Embed Size (px)
Citation preview

INFORME DE LABORATORIO DE ANÁLISIS INSTRUMENTAL
PRÁCTICA:
FOTOMETRÍA
PRESENTADO POR:
ALEXANDRA UBAQUE BEDOYA
CÓD. 1088328582
EDGAR ANDRÉS CARDONA DUQUE
CÓD. 1088334064
DOCENTE:
MARIBEL MONTOYA GARCÍA
UNIVERSIDAD TECNOLÓGICA DE PEREIRA
FACULTAD DE TECNOLOGÍAS
PROGRAMA DE TECNOLOGÍA QUÍMICA
JUNIO 2015

1. INTRODUCCIÓN
La espectrofotometría es uno de los métodos de análisis más usados, y se basa en la
relación que existe entre la absorción de luz por parte de un compuesto y su
concentración. Cuando se hace incidir luz monocromática (de una sola longitud de onda)
sobre un medio homogéneo, una parte de la luz incidente es absorbida por el medio y otra
transmitida, como consecuencia de la intensidad del rayo de luz sea atenuada desde Po a
P, siendo Po la intensidad de la luz incidente y P la intensidad del rayo de luz transmitido.
Dependiendo del compuesto y el tipo de absorción a medir, la muestra puede estar en fase
líquida, sólida o gaseosa. En las regiones visibles y ultravioleta del espectro
electromagnético, la muestra es generalmente disuelta para formar una solución.
ESPECTRO ELECTROMAGNÉTICO: Colores y rango de longitudes de onda
correspondiente
Color Violeta Azul Verde Amarillo Naranja Rojo
Rango de λ en
nm 380 - 450 450 - 495 495 - 570 570 - 590 590 - 620 620 - 720
Tabla 5. Rango de λ con su respectivo color

2. OBJETIVOS
Reconocimiento de los diferentes modelos de espectrofotómetros y de sus partes externas
e internas, distinguir los componentes básicos de un espectrofotómetro y su función.
Calibrar y manejar correctamente el espectrofotómetro. Estudiar algunas características
técnicas del instrumento. Definir las condiciones instrumentales óptimas para hacer un
análisis fotométrico. Estudiar el comportamiento de una sustancia en relación con la Ley
de Beer. Analizar cualitativa y cuantitativamente diferentes sustancias por medio de curvas
espectrales y gráficas de calibración. Aplicar la técnica fotométrica en la región del visible
en el control de calidad y procesos.
3. EQUIPOS, MATERIALES Y REACTIVOS
1 Espectrofotómetro.
Varias celdas (Vidrio, plástico) para el espectrofotómetro.
2 beaker de 100 mL.
1 beaker de 250 mL.
10 matraces aforados de 25 mL.
1 matraz aforado de 50 mL.
1 matraz aforado de 100 mL.
1 matraz aforado de 250 mL.
1 pipeta volumétrica de 5 mL.
1 pipeta volumétrica de 10 mL.
1 pipeta graduada de 10 mL.
1 probeta de 50 mL.
1 bureta de 25 mL graduación 1/20 mL
1 vidrio de reloj 100 mm de diámetro.
1 frasco lavador.
1 espátula acanalada
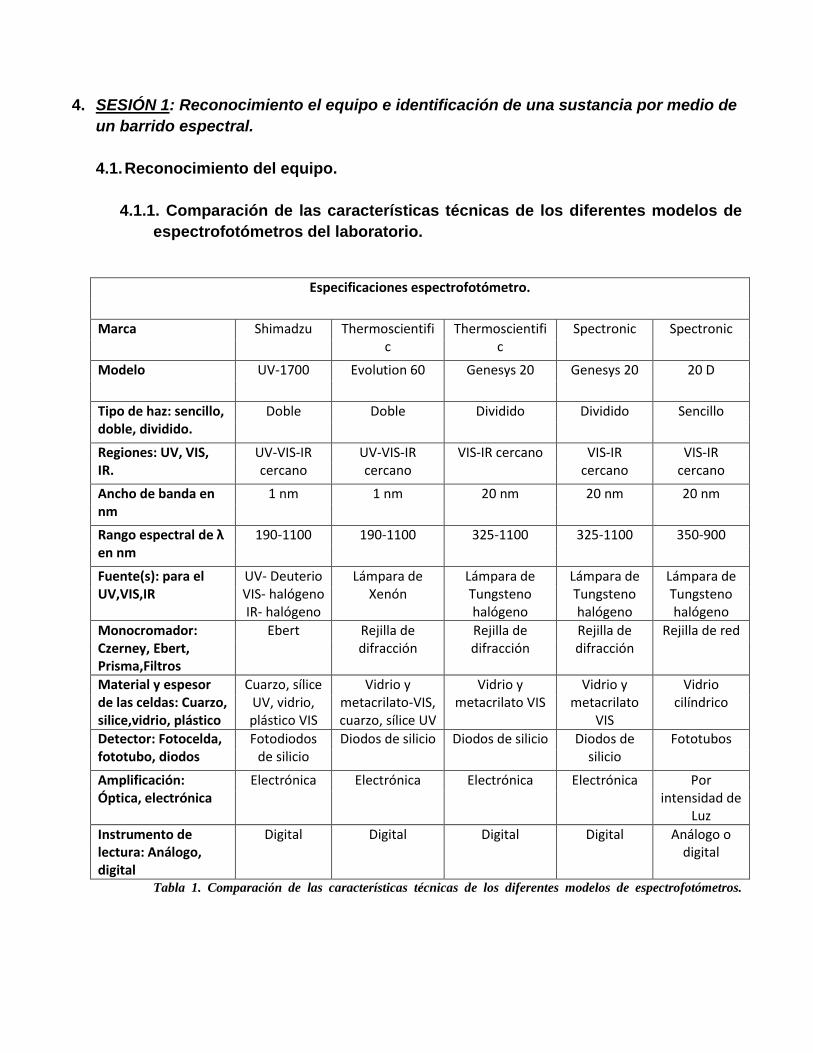
4. SESIÓN 1: Reconocimiento el equipo e identificación de una sustancia por medio de
un barrido espectral.
4.1. Reconocimiento del equipo.
4.1.1. Comparación de las características técnicas de los diferentes modelos de
espectrofotómetros del laboratorio.
Especificaciones espectrofotómetro.
Marca Shimadzu Thermoscientific
Thermoscientific
Spectronic Spectronic
Modelo UV-1700 Evolution 60 Genesys 20 Genesys 20 20 D
Tipo de haz: sencillo, doble, dividido.
Doble Doble Dividido Dividido Sencillo
Regiones: UV, VIS, IR.
UV-VIS-IR cercano
UV-VIS-IR cercano
VIS-IR cercano VIS-IR cercano
VIS-IR cercano
Ancho de banda en nm
1 nm 1 nm 20 nm 20 nm 20 nm
Rango espectral de λ en nm
190-1100 190-1100 325-1100 325-1100 350-900
Fuente(s): para el UV,VIS,IR
UV- Deuterio VIS- halógeno IR- halógeno
Lámpara de Xenón
Lámpara de Tungsteno halógeno
Lámpara de Tungsteno halógeno
Lámpara de Tungsteno halógeno
Monocromador: Czerney, Ebert, Prisma,Filtros
Ebert Rejilla de difracción
Rejilla de difracción
Rejilla de difracción
Rejilla de red
Material y espesor de las celdas: Cuarzo, silice,vidrio, plástico
Cuarzo, sílice UV, vidrio, plástico VIS
Vidrio y metacrilato-VIS, cuarzo, sílice UV
Vidrio y metacrilato VIS
Vidrio y metacrilato
VIS
Vidrio cilíndrico
Detector: Fotocelda, fototubo, diodos
Fotodiodos de silicio
Diodos de silicio Diodos de silicio Diodos de silicio
Fototubos
Amplificación: Óptica, electrónica
Electrónica Electrónica Electrónica Electrónica Por intensidad de
Luz
Instrumento de lectura: Análogo, digital
Digital Digital Digital Digital Análogo o digital
Tabla 1. Comparación de las características técnicas de los diferentes modelos de espectrofotómetros.

4.1.2. Reconocimiento de los componentes de los diferentes espectrofotómetros.
4.1.2.1. Thermo scientific Genesys 20.
Figura 1. Espectrofotómetro Thermo Scientific Genesys 20
Componentes:
1. Interruptor de Encendido/Apagado.
2. Pantalla digital.
3. Tapa del compartimiento de muestras.
4. Teclado.
5. Impresora interna opcional.
6. Puerta del compartimiento de la lámpara.
4.1.2.2. Spectronic 20 D
Figura 2. Espectrofotómetro Spectronic 20 D

Componentes:
1. Control de encendido y ajuste del cero.
2. Compartimiento para la muestra.
3. Piloto de encendido.
4. Selector de longitud de onda.
5. Control de ajuste del 100% de transmitancia, o de absorbancia.
6. Escala de longitud de ondas.
7. Instrumento de lectura, análogo, digital.
8. Indicador de estado. Selector de modo de operación.
9. Disminución de concentración.
10. Aumento de concentración.
4.1.2.3. Thermo scientific Evolution 60
Figura 3. EspectrofotómetroThermo Scinetific Evolution 60
Componentes:
1. Teclado.
2. Compartimiento para la celda.
3. Compartimiento para la impresora.
4.1.2.4. Shimadzu UV-1700
Figura 4. Espectroftómetro Shimadzu UV-1700

Componentes:
1. Pantalla.
2. Teclado.
3. Compartimiento para la celda.
4.1.2.5. Spectronic Genesys 20
Figura 5. Espectrofotómetro Spectronic Genesys 20
Componentes:
1. Interruptor de Encendido/Apagado.
2. Pantalla digital.
3. Tapa del compartimiento de muestras.
4. Teclado.
5. Impresora interna opcional.
6. Puerta del compartimiento de la lámpara.
4.2. Estudio de Celdas equivalentes
Se llenaron 6 celdas con agua destilada, la primera sería el blanco con la que se ajustó
a 100% de transmitancia y 0 de absorbancia para determinar las celdas equivalentes
en el espectrofotómetro Thermo scientific Genesys 20.
Celda A %T
1 -0,033 107,9
2 -0,025 105,9
3 -0,057 114
4 -0,033 107,9
5 0.001 99,7 Tabla 2. Estudio de celdas equivalentes.

Según la tabla anterior solo se obtuvo una celda equivalente (Celda #5) pues fue la
única cuyo valor de absorbancia se encontraba dentro del rango -0,003 a +0,003. Por
lo tanto solo se hicieron las medidas con esta celda y la celda del blanco.
4.3. Identificación de una sustancia por medio de un barrido espectral
Se procedió a determinar la absorbancia y el %T de una muestra problema con
concentración y nombre desconocido a diferentes longitudes de onda (λ) en el
espectrofotómetro Thermo scientific Genesys 20.
Λ (nm) %T A
380 72,7 0,138
400 90 0,046
420 96 0,018
440 92,1 0,036
460 80,6 0,094
480 63,3 0,198
500 45,7 0,340
520 38,8 0,411
540 38,9 0,410
560 46,4 0,334
580 63 0,201
600 83,2 0,080
620 86,5 0,063
640 89,1 0,050
660 91,2 0,040
680 93,6 0,029
700 95,6 0,020
720 97,2 0,013 Tabla 3. Barrido espectral de la sustancia problema 4
Según los datos de la tabla anterior, los picos de la muestra problema 4 están en la
longitud de onda 520 y 540.
Se procedió a realizar el barrido espectral de la muestra problema en el
espectrofotómetro Shimadzu UV-1700, el cual arrojó las siguientes graficas con unos
picos de 525,5 y 545,5:

Figura 6. Espectro de la sustancia problema 4
Figura 7. Espectro del permanganato de potasio
De acuerdo con los espectros de diferentes sustancias para comparar e identificar la
muestra problema y las gráficas anteriores se determinó que la muestra problema era
permanganato de potasio.
4.4. Valores de %T y A de la solución problema (permanganato de potasio) en
diferentes equipos.
Se procedió a determinar la A y el %T en los picos del barrido espectral en diferentes
modelos de espectrofotómetros:

A %T
λ (nm) Equipo
525,5 545,5 525,5 545,5
Thermo scientific
Genesys 20 0.410 0,389 38,9 40,8
Shimadzu
UV-1700 1,0741 1,0331 84,3 92,6
Thermo scientific
Evolution 60 1,079 1,039 83,4 91,4
Spectronic
20 D 1,720 0,806 1,91 15,63
Tabla 4. Valores de %T y A de la muestra problema en diferentes equipos
5. Sesión 2: Cuantificación de una sustancia absorbente
5.1. Celdas equivalentes
Se llenaron 6 celdas con agua destilada, la primera sería el blanco con la que se ajustó
a 100% de transmitancia y 0 de absorbancia para determinar las celdas equivalentes
en el espectrofotómetro Thermo scientific Genesys 20.
Celda A %T
1 0,015 96,6
2 -0,034 108,1
3 -0,039 114
4 -0,017 109,4
5 0.003 99,3 Tabla 5. Estudio de celdas equivalentes
Según la tabla anterior solo se obtuvo una celda equivalente (Celda #5), por lo tanto
solo se hicieron las medidas con esta celda y la celda del blanco.

5.2. Cuantificación de la muestra problema 1
Se procedió a identificar y cuantificar una muestra problema por medio de un barrido
espectral en el espectrofotómetro Shimadzu UV-1700.
Figura8. Espectro de la sustancia problema 1
Figura 9. Espectro del cloruro de cobalto
El barrido espectral de la sustancia problema 1 arrojó un pico 512 nm con una
absorbancia 0,6171.

De acuerdo con los espectros de diferentes sustancias para comparar e identificar la
muestra problema y las gráficas anteriores se determinó que la muestra problema era
cloruro de cobalto.
Se procedió a preparar los patrones con una solución de cloruro de cobalto 0,5 M.
Previamente se midió la absorbancia de esta solución (0,3823) y se hicieron los
cálculos con la ecuación A1V1=A2V2 (siendo A absorbancia y V volumen) para saber el
volumen necesario de dicha solución 0,5 M para preparar 25 mL de cada patrón de
0.2; 0.4; 0.6; 0.8 de Absorbancia.
Patrón A %T Concentración (M)
Volumen de sln 0,5 M utilizado
1 0,2 63,09 0,042 2,1
2 0,4 39,81 0,084 4,2
3 0,6 25,11 0,126 6,3
4 0,8 15,84 0,168 8,4
Solución 0,5 M 2,3823 0,4147 0,5 Tabla 6. Volúmenes de solución de cloruro de cobalto 0,5 M utilizados para preparar los patrones
Figura 10. Patrones de cloruro de cobalto
Se midieron las absorbancias de los patrones para construir la curva de calibración (Ver
figura 11) y de la muestra de cloruro de cobalto para determinar su concentración.
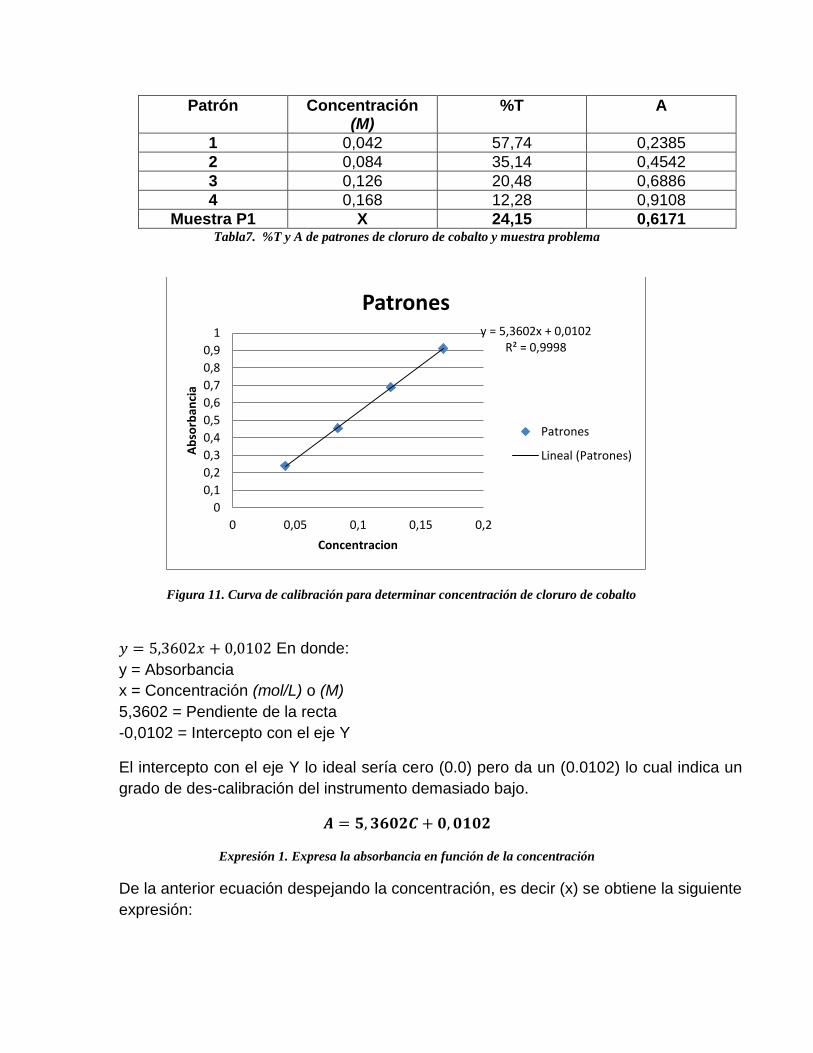
Patrón Concentración (M)
%T A
1 0,042 57,74 0,2385
2 0,084 35,14 0,4542
3 0,126 20,48 0,6886
4 0,168 12,28 0,9108
Muestra P1 X 24,15 0,6171 Tabla7. %T y A de patrones de cloruro de cobalto y muestra problema
Figura 11. Curva de calibración para determinar concentración de cloruro de cobalto
𝑦 = 5,3602𝑥 + 0,0102 En donde:
y = Absorbancia
x = Concentración (mol/L) o (M)
5,3602 = Pendiente de la recta
-0,0102 = Intercepto con el eje Y
El intercepto con el eje Y lo ideal sería cero (0.0) pero da un (0.0102) lo cual indica un
grado de des-calibración del instrumento demasiado bajo.
𝑨 = 𝟓, 𝟑𝟔𝟎𝟐𝑪 + 𝟎, 𝟎𝟏𝟎𝟐
Expresión 1. Expresa la absorbancia en función de la concentración
De la anterior ecuación despejando la concentración, es decir (x) se obtiene la siguiente
expresión:
y = 5,3602x + 0,0102 R² = 0,9998
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1
0 0,05 0,1 0,15 0,2
Ab
sorb
anci
a
Concentracion
Patrones
Patrones
Lineal (Patrones)

𝑪 =𝑨 − 𝟎, 𝟎𝟏𝟎𝟐
𝟓, 𝟑𝟔𝟎
Expresión 2. Expresa la concentración (M) en función de la absorbancia
Observando la tabla 7 se encuentra que la absorbancia de la muestra problema es
igual a 0,6171; reemplazando en la expresión #2 se obtiene:
𝑪𝒑𝒓𝒐𝒃𝒍𝒆𝒎𝒂 =𝟎. 𝟔𝟏𝟕𝟏 − 𝟎. 𝟎𝟏𝟎𝟐
𝟓, 𝟑𝟔𝟎= 𝟎. 𝟏𝟏𝟑𝟐 𝑴
Por este medio (curva de calibración) se determinó que la concentración de la (muestra
1) era de 0.1132 M
La concentración real era 0,125 M
Porcentaje de error:
%𝑬𝒓𝒓𝒐𝒓 =𝟎, 𝟏𝟏𝟑𝟐 − 𝟎, 𝟏𝟐𝟓
𝟎, 𝟏𝟐𝟓∗ 𝟏𝟎𝟎 = 𝟗, 𝟒𝟒%
Error instrumental:
%𝑬𝒓𝒓𝒐𝒓 𝒊𝒏𝒔𝒕𝒓𝒖𝒎𝒆𝒏𝒕𝒂𝒍 =∆𝑻
𝟐, 𝟑𝒍𝒐𝒈𝑻∗ 𝟏𝟎𝟎 =
𝟎, 𝟎𝟏
𝟐, 𝟑𝒍𝒐𝒈(𝟎, 𝟐𝟒𝟏𝟓)∗ 𝟏𝟎𝟎 = 𝟎, 𝟕𝟎𝟒𝟓%
Sensibilidad: Pendiente de la recta, 𝑦 = 5,3602𝑥 − 0,0102
𝑺 = 𝟓, 𝟑𝟔𝟎𝟐
Límite de detección (L.D): Tomando la expresión #2 con una A = 0,2385
𝑳. 𝑫 =𝟎, 𝟐𝟑𝟖𝟓 − 𝟎, 𝟎𝟏𝟎𝟐
𝟓, 𝟑𝟔𝟎= 𝟎, 𝟎𝟒𝟐𝟔 𝑴
Límite de cuantificación:
𝑳. 𝑪 = 𝑳. 𝑫 ∗ 𝟏𝟎 = 𝟎, 𝟎𝟒𝟐𝟔 ∗ 𝟏𝟎 = 𝟎, 𝟒𝟐𝟔 𝑴

6. Sesión 3; Determinación Fotométrica de Manganeso en un Acero
6.1. Clasificación del acero comercial
La clasificación del acero se puede determinar en función de sus características, las
más conocidas son la clasificación del acero por su composición química y por sus
propiedades o clasificación del acero por su uso; cada una de estas clasificaciones a la
vez se subdivide o hace parte de otro grupo de clasificación.
6.1.1. Clasificación de Acero por su composición química:
6.1.1.1. Acero al carbono Se trata del tipo básico de acero que contiene menos
del 3% de elementos que no son hierro ni carbono.
6.1.1.2. Acero de alto carbono El Acero al carbono que contiene más de 0.5% de
carbono.
6.1.1.3. Acero de bajo carbono Acero al carbono que contiene menos de 0.3%
de carbono.
6.1.1.4. Acero de mediano carbono Acero al carbono que contiene entre 0.3 y
0.5% de carbono.
6.1.1.5. Acero de aleación Acero que contiene otro metal que fue añadido
intencionalmente con el fin de mejorar ciertas propiedades del metal.
6.1.1.6. Acero inoxidable Tipo de acero que contiene más del 15% de cromo y
demuestra excelente resistencia a la corrosión.
6.1.2. Clasificación del acero por su contenido de Carbono:
6.1.2.1. Aceros extra suaves: el contenido de carbono varía entre el 0.1 y el 0.2
%
6.1.2.2. Aceros suaves: El contenido de carbono esta entre el 0.2 y 0.3 %
6.1.2.3. Aceros semi suaves: El contenido de carbono oscila entre 0.3 y el 0.4 %
6.1.2.4. Aceros semiduros: El carbono está presente entre 0.4 y 0.5 %
6.1.2.5. Aceros duros: la presencia de carbono varía entre 0.5 y 0.6 %
6.1.2.6. Aceros extramuros: El contenido de carbono que presentan esta entre el
0.6 y el 07 %
6.1.3. Clasificación del Acero por sus propiedades
6.1.3.1. Aceros especiales
6.1.3.2. Aceros inoxidables
6.1.3.3. Aceros inoxidables ferríticos
6.1.3.4. Aceros Inoxidables austeníticos

6.1.3.5. Aceros inoxidables martensíticos
6.1.3.6. Aceros de Baja Aleación Ultrarresistentes
6.1.3.7. Acero Galvanizado (Láminas de acero revestidas con Zinc)
6.1.4. Grados y composiciones químicas de aceros al carbono
Grado del Acero
Composición química, % Carbono
Equivalente
C Mn P, máx S, máx % Ceq (máx)
(1)
Gerdau 1006
SAE 1010
SAE 1015
SAE 1020
SAE 1045
(2)
A240ES
A270ES
A345ES
ASTM A36
Comercial
.
máx.0,08
0,08-0,13
0,13-0,18
0,18-0,23
0,43-0,50
< 0,22
< 0,23
< 0,24
< 0,26
0,28
.
0,30-0,50
0,30-0,60
0,30-0,60
0,30-0,60
0,60-0,90
< 1,15
< 1,25
< 1,45
0,60-0,90
0,30-0,80
.
0,030
0,030
0,030
0,030
0,030
0,040
0,040
0,040
0,030
0,050
.
0,035
0,050
0,050
0,050
0,050
0,050
0,050
0,050
0,050
0,060
(3)
0,23
0,34
0,39
0,44
0,76
0,48
0,48
0,48
0,52
0,55
Tabla 8. Grados y composición química de aceros al carbono
(1) Silicio: En los grados SAE, los siguientes límites serán empleados: SAE 1015 y SAE
1020,
0,10% máx ó los rangos de 0,10 - 0,20%, 0,15 - 0,30%, 0,20 - 0,40% ó 0,30 - 0,60%.
Para el SAE 1045, los rangos de Silicio son 0,10 - 0,20%, 0,15 - 0,30%, 0,20 - 0,40% ó
0,30 - 0,60%.
(2) En los aceros estructurales A240ES, A270ES, A345ES y Comercial, los valores de
Carbono y Manganeso corresponden a los máximos en el análisis de cuchara. En el
acero estructural ASTM A36, corresponde al análisis de cuchara.
(3) El Carbono equivalente (Ceq) es un índice de la soldabilidad del acero,
recomendándose un valor no superior a 0,48%.
El Ceq se determina por la siguiente expresión:

6.2. Celdas equivalentes
Se llenaron 6 celdas con agua destilada, la primera sería el blanco con la que se ajustó
a 100% de transmitancia y 0 de absorbancia para determinar las celdas equivalentes
en el espectrofotómetro Thermo scientific Genesys 20.
Celda A %T
1 0,021 95,28
2 0,005 98,85
3 0,002 99,54
4 -0,027 106,41
5 0.050 89,13 Tabla 9. Estudio de celdas equivalentes
Según la tabla anterior solo se obtuvo una celda equivalente (Celda #3), por lo tanto
solo se hicieron las medidas con esta celda y la celda del blanco.
6.3. Tratamiento simultáneo de la muestra y el estándar
Se pesaron 0,2292 g de una puntilla de acero de concentración de Mn desconocida y
0,2336 g de acero de concentración 0,37 % de Mn. Se transfirieron las muestras a
beakers de 250 mL y se llevaron a la vitrina de gases, se adicionaron 25 mL de ácido
nítrico diluído HNO3 (1:3). Se taparon los beakers con vidrios de reloj y se hirvieron
lentamente hasta que las muestras de acero se disolvieron. Se adicionaron 0,5 g de
persulfato amónico y se hirvió durante 10 minutos. Se adicionaron 5 mL de ácido
fosfórico concentrado y 0,2 g de peryodato potásico para asegurar la oxidación
completa del Mn. Se calentó a ebullición hasta la aparición de un color púrpura
persistente. Se dejaron enfriar las dos soluciones, y se transfirieron cuantitativamente
a matraces de 100 mL. Se obtuvieron 18 mL de solución con la puntilla y 16,9 mL de
solución con el acero estándar. Se aforaron las soluciones y se agitaron para
homogenizar. Los factores de dilución son 100/18 y 100/16,9 respectivamente. Se
tomaron 25 mL de cada solución y se llevaron a beakers de 100 mL y se adicionaron
gotas de ácido clorhídrico HCl diluído. Se calentó para reducir el permanganato y
decolorar las soluciones que fueron los blancos fotométricos para ajustar el 0 de A al
medir la A de la muestra de la puntilla y la A del estándar. Al medir las absorbancias de
las muestras se obtuvo 0,6176 A para la muestra de la puntilla y 0,3113 A para la
estándar.

Figura 12. Solución de la muestra de puntilla Figura 13. Solución de acero estándar
6.4. Preparación de patrones
Se calculó la masa de permanganato de potasio al 99 % de pureza necesaria para
preparar 100 mL de una solución de concentración de Mn de 100 mg / L o 100 ppm. La
masa que se debe pesar para preparar dicha solución es 29,2220 mg y se pesaron
28,8 mg. Se llevaron al matraz de 100 mL y se aforó con agua destilada.
Se hicieron los cálculos del volumen necesario de la solución anterior para preparar 25
mL de patrones de concentraciones de 2,5 - 5,0 - 10,0 - 15,0 y 20,0 mg/L o ppm.
Patrón Concentración (ppm) Volumen de solución 100 ppm utilizado (mL)
1 2,5 0,625
2 5,0 1,25
3 10,0 2,5
4 15,0 3,75
5 20,0 5,0 Tabla 10. Volúmenes de solución de Mn 100 ppm utilizados para preparar los patrones

Figura 14. Patrones de Permanganato de Potasio
Se midieron las absorbancias de los patrones ajustando el 0 de A con agua destilada
para construir la curva de calibración (Ver figura 15). Se midieron las A de la muestra
de la puntilla y del estándar ajustando el 0 de A con los blancos preparados para
determinar sus concentraciones.
Patrón Concentración (ppm)
A %T
1 2,5 0,0789 83,39
2 5,0 0,1377 72,83
3 10,0 0,3129 48,65
4 15,0 0,4646 34,31
5 20,0 0,6864 20,59
Muestra de la puntilla
x 0,6176 24,12
Muestra del acero estándar
x 0,3113 48,83
Tabla 11. A y %T de los patrones y de las muestras

Curva de calibración
Figura 15. Curva de calibración para determinar concentración de Mn
𝑦 = 0,0345𝑥 − 0,0258 En donde:
y = Absorbancia
x = Concentración (mg/L) o (ppm)
50,0345 = Pendiente de la recta
-0,0258 = Intercepto con el eje Y
El intercepto con el eje Y lo ideal sería cero (0.0) pero da un (-0.0258) lo cual indica un
grado de des-calibración del instrumento un poco bajo.
La ecuación anterior se pude expresar entonces así:
𝑨 = 𝟎, 𝟎𝟑𝟒𝟓𝑪 − 𝟎, 𝟎𝟐𝟓𝟖
Expresión 1. Expresa la absorbancia en función de la concentración
De la anterior ecuación despejando la concentración, es decir (x) se obtiene la siguiente
expresión:
𝑪 =𝑨 + 𝟎, 𝟎𝟐𝟓𝟖
𝟎, 𝟎𝟑𝟒𝟓
Expresión 2. Expresa la concentración (ppm) en función de la absorbancia
Observando la tabla 11 se encuentra que la absorbancia de la muestra de la puntilla es
igual a 0,6176; reemplazando en la expresión #2 se obtiene:
y = 0,0345x - 0,0258 R² = 0,9931
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0 5 10 15 20 25
A
Concentración (ppm)
Patrones
Patrones
Lineal (Patrones)

𝑪 𝒎𝒖𝒆𝒔𝒕𝒓𝒂 𝒑𝒖𝒏𝒕𝒊𝒍𝒍𝒂 =𝟎. 𝟔𝟏𝟕𝟔 + 𝟎. 𝟎𝟐𝟓𝟖
𝟎, 𝟎𝟑𝟒𝟓= 𝟏𝟖, 𝟔𝟒𝟗𝟐 𝒑𝒑𝒎
Por este medio (curva de calibración) se determinó que la concentración de la muestra
de la puntilla era de 18,6492 ppm
Ahora multiplicamos por el factor de dilución (100/18):
𝟏𝟖, 𝟔𝟒𝟗𝟐 𝒑𝒑𝒎 𝒙 𝟏𝟎𝟎
𝟏𝟖= 𝟏𝟎𝟑, 𝟔𝟎𝟔𝟕 𝒑𝒑𝒎
Esta concentración correspondería a los 18 mL de solución que se obtuvieron con la
puntilla. Entonces tenemos que:
𝟏𝟖 𝒎𝑳 𝒙 𝟏𝟎𝟑, 𝟔𝟎𝟔𝟕 𝒎𝒈 𝑴𝒏
𝟏𝟎𝟎𝟎 𝒎𝑳 𝒔𝒍𝒏𝒙
𝟏𝒈 𝑴𝒏
𝟏𝟎𝟎𝟎 𝒎𝒈 𝑴𝒏= 𝟏, 𝟖𝟔𝟒𝟗 𝒙𝟏𝟎−𝟑𝒈 𝑴𝒏
Ahora tenemos la masa de Mn que había en la muestra de puntilla que pesamos al
inicio (0,2292 g). Podemos calcular el % de masa de Mn en la puntilla así:
𝑴𝒂𝒔𝒂 𝒅𝒆 𝑴𝒏
𝑴𝒂𝒔𝒂 𝒅𝒆 𝒑𝒖𝒏𝒕𝒊𝒍𝒍𝒂𝒙 𝟏𝟎𝟎 =
𝟏, 𝟖𝟔𝟒𝟗 𝒙𝟏𝟎−𝟑𝒈 𝑴𝒏
𝟎, 𝟐𝟐𝟗𝟐 𝒈 𝒑𝒖𝒏𝒕𝒊𝒍𝒍𝒂𝒙 𝟏𝟎𝟎
= 𝟎, 𝟖𝟏𝟑𝟕 % 𝒅𝒆 𝑴𝒏 𝒆𝒏 𝒍𝒂 𝒑𝒖𝒏𝒕𝒊𝒍𝒍𝒂
Se obtuvo entonces que la concentración de Mn en la puntilla de acero es 0,8137 %
Observando la tabla 11 se encuentra que la absorbancia de la muestra de acero
estándar es igual a 0,3113; reemplazando en la expresión #2 se obtiene:
𝑪 𝒎𝒖𝒆𝒔𝒕𝒓𝒂 𝒆𝒔𝒕á𝒏𝒅𝒂𝒓 =𝟎. 𝟑𝟏𝟏𝟑 + 𝟎. 𝟎𝟐𝟓𝟖
𝟎, 𝟎𝟑𝟒𝟓= 𝟗, 𝟕𝟕𝟏𝟎 𝒑𝒑𝒎
Por este medio (curva de calibración) se determinó que la concentración de la muestra
de la puntilla era de 9,7710 ppm.
Ahora multiplicamos por el factor de dilución (100/16,9):
𝟗, 𝟕𝟕𝟏𝟎 𝒑𝒑𝒎 𝒙 𝟏𝟎𝟎
𝟏𝟔, 𝟗= 𝟓𝟕, 𝟖𝟏𝟔𝟔 𝒑𝒑𝒎

Esta concentración corresponde a los 16,9 mL de solución que se obtuvieron con la
muestra de acero estándar.
Entonces tenemos que:
𝟏𝟔, 𝟗 𝒎𝑳 𝒙 𝟓𝟕, 𝟖𝟏𝟔𝟔 𝒎𝒈 𝑴𝒏
𝟏𝟎𝟎𝟎 𝒎𝑳 𝒔𝒍𝒏𝒙
𝟏𝒈 𝑴𝒏
𝟏𝟎𝟎𝟎 𝒎𝒈 𝑴𝒏= 𝟗, 𝟕𝟕𝟏𝟎 𝒙𝟏𝟎−𝟒𝒈 𝑴𝒏
Ahora tenemos la masa de Mn que había en la muestra de acero estándar que
pesamos al inicio (0,2336 g). Podemos calcular el % de masa de Mn en el acero así:
𝑴𝒂𝒔𝒂 𝒅𝒆 𝑴𝒏
𝑴𝒂𝒔𝒂 𝒅𝒆 𝒂𝒄𝒆𝒓𝒐𝒙 𝟏𝟎𝟎 =
𝟗, 𝟕𝟕𝟏𝟎 𝒙𝟏𝟎−𝟒𝒈 𝑴𝒏
𝟎, 𝟐𝟑𝟑𝟔 𝒈 𝒂𝒄𝒆𝒓𝒐𝒙 𝟏𝟎𝟎 = 𝟎, 𝟒𝟏𝟖𝟑 % 𝒅𝒆 𝑴𝒏 𝒆𝒏 𝒆𝒍 𝒂𝒄𝒆𝒓𝒐 𝒆𝒔𝒕á𝒏𝒅𝒂𝒓
Se obtuvo entonces que la concentración de Mn en el acero es 0,4183 %
La concentración real de Mn en el acero estándar era 0,37 %
Porcentaje de error:
%𝑬𝒓𝒓𝒐𝒓 =𝟎, 𝟒𝟏𝟖𝟑 − 𝟎, 𝟑𝟕
𝟎, 𝟑𝟕∗ 𝟏𝟎𝟎 = 𝟏𝟑, 𝟎𝟓𝟒𝟎%
Error instrumental:
Para la puntilla
%𝑬𝒓𝒓𝒐𝒓 𝒊𝒏𝒔𝒕𝒓𝒖𝒎𝒆𝒏𝒕𝒂𝒍 =∆𝑻
𝟐, 𝟑𝒍𝒐𝒈𝑻∗ 𝟏𝟎𝟎 =
𝟎, 𝟎𝟏
𝟐, 𝟑𝒍𝒐𝒈(𝟎, 𝟐𝟒𝟏𝟐)∗ 𝟏𝟎𝟎 = 𝟎, 𝟕𝟎𝟒𝟎%
Para el acero estándar
%𝑬𝒓𝒓𝒐𝒓 𝒊𝒏𝒔𝒕𝒓𝒖𝒎𝒆𝒏𝒕𝒂𝒍 =∆𝑻
𝟐, 𝟑𝒍𝒐𝒈𝑻∗ 𝟏𝟎𝟎 =
𝟎, 𝟎𝟏
𝟐, 𝟑𝒍𝒐𝒈(𝟎, 𝟒𝟖𝟖𝟑)∗ 𝟏𝟎𝟎 = 𝟏, 𝟑𝟗𝟔𝟔%
Sensibilidad: Pendiente de la recta, 𝑦 = 0,0345𝑥 − 0,0258
𝑺 = 𝟎, 𝟎𝟑𝟒𝟓
Límite de detección (L.D): Tomando la expresión #2 con una A = 0,0789

𝑳. 𝑫 =𝟎, 𝟎𝟕𝟖𝟗 + 𝟎, 𝟎𝟐𝟓𝟖
𝟎, 𝟎𝟑𝟒𝟓= 𝟑, 𝟎𝟑𝟒𝟖 𝒑𝒑𝒎
Límite de cuantificación:
𝑳. 𝑪 = 𝑳. 𝑫 ∗ 𝟏𝟎 = 𝟑, 𝟎𝟑𝟒𝟖 ∗ 𝟏𝟎 = 𝟑𝟎, 𝟑𝟒𝟖 𝒑𝒑𝒎
7. Sesión 4: Aplicación de la técnica fotométrica en el control de calidad de un
producto comercial. Determinación de la concentración de hierro en una tableta
vitamínica.
7.1. Objetivo general
Aplicar la técnica fotométrica para determinar la concentración de hierro contenido en
una tableta
7.2. Objetivos específicos
• Aplicar las buenas prácticas de laboratorio.
• Aplicar normas de seguridad para el trabajo en el laboratorio.
• Calibrar y manejar correctamente el fotómetro.
• Realizar los cálculos estequiométricos para la preparación de los patrones.
• Medir la absorbancia de la muestra y de los patrones de concentraciones conocidas.
• Analizar los resultados obtenidos, para determinar su confiabilidad y reportarlos.
7.3. Fundamento
El hierro pese a encontrarse en cantidades muy pequeñas en nuestro organismo,
participa como cofactor en numerosos procesos biológicos indispensables para la vida,
tales como el transporte de oxígeno, fosforilación oxidativa, metabolismo de
neurotransmisores y la síntesis de ácido desoxirribonucleico.
La deficiencia de hierro es más prevalente a escala mundial y la principal causa de

anemia. En los países en vías de desarrollo los grupos más afectados son los niños
debido a los mayores requerimientos determinados por el crecimiento, y la mujer en
edad fértil por la pérdida de hierro debida al sangramiento menstrual o a las mayores
necesidades de este mineral durante el embarazo. Este aumento de las necesidades
no es cubierto por la dieta habitual la que tiene cantidades insuficientes de hierro y/o
presenta una baja biodisponibilidad de este nutriente. Por tal razón se hace necesario
el consumo de suplementos de hierro para contrarrestar las carencias de éste. El más
conocido por su bajo precio y sus buenos resultados es el sulfato ferroso.
La determinación de la concentración consumida de hierro en los suplementos
vitamínicos se realiza por la técnica fotométrica la cual se fundamenta en la interacción
de las radiaciones electromagnéticas con la materia. Un compuesto absorbe energía a
determinada longitud de onda que este caso corresponde para el visible desde 400 a
800 nm. La radiación absorbida por las moléculas en esta región del espectro provoca
transiciones electrónicas, las cuales pueden ser cuantificadas.
La Ley de Lambert-Beer declara que la cantidad de luz absorbida por una sustancia
depende de la concentración en la solución, de la absortividad específica y del espesor
del recipiente que la contiene. La cual la podemos simplificar de la siguiente manera: A
= acb, esta ley explica que hay una relación directa entre la absorbancia y la
concentración de esta:
• A : Absorbancia
• a : Absortividad específica (Característico de cada sustancia).
• b: Espesor de la celda (cm).
• C: Concentración (g/L).
El espectrofotómetro es un instrumento que permite medir la absorbancia de una
solución en una determinada longitud de onda (λ). Es por eso, que se hace posible
determinar la concentración de un soluto conocido siendo este, proporcional a la
absorbancia. En esta práctica se realizaran una serie de patrones de concentraciones
conocidas de Sulfato Ferroso Amoniacal ((NH4)2 Fe(SO4)2 6H2O), midiendo la
absorbancia para cada concentración, así se obtiene una gráfica de absorbancia
respecto a la concentración. Por interpolación de la absorbancia en la gráfica se puede
encontrar el valor de la concentración desconocida de la muestra.
El hierro de la tableta vitamínica es disuelto en ácido y reducido a Fe +2 con
hidroquinona y se forma un complejo colorido con o-fenantrolina medido a la longitud
de onda de máxima absorción (510nm).

7.4. Equipos
Espectrofotómetro Genesys 20
Estufa
7.5. Materiales
1 Beaker de 100 mL
Equipo de destilación al vació
1 Pipeta graduada de 5 mL
1 Pipeta volumétrica de 5 mL
1 Pipeta volumétrica de 10 mL
2 Matraces aforados de 100 mL
4 Matraces aforados de 25 mL
1 Barra de agitación de vidrio
1 Probeta
1 Celda de vidrio
1 Vidrio reloj
1 Espátula
1 Cronometro
Papel filtro banda roja
7.6. Reactivos
Sulfato Ferroso Amoniacal
Ácido Clorhídrico 6 M
Hidroquinona (0.5g/50mL)
o-Fenantrolina
(0.125g, 5 ml metanol/45 mL)
Carbón Activado
7.7. Procedimiento
7.7.1. Celdas equivalentes
Se llenaron 6 celdas con agua destilada, la primera sería el blanco con la que se
ajustó a 100% de transmitancia y 0 de absorbancia para determinar las celdas
equivalentes en el espectrofotómetro Thermo scientific Genesys 20.
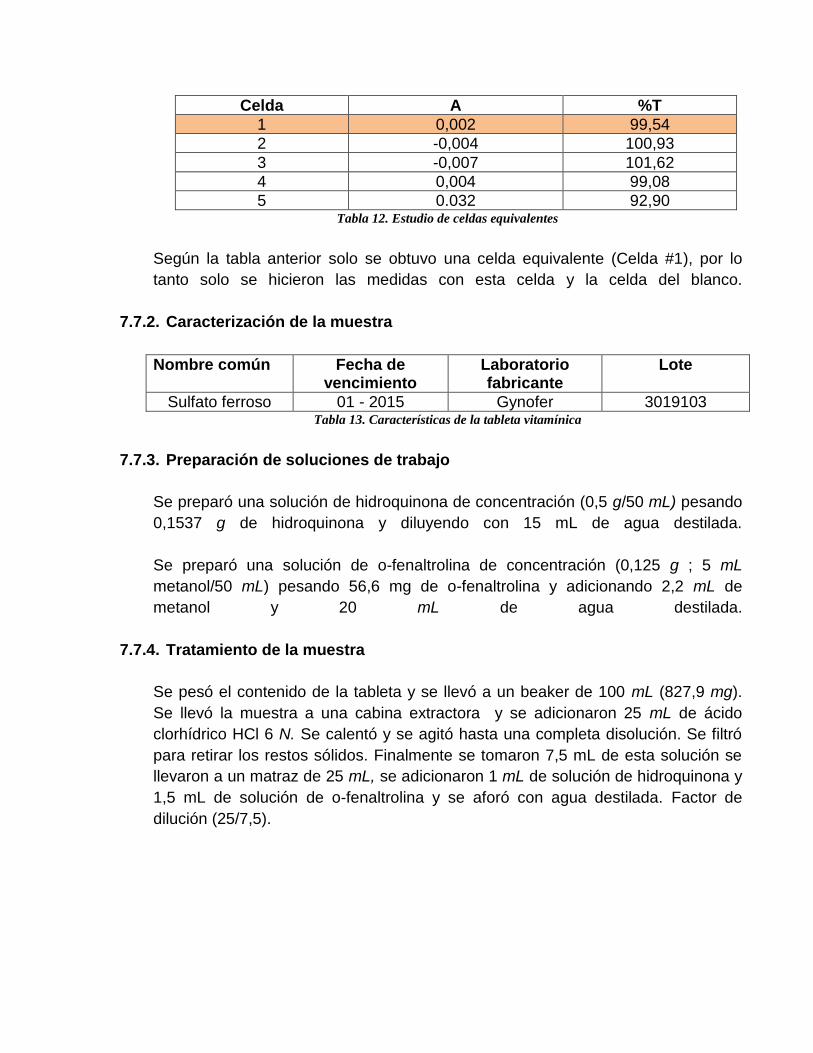
Celda A %T
1 0,002 99,54
2 -0,004 100,93
3 -0,007 101,62
4 0,004 99,08
5 0.032 92,90 Tabla 12. Estudio de celdas equivalentes
Según la tabla anterior solo se obtuvo una celda equivalente (Celda #1), por lo
tanto solo se hicieron las medidas con esta celda y la celda del blanco.
7.7.2. Caracterización de la muestra
Nombre común Fecha de vencimiento
Laboratorio fabricante
Lote
Sulfato ferroso 01 - 2015 Gynofer 3019103 Tabla 13. Características de la tableta vitamínica
7.7.3. Preparación de soluciones de trabajo
Se preparó una solución de hidroquinona de concentración (0,5 g/50 mL) pesando
0,1537 g de hidroquinona y diluyendo con 15 mL de agua destilada.
Se preparó una solución de o-fenaltrolina de concentración (0,125 g ; 5 mL
metanol/50 mL) pesando 56,6 mg de o-fenaltrolina y adicionando 2,2 mL de
metanol y 20 mL de agua destilada.
7.7.4. Tratamiento de la muestra
Se pesó el contenido de la tableta y se llevó a un beaker de 100 mL (827,9 mg).
Se llevó la muestra a una cabina extractora y se adicionaron 25 mL de ácido
clorhídrico HCl 6 N. Se calentó y se agitó hasta una completa disolución. Se filtró
para retirar los restos sólidos. Finalmente se tomaron 7,5 mL de esta solución se
llevaron a un matraz de 25 mL, se adicionaron 1 mL de solución de hidroquinona y
1,5 mL de solución de o-fenaltrolina y se aforó con agua destilada. Factor de
dilución (25/7,5).

Figura 16. Solución de la tableta vitamínica
7.7.5. Preparación de los patrones
Se calculó el peso necesario (0,018 g) de Sulfato Ferroso Amoniacal
hexahidratado ((NH4)2 Fe(SO4)2 6H2O) para preparar 100 mL de solución madre
con una concentración de 30 ppm. Se pesaron 18,4 mg y se aforó hasta 100 mL.
𝑔 (𝑁𝐻4)2 𝐹𝑒(𝑆𝑂4)26𝐻2𝑂 =
30𝑚𝑔 𝐹𝑒
1000 𝑚𝐿∗
1𝑔 𝐹𝑒
1000𝑚𝑔 𝐹𝑒∗
𝑎𝑡𝑚 𝐹𝑒
55,85𝑔 𝐹𝑒∗
𝑚𝑜𝑙 (𝑁𝐻4)2 𝐹𝑒(𝑆𝑂4)26𝐻2𝑂
𝑎𝑡𝑚 𝐹𝑒∗
336,12 𝑔 (𝑁𝐻4)2 𝐹𝑒(𝑆𝑂4)26𝐻2𝑂
𝑚𝑜𝑙(𝑁𝐻4)2 𝐹𝑒(𝑆𝑂4)26𝐻2𝑂∗ 100 𝑚𝐿
= 0,018𝑔 (𝑁𝐻4)2 𝐹𝑒(𝑆𝑂4)26𝐻2𝑂

Figura 17. Patrones de sulfato de hierro
Se calcularon los volúmenes requeridos de la solución de 30 ppm de Fe para
preparar 25 mL de cada patrón de concentraciones de 2,0 ; 3,0 ; 4,0 y 5,0 ppm.
Se adicionaron los volúmenes necesarios en cada matraz y no se aforaron hasta
el momento de realizar las mediciones.
Patrones Concentración (ppm)
Volumen de sln madre utilizado (mL)
1 2 1,6
2 3 2,5
3 4 3,3
4 5 4,2
Tabla 14. Volúmenes de solución madre utilizados para preparar los patrones
7.7.6. Preparación del blanco
Para el blanco de los patrones; se adicionaron 2 mL de solución de hidroquinona y
3 mL de solución de o-fenaltrolina en un matraz de 25 mL y se aforó con agua
destilada.
Para el blanco de la muestra: se adicionaron 2 mL de solución de hidroquinona, 3
mL de solución de o-fenaltrolina, 2 mL de ácido clorhídrico 6 N y se aforó con agua
destilada.
7.7.7. Medición
Se seleccionó en el fotómetro la longitud de onda de 510 nm para realizar las
mediciones. Se ajustó el 0 de A y 100 % T con el blanco para hacer las
mediciones de los patrones. Se adicionaron 2 mL de solución de hidroquinona y 3
mL de solución de o-fenaltrolina a cada matraz y se aforó 3 minutos antes de
realizar cada medición para construir la curva de calibración (Ver figura 16). De
igual manera se procedió con la muestra de sulfato ferroso pero en ese caso se
ajustó el blanco con el blanco preparado para esa muestra.
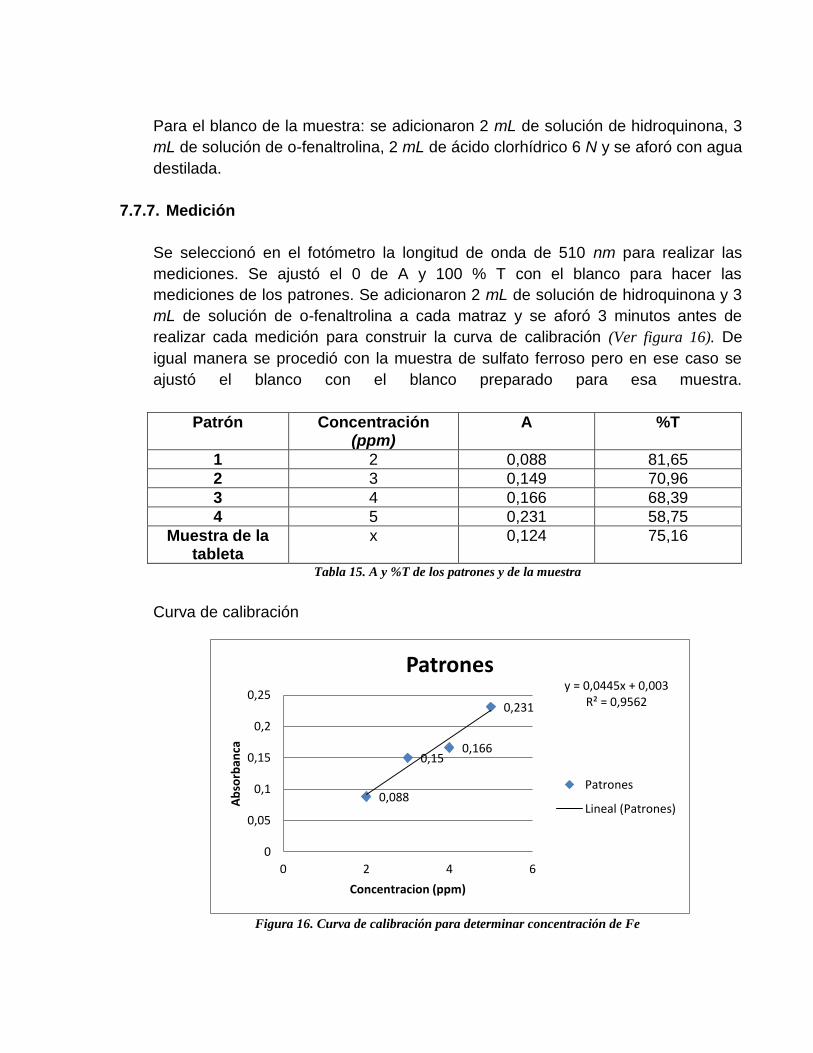
Patrón Concentración (ppm)
A %T
1 2 0,088 81,65
2 3 0,149 70,96
3 4 0,166 68,39
4 5 0,231 58,75
Muestra de la tableta
x 0,124 75,16
Tabla 15. A y %T de los patrones y de la muestra
Curva de calibración
Figura 16. Curva de calibración para determinar concentración de Fe
0,088
0,15 0,166
0,231
y = 0,0445x + 0,003 R² = 0,9562
0
0,05
0,1
0,15
0,2
0,25
0 2 4 6
Ab
sorb
anca
Concentracion (ppm)
Patrones
Patrones
Lineal (Patrones)

𝑦 = 0,0445𝑥 + 0,003 En donde:
y = Absorbancia
x = Concentración (mg/L) o (ppm)
0,0445 = Pendiente de la recta
0,003 = Intercepto con el eje Y
El intercepto con el eje Y lo ideal sería cero (0.0) pero da un (0.003) lo cual
indica un grado de des-calibración del instrumento un poco bajo.
La ecuación anterior se pude expresar entonces así:
𝑨 = 𝟎, 𝟎𝟒𝟒𝟓𝑪 − 𝟎, 𝟎𝟎𝟑
Expresión 1. Expresa la absorbancia en función de la concentración
De la anterior ecuación despejando la concentración, es decir (x) se obtiene la
siguiente expresión:
𝑪 =𝑨 − 𝟎, 𝟎𝟎𝟑
𝟎, 𝟎𝟒𝟒𝟓
Expresión 2. Expresa la concentración (ppm) en función de la absorbancia
Observando la tabla 15 se encuentra que la absorbancia de la muestra de la
puntilla es igual a 0,6176; reemplazando en la expresión #2 se obtiene:
𝑪 𝒎𝒖𝒆𝒔𝒕𝒓𝒂 𝒑𝒖𝒏𝒕𝒊𝒍𝒍𝒂 =𝟎. 𝟏𝟐𝟒 + 𝟎. 𝟎𝟎𝟑
𝟎, 𝟎𝟒𝟒𝟓= 𝟐, 𝟖𝟓𝟑𝟗 𝒑𝒑𝒎
Por este medio (curva de calibración) se determinó que la concentración de la
muestra de la tableta era de 2,8539 ppm
Ahora multiplicamos por el factor de dilución (25/7,5):
𝟐, 𝟖𝟓𝟑𝟗 𝒑𝒑𝒎 𝒙 𝟐𝟓
𝟕, 𝟓= 𝟗, 𝟓𝟏𝟑 𝒑𝒑𝒎
Esta concentración correspondería a los 25 mL de solución que se obtuvieron con la
tableta tratada con HCl 6 N. Entonces tenemos que:
𝟐𝟓 𝒎𝑳 𝒙 𝟗, 𝟓𝟏𝟑 𝒎𝒈 𝑭𝒆
𝟏𝟎𝟎𝟎 𝒎𝑳 𝒔𝒍𝒏= 𝟎, 𝟐𝟑𝟕𝟖 𝒎𝒈 𝑭𝒆
Según los datos obtenidos con las mediciones anteriores se determinó que la masa

de Fe contenida en una tableta vitamínica de sulfato ferroso Gynofer fol es 0,2378
mg.
Error instrumental:
%𝑬𝒓𝒓𝒐𝒓 𝒊𝒏𝒔𝒕𝒓𝒖𝒎𝒆𝒏𝒕𝒂𝒍 =∆𝑻
𝟐, 𝟑𝒍𝒐𝒈𝑻∗ 𝟏𝟎𝟎 =
𝟎, 𝟎𝟏
𝟐, 𝟑𝒍𝒐𝒈(𝟎, 𝟕𝟓𝟏𝟔)∗ 𝟏𝟎𝟎 = 𝟑, 𝟓𝟎𝟓𝟗%
Sensibilidad: Pendiente de la recta, 𝑦 = 0,0445𝑥 + 0,003
𝑺 = 𝟎, 𝟎𝟒𝟒𝟓
Límite de detección (L.D): Tomando la expresión #2 con una A = 0,0789
𝑳. 𝑫 =𝟎, 𝟎𝟖𝟖 − 𝟎, 𝟎𝟎𝟑
𝟎, 𝟎𝟒𝟒𝟓= 𝟐, 𝟎𝟒𝟒𝟗 𝒑𝒑𝒎
Límite de cuantificación:
𝑳. 𝑪 = 𝑳. 𝑫 ∗ 𝟏𝟎 = 𝟐, 𝟎𝟒𝟒𝟗 ∗ 𝟏𝟎 = 𝟐𝟎, 𝟒𝟒𝟗 𝒑𝒑𝒎

8. PREGUNTAS
a) ¿Qué factores afectaron las medidas de la A?
- No monocromaticidad: La ley de Beer presupone que se trabaje con luz
monocromática, pero al discutir el funcionamiento de los filtros y de los
monocromadores se observará que estos dispositivos no proporcionan luz
verdaderamente monocromática, sino que dan un ancho de banda más o menos
amplio según el diseño y calidad.
- Desviaciones químicas: Son debidas a los cambios en la absortividad específica.
- Efecto de la temperatura: El efecto es variable y puede ser de aumento o
disminución de la absortividad.
- Efecto del solvente: Aparecen variaciones en la absortividad de una especie según
el solvente en que se halle, debido a que las interacciones solvente-soluto dependen
del carácter polar o no polar del solvente.
- Efecto del tiempo: Es frecuente el cambio de la absortividad de una especie con el
tiempo, bien por efecto del oxígeno del aire, por efecto de la luz, por inestabilidad de las
especies, o debido a la cinética de la reacción cuando para hacer un análisis se debe
generar la especie absorbente.
b) ¿Qué factores afectan la medida de la absortividad molar o específica?
Por factores que afectan la química del sistema, en especial las variaciones no
controladas de la concentración efectiva de la especie a analizar, debidas a equilibrios
asimétricos.
Es indispensable conocer la química del sistema, porque si la especie a analizar está
involucrada en alguna reacción de equilibrio, se deben tomar las medidas necesarias
(de acuerdo con la ley de acción de masas) para evitar cambios en la concentración
debidos a alteraciones del equilibrio.
c) ¿Cómo puede determinar la confianza en los resultados analíticos obtenidos?
En el análisis de macrocomponentes, en general se requiere que el valor medido no
difiera significativamente del aceptado como referencia. Para determinarlo, se utiliza un
ensayo t de Student, efectuando varias determinaciones de la muestra de
concentración conocida y calculando el t experimental (tob) que se compara con
tablas t para n-1 grados de libertad en el nivel de confianza escogido, generalmente
p=0,05.

Si el tob es menor que el valor tabulado el método tiene la exactitud requerida para ese
ámbito de confianza. Si el tob resulta mayor que el valor tabulado, el método tiene un
error sistemático, del signo resultante, para ese ámbito de confianza.
d) ¿Cómo puede adaptar la técnica fotométrica en la región del visible para el
control de calidad y controlar algunos procesos industriales?
En general, cuando se miden espectros UV-visible, sólo es deseable que ocurra
absorbancia. Como la luz es una forma de energía, la absorción de la luz por la materia
causa que aumente el contenido de energía de las moléculas (o átomos). La energía
potencial total de una molécula, generalmente se representa como la suma de sus
energías electrónica, vibracional y rotacional: La cantidad de energía que una molécula
posee en cada forma no es un continuo, sino una serie de niveles o estados discretos.
La diferencias de energía entre los diferentes estados siguen el orden: En algunas
moléculas y átomos, los fotones de luz UV y visible tienen suficiente energía para
causar transiciones entre los diferentes niveles. La longitud de onda de la luz absorbida
es aquella que tiene la energía requerida para mover un electrón desde un nivel de
energía inferior a uno superior.
Las derivadas de los espectros pueden utilizarse para resaltar las diferencias entre
espectros, resolver bandas solapadas para el análisis cualitativo.
Hoy en día, varias compañías fabrican espectrómetros y con series de producción cada
vez más grandes. Otra gran ventaja de estos nuevos espectrómetros es que han salido
del laboratorio, y pueden utilizarse para control de calidad en instalaciones de campo o
en grandes plantas industriales. Gracias también al aumento de la resolución, la
industria se ha movido rápido y esta tecnología se utiliza ya con mucha frecuencia en
todo tipo de instalaciones industriales con el objetivo de controlar el color o la
composición química de un producto terminado. La determinación de la composición
química de los materiales, o el color, es de vital importancia en numerosos procesos
industriales, y esto ahora puede hacerse de forma eficiente y en tiempo real con
espectrómetros.
e) ¿Consultar en qué consisten las sondas fotométricas y los sensores
fotométricos y cuál es su utilidad?
Sondas Radiométricas y Fotométricas

LP471PHOT
Sonda fotométrica para la medición de la iluminancia con módulo SICRAM incluido.
Respuesta espectral de acuerdo con la visión fotopila estándar, difusora para la
corrección del coseno.
Rango de medida: 0,01lux.200-103.
LP471LUM2
Sonda fotométrica para la medida de la iluminancia con módulo SICRAM incluido.
Respuesta espectral de acuerdo con la visión fotopila estándar, Angulo de visión de 2º
Rango de medida: 0,1 cd/m2…2000 103 cd/m2.
LP471PAR
Sonda cuanto-radio métrica para la medida del flujo de fotones en el campo de la
clorofila PAR, fotosíntesis. Radiación activa 400nm…700nm) con módulo SICRAM
incluido Mide en umol/m2s, difusor para la corrección del coseno
Rango de medida: 0,01umol/m2s…10 103umol/m2s.
LP471UVA
Sonda radiométrica para la medida de la irradiación con módulo SICRAM incluido.
Campo espectral UVA 315nm…400nm. Pico a 360nm, difusor para la corrección del
coseno de cuarzo.
Rango de medida: 0,1 10-3W/m2…2000W/m2.
LP471UVB
Sonda radiométrica para la medida de la irradiación con módulo SICRAM incluido.
Campo espectral UVB 280nm…315nm. Pico a 305 nm, difusor para la corrección del
coseno de cuarzo.
Rango de medida: 0,1 10-3W/m2…2000W/m2.
LP471UVC
Sonda radiométrica para la medida de la irradiación con módulo SICRAM incluido.
Campo espectral UVC 220nm…280nm. Pico a 260 nm, difusor para la corrección del
coseno de cuarzo
Rango de medida: 0,1 10-3W/m2…2000W/m2.
LP471ERY
Sonda radiométrica para la medida de la irradiación total eficaz (W/m2) ponderada
según la curva de acción UV (CEI EN 603352-27) con módulo SICRAM incluido.
Campo espectral 250nm…400nm. Difusor para la corrección del coseno de cuarzo.
Rango de medida: 0,1 10-3W/m2…2000W/m2.

Sensores fotométricos digitales:
Sensor UV para la medida de SAC TOC DQO.
ViomaxCAS51D permite la medida in situ de SAC, también denominado
CAE: coeficiente de absorción espectral, del medio. Este equipo, que se
basa en el principio de absorción UV, utiliza la absorción que presenta la
sustancia a medir en el espectro UV para determinar la concentración
proporcional de esta. Sustancias como los nitratos o como la mayoría de
compuestos orgánicos absorben en la zona UV sin necesidad de reactivos.
Principales aplicaciones: monitorización de carga orgánica a salida o entrada
de depuradora, medida de nitratos en balsas de aireación o a salida de
planta.
9. CONCLUSIONES
- La fotometría es una técnica muy utilizada para la determinación de concentraciones o
identificación de sustancias a partir de sus barridos espectrales o absorbancias de sus
patrones.
- El espectrofotómetro Shimadzu arroja un valor de absorbancia y transmitancia más
preciso y confiable debido al ancho de banda tan reducido.
- Cuando se desea hacer una análisis a una sustancia mediante el método de fotometría
visible, se debe conocer la longitud de onda en la que la sustancia presenta su máximo de
absorción, debido a que esto nos da una idea de cuál es la sustancia, pues estos máximos
son característicos para cada especie, y además nos permite resultados óptimos.
- Para determinar concentraciones de determinado ión en una sustancia, es necesario
primero tratar la muestra adecuadamente para que este ión forme un complejo coloreado y
se pueda hacer la medición en el espectrofotómetro, pues para que una sustancia absorba
radiación, es necesario que tenga color.
- Para obtener buenos resultados al aplicar la técnia fotométrica, es necesario utilizar los
adecuados materiales volumétricos para la preparación de los patrones y de las muestras
necesarias para la aplicación.

10. BIBLIOGRAFÍA
Federmán Castro E, Análisis instrumental l, manual de prácticas de laboratorio, Cámara
colombiana del libro, Colombia 2014.
Federmán Castro E, Análisis instrumental Algunos métodos fotométricos y
espectrometricos, Apuntes de clase, Colombia.
Luisa F. Echeverri L. – Flor Ángela Henao – Francy J. Ramirez. Elaboración de nueve
guías complementarias para la ejecución de prácticas de aplicación analítica en la
asignatura de Laboratorio de Análisis Instrumental I. Trabajo de grado para optar el título
de Tecnólogo Químico. Universidad Tecnológica de Pereira.
11. WEBGRAFÏA
http://www.espectrometria.com/espectro_electromagntico.
http://www.interempresas.net/Medicion/FeriaVirtual/Producto-Sensores-fotometricos-
digitales-Endress+Hauser-Viomax-CAS51D-113996.html
http://www.ictsl.net/productos/instrumental/sondasradiometricasyfotometricas.html
http://www.fao.org/docrep/010/ah833s/ah833s15.htm
Fundamentos de la espectrofotometría UV-VIS moderna
http://www.chem.agilent.com/Library/primers/Public/5980-1397ES.pdf
http://www.chem.uci.edu/~dmitryf/manuals/Shimadzu%20UV-1700%20users%20guide.pdf
http://www.cienytec.com/PDFS/Espec_Genesys20_OpMan.pdf
http://www.equipar.com.mx/web2012/wp-
content/uploads/2012/info_cat/spectronic/Spanish-evolutions-60S.pdf
http://www.thermoscientific.es/product/evolution-60s-uv-visible-spectrophotometer.html
http://www.gerdau.cl/productos-y-servicios/124-grados-y-composiciones-quimicas-de-
aceros-al-carbono
http://allstudies.com/clasificacion-acero.html