Embed Size (px)
Citation preview

Profesor Argenis J. Sánchez Henríquez
1
I. INTRODUCCIÓN
Tales de Mileto, Demócrito, Aristóteles, entre otros grandes filósofos griegos
de la era antes de cristo, así como artesanos y grandes alquimistas de distintas
latitudes ocuparon gran parte de sus vidas en el estudio de la materia;
transmitiendo su conocimiento práctico y teórico a través de cientos de escritos,
que fueron posteriormente traducidos a las principales lenguas. Desde el primer
libro sistemático de química (Alchemia), escrito por Andreas Libavius en el siglo
XVI, hasta la más reciente publicación sobre química analítica cualitativa y/o
cuantitativa (libros, manuales, revistas, entre otros) de nuestra época, persigue el
fin de transmitir las ideas fundamentales del análisis químico, algunas veces de
forma abstracta y otras de manera simple. El presente trabajo fue realizado con el
propósito de servir como herramienta básica para el estudiante que se inicia en la
práctica del análisis químico cuantitativo y cualitativo. El análisis de materia
homogénea y heterogénea (muestras) en sus distintos estados de agregación es
una tarea que requiere de gran dedicación, técnica y conocimiento. Por tal razón,
es aconsejable que el estudiante en los primeros años de su aprendizaje logre el
dominio de las técnicas básicas del análisis químico, que le permitirán seguir
avanzando en este amplio campo de la química. En este sentido, el presente
manual toma en consideración aspectos prácticos y definiciones preliminares
importantes para la consecución de los objetivos planteados a lo largo del mismo;
sin olvidar un aspecto relevante, como es la seguridad dentro del laboratorio de
química. De igual manera, se enriquece el número de ilustraciones que
acompañan cada procedimiento analítico, para que el estudiante aferré mejor las
ideas y se le facilite el proceso de aprendizaje.
Hay que recordar que las distintas teorías y definiciones del análisis químico
clásico se mantienen hoy día, transcurridos ya muchos siglos; sin embargo el
análisis instrumental está en constante evolución, y nos exige cada día estar
atentos a estos cambios.

Profesor Argenis J. Sánchez Henríquez
2
II. PRELIMINARES
II.1. Normas de seguridad en los laboratorios de química
La actitud de todo el personal que ingresa a un laboratorio de química es de
vital importancia, ya que la mayoría de los accidentes que ocurren en los
laboratorios son originados por la apatía del personal que labora, recibe
instrucción o visita el lugar. Es por esto que todas las personas que ingresan a un
laboratorio de química deben tener una actitud de interés hacia las actividades que
se van a desarrollar, además de emplear el sentido común y seguir
cuidadosamente las normas de seguridad. La seguridad en los laboratorios de
química es responsabilidad tanto de los docentes, personal técnico, obreros,
estudiantes y visitantes, y por consiguiente es importante que todos tomen parte
activa en la prevención de accidentes. Para prevenir los accidentes en los
laboratorios de química se deben seguir las reglas de seguridad que a
continuación se enumeraran, éstas se refieren a la actitud y vestimenta que se
deben tener antes y durante nuestra permanencia en el laboratorio de química:
Antes de ingresar al laboratorio de química usted debe estar al tanto de
los peligros involucrados en la manipulación y exposición a las sustancias
químicas que va a emplear para el desarrollo de la actividad práctica.
Para ello puede referirse a un manual de reactivos, o para una búsqueda
completa a un Handbook de físico-química. Más adelante, en esta sección
de seguridad encontrará un par de figuras (1 y 2) que representan una
típica etiqueta de reactivo y pictogramas, que deben acompañar a los
reactivos en su presentación comercial, que le ayudaran a reconocer la
información más relevante para su correcta manipulación. De igual forma
en el apéndice de este manual, encontrará información completa
relacionada con las frases de riesgo (R) y seguridad (S), codificadas en
estas etiquetas de reactivos.
Antes de ingresar al laboratorio de química usted debe estar familiarizado
con el procedimiento que seguirá para el desarrollo de la actividad

Profesor Argenis J. Sánchez Henríquez
3
práctica que realizará. Recuerde que este procedimiento puede constar
de diferentes etapas, que a su vez pueden estar constituidas por diversas
operaciones. Es por ello que es recomendable hacer diagramas que
contengan a las distintas operaciones que están involucradas en cada
etapa del procedimiento.
Al ingresar al laboratorio de química usted debe llevar colocada la bata de
laboratorio. Ésta debe poseer ciertas características que le permitan
protegerlo de salpicaduras y derrames que puedan ocurrir durante el
desarrollo de sus actividades, es por tanto que el diseño de la misma
debe tomar en cuenta el largo apropiado en el cuerpo principal de la bata
y sus mangas. Usted debe vestir pantalones largos y evitar prendas cortas
como bermudas o faldas, ya que se expone de forma innecesaria a
sustancias altamente corrosivas e irritantes. Por otra parte, usted debe
utilizar calzados de cuero completamente cerrados y de tacón bajo. Evitar
el uso de joyería, ya que la misma puede ser dañada por las sustancias
químicas, además que permiten un contacto más prolongado entre su piel
y las sustancias químicas, al quedar éstas acumuladas en las prendas.
Por último, si tiene el cabello largo debe recogerlo.
Una vez que se encuentre en el laboratorio de química usted debe
familiarizarse con la ubicación de las salidas de emergencia, dispositivos
de seguridad como: lava ojos, duchas, extintores de fuego, anaquel de
primeros auxilios entre otros.
Una vez que se encuentre en el laboratorio de química usted debe
mantener una actitud responsable y seria durante su permanencia en el
mismo. No se aceptan juegos ni bromas, así como tampoco se permiten
cambios en los procedimientos establecidos en las prácticas sin previa
consulta con el profesor o el personal técnico que labora en el laboratorio.
Durante su permanencia en el laboratorio de química y a lo largo del
desarrollo de las actividades prácticas usted no debe preparar, consumir o
almacenar ningún tipo de alimento o bebida. Para ello existen áreas

Profesor Argenis J. Sánchez Henríquez
4
destinadas para tal fin. Tampoco debe masticar chicle, fumar cigarrillos o
utilizar cosméticos, ya que las sustancias químicas pueden absorberse en
estos.
Una vez que se encuentre en el laboratorio de química nunca desarrolle
prácticas si no está bajo la supervisión del profesor o del personal técnico
que allí labora.
Durante el desarrollo de las actividades prácticas en el laboratorio de
química, usted debe siempre estar atento al desplazarse por las áreas del
laboratorio y anticipar el movimiento de sus compañeros. Si llegase a
tropezar o caer mientras traslada material de vidrio o sustancias químicas,
procure siempre, si no logra equilibrarse, lanzarlas lejos de usted y de sus
compañeros de trabajo.
Durante el desarrollo de las actividades prácticas en el laboratorio de
química mantenga siempre el material de vidrio, equipos y sustancias
químicas lejos del borde de la mesa de trabajo.
Durante el desarrollo de sus actividades prácticas en el laboratorio de
química nunca pipetee sustancias químicas con la boca. Para ello utilice
una perilla de succión o pro-pipeta.
Durante el desarrollo de las actividades prácticas en el laboratorio de
química utilice los lentes de seguridad cuando se estén manipulando
sustancias químicas, o mezclas de ellas, que puedan generar vapores o si
está trabajando con algún sistema de baja presión. Ya sea usted la
persona que realiza la operación o algún compañero que se encuentre en
su cercanía.
Durante el desarrollo de las actividades prácticas en el laboratorio de
química utilice la campana extractora si va a realizar alguna operación
que genere gases, vapores, humos o partículas que puedan resultar
peligrosos al ser inhalados.
Durante el desarrollo de las actividades prácticas en el laboratorio de
química queda terminantemente prohibido descartar sustancias

Profesor Argenis J. Sánchez Henríquez
5
inflamables, tóxicas o corrosivas a través de los desagües de los
lavaderos. En cada caso se deben almacenar los residuos en
contenedores apropiados y seguir el protocolo de higiene y seguridad
avalado por las autoridades del ministerio del ambiente.
Durante el desarrollo de las actividades prácticas en el laboratorio de
química si llegase a romperse algún material de vidrio, éste no debe
descartarse en la papelera con los residuos comunes (papel, plásticos
entre otros). Dé aviso del accidente al profesor o personal técnico. Éstos
lo envolverán en papel y bolsas apropiadas, previo lavado del mismo, y lo
colocaran en una caja destinada para tal fin.
Una vez terminadas sus actividades en el laboratorio de química organice
su sitio de trabajo y antes de salir del laboratorio lave siempre sus manos
con jabón y abundante agua.
Las siguientes figuras de pictogramas y etiqueta son de uso obligatorio en
las presentaciones comerciales de las sustancias químicas o reactivos
(Chemdat.net, revisado Octubre 2007). El formato de las etiquetas puede variar
según las políticas de cada compañía productora de reactivos, pero los
pictogramas tienen un carácter universal. El objetivo que se persigue al
mostrarlas, es vincular a los usuarios de estos reactivos con la información mínima
para su correcta manipulación.
Materiales Explosivos
Materiales Oxidantes Materiales Nocivos o Comburentes (XN) o Irritantes (Xi)
Materiales o Altamente Materiales Inflamables (F) Inflamables (F+) Corrosivos
Materiales Peligroso para el Tóxicos (T) Medio Ambiente Muy Tóxicos
Figura 1. Pictogramas de Seguridad (Fuente: Chemdat.net, Octubre 2007)

Profesor Argenis J. Sánchez Henríquez
6
Seguidamente la figura 2 muestra una etiqueta de reactivo en la cual se han
enmarcado con óvalos los pictogramas de seguridad que identifican las
características peligrosas que supone la manipulación de este material; así como
también se han resaltado de la misma forma los códigos que se originan de las
combinaciones de frases de riesgo (R) y seguridad (S). (Ver Apéndice 5
Figura 2. Etiqueta de reactivo con pictogramas y códigos de frases de riesgo (R) y seguridad (S) (Fuente: www.Chemdat.net, Octubre 2007)
R:
11-2
0/2
1/2
2-3
6
S:
16-3
6/3
7R
: 11-2
0/2
1/2
2-3
6
S:
16-3
6/3
7R
: 11-2
0/2
1/2
2-3
6
S:
16-3
6/3
7R
: 11-2
0/2
1/2
2-3
6
S:
16-3
6/3
7

Profesor Argenis J. Sánchez Henríquez
7
II.2. Materiales de laboratorio
En el análisis químico cuantitativo y cualitativo se emplean una amplia
variedad de materiales o instrumentos elaborados con diferentes compuestos de
partida (vidrio, cuarzo, platino, plata, níquel, plástico, porcelana, entre otros). La
elección de los compuestos para la fabricación de estos instrumentos obedece,
por una parte al propósito para el cual se diseña el instrumento, y por otra parte a
las ventajas que el mismo le confiere al producto terminado. A continuación se
ofrecen algunas definiciones de los instrumentos de laboratorio más comunes.
Pipetas: Son cilindros de vidrio fabricados para
transferir líquidos de forma precisa. Básicamente
encontramos dos tipos de pipetas: 1) Pipetas
Volumétricas, las cuales tienen una marca o aforo
que les permite proporcionar un volumen de líquido
determinado bajo ciertas condiciones señaladas
por el fabricante; 2) Pipetas graduadas, las cuales
pueden suministrar diferentes volúmenes de
líquido a través de la graduación grabada en su
vástago, siempre bajo las condiciones
especificadas por el fabricante. Las pipetas
graduadas, a su vez, pueden ser de dos tipos: 2.1)
Pipetas de Mohr, y 2.2) Pipetas serológicas.
(1) (2.1) (2.2)
Buretas: Son cilindros largos de vidrio que
mantienen una calibración uniforme a lo largo de
su porción graduada, el extremo inferior de la
misma se cierra con una llave de vidrio o de teflón,
estos instrumentos están fabricados para emitir o
transferir volúmenes de líquido precisos. Las
buretas son instrumentos más precisos que las
0
1
2
3
4
5
6
7
8
9
10m
L 1
/10
0
1
2
3
4
5
6
7
8
9
10m
L 1
/10
0
1
2
3
4
5
6
7
8
9
0
1
2
3
4
5
6
7
8
9
10m
L 1
/10
0
1
2
3
4
5
6
7
8
9
10m
L 1
/10
10
0
1
2
3
4
5
6
7
8
9
10m
L 1
/10
10

Profesor Argenis J. Sánchez Henríquez
8
pipetas. La precisión de una bureta puede oscilar
entre 0.02 0.1mL, dependiendo de la capacidad
del instrumento.
Matraz volumétrico: Son recipientes de vidrio de
fondo plano, con forma de pera y cuello largo,
diseñados para contener un volumen específico a
una temperatura determinada, generalmente 20 C.
Tienen una línea delgada (línea de aforo) grabada
en el cuello del instrumento la cual indica el
volumen para el cual fue calibrado el recipiente. Su
capacidad oscila entre 1mL y 6000mL (6L), y su
precisión se incrementa entre 0.01 2mL, en
función de la capacidad del recipiente.
Cilindros graduados: Son recipientes cilíndricos
de vidrio relativamente grueso o de plástico,
graduados a lo largo de su cuerpo. Estos
recipientes están diseñados para transferir
volúmenes de líquidos aproximados, por tal razón
no deben emplearse para análisis que requieran
de una exactitud relativa. Su capacidad puede
variar entre 5mL y 4000mL (4L), con una precisión
que se incrementa desde 0.1mL hasta 29mL.
Matraz Erlenmeyer: Son vasos cónicos de vidrio o
plástico rematados en un cuello cilíndrico y corto,
con o sin tapa. Su capacidad puede variar entre
25mL y 6000mL (6L). Tienen numerosas
aplicaciones, siendo la más resaltante de ellas su
uso en el análisis volumétrico para la titulación de
soluciones.

Profesor Argenis J. Sánchez Henríquez
9
Vaso de precipitado o Berzelius: Son recipientes
cilíndricos de vidrio o plástico diferentes diámetros
y altura, con un pico que facilita la transferencia de
líquidos. Son de gran utilidad en la precipitación de
sólidos y en la evaporación de líquidos. Su
capacidad puede variar desde 10mL hasta 4000mL
(4L).
Vidrio de reloj: Es un circulo de vidrio cóncavo
que sirve para tapar vasos de precipitados,
cápsulas de porcelana o embudos. Se elaboran
con diferentes diámetros y en ocasiones pueden
emplearse para pesar sólidos usando una balanza.
Embudos cuantitativos: 1) Instrumento de vidrio,
plástico o porcelana, hueco de forma cónica
(ángulos de 58-60 ) o cilíndrica y cuello largo, o
corto, de diámetro muy pequeño en relación con su
parte superior. El diámetro de la boca de este
instrumento puede variar entre 6 y 15 cm y se
emplea para la filtración de líquidos y transferencia
de sólidos precipitados. 2) Los embudos cilíndricos
con placa de vidrio sinterizado se denominan
embudos de Buchner, y resultan muy útiles cuando
el líquido a ser filtrado ataca con facilidad al papel
filtrante.
(1) (2)
Pisetas o botellas de lavado: Son envases de
plástico con tapa de rosca en la parte superior y
con una salida para el líquido en forma de cuello
de cisne. Se elaboran con diferentes polímeros
dependiendo del tipo de solvente que vaya a
contener y su capacidad varia entre 125-1000mL.

Profesor Argenis J. Sánchez Henríquez
10
Crisoles: Son recipientes cónicos elaborados
generalmente de porcelana, sílice, óxido de
aluminio o platino. Básicamente los crisoles se
clasifican en dos tipos: 1) Crisoles comunes (sin
perforaciones en el fondo) y 2) Crisoles filtrantes
(como los crisoles de Gooch y Munroe). Los
crisoles se utilizan para el secado y la conversión
de los precipitados en formas más adecuadas para
su pesada.
(1)
(2)
Espátulas: Son Paletas metálicas (Nikel/Stainless
Steel), plásticas o de porcelana, con terminaciones
simples o en forma de cucharilla, que sirve para
tomar pequeñas porciones de sólidos para pesar o
ser transferidos de un recipiente a otro.
Pesa-filtros o pesa-sustancias: Son recipientes
generalmente de forma cilíndrica fabricados, en
algunos casos de polietileno, pero más
comúnmente de vidrio de borosilicato con tapa y
boca esmerilada para garantizar el cierre
hermético del recipiente. Los pesa filtros, con la
ayuda de un desecador, se emplean para
conservar muestras y reactivos, que han sido
sometidos a procesos de desecación, aislados de
la humedad atmosférica y de los gases como el
CO2 y el O2.
Varillas de vidrio: Barra de vidrio de 20 cm. de
largo aproximadamente y 3 - 4 mm de diámetro
con extremidades redondeadas, que sirve para
agitar las soluciones y transvasar líquidos y sólidos
precipitados.

Profesor Argenis J. Sánchez Henríquez
11
Mechero: Instrumento elaborado con distintas
aleaciones constituido básicamente de un tubo
cilíndrico y una base, que varían de diámetro
según el modelo. El tubo cilíndrico posee en su
parte inferior una llave o válvula aguja y un
mecanismo que permite regular la entrada del
carburante (gas) y el comburente,
respectivamente. Este instrumento se emplea para
calentar soluciones, así como para secar y calcinar
precipitados. Se pueden alcanzar temperaturas
variables en función del tipo de mechero
empleado. Existen básicamente tres modelos de
mecheros comúnmente utilizados en los
laboratorios: 1) Mechero Bunsen (a), 2) Mechero
Tirril, 3) Mechero Meker (b). Es de resaltar que a
través de los años se han modificado estos
modelos dando origen a otros mecheros como por
ejemplo; el mechero Fischer que es una
modificación del mechero Meker.
Desecador: Son recipientes semi-cilíndricos de
vidrio grueso Pyrex®, Phoenix® y más
recientemente de plástico, todos con tapas, con un
diámetro que puede oscilar entre 14 y 23 cm y una
altura comprendida entre 20 y 32 cm. El fondo del
recipiente contiene un material desecante (cloruro
de calcio anhidro, pentóxido de fósforo, entre
otros) separado por una placa porosa,
generalmente de cerámica, sobre la cual se
colocan las sustancias, contenidas en un crisol o
(1)
(2)

Profesor Argenis J. Sánchez Henríquez
12
en un pesa-filtro, que serán secadas o aisladas de
la humedad, oxígeno y dióxido de carbono. Los
hay básicamente de dos tipos: el de Scheibler (1) y
al vacío (2)
Pera de succión o pro-pipeta: Son dispositivos
empleados para llenar las pipetas de diferentes
capacidades, evitando así el uso de la boca. Estos
dispositivos pueden ser tan sencillos como una
simple pera de goma con una boquilla para
succión y otra para expulsión, o más complejos
con cuerpos de vinilo y mecanismos internos
automáticos que pueden proporcionar mayor
control en el volumen medido y durabilidad del
aparato.

Profesor Argenis J. Sánchez Henríquez
13
II.3. Técnicas comúnmente empleadas en el laboratorio
Seguidamente se ofrecen un conjunto de breves descripciones referidas al
uso y técnicas adecuadas para el manejo de ciertos materiales o instrumentos de
laboratorio.
Uso de equipo volumétrico
El material volumétrico esta fabricado y calibrado fundamentalmente con el
propósito de contener o transferir (verter) ciertos volúmenes de líquidos, esto se
indica generalmente en la superficie del material con las letras “TC” y “TD”,
respectivamente. Cuando se requiere la medición de volúmenes confiables (con
alta precisión) se emplean pipetas, buretas y balones aforados. Los vasos de
precipitados, cilindros graduados y Erlenmeyers son materiales fabricados con
otros propósitos tal como se describe en la sección anterior.
Pipetas. Para el análisis volumétrico de alta precisión se deben emplear
pipetas volumétricas (página 6). Antes de comenzar a describir el
procedimiento debemos recordar, atendiendo a las normas de
seguridad, que para aspirar sustancias químicas al interior de una pipeta
se emplea siempre una pro-pipeta o perilla de succión. Para el uso
correcto de este instrumento se deben cuidar las siguientes
instrucciones, estas son aplicables también a otros tipos de pipetas:
a) Se debe curar el interior de la pipeta limpia con una
pequeña porción (aproximadamente un 25% de la capacidad del
instrumento) de la solución que se va a transferir. Para ello, aspire una
porción del líquido en la pipeta y moje la superficie interior inclinando y
rotando la pipeta suavemente. Repita este proceso por lo menos dos
veces. Para terminar esta etapa aspire una porción del líquido y con
mucho cuidado hágala subir por la pipeta hasta unos milímetros por
encima de la marca de aforo del instrumento.
b) Una vez curada la pipeta puede proceder a transferir el
volumen del liquido para lo cual fue calibrada la pipeta. Para ello debe
tener a mano los materiales volumétricos desde donde y hacia donde va

Profesor Argenis J. Sánchez Henríquez
14
a realizar la operación. Aspire el volumen del líquido hasta la marca de
aforo, manteniendo para ello la pipeta en posición vertical (figura 3-a, b)
y a una altura apropiada para evitar errores de paralaje. Retire la punta
de la pipeta del interior del líquido y límpiela rápidamente con un papel
secante (figura 3-c)
Figura 3. Medición de una alicuota (Fuente: modificado del Skoog and West, 2001)
Para drenar el líquido contenido en la pipeta debe tomar en cuenta el
tipo de pipeta que esta utilizando, para las pipetas volumétricas el
drenado debe hacerse sosteniendo la pipeta en posición vertical
tocando suavemente las paredes del recipiente que contendrá el líquido
(figura 3-d), la columna de líquido debe hacerse descender por efecto de
la gravedad, es decir, vaciado libre, permitiendo al final del drenaje que
transcurra el tiempo de escurrimiento de la misma (aproximadamente 15
seg.).
Un tipo común de pipetas encontradas en muchos laboratorios
son las pipetas de Mohr y las pipetas serológicas (página 6). Para
utilizar estas pipetas se siguen las instrucciones descritas con
anterioridad, sólo que para drenar el volumen total para lo cual fueron
(
(c) (d)
(a) (b)
Lectura correcta
Error de paralaje
Error de paralaje
((
(c) (d)
(a) (b)
Lectura correcta
Error de paralaje
Error de paralaje
(a) (b)
Lectura correcta
Error de paralaje
Error de paralaje
Lectura correcta
Error de paralaje
Error de paralaje
Lectura correcta
Error de paralaje
Error de paralaje
(

Profesor Argenis J. Sánchez Henríquez
15
calibradas (ya que son graduadas para transferir distintos volúmenes
entre 0,1 y 10 mL) se debe permitir que la columna de líquido descienda
hasta la línea de aforo en la pipeta de Mohr, mientras que en la pipeta
serológica la columna de líquido debe descender totalmente y soplarse
la última gota. Esta última acción esta señalada en las pipetas de
fabricación reciente por un anillo esmerilado localizado cerca de la parte
superior de la pipeta.
Buretas. Para el uso correcto de la bureta, en análisis titrimétrico, se
deben seguir las instrucciones que se dan a continuación. Antes
debemos acotar que para medir volúmenes de soluciones alcalinas no
deben emplearse buretas con llaves de vidrio, en su lugar se emplean
llaves de teflón. Igualmente para las soluciones corrosivas o altamente
oxidables deben emplearse buretas con dispositivos de llenado
automáticos.
a) Asegúrese que la llave de drenaje de la bureta está
cerrada. Previamente a su utilización se debe curar el interior de la
bureta limpia con una pequeña porción (aproximadamente un 25% de la
capacidad del instrumento) de la solución que se va a transferir. Para
ello, llene la misma con el volumen antes señalado (puede utilizar un
embudo tallo corto en la parte superior de la bureta para tal fin, así evita
derrames por las paredes externas de la bureta) e incline y gire
lentamente la bureta de tal forma que el líquido pueda mojar en su
totalidad (incluso la máxima graduación) las paredes internas del
instrumento, repita esta operación por lo menos 2 veces, drenando
totalmente el volumen del líquido en cada proceso de curado (tome en
cuenta que el tiempo de escurrimiento para las buretas es de
aproximadamente 1 min.).
b) Una vez curada la bureta, asegúrese de cerrar la llave de
drenaje y colóquela en un soporte para buretas. Proceda a llenar la
bureta hasta unos milímetros por encima del nivel de aforo. Permita que

Profesor Argenis J. Sánchez Henríquez
16
se liberen las burbujas de aire introducidas en el proceso de llenado.
Proceda a liberar las burbujas de aire que se encuentran en la punta de
la bureta, esto se puede lograr girando rápidamente la llave de drenaje,
si la burbuja no logra salir, abra nuevamente la llave de forma rápida y
simultáneamente dé una sacudida vertical a la bureta. Repita este último
procedimiento tantas veces como sea necesario hasta lograr que no
haya burbujas en la punta del instrumento. Una vez libre de burbujas la
bureta, llene nuevamente el instrumento hasta unos milímetros por
encima del nivel de aforo y haga descender el menisco o curvatura del
líquido hasta la marca de aforo, evitando el error de paralaje. Limpie la
punta de la bureta con un papel secante.
c) Recuerde que este instrumento volumétrico se emplea para
el proceso de titulación o titrimétrico. Por tal razón, una vez curada y
enrasada la bureta para la titulación, usted debe manipular la llave de la
bureta como se muestra en la figura 4. Para ello, asegúrese que la
punta de la bureta esta dentro del recipiente hacia donde va realizar la
transferencia, sin llegar a tocar el líquido contenido en éste.
Figura 4. Manejo de la bureta (Fuente: Skoog and West, 2001)
En la medida que usted va adicionando volúmenes pequeños
(aproximadamente 1 mL) desde la bureta, también debe ir aplicando
suaves movimientos giratorios al recipiente que recibe el líquido, así
podrá homogenizar la mezcla. Estos volúmenes adicionados deben ser

Profesor Argenis J. Sánchez Henríquez
17
cada vez más pequeños cuando se acerca al momento de detener el
proceso de adición del líquido, hasta ir gota a gota en el momento que
usted juzgue necesario detener la acción. Procure lavar con un poco del
solvente las paredes del recipiente que recibe el líquido. Se pueden
lograr incrementos, en en los líquidos adicionados al recipiente,
menores a una gota dejando que se acumule líquido en la punta de la
bureta y tocando posteriormente las paredes del recipiente con la punta
de la bureta.
Matraz volumétrico o balones aforados. Un balón aforado debe estar
perfectamente limpio antes de su uso. De ser necesario que se
encuentre seco, puede colocar el mismo en posición invertida durante
algún tiempo. Si requiere agilizar el proceso de secado puede
enjuagarlo con un solvente volátil como la acetona o insertar un tubo de
vidrio unido a una línea de vacío. Nunca debe colocarlo en una estufa o
almacenarlo en una nevera, recuerde que estos instrumentos se
calibran para contener un volumen a una determinada temperatura. Los
balones aforados pueden emplearse para preparar soluciones a partir
de sólidos que se introducen directamente al balón o también para
preparar disoluciones a partir de líquidos transferidos a los balones por
medio de una pipeta volumétrica. Seguidamente se describe la forma
correcta de preparar estas soluciones empleando balones aforados:
a) Si se requiere preparar una solución a partir de un sólido
pesado que será transferido a un balón aforado, puede emplearse para
tal fin un embudo tallo largo. Se coloca el embudo en la boca del balón
aforado y se transfiere el sólido desde el pesa sustancia o vidrio de reloj,
con una pequeña porción del solvente. Se lava bien el recipiente que
contenía al sólido y las porciones de lavado se transfieren al balón
aforado empleando el mismo embudo, se retira el embudo y se procede
a enrasar el balón con el solvente. Se tapa el balón aforado y se

Profesor Argenis J. Sánchez Henríquez
18
homogeniza la solución invirtiendo el balón suavemente dos o tres
veces.
b) Si se requiere preparar una solución a partir de un sólido
pesado que necesite un calentamiento previo para su disolución o a
partir de un líquido cuya disociación libera o consume calor (exotérmico
o endotérmico), se debe pesar el sólido o medir el volumen del líquido y
trasferirlos a un vaso de precipitado. Una vez allí se adiciona solvente
caliente en el caso del sólido o el solvente a temperatura ambiente en el
caso del líquido, y se permite que las soluciones reposen hasta alcanzar
la temperatura ambiente. Posteriormente se transfieren estas soluciones
al balón aforado con ayuda de un embudo tallo largo y una varilla de
vidrio, y se enrasan con el solvente. Tome la precaución de lavar bien,
por lo menos dos veces, el vaso de precipitado donde se preparó
inicialmente la solución y trasvasar los líquidos de lavado al balón
aforado antes de enrasar el mismo.
c) Si se requiere preparar una solución diluida a partir de una
solución concentrada se debe tomar el volumen requerido de esta última
con una pipeta volumétrica, la solución concentrada debe encontrarse
en un vaso de precipitado para evitar la contaminación de la misma. Una
vez aspirado el volumen requerido éste debe transferirse al balón
aforado teniendo en cuenta que la punta de la pipeta debe ubicarse por
debajo de la marca de aforo y tocando suavemente la pared del balón
aforado, el cual debe estar ligeramente inclinado para agilizar la
transferencia del líquido. Recuerde que debe dejar transcurrir el tiempo
de escurrimiento de la pipeta y por último proceda a enrasar la misma
con el solvente, homogenizando la solución con una o dos inversiones
suaves del balón aforado.
Uso de equipo para pesar
La balanza analítica es uno de los instrumentos de laboratorio más
importantes para el analista que requiere hacer mediciones de masa con una

Profesor Argenis J. Sánchez Henríquez
19
elevada precisión. Ésta junto con otros instrumentos como los pesa-filtros o pesa-
sustancias, espátulas y desecadores conforman el conjunto de materiales de
laboratorio empleados en la operación de pesada. Por esta razón, es importante
que el analista conozca el empleo correcto de estos instrumentos.
Balanzas analíticas. Existen diversos tipos de balanzas analíticas,
estas se diferencian en función de los detalles de construcción y la
sensibilidad que pueden ofrecer. Así, basándonos en la forma en que se
construye la balanza encontramos balanzas analíticas mecánicas
(balanzas monoplatos, de cadena, entre otras) y balanzas analíticas
electrónicas, éstas últimas han sustituido a las primeras debido a la
rapidez y sencillez con la que se lleva a cabo la operación de pesada en
una balanza electrónica. Por otra parte, si nos basamos en la
sensibilidad que ofrece la balanza podemos clasificarlas en: i) balanzas
macro-analíticas cuya capacidad máxima de pesada oscila entre 160 y
200 g, con una precisión de ±0,1mg, ii) balanzas semi-microanalíticas
con una capacidad máxima de pesada en el intervalo de 10 a 30 g, y
una precisión de ±0,01mg, y iii) balanzas microanalíticas con capacidad
máxima de pesada entre 1 y 3 g, y una precisión de ±0,001mg. Para el
uso apropiado de la balanza analítica se deben tomar en cuenta las
siguientes recomendaciones:
a) Centrar la carga sobre el platillo lo mejor posible.
b) Proteger la balanza de la corrosión. Para ello coloque
sobre el platillo sólo metales y plásticos no reactivos, así como
materiales de vidrio o cuarzo.
c) Verifique que la balanza este bien nivelada. Si la misma
requiere de algún ajuste comuníquelo al docente o personal técnico del
laboratorio.
d) Permita que los objetos que se hayan calentado regresen a
la temperatura ambiente antes de pesarlo. Es recomendable que el

Profesor Argenis J. Sánchez Henríquez
20
objeto a ser pesado permanezca cerca de la balanza, al menos 20
minutos, en el interior de un desecador.
e) Utilice pinzas o guantes para evitar que los objetos secos
se humedezcan.
f) Mantenga la balanza limpia. Para ello puede emplear un
pincel de pelo de camello para limpiar cualquier residuo de material que
haya caído en el platillo o los alrededores se la balanza.
g) Procure que la balanza esté ubicada en un sitio
acondicionado para tal fin. Este sitio debe tener una humedad relativa
comprendida entre 50 y 60%, nunca inferior. Así mismo la temperatura
de la habitación debe variar en un intervalo máximo de ±1 C. La placa
sobre la cual descansa la balanza debe reposar sobre tacos de goma o
de plomo y los pilares que sostienen dicha placa no deben estar unidos
a la pared.
h) Durante la pesada debe usar exclusivamente las puertas
laterales de la balanza si desea pesar un sólido, y la puerta superior de
la misma si es un líquido no volátil lo que va a pesar.
i) No debe apoyarse sobre la mesa en la cual está colocada la
balanza.
Uso de equipo para secar
El desecador se emplea para mantener en su interior a algunos materiales
y reactivos en una atmósfera libre de humedad (seca), oxígeno, y dióxido de
carbono, constituyentes atmosféricos que pueden afectar la integridad de algunos
materiales y reactivos.
Los bordes esmerilados de las tapas y del recipiente, que conforman al
desecador, se cubren con una delgada capa de vaselina para conseguir un cierre
hermético al aire, una vez que se ha realizado el vacío. La vaselina o cualquier
otra grasa especial que se emplee en su lugar, no se deben colocar en mucha
cantidad, ya que se corre el riesgo de que la tapa resbale. El material empleado

Profesor Argenis J. Sánchez Henríquez
21
como agente desecante puede variar y con ello el grado de eficiencia en el
proceso de retención de la humedad, tal como se muestra en la siguiente tabla.
Tabla 1. Agentes desecantes
Agente desecante Agua Residual x Litro de Aire (mg)
Pentóxido difósforo 0,00002
Ácido Sulfúrico Concentrado 0,003
Sílica Gel 0,03
Cloruro de Calcio Anhidro puro 1,5
Sulfato de Calcio Anhidro 2,8
Una vez que el reactivo o material ha sido sacado de la estufa o de la
mufla, con ayuda de pinzas o guantes para objetos calientes, y se coloca en el
interior del desecador, se debe dejar transcurrir alrededor de 8 segundos para
permitir que el aire circundante en el recipiente se caliente y expanda.
Posteriormente se procede a colocar la tapa deslizándola suavemente hasta lograr
que el recipiente quede sellado herméticamente. Si se desea hacer un vacío
posterior de la atmósfera dentro del desecador (Desecador del tipo 2), se conecta
la llave de vidrio del desecador a una trompa o línea de vacío y luego de manera
gradual se abre la llave de vidrio del desecador, luego se cierra nuevamente la
llave de vidrio y se retira la línea de vacío.
Para abrir un desecador debe deslizarse suavemente la tapa para evitar la
entrada brusca de aire motivada a la depresión o diferencia de presión entre la
atmósfera externa y la atmósfera del interior del desecador, así evitamos pérdidas
de reactivos por bruscas corrientes de aire. Si se ha realizado un vacío adicional o
el desecador esta provisto de una llave para vacío, se debe primer lugar abrir
suavemente esta llave de tal forma de realizar la compensación de presiones, para
luego proceder a deslizar suavemente la tapa del recipiente.

Profesor Argenis J. Sánchez Henríquez
22
Uso de equipo para filtrar
Antes de indicar el uso adecuado de los distintos instrumentos empleados
para el proceso de filtración, debemos detenernos para hablar de forma breve
acerca de la técnica de filtración. La filtración de un precipitado es un proceso que
está comprendido en tres etapas; decantación, lavado y transvase. La etapa de
decantación, consiste en pasar a través del medio filtrante la mayor cantidad de
líquido sobrenadante que sea posible, sin perturbar al sólido precipitado. Para ello
debemos ayudarnos con una varilla de vidrio para dirigir el líquido sobrenadante
que será decantado, al centro del medio filtrante sin pérdidas o derrames del
mismo, tal como se muestra en la figura 5. La etapa de lavado, a su vez puede
constar de varios lavados en función de la naturaleza del precipitado obtenido.
Figura 5. Etapas de la filtración (Fuente: Skoog and West, 2001)
Para el lavado se adiciona al vaso de precipitado una porción del líquido o
solución de lavado, se mezcla bien con el precipitado y se permite que el sólido se
asiente antes de decantar esta solución de lavado; esta operación debe repetirse
tantas veces como la naturaleza del precipitado lo permita para su purificación.
Por último, encontramos la etapa de transvase, en la cual el precipitado obtenido y
(a)
(b)
(a)
(b)

Profesor Argenis J. Sánchez Henríquez
23
lavado se transfiere de forma cuantitativa al medio filtrante, con la ayuda de
chorros de solución de lavado (puede emplear una piseta para tal fin) dirigidos de
manera apropiada al vaso de precipitado, y la varilla de vidrio para canalizar la
mezcla sólido-solución de lavado y de esta forma minimizar las posibles pérdidas.
El método empleado para separar al precipitado de la solución madre que
lo originó, puede variar y va a depender, en cierto modo, de la disponibilidad del
laboratorio en el cual trabajemos. Básicamente, encontramos dos métodos para
filtrar: el primero de ellos por gravedad y el segundo al vacío. Éstos a su vez,
involucran a tres tipos de medios filtrantes: 1) Papel de filtro cuantitativo o no, en
embudos cuantitativos para filtrar por gravedad, 2) Mantos filtrantes, en crisoles de
Gooch o de Munroe, adaptado a una línea de vacío, y 3) Placas porosas filtrantes
de vidrio sinterizado, es decir, vidrio molido, en crisoles de vidrio Pyrex , adaptado
a una línea de vacío o tren de vacío como el mostrado en la figura 6.
Figura 6. Tren de filtración al vacío. (Fuente: Vogel A. I.,1960)
La selección y uso correcto de cada uno de estos métodos de filtración,
dependen principalmente de la naturaleza del precipitado que se desea filtrar, es
decir, del tamaño de partícula del precipitado formado. Si se desea filtrar por
gravedad haciendo uso de un embudo cuantitativo con papel de filtro, el papel de
filtro debe doblarse, ajustarse y asegurarse al embudo tomando en cuenta las
siguientes instrucciones: primero realice un dobles del papel justo a la mitad,
seguido de un segundo dobles que no haga coincidir ligeramente los extremos del
papel de filtro, de esta manera quedan dos secciones, una ligeramente superior y
A la línea de
vacío
A la línea de
vacío

Profesor Argenis J. Sánchez Henríquez
24
otra inferior, estos dos pasos pueden observarse en la figura 7, etapas a y b. Corte
un pequeño trozo de la esquina del papel que corresponde a la sección inferior,
como se muestra en la etapa c, luego abra en forma de cono el papel de filtro en el
centro de la sección que no fue cortada y ajuste el cono al embudo, tal como se
muestra en las etapas d y e. Finalmente, asegure el papel de filtro al embudo
humedeciendo ligeramente el papel con la solución de lavado, para que se adhiera
a las paredes del embudo.
Figura 7. Preparación del papel de filtro. (Fuente: Skoog and West, 2001)
Los crisoles de Gooch y Munroe utilizan los mantos filtrantes de amiento y
platino, respectivamente. Los embudos de Gooch pueden ser de porcelana,
platino y sílice, con perforaciones en el fondo del embudo donde reposa el manto
filtrante de amiento. Por otro lado, el crisol de Munroe es de platino íntegramente.
Anteriormente, el amianto debía ser preparado de forma cuidadosa en el
laboratorio, hoy día se puede adquirir en el comercio. Cuando el amianto se agita
en agua se separa en fibras muy pequeñas. Al emplear un crisol de Gooch para
filtrar un sólido a través de mantos filtrantes de amianto se debe utilizar un tren de
filtración como el mostrado en la figura 6, conectado a una línea de vacío. Luego
se prepara la suspensión de amianto en agua, y se agrega al crisol hasta alcanzar
la mitad de su capacidad, posteriormente se deja reposar entre 2-3 minutos para
(a) (b)
(c)
(d)
(e)
(a) (b)
(c)
(d)
(e)

Profesor Argenis J. Sánchez Henríquez
25
que las partículas de amianto de mayor tamaño se sedimenten en el fondo de
embudo, luego se aplica una ligera succión hasta que logre pasar el agua de la
suspensión, para finalmente llevar la succión al máximo y lograr así un manto
filtrante uniforme y de grosor apropiado (2-3 mm), bien adherido al fondo del
embudo. Para el lavado del manto de amianto emplea agua hasta no observar
fibras en el líquido filtrado. Nunca dirija un chorro de solución de lavado de la
piseta directamente al manto, utilice para ello la varilla de vidrio, tal como se indico
en la introducción a esta sección.
II.4. Concentración de las soluciones
En análisis químico las concentraciones se expresan, directa o
indirectamente, como el peso de soluto en una unidad de volumen de solución.
Recordemos que una solución es una mezcla homogénea de uno o varios solutos
en un solvente, en la cual no ocurre deposición o sedimentación de alguno de los
solutos. Pese a que las soluciones pueden incluir varias combinaciones de la
materia en sus diferentes estados: sólido, gas, líquido, plasma; normalmente en
química encontramos soluciones conformadas por solutos (constituyente en menor
proporción en la solución) sólidos o líquidos, en solventes (constituyente en mayor
proporción en la solución) líquidos.
Las unidades de concentración físicas (donde la unidad fundamental es el
gramo) y químicas (donde la unidad fundamental para establecer la cantidad de
materia es el Mol y el Equivalente) que emplearemos en el desarrollo de las
prácticas de laboratorio son:
Molaridad (M). Se define como el número de moles de soluto por litro (L)
de solución.
)(
º
LVsolución
solutomolesnM

Profesor Argenis J. Sánchez Henríquez
26
Normalidad (N). Se define como el número de equivalentes de soluto por
litro (L) de solución. A su vez, el número de equivalentes de soluto se define como
los gramos de soluto por peso equivalente-gramo del mismo, y el peso
equivalente-gramo del soluto es su masa molar o atómica entre los equivalentes.
Los equivalentes se definen en función del tipo de reacción química en la cual
participa el reactivo, como veremos más adelante. En las ecuaciones: M.M. (Masa
molar), M. At. (Masa atómica), Peq. (Peso equivalente gramo), eq. (equivalentes).
Partes por millón (ppm). Se define como la cantidad en masa de un soluto
en un volumen de solución. Esta masa puede ser microgramos ( g) o miligramo
(mg), y los volúmenes respectivos son mililitros (mL) y litros (L).
o
Tanto por ciento en volumen [%(v/v)]. Se define como la cantidad en
volumen del soluto en 100 partes en volumen de solución. Este volumen por lo
general se expresa en unidades de mililitro (mL).
Tanto por ciento masa en volumen [%(m/v)]. Se define como la cantidad
en masa de soluto en 100 partes en volumen de solución. Por lo general, la masa
se expresa en gramos (g) y el volumen en mililitros (mL).
)(
º
LVsolución
solutoesequivalentnN
PEq
solutogesequivalentnº
eq
AtMoMMPEq
...
)(mLVsolución
solutogppm
)(LVsolución
solutomgppm
100)/%(soluciónV
solutoVvv
100)/%(soluciónV
solutomasavm

Profesor Argenis J. Sánchez Henríquez
27
II.5. Soluciones tipo patrón
Los métodos volumétricos emplean soluciones titulantes cuya
concentración es conocida, estas soluciones pueden ser de tipo patrón primario o
de tipo patrón secundario. Las soluciones tipo patrón primario son soluciones
preparadas a través del método directo. El método directo consiste en pesar en
una balanza analítica la masa del reactivo patrón primario que se necesita para
que una vez disuelta esta masa, en un volumen final del solvente, se obtenga
directamente la concentración deseada. Por otra parte, las soluciones tipo patrón
secundario o estándar, se preparan a través del método indirecto. Este método
consiste en pesar en una balanza analítica una cantidad aproximada del reactivo
patrón secundario que se requiere para que al disolver esta masa, en un volumen
final del solvente, se obtenga una concentración aproximada del reactivo; luego
una porción o alícuota de este reactivo patrón secundario es estandarizada con un
reactivo patrón primario, de allí su nombre de solución estándar o solución patrón
secundario.
Ambos métodos de preparación de soluciones tipo patrón requieren el
empleo de reactivos denominados patrón primario. Para que un reactivo sea
considerado patrón primario debe cumplir con los siguientes requisitos:
1. Debe tener pureza absoluta (100%) en cuanto a su componente activo,
de no ser así se debe conocer exactamente el contenido de componente activo y
de las impurezas presentes, las cuales no deben ser mayores al 0,2% de la
composición total del reactivo.
2. De estar presentes impurezas en el reactivo, éstas deben ser inertes con
respecto a las especies químicas que participan en la reacción analítica de interés;
además estas impurezas deben ser susceptibles de ser identificadas a través de
ensayos simples.
3. El reactivo debe ser estable en su exposición al aire, es decir, que no sea
higroscópico, eflorescente y que no reaccione con el O2 y CO2 de la atmósfera.

Profesor Argenis J. Sánchez Henríquez
28
4. El reactivo debe ser estable a las temperaturas comunes de desecación
(110-120ºC), de tal forma que se conozca siempre su composición después del
proceso.
5. El peso equivalente-gramo del reactivo debe ser preferentemente
elevado.
6. Debe ser de fácil acceso comercial y de razonado costo.
II.6. Muestreo
El análisis químico cuantitativo clásico e instrumental abarca un amplio
espectro de métodos analíticos fundamentados en una, no menos, amplia
variedad de técnicas de análisis. Sin embargo, todos estos métodos de análisis
para lograr su objetivo final cumplen, por lo general, con las siguientes etapas: a)
toma de muestra o muestreo, b) tratamiento o acondicionamiento de la muestra, c)
determinación del analito, e d) interpretación de los resultados. Todas estas
etapas se complementan y representan parte importante del análisis químico. Sin
embargo, las etapas de muestreo y acondicionamiento de la muestra son cruciales
para el análisis; ya que de la realización satisfactoria, o no, del muestreo depende
la veracidad de los resultados obtenidos, amén de la minuciosidad, tecnificación y
precisión del análisis. Por su parte, el acondicionamiento de la muestra está
relacionado con la manera como se somete a la muestra para cambiar su estado
de agregación y pureza, en función de la técnica que se desea emplear para la
detección del analito. En todo caso hay que tener presente que el tipo de muestra
que se desea analizar, va a definir al método de muestreo, así como el tipo de
acondicionamiento que se debe aplicar.
El analista químico debe tener conocimiento de la procedencia de la
muestra, así como tener control sobre la manera en que se obtiene la misma. De
modo general, pueden presentarse dos casos en los cuales se requiera la
cuantificación de un analito presente en: 1) una muestra de composición
homogénea, y 2) una muestra voluminosa y composición heterogénea.

Profesor Argenis J. Sánchez Henríquez
29
Si la muestra es homogénea el muestreo es relativamente sencillo, ya que
al tomar una porción de la muestra que sea suficiente para el análisis, que
provenga de cualquiera de sus partes, ésta tendrá una misma composición físico-
química. Si por el contrario, la muestra a ser analizada presenta un gran volumen
o masa y heterogeneidad a lo largo de la misma, difícilmente cada porción tomada
tendrá la misma composición fisico-química. Este último caso plantea la búsqueda
de una forma de lograr obtener, un pequeño volumen o masa de la muestra, que
represente de forma fidedigna la composición fisico-química de la muestra original;
esto se conoce como muestra representativa. La representatividad en una muestra
se logra tomando el mayor número posible de porciones, de manera sistemática,
de diversos lugares de la muestra original, y luego mezclando éstas
sistemáticamente de tal forma de reducirlas a un volumen o masa suficiente para
el análisis. En función de los principales estados de agregación de la materia
(sólido, líquido, gas) podemos encontrar:
Muestreo de sólidos
Para el muestreo de sólidos se debe obtener una muestra bruta
representativa del material que se muestrea. Esto se logra recogiendo,
sistemáticamente, un cierto número de unidades de muestreo (porción del material
de volumen suficiente para contener aproximadamente la misma distribución de
tamaño de partícula que el material que se muestrea), mientras más unidades de
muestreo se recojan, más representativa será la muestra. Hay que tener presente,
que el número apropiado de unidades de muestreo a ser recogidas va a depender
de la heterogeneidad, en tamaño y composición, de la muestra, más que del
volumen o masa de la muestra sometida al muestreo. Posteriormente, la muestra
bruta debe ser sometida a una reducción de tamaño de partícula y cantidad, a
través de procesos de trituración y porfirización (pulverización a polvo muy fino);
para luego mezclarla sistemáticamente, la muestra final debe pasar por un tamiz
de 100 a 200 mallas. La muestra obtenida debe conservarse protegida de la
absorción o liberación de agua, dióxido de carbono, y la oxidación atmosférica.

Profesor Argenis J. Sánchez Henríquez
30
Asimismo la mayoría de los sólidos deben secarse a 110ºC antes de ser
analizados.
Muestreo de líquidos
Si el líquido es homogéneo y se encuentra inmóvil debe agitarse
suavemente antes de proceder a tomar la muestra. Si por el contrario, hay
partículas suspendidas en el líquido o existe una segunda fase no miscible, debe
homogenizarse la mezcla antes de la toma de muestra, o en el caso más simple,
tomar muestras del líquido a diferentes profundidades. Si el líquido que se desea
muestrear fluye a través de un cause, se deben tomar pequeñas porciones del
mismo en un tiempo programado.
Muestreo de gases
Los gases no representan por lo general una matriz de difícil muestreo, ya
que estos son comúnmente homogéneos. Si el gas se encuentra en “reposo” a la
presión atmosférica, puede muestrearse por desplazamiento de un líquido
saturado con el gas; estos líquidos pueden ser el mercurio o el agua. Otra forma
de muestrear gases en estas condiciones, es a través de matraces evacuados. Si
el gas se encuentra en otras condiciones de presión, temperatura y flujo se debe
recurrir a equipos especiales de muestreo.
No es el propósito de esta sección profundizar en las distintas teorías
acerca del muestreo, ya que es un tema sumamente amplio para el cual existe
una bibliografía igualmente extensa en función del tipo de material que se desea
analizar.
II.7. Limpieza del material de vidrio
El análisis químico volumétrico puede presentar baja reproducibilidad si el
material de vidrio empleado en la medición de volúmenes exactos, no se
encuentra perfectamente limpio y libre de grasa. Las partículas de suciedad y la
grasa adheridas a las superficies de estos instrumentos volumétricos, retienen
pequeñas gotas de los líquidos contenidos en ellos, derivando esto en medidas de

Profesor Argenis J. Sánchez Henríquez
31
volúmenes inexactos y por consiguiente afectando los resultados obtenidos. De
allí que se requiera un estricto control en la limpieza del material de vidrio
volumétrico como parte integral en el proceso de cuantificación de un analito.
Para comprobar si un material de vidrio está limpio, se debe llenar el mismo
con agua destilada y luego vaciarlo, si al escurrir el agua contenida en el material,
quedan pequeñas gotas o películas del líquido en las paredes del material, éste no
está limpio. Existen varios métodos para el lavado del material de vidrio, la
selección del más conveniente se hace principalmente en función del tipo de
suciedad que se desea eliminar, es decir, si el instrumental o material de vidrio ha
sido utilizado previamente para contener o transferir reactivos de naturaleza
orgánica o inorgánica. En el caso de material de vidrio volumétrico que ha sido
empleado para contener o transferir soluciones acuosas diluidas de compuestos
inorgánicos (sales, bases, ácidos), basta un método sencillo de limpieza, que
puede consistir en lavar el material con una solución al 5% de HNO3 o HCl, y
luego enjuagar repetidas veces con agua destilada, posteriormente se llena el
material con agua destilada y se deja escurrir para verificar ausencia de gotas o
películas de agua, de ocurrir lo contrario se procede a un método de lavado más
profundo. Un lavado más profundo del material de vidrio volumétrico implica la
utilización de soluciones ácidas o básicas más concentradas y/o soluciones
constituidas por mezclas de sales, ácidos y alcoholes. Estos métodos de lavado se
emplean generalmente cuando la suciedad es originada por restos grasosos
(reactivos de naturaleza orgánica) o impurezas que no logran limpiarse con un
método sencillo como el descrito anteriormente. Un método de limpieza profunda
muy empleado en la mayoría de los laboratorios de química, es el que utiliza
mezcla sulfocrómica como solución de lavado. La mezcla sulfocrómica es una
solución preparada a una concentración entre 2,5-5% de dicromato de potasio o
de sodio en ácido sulfúrico concentrado, en la preparación se prefiere emplear la
sal sódica debido a su mayor solubilidad y bajo costo en comparación con la sal
potásica. Esta mezcla tiene un carácter fuertemente oxidante y pierde su
efectividad luego de un prolongado uso. El método de limpieza con mezcla

Profesor Argenis J. Sánchez Henríquez
32
sulfocrómica implica llenar el material con esta solución o sumergirlo en ella, y
dejarlo reposando unos minutos (en función del grado de suciedad) en la mezcla,
luego enjuagar el material repetidas veces con abundante destilada para
finalmente dejarlo secar, no sin antes hacer el ensayo o prueba de limpieza con el
agua destilada. Otro método de limpieza profunda utiliza como agente de lavado
una solución de HNO3 al 5% en H2SO4 concentrado, también se puede emplear
agua regía como solución de lavado. El agua regía consiste en una mezcla en
proporción 1:3 de HNO3 en HCl.
Dos métodos de limpieza que resultan altamente eficientes cuando el
material de vidrio se encuentra muy engrasado, consisten en utilizar por una parte
una solución jabonosa (detergente industrial) caliente, siempre que el material lo
permita, y luego enjuagar a fondo con agua, seguido de un lavado con HCl
concentrado y finalmente con abundante agua destilada. Por otra parte, el
segundo método al que hacemos referencia emplea como solución de lavado una
mezcla de KOH en alcohol, esta solución de conoce con el nombre de potasa
alcohólica.
Posterior al proceso de lavado del material de vidrio volumétrico se debe
secar el mismo. Esto debe hacerse tomando en cuenta el tipo de material, es
decir, debemos recordar que el material volumétrico ha sido calibrado para
contener o transferir ciertos volúmenes. Este tipo de material incluye pipetas,
buretas y balones aforados, los cuales no deben ser secados en la estufa, ya que
perderían su calibración. Para ello se debe realizar el lavado con suficiente
antelación a la utilización del material para permitir que se sequen a la atmósfera,
de requerirse el material de forma más inmediata se puede proceder a realizar un
último enjuague con acetona pura y a insuflar (pasar una corriente de aire) el
material de vidrio para su pronto secado.

Profesor Argenis J. Sánchez Henríquez
33
II.8. Categorización de los reactivos
Los diferentes reactivos que pueden ser suministrados, por distintas casas
comerciales alrededor del mundo, a la amplia variedad de laboratorios que
realizan análisis químico orgánico, inorgánico y bioquímico, han sido catalogados
por diferentes organismos e instituciones internacionales consideradas como
autoridades en la materia, en función de la calidad y aplicación que poseen estas
sustancias, tal como se muestra en la siguiente tabla.
Tabla 2. Categorización de reactivos químicos
Fabricación de
tarjetas
electrónicas y de circuitos
Solventes fabricados para asegurar un
bajo nivel de contaminación de metales.
Cumple con los requerimientos del
Instituto de Equipos Semiconductores y
Materiales (SEMI). Incluye el análisis
del lote actual en la etiqueta.
Electrónica
Electroforesis,
biología
molecular,
secuenciación,
síntesis de
péptidos y oligonucleotidos
Solventes y reactivos que tienen que
ser especialmente purificados y
ensayados para aplicaciones en
biotecnología.
Biotecnología
Detectores de
longitud de onda
visible y
ultravioleta (UV-
Vis)
Solventes para uso en Espectrometría.
Cumple con especificaciones ACS.
Incluye análisis de lote actual en la
etiqueta.
Spectranalyzed*
GC con detector
de captura de
electrones
(ECD)
Solventes para uso en análisis de
residuos de pesticida. Cumple o excede
los estándares de pureza ACS para
análisis de residuos de pesticidas
Pesticida
HPLC y
procedimientos
espectrométricos
Solventes fabricados específicamente
para HPLC. Cumple todas las
especificaciones ACS. Filtrado en
submicrones.
HPLC
HPLC, GC,
Espectrometría
ICP/Plasma y
análisis de
residuos de
pesticidas
Ácidos y solventes de extremada alta
pureza. Los ácidos son analizados para
72 metales por ICP/MS; en niveles de
impurezas de ppt. Los niveles de
impurezas de los solventes en ppm.
Curvas de absorbancia de UV y
cromatogramas de muestra disponibles
a solicitud. Para los ácidos OPTIMA, un
análisis del lote típico es mostrado en el
catalogo.
OPTIMA*
Cromatografía
de Gas (GC)
Solventes con alto grado de pureza y
consistencia lote a lote. Libre de
contaminantes en nivel de ppt, Cumple
especificaciones ACS. Cromatogramas
disponibles a solicitud
GC Resolv*
AplicaciónDefinición Grado
Fabricación de
tarjetas
electrónicas y de circuitos
Solventes fabricados para asegurar un
bajo nivel de contaminación de metales.
Cumple con los requerimientos del
Instituto de Equipos Semiconductores y
Materiales (SEMI). Incluye el análisis
del lote actual en la etiqueta.
Electrónica
Electroforesis,
biología
molecular,
secuenciación,
síntesis de
péptidos y oligonucleotidos
Solventes y reactivos que tienen que
ser especialmente purificados y
ensayados para aplicaciones en
biotecnología.
Biotecnología
Detectores de
longitud de onda
visible y
ultravioleta (UV-
Vis)
Solventes para uso en Espectrometría.
Cumple con especificaciones ACS.
Incluye análisis de lote actual en la
etiqueta.
Spectranalyzed*
GC con detector
de captura de
electrones
(ECD)
Solventes para uso en análisis de
residuos de pesticida. Cumple o excede
los estándares de pureza ACS para
análisis de residuos de pesticidas
Pesticida
HPLC y
procedimientos
espectrométricos
Solventes fabricados específicamente
para HPLC. Cumple todas las
especificaciones ACS. Filtrado en
submicrones.
HPLC
HPLC, GC,
Espectrometría
ICP/Plasma y
análisis de
residuos de
pesticidas
Ácidos y solventes de extremada alta
pureza. Los ácidos son analizados para
72 metales por ICP/MS; en niveles de
impurezas de ppt. Los niveles de
impurezas de los solventes en ppm.
Curvas de absorbancia de UV y
cromatogramas de muestra disponibles
a solicitud. Para los ácidos OPTIMA, un
análisis del lote típico es mostrado en el
catalogo.
OPTIMA*
Cromatografía
de Gas (GC)
Solventes con alto grado de pureza y
consistencia lote a lote. Libre de
contaminantes en nivel de ppt, Cumple
especificaciones ACS. Cromatogramas
disponibles a solicitud
GC Resolv*
AplicaciónDefinición Grado
Profesor Argenis J. Sánchez Henríquez
34
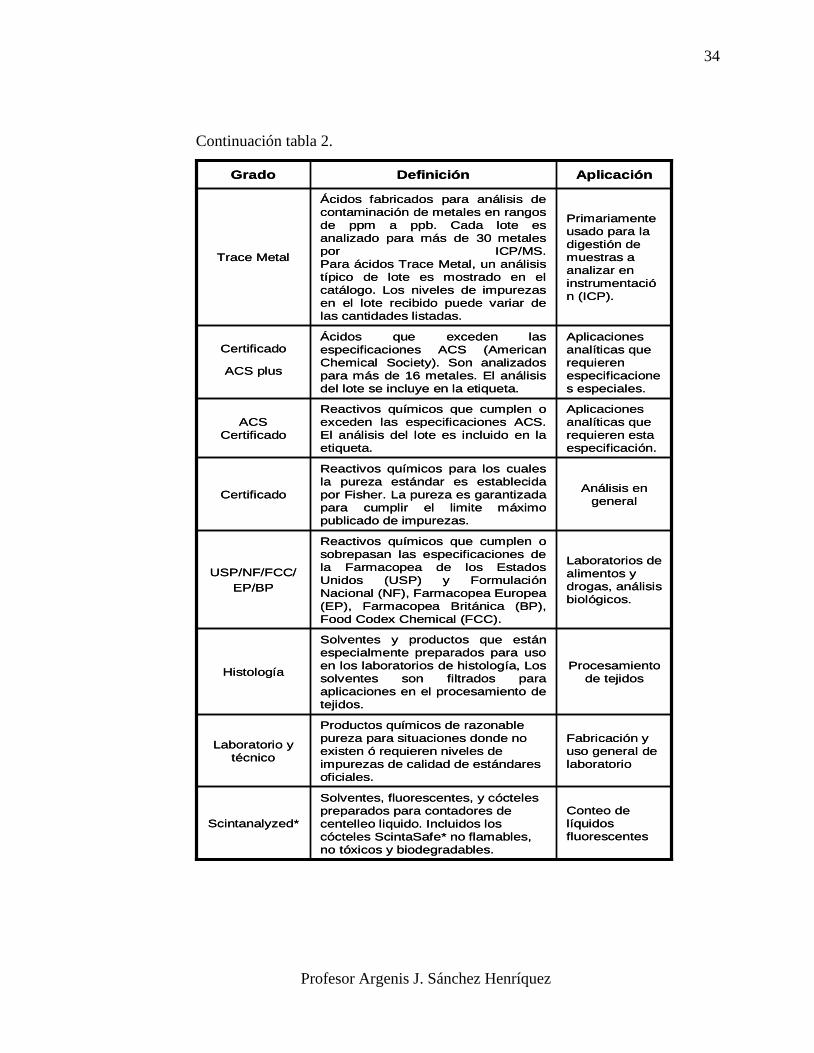
Continuación tabla 2.
Conteo de
líquidos
fluorescentes
Solventes, fluorescentes, y cócteles
preparados para contadores de
centelleo liquido. Incluidos los
cócteles ScintaSafe* no flamables,
no tóxicos y biodegradables.
Scintanalyzed*
Fabricación y
uso general de
laboratorio
Productos químicos de razonable
pureza para situaciones donde no
existen ó requieren niveles de
impurezas de calidad de estándares
oficiales.
Laboratorio y
técnico
Procesamiento
de tejidos
Solventes y productos que están
especialmente preparados para uso
en los laboratorios de histología, Los
solventes son filtrados para
aplicaciones en el procesamiento de
tejidos.
Histología
Laboratorios de
alimentos y
drogas, análisis
biológicos.
Reactivos químicos que cumplen o
sobrepasan las especificaciones de
la Farmacopea de los Estados
Unidos (USP) y Formulación
Nacional (NF), Farmacopea Europea
(EP), Farmacopea Británica (BP),
Food Codex Chemical (FCC).
USP/NF/FCC/
EP/BP
Análisis en
general
Reactivos químicos para los cuales
la pureza estándar es establecida
por Fisher. La pureza es garantizada
para cumplir el limite máximo
publicado de impurezas.
Certificado
Aplicaciones
analíticas que
requieren esta
especificación.
Reactivos químicos que cumplen o
exceden las especificaciones ACS.
El análisis del lote es incluido en la
etiqueta.
ACS
Certificado
Aplicaciones
analíticas que
requieren
especificacione
s especiales.
Ácidos que exceden las
especificaciones ACS (American
Chemical Society). Son analizados
para más de 16 metales. El análisis
del lote se incluye en la etiqueta.
Certificado
ACS plus
Primariamente
usado para la
digestión de
muestras a
analizar en
instrumentació
n (ICP).
Ácidos fabricados para análisis de
contaminación de metales en rangos
de ppm a ppb. Cada lote es
analizado para más de 30 metales
por ICP/MS.
Para ácidos Trace Metal, un análisis
típico de lote es mostrado en el
catálogo. Los niveles de impurezas
en el lote recibido puede variar de
las cantidades listadas.
Trace Metal
AplicaciónDefinición Grado
Conteo de
líquidos
fluorescentes
Solventes, fluorescentes, y cócteles
preparados para contadores de
centelleo liquido. Incluidos los
cócteles ScintaSafe* no flamables,
no tóxicos y biodegradables.
Scintanalyzed*
Fabricación y
uso general de
laboratorio
Productos químicos de razonable
pureza para situaciones donde no
existen ó requieren niveles de
impurezas de calidad de estándares
oficiales.
Laboratorio y
técnico
Procesamiento
de tejidos
Solventes y productos que están
especialmente preparados para uso
en los laboratorios de histología, Los
solventes son filtrados para
aplicaciones en el procesamiento de
tejidos.
Histología
Laboratorios de
alimentos y
drogas, análisis
biológicos.
Reactivos químicos que cumplen o
sobrepasan las especificaciones de
la Farmacopea de los Estados
Unidos (USP) y Formulación
Nacional (NF), Farmacopea Europea
(EP), Farmacopea Británica (BP),
Food Codex Chemical (FCC).
USP/NF/FCC/
EP/BP
Análisis en
general
Reactivos químicos para los cuales
la pureza estándar es establecida
por Fisher. La pureza es garantizada
para cumplir el limite máximo
publicado de impurezas.
Certificado
Aplicaciones
analíticas que
requieren esta
especificación.
Reactivos químicos que cumplen o
exceden las especificaciones ACS.
El análisis del lote es incluido en la
etiqueta.
ACS
Certificado
Aplicaciones
analíticas que
requieren
especificacione
s especiales.
Ácidos que exceden las
especificaciones ACS (American
Chemical Society). Son analizados
para más de 16 metales. El análisis
del lote se incluye en la etiqueta.
Certificado
ACS plus
Primariamente
usado para la
digestión de
muestras a
analizar en
instrumentació
n (ICP).
Ácidos fabricados para análisis de
contaminación de metales en rangos
de ppm a ppb. Cada lote es
analizado para más de 30 metales
por ICP/MS.
Para ácidos Trace Metal, un análisis
típico de lote es mostrado en el
catálogo. Los niveles de impurezas
en el lote recibido puede variar de
las cantidades listadas.
Trace Metal
AplicaciónDefinición Grado

Profesor Argenis J. Sánchez Henríquez
35
II.9. Presentación del informe científico de laboratorio
Un informe científico de laboratorio es un documento estructurado de tal
manera que permita informar de forma clara y precisa al lector sobre los
procedimientos seguidos y resultados obtenidos a lo largo del desarrollo de una
actividad práctica. Si bien es cierto que el informe científico de laboratorio se debe
elaborar siguiendo el orden lógico del método científico, no es menos cierto que
dentro de esos parámetros podemos encontrar una amplia diversidad de formatos
para llevar a cabo su presentación. Por esta razón y con el propósito de unificar el
formato del informe presentado para esta unidad curricular, se describen a
continuación los elementos básicos que deben conformar la estructura del informe
científico de laboratorio:
Elementos iniciales
1. Lugar donde se realizó el experimento
2. Nombre de la unidad curricular (asignatura)
3. Titulo de la práctica
4. Nombre completo del autor o autores del informe
5. Ciudad y fecha de realización
Cuerpo del informe
1. Introducción: esta debe suministrar al lector un breve marco teórico de
los principios que rigen la experiencia práctica que va a realizar.
Seguidamente se realiza una descripción sencilla del método que será
utilizado y por último se señalan los objetivos que se pretenden alcanzar
al final de la experiencia.
2. Materiales y Métodos: esta parte del informe debe contener una
descripción detallada de los materiales de laboratorio, reactivos y
equipos que se emplearán para el desarrollo de la práctica. De igual
manera debe describirse el procedimiento y la secuencia en que fue
llevado a cabo, pueden emplearse para ellos diagramas, esquemas,
dibujos de los arreglos experimentales entre otros.

Profesor Argenis J. Sánchez Henríquez
36
3. Resultados: los resultados involucran el tratamiento matemático
(cálculos) de los datos suministrados en el manual de práctica así como
de aquellos datos obtenidos en el desarrollo de la práctica de
laboratorio. Estos resultados deben presentarse en tablas y gráficos
numerados, que luego podrán ser citados en la discusión de los
resultados. Todos los datos y resultados analíticos deben ser tratados
estadísticamente según se exija en cada práctica. Tenga presente que
en algunos casos los cálculos pueden señalarse en el apéndice del
informe.
4. Discusión: en esta parte del informe se explican e interpretan los
resultados obtenidos en la experiencia práctica. Para el desarrollo de las
discusiones usted debe enfocarse en responder preguntas tales como:
¿qué resultados esperaba?, ¿qué resultados obtuve?, ¿Por qué discrepan
los resultados?, ¿qué método utilizo para comparar los resultados?, ¿qué
ventajas o desventajas tiene el método de análisis empleado?, ¿qué
puedo hacer para mejorar la experiencia práctica?, entre otras.
Recuerde, una buena discusión de resultados es el reflejo del
entendimiento de las actividades prácticas que realizó.
5. Conclusiones: las conclusiones deben estar relacionadas con los
objetivos que se pretenden alcanzar al finalizar la práctica. Se pueden
presentar en párrafos que reflejen ideas claras (de fácil lectura),
precisas (no debe existir ambigüedad) y concisas (breves) o pueden
numerarse para mostrar mejor orden en las ideas. Recuerde respaldar
sus conclusiones con argumentos lógicos, evidencia experimental
(observaciones o cifras) y referencias bibliográficas.
Elementos finales
1. Bibliografía: contiene las referencias bibliográficas (libros, revistas, entre
otros) que usted emplea para elaborar el informe. Esta referencias
deben numerarse siguiendo el orden de aparición o cita en el texto, y

Profesor Argenis J. Sánchez Henríquez
37
deben contener la información completa de la fuente (autores, título,
edición, volumen, número, editorial, año de publicación o edición)
2. Apéndices (opcional): se emplea en aquellos casos en que el informe
involucre gran cantidad de cálculos, información importante para el
entendimiento del problema (Normas COVENIN, AOAC, ASTM), tablas
extensas, entre otros, que de incorporarse al cuerpo del trabajo
dificultarían la lectura del mismo
II.10. Aspectos importantes en la evaluación
La evaluación que se llevará a cabo en la presente asignatura, unidad
curricular, estará apegada a la normativa interna de evaluación del Decanato de
Agronomía. Siendo que las actividades aquí programadas tienen carácter práctico,
se recuerda que la asistencia a las mismas es obligatoria, perdiéndose la
asignatura por inasistencia al 25% de las actividades programadas (Artículo 6).
Por otra parte, las pruebas empleadas para evaluar el rendimiento académico
estudiantil serán: a) Por su naturaleza: escritas, prácticas de observación y mixtas.
b) Por su estructura: objetivas, de trabajos de investigación, experimentación o
cualquier otro tipo que se juzgue adecuado para comprobar el logro de los
objetivos propuestos. c) Por el momento de su aplicación: periódicas y diferidas. d)
Por su duración: cortas. Las características de las pruebas antes descritas, se
extraen del artículo 21, de la misma normativa. La calificación numérica final del
laboratorio representa el 40% de la calificación de la asignatura, estando
representado el 60% restante de la calificación en las evaluaciones o pruebas
parciales que se realicen en la teoría. La distribución de estos porcentajes
calificativos (60% teoría y 40% prácticas) se adaptará a tres bloques o etapas, que
cubrirán el contenido programático de la asignatura:
Bloque 1 (18% teoría + 12 % práctica) (3,6 pts teoría + 2,4 práctica) = 6 pts.
Bloque 2 (21% teoría + 14% práctica) (4,2 pts teoría + 2,8 práctica) = 7 pts.
Bloque 3 (21% teoría + 14% práctica) (4,2 pts teoría + 2,8 práctica) = 7 pts.
20 pts.

Profesor Argenis J. Sánchez Henríquez
38
III. PARÁMETROS ESTADÍSTICOS BÁSICOS.
El análisis químico cuantitativo implica una serie de cálculos basados en
datos numéricos obtenidos a través de medidas cuidadosas de masas, volúmenes
u otros parámetros físico-químicos (Transmitancia, por ejemplo). Cuando un
determinado parámetro es medido varias veces, se impone un tratamiento
estadístico de la información obtenida; los resultados que se originan de estos
cálculos pueden o no tener un valor significativo para el analista, en función del
adecuado tratamiento estadístico de los datos y el uso correcto de las cifras
significativas. Dos parámetros estadísticos ampliamente empleados en el análisis
de confiabilidad de los resultados son: la precisión y la exactitud.
La pericia del analista y la calidad de los instrumentos que éste utiliza para
llevar a cabo las medidas, son dos factores importantes que se reflejan en los
resultados experimentales. Por ejemplo, el mismo analista al emplear distintos
instrumentos para medir el mismo parámetro, o distintos analistas midiendo el
mismo parámetro con el mismo instrumento, por lo general obtendrán en ambos
casos medidas diferentes. Es importante, para el buen desenvolvimiento de las
diferentes actividades prácticas que se llevaran a cabo a lo largo del presente
manual, un dominio de parámetros estadísticos básicos tales como; media de la
muestra de datos (medida), desviación estándar de la muestra de datos,
desviación estándar relativa, precisión, exactitud y cifras significativas.
Media de la muestra de datos ( x ). La media aritmética de un conjunto finito
de datos (n), es decir de un conjunto pequeño de datos, es la suma de todos los
datos individuales (Xi) dividida por el número total de datos sumados. El resultado
de esta operación algebraica puede constatarse en la tabla 3.
i
i nxx /
Desviación estándar de la muestra de datos (s). También conocida como
desviación cuadrática o desviación típica, es un indicador de la dispersión de un
conjunto finito de datos con respecto a su media ( x ). Ésta se define por la
ecuación:
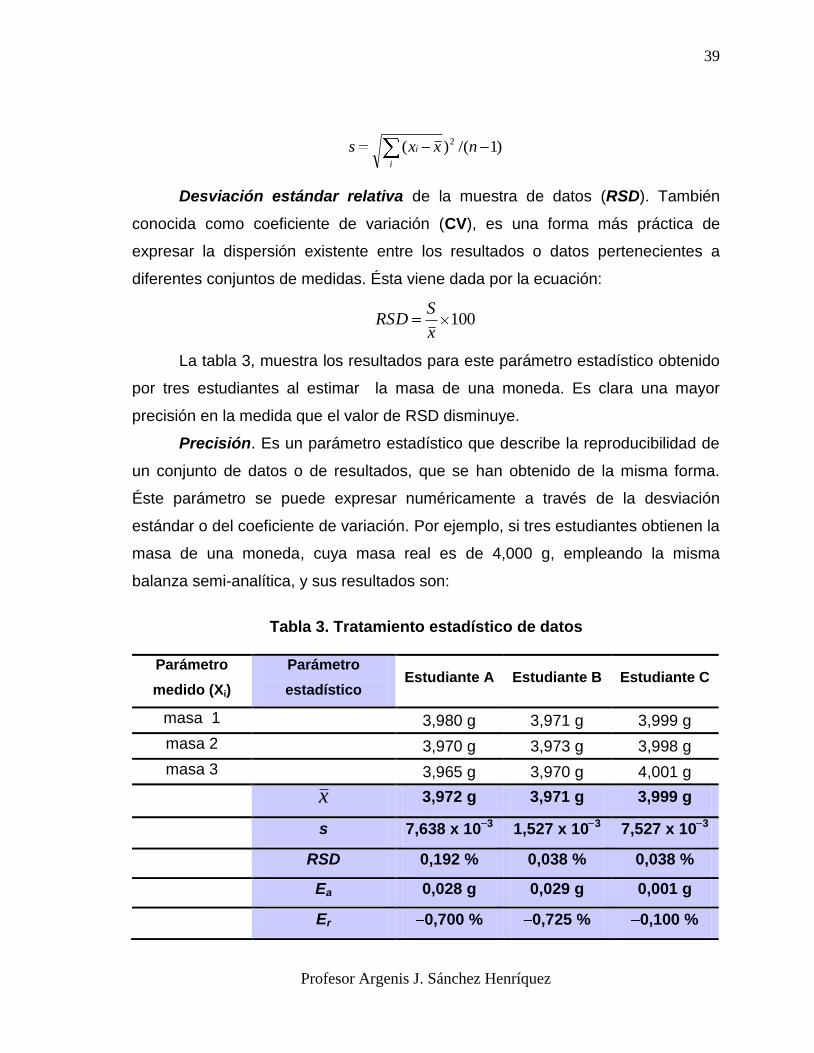
Profesor Argenis J. Sánchez Henríquez
39
Desviación estándar relativa de la muestra de datos (RSD). También
conocida como coeficiente de variación (CV), es una forma más práctica de
expresar la dispersión existente entre los resultados o datos pertenecientes a
diferentes conjuntos de medidas. Ésta viene dada por la ecuación:
100x
SRSD
La tabla 3, muestra los resultados para este parámetro estadístico obtenido
por tres estudiantes al estimar la masa de una moneda. Es clara una mayor
precisión en la medida que el valor de RSD disminuye.
Precisión. Es un parámetro estadístico que describe la reproducibilidad de
un conjunto de datos o de resultados, que se han obtenido de la misma forma.
Éste parámetro se puede expresar numéricamente a través de la desviación
estándar o del coeficiente de variación. Por ejemplo, si tres estudiantes obtienen la
masa de una moneda, cuya masa real es de 4,000 g, empleando la misma
balanza semi-analítica, y sus resultados son:
Tabla 3. Tratamiento estadístico de datos
Parámetro
medido (Xi)
Parámetro
estadístico Estudiante A Estudiante B Estudiante C
masa 1 3,980 g 3,971 g 3,999 g
masa 2 3,970 g 3,973 g 3,998 g
masa 3 3,965 g 3,970 g 4,001 g
x 3,972 g 3,971 g 3,999 g
s 7,638 x 10 3 1,527 x 10 3 7,527 x 10 3
RSD 0,192 % 0,038 % 0,038 %
Ea 0,028 g 0,029 g 0,001 g
Er 0,700 % 0,725 % 0,100 %
)1/()( 2 nxxsi
i

Profesor Argenis J. Sánchez Henríquez
40
Si la desviación estándar (S), es el parámetro que expresa la
reproducibilidad o precisión en las medidas realizadas de la masa de la moneda,
entonces el estudiante que obtuvo una menor desviación estándar como resultado
del procesamiento estadístico de las medidas de la masa de la moneda fue más
preciso. Queda claro que los estudiantes B y C fueron más precisos que A.
Exactitud. Es un parámetro que describe la veracidad de un resultado
experimental. La exactitud de un conjunto de medidas indica la aproximación de la
media ( x ) del conjunto de medidas al valor considerado como real o verdadero
(Xt) para esa medida. Es así, como la media ( x ) es empleada para expresar la
exactitud de un resultado, a través del error absoluto y del error relativo; como
puede observarse en las siguientes ecuaciones:
ta xxE 100t
t
r
x
xxE
Del procesamiento estadístico de los datos experimentales de la masa de la
moneda obtenidas por los tres estudiantes, tabla 3, podemos concluir entonces
que el estudiante C no solo fue más preciso que A e igual de preciso que B, sino
también fue más exacto que A y B, al tener sus medidas de la masa un menor
error absoluto que el de sus compañeros.
Cifras significativa. Un dígito es cualquiera de los siguientes caracteres: 0,
1, 2, 3, 4, 5, 6, 7, 8, 9, que solos o combinados expresan un número. Los dígitos
del 1 al 9 son siempre significativos, el dígito cero puede ser significativo o no. Los
ceros que forman parte de un número, como por ejemplo el número 2,008; son
significativos, en cambio aquellos que sólo tienen dígitos significativos a su
derecha no son significativos (sólo indican orden de magnitud), por ejemplo:
0,0015 o 0,015. Las cifras significativas de una medida son todos aquellos dígitos
que se conocen con certeza tomando en cuenta el primer dígito incierto, sin tomar
en cuenta la coma. Es así, como los siguientes números 103,69, 1,0369, 0,10369
tienen todos 5 cifras significativas, y como el número 2,008, así como los números
0,0015 y 0,015 tienen 4 y 2 cifras significativas respectivamente.

Profesor Argenis J. Sánchez Henríquez
41
Existen una serie de reglas sencillas que nos guían en la retención
adecuada de las cifras significativas, tanto de medidas realizadas con
instrumentos como de resultados derivados (suma, sustracción, multiplicación y
cociente):
1. Conservar en los datos obtenidos a través de medidas realizadas con
instrumento una sola cifra incierta. Por ejemplo, si un estudiante mide un volumen
de un líquido con una bureta que tiene una apreciación de 0,01 mL y obtiene 9,34
mL, el tercer dígito es incierto, ya que esta siendo estimado en una escala sin
graduación. Por lo tanto el número o valor del volumen reportado tendría tres
cifras significativas, de las cuales la última es incierta.
2. Para rechazar dígitos inexactos, se debe aumentar en cantidad de 1 el
último dígito que se conserva si el dígito siguiente rechazado es superior a 5. Es
así, que al rechazar el último dígito del número 35,576 el nuevo número es 35,58.
3. En la suma o en la sustracción de varios números la precisión de la suma
o de la sustracción no puede ser mayor que la de la medida o dato menos preciso;
esto es, los decimales de cada número sumado o restado deben ser iguales a los
que corresponden al número que tiene el dígito incierto más próximo a la coma. Es
así, que al sumar los tres números 0,0134, 35,70 y 2,06847 la operación y el
resultado será:
0,01 + 35,70 + 2,07 = 37,78
4. En la multiplicación y división de distintos números, debe mantenerse en
cada número multiplicado o dividido y en el resultado de la operación, una
cantidad de cifras significativas igual al del número que menor cantidad de cifras
significativas tenga. Es decir, si multiplicamos 0,0135, 35,71 y 2,06847 la
operación y el resultado será:
0,0135 x 35,7 x 2,07 = 0,998

Profesor Argenis J. Sánchez Henríquez
42
IV. ANÁLISIS VOLUMÉTRICO
El análisis químico volumétrico se enmarca dentro del análisis químico
clásico cuantitativo y tiene como propósito principal determinar las cantidades
relativas de un elemento, radical o compuesto que se encuentra presente en una
muestra, a través de la medida del volumen requerido de una solución de
concentración exactamente conocida que reaccionará de manera cuantitativa con
un volumen determinado de este elemento, radical o compuesto u otra especie
químicamente equivalente a éstos. Generalmente, la solución que contiene a la
especie química a ser determinada se le denomina solución titulada, y la solución
cuya concentración se conoce y se emplea para titular se le denomina solución
titulante. Es propicio el momento para aclarar ciertos términos que se han
generalizado dentro del análisis volumétrico, sin tener en cuenta que su significado
es más amplio. Cuando se habla de métodos volumétricos, no se refiere sólo a las
titulaciones de cualquier naturaleza, sino también a todos aquellos métodos que
implican la medición de un volumen de forma general; por ejemplo, la medición del
volumen de un gas en condiciones controladas da origen a un método volumétrico
conocido como gasometría. Es por ello, que en el estricto sentido de la palabra se
debe referir a los métodos volumétricos que implican la acción de titular como
métodos titrimétricos o isolumétricos. La titulación, por lo general, es un proceso
que implica la adición de volúmenes medidos, de una solución de concentración
conocida (solución titulante), desde una bureta, a una solución de concentración
desconocida (solución titulada); las soluciones titulantes a su vez, por tener una
concentración conocida, pueden ser de tipo patrón primario o de tipo patrón
secundario (ver sección II.5, Pág. 27).
En la naturaleza podemos encontrar una amplia gama de reacciones
químicas, sin embargo, sólo pueden ser empleadas como base para los métodos
volumétricos (titrimétricos) de análisis aquellas que cumplan con los siguientes
requisitos: deben ser rápidas, completas, sin interferentes y estequiométricas, es
decir, se debe conocer tanto la proporción en la que reaccionan las especies,
como la proporción en la que se encuentran los productos. Una última condición

Profesor Argenis J. Sánchez Henríquez
43
que tiene gran importancia, es que debe existir una forma de determinar el punto
estequiométrico o de equivalencia, definido como el punto de la titulación en el
cual los reaccionantes (solución titulada y titulante) se encuentran en proporciones
químicamente equivalentes, de tal manera que se debe detener el proceso de
titulación. Esto puede expresarse a través de la siguiente ecuación:
Esta última condición, nos conduce a la definición de punto final de la titulación y
al error de titulación.
El punto final de una titulación es el momento en el cual se detiene la
acción de titular, como consecuencia de un cambio físico-químico apreciable en el
seno de la mezcla reaccionante (solución titulada-solución titulante). Estos
cambios puede ser; la aparición o cambio de color, la formación de un precipitado,
cambios bruscos en el pH, la conductancia, entre otros. El punto o momento en el
cual se detiene la titulación, por lo general, no coincide con el punto de
equivalencia de la misma, estableciéndose entonces una pequeña diferencia que
puede ser por exceso o por defecto de la solución titulante adicionada; de tal
manera que se comete un error de titulación. Este error viene dado por la siguiente
ecuación:
Los métodos volumétricos de análisis se pueden clasificar en función al tipo
de reacción química que ocurre entre la especie titulante y la especie titulada, es
de esta forma que encontramos en la bibliografía:
1. Volumetrías de neutralización ácido-base.
2. Volumetrías de oxidación-reducción o redox.
tituladodelesEquivalentntitulantedelesEquivalentn ºº
100%titulaciónteóricoV
titulaciónteóricoVtitulaciónprácticoVTitulaciónError

Profesor Argenis J. Sánchez Henríquez
44
3. Volumetrías de precipitación.
4. Volumetrías de formación de complejos.
A su vez, cada una de estas volumetrías pueden llevarse a cabo de forma
directa o indirecta; es decir, en el primer caso la especie que será analizada
(especie titulada) se titula directamente, hasta el punto final, con una solución
patrón de la especie titulante. En el segundo caso, o forma indirecta, se adiciona
un exceso de la solución patrón titulante, y la parte de este exceso que queda sin
reaccionar con la especie titulada, se titula posteriormente con otra solución
patrón; a este procedimiento de análisis también se le denomina análisis por
retroceso. El siguiente diagrama ilustra el procedimiento descrito:
Figura 8. Diagrama de una volumetría por retroceso. (Fuente: Autor)
IV.1. VOLUMETRÍA DE NEUTRALIZACIÓN ÁCIDO-BASE
Los métodos titrimétricos fundamentados en las reacciones de
neutralización ácido-base consisten en la titulación de los iones hidronios (H3O+), o
de los iones hidróxilos (OH ), producidos de forma directa o indirecta por el analito
en el seno de una solución. Esto puede resumirse en la siguiente reacción:
Las volumetrías de neutralización ácido-base se emplean para la
determinación de un gran número de especies químicas inorgánicas, orgánicas y
bioquímicas que poseen propiedades ácidas o básicas en medios acuosos u
OHOHOH 23 2
Exceso sol. Patrón titulante A1
Especie tituladaParte sin reaccionar
Sol. Patrón titulante A2
Sol. Patrón titulante A1
Primera etapa de la titulación
Segunda etapa de la titulación
Titulación
reversa
Sobretitulación
Exceso sol. Patrón titulante A1
Especie tituladaParte sin reaccionar
Sol. Patrón titulante A2
Sol. Patrón titulante A1
Primera etapa de la titulación
Segunda etapa de la titulación
Titulación
reversa
Exceso sol. Patrón titulante A1
Especie tituladaParte sin reaccionar
Sol. Patrón titulante A2Sol. Patrón titulante A2
Sol. Patrón titulante A1
Primera etapa de la titulación
Segunda etapa de la titulación
Titulación
reversa
Sobretitulación

Profesor Argenis J. Sánchez Henríquez
45
orgánicos. Los métodos que conforman al análisis volumétrico de neutralización
ácido-base pueden ser clasificados, de forma general, como Alcalimetrías y
Acidimetrías. Las Alcalimetrías son titulaciones de neutralización ácido-base en la
cual se determina un ácido (especie titulada) a través del uso de una base de
concentración exactamente conocida o solución patrón básica (especie titulante), y
las Acidimetría son aquellas titulaciones de neutralización ácido-base cuyo
objetivo es determinar una base (especie titulada) a través del uso de un ácido de
concentración exactamente conocida o solución patrón ácida (especie titulante).
Las principales soluciones patrones empleadas en las titulaciones de
neutralización ácido-base son: HCl y H2SO4 para las acidimetrías, y NaOH,
Ba(OH)2 y KOH para las alcalimetrías; claro está, al no ser estos reactivos
patrones primarios, éstos deberán ser estandarizados previamente a través de un
reactivo que si lo sea; éstos reactivos patrones primarios pueden ser: carbonato
sódico (Na2CO3), biftalato de potasio (KHC8H4O4), ácido benzoico (HC7H5O2),
ácido adípico (H2C6H8O4), ácido clorhídrico de punto de ebullición constante.
Los ácidos y las bases que se titulan pueden ser monofuncionales o
polifuncionales, es decir, pueden generar un solo protón o hidróxido de forma
directa o indirecta (hidrólisis), o pueden generar más de un protón o hidróxido de
manera simultanea o secuencial. Las especies de naturaleza ácida o básica
pueden tener un carácter fuerte o débil, en función de este carácter se puede
establecer una sub-clasificación de los tipos de titulaciones de neutralización
ácido-base, en: a) Titulaciones ácido fuerte-base fuerte, b) Titulaciones ácido
fuerte-base débil y viceversa, c) Titulaciones ácido débil-base débil. Además, se
pueden encontrar en la naturaleza mezclas ácidas y mezclas alcalinas, que
pueden resolverse a través de alcalimetrías y acidimetrías, respectivamente.
Cuando dos o más especies ácidas se encuentran mezcladas se debe tener
presente el carácter de cada constituyente de la mezcla, para establecer su
resolución o nó a través de una titulación. En este sentido, si la mezcla se
encuentra conformada por especies ácidas de carácter fuerte, no se podrá
resolver, ya que se considera que los iones hidronios (H3O+) presentes en la

Profesor Argenis J. Sánchez Henríquez
46
solución provienen de la ionización total de los distintos ácidos fuertes presentes
en la mezcla, siendo posible entonces la neutralización total y simultánea de los
mismos a través de un álcali patrón (OH-). Esto puede resumirse en la siguiente
expresión:
[H3O+]= [H3O
+] ácido 1 + [H3O+] ácido 2 +...+ [H3O
+] ácido n
Cuando la mezcla esta constituida por ácidos fuertes y débiles, es factible
resolverla por titulación, si se toma en cuenta la magnitud de las constantes de
ionización de las especies débiles. Si el ácido es débil (Ka ≤ 1 x 10-4) y
monofuncional su ionización estará inhibida por el exceso de iones hidronios
(H3O+) provenientes del ácido fuerte, y en consecuencia primero se neutralizara
prácticamente sólo el ácido fuerte y posteriormente el ácido débil. Si el ácido es
débil y polifuncional, entonces hay que tomar en cuenta la magnitud de sus
constantes de ionización. Siempre que las magnitudes de estas constantes se
encuentren lo suficientemente separadas entre sí (Ka1 / Ka2 >10000, Ka2 / Ka3
>10000) cada etapa de ionización de este ácido puede ser considerada por
separado y tratada como si se encontrara en presencia de dos (si tiene Ka1 y
Ka2), o tres ácidos (si tiene Ka1, Ka2 y Ka3) débiles con diferentes fuerzas acídicas,
los cuales pueden ser neutralizados por etapas. Debido a la presencia del ácido
fuerte en esta mezcla se puede afirmar que las distintas ionizaciones del ácido
débil estarán inhibidas hasta tanto no se haya neutralizado prácticamente todo el
ácido fuerte, para luego dar paso a la neutralización por etapas del ácido débil. Si
el ácido débil posee una primera constante de ionización relativamente fuerte (Ka1
( 1 x10-3), entonces los iones hidronios (H3O+) provenientes de esta etapa de
ionización serán neutralizados paralelamente con los iones hidronios provenientes
de la ionización total del ácido fuerte en una primera etapa, y posteriormente serán
neutralizados los iones hidronios (H3O+) provenientes de las etapas sucesivas del
ácido débil, atendiendo a la magnitud de sus constantes. Por último, si se trata de
una mezcla de ácidos débiles monofuncionales con constantes de ionización
similares y con concentraciones analíticas (iniciales) parecidas, estos serán

Profesor Argenis J. Sánchez Henríquez
47
neutralizados como una mezcla de ácidos fuertes y por lo tanto no podrá
resolverse la mezcla por titulación. Si se trata de una mezcla de ácidos débiles
polifuncionales, una vez más, se debe tomar en cuenta la magnitud de sus
constantes de ionización para poder establecer como se llevará a cabo su
neutralización y resolución titulométrica.
El punto final en las volumetrías de neutralización ácido-base puede ser
señalado a través de dos métodos. El primero de ellos, y más empleado, es el
método visual, que utiliza indicadores orgánicos ácido-base los cuales se
adicionan en cantidad apropiada a la mezcla reaccionante (especie titulada-
especie titulante), con el propósito de que estos indicadores cambien su estructura
en función de la concentración de iones H3O+ en las cercanías del punto
estequiométrico de la titulación. Estos cambios estructurales en el indicador se
acompañan de cambios de color, que señalan el momento en el cual se debe
detener la titulación; en el apéndice de este manual encontrará una tabla con los
indicadores orgánicos ácido-base más empleados. Dentro de los métodos visuales
también se encuentran indicadores turbidimétricos, de adsorción, fluorescentes y
quimioluminiscentes.
El segundo método para determinar el punto final de una titulación implica
el uso de un electrodo de vidrio calomelanos para medir y registrar el pH, o la
conductancia de la mezcla reaccionante, en función del volumen de especie
titulante, a lo largo de toda la titulación; posteriormente se construye la curva de
titulación y se determina a través de métodos gráficos o examen visual el punto de
inflexión o de máxima pendiente en la curva, el cual se acepta como punto final de
la titulación.

Profesor Argenis J. Sánchez Henríquez
48
PRÁCTICA Nº 1
DETERMINACIÓN VOLUMÉTRICA DE ÁCIDOS MONO Y POLIFUNCIONALES
FUERTES Y DÉBILES
1. Objetivo General
Determinar a través del análisis volumétrico (Alcalimetrías) las
concentraciones de los diferentes ácidos mono y polifuncionales presentes en
muestras sintéticas y comerciales.
2. Objetivos Específicos
2.1. Desarrollar las destrezas del estudiante en las distintas etapas
involucradas en la preparación de soluciones alcalinas tipo patrón.
2.2. Establecer la concentración de ácido clorhídrico (fuerte-monofuncional)
en una muestra sintética, a través de una alcalimetría con solución de hidróxido de
sodio (NaOH) estándar.
2.3. Determinar la acidez total del vinagre comercial, en tanto por ciento
peso-volumen (%p/v), como ácido acético (débil-monofuncional), a través de una
alcalimetría con solución de hidróxido de sodio estándar.
2.4. Contrastar el valor teórico del pH al inicio, en un punto intermedio, y en
el punto final, de la neutralización de ácido acético comercial con NaOH, contra el
valor experimental del pH obtenido para los mismos puntos.
2.5. Establecer la concentración de ácido fosfórico (débil-polifuncional), en
tanto por ciento peso-volumen (%p/v), en una muestra sintética, a través de una
alcalimetría con solución de hidróxido de sodio (NaOH) estándar.
2.6. Redactar un informe científico de laboratorio siguiendo las pautas
establecidas al principio de este manual, donde se reflejen los resultados y
conocimientos adquiridos en el transcurso de la presente práctica.

Profesor Argenis J. Sánchez Henríquez
49
3. Actividades a Desarrollar
Antes de comenzar a desarrollar tus actividades de laboratorio mantén
siempre presente las normas de seguridad que se encuentran al principio de este
manual. Recuerda que la seguridad dentro del laboratorio es responsabilidad de
todos.
3.1. Preparación y estandarización de una solución de hidróxido de
sodio (KOH) aproximadamente 0,1N.
Fundamentos
Las soluciones tipo patrón secundario, como las de NaOH, deben ser
estandarizadas a través de un patrón primario. En este caso se debe preparar una
solución patrón de NaOH a través del método indirecto; y emplear una solución de
ácido clorhídrico Titrisol como patrón primario para su estandarización.
Material a Utilizar
1. Balanza analítica.
2. Espátula.
3. Matraz Volumétrico
4. Botella de lavado con agua destilada/desionizada.
5. Vaso de precipitado.
6. Pipeta Volumétrica.
7. Pro-pipeta
8.Matraz Erlenmeyer
9. Bureta
10. Soporte para bureta.
11. Soporte Universal.
Reactivos a Utilizar
1. Hidróxido de sodio
2. Ácido clorhídrico Titrisol
3. Fenolftaleína
Nota Importante: Es responsabilidad de
cada estudiante entregar al inicio de cada
práctica, en su cuaderno de laboratorio, una
tabla donde se resuman las siguientes
características de los reactivos arriba
señalados: Fórmula, % Pureza, Densidad,
Código de frases de riesgo (R) y seguridad
(S) para cada reactivo.

Profesor Argenis J. Sánchez Henríquez
50
Procedimiento
1. Pese ligeramente por exceso en un vaso de precipitado una masa
aproximada del reactivo (NaOH) que le permita obtener, en el volumen requerido,
una concentración de NaOH 0,1N. Para ello, debe realizar previamente el cálculo
teórico de la masa de NaOH necesaria. Donde: M.M. (Masa molar), M. At. (Masa
atómica), Peq. (Peso equivalente gramo), eq. (equivalentes)
2. Lave rápidamente la masa pesada de NaOH con agua destilada y
desionizada, y descarte la solución sobrenadante. De esta forma arrastrará con el
lavado la mayor cantidad de carbonatos posible.
3. Disuelva la masa de NaOH en agua destilada/desionizada previamente
hervida, tápela y deje enfriar por unos minutos hasta temperatura ambiente.
4. Transfiera cuantitativamente la solución de NaOH del vaso de precipitado
al matraz volumétrico, y enrase con agua destilada/desionizada. Invierta
suavemente dos o tres veces el matraz para homogenizar la solución.
5. Guarde la solución de NaOH en botellas de polietileno, ya que los álcalis
fuertes atacan el vidrio.
6. Estandarice por triplicado la solución de NaOH preparada. Para ello tome
tres alícuotas de 10 mL de ácido clorhídrico Titrisol y transfiéralas a tres matraces
Erlenmeyer.
(Alícuotas de HCl Titrisol )
)(
º
LVsolución
solutoesequivalentnN PEq
solutogsolutoesequivalentnº
eq
AtMoMMPEq
...

Profesor Argenis J. Sánchez Henríquez
51
7. Cure la bureta con una pequeña porción (15mL) de solución de NaOH
preparada y enrase la misma. Cuide de no dejar burbujas de aire atrapadas en la
punta de la bureta.
8. Adicione dos (2) gotas del indicador ácido-base fenolftaleína al matraz
Erlenmeyer que contiene la solución de ácido clorhídrico Titrisol , sólo en el
momento en que se disponga a titular.
9. Titule hasta el punto final y registre en su cuaderno de laboratorio el
volumen gastado de la solución de NaOH. Recuerde que tiene que repetir este
procedimiento de titulación dos veces más.
Vg (NaOH) = Y mL
10. Calcule la concentración de NaOH de la solución que preparó, la cual
servirá como patrón secundario para las demás actividades.
11. Rotule la botella de polietileno con la solución estandarizada de NaOH
con el nombre de la solución, su concentración y la fecha en que fue preparada.
tituladodelesEquivalentntitulantedelesEquivalentn ºº
ácidoácidobasebase VNVN

Profesor Argenis J. Sánchez Henríquez
52
3.2. Determinación de la concentración de HCl en una muestra
sintética por alcalimetría.
Fundamentos
La reacción química de neutralización de un ácido monofuncional fuerte con
una base monofuncional fuerte, tiene un solo punto final bien definido y
establecido por el indicador visual ácido-base fenolftaleína, pasando la mezcla de
reacción de incolora a rosado pálido cuando se alcanza la equivalencia de las
especies. Tomando como base la ecuación que define al punto estequiométrico o
de equivalencia de esta titulación, podemos establecer la concentración del ácido
titulado.
Material a Utilizar
1. Pipeta Volumétrica
2. Pro-pipeta
3. Matraz Erlenmeyer
4. Bureta
5. Soporte Universal
6. Soporte para bureta
7. Botella de lavado con agua destilada y desionizada.
8. Vaso de precipitado.
Reactivos a Utilizar
1. Hidróxido de sodio estándar 0,1N
2. Fenolftaleína
3. Ácido Clorhídrico
Nota Importante: Es responsabilidad de
cada estudiante entregar al inicio de cada
práctica, en su cuaderno de laboratorio, una
tabla donde se resuman las siguientes
características de los reactivos arriba
señalados: Fórmula, % Pureza, Densidad,
Código de frases de riesgo (R) y seguridad
(S) para cada reactivo.
Procedimiento
1. Transfiera con una pipeta volumétrica una alícuota de 10mL de la
muestra a un matraz Erlenmeyer limpio. Este procedimiento debe realizarse por
triplicado.

Profesor Argenis J. Sánchez Henríquez
53
2. Diluya, con agua destilada/desionizada, el contenido de cada Erlenmeyer
hasta aproximadamente 50mL.
3. Cure la bureta con una pequeña porción de solución estandarizada de
KOH 0,1N y enrase la misma. Cuide de no dejar burbujas de aire atrapadas en la
punta de la bureta.
4. Agregue dos (2) gotas ( 50 L) del indicador ácido-base fenolftaleína a
cada matraz Erlenmeyer, sólo en el momento que se disponga a titular.
5. Titule hasta el punto final y registre en su cuaderno de laboratorio el
volumen gastado de la solución estandarizada de NaOH 0,1N y el código de la
muestra para su posterior identificación. Recuerde que tiene que repetir este
procedimiento de titulación dos veces más.
Vg (NaOH) = Y mL
6. Calcule, con los datos obtenidos, la concentración de HCl presente en la
muestra sintética entregada.
tituladodelesEquivalentntitulantedelesEquivalentn ºº
ácidoácidobasebase VNVN

Profesor Argenis J. Sánchez Henríquez
54
3.3. Determinación de la acidez total del vinagre comercial por
alcalimetría.
Fundamentos
El vinagre comercial es una solución diluida de ácido acético (CH3COOH) al
5%. El ácido acético es un ácido orgánico soluble en agua que al ionizarse genera
por cada mol de ácido un mol de iones hidronios (H3O+):
CH3COOH + H2O CH3COO + H3O+ , Ka= 1,8 x 10 5
Estos iones hidronios son neutralizables empleando una solución alcalina
fuerte como el NaOH:
CH3COOH + NaOH CH3COONa + H2O
En la medida que avanza la neutralización y se forma la sal acetato de
sodio (CH3COONa), la sal forma con el ácido acético, aún sin neutralizar, un buffer
o solución tampón, que amortigua o regula los cambios brusco de pH del medio;
es por esta razón que el punto final en esta titulación no es tan nítido como en el
caso del ácido fuerte (HCl). Además, luego de la neutralización total del ácido, la
sal acetato de sodio sufre una disociación y posterior hidrólisis que hace al pH en
el punto de equivalencia ligeramente básico:
CH3COONa + H2O CH3COO + Na+ + H2O
CH3COO + H2O CH3COOH + OH , Khb= 5,5 x 10 10
Si empleamos fenolftaleína como indicador visual ácido-base, el punto final
de la titulación estará marcado por la aparición de un débil color rosado que
perdurará por algunos segundos antes de desvanecerse nuevamente. A través del
volumen de NaOH 0,1N gastado hasta el punto final, podemos establecer la
concentración en tanto por ciento peso-volumen (%p/v) del ácido acético presente
en la muestra de vinagre.

Profesor Argenis J. Sánchez Henríquez
55
Material a Utilizar
1. Pipeta Volumétrica
2. Pro-pipeta
3. Matraz Erlenmeyer
4. Bureta
5. Soporte Universal
6. Soporte para bureta
7. Botella de lavado con agua destilada y desionizada.
8. Vaso de precipitado.
9. Cilindro graduado
10. pH-metro
11. Agitador magnético
12. Plancha de agitación
Reactivos a Utilizar
1. Hidróxido de sodio estándar 0,1N
2. Fenolftaleína
3. Vinagre Comercial
Nota Importante: Es responsabilidad de
cada estudiante entregar al inicio de cada
práctica, en su cuaderno de laboratorio, una
tabla donde se resuman las siguientes
características de los reactivos arriba
señalados: Fórmula, % Pureza, Densidad,
Código R y S para cada reactivo.
Procedimiento
1. Transfiera con una pipeta volumétrica una alícuota de 10mL de la
muestra de vinagre a un matraz Erlenmeyer limpio. Este procedimiento debe
realizarse por duplicado.
2. Diluya, con agua destilada/desionizada, el contenido de cada Erlenmeyer
hasta aproximadamente 50mL.

Profesor Argenis J. Sánchez Henríquez
56
3. Cure la bureta con una pequeña porción de solución estandarizada de
KOH 0,1N y enrase la misma. Cuide de no dejar burbujas de aire atrapadas en la
punta de la bureta.
4. Agregue dos (2) gotas ( 50 L) del indicador ácido-base fenolftaleína a
cada matraz Erlenmeyer, sólo en el momento que se disponga a titular.
5. Titule hasta el punto final y registre en su cuaderno de laboratorio el
volumen gastado de la solución estandarizada de NaOH 0,1N y el código de la
muestra para su posterior identificación. Recuerde que tiene que repetir este
procedimiento de titulación una vez más.
Vg (NaOH) = Y mL
6. Calcule, con los datos obtenidos, la concentración en tanto por ciento
peso-volumen (%p/v), de CH3COOH presente en la muestra de vinagre entregada.
7. Transfiera 10 mL de la muestra de vinagre a un vaso de precipitado de
250 mL.
8. Mida 90 mL de agua destilada/desionizada con un cilindro graduado de
100 mL, y transfiera este volumen de agua al vaso de precipitado que contiene a
la muestra de vinagre (está diluyendo de 10 mL a 100 mL).
100/%muestra
basebase
V
PEqVNvp

Profesor Argenis J. Sánchez Henríquez
57
9. Coloque el agitador magnético en el interior del vaso de precipitado y
seguidamente coloque al vaso de precipitado encima de la plancha de agitación.
10. Coloque el electrodo del pH-metro, calibrado, en el interior del vaso de
precipitado, dispuesto de tal manera que el electrodo quede sumergido, al menos
en un 50%, en la solución diluida de vinagre; y cerca de las paredes del vaso, tal
como se muestra en la figura inferior. Permita que el pH de la solución se
estabilice y registre su valor en el cuaderno de laboratorio. (pH a volumen 0 mL de
NaOH)
pH ( 0 mL de NaOH) = Z
11. Cure la bureta con una pequeña porción de solución estandarizada de
NaOH 0,1N y enrase la misma. Cuide de no dejar
burbujas de aire atrapadas en la punta de la
bureta. Coloque la bureta en el espacio libre al
lado del electrodo, como se muestra en la
siguiente figura.
12. Agregue dos (2) gotas ( 50 L) del indicador ácido-base fenolftaleína al
vaso de precipitado; y encienda el sistema de agitación a una velocidad que
garantice la homogenización de la mezcla de reacción a medida que se adicione
titulante.

Profesor Argenis J. Sánchez Henríquez
58
13. Adicione, desde la bureta, 4 mL de NaOH 0,1N y espere que el pH de la
solución se estabilice, luego registre el valor de pH en su cuaderno de laboratorio
(pH a Volumen 4 mL de NaOH).
14. Continúe la titulación hasta alcanzar el punto final indicado por la
aparición del color rosado pálido; registre en su cuaderno de laboratorio el
volumen gastado de la solución estandarizada de NaOH 0,1N hasta el punto final
y el valor de pH en este punto.
15. Calcule, con el volumen gastado de solución estándar de NaOH 0,1N
hasta el punto final, la concentración molar inicial del ácido.
16. Calcule, con la concentración molar del ácido, los valores de pH
teóricos para la mezcla de reacción cuando ha adicionado los siguientes
volúmenes de NaOH 0,1N: 0mL, 4 mL y el volumen del punto final. Contrástelos
con los valores experimentales obtenidos a través del pH-metro.
, (Antes de comenzar a titular)
, (Henderson-Hasselbalch, al comenzar a titular)
, (En el punto final)
nMN
tituladodelesEquivalentntitulantedelesEquivalentn ºº
ácidoácidobasebase VNVN
iCOOHCHpKapH ][log2
1
2
13
e
e
COOHCH
COOCHpKapH
][
][log
3
3
14pOHpH
adicionadoe NaOHCOOCH ][][ 3
eCOOCHpKhbpOH ][log2
1
2
13

Profesor Argenis J. Sánchez Henríquez
59
3.4. Determinación del % p/v de ácido fosfórico (H3PO4) en una
muestra sintética.
Fundamentos
El ácido fosfórico es una especie triprótica de carácter débil que se ioniza
en tres etapas; este comportamiento se debe a la relación numérica que guardan
sus constantes de ionización (Ka1/Ka2 >>10000). Empleando los indicadores
visuales ácido-base apropiados, se pueden titular de forma secuencial (uno
después del otro) los dos primeros protones que genera este ácido, siendo el
tercero de ellos un protón que carece de relevancia analítica por estar muy poco
ionizado (constante muy pequeña, Ka3 4,8 x 10 13). Si empleamos como titulante
en la alcalimetría una solución de NaOH (base fuerte monobásica), consumiremos
1 equivalente de NaOH por cada protón titulado del ácido. El primero y segundo
punto final del ácido fosfórico se encuentran en zona ácida y básica,
respectivamente. Con el empleo de un indicador ácido-base cuya zona de viraje, o
cambio de color, se encuentre en la zona ácida apropiada, como el rojo de metilo,
se puede neutralizar de forma cuantitativa al primer protón; y si se coloca en la
mezcla de reacción un segundo indicador cuya zona de viraje se encuentre en la
región básica apropiada, como la fenolftaleína, se puede neutralizar
cuantitativamente al segundo protón. Cualquiera de los volúmenes gastados de la
solución titulante de NaOH para alcanzar los puntos finales puede ser empleado
para determinar la concentración del ácido.
Material a Utilizar
1. Pipeta Volumétrica
2. Pro-pipeta
3. Matraz Erlenmeyer
4. Bureta
5. Soporte Universal
Reactivos a Utilizar
1. Hidróxido de sodio estándar 0,1N
2. Fenolftaleína y rojo de metilo
3. Ácido Fosfórico
Nota Importante: Cada alumno es
responsable de entregar al inicio de práctica,
en su cuaderno de laboratorio, una tabla

Profesor Argenis J. Sánchez Henríquez
60
6. Soporte para bureta
7. Botella de lavado con agua destilada y desionizada.
8. Vaso de precipitado.
resumen de las siguientes características de
los reactivos arriba señalados: Fórmula, %
Pureza, Densidad, Código R y S de reactivos.
Procedimiento
1. Transfiera con una pipeta volumétrica una alícuota de 10mL de la
muestra a un matraz Erlenmeyer limpio. Este procedimiento debe realizarse por
triplicado. Registre en su cuaderno de laboratorio el código de la muestra para su
posterior identificación
2. Diluya, con agua destilada/desionizada, el contenido de cada Erlenmeyer
hasta aproximadamente 50mL.
3. Cure la bureta con una pequeña porción de solución estandarizada de
NaOH 0,1N y enrase la misma. Cuide de no dejar burbujas de aire atrapadas en la
punta de la bureta.
4. Agregue una (1) gota ( 25 L) del indicador ácido-base rojo de metilo y
dos (2) gotas del indicador ácido-base fenolftaleína a cada matraz Erlenmeyer,
sólo en el momento que se disponga a titular.
5. Titule hasta el primer punto final y registre en su cuaderno de laboratorio
el volumen gastado de la solución estandarizada de NaOH 0,1N (Volumen
gastado al RM= X mL). El cambio de color de la solución en este punto será de

Profesor Argenis J. Sánchez Henríquez
61
rojo púrpura a un color anaranjado claro. Continúe la titulación hasta el segundo
punto final y registre en su cuaderno el volumen gastado de NaOH (Volumen
gastado a la F= Y mL). El cambio de color en este punto será de amarillo a un
color anaranjado claro.
VgRM (NaOH) = X mL
VgF (NaOH) = Y mL
6. Calcule, con los datos obtenidos, la concentración de H3PO4 presente en
la muestra sintética en tanto por ciento peso-volumen (%p/v).
, (Para el 1er punto final)
, (Para el 2do punto final)
4. Post- Laboratorio
Redactar un informe científico de laboratorio siguiendo las pautas
establecidas al principio de este manual, donde se reflejen los resultados y
conocimientos adquiridos en el desarrollo de las actividades realizadas (3.1, 3.2,
3.3 y 3.4).
100/% 43
43
muestra
POHgRMbase
V
PEqVNPOHvp
100/% 43
43
muestra
POHgFbase
V
PEqVNPOHvp
eq
MMPEq
POH
POH43
43
.

Profesor Argenis J. Sánchez Henríquez
62
5. Cuestionario
1. ¿Cuál es el propósito del análisis volumétrico, y cómo se logra el mismo?
2. ¿Qué es una solución buffer? De un ejemplo.
3. Defina: a. Punto de equivalencia de una titulación.
b. Punto Final de una titulación.
c. Error de titulación.
4. Explique los métodos empleados para señalar el punto final en una
titulación ácido-base.
5. ¿Cómo se clasifican las volumetrías de neutralización ácido-base y
defina cada término?
6. Describa la preparación de las siguientes soluciones:
a. 500 mL de NaOH 0,25 M, a partir de NaOH sólido al 98% de
pureza. Masa molar (NaOH): 40,00 g/mol.
b. 250 mL de HCl 2 N, a partir de una solución de HCl 5 N.
c. 500 mL de HCl 3M, a partir de del ácido concentrado con una
pureza de 37% y densidad de 1,18 Kg/L. Masa molar (HCl): 36,46 g/mol.
d. 250 mL de ácido acético 2 M, a partir del ácido glacial con una
pureza de 98% y densidad de 1,05 Kg/L. Masa molar (CH3COOH): 60,05 g/mol.
6. Referencias Bibliográficas
Ayres Gilbert H. (1970). Análisis Químico Cuantitativo. (2a Ed.). Ediciones
del Castillo. Madrid (España).
Alexéiev Víctor N. (1976). Análisis Cuantitativo. Trad. Liminík E. Editorial
Mir. Moscú (Rusia).
Maritza Machado y Sorely Peraza. (2001). Manual de Práctica de
Laboratorio Química Analítica Instrumental. Universidad Centroccidental “Lisandro
Alvarado”. Barquisimeto (Venezuela).

Profesor Argenis J. Sánchez Henríquez
63
PRÁCTICA Nº 2
DETERMINACIÓN DE MEZCLAS DE ÁCIDOS Y SALES ÁCIDAS
POLIFUNCIONALES
1. Objetivo General
Determinación de pares compatibles en mezclas constituidas por ácidos
y sales ácidas polifuncionales, a través del uso de puntos finales sucesivos.
2. Objetivos Específicos
2.1. Establecer a través de la volumetría de neutralización ácido-base la
proporción en la que se encuentran presentes los constituyentes de una mezcla de
ácidos polifuncionales.
2.2. Establecer a través de la volumetría de neutralización ácido-base la
proporción en la que se encuentran presentes los constituyentes de una mezcla de
ácido polifuncional y su sal ácida.
2.3. Redactar un informe científico de laboratorio siguiendo las pautas
establecidas al principio de este manual, donde se reflejen los resultados y
conocimientos adquiridos en el transcurso de la presente práctica.
3. Actividades a Desarrollar
Antes de comenzar a desarrollar tus actividades de laboratorio mantén
siempre presente las normas de seguridad que se encuentran al principio de este
manual. Recuerda que la seguridad dentro del laboratorio es responsabilidad de
todos.

Profesor Argenis J. Sánchez Henríquez
64
3.1. Determinación de una mezcla de ácidos polifuncionales (H2SO4 +
H3PO4).
Fundamentos
El ácido sulfúrico es un ácido diprótico de carácter fuerte ya que sus dos
protones se ionizan en solución prácticamente de manera simultánea (Ka2 =
1x10 2) según la siguiente reacción:
H2SO4 + 2H2O SO42
+ 2H3O+, Ka2 = 1x10 2
Por otra parte, el ácido fosfórico posee una primera constante de ionización
(Ka1= 7,5x10-3) que es relativamente fuerte. Siendo así, en una mezcla conformada
por estos dos ácidos, los dos iones hidronios provenientes de la ionización total
del ácido sulfúrico pueden ser titulados, en el primer punto final, de forma paralela
con el ion hidronio proveniente de la primera ionización del ácido fosfórico, si se
emplea el indicador ácido-base apropiado. Esto quiere decir, que el segundo punto
final en la titulación de esta mezcla, corresponderá sólo a la neutralización del
segundo protón del ácido fosfórico (H2PO4 ). Si la titulación se lleva a cabo
empleando como solución titulante una base fuerte como el NaOH, se consumirá 1
equivalente de NaOH por cada protón neutralizado; lo que deriva en un volumen
consumido de la solución titulante mayor para el primer punto final, donde se
neutralizan tres protones, y menor para el segundo punto final. Esto puede
representarse matemáticamente de la siguiente manera:
VFN < 2VRM
Empleando el indicador ácido-base rojo de metilo para señalar el primer
punto final, y la fenolftaleína para señalar el segundo; se pueden establecer los
volúmenes de solución del titulante NaOH, que permiten evidenciar que se está en
presencia de una mezcla de ácidos polifuncionales y determinar la proporción en
la que se encuentran los constituyentes de la mezcla.
El siguiente diagrama esquematiza el comportamiento de los constituyentes
de esta mezcla ácida durante la titulación:

Profesor Argenis J. Sánchez Henríquez
65
Figura 9. Diagrama de neutralización mezcla de H2SO4/H3PO4. (Fuente: Autor)
Material a Utilizar
1. Pipeta Volumétrica
2. Pro-pipeta
3. Matraz Erlenmeyer
4. Bureta
5. Soporte Universal
6. Soporte para bureta
7. Botella de lavado con agua destilada y desionizada.
8. Vaso de precipitado.
Reactivos a Utilizar
1. Hidróxido de sodio estándar 0,1N
2. Fenolftaleína y rojo de metilo
3. Ácido Sulfúrico
4. Ácido Fosfórico
Nota Importante: Cada alumno es
responsable de entregar al inicio de práctica,
en su cuaderno de laboratorio, una tabla
resumen de las siguientes características de
los reactivos arriba señalados: Fórmula, %
Pureza, Densidad, Código R y S para cada
reactivo.
Procedimientos
1. Transfiera con una pipeta volumétrica una alícuota de 10mL de la
muestra seleccionada a un matraz Erlenmeyer limpio. Este procedimiento debe

Profesor Argenis J. Sánchez Henríquez
66
realizarse por triplicado. Registre en su cuaderno de laboratorio el código de la
muestra para su posterior identificación
2. Diluya, con agua destilada/desionizada, el contenido de cada Erlenmeyer
hasta aproximadamente 50mL.
3. Cure la bureta con una pequeña porción de solución estandarizada de
NaOH 0,1N y enrase la misma. Cuide de no dejar burbujas de aire atrapadas en la
punta de la bureta.
4. Agregue una (1) gota ( 25 L) del indicador ácido-base rojo de metilo y
dos (2) gotas del indicador ácido-base fenolftaleína a cada matraz Erlenmeyer,
sólo en el momento que se disponga a titular.
5. Titule hasta el primer punto final y registre en su cuaderno de laboratorio
el volumen gastado de la solución estandarizada de NaOH 0,1N (Volumen
gastado al RM= X mL). El cambio de color de la solución en este punto será de
rojo púrpura a un color anaranjado claro. Continúe la titulación hasta el segundo
punto final y registre en su cuaderno el volumen gastado de NaOH (Volumen
gastado a la FN= Y mL). El cambio de color en este punto será de amarillo a un
color anaranjado claro.
VRM (NaOH) = X mL
VFN (NaOH) = Y mL

Profesor Argenis J. Sánchez Henríquez
67
6. Verifique, con los datos obtenidos, si la mezcla seleccionada está
constituida por los ácidos H2SO4 y H3PO4, y Calcule la concentración de cada
ácido presente en la muestra, expresada en tanto por ciento peso-volumen (%p/v).
(Si se cumple la condición la mezcla está constituida por H2SO4 y H3PO4).
Volumen de NaOH estándar gastado para titular sólo H3PO4 (Puede
apreciarse del diagrama en los fundamentos)
Volumen de NaOH estándar gastado para titular sólo H2SO4 (Puede
apreciarse del diagrama en los fundamentos)
3.2. Determinación de una mezcla de ácido polifuncional y su sal ácida
(H3PO4 + NaH2PO4).
Fundamentos
La titulación del ácido fosfórico hasta su primer punto final con un
determinado volumen de un álcali (OH ), genera la neutralización del primer protón
ionizado, según las siguientes reacciones:
H3PO4 + H2O H2PO4 + H3O+ (Ka1= 7,5 x 10 3)
RMFN VV 2
RMFNNaOH VVPOHV )( 43
100/% 43
43
muestra
POHNaOHNaOH
V
PEqVNPOHvp
eq
MMPEq
POH
POH43
43
.
FNRMNaOH VVSOHV 2)( 42
100/% 42
42
muestra
SOHNaOHNaOH
V
PEqVNSOHvp
eq
MMPEq
SOH
SOH42
42
.

Profesor Argenis J. Sánchez Henríquez
68
H2PO4 + H3O+ + OH H2PO4 + 2 H2O
El pH de la mezcla reaccionante en este primer punto estequiométrico es de
4,67 pH, y podemos observar este primer punto final empleando rojo de metilo
como indicador visual ácido-base. Posteriormente, se produce la segunda
ionización del ácido, la cual estuvo inhibida por la primera ionización, seguida de
la neutralización del segundo protón con un volumen de álcali (OH ) similar al
gastado para alcanzar el primer punto final, según las reacciones:
H2PO4 + H2O HPO42
+ H3O+ (Ka2= 6,2 x 10 8)
HPO42
+ H3O+ + OH HPO4
2 + 2 H2O
El valor del pH para la mezcla reaccionante en este segundo punto
estequiométrico es de 9,77 pH, lográndose observar su punto final a través del
uso de la fenolftaleína como indicador visual ácido-base.
De lo anterior se deduce que para la titulación del ácido fosfórico el volumen
de álcali (NaOH) gastado para alcanzar el segundo punto final, es el doble del
volumen de álcali necesario para alcanzar el primer punto final, esto es:
VFN = 2 VRM
Si consideramos una solución conformada por la mezcla del ácido fosfórico
(H3PO4) con su sal ácida primaria (NaH2PO4), y tomamos en cuenta que:
i) La sal ácida primaria una vez en solución se disocia y genera iones H2PO4
ii) Este ion (H2PO4 ), es también generado por el ácido fosfórico en su primera
etapa de ionización y posterior neutralización.
Podemos entonces concluir que el volumen de álcali (NaOH) que se
consumirá en la titulación de la mezcla, hasta el segundo punto final, debe ser
mayor que el doble del volumen de álcali gastado para alcanzar el primer punto
final, debido a la contribución protónica de la sal ácida primaria al segundo punto
final. Esto es: VFN > 2 VRM

Profesor Argenis J. Sánchez Henríquez
69
El siguiente diagrama esquematiza el comportamiento de los constituyentes
de esta mezcla ácida durante la titulación:
Figura 10. Diagrama de neutralización mezcla de H3PO4/NaH2PO4. (Fuente: Autor)
Material a Utilizar
1. Pipeta Volumétrica
2. Pro-pipeta
3. Matraz Erlenmeyer
4. Bureta
5. Soporte Universal
6. Soporte para bureta
7. Botella de lavado con agua destilada y desionizada.
8. Vaso de precipitado.
Reactivos a Utilizar
1. Hidróxido de sodio estándar 0,1N
2. Fenolftaleína y rojo de metilo
3. Fosfato ácido de sodio
4. Ácido Fosfórico
Nota Importante: Cada alumno es
responsable de entregar al inicio de práctica,
en su cuaderno de laboratorio, una tabla
resumen de las siguientes características de
los reactivos arriba señalados: Fórmula, %
Pureza, Densidad, Código R y S de reactivos.

Profesor Argenis J. Sánchez Henríquez
70
Procedimientos
1. Transfiera con una pipeta volumétrica una alícuota de 10mL de la
muestra seleccionada a un matraz Erlenmeyer limpio. Este procedimiento debe
realizarse por triplicado. Registre en su cuaderno de laboratorio el código de la
muestra para su posterior identificación
2. Diluya, con agua destilada/desionizada, el contenido de cada Erlenmeyer
hasta aproximadamente 50mL.
3. Cure la bureta con una pequeña porción de solución estandarizada de
NaOH 0,1N y enrase la misma. Cuide de no dejar burbujas de aire atrapadas en la
punta de la bureta.
4. Agregue una (1) gota ( 25 L) del indicador ácido-base rojo de metilo y
dos (2) gotas del indicador ácido-base fenolftaleína a cada matraz Erlenmeyer,
sólo en el momento que se disponga a titular.
5. Titule hasta el primer punto final y registre en su cuaderno de laboratorio
el volumen gastado de la solución estandarizada de NaOH 0,1N
(Volumen gastado al RM= X mL). El cambio de color de la
solución en este punto será de rojo púrpura a un color
anaranjado claro. Continúe la titulación hasta el segundo punto
final y registre en su cuaderno el volumen gastado de NaOH
(Volumen gastado a la FN= Y mL). El cambio de color en este
punto será de amarillo a un color anaranjado claro.
VRM (NaOH) = X mL , VFN (NaOH) = Y mL

Profesor Argenis J. Sánchez Henríquez
71
6. Verifique, con los datos obtenidos, si la mezcla seleccionada está
constituida por el H3PO4 y NaH2PO4, y Calcule la concentración de cada especie
presente en la muestra, expresada en tanto por ciento peso-volumen (%p/v).
(Si se cumple la condición la mezcla está constituida por H3PO4 y NaH2PO4).
Volumen de NaOH estándar gastado para titular sólo H3PO4 (Puede
apreciarse del diagrama en los fundamentos)
Volumen de NaOH estándar gastado para titular sólo NaH2PO4 (Puede
apreciarse del diagrama en los fundamentos)
4. Post- Laboratorio
Redactar un informe científico de laboratorio siguiendo las pautas
establecidas al principio de este manual, donde se reflejen los resultados y
conocimientos adquiridos en el desarrollo de las actividades realizadas (3.1 y 3.2).
5. Cuestionario
1. ¿Qué factor debe tenerse en consideración al momento de resolver una
mezcla ácida o alcalina a través de una titulación?
RMFN VV 2
RMNaOH VPOHV )( 43
100/% 43
43
muestra
POHNaOHNaOH
V
PEqVNPOHvp
eq
MMPEq
POH
POH43
43
.
RMFNNaOH VVPONaHV 2)( 42
100/% 42
42
muestra
SOHNaOHNaOH
V
PEqVNSOHvp
eq
MMPEq
SOH
SOH42
42
.

Profesor Argenis J. Sánchez Henríquez
72
2. En presencia de una muestra que pueda estar constituida por alguna de
las siguientes mezclas compatibles:
a. H3PO4 y NaH2PO4 b. H3PO4 y H2SO4
3. Una solución contiene una mezcla compatible de dos de las siguientes
sustancias: Na2HPO4, NaH2PO4, H3PO4 y NaOH. Al titular con NaOH 0,125 N una
muestra de 25 mL de esta mezcla, en presencia de fenolftaleína se consumen
37,16 mL de la base. Con rojo de metilo, la misma muestra consume 15,40 mL del
NaOH. ¿Qué componentes y qué porcentaje de los mismos?
4. Una solución contiene una mezcla compatible de dos de las siguientes
sustancias: H2SO4, Na2HPO4, H3PO4 y NaOH. Al titular con NaOH 0,125 N una
muestra de 25 mL de esta mezcla, en presencia de fenolftaleína se consumen
44,5 mL de la base. Con rojo de metilo, la misma muestra consume 27,00 mL del
NaOH. ¿Qué componentes y qué porcentaje de los mismos?
6. Referencias Bibliográficas
Ayres Gilbert H. (1970). Análisis Químico Cuantitativo. (2a Ed.). Ediciones
del Castillo. Madrid (España).
Skoog Douglas A., Donald M. West, James F. Holler y Stanley R. Crouch.
(2001). Química Analítica. (7a Ed.). McGraw-Hill. México D.F. (México).
Skoog Douglas A. y Donald M. West. (1970). Introducción a la Química
Analítica. Editorial Reverté. Barcelona (España).
Maritza Machado y Sorely Peraza. (2001). Manual de Práctica de
Laboratorio Química Analítica Instrumental. Universidad Centroccidental “Lisandro
Alvarado”. Barquisimeto (Venezuela).

Profesor Argenis J. Sánchez Henríquez
73
IV.2. VOLUMETRÍA DE OXIDACIÓN-REDUCCIÓN (REDOX)
Las reacciones de oxidación-reducción (redox) pueden ocurrir de dos
formas: (i) Directa entre el agente oxidante y el agente reductor en el mismo
recipiente, o (ii) Indirecta en la cual el agente oxidante y el agente reductor se
encuentran en recipientes separados, que a su vez están unidos por un circuito
eléctrico externo y un puente salino. Las volumetrías de oxidación-reducción
ocurren de la primera forma; el agente oxidante se adiciona desde una bureta al
agente reductor que se encuentra en un matraz Erlenmeyer, o viceversa, y una
vez juntos en solución se produce la reacción redox. Debemos tener presente que
cuando ocurre una reacción de oxidación, simultáneamente, ocurre una reacción
de reducción; es decir, los electrones perdidos por una especie deben ser
ganados por otra.
Un agente oxidante es aquella especie química que al reducirse, causa la
oxidación de otra especie química; y un agente reductor es aquella especie
química que al oxidarse, causa la reducción de otra especie química. Por ejemplo:
(Agente oxidante) Fe3+ + 1e Fe2+ , EºFe(III)/Fe(II) = 0,77 volts
(Agente reductor) Sn2+ Sn4+ + 2 e , EºSn(II)/Sn(IV) = 0,15 volts
Aunque el proceso de oxidación-reducción tiene lugar de manera
simultánea y en un mismo recipiente, es conveniente considerar las reacciones de
oxidación-reducción por separado como dos semi-reacciones. Estas semi-
reacciones, una de oxidación y otra de reducción, combinadas dan origen a la
reacción redox global. Por ejemplo:
Semi-reacción de reducción: Ce4+ + 1e Ce3+ , EºCe(IV)/Ce(III) = +1,44 volts
Semi-reacción de oxidación: Fe2+ Fe3+ + 1e , EºFe(II)/Fe(III) = -0,77 volts Reacción global redox: Fe2+ + Ce4+ Ce3+ + Fe3+, Eºredox = 0,77 volts

Profesor Argenis J. Sánchez Henríquez
74
Una mirada exploratoria a una tabla de potenciales estándares normales de
oxidación o reducción, haría pensar que existen decenas de sistemas redox que
pueden emplearse como base para una volumetría de oxidación-reducción. Sin
embargo, la selección de los agentes oxidantes y reductores no es algo trivial.
Tanto el agente oxidante, como el reductor si fuera el caso, están sujetos a varias
condiciones. Primero, el oxidante debe ser lo suficientemente fuerte para que su
reacción con el agente reductor sea completa; esta condición se satisface si la
diferencia entre los potenciales normales de la semi-reacción de oxidación y
reducción son al menos 0,2 Voltios. Segundo, el oxidante no debe ser tan enérgico
que reaccione de forma rápida con cualquiera de los componentes presentes en la
mezcla de reacción, por ejemplo: el agua. Tercero, el oxidante debe ser selectivo y
reaccionar rápidamente con la especie reductora que se desea determinar; esta
última condición se refiere a que la reacción redox debe ser favorable, desde el
punto de vista termodinámico (potenciales redox globales positivos) y desde el
punto de vista del mecanismo de reacción.
Estas condiciones indican que no cualquier reacción química puede servir
como base para una volumetría redox. Por tal razón, existen una serie de
reactivos considerados como agentes oxidantes y agentes reductores estándares,
los cuales se muestran en la siguiente tabla:
Tabla 4. Agentes estándares redox.
Agentes oxidantes Agentes reductores
Permanganato de potasio (KMnO4) Tiosulfato de sodio (Na2S2O3)
Dicromato de potasio (K2Cr2O7) Oxalato de sodio (Na2C2O4)
Sulfato cérico [Ce(SO4)2] Ácido oxálico (H2C2O4)
Yodo (I2) Sales Ferrosas
Yodato de potasio (KIO3) Sulfato o Cloruro titanoso
Bromato de potasio (KBrO3) Sulfato o Cloruro cromoso
Cloroamina T Sulfato o Cloruro vanadoso
Peróxido (H2O2) Tetra oxalato potásico

Profesor Argenis J. Sánchez Henríquez
75
Una condición de vital importancia para cualquier titrimetría (titulación), es
poder establecer de manera clara y sencilla el punto estequiométrico. En la
volumetría de oxidación-reducción esta condición se satisface, comúnmente, a
través del empleo de indicadores visuales de oxidación-reducción. Los indicadores
de oxidación-reducción pueden ser básicamente de tres tipos. En primer lugar,
encontramos los reactivos auto-indicadores; estos reactivos empleados como
agentes titulantes presentan una fuerte coloración, resultando sus productos de
reacción incoloros o débilmente coloreados. Este es el caso del permanganato de
potasio en soluciones ácidas que pasa de violeta intenso a rosado tenue en el
punto final, y el ion cerio (IV) que pasa de amarillo a incoloro. Otra modalidad
enmarcada en los reactivos auto-indicadores son las soluciones tituladas que
presentan coloración como el caso de la solución de Fe(SCN)2+ de color roja, la
cual pasa a incolora al ser titulada con titanio (III). En segundo lugar, tenemos a
los indicadores redox reversibles; los cuales son compuestos orgánicos que
cambian su color de forma reversible al oxidarse o reducirse, su semi-reacción
puede resumirse en la siguiente expresión general:
InOx + ne InRd , EºOx/Rd = X volts
Al igual que todo sistema redox esta semi-reacción posee un potencial
estándar, este potencial estándar (EºOx/Rd) permite seleccionar el indicador redox
reversible más apropiado, en función del cambio de potencial alrededor del punto
de equivalencia, para llevar a cabo una determinada titulación. Y en tercer lugar,
están los indicadores redox irreversibles; que consisten en compuestos orgánicos
intensamente coloreados que sufren oxidación o reducción de manera irreversible,
cambiando su color con un ligero exceso de solución titulante. Dentro de estos
reactivos indicadores tenemos al rojo de metilo, la heliantina, y el azul negro de
naftol.
Por otra parte, los métodos potenciométricos representan la alternativa
instrumental para la señalización del punto estequiométrico de una reacción redox.
Hay que recordar que este método permite hallar el punto de equivalencia
gráficamente a través de la elaboración de una curva de titulación potenciométrica.

Profesor Argenis J. Sánchez Henríquez
76
Las volumetrías de oxidación-reducción pueden clasificarse, de manera
general, atendiendo al tipo de agente titulante que se encuentra en la bureta; es
decir, si el titulante es un agente oxidante, entonces se denomina al método
oxidimetría, y si se trata de un agente reductor, se denomina reductimetría.
Entre los agentes titulantes más empleados en los métodos volumétricos
redox, encontramos al permanganato de potasio, dicromato de potasio, cerio (IV) y
yodo (ion triyoduro). En este manual nos dedicaremos a las aplicaciones
relacionadas con el uso del permanganato de potasio (KMnO4) como agente
oxidante y titulante; es decir, a la permanganimetría.
El permanganato de potasio tiene la ventaja, por su intenso color violeta, de
servir como reactivo auto-indicador en la mayoría de las titulaciones donde
participa. Sin embargo, el empleo de este agente oxidante involucra algunas
desventajas prácticas. En primer lugar, es difícil obtener el reactivo completamente
puro; ya que generalmente ésta impurificado con dióxido de manganeso. Además,
las soluciones acuosas de este oxidante no son lo suficientemente estables en el
tiempo, debido a que las trazas de sustancias reductoras presentes en el agua
destilada/desionizada reducen al ion permanganato (MnO4 ) a dióxido de
manganeso (MnO2), el cual cataliza la auto-descomposición de la solución de
permanganato, por catálisis fotoquímica, en el transcurso del tiempo. Por esta
razón, se filtran las soluciones de permanganato de potasio a través de medios
filtrantes no reductores para eliminar el MnO2.
Queda claro que el permanganato de potasio no es un patrón primario, por
lo que se requiere estandarizar sus soluciones contra un patrón primario para
determinar con exactitud su concentración. Los patrones primarios, generalmente,
empleados para esta estandarización son: oxalato de sodio (Na2C2O4), yoduro de
potasio (KI), oxido arsenioso (As2O3), entre otros.
La reacción de oxidación-reducción que ocurra entre el ion permanganato y
el agente reductor que se desea determinar, puede ocurrir en medio ácido, neutro
o alcalino; estableciéndose de esta forma los dos equilibrios redox principales que
puede tener el ion permanganato:

Profesor Argenis J. Sánchez Henríquez
77
Medio ácido: MnO4 + 8H+ + 5e– Mn2+ + 4H2O
Medio neutro y alcalino: MnO4 + 2H2O + 3e MnO2 + 4OH

Profesor Argenis J. Sánchez Henríquez
78
PRÁCTICA Nº 3
DETERMINACIÓN DE CALCIO POR PERMANGANIMETRÍA
1. Objetivo General
Determinar la concentración de calcio, en tanto por ciento peso-volumen
(%p/v), en una muestra de alimento a través de una permanganimetría.
2. Objetivos Específicos
2.1. Desarrollar las destrezas del estudiante en las distintas etapas
involucradas en la preparación de una solución de permanganato de potasio
(KMnO4).
2.2. Establecer la concentración de una solución de KMnO4 a través de su
estandarización con el patrón primario oxalato de sodio (Na2C2O4).
2.3. Determinar a través de una permanganimetría el contenido de calcio
presente en una solución de cenizas, obtenida a partir de una muestra de
alimento.
2.4. Redactar un informe científico de laboratorio siguiendo las pautas
establecidas al principio de este manual, donde se reflejen los resultados y
conocimientos adquiridos en el transcurso de la presente práctica.
3. Actividades a Desarrollar
Antes de comenzar a desarrollar tus actividades de laboratorio mantén
siempre presente las normas de seguridad que se encuentran al principio de este
manual. Recuerda que la seguridad dentro del laboratorio es responsabilidad de
todos.

Profesor Argenis J. Sánchez Henríquez
79
3.1. Preparación de una solución de permanganato de potasio (KMnO4)
aproximadamente 0,05N (0,01M). (Para medio ácido)
Fundamentos
El permanganato de potasio da origen a soluciones de tipo patrón
secundario y se ha mencionado con anterioridad, que estas soluciones deben ser
preparadas a través del método indirecto. Tomando en consideración algunas
medidas, se puede preparar una solución de KMnO4 en un tiempo razonablemente
rápido y de tal forma, que se garantice la estabilidad y concentración del reactivo;
como veremos a continuación.
Material a Utilizar
1. Balanza analítica.
2. Espátula.
3. Matraz Erlenmeyer
4. Botella de lavado con agua destilada y desionizada.
5. Vaso de precipitado.
6. Matraz Volumétrico
7. Mechero Meker
8. Rejilla para calentamiento
9. Trípode
10. Embudo de Buchner
11. Vidrio de reloj
12. Soporte para embudo
13. Soporte universal
14. Cilindro graduado
Reactivos a Utilizar
1. KMnO4
Nota Importante: Es responsabilidad de
cada estudiante entregar al inicio de cada
práctica, en su cuaderno de laboratorio, una
tabla donde se resuman las siguientes
características de los reactivos arriba
señalados: Fórmula, % Pureza, Densidad,
Código de frases de riesgo (R) y seguridad
(S) para cada reactivo.

Profesor Argenis J. Sánchez Henríquez
80
Procedimiento
1. Pese ligeramente por exceso en un pesa sustancia o vidrio de reloj una
masa aproximada del reactivo (KMnO4) que le permita obtener, en un volumen de
1L, una concentración de KMnO4 0,05N (Para medio ácido). Para ello, debe
realizar el cálculo teórico de la masa de KMnO4 necesaria.
2. Mida con un cilindro graduado 1L de agua
destilada/desionizada, y con la misma, transfiera la
masa pesada de reactivo KMnO4 a un vaso de
precipitado de 2L. Ver figura adjunta.
3. Coloque el vaso de precipitado con el reactivo
disuelto en el trípode, tal como muestra la figura adjunta,
puede ayudarse con una varilla de vidrio para revolver la
mezcla y disolver restos del reactivo, tápelo con un vidrio de
reloj y sin sacar la varilla de vidrio proceda a calentar la
solución hasta ebullición.
4. Una vez que la solución llegue a ebullición, baje la intensidad de la llama
y permita una ebullición suave durante 30 minutos. El calentamiento de la solución
)(
º
LVsolución
solutoesequivalentnN
PEq
solutogsolutoesequivalentnº
eq
AtMoMMPEq
...

Profesor Argenis J. Sánchez Henríquez
81
cataliza la oxidación de cualquier agente reductor presente en el medio y permite
aligerar la preparación, de lo contrario se tendría que esperar 6 o 7 días
aproximadamente para poder filtrar la solución.
5. Apague el mechero y permita que la solución permanezca tapada en
reposo hasta alcanzar la temperatura ambiente.
6. Filtre la solución fría a través de un embudo de
Buchner y recoja la solución filtrada de KMnO4 en el matraz
volumétrico de 1 L. Tal y como se muestra en la figura anexa.
7. Enrase la solución de permanganato de potasio con
agua destilada/desionizada, homogenice y transfiérala a un
frasco color ámbar. Donde permanecerá guardada en la
oscuridad hasta el momento de su estandarización.
3.2. Estandarización de una solución de permanganato de potasio
(KMnO4) con oxalato de sodio (Na2C2O4).
Fundamentos
La solución patrón secundario de KMnO4 puede ser estandarizada a través
del uso de Na2C2O4 anhidro como patrón primario, en medio ácido. El oxalato de
sodio reacciona con ácido sulfúrico, en medio acuoso, para generar ácido oxálico
muy poco ionizado (exceso de ácido sulfúrico); la estequiometría de la reacción
puede representarse como:
Na2C2O4 + H2SO4 H2C2O4 + Na2SO4

Profesor Argenis J. Sánchez Henríquez
82
Posteriormente, ocurre una reacción de oxidación-reducción entre el ácido
oxálico y el ión permanganato (MnO4 ); este mecanismo de reacción es muy
complicado, ya que presupone la participación de especies intermediarias
transitorias del manganeso (IV) y (VI). Sin embargo, la reacción analítica redox
global de interés puede representarse como:
2 MnO4 + 5 H2C2O4 + 6 H+ 2 Mn2+ + 10 CO2 + 8 H2O
Donde el ion permanganato reacciona muy lentamente al principio de la
titulación, requiriéndose la catálisis térmica; a medida que la titulación prosigue la
presencia de ion manganeso (II) cataliza la reducción del ion permanganato y la
reacción se hace casi instantánea, esto se evidencia en la desaparición inmediata
del color púrpura del permanganato. Una vez alcanzado el punto estequiométrico
de la reacción redox; y al caer el primer exceso de la solución titulante de
permanganato, la mezcla de reacción queda teñida de color rosa pálido
determinando de esta manera el punto final de la titulación.
Material a Utilizar
1. Balanza analítica.
2. Espátula.
3. Botella de lavado con agua destilada y desionizada.
5. Vaso de precipitado.
7. Mechero Meker.
8. Rejilla para calentamiento.
9. Trípode
11. Vidrio de reloj
12. Soporte universal
13. Cilindro graduado
14. Bureta
15. Termómetro
Reactivos a Utilizar
1. Na2C2O4 anhidro
2. Solución de KMnO4
3. H2SO4
Nota Importante: Es responsabilidad de
cada estudiante entregar al inicio de cada
práctica, en su cuaderno de laboratorio, una
tabla donde se resuman las siguientes
características de los reactivos arriba
señalados: Fórmula, % Pureza, Densidad,
Código de frases de riesgo (R) y seguridad
(S) para cada reactivo.

Profesor Argenis J. Sánchez Henríquez
83
Procedimiento
1. Pese exactamente, al 0,1 mg, en un pesa sustancia o vidrio de reloj una
masa de 0,1500 g de Na2C2O4 anhidro, previamente secado en estufa a 110 C por
espacio de 1 hora y enfriado en desecador. Registre la masa en su cuaderno de
laboratorio.
Masa de N2C2O4= Y g
2. Mida con un cilindro graduado 250 mL
de agua destilada/desionizada, y con la misma,
transfiera la masa pesada del reactivo Na2C2O4 a
un vaso de precipitado de 500 mL, como muestra
la figura adjunta.
3. Mida con una pipeta serológica o de Mohr, con ayuda de
una pro-pipeta, 7 mL de ácido sulfúrico (H2SO4) concentrado, y
adiciónelo lentamente al vaso de precipitado que contiene a la
solución de oxalato de sodio, como se indica en la figura adjunta.
4. Coloque el vaso de precipitado con la solución en el
trípode, ayúdese con un termómetro para agitar la mezcla y
homogenizarla, mantenga el termómetro dentro de la solución;
de lo contrario perderá parte del agente reductor. La figura
anexa muestra el montaje.
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
9
8
7
10
9
8
7
10
9
8
7
10
9
8
7
10

Profesor Argenis J. Sánchez Henríquez
84
5. Caliente la solución hasta alcanzar 80 C (método McBride), mientras
esto ocurre, cure y enrase una bureta de 50 mL con la solución de permanganato
de potasio (KMnO4) que será estandarizada; cuide de no dejar burbujas en la
punta de la bureta.
6. Adicione desde la bureta 40 mL de la solución de KMnO4 (método
Fowler-Brigth) a la solución reductora del vaso de precipitado mientras agita con el
termómetro esta última, cuide que la temperatura de la solución no baje de 70ºC,
si es necesario calentar nuevamente hágalo. La siguiente figura ilustra el
procedimiento.
(KMnO4)
7. Una vez que el color púrpura haya desaparecido, continúe revolviendo la
mezcla reaccionante y titulando, gota a gota, hasta la aparición del color rosado
pálido que señala el punto final de la titulación. Registre en su cuaderno de
laboratorio el volumen gastado de KMnO4.
Vg (KMnO4) = X mL
8. Calcule con los datos obtenidos y registrados a lo largo de esta actividad,
la concentración de la solución de permanganato de potasio (KMnO4). Rotule el
frasco ámbar, donde conserva esta solución, con la concentración y fechas en que
fue preparada y estandarizada la misma; guárdelo en un lugar oscuro y libre de
polvo.
9
8
7
10
9
8
7
10
9
8
7
10
9
8
7
10

Profesor Argenis J. Sánchez Henríquez
85
(semi-reacción reducción sin balanceo) H2C2O4 CO2
(semi-reacción oxidación sin balanceo) MnO4 Mn2+
3.3. Determinación de calcio en una muestra de alimento.
Fundamentos
La determinación de calcio por volumetría redox es un ejemplo de una
titulación donde la determinación del analito, calcio, no se realiza por reacción
directa entre el agente titulante (KMnO4) y el analito, sino a través de la reacción
del agente titulante y una especie reductora (C2O42 ) que es químicamente
equivalente al calcio. La muestra de alimento se somete a un tratamiento físico-
químico para la destrucción progresiva de la materia orgánica; luego del secado,
carbonización y calcinación de la muestra, se obtienen las cenizas contentivas de
una muestra representativa de los minerales presentes en el alimento. Estas
cenizas son disueltas convenientemente y una alícuota de su solución es
analizada. El calcio presente en la alícuota se precipita como oxalato de calcio
monohidratado (CaC2O4H2O), se hace reaccionar con ácido sulfúrico, y su
producto se titula contra el agente oxidante permanganato de potasio hasta el
punto final.
)(
º
LV
solutoesequivalentnN
PEq
solutogsolutoesequivalentnº
eq
AtMoMMPEq
...
tituladodelesEquivalentntitulantedelesEquivalentn ºº
reductorreductoroxidanteoxidante VNVN

Profesor Argenis J. Sánchez Henríquez
86
Material a Utilizar
1. Pipeta volumétrica 25 mL
2. Botella de lavado con agua destilada y desionizada.
3. Vaso de precipitado.
4. Mechero Meker.
5. Rejilla para calentamiento.
6. Trípode
7. Vidrio de reloj
8. Soporte universal
9. Cilindro graduado
10. Bureta
11. Termómetro
12. Embudo cuantitativo.
13. Soporte para embudo.
14. Papel de filtro cuantitativo.
Reactivos a Utilizar
1. Solución de cenizas
2. Solución de KMnO4 estandarizada
3. H2SO4
4. (NH4)2C2O4 al 3,5 % p/v.
5. Rojo de metilo
6. NH4OH 0,5 N.
Nota Importante: Es responsabilidad de
cada estudiante entregar al inicio de cada
práctica, en su cuaderno de laboratorio, una
tabla donde se resuman las siguientes
características de los reactivos arriba
señalados: Fórmula, % Pureza, Densidad,
Código de frases de riesgo (R) y seguridad
(S) para cada reactivo.
Procedimiento
1. Transfiera, con una pipeta volumétrica y usando pro-pipeta, 25 mL de la
solución de cenizas (muestra) a un vaso de precipitado de 250 mL.
2. Diluya la muestra hasta 50 mL con agua destilada/desionizada.
3. Caliente la solución de la muestra hasta
ebullición, retírela de la llama y agregue
lentamente desde una pipeta 10 mL de NH4C2O4
al 3,5% p/v. Tal como se muestra en la figura
anexa.

Profesor Argenis J. Sánchez Henríquez
87
4. Agregue a la solución de la muestra, aún caliente, dos (2) gotas del
indicador visual ácido-base rojo de metilo al 0,02% p/v; si la solución se torna roja
adicione, gota a gota, desde un frasco gotero, solución de NH4OH 0,5 N hasta el
cambio de color de la solución a amarillo.
5. Caliente nuevamente la solución de la muestra y permita que alcance la
ebullición; luego de 3 minutos retírela de la llama y déjela reposar, tapada con un
vidrio de reloj, durante 12 horas, tal como se muestra en la figura anexa.
6. Separe cuantitativamente el precipitado de oxalato de calcio formado
filtrando la solución madre a través de un papel de filtro cuantitativo (A); lave al
precipitado, consecutivamente, con 3 porciones aproximadas de 7 mL de agua
destilada/desionizada (B). Para ello, lave con cada porción de agua el vaso de
precipitado donde se llevo a cabo la precipitación, y posteriormente vierta el
lavado sobre el precipitado en el papel de filtro (C), asegurándose de transvasar
todo el precipitado al papel de filtro.
(C)
(A)
(B)

Profesor Argenis J. Sánchez Henríquez
88
7. Coloque el papel de filtro (con el
precipitado) en el mismo vaso de
precipitado en el que se realizó la
precipitación; y adicione desde una pipeta
volumétrica 10 mL de solución de H2SO4
al 7,7 % v/v, seguido de 50 mL de agua
destilada/desionizada; la figura adjunta
ilustra el proceso.
8. Caliente la solución hasta alcanzar 80 C (método
McBride), mientras esto ocurre, cure y enrase una bureta de 50
mL con la solución de permanganato de potasio (KMnO4)
estandarizada; cuide de no dejar burbujas en la punta de la
bureta.
9. Retire la solución de la llama una vez alcance la temperatura señalada; y
titúlela con la solución de KMnO4 estandarizada, agitando con el termómetro la
solución, y cuidando que la temperatura no baje de 70ºC. Tal como se muestra en
la siguiente figura.
9
8
7
10
9
8
7
10
9
8
7
10
9
8
7
10
9
8
7
10
9
8
7
10
9
8
7
10

Profesor Argenis J. Sánchez Henríquez
89
10. Continúe revolviendo la mezcla reaccionante y titulando, hasta la
aparición del color rosado pálido que señala el punto final de la titulación. Registre
en su cuaderno de laboratorio el volumen gastado de KMnO4 estándar.
Vg (KMnO4) = X mL
11. Calcule con los datos obtenidos la concentración de Calcio (Ca2+), en
tanto por ciento peso-volumen (%p/v).
(semi-reacción reducción sin balanceo) H2C2O4 CO2
(semi-reacción oxidación sin balanceo) MnO4 Mn2+
4. Post- Laboratorio
Redactar un informe científico de laboratorio siguiendo las pautas
establecidas al principio de este manual, donde se reflejen los resultados y
conocimientos adquiridos en el desarrollo de las actividades realizadas (3.1, 3.2 y
3.3).
5. Cuestionario
1. Defina y ejemplifique:
a. Agente Oxidante
b. Agente Reductor
2. ¿Qué condiciones deben cumplir los agentes oxidantes, o reductores,
para ser seleccionados para una volumetría redox?
3. ¿Qué tipo de indicadores emplea la volumetría redox para señalar el
punto estequiométrico en una titulación?
100/%22
muestra
Caoxidanteoxidante
V
PEqVNCavp
eq
AtMPEq Ca
Ca
2
2
.

Profesor Argenis J. Sánchez Henríquez
90
4. Escriba algunas ventajas y desventajas del uso de KMnO4 como agente
titulante.
5. Escriba los dos equilibrios químicos redox principales que puede tener el
ion permanganato (MnO4 ).
6. Describa la preparación de 1 L de solución de KMnO4 0,25 N, que será
empleada como agente titulante en medio ácido. El reactivo tiene una pureza del
99% y su masa molar es de 158,04 g/mol.
7. Balancee el siguiente sistema redox global en medio ácido:
MnO4 + C2O42 Mn2+ + CO2
7. Referencias Bibliográficas
Skoog Douglas A., Donald M. West, James F. Holler y Stanley R. Crouch.
(2001). Química Analítica. (7a Ed.). McGraw-Hill. México D.F. (México).
Skoog Douglas A. y Donald M. West. (1970). Introducción a la Química
Analítica. Editorial Reverté. Barcelona (España).
Alexéiev Víctor N. (1976). Análisis Cuantitativo. Trad. Liminík E. Editorial
Mir. Moscú (Rusia).
COMISIÓN VENEZOLANA DE NORMAS INDUSTRIALES. (1982).
Determinación de Calcio. Alimentos. Fondo Normas. COVENIN 1178-82. Caracas
(Venezuela).
Vogel Arthur I. (1960). Química Analítica Cuantitativa (Teoría y Práctica).
(2a Ed.). Editorial Kapelusz. Buenos Aires (Argentina).

Profesor Argenis J. Sánchez Henríquez
91
IV.3. VOLUMETRÍA DE PRECIPITACIÓN
La volumetría de precipitación involucra a todos aquellos métodos de
titulación que se fundamentan en las reacciones químicas de formación de
compuestos poco solubles, precipitados. A pesar que se conocen muchos
sistemas químicos capaces de reaccionar para producir precipitados, la volumetría
de precipitación ha encontrado menor aplicación que la volumetría de
neutralización ácido-base y redox. Esto obedece, principalmente, a las
condiciones que deben satisfacer estos sistemas químicos reactivos para poder
ser considerados punto de partida de una volumetría de precipitación. Las
condiciones mínimas que deben cumplir estos sistemas son:
1.- El precipitado formado en el curso de reacción debe ser prácticamente
insoluble, es decir, debe tener una solubilidad molar (mol/L) baja.
2.- La reacción de precipitación debe ocurrir con suficiente rapidez, de esta
manera se mejora el reconocimiento del punto final. Además, si son rápidos; tanto
la velocidad de reacción entre el agente precipitante y la especie química que se
desea precipitar, así como el equilibrio de solubilidad entre el precipitado y los
iones en solución que lo conforman, se podrá seguir el progreso de la titulación de
manera más exacta.
3.- Los resultados de la titulación no deben ser afectados de manera notoria
por fenómenos de coprecipitación (adsorción, inclusión iso y no isomórfica,
oclusión). Es decir, la reacción de precipitación debe transcurrir de forma
cuantitativa y con una relación estequiométrica bien definida entre el agente
precipitante y la especie a ser precipitada; de esta manera se puede tener la
certeza de que el sólido obtenido tiene una pureza muy cercana al 100%.
4.- Por último, debe existir una forma sencilla y cómoda de establecer el
punto estequiométrico en la reacción entre el agente precipitante y la especie que
se precipita.
Al igual que en las volumetrías de neutralización ácido-base y de oxidación-
reducción, las volumetrías de precipitación se pueden clasificar en base a la

Profesor Argenis J. Sánchez Henríquez
92
especie química empleada como agente titulante o agente precipitante; así es
como encontramos, por ejemplo, entre otras:
Argentometrías: Volumetrías de precipitación que emplean al catión plata
(Ag+) como agente precipitante. Este ion se incorpora a la solución generalmente
como una sal de nitrato de plata (AgNO3).
Mercurometrías: Volumetrías de precipitación que emplean a los cationes
Hg+1 y Hg+2 como agentes precipitantes. Estos iones son introducidos al medio de
reacción en forma de sales de nitrato mercurioso [Hg2(NO3)2] y cloruro mercúrico
(HgCl2), respectivamente.
La tabla que se presenta a continuación muestra un resumen de los
métodos de precipitación más empleados y sus aplicaciones.
Tabla 5. Métodos Volumétricos de Precipitación (Fuente: Skoog and West, 1970)
Reactivo
Precipitante
Elemento
Determinado
Indicación Punto
Final Notas
AgNO3 AsO43, Br , I ,
CNO , SCN
Volhard – Alumbre
Férrico
----------------------
AgNO3 CrO4
2, S
2, Cl ,
PO43
CO32, CN , C2O4
2
Volhard – Alumbre
Férrico
Es preciso separar la sal de
plata precipitada antes de
valorar por retroceso el exceso
de plata
AgNO3 BH4 Volhard – Modificado
Fundamento: valoración del
exceso de plata después de:
BH4 + 8Ag+ + 8OH
8Ag + H2BO3 + 5H2O
AgNO3 Epóxido Volhard – Alumbre
Férrico
Fundamento: valoración del
exceso de Cl después de
hidrohalogenación
AgNO3 K+
Volhard – Modificado
Fundamento: precipitación de
K+ con exceso conocido de
B(C6H5)4 , adición de un
exceso de Ag+ que precipita Ag
B(C6H5)4 y valoración por
retroceso de este exceso.
AgNO3 Cl , Br Mohr – K2CrO4 ---------------------
AgNO3 I Punto claro ---------------------
Profesor Argenis J. Sánchez Henríquez
93
Continuación tabla 5.
AgNO3 SeO3
2, Br , I ,
Cl
Fajans – Indicadores
de Adsorción ------------------------
AgNO3 V(OH)4
+, ácidos
grasos, mercaptanos Electroanalítico Valoración directa con Ag
+
K4Fe(CN)6 Zn, In, Ga, Ag, Co,
Ni, Mn, Hg(II)
Difenilamina
electroanalítico o iodo-
almidón
electroanalítico
Valoración inversa también
posible.
Hg2(NO3)2 Fe(CN)63, Br , Cl
Difenilcarbazona
electroanalítico Valorar en alcohol al 20%
HgCl2 I Yodo-almidón
Pb(NO3)2 SO42
Fajans – Indicadores
de Adsorción Indicador: Eritrosina B
Pb(OAc)2 PO43, C2O4
2
Fajans – Indicadores
de Adsorción
Dibromofluoresceína para
fosfatos; fluoresceína para
oxalatos
Th(NO3)4 F , C2O42 Fajans – Indicadores
de Adsorción Alizarinsulfato sódico para F y
rojo de alizarina para C2O42.
Por los momentos, son de nuestro especial interés los métodos
argentométricos (AgNO3), por ser éstos los más utilizados en la determinación de
haluros en relación a los demás métodos de precipitación.
Las curvas de titulación en volumetría de precipitación son una
representación gráfica de la concentración de la especie titulada o del titulante,
expresada en concentración molar [A] o pA, en función del volumen gastado de la
especie titulante (figura 8). Estas curvas al igual que las curvas de otros métodos
volumétricos nos proporcionan, a través de cálculos teóricos o datos
experimentales, las variaciones que experimentan las concentraciones de las
especies reaccionantes durante el transcurso de la titulación; sirviendo de esta
manera como un herramienta valiosa para determinar si una especie química es
titulable, o no, a través de un método volumétrico particular. En la volumetría de
precipitación existen dos factores importantes que afectan la forma de la curva de
titulación. El primero de ellos es la concentración de las especies reaccionantes,
mientras mayor sea la concentración de los reaccionantes, mayor será el salto
reflejado en la curva de titulación en el eje relacionado con la concentración ([A] o

Profesor Argenis J. Sánchez Henríquez
94
pA) de la especie medida (figura 11-A). El segundo factor se refiere a la
cuantitatividad de la reacción de precipitación; es decir, que tan desplazado se
encuentra el equilibrio hacia la formación del precipitado. En lo referente a este
factor encontramos un comportamiento inverso, mientras más soluble es el
precipitado formado (Kps muy grande) menor es el salto de la curva de titulación
en las cercanías del punto de equivalencia. (figura 11-B).
(A) (B)
Figura 11. Factores que afectan la curva de titulación en volumetría de precipitación. (A)Curva teórica de valoración del ion cloruro a diferentes concentraciones con ion
plata. (B)Curva de valoración de varios aniones con ion plata. Representación de los valores de pAg , agregado a 50 mL de una solución en la que la concentración del anión es 0,1 M. (Fuente: del Skoog D. and West D., 1970)
Existen varios métodos para establecer el punto final en una volumetría de
precipitación, todos ellos tienen como objeto señalar, en el mejor de los casos,
exactamente el punto de equivalencia teórico entre los reaccionantes, o al menos
el punto más cercano a éste. El primer método para establecer el punto final en
volumetría de precipitación, fue establecido por Gay-Lussac (1832); el mismo se
conoce como el método de igual enturbiamiento. Este método es muy exacto, pero
requieren de mucha pericia y laboriosidad; por esta razón hoy día no es empleado,
aunque resulta una alternativa para aquellos laboratorios que no disponen de
reactivos específicos (indicadores).

Profesor Argenis J. Sánchez Henríquez
95
Luego tenemos el uso de indicadores externos, los cuales se emplean fuera
de la solución estudiada, en placas de toque; estos indicadores no introducen
errores significativos en la titulación, aunque sólo se recomienda su uso en
aquellos casos en los que no se disponga de indicadores internos.
Por otra parte, se encuentra los indicadores internos; éstos son
compuestos orgánicos e inorgánicos que en solución pueden formar un complejo
coloreado soluble que imparte color a la solución, o un precipitado coloreado que
se produce con el primer exceso de la solución titulante. Estos indicadores,
también pueden presentarse en forma de un complejo coloreado que se adsorbe
sobre la superficie del precipitado coloidal principal, por efecto de atracción
electrostática impartiendo de esta manera un color al precipitado.
Si la reacción de precipitación va acompañada del consumo o liberación de
iones hidronios o hidróxidos, o en los casos en que el agente precipitante es un
ácido o una base, el punto final puede detectarse a través de un indicador ácido-
base. Análogamente, en los casos en los que el agente precipitante posea
propiedades oxidantes o reductoras, se pueden emplear indicadores redox.
Todos los métodos mencionados hasta el momento pertenecen a los
procedimientos visuales para la determinación del punto final. Por último, y no
menos importante, se ha encontrado el uso de potenciómetros, conductímetros, y
amperímetros para determinar el punto final de una titulación de precipitación.
Enmarcados en el análisis argentométrico, se estudiarán dos métodos para
la determinación de punto final con indicadores visuales internos: Mohr y Volhard.

Profesor Argenis J. Sánchez Henríquez
96
PRÁCTICA Nº 4
DETERMINACIÓN DE CLORUROS POR EL MÉTODO DE MOHR
1. Objetivo General
Establecer la concentración de cloruros solubles presentes en una
muestra sintética.
2. Objetivos Específicos
2.1. Desarrollar las destrezas del estudiante en las distintas etapas
involucradas en la preparación de una solución tipo patrón primario de nitrato de
plata (AgNO3).
2.2. Determinar la concentración de cloruros solubles, en tanto por ciento
peso-volumen (%p/v), por argentometría con indicador visual interno de K2CrO4.
2.3. Redactar un informe científico de laboratorio siguiendo las pautas
establecidas al principio de este manual, donde se reflejen los resultados y
conocimientos adquiridos en el transcurso de la presente práctica.
3. Actividades a Desarrollar
Antes de comenzar a desarrollar tus actividades de laboratorio mantén
siempre presente las normas de seguridad que se encuentran al principio de este
manual. Recuerda que la seguridad dentro del laboratorio es responsabilidad de
todos.
3.1. Preparación de una solución tipo patrón primario de nitrato de
plata (AgNO3) 0,1N.
Fundamento
El nitrato de plata es un reactivo que puede adquirirse con un alto grado de
pureza (99,8-100,5%), además de encontrarse en ampollas comerciales listas
para su dilución, a 1 o 4 L, y obtener concentraciones que oscilan entre 0,1 y 0,25
N. Este reactivo por reunir las características de un patrón primario debe

Profesor Argenis J. Sánchez Henríquez
97
prepararse, a partir de su sal sólida, a través del método directo. Considerando el
costo del reactivo, y por supuesto, la exactitud de los resultados analíticos, se
deben tomar ciertas medidas preventivas para conservar en buenas condiciones,
tanto el reactivo sólido como sus soluciones; ya que ambos son susceptibles a la
luz, el polvo y las materias orgánicas, produciéndose como resultado de su
interacción la reducción del ion plata (Ag+) a plata metálica (Ag ).
Material a Utilizar
1. Balanza analítica.
2. Espátula.
3. Pesa-sustancia.
3. Matraz Volumétrico
4. Botella de lavado con agua destilada y desionizada.
5. Vaso de precipitado.
6. Vidrio de reloj
7. ganchos de vidrio
8. Estufa
Reactivos a Utilizar
1. Hidróxido de sodio
2. Ácido clorhídrico Titrisol
3. Fenolftaleína
Nota Importante: Es responsabilidad de
cada estudiante entregar al inicio de cada
práctica, en su cuaderno de laboratorio, una
tabla donde se resuman las siguientes
características de los reactivos arriba
señalados: Fórmula, % Pureza, Densidad,
Código de frases de riesgo (R) y seguridad
(S) para cada reactivo.
Procedimiento
1. Pese ligeramente por exceso en un pesa-sustancia, limpio y seco, una
masa aproximada del reactivo (AgNO3) que le permita obtener, en 1 L de solución,
una concentración de AgNO3 0,1N. Para ello, debe realizar el cálculo teórico de la
masa de AgNO3 necesaria.
)(
º
LV
solutoesequivalentnN
PEq
solutogsolutoesequivalentnº
eq
AtMoMMPEq
...

Profesor Argenis J. Sánchez Henríquez
98
2. Coloque el pesa-sustancia, contentivo del reactivo, dentro de un vaso de
precipitado y tápelo con un vidrio de reloj, colocando tres ganchos de vidrio para
permitir la salida de vapores. Tal como se ilustra en la figura mostrada a
continuación:
3. Introduzca el conjunto (vaso de precipitado-pesa-sustancia) en una
estufa pre-calentada a 110ºC, por periodo de 1 hora.
4. Saque el conjunto de la estufa, tomando las medidas apropiadas para
manipulación de objetos calientes, y déjelo enfriar por varios minutos; luego
introduzca el pesa-sustancia con el reactivo en un desecador por espacio de 45
minutos.
5. Pese, con precisión de 1 mg, en un pesa-sustancia la masa exacta
(según cálculo anterior), que requiere para la preparación de la solución patrón
primario de AgNO3 0,1 N.
6. Transfiera la masa pesada a un matraz aforado de
1 L, con ayuda de un embudo cuantitativo de
cuello largo, procurando no tocar el pesa-
sustancia con los dedos desnudos. (Utilice
tiras de papel, ver ilustración)

Profesor Argenis J. Sánchez Henríquez
99
7. Disuelva el AgNO3 en el embudo, empleando para ello una
piseta con agua destilada/desionizada. Lave bien el embudo
antes de retirarlo, así como el cuello del matraz; tal como se
muestra en la figura adjunta.
8. Disuelva completamente la masa de AgNO3 transferida antes de enrasar;
para ello revuelva bien la solución y adicione, sólo, el volumen mínimo requerido
de agua destilada/desionizada. Luego proceda a enrasar y homogenizar bien la
solución, por inversión del matraz aforado. Y finalmente, transfiera la solución a un
frasco limpio rotulado, para su almacenamiento en un lugar protegida de la luz.
3.2. Determinación de la concentración de cloruros solubles en una
muestra sintética.
Fundamentos
El método de Mohr es una excelente ejemplificación de una argentometría
en la cual el punto final de la titulación es señalado por la aparición, en el seno de
la mezcla reaccionante, de un segundo precipitado coloreado. Este método se
fundamenta en la determinación directa de iones cloruros, a través del uso de
nitrato de plata como agente precipitante (titulante); la reacción cuantitativa
obedece a la siguiente estequiometría:
Ag + + Cl AgCl (s) (color blanco)
El punto final en el método de Mohr se observa por adición del indicador
interno visual cromato de potasio (K2CrO4); este indicador reacciona, una vez

Profesor Argenis J. Sánchez Henríquez
100
precipitados todos los iones cloruros, con el primer exceso de nitrato de plata para
formar un segundo precipitado de cromato de plata de color pardo – rojizo, según
la estequiometría:
2 Ag + + CrO4
2 Ag2CrO4 (s) (color pardo – rojizo)
En el método de Mohr existen varios factores importantes que pueden
afectar la veracidad de los resultados, estos son:
1.- El pH de la muestra a analizar
Si la muestra es ácida (pH<6,5), el punto final de la valoración puede
verse afectado por la formación de HCrO4 , según la siguiente reacción:
2 H+ + 2 CrO42 2 HCrO4
En caso de tener una muestra ácida, debe neutralizarse con una
solución de carbonato de calcio, bicarbonato de sodio o bórax (libres de cloruros).
Si la muestra es fuertemente básica (pH > 10,5), se produce hidróxido de plata con
posterior formación de su oxido (Ag2O) de color pardo oscuro, según la siguiente
estequiometría:
2 Ag + + 2 OH 2 AgOH(s) Ag2O(s) + H2O
En caso de tener una muestra alcalina, debe neutralizarse con una
solución de ácido nítrico libre de cloruros, utilizando un indicador visual ácido-base
como la fenolftaleína o el rojo de fenol, para señalar la neutralización.
2.- Cantidad y concentración del indicador interno
La concentración de iones cromato, CrO42 , en la solución que se titula
debe estar comprendida entre 3x10 3 y 5x10 3 mol/L, para un volumen final de
solución titulada comprendido entre 50 y 100 mL; para asegurar de esta forma el
rango de concentración requerida del ion cromato en el punto final.

Profesor Argenis J. Sánchez Henríquez
101
3.- La presencia de sales y cationes
En la determinación de cloruros por el método de Mohr la presencia de
sales como: fosfatos, sulfitos, y arseniatos interfieren en la titulación; ya que
reaccionan con el nitrato de plata, para formar precipitados, produciendo un mayor
gasto del titulante.
De igual manera, la presencia de cationes como: Ba2+, Pb2+ y Bi3+,
interfieren; ya que forman precipitados con el ion cromato del indicador,
requiriéndose mayor cantidad del titulante para señalar el punto final.
4. Temperatura
Aumentos de temperatura solubilizan al precipitado Ag2CrO4, lo cual
impide la observación del punto final una vez alcanzado el punto estequiométrico;
esto trae como consecuencia resultados altos en la determinación.
Material a Utilizar
1. Pipeta volumétrica 10 mL
2. Botella de lavado con agua destilada y desionizada.
3. Vaso de precipitado.
4. Soporte universal
5. Bureta
6. Matraz Erlenmeyer
7. Cilindro graduado.
Reactivos a Utilizar
1. Solución patrón primario de AgNO3 0,1 N.
2. Solución de HNO3 0,1 N.
3. Solución de NaHCO3 0,1 N.
4. Solución indicadora de K2CrO4 al 5% p/v.
5. Solución indicadora ácido-base de fenolftaleína.
6. CaCO3
Nota Importante: Es responsabilidad de
cada estudiante entregar al inicio de cada
práctica, en su cuaderno de laboratorio, una
tabla donde se resuman las siguientes
características de los reactivos arriba
señalados: Fórmula, % Pureza, Densidad,
Código de frases de riesgo (R) y seguridad
(S) para cada reactivo.

Profesor Argenis J. Sánchez Henríquez
102
Procedimiento
1. Coloque en dos (2) matraces Erlenmeyer 80 mL de agua
destilada/desionizada y vierta en cada uno de ellos 1 mL de la solución indicadora
(K2CrO4) al 5 % p/v.
2. Introduzca en cada matraz Erlenmeyer 300 mg de CaCO3 sólido
pulverizado, esto simulará el precipitado de AgCl . Conserve una de estas
soluciones como testigo para comparar el color antes del punto final.
3. Cure la bureta, limpia, con una pequeña porción (5mL) de solución de
AgNO3 estándar. Enrase la misma hasta 2 mL después de la última graduación del
instrumento. Cuide de no dejar burbujas de aire atrapadas en la punta de la
bureta.
4. Titule con la solución estándar el segundo matraz Erlenmeyer, gota a
gota, hasta la aparición del precipitado color rojo-pardo de Ag2CrO4. Registre en
su cuaderno de laboratorio este volumen de solución titulante de AgNO3 estándar,
como volumen del blanco o testigo. Conserve la solución como referencia del color
para el punto final de la titulación. Recuerde emplear la técnica correcta para la
titulación, observe la figura.
Vblanco (AgNO3) = X mL
5. Cure una pipeta volumétrica de 10 mL con una pequeña porción (3mL)
de la solución de muestra, posteriormente transfiera 10 mL de la muestra sintética
a los tres (3) matraces Erlenmeyer limpios de 250 mL.

Profesor Argenis J. Sánchez Henríquez
103
6. Determine, cualitativamente, el pH de las muestras. Para ello, agregue 2
gotas de fenolftaleína, y dependiendo de su color realice la neutralización de la
siguiente forma: Con solución 0,1 N de bicarbonato de sodio (NaHCO3) si la
muestra es ácida, o con solución de ácido nítrico (HNO3) 0,1 N si la muestra es
alcalina.
7. Agregue a cada matraz, una vez ajustado el pH de las muestras, 1 ml de
cromato de potasio (K2CrO4) al 5% p/v.
8. Enrase la bureta con la solución estándar de AgNO3 0,1 N. Cuide de no
dejar burbujas de aire atrapadas en la punta de la bureta.
9. Titule con la solución estándar y agitación
permanente, cada muestra, hasta la aparición del precipitado
color rojo-pardo de Ag2CrO4. Registre en su cuaderno de
laboratorio estos volúmenes de solución titulante de AgNO3
estándar. (Puede colocar un fondo blanco, hoja de papel,
para ver con mayor claridad el cambio de color de la
solución).
V1 (AgNO3) = X mL
V2 (AgNO3) = Y mL
V3 (AgNO3) = Z mL
10. Calcule, con los volúmenes obtenidos, la concentración de cloruros
solubles presente en la muestra analizada, expresando esta concentración en
tanto por ciento peso-volumen (%p/v).

Profesor Argenis J. Sánchez Henríquez
104
4. Post-Laboratorio
Redactar un informe científico de laboratorio siguiendo las pautas
establecidas al principio de este manual, donde se reflejen los resultados y
conocimientos adquiridos en el desarrollo de las actividades realizadas (3.1 y 3.2).
5. Cuestionario
1. ¿Qué es una argentometría?
2. ¿Cuáles son las condiciones mínimo que deben cumplir los sistemas
reactivos para ser empleados en volumetría de precipitación?
3. ¿Qué factores afectan la curva de titulación en una volumetría de
precipitación? Explique.
4. Describa los diferentes métodos empleados para señalar el punto
estequiométrico en una volumetría de precipitación.
5. Describa la preparación de 250 mL de una solución de K2CrO4 al 5% p/v.
6. Describa la preparación de 200 mL de una solución 0,2 N en AgNO3, a
partir del reactivo sólido con una pureza de 99,7% y una masa molar de 169,87
g/mol.
7. ¿En qué consiste el método de Mohr?
8. ¿Qué factores pueden afectar los resultados en el método de Mohr?
Explique.
100)(
/% 33
muestra
ClblancoAgNOAgNO
V
PEqVVNClvp
eq
AtMPEq Cl
Cl
.
tituladodelesEquivalentntitulantedelesEquivalentn ºº

Profesor Argenis J. Sánchez Henríquez
105
6. Referencias Bibliográfícas
Skoog Douglas A., Donald M. West, James F. Holler y Stanley R. Crouch.
(2001). Química Analítica. (7a Ed.). McGraw-Hill. México D.F. (México).
Skoog Douglas A. y Donald M. West. (1970). Introducción a la Química
Analítica. Editorial Reverté. Barcelona (España).
Vogel Arthur I. (1960). Química Analítica Cuantitativa (Teoría y Práctica).
(2a Ed.). Editorial Kapelusz. Buenos Aires (Argentina).
Ayres Gilbert H. (1970). Análisis Químico Cuantitativo. (2a Ed.). Ediciones
del Castillo. Madrid (España).
Fischer Robert B. y Dennos G. Peters. (1970). Análisis Químico
Cuantitativo. (3a Ed.). Editorial Interamericana. Trad. Toral M. Teresa. México D.
F. (México).

Profesor Argenis J. Sánchez Henríquez
106
PRÁCTICA Nº 5
DETERMINACIÓN INDIRECTA DE CLORUROS POR EL MÉTODO DE
VOLHARD
1. Objetivo General
Establecer la concentración de cloruros solubles presentes en una
muestra sintética.
2. Objetivos Específicos
2.1. Desarrollar las destrezas del estudiante en las distintas etapas
involucradas en la preparación y estandarización de una solución tipo patrón de
tiocianato de potasio (KSCN).
2.2. Determinar la concentración de cloruros solubles, en tanto por ciento
peso-volumen (%p/v), por argentometría con indicador visual interno de alumbre
férrico [Fe(NH4)(SO4)212H2O].
2.3. Redactar un informe científico de laboratorio siguiendo las pautas
establecidas al principio de este manual, donde se reflejen los resultados y
conocimientos adquiridos en el transcurso de la presente práctica.
3. Actividades a Desarrollar
Antes de comenzar a desarrollar tus actividades de laboratorio mantén
siempre presente las normas de seguridad que se encuentran al principio de este
manual. Recuerda que la seguridad dentro del laboratorio es responsabilidad de
todos.

Profesor Argenis J. Sánchez Henríquez
107
3.1. Preparación y estandarización de una solución de tiocianato de
potasio (KSCN) 0,1N.
Fundamentos
Por su naturaleza el tiocianato de potasio no puede considerarse un
reactivo tipo patrón primario; por lo cual, las soluciones preparadas de este
reactivo por el método indirecto, deben ser estandarizadas a través del uso de un
reactivo patrón primario, como el nitrato de plata (AgNO3).
Material a Utilizar
1. Balanza analítica.
2. Espátula.
3. Vaso de precipitado 250 mL
3. Matraz Volumétrico 1 L
4. Botella de lavado con agua destilada y desionizada.
5. Varilla de vidrio.
6. Embudo cuantitativo.
7. Bureta 25 mL.
8. Matraz Erlenmeyer 250 mL
9. Pipeta volumétrica.
10. Pro-pipeta.
11. Soporte Universal.
12. Soporte para bureta.
Reactivos a Utilizar
1. KSCN grado analítico
2. AgNO3 0,1 N
3. Fe(NH4)(SO4)212H2O solución Saturada en HNO3 1M.
4. HNO3 1 y 6 M.
Nota Importante: Es responsabilidad de
cada estudiante entregar al inicio de cada
práctica, en su cuaderno de laboratorio, una
tabla donde se resuman las siguientes
características de los reactivos arriba
señalados: Fórmula, % Pureza, Densidad,
Código de frases de riesgo (R) y seguridad
(S) para cada reactivo.
Procedimiento
1. Pese en un vaso de precipitado, limpio y seco, una masa aproximada del
reactivo (KSCN) que le permita obtener, en 1 L de solución, una concentración de
KSCN 0,1 N. Para ello, debe realizar previamente el cálculo teórico de la masa
requerida de KSCN.

Profesor Argenis J. Sánchez Henríquez
108
2. Disuelva el reactivo, en el interior del vaso de precipitado, con una
porción de agua destilada/desionizada, y transfiéralo posteriormente al matraz
volumétrico empleando para ello una varilla de vidrio y un embudo cuantitativo
cuello largo. Lave bien, el vaso de precipitado, la varilla y el embudo antes de
retirarlo.
3. Afore la solución del matraz, tápelo y homogenice por agitación e
inversión del mismo.
4. Prepare una solución saturada de Fe(NH4)(SO4)212H2O en HNO3 1M.
Para ello, prepare 50 mL de HNO3 1M, y disuelva la máxima cantidad posible de la
sal férrica. Luego filtre la solución a través de papel cuantitativo y guárdela hasta
el momento de uso.
5. Prepare 100 mL de solución de HNO3 6 M.
)(
º
LV
solutoesequivalentnN
PEq
solutogsolutoesequivalentnº
eq
AtMoMMPEq
...

Profesor Argenis J. Sánchez Henríquez
109
6. Estandarice por triplicado la solución de KSCN preparada. Para ello,
tome tres alícuotas de 15 mL, cada una, de solución 0,1 N de AgNO3 tipo patrón
primario y transfiéralas a tres (3) matraces Erlenmeyer. (La preparación de la
solución tipo patrón primario de AgNO3 0,1 N se explica en la práctica Nº 4,
actividad 3.1).
7. Cure la bureta con una pequeña porción (10 mL) de solución de KSCN
preparada y enrase la misma. Cuide de no dejar burbujas de aire atrapadas en la
punta de la bureta.
8. Adicione a cada matraz Erlenmeyer, sólo en el momento en que se
disponga a titular, 3 mL de HNO3 6 M, 1 mL de la solución indicadora saturada de
alumbre férrico y 10 mL de agua destilada/desionizada.
9. Titule la solución manteniendo una agitación continua,
hasta el momento en que un color rojo pardo débil aparezca de
forma permanente; este instante señala el punto final. Registre
en su cuaderno de laboratorio el volumen gastado de la
solución de KSCN. Recuerde que tiene que repetir este
procedimiento de titulación dos veces más.
Vg (KSCN) = a mL, Vg (KSCN) = b mL y Vg(KSCN) = c mL
10. Calcule la concentración de la solución de KSCN que preparó, a través
de los volúmenes gastados de solución de KSCN, y la concentración y volumen de
la solución tipo patrón primario. Rotule el recipiente que contendrá a la solución de

Profesor Argenis J. Sánchez Henríquez
110
KSCN, la cual servirá como solución tipo patrón secundario o estándar para las
demás actividades.
3.2. Determinación de la concentración de cloruros solubles en una
muestra sintética.
Fundamentos
El método de Volhard se fundamenta en el análisis volumétrico indirecto de
haluros solubles, en este caso particular cloruros. La titulación emplea la
formación de complejos solubles estables de Fe3+ intensamente coloreados, para
destacar de forma apropiada el punto final de la misma; de estos complejos se
reconoce como principal responsable del color rojo pardo que señala el punto final
al [Fe(SCN)]2+. La aplicación que tiene el método de Volhard en la determinación
de haluros, en comparación con Mohr, es más amplia; esto se debe
principalmente a que el primero está menos interferido que el segundo. En este
sentido, las restricciones relacionadas con la acidez del medio reaccionante en
Mohr (6,5<pH<10), en Volhard quedan simplificadas por la presencia de un medio
netamente ácido que lejos de perjudicar a la reacción principal (titulado-titulante),
la favorece con resultados más precisos al evitar la hidrólisis del Fe3+. Por otra
parte, sólo interfieren en la titulación sales de mercurio, fuertes oxidantes y F ; ya
que los primeros precipitan y oxidan al ion SCN , respectivamente, y el último
quela o acompleja al Fe3+. Durante el desarrollo del método de Volhard se
formaran dos precipitados con distintas características de solubilidad; el primero
de ellos, entre el agente precipitante AgNO3 y el analito (ion cloruro), obedece a la
siguiente estequiometría de reacción:
tituladodelesEquivalentntitulantedelesEquivalentn ºº
oprecipitadoprecipitadteprecipiteprecipi VNVN tantan

Profesor Argenis J. Sánchez Henríquez
111
Ag+ + Cl AgCl (s) (color blanco)
Esta precipitación se lleva a cabo en presencia de un exceso del agente
precipitante AgNO3, para asegurar la separación cuantitativa del ion cloruro del
seno de la solución y disminuir la solubilidad de precipitado (AgCl(s)). El segundo
precipitado, es el producto de la reacción entre el exceso del primer agente
precipitante (AgNO3) y la solución estándar de KSCN, la cual contiene al segundo
ion precipitante, SCN . Esta reacción sigue la estequiometría:
Ag+exceso + SCN AgSCN (s) (color blanco)
Este segundo precipitado, tiene la particularidad de presentar una menor
solubilidad que el primero (KpsAgSCN = 1x10 12, KpsAgCl = 1x10 10). Esto hecho
incide en dos aspectos importantes en la titulación, los cuales pueden conllevar a
errores de titulación por exceso o sobre-titulación de la solución. El primer
aspecto, está relacionado con la etapa de retro titulación o titulación inversa del
exceso del primer agente precipitante (AgNO3) con el segundo ion precipitante
SCN , en presencia del precipitado AgCl (s). En función del valor de sus Kps, se
puede predecir que habrá metátesis (conversión) del precipitado AgCl (s) por la
adición del ion SCN al medio de reacción. Esto obedece a la siguiente
estequiometría:
AgCl (s) + SCN AgSCN (s) + Cl
El segundo aspecto, compromete la nitidez del punto final en la retro
titulación, ya que el primer exceso de ion SCN que reacciona con el indicador
Fe3+ para formar el complejo que señala la culminación de la titulación, se ve
comprometido con la formación del precipitado AgSCN (s), según la
estequiometría:
[Fe(SCN)]2+ + AgCl (s) Fe3+ + Cl + AgSCN (s)
Estos inconvenientes pueden evitarse filtrando al precipitado de AgCl (s),
previamente a la titulación del exceso de Ag+ con SCN .

Profesor Argenis J. Sánchez Henríquez
112
Material a Utilizar
1. Pipeta volumétrica.
2. Botella de lavado con agua destilada y desionizada.
3. Vaso de precipitado.
4. Soporte universal.
5. Soporte para bureta.
6. Bureta.
7. Matraz Erlenmeyer.
8. Embudo cuantitativo cuello largo.
9. Papel de filtro cuantitativo.
10. Varilla de vidrio.
11. Soporte para embudo.
Reactivos a Utilizar
1. Solución tipo patrón primario de AgNO3 0,1 N.
2. Solución de KSCN 0,1 N.
3. Solución de HNO3 1 y 6 N.
4. Solución saturada indicadora de Fe(NH4)(SO4)212H2O
Nota Importante: Es responsabilidad de
cada estudiante entregar al inicio de cada
práctica, en su cuaderno de laboratorio, una
tabla donde se resuman las siguientes
características de los reactivos arriba
señalados: Fórmula, % Pureza, Densidad,
Código de frases de riesgo (R) y seguridad
(S) para cada reactivo.
Procedimiento
1. Cure una pipeta volumétrica de 10 mL con una
pequeña porción (3mL) de la solución de muestra,
posteriormente transfiera 10 mL de la muestra sintética a los
tres (3) vasos de precipitado limpios de 250 mL.
2. Adicione 5 mL de HNO3 6 N a cada vaso de
precipitado.
3. Adicione lentamente, desde una bureta, a cada vaso de precipitado 50
mL de la solución tipo patrón primario de AgNO3 0,1 N. Mantenga el vaso de
precipitado en movimiento circular suave mientras realiza la adición.

Profesor Argenis J. Sánchez Henríquez
113
4. Prepare un sistema de filtración por gravedad y
emplee como recipiente recolector un matraz Erlenmeyer
limpio de 500 mL. Debe disponer de 1 matraz Erlenmeyer y
1 embudo para cada muestra.
5. Filtre el precipitado coagulado de AgCl(s), recordando
las distintas etapas involucradas en la filtración: decantación,
lavado y transvase. Deben ser realizados 5 lavados con
porciones de 10 mL de la solución de HNO3 1 N. Cada lavado
debe pasarse por el papel de filtro y ser recolectado en el
mismo matraz Erlenmeyer, a fin de recuperar cuantitativamente
el exceso adicionado de AgNO3.
6. Adicione posteriormente, al líquido filtrado y los lavados, 4 mL de la
solución indicadora interna de alumbre férrico y homogenice removiendo
suavemente el matraz.
7. Titule esta solución con el KSCN 0,1 N estandarizado en
la actividad anterior. Recuerde mantener una agitación continua,
el color rojo pardo desaparecerá al principio rápidamente,
continúe la titulación hasta que observe que el color desaparece
relativamente despacio; en ese instante habrá alcanzado el punto
final. Registre en su cuaderno de laboratorio el volumen gastado
de la solución de KSCN. Recuerde hacerlo por triplicado
Vg (KSCN) = a mL, Vg (KSCN) = b mL y Vg(KSCN) = c mL

Profesor Argenis J. Sánchez Henríquez
114
8. Calcule la concentración de cloruros presente en la muestra, a través de
los volúmenes gastados de solución de KSCN estándar, el volumen adicionado de
la solución tipo patrón de AgNO3 y el volumen de la muestra utilizado. Exprese los
resultados en tanto por ciento peso-volumen (%p/v) de cloruros en la muestra.
4. Post-Laboratorio
Redactar un informe científico de laboratorio siguiendo las pautas
establecidas al principio de este manual, donde se reflejen los resultados y
conocimientos adquiridos en el desarrollo de las actividades realizadas (3.1 y 3.2).
5. Cuestionario
1. Describa el fundamento del método de Volhard.
2. ¿Cuáles son las ventajas que presenta el método de Volhard con
respecto al método de Mohr para la determinación de haluros?
3. ¿Qué indicador emplea el método de Volhard para señalar el punto
estequiométrico?
4. ¿Qué complicación surge en el método de Volhard, si se lleva a cabo la
titulación inversa del ion Ag+, en exceso, en presencia del precipitado AgCl?
5. Describa la preparación de 500 mL de una solución de KSCN 0,25 N, a
partir del reactivo sólido con una pureza 99% y una masa molar 97,18 g/mol.
AgSCNAgCltotales AgesEquivalentnAgesEquivalentnAgesEquivalentn ººº
ClesEquivalentnAgesEquivalentn AgCl ºº
100º
/%muestra
Cl
V
PEqClesEquivalentnClvp
eq
AtMPEq Cl
Cl
.

Profesor Argenis J. Sánchez Henríquez
115
6. Referencias Bibliográficas
Vogel Arthur I. (1960). Química Analítica Cuantitativa (Teoría y Práctica).
(2a Ed.). Editorial Kapelusz. Buenos Aires (Argentina).
Skoog Douglas A. y Donald M. West. (1970). Introducción a la Química
Analítica. Editorial Reverté. Barcelona (España).
Fischer Robert B. y Dennos G. Peters. (1970). Análisis Químico
Cuantitativo. (3a Ed.). Editorial Interamericana. Trad. Toral M. Teresa. México D.
F. (México).

Profesor Argenis J. Sánchez Henríquez
116
V. ANÁLISIS GRAVIMÉTRICO
El análisis químico gravimétrico esta enmarca dentro del análisis químico
clásico cuantitativo y tiene como propósito principal determinar las cantidades
relativas de un elemento, radical o compuesto que se encuentra presente en una
muestra, a través de la transformación de este elemento, radical o compuesto en
una nueva especie química de proporción bien definida que puede ser pesada. Al
igual que los métodos volumétricos, los gravimétricos también pueden ser
clasificados; esta clasificación obedece principalmente al procedimiento empleado
para llevar a cabo la separación del analito, o su derivado, de la matriz. Así
encontra: 1) Análisis gravimétrico por precipitación y, 2) Análisis gravimétrico por
volatilización. Los análisis basados en volatilización son menos aplicados, ya que
necesitan que el analito o su derivado sean volátiles para poder ser extraídos de la
matriz. Estos métodos por volatilización a su vez pueden ser directos o indirectos,
en los primeros el analito volatilizado se recolecta en una especie absorbente,
previamente pesada, por el aumento de la masa de la especie absorbente se
determina la cantidad de analito presente en la muestra. En los métodos indirectos
el analito se volatiliza de una porción pesada de la muestra, y por diferencia de
masa se obtiene la cantidad de analito.
Los métodos que se estudiarán en este manual se basan en el análisis
gravimétrico por precipitación del analito. En estos métodos la etapa que concreta
al análisis es la acción de pesar un precipitado que puede ser el analito puro o un
derivado de éste. Un precipitado es un compuesto muy poco soluble, formado por
la fuerte atracción entre los cationes y aniones que se encuentran en solución, que
una vez unidos se separan de la misma, formando así una solución heterogénea.
En este punto se hará una pausa para aclarar la terminología empleada en
análisis gravimétrico para designar a las especies o compuestos químicos que se
obtienen; el precipitado que se separa de la solución madre es conocido como
forma precipitada o forma aislada, por lo general, esta forma aislada de debe ser
sometida a un proceso de secado y/o calcinación antes de ser pesada, en este

Profesor Argenis J. Sánchez Henríquez
117
proceso térmico la forma aislada puede o no transformarse en otro compuesto con
estequiometría bien definida y estabilidad apropiada, a este nuevo compuesto se
le conoce como la forma ponderable. En ocasiones la forma aislada y la forma
ponderable responden a la misma formula química. La forma aislada debe reunir
ciertas características para servir como base al método gravimétrico desarrollado.
Estas características, a su vez, están condicionadas y de no satisfacerse las
condiciones los resultados obtenidos carecerían de toda veracidad. Por esta
razón, estos precipitados deben tener características de solubilidad, pureza, y
filtrabilidad, que cumplan con las siguientes condiciones:
Solubilidad
El precipitado obtenido debe ser prácticamente insoluble a fin de minimizar
las posibles pérdidas por solubilidad del precipitado. Se ha demostrado que en el
caso de electrolitos binarios (cada mol de la molécula al ionizarse libera 1 mol de
cada ion, 1:1) que posean Kps < 1x10 8, la precipitación es prácticamente
completa y las perdidas por solubilidad son desestimables. Por esta razón, en
análisis gravimétrico no se emplean, por lo general, formas precipitadas que
tengan Kps > 1x10 8.
Pureza
El precipitado obtenido debe tener una pureza casi absoluta. Debido a que
existen especies químicas solubles en el seno de la solución madre de la cual se
obtiene el precipitado, es inevitable que algunas de estas especies químicas co-
precipiten con la forma aislada de interés. En este caso, el precipitado debe
poseer una naturaleza tal que permita, a través de procedimientos sencillos,
eliminar las impurezas presentes en él.
Filtrabilidad
Esta característica esta relacionada al tamaño de partícula obtenido en el
precipitado o forma aislada. El precipitado debe poseer un tamaño de partícula
que permita su separación cuantitativa del seno de la solución, a través de
métodos sencillos y razonablemente rápidos. Los precipitados de cristales grandes

Profesor Argenis J. Sánchez Henríquez
118
(tamaños > 1x10 6 cm.) son de fácil filtración, además que sus superficies poco
desarrolladas absorben y adsorben menos impurezas del seno de la solución.
Así como existen características deseables condicionadas para la forma
aislada, también la forma ponderable debe cumplir con ciertas condiciones que
permitan un análisis más confiable, estas son:
1. Debe existir una correspondencia exacta entre la composición
(elementos) que presenta la especie química ponderable y su formula.
2. Esta forma ponderable debe ser químicamente estable, es decir, no debe
reaccionar con el H2O, CO2 u O2 del medio ambiente; así como sufrir oxidación o
reducción durante los procesos térmicos a los que sea sometida. En algunas
ocasiones, la forma ponderable debe ser tratada química y térmicamente, para
transformarla a una forma ponderable más conveniente para concretar el análisis.
Por ejemplo:
(f. Ponderable 1) (f. Ponderable 2)
3. Es deseable que el contenido de la especie que será analizada se
encuentre en baja proporción estequiométrica dentro de la forma ponderable, de
esta manera los errores por pesada, traspaso, solubilidad, entre otros, no
afectaran de forma significativa el resultado final del análisis. Por ejemplo, si en un
análisis se tuviese una pérdida neta de 2 mg para cada una de las siguientes
formas ponderables del cromo (Cr elemental), BaCrO4 y Cr2O3, los errores
cometidos al determinar la masa de cromo presente, a partir de las especies
ponderadas, serían:
422442 SOHOHCaSOSOHCaO
CrdemgBaCrOdemgBaCrOmg
Crdemg4,02
3,253
524
4
CrdemgOCrdemgOCrmg
Crdemg4,12
152
10432
32

Profesor Argenis J. Sánchez Henríquez
119
Como pueden observar las características y condiciones exigidas a los
precipitados o formas aisladas, presuponen un conocimiento de las condiciones
apropiadas para llevar a cabo una precipitación. Por tal razón, es de vital
importancia conocer la teoría relacionada con la formación, tamaño, pureza y
solubilidad de estas especies poco solubles.
Los precipitados se forman una vez que su producto iónico (P.I) supera al
valor de su Kps. El P.I se define como las concentraciones molares de los iones
que constituyen al precipitado, elevadas a sus respectivos coeficientes
estequiométricos, en un instante dado fuera del equilibrio. Por ejemplo:
Ag+ + Cl AgCl (s)
Al3+ + 3OH Al(OH)3 (s)
Es así, como en función del valor del P.I, se define cuando una solución
está insaturada (P.I < Kps), saturada (P.I = Kps) o sobre-saturada (P.I > Kps).
Bien, sabemos que si el valor del P.I supera al valor del Kps la solución
estará sobre-saturada y se formaran partículas que posteriormente precipitaran.
Existen factores físico-químicos experimentales que van a definir el tamaño de las
partículas que se forman, y por ende la pureza del precipitado:
11][][. ii ClAgIP
313 ][][. ii OHAlIP
Factores que afectanel tamaño de partícula
1. Concentración de los reactivos
2. Velocidad de mezcla de los reactivos
3. Solubilidad
1. Efecto de ion común
2. Efecto salino
3. pH
4. Naturaleza del solvente
5. Temperatura
Factores que afectanel tamaño de partícula
1. Concentración de los reactivos
2. Velocidad de mezcla de los reactivos
3. Solubilidad
1. Efecto de ion común
2. Efecto salino
3. pH
4. Naturaleza del solvente
5. Temperatura
1. Efecto de ion común
2. Efecto salino
3. pH
4. Naturaleza del solvente
5. Temperatura
1. Efecto de ion común
2. Efecto salino
3. pH
4. Naturaleza del solvente
5. Temperatura

Profesor Argenis J. Sánchez Henríquez
120
El efecto total de las variables relacionadas con el tamaño de partícula se
puede explicar a través de una propiedad conocida como sobre-saturación relativa
de la solución, definida por:
Donde: Q, es la relación de concentraciones molares de los reaccionantes y
productos; S, la solubilidad del precipitado; y Q S, la sobre-saturación de la
solución al comienzo de la precipitación.
Se puede decir que el tamaño de partícula para un precipitado es
inversamente proporcional a la magnitud de esta propiedad; así para magnitudes
grandes de SSR se formaran partículas pequeñas (tamaño<1x10 6 cm.), y para
magnitudes pequeñas se formaran partículas grandes (tamaño>1x10 6 cm.). Estos
tamaños de partícula a su vez definen el tipo de precipitado que se formará, es
decir, partículas con tamaño > 1x10 6 cm. forman precipitados cristalinos, mientras
que tamaños inferiores forman suspensiones coloidales que luego precipitan, por
coagulación, formando así precipitados coloidales. Relacionado al tamaño de
partícula se encuentra el grado de pureza del precipitado formado, ya que los
precipitados cristalinos al poseer una menor superficie irregular tienden a sufrir
menos los efectos de co-precipitación (adsorción, inclusión isomórfica y no
isomórfica, oclusión) lográndose una purificación suficiente, para efectos del
análisis cuantitativo, a través de simples lavados. En cambio, las suspensiones
coloidales coaguladas presentan una gran superficie efectiva amorfa propicia para
los efectos de co-precipitación, siendo además difíciles para su purificación a
través de métodos sencillos de lavado; ya que pueden peptizarse con facilidad, y
volver a su estado de suspensión. Seguidamente se realiza un análisis breve de
los distintos factores señalados en el esquema anterior.
S
SQSSRrelativasaturaciónsobre )(

Profesor Argenis J. Sánchez Henríquez
121
Concentración de los reactivos
Este factor afecta el tamaño de las partículas, al emplear soluciones de
reactivos diluidas e ir adicionando volúmenes pequeños, el producto de solubilidad
(Kps) del precipitado se supera sólo ligeramente; de esta forma se evita una
sobre-saturación relativa grande y por ende se obtienen cristales pequeños.
Velocidad de mezcla de los reactivos
Al mezclar los reactivos diluidos y de manera lenta en porciones pequeñas,
se permite que las primeras partículas, o núcleos, del precipitado que se formen
en el seno de la solución puedan crecer a expensas de los nuevos iones
adicionados. Así se obtiene cristales de mayor tamaño.
Solubilidad del precipitado
Aumentando la solubilidad del precipitado en el momento de su formación
se puede lograr aumentar el tamaño de partícula del mismo, como consecuencia
de una disminución de la sobre-saturación relativa. Esto se puede conseguir de
distintas maneras, por ejemplo; llevando a cabo la precipitación a temperaturas
elevadas y dejando enfriar la mezcla de reacción de forma paulatina.
Alterando el pH del medio de tal manera que el precipitado se forme en
condiciones de pH que favorezcan su solubilidad, y luego se varía lentamente este
parámetro hasta lograr condiciones que favorezcan nuevamente su precipitación.
Hay que destacar que estos factores no siempre afectan la solubilidad de
todos los precipitados en la misma forma, es decir, para algunos podría
aumentarla y para otros disminuirla.
Siguiendo con los factores que afectan la solubilidad de un precipitado, se
encuentra que si se lleva a cabo la precipitación del analito en presencia de un
exceso estequiométrico, no mayor del 50%, del reactivo precipitante, el cual tiene
un ion común con el precipitado. Esto disminuiría la concentración del analito en
equilibrio con el precipitado. Se debe tener cuidado de los grandes excesos de

Profesor Argenis J. Sánchez Henríquez
122
reactivo precipitante que podrían tener un efecto contrario por formación de
complejos solubles con el precipitado.
Por otra parte, la adición de electrolitos que tengan iones no comunes con
el precipitado aumenta la fuerza iónica de la solución, por disminución de la
actividad de los iones en solución; esto deriva en un aumento de la solubilidad del
precipitado. Esto se conoce como efecto salino.
Por último, es sabido que la naturaleza del solvente en el cual se lleva a
cabo la precipitación juega un papel importante en la solubilidad del precipitado;
así alterando la polaridad del medio de reacción, a través de la adición de
solventes orgánicos se puede aumentar o disminuir la solubilidad de un
precipitado.

Profesor Argenis J. Sánchez Henríquez
123
PRÁCTICA Nº 6
EQUILIBRIO DE SÓLIDOS POCOS SOLUBLES
1. Objetivo General
Desarrollar las competencias del alumno dentro del área experimental a
través del estudio de los diversos factores físico-químicos que determinan el tipo
de precipitado formado en el curso de una reacción, y su solubilidad.
2. Objetivos Específicos
2.1. Desarrollar la habilidad del estudiante en el manejo de instrumentos y
técnicas para ensayos cualitativos.
2.2. Desarrollar la capacidad de observación del estudiante a través de la
ejecución de diversas experiencias relacionadas con la teoría de formación de
sólidos pocos solubles: BaCrO4, AgCl, MgCO3, Al(OH)3, PbI2.
2.3. Desarrollar el sentido crítico del estudiante a través de la relación de la
teoría del equilibrio químico con los hechos observados experimentalmente.
2.4. Redactar un informe científico de laboratorio siguiendo las pautas
establecidas al principio de este manual, donde se refleje el análisis de sus
observaciones y los conocimientos adquiridos en el transcurso de la presente
práctica.
3. Actividades a Desarrollar
Antes de comenzar a desarrollar tus actividades de laboratorio mantén
siempre presente las normas de seguridad que se encuentran al principio de este
manual. Recuerda que la seguridad dentro del laboratorio es responsabilidad de
todos. Durante el desarrollo de las diferentes actividades toma nota detallada, en
tu cuaderno de laboratorio, de las observaciones experimentales que pueden
surgir de cada paso o etapa del ensayo. Es importante resaltar color, o cambios de
color, de las soluciones y los precipitados, aspecto de los sólidos formados y
soluciones sobre-nadantes, entre otros.

Profesor Argenis J. Sánchez Henríquez
124
3.1. Equilibrios químicos del cromato de bario (BaCrO4).
Fundamentos
El ion cromato (CrO42 ) es un cromóforo ínterconvertible que cambia su
color en función del pH del medio según la siguiente estequiometría:
2 CrO42 + 2 H+ Cr2O7
2 + H2O (Amarillo) (Anaranjado)
Si el medio es netamente básico, predomina el ion cromato (CrO42 ) de color
amarillo; si el medio en cambio es netamente ácido, entonces el ion que se
encontrará en mayor concentración en la solución es el dicromato (Cr2O72) de
color anaranjado.
Por otra parte, el anión cromato en medio básico forma con el catión bario
un precipitado que posee una constante de equilibrio, o Kps, igual a 3x10 10;
según la estequiometría de reacción:
CrO42 + Ba2+ BaCrO4 (s) Kps = 3x10 10
Material a Utilizar
1. Tubo de ensayo.
2. Botella de lavado con agua destilada/desionizada.
3. Vaso de precipitado.
4. Frascos gotero
Reactivos a Utilizar
1. K2CrO4 al 5 % p/v
2. HCl 2 N
3. NaOH 2 N
4. Ba(NO3)2 al 10 % p/v
Nota Importante: Es responsabilidad del
estudiante entregar al inicio de práctica, en su
cuaderno de laboratorio, una tabla donde se
resuman las siguientes características de los
reactivos arriba señalados: Fórmula, %
Pureza, Densidad, Código de frases de riesgo
(R) y seguridad (S) para cada reactivo.

Profesor Argenis J. Sánchez Henríquez
125
Procedimiento
1. Coloque en un tubo de ensayo 10 gotas de
solución de K2CrO4 al 5 % p/v, tome nota del color de la
solución.
2. Adicione 2 gotas de HCl 2 N, y homogenice la
solución con giros ligeros, observe si hay cambios en la
solución, tome nota.
3. Agregue 5 gotas de NaOH 2 N, observe cuidadosamente la solución y
registre si hay cambios perceptibles en la misma, en la medida que adiciona gota
a gota el NaOH.
4. Añada 3 gotas de Ba(NO3)2 al 10 % p/v, y continúe registrando sus
observaciones.
5. Adicione 5 gotas de HCl 2 N, y mueva suavemente de forma circular el
tubo de ensayo y su contenido. Tome nota detallada de lo observado para su
posterior interpretación.
3.2. Equilibrios químicos del cloruro de plata (AgCl).
Fundamentos
El catión plata (Ag+), junto con el Hg+ y el Pb2+, son los únicos cationes que
forman cloruros insolubles. Si adicionamos a una solución que contiene iones Ag+
un volumen de HCl, se formara un precipitado de aspecto cuajoso y color blanco,
que obedece a la siguiente estequiometría:
Ag+ + Cl AgCl (s) Kps = 1x10 10

Profesor Argenis J. Sánchez Henríquez
126
Este precipitado posee una constante de equilibrio, o Kps, igual a 1x10 10; y
si se expone a la luz sufre foto-descomposición, tomando el precipitado un matiz
púrpura, esta reacción sigue la estequiometría:
2 AgCl 2 Ag0 + Cl2 (g)
Por otra parte, el ion plata forma un ion complejo soluble estable con el
amoniaco (NH3), esta prueba sirve para identificarlo con respecto a los otros dos
cloruros insolubles. La formación del ion complejo diaminoplata (I) sigue la
estequiometría:
AgCl (s) + 2 NH3 [Ag(NH3)2]+ + Cl Kf = 1,7x107
El precipitado de cloruro de plata puede ser regenerado, por desplazamiento del
equilibrio químico, a través de la eliminación del amoniaco con HNO3, según:
[Ag(NH3)2]+ + Cl + 2 HNO3 AgCl (s) + 2 NH4
+ + 2 NO3
Material a Utilizar
1. Tubo de ensayo.
2. Botella de lavado con agua destilada/desionizada.
3. Vaso de precipitado.
4. Frascos gotero.
5. Pipeta
6. Pro-pipeta
Reactivos a Utilizar
1. AgNO3 0,1 M
2. HCl 2 N
3. NH3 2 N
4. HNO3 4 N
Nota Importante: Es responsabilidad del
estudiante entregar al inicio de práctica, en su
cuaderno de laboratorio, una tabla donde se
resuman las siguientes características de los
reactivos arriba señalados: Fórmula, %
Pureza, Densidad, Código de frases de riesgo
(R) y seguridad (S) para cada reactivo.
hluz ,

Profesor Argenis J. Sánchez Henríquez
127
Procedimiento
1. Coloque en un tubo de ensayo 2 gotas de solución de AgNO3 0,1 M y 1
mL de agua destilada/desionizada.
2. Adicione 4 gotas de HCl 2 N, y homogenice la
solución con giros ligeros, observe si hay cambios en la
solución, tome nota.
3. Agregue 4 gotas de NH3 2 N, homogenice la
solución con suaves giros, y observe cuidadosamente la
mezcla reaccionante. Registre si hay cambios perceptibles
en la misma.
4. Añada 3 gotas de HNO3 4 N, homogenice y registre en su cuaderno de
laboratorio lo observado, detalladamente, para su posterior interpretación.
3.3. Equilibrios químicos del carbonato básico de magnesio.
Fundamentos
El ion magnesio (Mg2+) incoloro, en medio acuoso, al combinarse con
carbonatos de metales alcalinos como el sodio y potasio, forma un precipitado de
carbonato básico de magnesio de color blanco semi-gelatinoso. Esta reacción
sigue la estequiometría:
4 CO32 + H2O 3 CO3
2 + HCO3 + OH 3CO32 + 2OH + CO2
4 Mg2+ + 4 CO32 + H2O Mg4(CO3)3(OH)2 (s)↓ + CO2 ↑

Profesor Argenis J. Sánchez Henríquez
128
Esta reacción se encuentra condicionada por el pH del medio, de tal manera que si
se introduce al seno de la solución una especie química que altere el equilibrio
amortiguador establecido en la primera etapa de la reacción, el producto iónico (P.I) de las
especies que conforman al precipitado se verá afectado. Por ejemplo:
�NH4Cl + H2O NH3 + Cl( + H3O+
H3O+ + CO3
2 HCO3 + H2O
H3O+ + OH 2 H2O
Material a Utilizar
1. Tubo de ensayo.
2. Botella de lavado con agua destilada/desionizada.
3. Vaso de precipitado.
4. Frascos gotero.
5. Pipeta
6. Pro-pipeta
7. Mechero.
8. Lentes de seguridad.
9. Pinza para tubo de ensayo.
10. Espátula.
Reactivos a Utilizar
1. Mg(NO3)2 0,1 M
2. Na2CO3 0,1 M
3. NH4Cl Sólido
Nota Importante: Es responsabilidad del
estudiante entregar al inicio de práctica, en su
cuaderno de laboratorio, una tabla donde se
resuman las siguientes características de los
reactivos arriba señalados: Fórmula, %
Pureza, Densidad, Código de frases de riesgo
(R) y seguridad (S) para cada reactivo.
Procedimiento
1. Coloque en un tubo de ensayo 2 gotas de solución
de Mg(NO3)2 0,1 M, y diluya con 1 mL de agua destilada
/desionizada.

Profesor Argenis J. Sánchez Henríquez
129
2. Adicione 5 gotas de Na2CO3 0,1 M, y homogenice la solución con giros
ligeros, observe si hay cambios en la solución, tome nota.
3. Caliente la mezcla reaccionante directamente a la
llama. Para ello, siga las instrucciones del personal del
laboratorio y tome las debidas precauciones. Registre si hay
cambios perceptibles en la solución.
4. Coloque en un nuevo tubo de ensayo 2 gotas de
solución de Mg(NO3)2 0,1 M, y diluya con 1 mL de agua
destilada /desionizada.
5. Sature la solución con una porción de NH4Cl sólido y
posteriormente añada 5 gotas de Na2CO3 0,1 M, homogenice
la solución con giros suaves. Observe si hay cambios
perceptibles en la mezcla reaccionante.
NH4ClNH4Cl

Profesor Argenis J. Sánchez Henríquez
130
6. Caliente la mezcla reaccionante directamente a la llama. Para ello, siga
las instrucciones del personal del laboratorio y tome las debidas precauciones.
Registre de forma detallada en su cuaderno de laboratorio lo observado para su
posterior interpretación.
3.4. Equilibrios químicos del hidróxido de aluminio, Al(OH3)(s).
Fundamentos
El catión aluminio (Al3+), al igual que el Cr3+, Zn2+ y Sn2+, forma en medios
ligeramente básicos, óxidos e hidróxidos anfóteros insolubles. El precipitado
hidratado de aluminio, Al(OH)3 o Al(H2O)3(OH)3, presenta un color blanco grisáceo
y aspecto gelatinoso; el mismo obedece a la estequiometría:
Al3+ + 3OH Al(OH)3 (s) Kps = 3,7x10 15
o
Al(H2O)63+ + 3 OH Al(H2O)3(OH)3 (s) + 3H2O
La propiedad anfotérica de estos óxidos e hidróxidos metálicos radica en
que son solubles tanto en soluciones ácidas como en básicas. Queda claro el

Profesor Argenis J. Sánchez Henríquez
131
comportamiento de la solubilidad de estos precipitados en medios ácidos, si
tenemos en cuenta el producto iónico del compuesto y la conversión del ion básico
del mismo al electrolito débil, H2O. En cambio al adicionar un exceso de base
(OH ), el comportamiento de la solubilidad responde a la formación de un nuevo
complejo, esta vez soluble, entre el precipitado de aluminio y el exceso de
hidróxidos (OH ), el ion complejo tetrahidroxoaluminato, esto puede apreciarse en
la siguiente reacción:
Al(OH)3 (s) + OH [Al(OH)4]
Material a Utilizar
1. Tubo de ensayo.
2. Botella de lavado con agua destilada/desionizada.
3. Vaso de precipitado.
4. Frascos gotero.
Reactivos a Utilizar
1. Al(NO3)3 0,1 M.
2. NaOH 2 N.
3. HCl 2 N.
Nota Importante: Es responsabilidad del
estudiante entregar al inicio de práctica, en su
cuaderno de laboratorio, una tabla donde se
resuman las siguientes características de los
reactivos arriba señalados: Fórmula, %
Pureza, Densidad, Código de frases de riesgo
(R) y seguridad (S) para cada reactivo.
Procedimiento
1. Coloque en un tubo de ensayo 8 gotas de solución
de Al(NO3)3 0,1 M.
2. Adicione por las paredes de tubo de ensayo 2
gotas de NaOH 2 N, observe si hay cambios en la solución,
tome nota.

Profesor Argenis J. Sánchez Henríquez
132
3. Agregue 3 gotas de NaOH 2 N, homogenice la solución con suaves giros
y golpes con la palma de la mano. Observe cuidadosamente la mezcla
reaccionante. Registre si hay cambios perceptibles en la misma.
4. Añada 3 gotas de HCl 2 N, homogenice la solución con suaves giros,
observe si hay cambios en la solución, tome nota.
5. Adicione 3 gotas más de HCl 2 N, y observe cuidadosamente la mezcla
reaccionante. Registre en su cuaderno de laboratorio lo observado, de forma
detallada, para su posterior interpretación.
3.5. Equilibrio químico del ioduro de plomo, PbI2 (s).
Fundamentos
Las sales de plomo solubles, como el nitrato de plomo Pb(NO3)2, al
reaccionar con halogenuros (Cl , F y I ) de metales alcalinos, como el Na+ y K+,
precipitan formando un compuesto de solubilidad moderada para la sal de cloruro
y menos soluble para las sales de yoduro y fluoruro, respectivamente. La
estequiometría seguida en el curso de la reacción entre el Pb(NO3)2 y el KI para
generar el respectivo precipitado es:
Pb(NO3)2 + 2 KI PbI2 (s) + 2 KNO3 Kps = 2,4x10 8
Estos precipitados de plomo se pueden purificar moderadamente a través
de su disolución, en la solución madre, por aumento de la temperatura del medio.
Es así, como se originan cristales de PbI2 de mayor tamaño y pureza en la medida
que se ve favorecido un crecimiento de partícula, en lugar del proceso de
formación de nuevos núcleos, producto de una disminución paulatina de la
temperatura del medio.

Profesor Argenis J. Sánchez Henríquez
133
Material a Utilizar
1. Tubo de ensayo.
2. Botella de lavado con agua destilada/desionizada.
3. Vaso de precipitado.
4. Frascos gotero.
5. Mechero.
6. Lentes de seguridad.
7. Pinza para tubo de ensayo.
Reactivos a Utilizar
1. Pb(NO3)2 0,1 M
2. KI 0,1 M
Nota Importante: Es responsabilidad del
estudiante entregar al inicio de práctica, en su
cuaderno de laboratorio, una tabla donde se
resuman las siguientes características de los
reactivos arriba señalados: Fórmula, %
Pureza, Densidad, Código de frases de riesgo
(R) y seguridad (S) para cada reactivo.
Procedimiento
1. Coloque en un tubo de ensayo 2 gotas de solución de
Pb(NO3)2 0,1 M y 1 mL de agua.
2. Adicione 2 gotas de KI 0,1 M, y homogenice la
solución con giros ligeros, observe si hay cambios en la
solución, tome nota.
3. Caliente la mezcla reaccionante directamente a la
llama. Para ello, siga las instrucciones del personal del
laboratorio y tome las debidas precauciones. Registre si hay
cambios perceptibles en la solución.

Profesor Argenis J. Sánchez Henríquez
134
4. Post- Laboratorio
Redactar un informe científico de laboratorio siguiendo las pautas
establecidas al principio de este manual, donde se refleje el análisis de sus
observaciones y los conocimientos adquiridos en el desarrollo de las actividades
realizadas (3.1, 3.2, 3.3, 3.4 y 3.5).
5. Cuestionario
1. ¿Qué es un precipitado?
2. ¿Qué es el producto iónico (P.I) de un precipitado?
3. Defina:
a. Solución insaturada
b. Solución saturada
c. Solución sobresaturada
4. ¿Qué factores físico-químicos pueden definir el tamaño y la pureza de los
precipitados?
5. ¿Cómo afecta la sobre-saturación relativa al tamaño de partícula de los
precipitados?
6. ¿Qué es un precipitado cristalino y qué un precipitado coloidal?
7. ¿Qué es la peptización?
8. Explique los diferentes factores que afectan la solubilidad de los
precipitados.
6. Referencias Bibliográficas
Skoog Douglas A., Donald M. West, James F. Holler y Stanley R. Crouch.
(2001). Química Analítica. (7a Ed.). McGraw-Hill. México D.F. (México).
Alexéiev Víctor N. (1976). Análisis Cuantitativo. Trad. Liminík E. Editorial
Mir. Moscú (Rusia).
Vogel Arthur I. (1960). Química Analítica Cuantitativa (Teoría y Práctica).
(2a Ed.). Editorial Kapelusz. Buenos Aires (Argentina).

Profesor Argenis J. Sánchez Henríquez
135
PRÁCTICA Nº 7
DETERMINACIÓN GRAVIMÉTRICA DE CALCIO
1. Objetivo General
Determinar la concentración de calcio presente en una muestra sintética
a través de un método gravimétrico por precipitación.
2. Objetivos Específicos
2.1. Desarrollar las destrezas del estudiante en las distintas etapas
experimentales involucradas en la obtención de oxalato de calcio hidratado en
condiciones acordes para el análisis gravimétrico.
2.2. Instruir al estudiante en las etapas de secado, carbonización y
calcinación de oxalato de calcio hidratado para obtener la forma ponderable más
conveniente.
2.3. Determinar la concentración de calcio, en tanto por ciento peso-peso
(%p/p), presente en la muestra.
2.4. Redactar un informe científico de laboratorio siguiendo las pautas
establecidas al principio de este manual, donde se reflejen los resultados y
conocimientos adquiridos en el transcurso de la presente práctica.
3. Actividades a Desarrollar
Antes de comenzar a desarrollar tus actividades de laboratorio mantén
siempre presente las normas de seguridad que se encuentran al principio de este
manual. Recuerda que la seguridad dentro del laboratorio es responsabilidad de
todos.

Profesor Argenis J. Sánchez Henríquez
136
3.1. Precipitación de calcio (Ca2+) como oxalato de calcio mono-
hidratado (CaC2O4H2O).
Fundamento
El calcio forma con el ion oxalato, en medio acuoso, un precipitado
hidratado de color blanco que sigue la estequiometría de reacción:
Ca2+ + C2O42 + H2O CaC2O4H2O (s) Kps = 2,6x10 9
Este precipitado o forma aislada, se caracteriza por separarse del seno de
soluciones neutras con un tamaño muy pequeño de partícula, lo cual por un lado;
hace casi imposible su filtración sin tener pérdidas en el proceso y conlleva a una
mayor contaminación del precipitado por fenómenos de co-precipitación. Por estas
razones, es conveniente establecer las condiciones adecuadas de precipitación
para lograr la formación de partículas de CaC2O4H2O de mayor tamaño. Esto se
logra estableciendo una sobre-saturación relativa (SSR) pequeña al momento de
la precipitación. Para ello, se precipita el CaC2O4H2O en un medio relativamente
ácido, donde éste es soluble, y se disminuye la acidez de forma paulatina hasta
alcanzar valores de pH comprendidos entre 3,5 y 4. El principio de esta baja SSR
se encuentra en el aumento de la solubilidad del precipitado por disminución de la
concentración del ion C2O42 en el seno de la solución en medios muy ácidos:
C2O42 + H+ HC2O4
HC2O4 + H+ H2C2O4
Posteriormente, al ir haciendo el medio de reacción paulatinamente menos
ácido (3,5 pH 4) la concentración de ion C2O42 aumenta gradualmente,
permitiendo así el crecimiento de partículas de CaC2O4H2O.
H

Profesor Argenis J. Sánchez Henríquez
137
Material a Utilizar
1. Balanza analítica.
2. Espátula.
3. Vidrio de reloj.
4. Botella de lavado con agua destilada y desionizada.
5. Vaso de precipitado.
6. Mechero Meker
7. Rejilla para calentamiento.
8. Trípode.
10. Cilindro graduado.
11. Soporte universal
12. Papel de filtro cuantitativo Whatman Nº540.
13. Soporte para embudo.
14. Embudo cuantitativo.
15. Tubo de ensayo.
Reactivos a Utilizar
1. Ca(NO3)24H2O
2. (NH4)2C2O4 al 4 y 0,2 % p/v.
3. AgNO3 0,1 M.
4. HNO3 6 M.
5. NH3 1:1
6. HNO3 1:1
7. Rojo de metilo.
Nota Importante: Es responsabilidad de
cada estudiante entregar al inicio de cada
práctica, en su cuaderno de laboratorio, una
tabla donde se resuman las siguientes
características de los reactivos arriba
señalados: Fórmula, % Pureza, Densidad,
Código de frases de riesgo (R) y seguridad
(S) para cada reactivo.
Procedimiento
1. Pese en un vidrio de reloj, al 0,1 mg, una masa de muestra comprendida
entre 0,7000 y 0,8600 g.
2. Transfiera cuantitativamente esta masa a un vaso de
precipitado de 400 mL. Para ello, puede ayudarse con una
porción de 10 mL de agua destilada/desionizada.

Profesor Argenis J. Sánchez Henríquez
138
3. Adicione 15 mL de HNO3 (1:1) a la solución de la muestra, tápela con el
vidrio de reloj y caliente hasta ebullición; gradúe luego la llama para permitir una
ebullición suave de la mezcla reaccionante durante 3 minutos. El montaje es
mostrado en la figura adjunta.
4. Diluya el contenido del vaso de precipitado con agua
destilada/desionizada hasta alcanzar aproximadamente 200 mL de solución. Para
ello, con ayuda de una piseta agregue el agua de tal forma de lavar las paredes
del vaso de precipitado y el vidrio de reloj por la cara expuesta al vapor. Tal como
se muestra en siguiente figura:
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
HNO30
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
HNO30
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
HNO30
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
HNO3

Profesor Argenis J. Sánchez Henríquez
139
5. Caliente nuevamente la solución hasta ebullición, retírela del fuego y aún
caliente adicione lentamente 75 mL de (NH4)2C2O4 al 4 % p/v, previamente
medidos con un cilindro graduado.
6. Adicione a la mezcla reaccionante 6 gotas del
indicador visual ácido-base anaranjado de metilo y
seguidamente, desde un frasco gotero, agregue gota a gota
la solución de NH3 (1:1) hasta observar el cambio de color
de la solución de rosado a amarillo.
7. Permita que el precipitado formado se sedimente por completo antes de
su filtración. Para ello, es necesario dejar la solución tapada y en reposo durante
al menos 12 horas.
8. Ensaye la cuantitatividad en la precipitación del Ca2+ a través de la
adición de gotas de (NH4)C2O4 al 4 % p/v; observe con detenimiento el punto de
contacto de cada gota adicionada, con la solución, para ver si se forma turbidez, lo
cual es indicativo de una precipitación incompleta.
9. Filtre el precipitado o forma aislada, CaC2O4H2O. Si la forma aislada
será sometida a tratamiento térmico para transformarla a la forma ponderable
CaCO3, se debe realizar la filtración en embudo de Gooch de sílice con manto
filtrante de amianto o disco de fibra de vidrio empleado por pares, además de un
tren de filtración como el mostrado en la figura 6 (página 23). Si se desea
transformar a la forma aislada a la forma ponderable CaO, se debe emplear papel
de filtro de porosidad apropiada (7 mm diámetro) y un embudo cuantitativo.
NH3NH3

Profesor Argenis J. Sánchez Henríquez
140
Por razones técnicas y logísticas, emplearemos la filtración por gravedad a
través de papel de filtro cuantitativo.
10. Disponga el equipo de filtración y proceda a filtrar el precipitado de
CaC2O4H2O respetando las etapas de: A) decantación del líquido sobrenadante,
B) lavado del precipitado con 5 porciones de 5 mL de agua fría, y C) transvase
cuantitativo del precipitado al papel de filtro.
11. Recoja en un tubo de ensayo un último lavado de 5 mL directamente del
vástago del embudo cuantitativo, y ensaye la ausencia de Cl en el lavado. Para
ello, adicione a la solución de lavado recogida en el tubo de ensayo
aproximadamente 1 mL de HNO3 6 M y 3 gotas de AgNO3 0,1 M. De estar
presente el ion Cl se formará turbidez, y por lo tanto se continuará la etapa de
lavado hasta que el ensayo resulte negativo.
Una vez concluida esta etapa el precipitado, o forma aislada, contenido en
el papel de filtro está listo para su transformación térmica a la forma ponderable
más conveniente.
(A y C)
(B)
(A y C)
(B)
AgNO3AgNO3AgNO3

Profesor Argenis J. Sánchez Henríquez
141
3.2. Secado, carbonización y calcinación de oxalato de calcio mono-
hidratado (CaC2O4H2O).
Fundamento
El oxalato de calcio mono-hidratado puede ser transformado a través de un
tratamiento térmico controlado es tres diferentes formas ponderables, como puede
observarse en la curva de pirólisis o termograma de la figura 12. De estas tres
formas ponderables, el carbonato cálcico (CaCO3) formado entre 420 y 600 ºC
representa la forma más conveniente para la determinación gravimétrica de calcio.
CaC2O4 CaCO3 + CO (g)
Esto se obedece principalmente a que el oxalato de calcio anhidro, CaC2O4,
obtenido entre 100 y 226 ºC, y estable hasta 390ºC, es higroscópico y gana
rápidamente agua del medio ambiente. Por otra parte, el óxido de calcio (CaO)
que se comienza a formar a partir de 600 ºC, según la estequiometría:
CaCO3 CaO + CO2 (g)
Presenta un equilibrio con el dióxido de carbono liberado (CO2) que debe
ser controlado, ya que una mayor presión parcial de CO2 en la atmósfera
circundante durante la calcinación conllevará a una baja tasa de conversión del
carbonato a óxido de calcio, además de que el óxido es higroscópico. Para evitar
estos problemas se realiza la calcinación en crisol de platino semi abierto en una
mufla a temperaturas entre 1100 y 1200 ºC durante 2 horas.
C25500,
C1100,

Profesor Argenis J. Sánchez Henríquez
142
Figura 12. Termogramas. A, Oxalato cálcico; B, Oxalato magnésico. (Tomado de Ayres H. G., 1970)
Material a Utilizar
1. Balanza analítica.
2. Mufla eléctrica.
3. Crisol de porcelana con tapa.
4. Mechero Bunsen.
5. Triángulo para calentamiento.
6. Soporte universal.
7. Pinza para crisol.
8. Desecador.
9. Aro metálico.
Reactivos a Utilizar
1. (NH4)2CO3 solución saturada
Nota Importante: Es responsabilidad de
cada estudiante entregar al inicio de cada
práctica, en su cuaderno de laboratorio, una
tabla donde se resuman las siguientes
características de los reactivos arriba
señalados: Fórmula, % Pureza, Densidad,
Código de frases de riesgo (R) y seguridad
(S) para cada reactivo.
Procedimiento
1. Transfiera el papel de filtro a un crisol de porcelana con tapa,
previamente llevado a peso constante a través de un programa de calcinación a
525 ºC en una mufla eléctrica durante 2 horas. Tenga la precaución de permitir
que el papel de filtro se seque parcialmente antes de manipularlo con delicadeza,
siguiendo las instrucciones gráficas que se muestran a continuación; el papel de

Profesor Argenis J. Sánchez Henríquez
143
filtro doblado en el paso 3 debe quedar con la mayor masa de precipitado cercana
al fondo del crisol, paso 4. La figura adjunta muestra el proceso.
2. Carbonice el papel de filtro dentro del crisol
empleando para ello el mechero Bunsen con una llama
muy suave al principio, para evitar pérdidas mecánicas
del precipitado por expulsión rápida de la humedad.
3. Aumente la intensidad de calentamiento del crisol, para eliminar el
carbón residual, bajando el mismo hacia la llama, en la medida que la columna
ligera de humo desaparece.
(1)
(3)
(2)
(4)
(1)
(3)
(2)
(4)

Profesor Argenis J. Sánchez Henríquez
144
4. Introduzca el crisol semi tapado, en una mufla eléctrica, una vez
finalizada la etapa previa de carbonización del papel de filtro. Calcine su contenido
a 525 ºC durante 2 horas.
5. Enfríe el crisol con su contenido en un desecador y luego se pesa.
Humedezca la forma ponderable, CaCO3, dentro del crisol con unas gotas de
solución saturada de (NH4)2CO3 y seque en una estufa a 110 ºC durante 10
minutos; deje enfriar en el desecador y pese nuevamente. Un aumento de peso en
relación con la primera pesada, indica presencia de CaO.
6. Calcule con la diferencia de masa, entre el crisol tarado inicialmente y
después del calcinado conteniendo a la forma ponderable, el contenido de calcio
presente en la muestra sintética, expresado en tanto por ciento peso-peso.
4. Post- Laboratorio
Redactar un informe científico de laboratorio siguiendo las pautas
establecidas al principio de este manual, donde se reflejen los resultados y
conocimientos adquiridos en el desarrollo de las actividades realizadas (3.1 y 3.2).
crisolPesocalcinadocrisolPesoCaCOPeso )(3
100/% 3
2
3
2
muestraPeso
CaCOMolarMasa
CaAtómicaMasaCaCOPeso
Capp

Profesor Argenis J. Sánchez Henríquez
145
5. Cuestionario
1. ¿Cuál es el propósito del análisis gravimétrico y cómo se logra?
2. ¿Cómo se clasifican los métodos gravimétricos de análisis?
3. ¿Qué es la forma aislada y qué la forma ponderable?
4. ¿Qué características condicionadas debe cumplir la forma aislada para
servir como base a un método gravimétrico?
5. ¿Qué condiciones debe cumplir la forma ponderable para permitir
resultados confiables?
6. Escriba cuales son las formas ponderables a las que pueden
transformarse el CaC2O4H2O aislado. ¿Cuál de ellas es más conveniente y a qué
temperatura se obtiene? Argumente su respuesta.
7. Determine los siguientes factores gravimétricos para convertir:
a. BaSO4 en Ba.
b. Nb2O5 en Nb.
c. Mg2P2O7 en MgO.
d. KClO4 en K2O.
e. (NH4)2PtCl6 en NH3.
6. Referencias Bibliográficas
Fischer Robert B. y Dennos G. Peters. (1970). Análisis Químico
Cuantitativo. (3a Ed.). Editorial Interamericana. Trad. Toral M. Teresa. México D.
F. (México).
Skoog Douglas A. y Donald M. West. (1970). Introducción a la Química
Analítica. Editorial Reverté. Barcelona (España).
Alexéiev Víctor N. (1976). Análisis Cuantitativo. Trad. Liminík E. Editorial
Mir. Moscú (Rusia).
Vogel Arthur I. (1960). Química Analítica Cuantitativa (Teoría y Práctica).
(2a Ed.). Editorial Kapelusz. Buenos Aires (Argentina).

Profesor Argenis J. Sánchez Henríquez
146
VI. ANÁLISIS INSTRUMENTAL
El análisis químico instrumental se fundamenta en todas aquellas técnicas
analíticas, que emplean la medición de alguna propiedad física de la materia para
su cuantificación, o aprovechan estas propiedades físicas para su separación y
posterior cuantificación. Estas propiedades físicas pueden ser: conductividad
eléctrica, absorción de radiación, emisión de radiación, difracción de radiación,
razón masa a carga, adsorción, intercambio iónico, distribución entre dos fases,
entre otras. Fundamentalmente pueden distinguirse cuatro tipos de métodos
instrumentales de análisis: 1) métodos espectroscópicos, 2) métodos
electroanalíticos, 3) métodos cromatográficos, y 4) métodos térmicos. Éstos a su
vez engloban una amplia variedad de técnicas, por ejemplo; dentro de los métodos
espectroscópicos podemos encontrar las técnicas de espectroscopia de absorción
molecular UV-Visible, espectroscopia de absorción atómica, espectroscopia de
masa, entre otras.
Las actividades prácticas que se desarrollaran en esta parte del manual se
fundamentan en la técnica de espectroscopia de absorción molecular UV-Visible,
por esta razón se revizarán de forma breve algunas definiciones que nos serán de
utilidad.
La espectroscopia de absorción molecular UV-Visible (EAM UV-Vis) es una
técnica que estudia el comportamiento de absorción de radiación
electromagnética, en la región espectral del ultravioleta y visible, por parte de las
moléculas que constituyen una muestra. La región UV-Vis apenas abarca una
pequeña parte del espectro electromagnético de la radiación, esta parte se
extiende desde las longitudes de onda de 10 nm hasta 900 nm; dividiéndose en:
UV de vacío (10-190 nm), UV (191-350 nm) y Vis (351-900 nm).
El comportamiento de absorción de radiación por parte de las moléculas
que constituyen a la muestra, se debe principalmente a la transferencia de energía

Profesor Argenis J. Sánchez Henríquez
147
desde la radiación electromagnética (c
hhE ) hacia los electrones de las
moléculas, localizados éstos en los distintos orbitales moleculares (n, , );
lográndose con esta transferencia de energía la excitación y transición de los
electrones a orbitales moleculares de mayor energía. En algunos casos, esta
transferencia de energía ocurre entre la radiación y los iones hidratados de los
metales de transición, lantánidos y actínidos, en cuyo caso los electrones se
localizan en orbitales 3d y 4d, y 4f y 5f, respectivamente. Los grupos funcionales
que pueden proporcionar orbitales del tipo , de tal forma de facilitar las
transiciones electrónicas del tipo n * y *, son conocidos como grupos
cromóforos.
Los instrumentos para EAM UV-Vis se constituyen fundamentalmente de 5
componentes: 1) fuente o lámpara, 2) selector de longitud de onda ( ), 3) celda o
cubeta para la muestra, 4) detector, y 5) transductor o registrador. La fuente
suministra el espectro continuo de radiación electromagnética que será
discriminado posteriormente por el selector de longitud de onda, el haz de
radiación aislado por el selector interaccionará luego con las moléculas de la
muestra contenidas en la celda, y la parte de la radiación que no se absorbe
continua su trayectoria hacia el detector, éste convierte la energía radiante que
llega a él en una señal eléctrica que el transductor puede mostrar en una escala
numérica analógica o digital. La figura 13, muestra un diagrama óptico del
espectrofotómetro Spectronic 20, instrumento muy empleado en la mayoría de los
laboratorios de análisis químico cuantitativo por su sencillez, robustez y bajo costo.
El comportamiento de absorción de radiación electromagnética, por parte
del analito (molécula o ion que se desea determinar) en la matriz (muestra),
medido a través de la absorbancia de la muestra y en función de la concentración
del analito, obedece a una linealidad, cuando se trabaja bajo ciertas condiciones
de concentración. Este comportamiento no sólo puede reflejarse a través del

Profesor Argenis J. Sánchez Henríquez
148
parámetro absorbancia de la solución, sino también por medio del parámetro
transmitancia.
Figura 13. Diagrama óptico del Spectronic 20. (Fuente: Modificado del Skoog and Leary, 1994)
La absorbancia se define como la fracción de la radiación electromagnética
que es absorbida por la muestra, en cambio la transmitancia es la fracción de esta
radiación que es transmitida por la misma. Ambos parámetros encuentran
representación matemática a través de la Ley de Beer-Lambert:
(Ley de Beer-Lambert)
Donde: K, es una constante que depende de la temperatura, índice de
refracción de la solución, naturaleza de la especie absorbente, entre otros. b, es el
CbKP
PTA 0loglog
0P
PT
CbKA

Profesor Argenis J. Sánchez Henríquez
149
paso óptico de la celda o distancia que recorre el haz de radiación a través de la
muestra. Y C, la concentración de la especie absorbente (analito) en la muestra.
En las otras expresiones matemáticas, P0 y P representan la potencia del haz de
radiación antes y después de atravesar la muestra, respectivamente.
La Ley combinada de Beer-Lambert permite llevar a cabo la cuantificación
del analito presente en la matriz, siempre que no se presenten serias
interferencias por parte de otras especies químicas que coexisten con el analito en
la muestra.

Profesor Argenis J. Sánchez Henríquez
150
PRÁCTICA Nº 8
DETERMINACIÓN ESPECTROFOTOMÉTRICA DE FÓSFORO
1. Objetivo General
Determinar la concentración de fósforo inorgánico (PO43 ) presente en
una muestra de alimento a través de un método fundamentado en la
espectroscopia de absorción molecular UV-Vis.
2. Objetivos Específicos
2.1. Desarrollar las destrezas del estudiante en las distintas etapas
experimentales involucradas en la preparación de cenizas a partir de una muestra
sólida de alimento homogenizada.
2.2. Adiestrar al estudiante en el procedimiento seguido para la obtención
de un extracto a partir de cenizas.
2.3. Instruir al estudiante en las diferentes etapas que constituyen la
elaboración y caracterización de una curva de calibración.
2.4. Determinar la concentración de fósforo inorgánico presente en el
extracto de cenizas a través de la curva de calibración y la Ley combinada de
Beer-Lambert.
2.5. Redactar un informe científico de laboratorio siguiendo las pautas
establecidas al principio de este manual, donde se reflejen los resultados y
conocimientos adquiridos en el transcurso de la presente práctica.
3. Actividades a Desarrollar
Antes de comenzar a desarrollar tus actividades de laboratorio mantén
siempre presente las normas de seguridad que se encuentran al principio de este
manual. Recuerda que la seguridad dentro del laboratorio es responsabilidad de
todos.

Profesor Argenis J. Sánchez Henríquez
151
3.1. Preparación de cenizas a partir de una muestra sólida de alimento
homogenizada.
Fundamento
La obtención de la fracción mineral (Calcio, potasio, hierro, fósforo, sodio,
yodo, magnesio, entre otros) de un alimento, tejido vegetal o animal, se realiza a
través de la incineración de la materia orgánica a la cual se encuentran ligados
estos minerales. De esta forma el residuo final o cenizas contendrá el porcentaje
total de los diferentes elementos minerales que constituían a la muestra.
Material a Utilizar
1. Balanza analítica.
2. Mufla eléctrica.
3. Crisol de porcelana con tapa.
4. Espátula.
5. Estufa eléctrica.
6. Pinza para crisol
7. Desecador.
8. Plancha de calentamiento.
Reactivos a Utilizar
1. Aceite de oliva o ajonjolí
2. H2O destilada/desionizada.
Nota Importante: Es responsabilidad de
cada estudiante entregar al inicio de cada
práctica, en su cuaderno de laboratorio, una
tabla donde se resuman las siguientes
características de los reactivos arriba
señalados: Fórmula, % Pureza, Densidad,
Código de frases de riesgo (R) y seguridad
(S) para cada reactivo.
Procedimiento
1. Pese en un crisol de porcelana con tapa, previamente llevado a peso
constante a través de un programa de calcinación a 525 ºC en una mufla durante
un periodo de 16 horas, la masa adecuada de muestra homogenizada que será
transformada a cenizas. Esta masa va a depender del tipo de muestra estudiada
(ver normas COVENIN 1158-82). Tome nota en su cuaderno de laboratorio del
peso del crisol tratado y el peso de la muestra.

Profesor Argenis J. Sánchez Henríquez
152
2. Coloque el crisol con la muestra en el interior de una estufa eléctrica que
se encuentre a 100 10 ºC, durante 12 horas, a fin de deshidratarla.
3. Adicione a la muestra, posteriormente al periodo
de deshidratación, unas gotas de aceite de oliva o ajonjolí,
suficientes para formar una delgada capa sobre ésta. Y se
procede a carbonizarla con el mechero empleando una
llama suave al principio e intensificando la llama hacia el
final de la carbonización.
4. Tape el crisol una vez finalizada la carbonización e introdúzcalo en una
mufla eléctrica a 525 ºC por espacio de 16 horas. Así se eliminará la materia
orgánica y se obtendrá un residuo (cenizas) blanco, gris pálido o amarillo. Guarde
las cenizas en el crisol tapado en un desecador mientras se enfrían.
5. Humedezca las cenizas en el crisol con unas gotas de agua
destilada/desionizada y séquelas en plancha de calentamiento.
6. Tape el crisol una vez finalizada la etapa de secado e introdúzcalo
nuevamente en una mufla eléctrica a 525 ºC por espacio de 4 horas. Déjelo enfriar
en un desecador y pese el crisol con las cenizas. Registre en su cuaderno de
laboratorio el peso del crisol + cenizas.

Profesor Argenis J. Sánchez Henríquez
153
3.2. Preparación del extracto a partir de las cenizas del alimento.
Fundamento
Las cenizas obtenidas a través de la calcinación son sometidas a procesos
de digestión humeda con ácido clorhídrico, con la finalidad de deshidratar silicatos
e insolubilizarlos, así como hidrolizar pirofosfatos a ortofosfatos. De esta manera
se obtiene un extracto o solución de la muestra lista para ser analizada.
Material a Utilizar
1. Crisol de porcelana con tapa.
2. Plancha de calentamiento.
3. Papel de filtro.
4. Embudo cuantitativo.
5. Soporte para embudo.
6. Piseta
7. Matraz aforado de 100 mL.
8. Soporte universal.
9. Aro metálico.
10. Triángulo para calentamiento.
11. Mechero Bunsen.
Reactivos a Utilizar
1. HCl (1:1)
2. H2O destilada/desionizada.
Nota Importante: Es responsabilidad de
cada estudiante entregar al inicio de cada
práctica, en su cuaderno de laboratorio, una
tabla donde se resuman las siguientes
características de los reactivos arriba
señalados: Fórmula, % Pureza, Densidad,
Código de frases de riesgo (R) y seguridad
(S) para cada reactivo.
Procedimiento
1. Humedezca con 5 mL de HCl
(1:1) las cenizas obtenidas en la actividad
anterior, en el mismo crisol en que se
calcinaron. Y Séquelas empleando una
plancha de calentamiento.
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
HCl (1:1)0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
HCl (1:1)

Profesor Argenis J. Sánchez Henríquez
154
2. Adicione 5 mL más de HCl (1:1) al crisol y colóquelo nuevamente en la
plancha por espacio de 30 minutos.
3. Deje enfriar la solución en el crisol y fíltrela a
través de papel de filtro, recogiendo el filtrado en un matraz
aforado de 100 mL. Como se muestra en la figura adjunta.
4. Incinere el papel de filtro con las cenizas que no
se solubilizaron en el mismo crisol donde se llevó a cabo la
calcinación y digestión. Luego humedezca estas cenizas
con 5 mL de HCl (1:1) y caliente en una plancha durante 5 minutos. El proceso se
muestra en la figura adjunta.
5. Deje enfriar la solución en el crisol y fíltrela a través de papel de filtro,
empleando el mismo embudo cuantitativo y recogiendo el filtrado y los lavados con
agua destilada/desionizada en el mismo matraz aforado de 100 mL. Enrase la
solución con agua destilada/desionizada, rotúlela y guárdela para el posterior
análisis de fósforo (P).
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
HCl (1:1)0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
HCl (1:1)

Profesor Argenis J. Sánchez Henríquez
155
3.3. Preparación y caracterización de la curva de calibración para la
determinación de fósforo.
Fundamento
Una curva de calibración es una representación gráfica en un eje cartesiano
(x, y), de la concentración de varias soluciones estándares (x), contra sus
respectivas absorbancias (y), medidas a una longitud de onda dada ( ). Una
representación de este tipo sigue un comportamiento lineal a lo largo de cada par
de puntos graficados, siempre que se cumpla la Ley combinada de Beer-Lambert.
Sabemos que una solución estándar es aquella cuya concentración, con
respecto al analito, se conoce exactamente; las soluciones estándares para la
preparación de una curva de calibración se preparan, por lo general, por dilución a
partir de una solución de mayor concentración denominada solución madre. La
solución madre, y por ende, las soluciones estándares contienen al analito que se
desea determinar. Una solución preparada sin la adición intencional del analito por
parte del analista se denomina solución blanco. La solución blanco se preparan
con la finalidad de corregir valores de absorbancia que se presentan por parte de
algunos reactivos empleados en el análisis, así como de posible presencia de
cantidades trazas (muy pequeñas) del analito en alguno de estos reactivos o en el
solvente utilizado.
Una curva de calibración posee varios parámetros analíticos y estadísticos
que la caracterizan, básicamente estos parámetros son: 1) Concentración del
límite de detección (CLD) del analito en la muestra a través de la curva de
calibración diseñada para el método analítico empleado. 2) Concentración mínima
del analito (Cmín) que puede ser determinada a través de la curva de calibrado
diseñada para el método de análisis empleado. 3) Sensibilidad de calibración (S)
que representa la capacidad que tiene el método o el instrumento para diferenciar
entre pequeñas variaciones de la concentración, este parámetro está
representado por la pendiente de la curva de calibración (m). 4) Concentración
máxima (Cmáx) que puede determinarse del analito a través de la curva de

Profesor Argenis J. Sánchez Henríquez
156
calibración diseñada para el método analítico empleado. Todos estos parámetros
se derivan de la curva de calibración y un simple tratamiento estadístico de los
valores de absorbancia de la solución blanco.
Hasta el momento se ha supuesto que el analito presente en las soluciones
estándares preparadas presenta una absorbancia natural a la longitud de onda
seleccionada para el análisis, esto no siempre es así. Por lo general, hay que
llevar a cabo reacciones químicas entre el analito y un ligando, para formar
complejos, que además de conferirle propiedades de absorción al complejo
(analito-ligando) en la región espectral deseada (UV-Vis), le otorgan al método
analítico mayor selectividad y sensibilidad.
En el caso que nos concierne, tendremos que llevar a cabo estas
reacciones para lograr la cuantificación del fósforo inorgánico presente en la
muestra. Las reacciones que se presentan entre el fósforo inorgánico y los
diferentes reactivos selectivos empleados en el método analítico seleccionado
son:
(NH4)6Mo7O24 4H2O + 3 H2SO4 7 H2MoO4 + 3 (NH4)2SO4
PO43 + 12 H2MoO4 P(Mo3O10)4
3 + 12 H2O
(analito) (ligando) (heteropoli-complejo)
(color amarillo)
P(Mo3O10)43 + C6H8O6 Complejo azul de molibdeno
(reductor)
De esta manera, el fósforo presente en la muestra sirve como centro de
coordinación del heteropoli-complejo que es posteriormente reducido por el ácido
ascórbico (C6H8O6).

Profesor Argenis J. Sánchez Henríquez
157
Material a Utilizar
1. Balanza analítica.
2. Espátula.
3. Pesa sustancia o vidrio de reloj.
4. Matraces aforado de 250 y 100 mL.
5. Piseta
6. Pipetas volumétricas de 1, 2, 3, 4 y 5 y 10 mL.
7. Vaso de precipitado.
8. Spectronic 20D+.
9. Celdas de vidrio.
10. Embudo cuantitativo.
Reactivos a Utilizar
1. KH2PO4 1000 ppm.
2. Ácido ascórbico al 1 % p/v.
3. (NH4)6Mo7O24 4H2O al 1 % p/v.
4. H2SO4 concentrado.
5. CH3COONa3H2O
6. CH3COOH glacial.
Nota Importante: Es responsabilidad de
cada estudiante entregar al inicio de cada
práctica, en su cuaderno de laboratorio, una
tabla donde se resuman las siguientes
características de los reactivos arriba
señalados: Fórmula, % Pureza, Densidad,
Código de frases de riesgo (R) y seguridad
(S) para cada reactivo.
Procedimiento
1. Pese, al 0,1 mg, en un pesa sustancia o vidrio de
reloj una masa de reactivo de fosfato ácido de potasio
(KH2PO4) igual a 1,0970 g, y transfiérala cuantitativamente a
un matraz aforado de 250 mL, empleando para ello una
porción aproximada de 30 mL agua destilada/desionizada.

Profesor Argenis J. Sánchez Henríquez
158
2. Adicione, con una pipeta serológica, 10 mL de ácido sulfúrico (H2SO4)
10 N a la solución en el matraz aforado. Y posteriormente enrase con agua
destilada/desionizada. Esta solución resulta 1000 ppm en fósforo (P).
3. Tome, con una pipeta volumétrica, 5 mL
de la solución madre preparada
anteriormente y transfiéralos a un matraz
aforado de 100 mL, y enrase con agua
destilada/desionizada. Esta solución
intermedia resulta con una concentración de
50 ppm en fósforo (P).
4. Pese, en un vaso de precipitado de 500 mL, 34 g de CH3COONa3H2O.
Disuélvalo con la menor cantidad posible de agua destilada/desionizada; y
adicione a la solución, con un cilindro graduado, 57 mL de CH3COOH glacial.
Homogenice la solución y
transfiérala a un matraz aforado
de 1L para su posterior aforo con
agua destilada/desionizada. Esta
solución buffer tiene un pH de 4.
soluciónLV
PmgPppm
)(42
42.
.
POKHMM
PAtMPOKHgPg
.... dildilconcconc VppmVppm
CH3COOHCH3COOHCH3COOH

Profesor Argenis J. Sánchez Henríquez
159
5. Mida, con una pipeta serológica, 1,33 mL de H2SO4 concentrado y
transfiéralos a un vaso de precipitado de 250 mL, al cual se le ha adicionado
previamente 30 mL de agua destilada/desionizada. Posteriormente, pese en un
vidrio de reloj o pesa sustancia, 10 g de heptamolibdato de amonio tetrahidratado
[(NH4)6Mo7O244H2O], y disuélvalos en el vaso de precipitado que contiene la
solución de H2SO4, con una porción adicional de agua destilada/desionizada.
Transfiera la solución a un matraz aforado de 1L para su aforo. De esta solución al
1 % p/v de molibdato de amonio, en ácido sulfúrico, se origina el ácido
molibdénico.
6. Pese, al 0,1 mg, en un pesa sustancia o vidrio de reloj
una masa de reactivo de ácido ascórbico (C6H8O6) igual a
0,1000 g, y transfiérala cuantitativamente a un matraz aforado
de 10 mL, empleando para ello una porción agua
destilada/desionizada. Enrase y homogenice la solución por
inversión del matraz. La solución resultante es 1% p/v en ácido
ascórbico.
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
H2SO4 (conc.)
(NH4)6Mo7O244H2O
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
H2SO4 (conc.)
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
H2SO4 (conc.)
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
0
1
2
3
4
5
6
7
8
9
0
1
2
3
4
5
6
7
8
9
10
mL
1/1
0
H2SO4 (conc.)
(NH4)6Mo7O244H2O(NH4)6Mo7O244H2O

Profesor Argenis J. Sánchez Henríquez
160
7. Prepare soluciones estándares para la curva de calibración, con
concentraciones comprendidas entre 0 y 2,5 ppm en fósforo (P) e incrementos de
0,5 ppm entre cada estándar. Para ello, siga el esquema de trabajo que se
presenta a continuación:
8. Calibre el Spectronic 20D+ a 100% de transmitancia y 0 de absorbancia,
a la longitud de onda de trabajo de 620 nm, luego de un previo calentamiento de la
lámpara del instrumento durante 20 minutos.
9. Mida, con el Spectronic 20D+ a la longitud de onda de 620 nm, la
absorbancia de la solución blanco al menos 15 veces. Registre los valores en su
cuaderno de laboratorio.
5 mL
1 mL
4 mL
3 mL
2 mL
Sol. Int. 50 ppm en P
10
1
10
1 mL Sol. C6H8O6 al 1%
10 mL Sol. Buffer de Acetato
10 mL Sol. H2MoO4
Sol. Blanco
5 mL
1 mL
4 mL
3 mL
2 mL
Sol. Int. 50 ppm en P
10
1
10
1 mL Sol. C6H8O6 al 1%
10 mL Sol. Buffer de Acetato
10 mL Sol. H2MoO4
5 mL
1 mL
4 mL
3 mL
2 mL
Sol. Int. 50 ppm en P
5 mL
1 mL
4 mL
3 mL
2 mL
5 mL
1 mL
4 mL
3 mL
2 mL
5 mL
1 mL
4 mL
3 mL
2 mL
Sol. Int. 50 ppm en P
10
1
10
1 mL Sol. C6H8O6 al 1%
10 mL Sol. Buffer de Acetato
10 mL Sol. H2MoO4
101010
1111
101010
1 mL Sol. C6H8O6 al 1%1 mL Sol. C6H8O6 al 1%
10 mL Sol. Buffer de Acetato10 mL Sol. Buffer de Acetato
10 mL Sol. H2MoO410 mL Sol. H2MoO4
Sol. BlancoSol. Blanco

Profesor Argenis J. Sánchez Henríquez
161
10. Mida por triplicado, con el Spectronic 20D+ a la longitud de onda de 620
nm, las absorbancias del complejo azul de molibdeno que se forma para cada una
de las soluciones estándares que conforman la curva de calibración. Calibrando el
instrumento cada 10 lecturas de absorbancia realizadas. Y no realice las replicas
de forma secuencial, sino cíclica. Registre los valores en su cuaderno de
laboratorio.
11. Obtenga la media aritmética ( blA ) y la desviación estándar ( blS ) de los
valores de absorbancia para la solución blanco. Registre estos resultados en su
cuaderno de laboratorio.
12. Grafique, en papel milimetrado o algún “software” apropiado, los valores
medios de absorbancias, obtenidos para cada solución estándar preparada para la
curva de calibración, contra sus respectivos valores de concentración, en un eje
cartesiano (x, y); donde el eje y, es la ordenada representada por la absorbancia, y
el eje x, es la abscisa representada por la concentración de fósforo en unidades de
parte por millón (ppm). Recuerde corregir cada lectura de absorbancia de las
soluciones estándares con la lectura de absorbancia media obtenida para la
solución del blanco.
i
i nxx / )1/()( 2 nxxSi
i
blstdstdcorregida AAA 11, C (ppm de P)
0,0
0,5
1,0
1,5
2,0
0,000
0,060
0,118
0,173
0,231
Acorregida,stdn
0
1
2
3
4
C (ppm de P)
0,0
0,5
1,0
1,5
2,0
0,000
0,060
0,118
0,173
0,231
Acorregida,stdAcorregida,stdn
0
1
2
3
4
Cstd1 Cstd2 Cstd3 Cstd40
Astd1
Astd2
Astd3
Astd4
Concentración P ppm
Absorb
ancia
Cstd1 Cstd2 Cstd3 Cstd40
Astd1
Astd2
Astd3
Astd4
Concentración P ppm
Absorb
ancia
Cstd1 Cstd2 Cstd3 Cstd40
Astd1Astd1
Astd2Astd2
Astd3Astd3
Astd4Astd4
Concentración P ppm
Absorb
ancia

Profesor Argenis J. Sánchez Henríquez
162
13. Caracterice, empleando la pendiente de la curva de calibración (m), el
corte (b) y los parámetros estadísticos para la absorbancia de la solución blanco
( blA , blS ), la curva de calibración diseñada para la determinación de fósforo.
Concentración del límite de detección:
Concentración mínima del analito (P):
Sensibilidad de calibración:
Sensibilidad = Pendiente curva de calibración = m
Concentración máxima de analito:
Determinada por el punto de inflexión en la curva de calibración, es
decir, el par de valores x, y, donde la recta se aleja de la linealidad o la regresión
lineal de la curva de calibración comienza a alejarse de la unidad (1).
3.4. Determinación de la concentración de fósforo inorgánico en el
extracto de cenizas.
Fundamento
La absorbancia que presenta el complejo azul de molibdeno a la longitud de
onda analítica de trabajo, 620 nm, es directamente proporcional a la concentración
de fósforo inorgánico que se encuentra presente en cada una de las soluciones de
cenizas o muestras, y que sirve como centro de coordinación del complejo. Las
LDLD CmbAblblLD SAA 3
mínmínCmbA
blblmínSAA 10

Profesor Argenis J. Sánchez Henríquez
163
absorbancias, del complejo azul de molibdeno, en las soluciones estándares
preparadas obedecen la Ley de Beer-Lambert en el intervalo de concentraciones
de la curva de calibración diseñada; por lo que si las lecturas de absorbancias
medias obtenidas para el complejo en las diferentes muestras o soluciones de
cenizas entran en el intervalo de concentraciones estudiado por la curva de
calibración, se puede determinar la concentración de fósforo presente en estas
muestras por interpolación en la curva de calibración o por simple despeje de la
Ley de Beer-Lambert.
Material a Utilizar
1. Matraces aforado de 100 mL.
2. Piseta
3. Pipeta volumétricas de 1, 2, y 10 mL 4. Vasos de precipitado.
5. Spectronic 20D+.
6. Celdas de vidrio.
Reactivos a Utilizar
1. Solución de cenizas
2. Ácido ascórbico al 1 % p/v.
3. (NH4)6Mo7O24 4H2O al 1 % p/v.
4. Buffer acetato/ácido acético
Nota Importante: Es responsabilidad de
cada estudiante entregar al inicio de cada
práctica, en su cuaderno de laboratorio, una
tabla donde se resuman las siguientes
características de los reactivos arriba
señalados: Fórmula, % Pureza, Densidad,
Código de frases de riesgo (R) y seguridad
(S) para cada reactivo.
Procedimiento
1. Prepare la solución de cenizas, obtenida en la actividad 3.2, para la
lectura de la absorbancia que generará el fósforo contenida en ella, al reaccionar
con los diferentes reactivos específicos empleados para la formación del complejo
azul de molibdeno. Para ello, siga el esquema de trabajo que se presenta a
continuación:

Profesor Argenis J. Sánchez Henríquez
164
2. Mida por triplicado, con el Spectronic 20D+ a la longitud de onda de 620
nm, la absorbancia del complejo azul de molibdeno generado para cada una de
los extractos de cenizas que se analizan. Registre estos valores en su cuaderno
de laboratorio.
3. Halle el valor de la concentración de fósforo (P) en las muestras de
extracto de cenizas, por interpolación de las lecturas de absorbancia media de
cada muestra en la curva de calibración, y a través de la Ley combinada de Beer-
Lambert.
4. Post- Laboratorio Aquí
Redactar un informe científico de laboratorio siguiendo las pautas
establecidas al principio de este manual, donde se reflejen los resultados y
10
1
10
1 mL Sol. C6H8O6 al 1%
10 mL Sol. Buffer de Acetato
10 mL Sol. H2MoO4
Sol. de Cenizas
2 mL
2 2 mLSol. de Cenizas
10
1
10
1 mL Sol. C6H8O6 al 1%
10 mL Sol. Buffer de Acetato
10 mL Sol. H2MoO4
101010
1111
101010
1 mL Sol. C6H8O6 al 1%1 mL Sol. C6H8O6 al 1%
10 mL Sol. Buffer de Acetato10 mL Sol. Buffer de Acetato
10 mL Sol. H2MoO410 mL Sol. H2MoO4
Sol. de Cenizas
2 mL
2 2 mLSol. de Cenizas
Sol. de Cenizas
2 mL
Sol. de Cenizas
2 mL2 mL
22 2 mLSol. de Cenizas
2 mLSol. de Cenizas
muestramuestra CmbA

Profesor Argenis J. Sánchez Henríquez
165
conocimientos adquiridos en el desarrollo de las actividades realizadas (3.1, 3.2,
3.3 y 3.4).
5. Cuestionario
1. ¿Cuáles son los métodos instrumentales de análisis?
2. ¿Qué es la EAM UV-Vis?
3. ¿Qué longitudes de onda abarca región espectral de la radiación UV-Vis,
y cómo se sub-divide la misma?
4. ¿Cómo se explica el comportamiento de absorción de radiación
electromagnética por parte de la materia?
5. ¿Qué es un cromóforo?
6. ¿Cuáles son los componentes fundamentales de un espectrofotómetro
para EAM UV-Vis?
7. Defina y escriba las distintas expresiones matemáticas para la Ley de
Beer-Lambert.
8. ¿Qué finalidad tiene la digestión ácida en la preparación del extracto de
cenizas?
9. ¿Qué es una solución blanco?
10. ¿Qué es una curva de calibración?
11. Describa la preparación de 500 mL de una solución de 750 ppm en
Cobre (Cu2+), a partir de la sal hidratada CuCl22H2O, que tiene una pureza de
99% y una masa molar de 170,48 g/mol.
6. Referencias Bibliográficas
Skoog Douglas A. Y James A. Leary. (1994). Análisis Instrumental. (4a Ed.).
McGraw-Hill. Madrid (España).
COMISIÓN VENEZOLANA DE NORMAS INDUSTRIALES. (1983).
Determinación de Fósforo. Alimentos. Fondo Normas. COVENIN 1178-83.
Caracas (Venezuela).

Profesor Argenis J. Sánchez Henríquez
166
Maritza Machado y Sorely Peraza. (2001). Manual de Práctica de
Laboratorio Química Analítica Instrumental. Universidad Centroccidental “Lisandro
Alvarado”. Barquisimeto (Venezuela).
Rouessac Francis y Annick Rouessac. (2003). Análisis Químico-Métodos y
Técnicas Instrumentales Modernas. (1a. Ed.). McGraw-Hill. Madrid (España).

Profesor Argenis J. Sánchez Henríquez
167
VII. BIBLIOGRAFÍA
Alexéiev Víctor N. (1976). Análisis Cuantitativo. Trad. Liminík E. Editorial
Mir. Moscú (Rusia).
Ayres Gilbert H. (1970). Análisis Químico Cuantitativo. (2a Ed.). Ediciones
del Castillo. Madrid (España).
Burguera P. Marcela. ( ). Química Analítica Cuantitativa-Manual Práctico
de Laboratorio. Universidad de Los Andes. Mérida (Venezuela).
Chemdat.net. (2007). Disponible en red: http://www.merck.cl. Consultado:
12 Oct 2007.
COMISIÓN VENEZOLANA DE NORMAS INDUSTRIALES. (1983).
Determinación de Fósforo. Alimentos. Fondo Normas. COVENIN 1178-83.
Caracas (Venezuela).
COMISIÓN VENEZOLANA DE NORMAS INDUSTRIALES. (1982).
Determinación de Calcio. Alimentos. Fondo Normas. COVENIN 1178-82. Caracas
(Venezuela).
Fischer Robert B. y Dennos G. Peters. (1970). Análisis Químico
Cuantitativo. (3a Ed.). Editorial Interamericana. Trad. Toral M. Teresa. México D.
F. (México).
Frases de Riesgo y Seguridad. (2007). Disponible en red:
http://www.unizar.es/guiar/1/Accident/An_riesgo/FrasesR.htm. Consultado: 26
Octubre 2007.

Profesor Argenis J. Sánchez Henríquez
168
Maritza Machado y Sorely Peraza. (2001). Manual de Práctica de
Laboratorio Química Analítica Instrumental. Universidad Centroccidental “Lisandro
Alvarado”. Barquisimeto (Venezuela).
Rouessac Francis y Annick Rouessac. (2003). Análisis Químico-Métodos y
Técnicas Instrumentales Modernas. (1a. Ed.). McGraw-Hill. Madrid (España).
Skoog Douglas A. y Donald M. West. (1970). Introducción a la Química
Analítica. Editorial Reverté. Barcelona (España).
Skoog Douglas A. y James A. Leary. (1994). Análisis Instrumental. (4a Ed.).
McGraw-Hill. Madrid (España)
Skoog Douglas A., Donald M. West, James F. Holler y Stanley R. Crouch.
(2001). Química Analítica. (7a Ed.). McGraw-Hill. México D.F. (México).
Vogel Arthur I. (1960). Química Analítica Cuantitativa (Teoría y Práctica).
(2a Ed.). Editorial Kapelusz. Buenos Aires (Argentina).
![Handbook esp[1]](https://img.pdfslide.es/doc/110x75/559a7e691a28aba2648b479e/handbook-esp1.jpg)




























