Embed Size (px)
Citation preview

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
MINISTERIO DE SALUD PUBLICA Y ASISTENCIA SOCIAL
PROGRAMA NACIONAL DE FARMACOVIGILANCIA
FARMACOVIGILANCIA
1

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
INTRODUCCION
La Farmacovigilancia es una actividad de Salud Publica destinada a la
Identificación, Evaluación y Prevención de los riesgos asociados a los
Medicamentos una vez comercializados. Como tal está orientada inevitablemente
a la toma de decisiones que permitan mantener la relación Beneficio-Riesgo de
los medicamentos en una situación favorable, o bien suspender su uso cuando
esto no sea posible.
Para cumplir este cometido la Farmacovigilancia obtiene la información de
diversas fuentes, pero su principal soporte científico la constituye la
Farmacoepidemiología, ciencia que utiliza el conocimiento, método y
razonamiento epidemiológicos para el estudio del uso y de los efectos del uso de
los medicamentos. A veces se produce cierta confusión entre ambas disciplinas,
pero no hay solapamiento posible: Farmacoepidemiología y
Framacovigilancia están situadas a distinto nivel. Entre ellas existe la misma
relación que hay entre la Epidemiología y la Salud Publica: la primera dedicada a
la formulación de Hipótesis y su refutación empírica, y la segunda orientada a la
acción.
Los procesos que integran la Farmacovigilancia pueden agruparse desde un
punto de vista operativo en dos fases: 1) El análisis de riesgos y 2) La gestión de
riesgos.
2

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
1. EL ORIGEN DE LA FARMACOVIGILANCIA
Los medicamentos son venenos útiles. De esta manera tan simple como efectiva
describe el farmacólogo ingles James W. Black, premio Novel de Medicina, las
dos caras indivisibles que poseen todos los medicamentos, capaces de aliviar o
curar enfermedades, pero también de causar daño si concurren circunstancias
que lo favorezcan. Esta dualidad del medicamento se conoce, en realidad, desde
muy antiguo, pero no es hasta bien avanzado el presente siglo, que tanto la
sociedad como los científicos y los gobiernos toman verdadera conciencia de los
efectos nocivos de los medicamentos como un problema de Salud Publica,
precisamente cuando la industrialización de los medicamentos permite la
difusión de los mismos a amplias capas de la sociedad como un producto mas de
consumo.
La primera advertencia seria sobre los riesgos de los medicamentos tiene lugar
en los Estados Unidos en 1937, cuando un elixir de Sulfonamida produce la
muerte de 107 personas, en su mayoría niños, debido al Dietilenglicol que se
utilizaba como excipiente en su preparación. Este episodio provoca que se dicten
normas legales para supervisar la seguridad de los medicamentos antes de su
distribución, fin para el que se crea en los Estados Unidos la Food and Drug
Administartion, la Primera agencia reguladora de medicamentos que aparece
en el mundo.
La segunda advertencia tiene a Europa como escenario. Estamos a comienzo de
la década de los sesenta, cuando la llamada “Revolución de los
medicamentos” se encuentra en pleno apogeo y la confianza en ellos y en las
posibilidades del hombre para combatir las enfermedades paracen ilimitadas.
Nada menos que unos 70 principios activos nuevos se introducían cada año ( hoy
no son mas de 30) En este clima de euforia tiene lugar en Alemania un brote de
una malformación congénita que hasta entonces era extraordinariamente rara,
llamada Meromelia o Focomelia. En 1958 se describía el primer caso, en 1959
eran ya 17, 129 en 1960 y 477 en 1961. El brote no parecía confinado a
3

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
Alemania y empezaron a describirse casos también en Gran Bretaña y Australia.
Inicialmente se pensó en factores hereditarios, pero su carácter epidémico
indujo a pensar en la intervención de factores externos: infecciones víricas,
radiaciones, alimentos... En Noviembre de 1961, W. Lenz, en una reunión de la
Sociedad de Pediatría de Rumania, sugirió la asociación entre la malformación y
el uso de un medicamento durante el embarazo. Era este fármaco un hipnótico
no barbitúrico que había alcanzado por la época una enorme popularidad debido
a que presentaba en margen de seguridad en sobredosificacion mucho mas
elevado que los barbitúricos. Tan seguro parecía que se recomendaba su uso
incluso en niños y también en mujeres embarazadas, aprovechando en ellas un
cierto efecto antiemético. Lenz llegó a la conclusión de la asociación después de
un estudio en el que entrevistó a 46 mujeres que habían tenido niños
malformados (casos), y ha 300 mujeres con niños normales (controles) y les
pregunto sobre la ingestión de Talidomida entre la cuarta y la novena semana
de embarazo. De los casos, 41 contestaron afirmativamente y ninguna entre los
controles. La asociación parecía por tanto fuerte. El 27 de Noviembre de 1961
se retiraba el medicamento de Alemania y sucesivamente después en otros
países. No se conoce con precisión las consecuencias de este accidente pero se
estima que nacieron en todo el mundo mas de 10.000 niños malformados la
mitad de los cuales murieron por malformaciones incompatibles con la vida. El
Desastre de la Talidomida, como ha quedado acuñado este trágico episodio
para la historia, tuvo otras consecuencias, esta vez positivas: 1) Los gobiernos
empezaron a exigir a las compañas farmacéuticas pruebas de toxicidad en
animales más exhaustivas; 2) Los ensayos clínicos controlados se propusieron
como herramienta básica para que los nuevos medicamentos demostraran
eficacia y seguridad; 3) Se propusieron diversas estrategias para evitar
accidentes similares que tomaron cuerpo en lo que hoy se conoce como
FARMACOVIGILANCIA. Otros episodios de menor magnitud confirmaron la
importancia de continuar el estudio de la seguridad de los fármacos
después de su comercialización.
2. EL RIESGO DE LOS MEDICAMENTOS
4

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
Cuando un medicamento se autoriza hoy día para su comercialización se sabe
mucho de él. Su actividad farmacológica y su toxicidad potencial se han probado
exhaustivamente en diversas especies de animales, con distintas dosis y tras
diversos tipos de exposición. Asimismo se ha ensayado en un promedio de 1,500
de seres humanos para demostrar su eficacia a corto y medio plazo ( < 1 año) en
indicaciones médicas concretas y habitualmente a diferentes dosis. Estos
ensayos han permitido también identificar y cuantificar una buena parte de sus
efectos adversos:
aquellos que ocurren con una frecuencia superior a 1 de cada 500 pacientes
expuestos y que se presentan tras periodos de exposición cortos. Pero estos
datos tienen también otra lectura, y es que aquellos efectos adversos que
ocurren con una frecuencia inferior a 1:500 o tienen una incidencia basal alta o
requieren para producirse periodos de exposición o inducción prolongados, o bien
ocurren especialmente en subpoblaciones que habitualmente no participan en los
ensayos clínicos, son esencialmente desconocidos cuando el fármaco alcanza el
mercado y, sin embargo, pueden ser lo suficientemente graves como para
desequilibrar su relaci6n beneficio-riesgo. Así se explica que alrededor del 3% de
los nuevos principios activos que se autorizan tengan después que ser retirados
por razones primarias de seguridad y que una proporción aun mayor sufra
modificaciones en sus condiciones de autorización.
2.1. Reacciones adversas a medicamentos: definición y tipos
Una reacción adversa es un efecto nocivo y no deseado que ocurre en el
hombre a dosis empleadas para el diagnóstico, la profilaxis o
terapéutica ; se excluyen por tanto las sobredosificaciones, ya sean accidentales
o con intención suicida. Esta es la definición del Programa Internacional de
Farmacovigilancia de la Organización Mundial de Salud, sin duda la más
aceptada. Los términos afectos adversos y efectos indeseables se pueden
considerar sinónimos del primero. No lo son, en cambio, los términos efecto
colateral, efecto secundado o efecto tóxico y debe rechazarse su uso. El término
acontecimiento (que no "evento") adverso hace referencia a cualquier suceso que
sea nocivo para el paciente y que ocurre una vez iniciada la administración de un
5

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
fármaco, tenga o no relación causal con el es una practica habitual recoger
acontecirnientos adversos en los ensayos clínicos, pero su utilización fuera de un
estudio formal no es una práctica aconsejable.
Hoy día es ya una costumbre distinguir entre dos grandes tipos de reacciones
adversas, siguiendo la clasificacion propuesta por Rawlins y Thompson en 1977:
las reacciones adversas de tipo A (dcl ingles Augmented) y las de tipo B (del
ing1es Bizarre). En la tabla 2 se recogen sus caracteristicas mas importantes.
Las de tipo A: serian aquellas reacciones adversas explicables por el mecanismo
de acción farmacológica del medicamento y que. por tanto, tendrían una relación
directa con la dosis; un ejemplo podría ser la hipotensión ortostatica producida
por algunos antihipertensivos.
Las de tipo B: en cambio, no serian explicables por el mecanismo de acción del
farmaco y aparecerian con independencia de las dosis administradas, siendo por
lo tanto atribuibles a una respuesta idiosincrasica del organismo; sirvan como
ejemplo las reacciones anafilácticas y, en general, las mediadas por mecanismos
inmunológicos.
Reacción adversa de tipo A Reacción adversa de tipo B● Relacionada con el mecanismo de
acción del fármaco.
● Dependiente de la dosis
● Predecible
● Conocida antes de la
comercialización
● Frecuente
● Menos grave (rara vez monartal)
● No relacionada con el mecanismo
de acción dcl fármaco
● No dependiente de la dosis
● Impredecible
● Suele ser desconocida antes de la
comercialización
● Suele ser infrecuente
● Grave (ocasionalmente mortal)
Tabla 2.Tipos de reacción adversa y Sus caracteristicas, según la clasificación de
Rawlins y Thompson modificada por los autores.
6

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
Esta clasificación es interesante porque explica de forma sencilla el problema de
la predicción de los efectos adversos. Es fácil ver, por ejemplo, que las pruebas
de toxicidad en animales básicamente pondrán de manifiesto las reacciones
adversas de tipo A, y no todas, sino aquellas que sean objetívales; reacciones de
carácter subjetivo como las alucinaciones o la alteración del gusto, pasaran
desapercibidas. Para las reacciones adversas de tipo B su capacidad predictiva es
aún menor, de tal manera que son frecuentes los falsos negativos y los falsos
positivos (notese que si los primeros conducen a probar en el hombre
rnedicamentos que despues se demuestran inseguros, los ultimos llevan a
rechazar un numero incalculable de farmacos que podrian ser eficaces y seguros
para el hombre.
Siguiendo el mismo razonamiento, durante el desarrollo clinico del medicamento
lo que debe esperarse es poner de manifiesto la mayoria de las reacciones
adversas de tipo A, pero solo aquellas de tipo B que sean frecuentes, que ocurran
tras periodos cortos de exposicion y que no sean especificas de subpoblaciones
con escasa o ninguna representacion en los ensayos clinicos.
Esta clasificacion de las reacciones adversas es eminentemente práctica, y no
tiene pretensiones de exhaustividad; por ella es relativamente fácil encontrar
reacciones adversas que no se ajustan estrictamente a ninguno de los dos tipos o
que tengan caracteristicas de ambos o, incluso, que siendo primero etiquetadas
de tipo B pasen a ser de tipo A cuando se conoce su naturaleza. Algunos autores
queriendo dotar de academicismo a esta clasificacion han ido añadiendo letras a
la misma: C (del ingles Cumulated, reacciones que se producen con dosis
acumulativas), D (del ingles Delayed, reacciones que aparecen con un decalaje
tras la exposicion), pero restan claridad a la original, aunque le añaden mas
precision.
Desde el punto de vista de la salud publica tiene interes distinguir entre
reacciones adversas evitables y no evitables . Las primeras ocurren por
mediacion de un error que puede estar situado en alguno de los siguientes
7

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
pasos: fabricación, suministro, prescripción, dispensación o administración del
medicamento, o bien por la no aplicacion de medidas preventivas disponibles,
por ejemplo la solicitud del nivel plasmatico de un farmaco o el seguimiento
frecuente de recuentos hematologicos cuando tales pruebas se hayan
demostrado utiles para prevenir o reducir la toxicidad. Algunas reacciones
adversas consideradas inicialmente como no evitables, se convierten en evitables
a lo largo de la vida del farmaco, gracias al mayor conocimiento que se posee de
su naturaleza y de sus factores de riesgo.
2.2 El costo sociosanitario de las reacciones adversas a medicamentos
Se estima que las reacciones adversas a medicamentos son responsables del 1-
3% de las consultas de atencion primaria, el 3-4% el las consultas a los servicios
de urgencias de los hospitales y el 4-6% de todos los ingresos hospitalarios.
Entre los pacientes hospitalizados, una proporcion que oscila segun los estudios
entre el 10 y el 30%, presenta reacciones adversas durante su estancia en el
hospital, lo que con frecuencia tiende a prolongarla. Asimismo entre el 18 y el
29% de las readmisiones hospitalarias precoces son causadas por reacciones
adversas. En la población general los datos son menos precisos y varian mucho
de unos estudios a otros, pero podemos asumir una incidencia de reacciones
adversas entre los individuos expuestos a medicamentos de entre el 5 y el 20%,
en su mayor parte de caracter leve.
Muy pocos estudios han tratado de estimar la morbilidad evitable causada por
reacciones adversas; los datos disponibles apuntan a que al menos un tercio de
las reacciones adversas que ocurren serian evitables, en su mayoria debidas a
errores de prescripcion, lo cual subraya la importancia de la educacion en
farmacologia clinica y terapeutica durante el pregrado y el postgrado de
medicina. Otra forma de medir el impacto sociosanitario de las reacciones
adversas es a traves de lo que le cuestan a los servicios de salud en teminos
economicos. Se ha estimado, por ejemplo, que en un hospital de tercer nivel de
700 camas las lesiones debidas al uso correcto e incorrecto de los medicamentos
8

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
tienen un costo anual en los Estados Unidos de 3,8 millones de dólares, de los
cuales 1 millon se deberian a lesiones evitables. En la población general, Johnson
y Bootman han calculado que los problemas relacionados con medicamentos (y
aqui incluyen, ademas de las reacciones adversas, la morbilidad derivada del uso
inapropiado en general, del no tratamiento farrnacologico cuando esta indicado,
de la sobredosis y de las interacciones con otros rnedicamentos, con alimentos o
con pruebas de laboratorio) suponen en los Estados Unidos unos 76.600 millones
de dolares al año en gastos directos (Sin contar, pues, la perdidia de
productividad y castos intangibles), lo cual les situa entre los problemas medicos
que mas recursos consumen, como las enfermedades cardiovasculares, la
diabetes o la obesidad.
3. EL ANALISIS DE RIESGOS EN FARMACOVIGILANCIA
El termino riesgo se presta a cierta confusion. En epidemiología se denomina
r i esgo a la probabilidad de un acontecimiento (un daño, por ejemplo) tras la
exposición a un determinado agente. Sin embargo, en el lenguaje coloquial
riesgo tiene ademas otra dimensión: la magnitud del daño (su gravedad y
duración), de tal manera que salemos aceptar que el riesgo aumenta no solo
cuando aumenta su probabilidad, sino tambien cuando aumenta su magnitud. En
esta revisión se entenderá riesgo en este sentido amplio, aun reconociendo que
el componente científicamente más relevante es su probabilidad.
El análisis de riesgos que se realiza en farmacovigilancia no difiere esencialmente
del que se realiza en otras áreas donde se incorporan nuevas tecnologías que
inciden de un modo u otro sobre la salud humana (figura 1). El primer paso del
análisis consiste en la identificación del riesgo, él segundo en su
estimación o cuantificación y el tercero en la evaluación de su
aceptabilidad social.
Habitualmente el análisis de riesgos es realizado íntegramente por expertos, pero
hay una demanda cada vez mas insistente de incorporar en el tercer paso del
analisis la valoración de los propios ciudadanos afectados.
9

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
3.1 La identificación del riesgo
Por identificación de un riesgo se entiende aquí la detección de un nuevo
problema de seguridad desconocido antes de la comercialización del
medicamento, o al menos, la sospecha razonable de su existencia. Son diversas
las fuentes de información que ayudan a identificar nuevos riesgos de los
medicamentos una vez comercializados. En general, cualquier estudio puede
aportar información relevante, desde pruebas realizadas en animales como las de
cancerogénesis hasta estudios fomales en grandes poblaciones; pero sin duda, el
procedimiento mas habitual de identificación de nuevos riesgos tras la
comercialización es la detección clinica de casos individuales o series de casos en
los que se sospecha que la enfermedad pudiera estar asociada al uso de un
farmaco. Encauzar de forma estructurada esta fuente de información tan
relevante es lo que se proponen los programas de notificación espontánea.
3.1.1 Notificación espontanea de casos individuales
La notificación espontánea de sospechas de reacciones adversas por parte del
profesional sanitario ha demostrado ser el metodo más eficiente para la
identificación de riesgos previamente no conocidos de los medicamentos. Por
ello, en muchos países se han creado sistemas permanentes de información que
pretenden: (a) facilitar al profesional Sanitario la notificación de sospechas de
reacciones adversas a través de un sencillo formulario, (b) recoger y validar
dicha información y (c) registrarla en una base de datos común que posibilite la
generación de "señales", manteniendo siempre la confidencialidad del paciente y
del notificador.
Las "señales" eran constituidas por un grupo mas o menos numeroso de sospechas de
una reacción adversa cuya asociación con el medicamento no es conocida en su
naturaleza o gravedad. En ocasiones una frecuencia de notificación mayor a la
esperada puede ser origen también de una señal.
Sospechar de un medicamento como causa de una enfermedad es una tarea no
exenta de dificultades. La mayor parte de las reacciones adversas que se
10

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
detectan como nuevas tras la comercialización son de tipo B. En ocasiones,
cuando la enfermedad que aparece es típicamente inducida por medicamentos,
como algunas reacciones cutáneas graves (vgr. Síndrome de Stevens-Iohnson,
necrolisis epidermica toxica), o hematológicas (vgr. agranulocitosis), el grado de
sospecha del medico es elevado. Sin embargo, con cierta frecuencia las
reacciones adversas constituyen cuadros o enfermedades que no son típicamente
inducidas por fármacos ni distinguibles de las que aparecen por otro tipo de
causas. Es aquí donde la cuidadosa observación, la perspicacia clínica y la mente
critica del médico juegan un papel fundamental. Tras este primer paso de
considerar la etiología iatrogénica cuando se realiza el diagnostico diferencial de
cualquier cuadro clínico y de valorarla como plausible, es necesario que él medico
notifique dicha sospecha.
Las ventajas y limitaciones de la notificación espontánea se muestran en la tabla 3.
Los principales valores de este método son su sencillez y su carácter universal,
ya qué potencialmente abarca a toda la población, a todas las reacciones
adversas y a todos los medicamentos desde el comienzo mismo de la
comercialización. La infranotificación, por otra parte, es su talón de Aquiles.
Ventajas Limitaciones● Método sencillo
● Abarca toda la población
● Abarca a todos los medicamentos
desde el comienzo de su
comercialización
● No interfiere con los hábitos de
prescripción
● Permite detectar reacciones
adversas poco frecuentes.
● La infranotificación disminuye la
sensibilidad
● La tasa de notificación no es
constante
● Dificil detección de reacciones
adversas de aparición retardada
● No se puede cuantificar incidencias
Tabla 3. Ventajas y limitaciones de Ia notificación espontánea
a) El problema de Ia infranotificación
11

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
La baja tasa de notificación por parte del profesional sanitario es una realidad.
Baste decir que en los paises con más tradición en farmacovigilancia, como el
Reino Unido, el número de médicos que notifican no supera el 10%. La
proporción de reacciones adversas que se notifican suele compararse con la
zonas visibles de un iceberg, que sólo representa una pequeña parte de lo que
realmente existe.
Las causas de la infranotificación han sido objeto de diversos estudios y
encuestas realizadas a los profesionales sanitarios. La tabla 4 muestra los
motivos identificados por Inman hace ya algunos años, quien los describe como
"los siete pecados capitales del potencial notificador". Se han ideado estrategias
para que el profesional sanitario tenga a mano tarjetas amarillas y se anime a
notificar, incluyéndolas por ejemplo en talonarios de recetas o en el Vademecum.
Más recientemente se tiende a facilitar la notificación a través del telefono e
incluso por vía electrónica haciendo uso de las nuevas tecnologías de la
información.
Es importante que el profesional sanitario conozca que toda la información que
envía es de utilidad, y que sólo la sospecha de que el medicamento ha podido
participar en la aparición de cualquier cuadro clínico, es suficiente para
notificarla. El centro de farmacovigilancia donde llega dicha notificación
se encargará de evaluar el grado de relación causal, sin entrar a valorar
ni enjuiciar la actuación medica.
12

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
⮚ Complacencia o falsa idea de que únicamente se comercializan
Medicamentos seguros.● Miedo de sufrir denuncias por parte de los pacientes.
● Culpabilidad al pensar que el daño del paciente es debido almedicamento prescrito.
● Ambición de recoger y publicar series de casos.
● Ignorancia del procedimiento.
● Vergüenza a notificar meras sospechas.
● Pereza, una mezcla de falta de tiempo, falta de interés, falta de tarjetasamarillas.
Tabla 4. - Los siete pecados capitales del potencial notificador, segunInman
La notificación espontanea de una sospecha de reacción adversa es totalmente
compatible con la publicación del caso. Cada vez es más frecuente notificar antes
de remitir el caso para su publicación. Algunas revistas exigen incluso al autor un
contacto previo con el centro de farmacovigilancia.
Conscientes de la sobrecarga de trabajo en todos los ámbitos asistenciales, los
centros de farmacovigilancia hacen hincapié en que el médico priorice la
notificación de sospechas de reacciones adversas graves, y las que involucran a
medicamentos comercializados en los útimos cinco años.
b) Evaluación de la relación de causalidad de casos individuales
Mientras que para confirmar otras etiologías se suele disponer de pruebas
histológicas, serológicas o bacteriológicas, la etiología medicamentosa
normalmente no puede confirmarse en los casos individuales; la mayor
certidumbre se obtiene cuando el paciente es reexpuesto al medicamento y
presenta el mismo cuadro. Por razones obvias, esta información solo está
disponible en una minoría de ocasiones.
Para facilitar el análisis de la relación causal entre el fármaco sospechoso y la
13

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
reacción adversa y al mismo tiempo para lograr una mayor concordancia entre
los evaluadores, se han ideado una serie de algoritmos que pretenden "etiquetar"
cada sospecha de reacción adversa con un determinado grado de probabilidad.
Aunque existen publicados en la literatura más de 20 algoritmos diferentes, en
todos ellos se valoran al menos los siguientes parámetros:
Secuencia temporal: Tiempo transcurrido entre el inicio del tratamiento y la
aparición de la reacción adversa. Se analiza si dicha secuencia es compatible con
la fisiopatología de la enfermedad y el mecanismo de acción del fármaco. Por
ejemplo, no estaría a favor de una relación causal una cirrosis hepática que
apareciera a los dos días de haber comenzado el tratamiento.
Conocimiento previo: Descripción de la asociación fármaco-reacción en la
bibliografía o en bases de datos de reacciones adversas consultadas “ad hoc” El
hecho de que la reacción haya sido descrita previamente apoyaría la relación
causal; si es el primer caso descrito, la relación causal estaría "condicionada" a la
aparición de nuevos casos.
Efecto de la retirada del medicamento: La relación farmaco-reacción adversa
séra más probable si esta última mejora al retirar la medicación, En reacciones
adversas irreversibles, este parámetro no podrá valorarse.
Efecto de la reexposición al fármaco sospechoso: La reexposición será
positiva si la reacción adversa reaparece, o negativa en caso contrario. En
muchas de las ocasiones no es valorable al no haber reexposición.
Existencia de una causa alternativa: Cuando haya una enfermedad, otro
fármaco u otros factores que sean tanto a más verosímiles como causa de la
reacción que la exposición al farmaco sospechoso.
14
Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
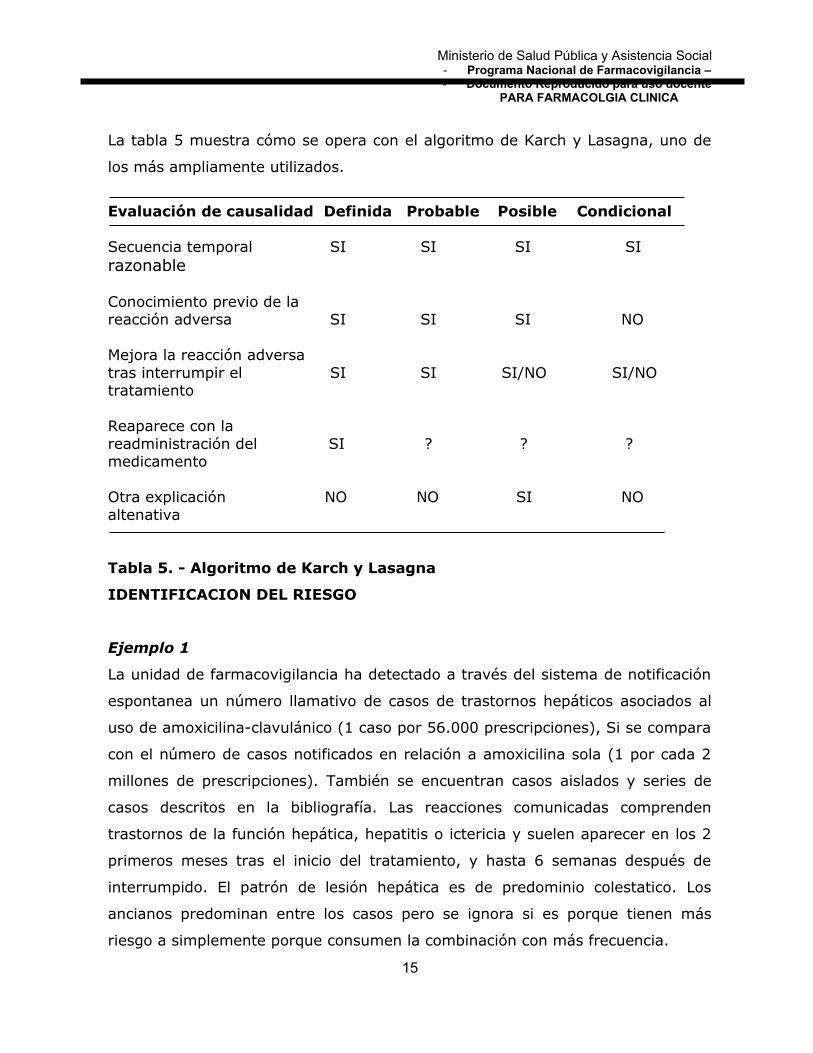
La tabla 5 muestra cómo se opera con el algoritmo de Karch y Lasagna, uno de
los más ampliamente utilizados.
Evaluación de causalidad Definida Probable Posible Condicional
Secuencia temporal SI SI SI SIrazonable
Conocimiento previo de lareacción adversa SI SI SI NO
Mejora la reacción adversatras interrumpir el SI SI SI/NO SI/NOtratamiento
Reaparece con lareadministración del SI ? ? ?medicamento
Otra explicación NO NO SI NOaltenativa
Tabla 5. - Algoritmo de Karch y Lasagna
IDENTIFICACION DEL RIESGO
Ejemplo 1
La unidad de farmacovigilancia ha detectado a través del sistema de notificación
espontanea un número llamativo de casos de trastornos hepáticos asociados al
uso de amoxicilina-clavulánico (1 caso por 56.000 prescripciones), Si se compara
con el número de casos notificados en relación a amoxicilina sola (1 por cada 2
millones de prescripciones). También se encuentran casos aislados y series de
casos descritos en la bibliografía. Las reacciones comunicadas comprenden
trastornos de la función hepática, hepatitis o ictericia y suelen aparecer en los 2
primeros meses tras el inicio del tratamiento, y hasta 6 semanas después de
interrumpido. El patrón de lesión hepática es de predominio colestatico. Los
ancianos predominan entre los casos pero se ignora si es porque tienen más
riesgo a simplemente porque consumen la combinación con más frecuencia.
15

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
Ejemplo 2
Hoy día es bien conocido que el riesgo de tromboembolismo venoso aumenta en
usuarias de anticonceptivos orales. Se plantea entonces la pregunta de si con la
terapia hormonal de sustitución también existe un aumento del riesgo, a pesar
de que la dosis de estrógenos que se utilizan en dichos preparados son más
cercanos a los niveles fisiológicos, a diferencia de las que se utilizan en la
contracepción hormonal. Tanto en la bibliografía como en las bases de datos de
reacciones adversas aparecen casos descritos.
3.1.2 Otras estrategias para la identificación del riesgo
Las limitaciones de la notificación espontanea, básicamente el bajo grado de
sospecha de los profesionales sanitarios y la infranotificación, han forzado la
puesta en marcha de otros procedimientos que ayuden identificar riesgos de
forma precoz. En principio, caben dos alternativas genéricas: el seguimiento de
pacientes expuestos a un medicamento intentando registrar las reacciones
adversas que puedan aparecer con el tiempo, o bien de forma inversa, la
detección de pacientes con ciertas enfermedades e investigar su asociación con
la exposición previa a medicamentos.
La primera estrategia, el seguimiento de pacientes expuestos, es frecuentemente
utilizada por las compañías farmacéuticas en los llamados "estudios de
farmacovigilancia o de fase IV". Lamentablemente, estos estudios han servido en
muy contadas ocasiones para identificar nuevos riesgos y son frecuentemente
utilizados de forma torticera para inducir la prescripción del medico:
normalmente se siguen entre 1.000 y 5.000 pacientes en tratamiento a lo largo
de varios meses, lo que suele añadir poco a la información que se dispone en el
momento de la autorización. Se estima que estos estudios deberían multiplicar al
menos por 5 la experiencia de exposición (número de pacientes y tiempo de
observación) obtenida durante el desarrollo clínico para tener posibilidades reales
de detectar nuevos riesgos, si existen. El problema sigue siendo su ineficiencia:
16

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
mucho costo y poca sensibilidad para detectar problemas nuevos. Por otra parte,
la ausencia de un grupo control dificulta la atribución de las posibles reacciones
adversas al medicamento.
La segunda estrategia, esto es, la identificación de casos, parece más razonable
si las reacciones adversas son muy infrecuentes (< 1 caso por cada 10.000
pacientes expuestos). Junto a los casos pueden recogerse uno o varios controles
(estrategia que se denomina "vigilancia caso-control") lo cual permite no solo
identificar riesgos sino también cuantificarlos (la fuerza de la asociación al
menos, véase más adelante). La recogida de controles, no obstante, pierde
eficiencia si la prevalencia de uso de los medicamentos implicados no es alta. La
limitación más importante de esta estrategia es que no permite detectar,
obviamente, reacciones adversas no incluidas en la vigilancia.
3.2.La estimación del riesgo
Una vez que un presumible nuevo riesgo de un medicamento ha sido
identificado, el siguiente paso consiste en intentar cuantificar la fuerza de la
asociación entre la reacción adversa y el fármaco y su probabilidad de aparición
(su incidencia).
Si bien la notificación espontanea ofrece a menudo una aproximación razonable
al problema de la relación de causalidad entre el fármaco y la reacción adversa,
no permite cuantificar la fuerza de la asociación. Tampoco permite estimar la
incidencia con la que aparece la reacción adversa debido, por un lado, a la
infranotificación, que impide conocer un número real de casos (el numerador) y,
por otro, a que no proporciona una estimación de la población expuesta (el
denominador). Los datos de consumo de medicamentos se utilizan a menudo
como una aproximación del denominador (expresándolo en meses o años de
tratamiento a partir de la dosis diaria media), pero el valor de la incidencia así
estimada es muy limitado. En la mayoría de las ocasiones este segundo paso del
análisis de riesgos solo podrá hacerse con rigor a través de estudios
epidemiológicos analíticos.
3.2.1 Estudios epidemiológicos
17

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
El ensayo clínico controlado es el paradigma de la investigación clínica y la
herramienta básica para evaluar la eficacia de los medicamentos. Su aplicación
en la evaluación de la seguridad después de la comercialización, sin embargo, se
suele considerar poco eficiente, salvo en aquellos casos en los que el problema
de seguridad constituya un objetivo muy definido, suficientemente frecuente y,
sobre todo, cuando concurran factores de confusión de dificil ajuste
(especialmente la confusión por indicación, véase más adelante). Por otra parte,
debe tenerse en cuenta que lo que se pretende es valorar los efectos del
medicamento en sus condiciones reales de uso, por lo que no parece apropiado
intervenir en la asignación del tratamiento. Todo ello lleva a considerar a los
estudios epidemiológicos observacionales como los más aconsejables en general.
A diferencia de los estudios experimentales, en los que el investigador determina
la asignación de la exposición de forma aleatoria, en los estudios observacionales
el investigador no interviene en el proceso. Es más probable, por tanto, que los
factores relevantes para el objetivo del estudio no se distribuyan
homogéneamente en los grupos de comparación y por tanto estén más
expuestos a errores sistemáticos, lo cual deberá tenerse en cuenta tanto en la
fase de diseño y análisis como en la interpretación de los resultados.
Los estudios analíticos observacionales, se clasifican en dos grandes tipos
atendiendo al criterio de selección de los pacientes: los estudios de cohorte y los
de casos y controles.
a) Estudios de cohorte
Los pacientes se seleccionan atendiendo a la exposición al medicamento
(expuestos y no expuestos) y se siguen a lo largo del tiempo con el objeto de
detectar la aparición de la(s) reacción(es) adversa(s) de interés. A menudo, el
periodo de observación debe prolongarse durante varios años, para registrar un
número suficiente de pacientes con el desenlace de interés. El procedimiento de
detección de la reacción adversa dependerá del tipo de enfermedad objeto de
estudio pudiéndose realizar a través de entrevistas periódicas, registros de
mortalidad, diagnósticos de alta hospitalaria, historias clínicas, etc. Los estudios
18
Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
de cohorte permiten estudiar más de una reacción adversa. Sin embargo, el
trabajo de campo que precisan es costoso y son poco eficientes para investigar
reacciones adversas infrecuentes o cuyo cuadro clínico tenga una incidencia basal
alta. Tampoco son apropiados para investigar aquellas reacciones que aparecen
tras períodos de exposición o de inducción muy prolongados. Su principal
problema metodológico son las pérdidas de los pacientes a lo largo del
seguimiento; perdidas superiores a un 30% pueden ser suficientes para invalidar
el estudio, al no poderse (pasa a pag. 20)
ESTIMACION DEL RIESGO
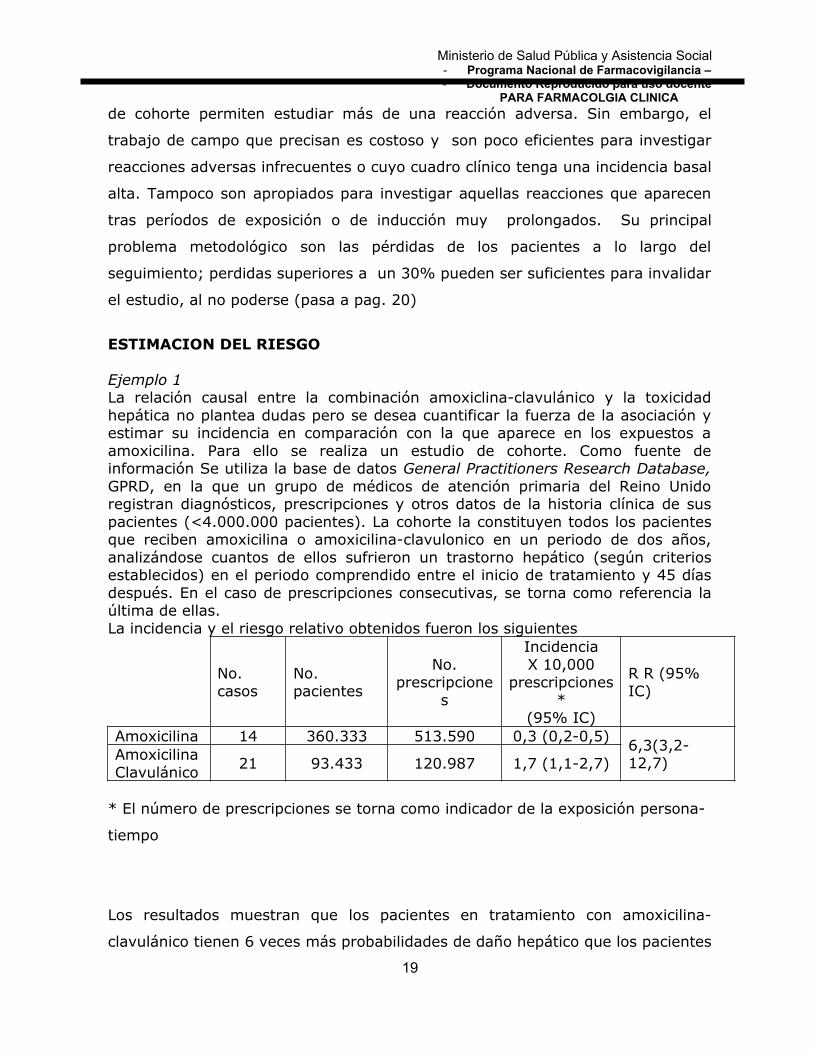
Ejemplo 1La relación causal entre la combinación amoxiclina-clavulánico y la toxicidadhepática no plantea dudas pero se desea cuantificar la fuerza de la asociación yestimar su incidencia en comparación con la que aparece en los expuestos aamoxicilina. Para ello se realiza un estudio de cohorte. Como fuente deinformación Se utiliza la base de datos General Practitioners Research Database,GPRD, en la que un grupo de médicos de atención primaria del Reino Unidoregistran diagnósticos, prescripciones y otros datos de la historia clínica de suspacientes (<4.000.000 pacientes). La cohorte la constituyen todos los pacientesque reciben amoxicilina o amoxicilina-clavulonico en un periodo de dos años,analizándose cuantos de ellos sufrieron un trastorno hepático (según criteriosestablecidos) en el periodo comprendido entre el inicio de tratamiento y 45 díasdespués. En el caso de prescripciones consecutivas, se torna como referencia laúltima de ellas.La incidencia y el riesgo relativo obtenidos fueron los siguientes
No. casos
No. pacientes
No.prescripcione
s
IncidenciaX 10,000
prescripciones*
(95% IC)
R R (95% IC)
Amoxicilina 14 360.333 513.590 0,3 (0,2-0,5)6,3(3,2-12,7)Amoxicilina
Clavulánico21 93.433 120.987 1,7 (1,1-2,7)
* El número de prescripciones se torna como indicador de la exposición persona-
tiempo
Los resultados muestran que los pacientes en tratamiento con amoxicilina-
clavulánico tienen 6 veces más probabilidades de daño hepático que los pacientes
19
Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
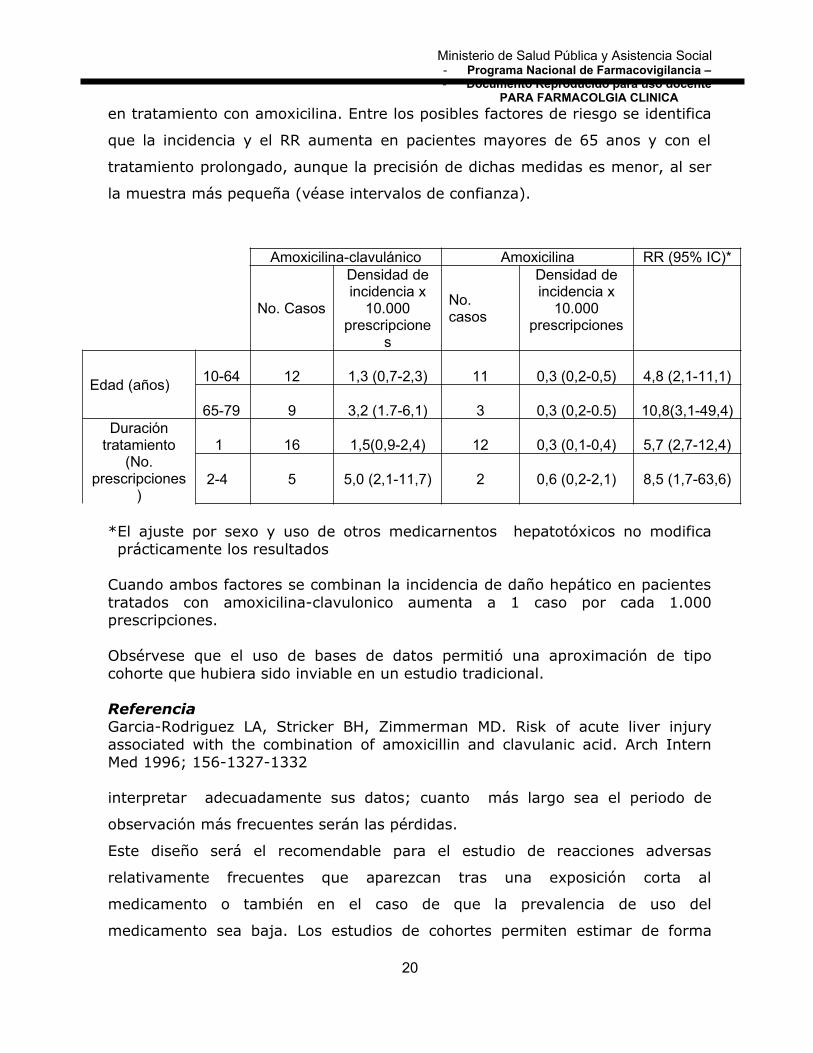
en tratamiento con amoxicilina. Entre los posibles factores de riesgo se identifica
que la incidencia y el RR aumenta en pacientes mayores de 65 anos y con el
tratamiento prolongado, aunque la precisión de dichas medidas es menor, al ser
la muestra más pequeña (véase intervalos de confianza).
Amoxicilina-clavulánico Amoxicilina RR (95% IC)*
No. Casos
Densidad deincidencia x
10.000prescripcione
s
No. casos
Densidad deincidencia x
10.000prescripciones
Edad (años)10-64 12 1,3 (0,7-2,3) 11 0,3 (0,2-0,5) 4,8 (2,1-11,1)
65-79 9 3,2 (1.7-6,1) 3 0,3 (0,2-0.5) 10,8(3,1-49,4)Duración
tratamiento(No.
prescripciones)
1 16 1,5(0,9-2,4) 12 0,3 (0,1-0,4) 5,7 (2,7-12,4)
2-4 5 5,0 (2,1-11,7) 2 0,6 (0,2-2,1) 8,5 (1,7-63,6)
*El ajuste por sexo y uso de otros medicarnentos hepatotóxicos no modificaprácticamente los resultados
Cuando ambos factores se combinan la incidencia de daño hepático en pacientestratados con amoxicilina-clavulonico aumenta a 1 caso por cada 1.000prescripciones.
Obsérvese que el uso de bases de datos permitió una aproximación de tipocohorte que hubiera sido inviable en un estudio tradicional.
ReferenciaGarcia-Rodriguez LA, Stricker BH, Zimmerman MD. Risk of acute liver injuryassociated with the combination of amoxicillin and clavulanic acid. Arch InternMed 1996; 156-1327-1332
interpretar adecuadamente sus datos; cuanto más largo sea el periodo de
observación más frecuentes serán las pérdidas.
Este diseño será el recomendable para el estudio de reacciones adversas
relativamente frecuentes que aparezcan tras una exposición corta al
medicamento o también en el caso de que la prevalencia de uso del
medicamento sea baja. Los estudios de cohortes permiten estimar de forma
20

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
directa tanto medidas de asociación (riesgo relativo, RR) como de frecuencia
(riesgo absoluto). También es posible estimar el llamado riesgo atribuible
(diferencia de incidencias de expuestos y no expuestos), medida que tiene un
gran interés desde el punto de vista de la salud pública.
b) Estudios de casos y controles
En este tipo de estudios, los pacientes son seleccionados según presenten o no
una enfermedad determinada. Los casos serán pacientes con la enfermedad y los
controles pacientes seleccionados aleatoriamente de la misma población fuente
de la que surgen los casos y que no presentan la enfermedad en el momento de
su selección. En ambos grupos se estudia la exposición a los medicamentos de
interés en un intervalo de tiempo (ventana de exposición) previo al inicio de la
reacción adversa (día índice) para los casos o el día correspondiente para los
controles. La determinación del día índice y de la ventana de exposición es
crucial, y debe obedecer a criterios clínicos y epidemiológicos.
La exposición previa a los medicamentos, se puede obtener mediante entrevistas
al paciente a través de un cuestionario estructurado o bien a través de la revisión
de su historia clínica. El método de obtención de dicha información deberá ser en
todo igual en los casos que en los controles, para evitar la aparición de sesgos de
información.
Este diseño es especialmente útil cuando se quiere estudiar reacciones adversas
poco frecuentes o que requieren periodos largos de exposición o inducción para
producirse, ya que se garantiza la inclusión de un número de casos suficientes
sin que la muestra tenga que ser rnuy grande, como ocurriría si se eligiera un
diseño de tipo cohorte. Por ejemplo, en el estudio de la relación entre el cáncer
vaginal de celu1as claras en mujeres jóvenes y su exposición intraútero a
dietilestilbestrol, bastó un estudio de 8 casos y 32 controles; de los 8 casos, 7
habían estado expuestas a dietilestilbestrol, mientras que no se identificó dicha
exposición para ninguno de los controles. Otra ventaja de los estudios de casos y
controles es que permiten analizar la asociación de la enfermedad con diversos
factores simultáneamente.
Su principal dificultad estriba en la selección adecuada del grupo control. Como
21

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
se ha dicho los controles deben ser una muestra de la población de donde
provienen los casos, pero lo difícil, a veces, es trasladar esta idea a un
procedimiento operativo de selección. Es curioso comprobar que la semejanza
entre los estudios de casos y controles y los de cohortes es mayor de la que a
primera vista parece. De hecho los estudios de casos y controles se pueden
conceptualizar como un estudio de cohorte en el que la experiencia de exposición
persona-tiempo de los denominadores de incidencia se ha muestreado en vez de
haberla contabilizado en su totalidad. Si la distribución de la exposición entre los
controles es representativa de la distribución de la exposición en toda la
población se puede hacer una estimación no sesgada del riesgo relativo sin
conocer la incidencia dado que la fracción de muestreo será la misma tanto en la
población de expuestos como en la de no expuestos. Es frecuente utilizar en los
estudios de casos y controles una medida de asociación conocida como razón de
ventaja, o más comúnmente por su término inglés odds ratio (OR), pero si los
controles se han muestreado de la población fuente como decirnos, se demuestra
fácilmente que OR y RR coinciden.
RR = Ic = a / Pc = a / Pc . f = a / b = a . d = OR Io c / Po c / Po. f c /d c. b
donde a = casos expuestos y c = casos no expuestos; Pc = Población fuente
expuesta;
Po =P oblación fuente no expuesta; f = Fracción de muestreo; b = controles
expuestos y d = controles no expuestos. (pasa a pagina 23)
ESTIMACION DEL RIESGO
Ejemplo 2.
Con el fin de cuantificar la magnitud de la asociación entre tromboembolismovenoso y el uso de terapia hormonal de sustitución (THS) se realiza un estudio decasos y controles ya que el riesgo se presume pequeño. Se recogen todos loscasos de tromboembolismo diagnosticados en mujeres de 45 a 64 años en loshospitales de una región determinada a lo largo de casi dos años (n=103); comocontroles se identifican mujeres ingresadas en los mismos hospitales condiagnósticos quo no tienen ninguna relación con el uso de terapia hormonalsustitutiva: trastornos: oculares, óticos, dermatológicos, respiratorios, etc.(n=178) Las controles se aparean con los casos por grupo de edad, distrito de
22

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
procedencia y fecha de admisión ya que se piensa que estos factores pueden serde confusión. Se entrevista en el hospital a todas las mujeres incluidas en elestudio, preguntando sobre su historia médica, uso de THS y otros fármacostomados en los últimos tres meses. Se definen usuarias actuales de THS a lasmujeres que refieren haber tornado esta medicación en el mes previo al ingreso(ventana de exposición).
Los resultados obtenidos fueron los siguientes:
Casos Controles O R (95%IC) OR(95%IC)No usuarias de THS* 59 134
3,0(1,6-5,6)3,5 (1,8-7,0)
Usuarias actuales de THS
44 44
* Mujeres que no han tornado THS en el últimos mes
El OR ajustado se obtiene al tener en cuenta en el análisis ciertos parámetros de
los que se recogió información que podrían actuar como factores de confusión
(índice de masa corporal, historia previa de varices, grupo socioeconómico). El
riesgo de tromboembolismo se multiplica por tres en las pacientes que recibieron
THS en el último mes. En otros estudios se han obtenido resultados similares. A
partir de datos poblacionales de tromboembolismo pulmonar se estima que el
riesgo atribuible a THS oscila entre 1,6 y 2,3 casos por 10.000 mujeres por año.
Referencía
Daly E, Vessey MP, Hawkins MM et al. Risk of venous tromboembolism in users of hormone
replacement therapy. Lancet 1996, 348: 977-80.
Como se deduce de la discusión precedente, los estudios de casos y controles, no
permiten estimar medidas de frecuencia (riesgo absoluto) de forma directa ya
que se desconoce el denominador.
c)Uso de bases de datos sanitarias informatizadas
El trabajo de campo que requieren los estudios observacionales en general
consumen muchos recursos (materiales y humanos) y mucho tiempo. La
utilización de bases de datos informatizadas ha permitido mejorar la eficiencia de
la recogida de información abaratando el costo de estos estudios y reduciendo el
23

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
tiempo necesario para obtener resultados. Este último factor ha sido
especialmente importante ya que ante riesgos graves, a menudo no era posible
esperar a la finalización de estudios formales tradicionales para tomar decisiones
reguladoras.
Las bases de datos sanitarias informatizadas recogen de forma sistemática
información individualizada sobre el uso de recursos sanitarios de grandes grupos
de población (prescripción de medicamentos, visitas ambulatorias, ingresos
médicos con sus diagnósticos de alta). Los responsables de su gestión son
compañías de seguros médicos, sistemas nacionales de salud o colectivos
médicos. Esta información permite la identificación de cohortes de expuestos y
no expuestos, de casos y controles, o bien adoptar estrategias de selección
híbridas como ocurre en los estudios de casos y controles anidados en una
cohorte (nested case-control studies). Al investigador se le proporciona siempre
la información de forma anónima preservando de este modo la confidencialidad
de los datos del paciente.
Existen dos tipos de sistemas: bases de datos múltiples enlazadas por un identificadorpersonal único, y bases de datos globales que registran todos los datos de unmismo paciente, y que generalmente son gestionadas por el médico decabecera. Aunque, como cualquier herramienta, no carece de problemas,también permite superar otros, siendo el balance francamente favorable. Porejemplo, la información sobre la exposición a los medicamentos de interéssuele ser más completa y fiable que en los estudios tradicionales ya que loque se registra en la base de datos es su prescripción o su dispensación envez De confiar en la memoria del paciente. Las bases de datos han permitidorealizar estudios de cohortes con cientos de miles de individuos, algorealmente impensable con trabajos de campo (véase ejemplo 1). Por otraparte, simplifican de forma muy notable el procedimiento de selección decontroles en los estudios de casos y controles.
3.2.2 Precisión y validez de la estimación
La precisión de la estimación está inversamente relacionada con el papel que el azarjuega en los resultados obtenidos. El error aleatorio viene medido en laspruebas de significación estadística por el valor de la “P”. El problema de esteparámetro es que no refleja la influencia del tamaño de la muestra: un RRirrelevante (1,10) puede ser estadísticamente significativo si la muestra esrnuy grande y al contrario, un RR importante (5,20) puede no alcanzar lasignificación estadística debido a quc la muestra es pequeña. Para tener una
24

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
idea de como el tamaño de la muestra está afectando a los resultados, esdecir, cuan precisa es la medida, es fundamental calcular el intervalo deconfianza de la estimación puntual para un nivel de seguridad dado(habitualmente 95%). Cuanto menor sea dicho intervalo, más precisa será lamedida, lo que refleja que la muestra es suficientemente grande. Por otraparte, también se deduce que cuanto menor sea la asociación que se busca,más precisión se necesitara para detectarla.
La validez de la estimación se relaciona inversamente con la presencia de erroressistemáticos: sesgos y factores de confusión. De forma muy general, lossesgos se clasifican en sesgos de selección y de información Los sesgos deselección ocurren cuando el procedimiento empleado para obtener los gruposde comparación introduce diferencias entre ambos que distorsionan la medidade efecto. Los sesgos de información aparecen cuando las diferencias entrelos grupos de comparación Son debidas a los procedimientos de obtención dela información, por ejemplo cuando en los estudios de casos y controles sehace más énfasis en la obtención de los datos sobre exposiciones previas enlos casos que en los controles. Finalmente, se dice que un factor es deconfusión cuando se asocia simultáneamente
con la exposición y con la enfermedad, de tal manera que podemos atribuir a la
exposición (al medicamento) una asociación espúrea con la enfermedad ( piense
el lector en la siguiente broma: la cama es el lugar más peligroso del mundo,
porque es donde se muere más gente). En farmacoepidemiología la llamada
confhsión por indicación es una de las más frecuentes y difíciles de resolver.
Resulta de la prescripción selectiva de los medicamentos en función de factores
de riesgo de la enfermedad cuya asociaci6n con el medicamento se desea
cuantificar. Por ejemplo, cuando se quiere estudiar si el uso de dosis altas de
bloqueantes de canales de calcio como agentes antihipertensivos se asocia a
infarto agudo de miocardio, habrá que valorar en que medida el uso de dosis
elevadas se debe a una mayor gravedad del cuadro hipertensivo y por tanto el
que dichos pacientes tengan mayor riesgo de padecer un infarto agudo de
miocardio.
Los sesgos no pueden corregirse en el análisis de los datos, por lo que un
cuidadoso diseño del estudio será la mejor manera de prevenirlos. La distorsión
25

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
que introducen los factores de confusión, en cambio, suele poderse corregir o
ajustar en el análisis (mediante análisis estratificados o análisis multivaríados) Si
se ha recogido la información necesaria.
3.2.3 De la asociación a la relación causal
Una vez que las medidas de asociación (RR, OR o riesgo atribuible) se consideran
suficientemente validas y precisas el siguiente paso es considerar si dicha
asociación es de tipo causal. Para ello, deberá tenerse en cuenta toda la
información disponible y valorar los resultados del estudio en su contexto. Los
parámetros que apoyan la relación causal son los siguientes:
⮚ Fuerza de la asociación: Cuanto mayor sea la magnitud de la asociación,
menor será la posibilidad de que el hallazgo sea debido a otros factores.
⮚ Posibilidad o coherencia biológica: La relación causal cobrará fuerza Si existe
algún mecanismo biológico que la explique.
⮚Consistencia con los resultados de otros trabajos: La existencia de resultados
similares d
diferentes estudios apoyaría la relación causal.
⮚ Secuencia temporal: La causa tiene que preceder al efecto. Aunque parece
obvio, en ocasiones esto no es tan fácil de establecer, especialmente en los
estudios de casos y controles (de aquí la importancia de determinar
apropiadamente el día índice).
⮚ Relación dosis-respuesta: El riesgo aumenta con una exposición mis intensa
(sólo valido para reacciones adversas do tipo A).
3.3 Evaluación del riesgo
El tercer paso del análisis es juzgar si el riesgo identificado y cuantificado es
aceptable pra la sociedad y en que condiciones. Además de los datos sobre el
riesgo del medicamento, debe considerarse su beneficio potencial y los riesgos y
beneficios de las alternativas terapéuticas cuando existan. También conviene
realizar el esfuerzo de situar el riesgo del medicamento en el contexto de los
26

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
otros riesgos que solernos aceptar en nuestra vida cotidiana. Los resultados
pueden ser sorprendentes; mucha gente se alarmaría si le dijeran que una
determinada tecnología causa una cantidad de muertos equivalente a si se
estrellaran 3 aviones Jumbo ¡ todos los días del año!. Desde luego ningún
medicamento saldría airoso. Pues bien, estas muertes son las que se atribuían al
consumo de tabaco en los Estados Unidos en la década de los ochenta.
Una cuestión que aun queda por resolver es quien debe realizar la evaluación. Lo
habitual es que sean expertos o comités de expertos (como en España la
Comisión Nacional de Farmacovigilancia), lo cual es al menos discutible. Así
corno la identificación y la estimación del riesgo pertenecen fundamentalmente al
ámbito científico, ya que en ambos pasos del análisis se dirimen cuestiones de
hecho, en la evaluación entran en juego además cuestiones de valor, las cuales
exceden los márgenes de la ciencia. El problema estriba en que los valores no se
pueden objetivar como los hechos, no se perciben sino que se estiman
subjetivamente (aún cuando sobre muchos de ellos pueda haber un gran
consenso). Es claro que un mismo riesgo puede ser aceptable para un individuo y
no para otro. Este argumento nos lleva a considerar que si bien los expertos se
bastan por sí solos para las dos primeras etapas del análisis de riesgos (las
cuestiones de hecho), su criterio resulta necesario pero insuficiente para la
tercera (las cuestiones de valor), so pena de caer en un paternalismo flagrante a
comienzos de la década de los noventa se retiro del mercado mundial, a poco de
comercializarse, Un fármaco para el tratamiento de la incontinencia urinaria: la
terodílina. Tal medida se debió a que el medicamento se asociaba a un riesgo
elevado de muerte subita por arritmia, que en algunos subgrupos podía tener
una probabilidad de hasta 1 por cada 1.000 expuestos. Ocurri6 entonces la
siguiente anécdota. Una viejecita Sueca de 80 años llamo al centro de
farmacovigilancia de su país para pedir explicaciones sobre la retirada del
fármaco que para ella había sido muy eficaz (se quedó sin alternativa, como no
fuera el uso de pañales). Cuando el director del centro le explico los hechos
irrebatibles que habían conducido a la decisión, la viejecita le interpeló:
'tMuchacho - por el director - desde luego prefiero morir súbitamente pero seca,
27

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
en vez de vivir mojando mi ropa interior todo el día, y por cierto, ¿quien le ha
dado a ud. el derecho a decidir de qué tengo yo que morir?" (anécdota referida a
log autores por el Dr. B.E. Wiholm). He aquí, pues, un problema que no quedara
resuelto hasta que se de un procedimiento apropiado que permita incorporar los
valores de los potencialmente afectados en el proceso de evaluación de la
aceptabilidad social del riesgo.
4. LA GESTION DEL RIESGO EN FARMACOVIGILANCIA
Concluida a fase de análisis todo queda dispuesto para llevar a cabo las acciones
oportunas, lo que globalmente denominamos como gestión del riesgo (risk
managemet)). Desde el punto de vista especifico de la farmacovigilancia tres son
las acciones relevantes: 1) Adoptar medidas administrativas de reducción del
riesgo; 2) Comunicar a los profesionales sanitarios y a los pacientes la existencia
del riesgo, las medidas adoptadas y las recomendaciones al respecto; y 3)
Establecer estrategias especificas de prevención.
4.1 Medidas administrativas de reducción del riesgo
La administración sanitaria y las compañías farmacéuticas, como responsables de la
autorización y de la comercialización del medicamento respectivamente, son las
encargadas de tomar las medidas necesarias para reducir el riesgo que pueda
presentar su uso. La decisión de tomar una medida de carácter regulador no es
fácil. Debe tener básicamente en cuenta la aceptabilidad social del riesgo en
función del beneficio que procura, si bien otros factores de variada índole
(económicos, industriales...) pueden entrar en juego cuando la información de
que se dispone es insuficiente o dudosa. Las medidas pueden ser diversas,
oscilando entre únicamente informar del nuevo riesgo, hasta la retirada
inmediata del medicamento del mercado (tabla 6). Un elemento consustancial a
la adopción de una medida reguladora es el seguimiento de su impacto en el
uso del medicamento.
Aceptabilidad del riesgo Medidas reguladoras
⮚ Riesgo aceptable en las condiciones de
uso autorizadas
⮚ Restricción de indicaciones y medidas
para prevenirla (si se conoce)
28

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
⮚ Riesgo aceptable en ciertas condiciones
⮚ Riego inaceptable en cualquier situación
⮚ Introducción de contraindicaciones
⮚ Restricción a ciertos grupos de
población
⮚ Realización de pruebas clínicas o
analíticas
⮚ Restricción del ámbito de la
prescripción => Diagnóstico hospitalario =>Uso hospitalario =>Prescripción por especialista =>Restricción de ciertas presentaciones
⮚ Retirada
=> Inmediata => Progresiva
Tabla 6.- Medidas administrativas de reducci6n del riesgo
4.2. Comunicación del riesgo
Los individuos tienen derecho a ser informados verazmente y de forma completa
sobre los riesgos quo para su salud comportan las nuevas tecnologias y solo
excepcionalmente, para evitar un mal mayor, podria justificarse la no
cornunicación total o parcial de la información. Este planteamiento, que es
radicalmente etico, constituye tambien el modo mas eficaz de gestionar las
situaciones de riesgo. Es un hecho bien conocido que los seres humanos
aceptamos niveles de riesgo mas altos cuando hay una elección voluntaria dcl
mismo que cuando es impuesto. Compartir la infoemación y hacer participe a los
ciudadanos en el proceso de evaluación y toma de decisiones consigue,
precisamente, esto, transformar un riesgo involuntario, muy mal aceptado, en un
riesgo autonornamente asumido.
EVALUACION DEL RIESGO Y MEDIDAS A TOMAR
Tanto en EL ejemplo de trastornos hepáticos y amoxicilina-clavulonico como en elde tromboembolismo venoso y terapia hormonal sustitutiva, los expertosconsideran que los beneficios que aportra el farmaco superan los riesgosestimados por lo que, en general, son aceptables, excepto para algunossubgrupos de población o condiciones de uso.
29

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
Ejemplo 1Para amoxicilina-clavulonico, se incluyó en la ficha tecnica información de lareacción adversa y de sus factores de riesgo (edad superior a 65 años yprolongación del tratamiento). Se recornienda que la duración del tratamiento nosea superior a 14 dias, salvo expresa indicación medica. Tambien se informa alpaciente a traves del prospecto.
Ejemplo 2El riesgo de padecer tromboembolismo debido a la exposición a THS Cs do 2-3/10.000 mujeres por año (el riesgo absoluto de la enfermedad en la poblaciónde 1/10.000 mujeres por año). El riesgo además parece concentrarse en elprimer año de tratamiento y es reversible al retirar el medicamento; la letalidadde esta enfermedad oscila entre el 1% y el 2%. Tanto en la ficha tecnica comoen el prospecto para el paciente se informa del riesgo y se contraindican dichospreparados en pacientes con antecedentes de tromboembolismo; ademas seadvierte que se utilicen con precaución en mujeres con factores de riesgo detromboembolismo (varices, obesidad, inmovilización igual o superior a 3semanas)
Que grado de información dar, como hacerlo y cuando son las tres cuestiones clave quedeben responderse toda vez que se haya de comunicar un riesgo. Quizá cabedistinguir de partida dos situaciones diferentes: a) cuando se trata de un riesgoconocido, o b) cuando es un riesgo emergente. La primera debe formar parte dela rutina de la practica clinica diaria. La cuestión más ardua de resolver en estecontexto, es que grado de información suministrar. El debate etico y juridico aeste respecto es muy amplio y excede las pretensiones de este capitulo. Comonorma se puede decir que la información deberia ser lo más completa posible,dentro de lo que el paciente sea capaz de ir asurniendo, paniendo desde luego deunos minimos quo serian los riesgos considerados evitables (vgr. interaccionesde los IMAO con otros medicamentos 0 alimentos) y los graves impredecibies. Lainformación complementaria por escrito, especialmente cuando no exista unprospecto genuino dirigido al paciente, puede ayudar mucho en esta labor.
En relación con la segunda situación, un riesgo emergente, se ha discutido mucho sobreel modo de infomar a los ciudadanos para que tomen decisiones consecuentes sincrear innecesarias situaciones de panico y alarma social, pero de momento noexisten unas directrices asurnidas por todos que sirvan de guia y eviten lasimprovisaciones, siendo todavia una asignatura pendiente para la mayoria de lasagendas reguladoras de medicamentos. El sentido de la responsabilidad y laprudencia son los parametros maestros que deben orientar a los diferentesagentes. En farmacovigilancia existe un cierto consenso en considerar que elprocedimiento mas apropiado, salvo en casos de verdadera urgencia, es el queinvolucra a los profesionales sanitarios como receptores primarios de lainformación, lo que les permite actuar como referencia para el ciudadanopotencialmente afectado Solo despues do esta primera fase es cuando la noticiadcl riesgo debiera, en su caso, ponerse en conocimfento de la poblaci6n, bien seaa trave's de los medios de comunicacidn de masas 0 de otros procedirnientos.
30

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA
4.3. Estrategias de prevencion del riesgoUna proporción nada desdeñable de reacciones adversas se consideran evitables.Su prevención es el objetivo ultimo de la farmacovigilancia. La prevencion deberealizarse de forma rutinaria extremando la vigilancia para evitar la comisión deerrores. Los profesionales sanitarios (médicos, farmaceuticos, enfermeros), losusuarios, las compañias y las autoridades sanitarias tienen su parcela deresponsabilidad. La comunicación franca entre estos agentes juega un papelclave para evitar o detectar precozmente los errores. Ademas de esta prevenciónrutinaria cabe hablar de una prevención ad hoc que se ejerce medianteprogramas especificos sobre determinados farmacos (ej. el programa deseguimiento de clozapina) o bien sobre determinados grupos de riesgo (ej.mujeres embarazadas).Con respecto a las reacciones adversas consideradas como no evitables Se debepretender al menos su detecci6n precoz, lo que no deja de ser una medida deprevención de la magnitud del daño. La información tanto a los profesionalessanitarios coma a los pacientes constituye sin duda la mejor estrategia. Losfarmacologos clinicos integrados en los equipos asistenciales tanto en atenciónprimaria como en los hospitales pueden jugar un papel crucial en la prevenciónde riesgos.
5.CONCLUSIONLa farmacovigilancia surgió históricamente coma respuesta a una tragedia. Se
reconocía así la limitación para predecir los riesgos que conllevan losmedicamentos, pero al mismo tiempo se hacía una apuesta decidida porafrontarlos. Nuestra sociedad está hoy dia lejos de las. actitudes temerarias quepretenden el progreso sin control pero tambien esta muy distante de lasactitudes timoratas que le huyen. En el libro Against the gods, el economistanorteamericano Peter Bernstein afirmaba con elocuencia: "La idea revolucionariaque determina la frontera entre los tiempos modernos y el pasado es ladominación del riesgo: la noción de que el futuro es más que un capricho de losdioses y que los hombres y mujeres no son sujetos pasivos frente a lanaturaleza... Entendiendo el riesgo, midiéndolo y sopesando sus consecuencias,hemos convertido su desafío en el principal catalizador de la sociedad occidental ”.Vista desde esta perspectiva la farmacovigilancia no debe considerarsemeramente como un procedimiento de control del progreso en el mundo delmedicamento, sino sino más bien como uno de sus más decididos promotores, lavertiente positiva que desde estas páginas se propugna.
Bibliografía recomendada
Abraham S. Science, politics and the pharmaceutical industry. UCL Press Limited, London, 1995.
Asschcr AW, Parr GD, Whitmarsh VB. Towards the safer use of medicines. BMJ 1995; 311:
31

Ministerio de Salud Pública y Asistencia Social- Programa Nacional de Farmacovigilancia –- Documento Reproducido para uso docente
PARA FARMACOLGIA CLINICA1003-1005.
Baicke OM, Manocchia M, De Abajo PJ, Kaitin K, Lasagna L. Drug safety discontinuations in theUnited Kingdom, the United States and Spain from 1974 through 1993: a regulatory perspective.Clin Pharmacol Ther 1995; 58:108-117.
Bernstein PL. Against the gods - the remarkable story of risk. John Wiley and Sons, New York,1996.
Carvajal A. Farmacoepidemiología. Valladolid, Universidad de Valiadolid, 1993. Davis DM. Textbookof adverse drug reactions 2nd ed, Oxford University Press, Oxford, 1991.
Elguero J, Perez Gutthann S. Bases de datos en farmacologia y terapeutica. Monografias Dr. Antonio Esteve (no. 18). Ediciones Doyma, Barcelona, 1996.
Hartzema AG, Porta MS, TilsonHH. Pharmacoepidemiology. an introduction. Harvey Whitney Books,Cincinnaty, 1991.
Inman WHW. Monitoring for drug safety, MTD Press Limited, Lancaster, England, 1996.
Hennekens CH, Buring JE. Epidemiology in Medicine.. Little Brown and Company, Boston/Toronto, 1987.
Laporte JR, Tognoni G. Principios de epidemiología del medicamento. Editorial Masson-Saivat,1993.
Rothman KJ. Epidemiología Moderna. Ediciones Diaz de Santos, S.A, 1987.
Strom BL. Pharmacoepidemiology, 2nd ed, Chichester: John Wiley and Sons, 1994.
ValIvé C. Seguridad y medicamentos - reacciones adversas a los medicamentos: métodos y problemas de la farmacovigilancia. JR Prous, S.A. Barcelona, 1987.
32









![[Pecha Kucha] Augmented Vision](https://img.pdfslide.es/doc/110x75/54bda1384a7959ad088b45db/pecha-kucha-augmented-vision.jpg)


















