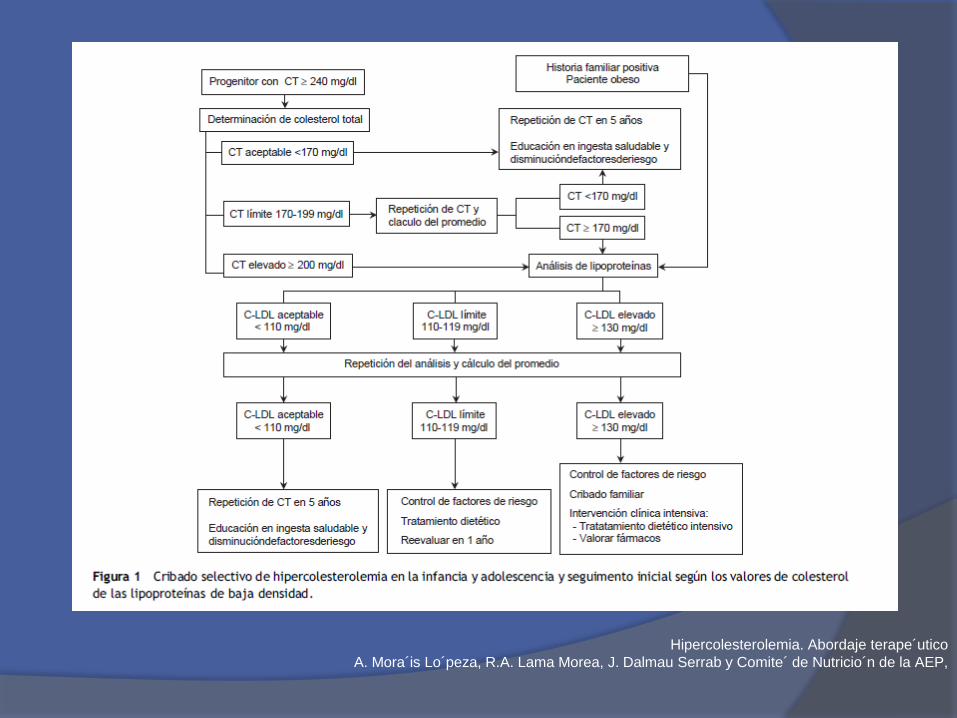
Hipercolesterolemia. Abordaje terape´utico
A. Mora´is Lo´peza, R.A. Lama Morea, J. Dalmau Serrab y Comite´ de Nutricio´n de la AEP,

GRACIAS





























