Embed Size (px)
Citation preview

PATOLOGIA HEPATICA EN EL PACIENTE CRITICO
El presente artículo es una actualización al mes de enero del 2006 del Capítulo del Dr. Carlos Lovesio,
del Libro Medicina Intensiva, Dr. Carlos Lovesio, Editorial El Ateneo, Buenos Aires (2001)
Se ha comprobado que hasta el 60% de los pacientes críticos pueden presentar alteraciones de la función hepática. Sólo una minoría presenta una enfermedad hepática primaria como causa de tales alteraciones, siendo las causas más comunes la repercusión hepática de la enfermedad de base, los efectos de las medidas implementadas en la unidad para revertir el trastorno fisiológico primordial, y los efectos indeseables de las drogas o asociaciones de drogas utilizadas. En otras ocasiones, la enfermedad hepática produce repercusiones sistémicas que afectan la evolución y el pronóstico de la enfermedad de base.
En el presente capítulo se analizarán situaciones particulares que por su frecuencia deben ser adecuadamente reconocidas y tratadas por los médicos intensivistas, incluyendo la respuesta hepática a la sepsis, el síndrome hepatopulmonar, la hipertensión pulmonar en las enfermedades hepáticas, la hepatitis isquémica y la enfermedad veno-oclusiva hepática.
EL HIGADO EN EL PACIENTE SEPTICO
INTRODUCCION
El hígado desempeña un rol clave durante los procesos sépticos. “Está involucrado como actor y como víctima” (Dhainaut y col.). Dos hechos fundamentales justifican el rol doble del hígado: el primero es la importancia del aporte sanguíneo al órgano, que representa alrededor del 25% del total del volumen minuto cardiaco. De particular interés es el flujo portal, que recoge la sangre de todo el lecho esplacno-mesentérico, una región especialmente sujeta a cambios vasomotores y receptora de la traslocación bacteriana durante la sepsis. El segundo es la heterogeneidad celular del hígado, que contiene la mayor parte de los macrófagos del organismo (células de Kupffer) capaces de eliminar las endotoxinas y bacterias que pueden estimular la respuesta inflamatoria sistémica. Los hepatocitos, por su parte, sintetizan las proteínas de fase aguda y las enzimas requeridas para modular la respuesta inflamatoria. Adicionalmente, durante la traslocación bacteriana desde el intestino, el hígado limita el acceso de sustancias proinflamatorias hacia la circulación sistémica.
Como actor, el hígado es el órgano fundamental para la eliminación bacteriana, inactivación de productos bacterianos, y eliminación y producción de mediadores inflamatorios.

Los hepatocitos, que exhiben receptores para múltiples mediadores (Ej. Factor de necrosis tumoral, interleuquina 1 y 6), modifican sus vías metabólicas hacia la captación de aminoácidos y gluconeogénesis, así como al incremento de la síntesis y liberación de factores de la coagulación, factores de complemento y enzimas antiproteolíticas (proteínas de fase aguda). Por distintos mecanismos, las proteínas de fase aguda contribuyen al estado procoagulante y a la inhibición de la fibrinolisis observada en la sepsis.
Como víctima, el hígado puede ser lesionado y ver alteradas sus funciones. La disfunción hepática puede ser la consecuencia de una alteración primaria que ocurre en las primeras horas que siguen a la lesión inicial. Esto está relacionado con el shock y la hipoperfusión del órgano, produciendo distintas alteraciones funcionales, incluyendo trastornos de coagulación. Esta disfunción hepática temprana se puede revertir con un tratamiento de soporte adecuado. La disfunción hepática también puede presentarse como una alteración secundaria silente, y en ocasiones imperceptible, que se caracteriza por la incapacidad de controlar bacterias, endotoxinas y mediadores inflamatorios, que pasan a la circulación. Esta disfunción secundaria puede promover y o agravar la disfunción orgánica múltiple, como consecuencia del fallo del eje intestino-hígado.
ALTERACIONES DE LA FUNCION HEPATICA INDUCIDAS POR LA SEPSIS
Las alteraciones de la función hepática inducidas por la sepsis involucran a varios tipos celulares y sus respectivas interacciones, así como a varios mediadores secretorios en forma local o sistémica. Tres tipos principales de células contribuyen a la respuesta hepática en la sepsis: las células de Kupffer, los hepatocitos y las células endoteliales sinusoidales. En adición, los neutrófilos activados, que son reclutados en el hígado y producen enzimas potencialmente destructivas, pueden producir lesión hepática directa.
Células de Kupffer. Las células de Kupffer constituyen el 70% de los macrófagos totales del organismo. Estas células se presume que constituyen la defensa primaria contra la bacteriemia y endotoxemia de origen portal. Previenen que las bacterias y endotoxinas accedan a la circulación sistémica a través de su remoción de la sangre venosa portal. Una vez activadas, las células de Kupffer producen citoquinas que a su vez regulan las funciones de las células hepáticas y endoteliales a través de una interacción paracrina, o bien se liberan a la circulación sistémica. Por otra parte, las células de Kupffer son potentes eliminadores de mediadores inflamatorios, productos tóxicos y citoquinas sistémicos y derivados del intestino, y de este modo, desempeñan un rol principal en la limitación de la magnitud de la respuesta inflamatoria sistémica.
La disfunción hepática puede promover el pasaje a la circulación de bacterias y endotoxinas, pero no es simple demostrar que este mecanismo esté involucrado en la patogénesis de la respuesta inflamatoria sistémica y de la falla orgánica múltiple.
Las células de Kupffer comparten con otros macrófagos la capacidad de iniciar y regular las respuestas inmunes y de producir y liberar mediadores inmunomodulatorios. Luego de la estimulación de las células de Kupffer por endotoxinas, se liberan moléculas proinflamatorias incluyendo el TNFα , IL-1α y β, IL-8, G-CSF, IL-12, IL-18 y GM-CSF, cuyos efectos son

contrabalanceados por las acciones antiinflamatorias del receptor soluble del TNFα, antagonistas del receptor de IL-1, IL-4, IL-6, IL10 y factor de crecimiento beta.
Hepatocitos. Durante la sepsis, las células del parénquima hepático están involucradas tanto en la respuesta inmune, para promover la defensa del huésped, como en la desviación metabólica hacia la gluconeogénesis, para priorizar la síntesis proteica y asegurar de este modo la reparación celular. En este sentido, los hepatocitos exhiben receptores para endotoxinas, citoquinas, mediadores inflamatorios y sustancias vasoactivas, que actúan modificando sus vías metabólicas hacia la captación de aminoácidos, ureagénesis y gluconeógenesis, así como al incremento de la síntesis y liberación de factores de coagulación, factores del complemento y proteínas de fase aguda.
El rol de las proteínas de fase aguda es aumentar las defensas del huésped y las funciones protectoras modulando una serie de procesos que lesionan a los tejidos orgánicos en el proceso de destruir a las bacterias. Existe un aumento de la síntesis de las denominadas proteínas de fase aguda positivas y una disminución de la producción de las consideradas negativas. Las concentraciones de proteína C reactiva, antitripsina, fibrinógeno, protrombina, haptoglobina, ceruloplasmina, ferritina, proteína de unión a lipopolisacárido y fibronectina están aumentadas durante la respuesta de fase aguda. A la inversa, los niveles de albúmina, properdina, lipoproteína de alta densidad, proteína C y antitrombina disminuyen. Esta regulación diferencial sugiere una repriorización para la síntesis y liberación sistémica de proteínas específicas durante la sepsis.
La respuesta de fase aguda contribuye significativamente al proceso procoagulante: a) aumentando la inhibición de la proteína C (α1-antitripsina y α2-macroglobulina); b) aumentando la proteína de unión a C4, que disminuye los niveles de proteína S activa; c) disminuyendo la síntesis hepática de antitrombina y proteína C; d) aumentando la expresión del factor tisular; y c) inhibiendo la fibrinosis. Todos estos fenómenos inducen un proceso procoagulante desfavorable en el curso del proceso séptico.
Células endoteliales. Las células endoteliales del hígado normal producen IL-1 e IL-6. Esta producción está aumentada en las células tratadas con endotoxinas. Las células endoteliales también pueden contribuir al estado procoagulante y proinflamatorio observado en la sepsis.
La producción de óxido nítrico por las células endoteliales puede aumentar la defensa orgánica a través de la promoción de actividad antimicrobiana y limitando la destrucción tisular eliminando aniones superóxido e inhibiendo la adhesión leucocitaria a las células del endotelio hepático.
Interacción neutrófilos-hepatocito. El clearance por parte de las células de Kupffer de material dañino puede proteger a otras células de la injuria, pero la liberación de proteasas o de radicales libres de oxígeno luego de la fagocitosis puede dañar a los hepatocitos adyacentes. La atracción y migración de los neutrófilos puede desempeñar un rol mayor en la protección contra la infección, pero también desempeña un rol significativo en el daño hepatocitario. Esta última observación está soportada por el hecho que el pretratamiento con anticuerpos antineutrófilos, que depletan de neutrófilos la sangre periférica, protege contra la disfunción hepática en animales de

experimentación. Los neutrófilos se consideran las células efectoras principales responsables del daño hepático en la sepsis.
DISFUNCION Y FALLO HEPATICO INDUCIDOS POR LA SEPSIS
La disfunción hepática primaria hace referencia a la disfunción inducida por la sepsis en el periodo inmediato al episodio de shock y resucitación; esta disfunción frecuentemente conduce a una coagulación intravascular diseminada y sangrado. El clearance de lactato y de aminoácidos, así como la síntesis de proteínas, están reducidos. La gluconeogénesis y la glicogenólisis están disminuidas, y se puede producir hipoglucemia. El aumento de las aminotransferasas es característico de esta lesión y refleja la lesión aguda celular y mitocondrial. Estas enzimas habitualmente retornan a niveles normales pocos días después de la resucitación.
A pesar de la opinión clásica sobre la existencia de hipoperfusión esplácnica durante la sepsis, estudios recientes han comprobado que el flujo sanguíneo esplácnico, el aporte y el consumo de oxígeno están aumentados tanto en la insuficiencia hepática aguda como en la sepsis. La capacidad del hígado de extraer oxígeno, aun bajo condiciones extremas, hace que el órgano sea poco afectado por la hipoxia. Existen evidencias crecientes de que tanto la insuficiencia hepática aguda como la sepsis se acompañan de un estado hipermetabólico en el área hepatoesplácnica, caracterizado por un aumento de la glicólisis e hiperlactacidemia. Esto no debe ser interpretado en forma rigurosa como una indicación de hipoxia. En efecto, la hipoxia esplácnica clínicamente significativa parece ser un fenómeno relativamente infrecuente en estos pacientes.
Se produce una disfunción silenciosa y en ocasiones imperceptible en forma secundaria al pasaje de bacterias, endotoxinas y mediadores inflamatorios, que superan el filtro hepático y pasan a la circulación sistémica, en un momento en que la mayoría de las funciones hepáticas están intactas. Areas hepáticas locales, inflamadas como consecuencia de la detoxificación de productos bacterianos por las células de Kupffer, inducen la activación de la coagulación y de las cascadas del complemento y de las quininas. Esta activación se produce como consecuencia de la liberación local de eicosanoides, óxido nítrico, endotelina, productos de la activación de las células cebadas y otros mediadores inflamatorios.
Los neutrófilos también participan en la lesión de las células endoteliales y de los hepatocitos. Una teoría propone que el aumento de la concentración de endotoxinas en la vena porta conduce a la estimulación del sistema de macrófagos hepáticos (Fig. 1). Las células de Kupffer activadas liberan leucotrieno B4 y TNFα, los que atraen neutrófilos al hígado y los activan in situ. Los neutrófilos sobreregulan sus moléculas de adhesión y se unen a las células sinusoidales. La sobreregulación de las integrinas endoteliales promueve la migración de neutrófilos y trombosis microvascular. Luego de la migración en el parénquima, los neutrófilos activados producen radicales libres de oxígeno y proteasas que pueden producir necrosis de los hepatocitos.

Fig. 1.- El rol de los neutrófilos en el daño endotelial y hepatocítico.
Una lesión adicional tanto a las células endoteliales como a los hepatocitos se produce cuando se inicia la resucitación. Se producen y liberan una serie de mediadores de reperfusión que aumentar la injuria tisular.
Las alteraciones precedentes pueden promover y o agravar la disfunción orgánica múltiple. La disfunción hepática puede ser una consecuencia de las infecciones repetidas o del shock, la sobreactivación de la respuesta sistémica, la persistencia del fallo de la microcirculación, o aun de los efectos indeseables de los tratamientos administrados.
CLINICA DE LA DISFUNCION HEPATICA EN LA SEPSIS
La lesión hepática ha sido investigada en pacientes críticos, pero pocos estudios han incluido sólo pacientes sépticos. Los criterios utilizados para definir la lesión hepática son la ictericia, hiperbilirrubinemia, aumento de la concentración plasmática de aminotransferasas, fosfatasa alcalina o lactodeshidrogenasa, y disminución de la concentración de albúmina sérica. Estos criterios varían en los distintos estudios. Un aumento desproporcionado de la concentración plasmática de bilirrubina total, en comparación con el aumento de las aminotransferasas, es característico de los pacientes sépticos. El tiempo de protrombina ha sido propuesto por Le Gall y col. como un criterio precoz de lesión hepática. Para cuantificar el grado de lesión hepática se han propuesto distintos escores con una gradación de severidad (Tabla 1). Estos escores miden el valor
más alterado observado durante la enfermedad. Una limitación importante es que no toman en cuenta la duración de la lesión hepática.
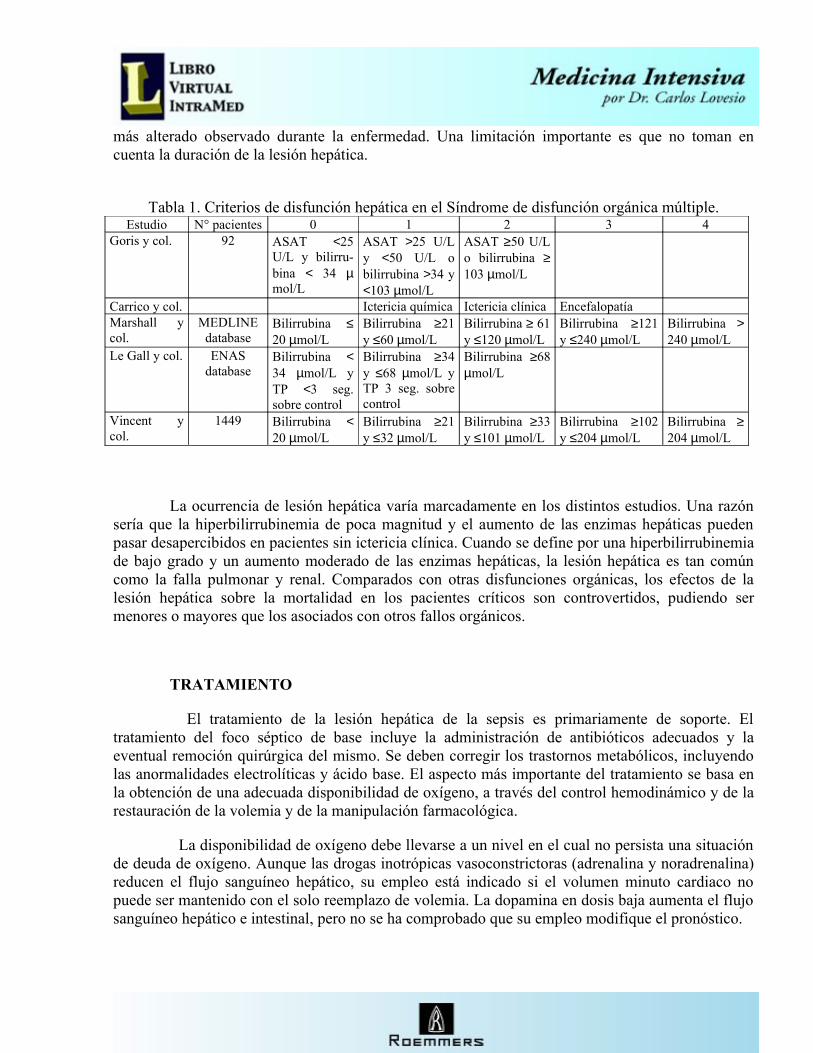
Tabla 1. Criterios de disfunción hepática en el Síndrome de disfunción orgánica múltiple.Estudio N° pacientes 0 1 2 3 4
Goris y col. 92 ASAT <25 U/L y bilirru-bina < 34 µmol/L
ASAT >25 U/L y <50 U/L o bilirrubina >34 y <103 µmol/L
ASAT ≥50 U/L o bilirrubina ≥103 µmol/L
Carrico y col. Ictericia química Ictericia clínica EncefalopatíaMarshall y col.
MEDLINE database
Bilirrubina ≤20 µmol/L
Bilirrubina ≥21 y ≤60 µmol/L
Bilirrubina ≥ 61 y ≤120 µmol/L
Bilirrubina ≥121 y ≤240 µmol/L
Bilirrubina >240 µmol/L
Le Gall y col. ENAS database
Bilirrubina <34 µmol/L y TP <3 seg. sobre control
Bilirrubina ≥34 y ≤68 µmol/L y TP 3 seg. sobre control
Bilirrubina ≥68 µmol/L
Vincent y col.
1449 Bilirrubina <20 µmol/L
Bilirrubina ≥21 y ≤32 µmol/L
Bilirrubina ≥33 y ≤101 µmol/L
Bilirrubina ≥102 y ≤204 µmol/L
Bilirrubina ≥204 µmol/L
La ocurrencia de lesión hepática varía marcadamente en los distintos estudios. Una razón sería que la hiperbilirrubinemia de poca magnitud y el aumento de las enzimas hepáticas pueden pasar desapercibidos en pacientes sin ictericia clínica. Cuando se define por una hiperbilirrubinemia de bajo grado y un aumento moderado de las enzimas hepáticas, la lesión hepática es tan común como la falla pulmonar y renal. Comparados con otras disfunciones orgánicas, los efectos de la lesión hepática sobre la mortalidad en los pacientes críticos son controvertidos, pudiendo ser menores o mayores que los asociados con otros fallos orgánicos.
TRATAMIENTO
El tratamiento de la lesión hepática de la sepsis es primariamente de soporte. El tratamiento del foco séptico de base incluye la administración de antibióticos adecuados y la eventual remoción quirúrgica del mismo. Se deben corregir los trastornos metabólicos, incluyendo las anormalidades electrolíticas y ácido base. El aspecto más importante del tratamiento se basa en la obtención de una adecuada disponibilidad de oxígeno, a través del control hemodinámico y de la restauración de la volemia y de la manipulación farmacológica.
La disponibilidad de oxígeno debe llevarse a un nivel en el cual no persista una situación de deuda de oxígeno. Aunque las drogas inotrópicas vasoconstrictoras (adrenalina y noradrenalina) reducen el flujo sanguíneo hepático, su empleo está indicado si el volumen minuto cardiaco no puede ser mantenido con el solo reemplazo de volemia. La dopamina en dosis baja aumenta el flujo sanguíneo hepático e intestinal, pero no se ha comprobado que su empleo modifique el pronóstico.

Si bien la recomposición de la disponibilidad de oxígeno sistémica es fundamental en el tratamiento de la sepsis, la misma no asegura la adecuada disponibilidad a lechos específicos, en particular el eje intestino-hepático. La determinación de la perfusión de órganos específicos se puede realizar midiendo su diferencia arterio-venosa de oxígeno, o por métodos indirectos, tal la medición del pH de la mucosa gástrica por tonometría intragástrica a los fines de inferir datos sobre la perfusión esplácnica. La corrección precoz de los trastornos de la perfusión ha demostrado ser útil para evitar la repercusión sistémica de la sepsis y para aumentar la sobrevida (Rivers y col.).
Han sido destacadas por muchos autores las ventajas del soporte nutricional específico en la sepsis y en la falla pluriparenquimatosa, aunque no se ha descrito ningún régimen nutricional particular capaz de aumentar la sobrevida. Los regimenes de nutrición parenteral no deben incluir un exceso de glucosa, recomendándose administrar hasta el 25% de las calorías no proteicas bajo la forma de lípidos. El valor específico de los aminoácidos de cadena ramificada en el tratamiento del hígado de sepsis no ha sido investigado. Todos los estudios coinciden en la ventaja de la nutrición enteral sobre la parenteral, siempre que pueda ser utilizada.
SINDROME HEPATOPULMONAR
DEFINICION
El síndrome hepatopulmonar consiste en una triada de disfunción hepática, dilatación vascular intrapulmonar e hipoxia. A continuación se definen los componentes requeridos para el diagnóstico del síndrome:
1. Disfunción hepática
a. Cirrosis
b. Hipertensión portal de origen no cirrótico
c. Hepatitis aguda fulminante
d. Rechazo de injerto
2. Hipoxemia
a. Aumento del gradiente alvéolo-arterial (>20 mm Hg) en reposo, en la posición supina o en posición erecta (ortodeoxia)
3. Dilatación vascular intrapulmonar
a. Demostrada por ecocardiografía contrastada, centellografía de perfusión pulmonar o angiografía pulmonar

La vasodilatación en la microcirculación pulmonar puede ser detectada y cuantificada por una serie de técnicas de diagnóstico por imágenes (ver más adelante), reconociéndose en la actualidad que el síndrome hepatopulmonar se desarrolla en el 15 al 20% de los pacientes con cirrosis.
FISIOPATOLOGIA
La fisiopatología del síndrome hepatopulmonar no es bien conocida, aunque los cambios estructurales han sido bien descriptos. En el año 1966, Berthelot y col. fueron los primeros en demostrar la presencia de marcada dilatación precapilar (por encima de 500 µm de diámetro). Describieron esta alteración como “nevus arácnidos” del pulmón. Los cambios estructurales hallados en el síndrome hepatopulmonar en la autopsia sugieren la existencia de dilataciones vasculares pulmonares y pleurales. En conjunto, estos cambios se considera que constituyen las dilataciones vasculares intrapulmonares que caracterizan el síndrome hepatopulmonar.
Las anormalidades vasculares que producen alteraciones del intercambio gaseoso en el síndrome hepatopulmonar asumen dos formas fisiológicas. Las dilataciones difusas precapilares y capilares producen anormalidades de la relación ventilación/perfusión en las cuales sangre venosa no saturada pasa a través de vasos dilatados y es inadecuadamente saturada con oxígeno debido a la combinación de un alto flujo de perfusión y un aumento de la distancia para la difusión. La hipoxia observada en estas circunstancias puede ser revertida respirando oxígeno al 100%. Ello permite distinguirla de la segunda forma fisiológica, en la cual se producen comunicaciones arteriovenosas directas que crean un shunt anatómico verdadero y no se evidencia mejoría con la respiración con oxígeno puro. Teniendo en cuenta el estado hiperdinámico de los pacientes con enfermedad hepática avanzada, la hipoxemia puede ser agravada por el disminuido tiempo de tránsito de los glóbulos rojos a nivel pulmonar.
Otro problema a resolver es el mecanismo que produce la dilatación vascular intrapulmonar. El síndrome hepatopulmonar afecta primariamente las arteriolas precapilares y los capilares en las bases pulmonares, produciendo dilatación de los vasos. Normalmente, estos vasos miden aproximadamente 8 a 15 µm; en presencia de un síndrome hepatopulmonar, pueden exceder los 500 µm de diámetro. La pérdida del tono vascular en los pacientes con dilatación vascular intrapulmonar no ha sido explicada. Muchos admiten que existe un disbalance entre vasodilatadores y vasoconstrictores pulmonares, produciendo un predominio de la vasodilatación en pacientes con síndrome hepatopulmonar. En el extremo opuesto se encuentran los enfermos en que predomina el tono vasoconstrictor y presentan hipertensión pulmonar.
En la actualidad se asume que la producción vascular excesiva de vasodilatadores, en particular de óxido nítrico, es responsable de la vasodilatación en el síndrome hepatopulmonar. Las evidencias que soportan esta asunción surgen de la observación que el nivel de óxido nítrico exhalado está aumentado en pacientes con el síndrome y se normaliza luego del trasplante en la medida en que el síndrome hepatopulmonar se resuelve. En adición, la administración aguda de azul

de metileno, un inhibidor de la acción del óxido nítrico, o de L-NAME, un inhibidor de la óxido nítrico sintetasa, mejora transitoriamente la oxigenación. A pesar de estos hallazgos, no siempre se encuentra una relación entre los niveles de óxido nítrico y la magnitud de la vasodilatación pulmonar.
En los últimos años, una serie de estudios experimentales se han focalizado en el rol de la ET-1 como mediador inicial de las alteraciones endoteliales que ocurren en el síndrome hepatopulmonar. Se ha comprobado que existe un aumento en el nivel del receptor vascular endotelial de la endotelina (ETB) en la vasculatura pulmonar; la estimulación de este receptor por la ET-1 aumenta la producción local de óxido nítrico.
CUADRO CLINICO
Los pacientes con síndrome hepatopulmonar generalmente se presentan con manifestaciones hepáticas más que pulmonares. El 82% de los pacientes se presentan con síntomas o signos relacionados con su enfermedad hepática, y el resto se presenta con disnea. La duración de los síntomas respiratorios es de hasta cinco años antes de que se haga evidente el diagnóstico de síndrome hepatopulmonar.
La historia natural del síndrome hepatopulmonar es desconocida. Los pacientes con enfermedad hepática que presentan dilatación intrapulmonar por ecografía contrastada pueden o no presentar alteraciones del intercambio gaseoso.
Las manifestaciones clínicas del síndrome hepatopulmonar son características del síndrome pero no exclusivas. Los síntomas pulmonares incluyen disnea de ejercicio y platipnea, que es la sensación de respiración dificultosa cuando el paciente pasa de la posición supina a la erecta. Los hallazgos físicos incluyen dedos en palillo de tambor y cianosis de las extremidades en aquellos con hipoxemia significativa, aunque pacientes con significativa hipoxemia pueden no tener cianosis. En adición se encuentran los hallazgos de la enfermedad hepática crónica, incluyendo hepatomegalia, ascitis y nevus cutáneos.
Se debe destacar que el síndrome hepatopulmonar generalmente se reconoce en pacientes con una función de síntesis hepática bien conservada, no existiendo una relación entre los índices bioquímicos de disfunción hepática o la clasificación de Child-Pugh y la severidad de la hipoxemia o la magnitud del shunt vascular intrapulmonar.
Un hallazgo característico del síndrome hepatopulmonar es la ortodeoxia, que sin embargo, no es exclusivo del síndrome. La ortodeoxia se define por una disminución en la PaO2
mayor de 3 mm Hg, cuando el paciente se mueve de la posición supina a la erecta. Este hallazgo, responsable de la platipnea, probablemente refleja el grado y localización de las dilataciones vasculares pulmonares. Teóricamente, debido a que las dilataciones vasculares tienden a localizarse en las bases de los pulmones, la posición erecta produce un aumento del flujo sanguíneo por gravedad, resultando en una agravación del shunt. La ortodeoxia se produce en aproximadamente el 88% de los pacientes con síndrome hepatopulmonar que respiran aire u oxígeno al 100%.

La platipnea-ortodeoxia no es privativa del síndrome hepatopulmonar, habiéndose descrito en pacientes con shunts intracardiacos de derecha a izquierda (foramen oval permeable o defectos atriales), luego de resecciones pulmonares, asociada a deformidades esqueléticas y enfermedades pulmonares (EPOC, embolismo pulmonar, tumores de la vía aérea superior y SDRA).
DIAGNOSTICO
El diagnóstico del síndrome hepatopulmonar incluye la medida de los gases en sangre arterial, los estudios del shunt por la determinación de los gases en sangre, centellograma pulmonar, ecocardiografia contrastada y angiografía pulmonar.
La determinación de gases en sangre debe ser la primera medida destinada a la evaluación de la disnea en pacientes con enfermedad hepática. Se deben destacar dos puntos. Primero, la determinación de la saturación arterial de oxígeno puede ser utilizada como método de descarte, pero el valor informado puede ser falsamente bajo en pacientes con hiperbilirrubinemia. Segundo, la determinación de la saturación de oxígeno debe ser realizada en pacientes respirando aire ambiente en la posición erecta. Ello permite reconocer la ortodeoxia.
En conjunto con la PaO2 anormal, se debe realizar un estudio del shunt a partir de la saturación obtenida con el paciente respirando aire y una concentración de oxígeno al 100%, en posición supina y erecta. El test de suplementación con oxígeno al 100% permite la distinción entre el síndrome tipo I, con un predominio de dilataciones precapilares y capilares y una respuesta casi normal a la administración de oxígeno al 100% (PaO2 >400 mm Hg), y el tipo II, con la presencia de cambios similares a aquellos de las malformaciones arteriovenosas y una inadecuada respuesta al oxígeno al 100%. Se ha recomendado que en los pacientes con respuestas de menos de 300 mm Hg se realice una arteriografía para la determinación de la presencia de un síndrome hepatopulmonar tipo II y para seleccionar pacientes para ser tratados con embolización.
Otro método para cuantificar el grado de dilatación vascular es el centellograma de cuerpo entero con macroagregados de albúmina marcados con technetium-99. La mayor parte de la albúmina marcada normalmente es atrapada en la vasculatura pulmonar y el pulmón capta la mayor parte del radioisótopo. En presencia de un shunt intrapulmonar o intracardiaco, el isótopo no es atrapado en el lecho capilar pulmonar y pasa al cerebro, hígado y riñones. La cantidad de radioisótopo que escapa a la circulación pulmonar puede ser cuantificada.
La ecocardiografía contrastada se ha convertido en el gold standard para demostrar la presencia de dilataciones vasculares intrapulmonares. Esta técnica utiliza solución salina agitada, creando microburbujas de al menos 15 µm de diámetro. Cuando se inyectan por vía periférica, estas burbujas son atrapadas durante el primer pasaje en los capilares pulmonares y luego reabsorbidas. En pacientes con dilataciones vasculares intrapulmonares, las burbujas son visualizadas en la aurícula izquierda por ecocardiografia transtorácica o trasesofágica dentro de los tres o seis ciclos cardiacos que siguen a la observación en las cavidades derechas.
La tomografía de tórax de alta resolución en pacientes con síndrome hepatopulmonar puede mostrar arterias pulmonares periféricas agrandadas. En los pacientes normales, las arterias pulmonares en las proximidades de la superficie pleural son muy pequeñas como para ser visualizadas, pero en pacientes con síndrome hepatopulmonar, las dilataciones vasculares en general son fácilmente identificadas en las regiones subpleurales del pulmón.
La angiografía pulmonar es útil para delinear las anormalidades pulmonares vasculares en el síndrome hepatopulmonar. Es útil para excluir la enfermedad tromboembólica crónica como causa de hipoxemia en estos pacientes. El estudio arteriográfico ha permitido clasificar el síndrome hepatopulmonar en distintos grados, de acuerdo a la magnitud de las lesiones observadas.
PRONOSTICO
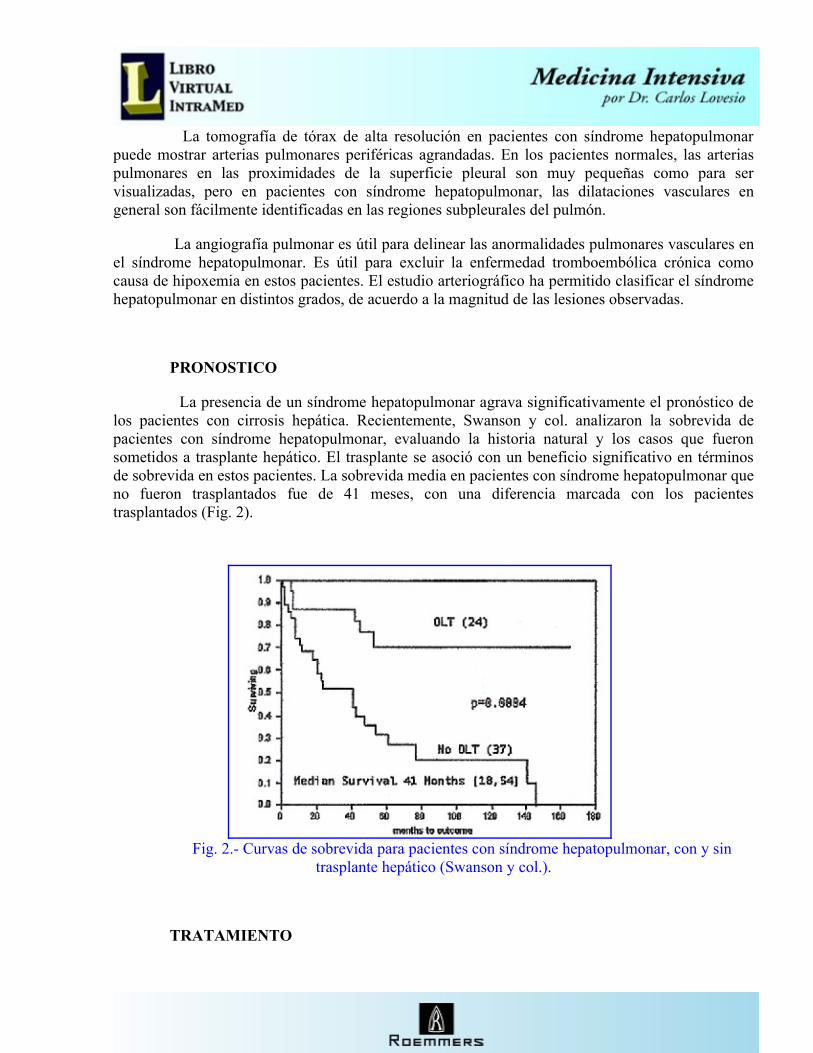
La presencia de un síndrome hepatopulmonar agrava significativamente el pronóstico de los pacientes con cirrosis hepática. Recientemente, Swanson y col. analizaron la sobrevida de pacientes con síndrome hepatopulmonar, evaluando la historia natural y los casos que fueron sometidos a trasplante hepático. El trasplante se asoció con un beneficio significativo en términos de sobrevida en estos pacientes. La sobrevida media en pacientes con síndrome hepatopulmonar que no fueron trasplantados fue de 41 meses, con una diferencia marcada con los pacientes trasplantados (Fig. 2).
Fig. 2.- Curvas de sobrevida para pacientes con síndrome hepatopulmonar, con y sin trasplante hepático (Swanson y col.).
TRATAMIENTO

Se han propuesto una serie de agentes terapéuticos para el tratamiento del síndrome hepatopulmonar, sin mejoría sustancial en la evolución. Teóricamente, estos agentes actuarían produciendo vasoconstricción (tal como la almitrina) o inhibiendo la vasodilatación (tal como el azul de metileno). El octreótido administrado en pacientes severamente hipoxémicos disminuye significativamente la fracción de shunt, pero investigaciones recientes no han podido confirmar una respuesta positiva reproducible. Desgraciadamente, ninguno de estos agentes ha modificado la historia natural de la enfermedad.
La radiología intervencionista ofrece otra opción terapéutica. Poterucha y col. recientemente informaron una embolización vascular exitosa en un paciente con lesiones severas responsables de síndrome hepatopulmonar, cuya PaO2 en posición erecta era de 45 mm Hg. Luego de la embolización, la PaO2 alcanzó a 59 mm Hg. Se admite que la embolización sería una opción adecuada en pacientes con gran shunt de derecha a izquierda intrapulmonar que no mejora con la respiración con oxígeno al 100%.
En casos de severa hipoxemia, el trasplante hepático puede ser riesgoso o estar contraindicado y no siempre se asocia con una reversión completa de las anormalidades de oxigenación. La colocación de un shunt portosistémico intrahepático (TIPS), reduciendo la presión portal y sus consecuencias, puede revertir la fisiopatología del síndrome. También puede inducir un aumento en el volumen minuto cardiaco, produciendo una redistribución del flujo sanguíneo pulmonar y mejorando la relación ventilación/perfusión. En la literatura existen ejemplos aislados de empleo del TIPS en estas circunstancias, como tratamiento paliativo a largo tiempo o como puente para la realización de un trasplante hepático.
Una serie de informes recientes han documentada una reversión completa de las dilataciones vasculares intrapulmonares y mejoría de la oxigenación luego del trasplante hepático hasta en el 70% de los pacientes, a pesar de la presencia de una severa hipoxemia preoperatoria. Castro y col., sostienen que el síndrome hepatopulmonar constituye una indicación para el transplante hepático en el contexto de una hipoxemia progresiva o al menos antes del desarrollo de una hipoxemia severa en pacientes apropiados.
El mayor riesgo de mortalidad postrasplante en pacientes con síndrome hepatopulmonar se ha descrito en aquellos con una PaO2 pretrasplante de menos de 50 mm Hg y en aquellos con una captación de technetium marcado por el cerebro mayor del 30%. La normalización de la PaO2 y la resolución de las dilataciones vasculares se han descrito en el 62 al 82% de los pacientes trasplantados. Sin embargo, la resolución puede ser lenta, requiriendo hasta 15 meses de oxígeno suplementario en el postoperatorio.
SINDROME DE HIPERTENSION PORTOPULMONAR

DEFINICION
La hipertensión portopulmonar (HPP) es el desarrollo de hipertensión arterial pulmonar causado por la obstrucción vascular al flujo sanguíneo dependiente de la proliferación anormal de las células endoteliales y musculares lisas, así como de la vasoconstricción y de la trombosis in situ. En individuos susceptibles genéticamente, este síndrome sigue al desarrollo de hipertensión portal. Como en el caso del síndrome hepatopulmonar, su presencia tiene implicancias significativas cuando se considera la realización de un trasplante ortotópico de hígado.
EPIDEMIOLOGIA
La HPP es relativamente infrecuente. En la era del trasplante hepático, los informes de grandes centros de trasplante en el mundo sugieren una frecuencia del 4 al 14,5%. El aumento de la presión pulmonar sugerido por la ecocardiografia Doppler es relativamente común (30-40%), pero la razón más frecuente incluye los efectos de la circulación hiperdinámica y el exceso de volemia. Ninguna de estas condiciones se asocia con el pronóstico ominoso que se documenta cuando existe una HPP verdadera.
FISIOPATOLOGIA
No existe un modelo animal que simule la patología de la HPP, que incluye vasoconstricción, proliferación endotelial y del músculo liso, trombosis in situ, agregación plaquetaria y dilatación vascular con recanalización por estructuras plexiformes. Se admite actualmente que la hipertensión arterial pulmonar es mucho más que una simple vasoconstricción que puede ser modificada con la utilización de un vasodilatador. Aunque se han implicado mediadores específicos tales como la endotelina-1, estarían involucrados otros factores tales como el efecto del alto flujo y la injuria por estrés de estiramiento sobre el endotelio pulmonar, además de la liberación de productos plaquetarios y una susceptibilidad genética.
CUADRO CLINICO Y DIAGNOSTICO
El hallazgo cardinal de la HPP es la disnea de esfuerzo, la cual no es un síntoma específico en el contexto de la enfermedad hepática avanzada. A medida que la hipertensión progresa, habitualmente en meses o años, el paciente desarrolla dolor torácico, disnea de reposo, palpitaciones y síncope. Este último síntoma es particularmente alarmante. La muerte súbita no es infrecuente, afectando al 25% de los pacientes con cualquier forma de hipertensión pulmonar severa.
La hipertensión arterial pulmonar que caracteriza a la HPP es mejor definida por un procedimiento invasivo, la cateterizacion cardiaca derecha. En la Tabla 2 se indican los criterios de

una reunión de consenso de la European Respiratory Society-European Study for Advanced Liver Disease para el diagnóstico de la HPP. El aumento de la resistencia vascular pulmonar (RVP) es una característica clave de la HPP. Los valores normales de RVP pueden variar. Valores entre 120-240 dinas/seg/cm-5 se consideran anormales por algunos investigadores. Los valores de RVP >240 dinas/seg/cm-5 son considerados anormales por todos los investigadores.
Tabla 2.- Criterios diagnósticos para el síndrome de hipertensión portopulmonar
1. Presencia de hipertensión portal (diagnóstico clínico)
2. Presión arterial pulmonar media (PAPM) >25 mm Hg
3. Presión de oclusión arterial pulmonar (POAP) <15 mm Hg
4. Resistencia vascular pulmonar (RVP) >240 dinas/seg/cm-5
El método óptimo de primera evaluación es el ecocardiograma Doppler trastorácico, con el cual se puede realizar una estimación de la presión sistólica en la arteria pulmonar y del tamaño y la función del ventrículo derecho. El Doppler provee una estimación de la presión sistólica del ventrículo derecho, la cual es una estimación razonable de la presión pulmonar arterial sistólica. En presencia de una presión pulmonar aumentada, se debe realizar una cateterización del ventrículo derecho. En la Tabla 3 se indica un esquema de clasificación de la hipertensión portopulmonar.
Tabla 3.- Clasificación de la Mayo Clinic de la hipertensión pulmonar en el contexto de la hipertensión portal.Tipo PAPM POP VMC RVPEstados de alto flujo en arteria pulmonar ↑ No o ↓ ↑↑ ↓Exceso de volumen venoso pulmonar ↑ ↑ ↑ ↓Hipertensión portopulmonar a. Obstrucción vascular pulmonar b. Obstrucción vascular pulmonar con exceso de volumen
↑↑↑↑↑
No o ↓↑
↑↑
↑↑↑↑
PAPM: presión media en arteria pulmonar; POP: presión de oclusión pulmonar; VMC: volumen minuto cardiaco; RVP: resistencia vascular pulmonar
TRATAMIENTO
En los últimos años se han descrito una serie de sustancias capaces de mejorar la hemodinamia pulmonar en las formas moderadas a severas de HPP, incluyendo prostaciclinas

intravenosas e inhaladas, un antagonista dual del receptor de endotelina por vía oral (bosentan) y un inhibidor oral de la fosfodiesterasa (sildenafil). El tratamiento recomendado por la Mayo Clinic en pacientes con PAMP >35 mm Hg y con una buena opción de trasplante hepático es la administración continua de una prostaciclina, el epoprosterenol, por infusión intravenosa.
A diferencia del síndrome hepatopulmonar, la HPP no se considera una razón justificable para proceder a un trasplante hepático. Las razones son dos. Primero, aun con la mejoría pretrasplante en la hemodinamia pulmonar y un trasplante exitoso, algunos pacientes no experimentan una cura para la HPP; la hemodinamia pulmonar puede mejorar, pero no normalizarse. Segundo, aun con una adecuada selección de pacientes y el empleo de prostaciclinas y óxido nítrico inhalado, muchos pacientes mueren o presentan una larga estadía hospitalaria luego del trasplante hepático. Por estas razones, cada centro de trasplante debe fijar su propia política respecto a la conducta terapéutica en esta grave patología.
HEPATITIS ISQUEMICA
En el año 1979, Bynum y col. acuñaron el término “hepatitis isquémica” para hacer referencia a la lesión hepática caracterizada por necrosis celular centrilobular, y evidenciada por un incremento brusco y significativo en la actividad de aminotransferasas en el suero en el contexto de una insuficiencia cardiaca. Los autores propusieron la expresión hepatitis debido a ciertas similitudes clínicas (anorexia, mal estado general, ictericia, hepatomegalia dolorosa) con la hepatitis infecciosa, y la expresión isquémica debido a que asumieron que la necrosis hepática se producía como consecuencia de la hipoperfusión del órgano.
Henrion y col., recogiendo una amplia experiencia sobre esta patología, consideran que la isquemia hepática, esto es la disminución en el flujo sanguíneo hepático, no es el único mecanismo hemodinámico responsable de la necrosis hepática, y por ello han propuesto denominar al síndrome “hepatitis hipóxica”. Los autores consideran que la hepatitis hipóxica no es una condición rara, afectando al 0,9% de los pacientes admitidos a terapia intensiva.
CONCEPTO
La hepatitis isquémica se caracteriza por los siguientes criterios: 1) aparición en un contexto clínico de fallo cardiaco, circulatorio o respiratorio; 2) presencia de un aumento brusco y significativo pero transitorio en la actividad de aminotransferasas en el suero, alcanzando al menos 20 veces el límite superior de lo normal; y 3) exclusión de otras causas posibles de necrosis celular hepática, en particular la hepatitis viral o inducida por drogas. No se requiere la biopsia hepática para el diagnóstico de hepatitis isquémica, siempre que se reúnan los criterios anteriores.

ETIOLOGIA
Henrion y col. pudieron agrupar a los pacientes que desarrollaron una hepatitis isquémica en cuatro grupos bien definidos. La condición basal más frecuente fue la insuficiencia cardiaca (70%), y particularmente la insuficiencia cardiaca congestiva (56%). Esto coincide con el resto de la literatura, en la cual se encuentra una prevalencia del 81% para la enfermedad cardiaca y un 63% para la insuficiencia cardiaca congestiva. Los episodios de hepatitis hipóxica relacionados con insuficiencia cardiaca congestiva en general están precedidos por un periodo de deterioro progresivo de la función cardiaca y son precipitados por un evento agudo, incluyendo arritmias, edema agudo de pulmón o embolismo pulmonar.
En el estudio de los autores, el 16% de los episodios de hepatitis isquémica fueron atribuidos a una insuficiencia respiratoria severa crónica, situación que no se repite en otros estudios y que se ha atribuido a la alta incidencia de neumoconiosis en la experiencia de Henrion y colaboradores.
Un tercer grupo de pacientes está constituido por aquellos con insuficiencia cardiaca aguda (12,5% a 16% según los autores). El proceso más frecuente en este grupo es el infarto agudo de miocardio. Por último, la causa menos frecuente de hepatitis isquémica es el shock circulatorio, incluyendo el shock séptico, tóxico, traumático, hemorrágico e hipovolémico. Es llamativo que en la mayoría de los estudios la hepatitis isquémica producida por el shock hemorrágico sea excepcional, aun en pacientes con hemorragias digestivas severas asociadas con cirrosis hepática.
FISIOPATOLOGIA
A partir del estudio de Dunn y col. se ha propuesto que tres mecanismos hemodinámicos teóricamente pueden resultar en una injuria hipóxica del hígado: la isquemia debido a la disminución del flujo sanguíneo hepático, la congestión venosa debido a la falla cardiaca derecha, y la hipoxemia debida a la disminución del contenido de oxígeno en la sangre aferente. Los estudios actuales demuestran que la isquemia hepática no es el único determinante hemodinámico de la hepatitis hipóxica; que el rol de la congestión venosa es importante en la mayoría de los casos relacionados con falla cardiaca; que el rol de la hipoxemia arterial es crucial en los casos relacionados con la insuficiencia respiratoria crónica; y que otros mecanismos pueden interactuar en los casos relacionados con shock circulatorio.
Cuando la insuficiencia cardiaca es la causa de base de la hepatitis hipóxica, el rol de la isquemia está sugerido por la disminución de la presión arterial media, y está directamente soportado por las mediciones de la disponibilidad de oxígeno y del flujo sanguíneo hepático. El aporte de oxígeno, que depende del volumen minuto cardiaco y del contenido de oxígeno de la sangre arterial está próximo al valor crítico como consecuencia de la disminución del índice

cardiaco. El flujo sanguíneo hepático está deprimido en la insuficiencia cardiaca descompensada con y sin hepatitis hipóxica, pero está significativamente más deprimido cuando está presente la hepatitis hipóxica. El rol del estasis venoso fue sugerido por la elevada prevalencia de signos clínicos de falla cardiaca derecha, y está soportado por la presión venosa central elevada observada en todos los pacientes con hepatitis hipóxica relacionada con falla cardiaca. Se ha comprobado que en la insuficiencia cardiaca descompensada, la presión venosa central está significativamente más elevada cuando existe una hepatitis hipóxica. Se ha postulado que en casos de hepatitis hipóxica relacionada con insuficiencia cardiaca congestiva, el hígado esta crónicamente expuesto a cierto grado de hipoxia por la congestión venosa crónica. En el momento de un evento cardiogénico agudo, aun de carácter moderado y breve, un descenso de la presión arterial, aun no reconocido, agrava la hipoxia de modo suficiente como para inducir necrosis hepática. Esto puede explicar porque no se reconoce un episodio franco de shock en muchos casos de hepatitis hipóxica.
Cuando la insuficiencia respiratoria crónica es la causa de base de la hepatitis hipóxica, el grado de hipoxemia arterial es considerable, con niveles de PaO2 por debajo de 40 mm Hg en la mayoría de los casos. En esta situación, por otra parte, es habitual que una descompensación cardiaca precipite el episodio de hepatitis isquémica, habiéndose comprobado que la hepatitis hipóxica generalmente resulta de la asociación de hipotensión sistémica con aumento de la presión venosa central, mientras que la hipoxemia arterial no es un prerequisito para el desarrollo de esta complicación.
La patente hemodinámica de la hepatitis hipóxica que se produce en el contexto de un shock circulatorio es diferente. En este grupo, es constante la presencia de un estado de shock. Como es de esperar, la presión venosa central es baja y la PaO2 es normal. Por ello, y de acuerdo a una evaluación hemodinámica elemental, la hepatitis hipóxica relacionada con el shock séptico parece obviamente ligada con el estado de shock y por tanto producida exclusivamente por la isquemia hepática. Sin embargo, cuando se realiza una evaluación hemodinámica más profunda, se comprueba que lo que en realidad ocurre es que a pesar de un adecuado índice cardiaco y disponibilidad de oxígeno, la tensión de oxígeno intraparenquimatosa permanece baja, probablemente por una incapacidad celular para la extracción de oxígeno (hipoxia citopática). En resumen, la hepatitis isquémica que ocurre en el shock séptico es fundamentalmente la consecuencia de la incapacidad del hígado para extraer y utilizar el oxígeno.
CUADRO CLINICO
Los hallazgos clínicos se refieren fundamentalmente a la enfermedad de base. Un estado de shock se observa en el 55% de los pacientes, pero se reconocen signos de hipoporfusión sistémica en el 86% de los casos a la admisión. En alrededor del 50% de los casos se reconocen signos clínicos de insuficiencia cardiaca derecha, incluyendo hepatomegalia dolorosa, edema maleolar y reflujo hepatoyugular. Rara vez se reconocen signos evidentes de fallo cardiaco agudo. La ictericia es rara, afectando al 16-20% de los pacientes y se produce tardíamente en el curso de la hepatitis isquémica. Cierto grado de deterioro intelectual se observa en más de la mitad de los casos, pero dada la existencia de hipoxia generalizada, la misma puede afectar al cerebro independientemente de las otras connotaciones del proceso.

La hepatitis isquémica se caracteriza por un aumento marcado de las enzimas ASAT y ALAT, así como de la LDH, que rápidamente se recuperan en un periodo de 7 a 14 días. Las aminotransferasas y la lactodehidrogenasa están elevadas significativamente desde las mediciones iniciales. El aumento de las enzimas hepáticas no difiere significativamente en los cuatro grupos etiológicos analizados. Otro hallazgo bioquímico importante de la hepatitis hipóxica es la caída de la actividad de protrombina. El nadir de la actividad de protrombina se observa en los primeros días, recuperándose completamente alrededor de la semana. Un hallazgo común es una elevación moderada de la bilirrubina sérica, pero rara vez progresa a una ictericia franca. La creatinina sérica aumenta por encima de 2 mg/dL en el 60% de los casos. El deterioro renal es significativamente más severo en la hepatitis hipóxica en el contexto del shock hipovolémico. La hipoglucemia que ha sido destacada por algunos investigadores, rara vez se observa. Gitlin y col. ha destacada el hallazgo común de hiperglucemia.
En los casos en que se ha realizado una evaluación histopatológica de la lesión, lo que se observa habitualmente es una necrosis hepática centrilobular.
PRONOSTICO
La mortalidad en los pacientes con hepatitis isquémica oscila entre el 40 y el 70%, siendo la muerte consecuencia del shock o de la causa desencadenate de éste, mas que de la insuficiencia hepática. No se conoce en que medida la lesión hepática contribuye al desarrollo de falla pluriparenquimosa o muerte.
TRATAMIENTO
Los principios de tratamiento de los pacientes con hepatitis isquémica son la reversión del shock, el control del proceso de base y el soporte de otros órganos que puedan fallar debido al estado de hipoperfusión, tales como pulmones y riñones.
El empleo de dobutamina en pacientes con shock cardiogénico mejora la función cardiaca y reduce los marcadores de hepatitis isquémica, y es posible que otras medidas terapéuticas que mejoran la perfusión tisular puedan tener efectos beneficiosos similares.
ENFERMEDAD VENO-OCLUSIVA HEPATICA
La enfermedad veno-oclusiva hepática (VOD) es una entidad clínica característica descrita originalmente en Sudáfrica y relacionada con la ingesta de un alcaloide específico (monocrataline) contenido en un té de la región. En el año 1979 el VOD fue descrito en asociación con el trasplante de médula ósea (TMO), constituyéndose este método terapéutico en la causa más frecuente de VOD en el hemisferio oeste. El VOD también ha sido descrito en asociación con

agentes quimioterápicos tales como la actinomicina D, mitramicina, dacarbazina, citosina arabinosa y 6-tioguanina, utilizadas en dosis convencionales, y con el empleo por tiempo prolongado de la azatioprina. Más recientemente, el VOD se ha reconocido luego de la terapéutica de la leucemia mieloide aguda con el anticuerpo monoclonal anti-CD33 gemtuzumab oxogamicina (Mylotarg).
INCIDENCIA
El VOD es una complicación bien reconocida del TMO, tanto alogénico como autólogo, y se produce como consecuencia de la toxicidad de los regimenes ablativos utilizados. La incidencia de esta condición varía desde menos del 5% hasta más del 70% en diferentes informes, dependiendo del criterio diagnóstico utilizado, de la población estudiada (pediátrica o adulta) y de las diferentes terapéuticas ablativas utilizadas.
DEFINICION
El síndrome clínico de enfermedad veno-oclusiva hepática luego del TMO se caracteriza por la presencia de dolor y aumento del tamaño del hígado, retención de fluidos, ganancia de peso e ictericia. Comienza en forma característica antes del día 30 del trasplante, aunque se han descrito casos más tardíos.
ANATOMIA PATOLOGICA
El endotelio sinusoidal en el hígado está recubierto por numerosos pequeños poros y fenestraciones que crean una arquitectura microvascular única. La sangre aferente que entra al acino hepático a través de la arteria hepática o de la vena porta atraviesa estos sinusoides hepáticos y drena en las vénulas centrolobulillares. La injuria de las vénulas hepáticas representa el primer cambio histológico en el VOD. Este se caracteriza histológicamente por edema subendotelial, extravasación de glóbulos rojos, deposición de fibrina, y la expresión del factor VIII/von Willebrand dentro de la pared venular. A continuación se produce la dilatación de los sinusoides y la necrosis de los hepatocitos, seguido por una esclerosis de las paredes venulares e intensa deposición de colágeno en los sinusoides y vénulas, tanto dentro como fuera de la luz vascular. Este proceso conduce a una obliteración venular completa, extensa necrosis hepatocelular y reemplazo del tejido hepático normal por fibrosis extensa, simulando en un todo al proceso de cirrosis hepática.
El rol de las vías de coagulación en la fisiopatología del VOD es un área de controversia. Aunque generalmente se considera al VOD como una enfermedad vascular no trombótica del hígado, algunas evidencias exigen considerar la contribución del sistema hemostático en la fisiopatología del síndrome. Los datos más llamativos surgen de los informes sobre el tratamiento efectivo del VOD con el uso de agentes trombolíticos y de la obtención de cierto beneficio con el

empleo de heparina profiláctica para prevenir el mismo. Una serie de estudios han demostrado que los niveles de proteínas anticoagulantes tales como la proteína C, proteína S y antitrombina están considerablemente descendidos en pacientes con VOD en comparación con aquellos sin VOD. Se desconoce si estos cambios son secundarios a la enfermedad o si los mismos contribuyen a la oclusión trombótica de los sinusoides hepáticos.
FACTORES PREDISPONENTES
Han sido implicadas varias características de los pacientes en las etapas de pretrasplante y de trasplante en la patogénesis del VOD. El VOD ha sido observado en pacientes sometidos a trasplante, independientemente si se trata de alogénico o autólogo, si corresponde a células periféricas o de la médula, y del tipo de donante y del tipo de método ablativo (Tabla 4).
Los niveles elevados de drogas citotóxicas utilizadas en el TMO, tales como el busulfan o los metabolitos de la ciclofosfamida, se asocian con un aumento del riesgo de VOD. El VOD ocurre más frecuentemente en pacientes cuya área bajo la curva de concentración versus el tiempo (AUC) del busulfan es elevada. El riesgo de VOD puede ser reducido ajustando la dosis de busulfan en pacientes cuya AUC luego de la primera dosis es elevada. Otros estudios han confirmado la importancia de la ciclodfosfamida y sus metabolitos en la disfunción hepática que sigue al TMO.
Tabla 4.- Factores de riesgo para la enfermedad veno-oclusiva.Factores pretrasplante
Enfermedad hepática preexistentePresencia de metástasis hepáticasEdad avanzadaIrradiación previa del hígadoUtilización de vancomicina o aciclovir en el periodo pretrasplanteTrasplante previoTerapia previa con gentuzumab ozogamicinaHepatitis viral C?Disminución de proteína C, mutación del Factor V Leiden, Protrombina 20210?
Factores relacionados con el trasplanteAltas dosis en los regimenes ablativosTrasplante alogénico en comparación con autólogoEmpleo de busulfan, en particular en combinación con ciclofosfamidaIrradiación corporal total, en particular en combinación con ciclofosfamidaMetotrexate como parte de profilaxis de enfermedad injerto vs huéspedInfección por citomegalovirus?
CUADRO CLINICO
Las manifestaciones clínicas del VOD generalmente aparecen hacia el final de la primera semana o comienzos de la segunda que siguen al trasplante, y muchos pacientes que desarrollan esta

complicación lo hacen dentro de las primeras tres semanas que siguen al trasplante. Algunos autores han descrito una forma de comienzo tardío, hasta 50 días después del trasplante.
El primer signo en la mayoría de los pacientes es un aumento de peso asintomático, el cual se produce como consecuencia de la retención de agua y sal por el riñón. Este hallazgo es desapercibido en estos pacientes debido a que reciben distintas preparaciones intravenosas, y su ganancia de peso es adscripta a estas infusiones. Algunos días después, aparece una hiperbilirrubinemia directa aislada, que progresa en los días sucesivos. La presencia de niveles elevados de bilirrubina y un aumento rápido de la bilirrubina directa generalmente indica una enfermedad grave y de mal pronóstico, y se acompaña o es seguida por un aumento de la fosfatasa alcalina y de las transaminasas, que varían en grado de anormalidad.
El primer síntoma informado en pacientes con VOD y en muchos casos el único es un dolor en el cuadrante superior derecho del abdomen, que se hace cada vez más intenso y puede requerir analgesia profunda. El examen físico revela un hígado aumentado de tamaño y ascitis. La ascitis y la ganancia de peso tienden a ser refractarios al tratamiento con diuréticos. En la mitad de los casos aparecen manifestaciones de disfunción renal, y un 50% de estos pacientes requieren diálisis. Un hallazgo característico observado en muchos de estos pacientes es la trombocitopenia refractaria a la transfusión de plaquetas, aunque es infrecuente el sangrado significativo. La declinación progresiva de la función hepática puede conducir a un déficit de factores de coagulación y a una prolongación del tiempo de protrombina. A medida que la enfermedad progresa, algunos pacientes pueden desarrollar una encefalopatía y pueden entrar en coma. Muchos de estos pacientes presentan otras manifestaciones de fallo orgánico, tales como hemorragia alveolar difusa y neumonitis intersticial, en particular en el contexto de un trasplante alogénico.
DIAGNOSTICO
El diagnóstico diferencial del VOD durante el periodo postrasplante exige descartar una serie de afecciones, tales como las descriptas en la Tabla 5.
Tabla 5. Diagnóstico diferencial de la enfermedad veno-oclusiva hepática.Enfermedad de injerto contra huésped hepática agudaHepatotoxicidad inducida por ciclosporinaInfiltración micóticaHepatitis viral, incluyendo citomegalovirusColestasis relacionada con la sepsisHepatitis colestática inducida por drogas (fluconazol, itraconazol, trimetoprin)Colestasis relacionada con nutrición parenteralInfiltración tumoral del hígadoInsuficiencia cardiaca congestivaColitis neutropénica

El gold standard para el diagnóstico de VOD es el examen histológico del hígado. Sin embargo, debido al riesgo de realizar una biopsia hepática en pacientes con trombocitopenia generalmente refractaria a las trasfusiones de plaquetas, el diagnóstico se basa primariamente en los hallazgos clínicos. En caso de considerarse imprescindible la realización de una biopsia, la misma se realizará por vía trasyugular a través de un catéter percutáneo. La presencia de hiperbilirubinemia, ganancia de peso y signos y síntomas de congestión hepática forman la base del diagnóstico. En la Tabla 6 se indican los criterios diagnósticos propuestos por distintos grupos de investigadores.
Tabla 6. Criterios diagnósticos para enfermedad veno-oclusiva.Criterios de SeattleDesarrollo de al menos dos de los siguientes tres hallazgos clínicos antes del día 30 después del trasplanteIctericiaHepatomegalia con dolor en el cuadrante superior derechoAscitis y o ganancia inexplicable de pesoCriterios de BaltimoreDesarrollo de hiperbilirubinemia con bilirrubina sérica >2 mg/dL dentro de los 21 días ulteriores al trasplante y al menos dos de los siguientes signos y síntomas clínicosHepatomegalia que puede ser dolorosaGanancia de peso >5% del peso de baseAscitisCriterios de Seattle modificadosDesarrollo de al menos dos de los siguientes tres hallazgos clínicos antes del día 20 después del trasplanteHiperbilirubinemia con bilirrubina sérica >2 mg/dL Hepatomegalia con dolor en el cuadrante superior derechoGanancia de peso >5% del peso de base debido a la acumulación de fluidos
La ultrasonografía y la tomografía computada del abdomen son útiles para confirmar la hepatomegalia, presencia de ascitis y con los estudios de Doppler, determinar si existe una atenuación o reversión del flujo venoso o una trombosis de la vena porta. Tanto la ultrasonografia como la TAC son útiles para excluir el derrame pericárdico, pericarditis constrictiva, síndrome de Budd-Chiari y lesiones expansivas en el hígado. El Doppler es un método no invasivo y puede ser realizado a la cabecera del paciente, pero se debe tener en cuenta que un flujo venoso hepático pulsátil puede ser observado en otras causas de congestión hepática, y la reversión del flujo portal es un hallazgo tardío del VOD.
La biopsia hepática transvenosa y la medición del gradiente de presión venosa hepático enclavado constituyen el método de elección para el diagnóstico patológico del VOD. Un gradiente de más de 10 mm Hg se asocia con una especificidad del 91% y un valor predictivo positivo del 86% para el VOD, con una sensibilidad del 53%.

PRONOSTICO
En la mayoría de los pacientes (50-80%) existe una resolución gradual de los síntomas y signos en un periodo de dos a tres semanas luego del inicio de la enfermedad. La mortalidad total varía entre el 20 y el 50% en diferentes series. Se ha propuesto un sistema de clasificación de la severidad del VOD (Tabla 7) en base al grado de disfunción hepática, la necesidad de terapéutica y la evolución. Desgraciadamente, este modelo sólo brinda un análisis retrospectivo de la severidad y no es útil para establecer conductas terapéuticas.
Tabla 7. Clasificación de la severidad de la enfermedad veno-oclusiva.LeveLos pacientes no tienen efectos adversos por la enfermedad hepáticaLos pacientes no requieren tratamiento de la VODLa enfermedad es auto limitadaModeradaLos pacientes tienen efectos adversos por la enfermedad hepáticaLos pacientes requieren tratamiento de la VOD, incluyendo diuréticos o medicaciones analgésicasGraveSignos y síntomas de enfermedad veno-oclusiva que no se resuelven en el día 100Los pacientes mueren de complicaciones atribuibles directamente a la enfermedad veno-oclusiva
Varios factores pronósticos ayudan a identificar a los pacientes que pueden evolucionar mal. El grado de elevación de la bilirrubina y la velocidad de aumento parecen ser los dos predictores más importantes. Los pacientes con VOD severo desarrollan falla orgánica múltiple y habitualmente mueren por causas distintas de la falla hepática (Tabla 8). La falla renal es frecuente; el compromiso pulmonar, que en ocasiones requiere ventilación mecánica, también es común, como la insuficiencia cardiaca que requiere soporte inotrópico. La bacteriemia se desarrolla en un número considerable de pacientes y puede contribuir a la alta mortalidad observada. Los pacientes que requieren un soporte de múltiples órganos y sistemas tienen un mal pronóstico.
Tabla 8. Causas de muerte en la enfermedad veno-oclusiva severaFalla hepática atribuible en forma directa a la enfermedad veno-oclusivaFallo renal debido al síndrome hepatorenalFalla respiratoria debida a: enfermedad pulmonar veno-oclusiva, neumonitis intersticial, hemorragia pulmonarHemorragia gastrointestinalInsuficiencia cardiaca congestiva

PREVENCION
Una práctica establecida en la prevención del VOD ha sido el empleo de estudios farmacocinéticos para monitorizar los niveles de drogas con el intento de minimizar la lesión hepática. Este método se ha utilizado con éxito controlando los niveles de busulfan.
La administración profiláctica de ácido ursodeoxicólico, un ácido biliar hidrofílico soluble en agua, ha sido estudiada en una serie de estudios randomizados controlados, aunque un gran estudio de fase III del Nordic Bone Marrow Transplantation Group no demostró beneficios significativos.
En modelos experimentales se ha evaluado el rol del glutation, pero esto ha sido difícil de trasladar a pacientes debido a la dificultad en obtener niveles terapéuticos en humanos. Otras drogas evaluadas, todas sin resultado satisfactorio, han sido los corticoides, dosis bajas de heparina, concentrados de antitrombina, prostaglandina E1 (PGE1).
TERAPEUTICA
El tratamiento de sostén en pacientes con VOD establecido incluye el evitar drogas potencialmente hepatotóxicas (ciclosporina) y drogas nefrotóxicas (aminoglucósidos, anfotericina), limitar la ingesta de sodio, inducción de diuresis para eliminar la retención hídrica, analgesia para el dolor, paracentesis en caso de ascitis tensa que dificulte la función respiratoria, y corrección de la coagulopatía. La infección es un desafío clínico mayor en la VOD severa, dado que la falla hepática y el edema de la pared intestinal producen traslocación de organismos desde el intestino a la circulación como resultado de la hipertensión portal.
Basado en la observación histológica de microtrombosis y deposición de fibrina, así como una intensa actividad de factor VIII/von Willebrand en el órgano, se han desarrollado tratamientos destinados a promover la fibrinolisis. El empleo de tPA/heparina no ha sido recomendado en forma rutinaria, y en caso de utilizarse deberá ser hecho en la etapa inicial de la enfermedad. La administración de ATIII y de proteína C activada no ha demostrado ser efectiva en una serie de estudios recientes. La infusión de PGE1 también ha fracasado con este objeto.
El defibrotide es un oligonucleótido de PM 23kD, que se ha identificado como un agente que modula la lesión celular endotelial sin aumentar el sangrado y protege el endotelio sinusoidal sin comprometer los efectos antitumor de la terapéutica citotóxica. Una serie de estudios han demostrado la utilidad de su empleo en el VOD. La respuesta se hace evidente dentro de los primeros siete días, y la dosis activa parece ser de aproximadamente 25 mg/kg/día. Se ha sugerido que la intervención precoz es más efectiva.
BIBLIOGRAFIA

Anderson G., Johnson D.: Splanchnic resucitation: a strategy for preventing liver failure in sepsis. New Horizons 1:353-1993
Banks J., Foulis A.: Liver function in septic shock. J Clin Pathol 35:1249-1982Blendis L., Wong F.: Portopulmonary hypertension: an increasingly important complication of
cirrhosis. Gastroenterology 126:622-2003Bynun T., Boitnott J.: Ischemic hepatitis. Dig Dis Sci 24:129-1979Carrico C.: Multiple organ failure syndrome. Arch Surg 121:196-1986Castro M., Krowka M.: Hepatopulmonary syndrome: a pulmonary vascular complication of liver
disease. Clin Chest Med 17:35-1996Castro M., Krowka M., Schroeder D.: Frequency and clinical implications of increased pulmonary
artery pressures in liver transplant patients. Mayo Clin Proc 71:543-1996Chevalier P., Novelli L., Motamedi J.: Hepatopulmonary syndrome successfully treated with TIPS:
a three year follow-up. J Vasc Int Radiol 15:647-2004Chopra B., Eaton J., Grassi A.: Defibrotide for the treatment of hepatic veno-occlusive disease:
results of the European compassionate use study. Brit J Haemat 111:1122-2000Clemmesen O., Ott P., Larsen F.: Splanchnic metabolism in acute liver failure and sepsis. Curr
Opin Crit Care 10:152-2004Dhainaut J., Marin N., Mignon A.: Hepatic response to sepsis: interaction between coagulation and
inflammatory processes. Crit Care Med 29:(Supp.):S42-2001Dahn M.: Hepatic dysfunction in the critically ill and injured. Intensive Care World 11:9-1994Fallon M.: Mechanisms of pulmonary vascular complications of liver disease: hepatopulmonary
syndrome. J Clin Gastroenterol 39:(Supp.2):S138-2005Fuchs S., Bogomolski Y., Paltiel O.: Ischemic hepatitis: clinical and laboratory observations of 34
patients. J Clin Gastroent 26:183-1998Gibson P., Dudley F.: Ischemic hepatitis: clinical features, diagnosis and prognosis. Aust and New
Zealand J Med 14:822-1984Gimson A.: Hepatic dysfunction during bacterial sepsis. Intensive Care Med 13:162-1987Gitlin N., Serio K.: Ischemic hepatitis: widening horizons. Am J Gastroenterol 87:831-1992Goris R., Boekhorst T.: Multiple organ failure: generalized autodestructive inflammation. Arch
Surg 120:1109-1985Gotway M., Dotosn R., Dawn S.: Hypoxemia in a patient with end-stage liver disease. Clin Pulm
Med 12:61-2005Hussain S., Mekan S.: Platypnea-orthodeoxia: report of two cases and review of the literature.
South Med J 97:657-2004Henrion J., Schapira M., Luwaert R.: Hypoxic hepatitis: clinical and hemodynamic study in 142
consecutive cases. Medicine 82:392-2003Koksal D., Kacar S., Koksal A.: Evaluation of intrapulmonary vascular dilatations with high
resolution computed thorax tomography in patients with hepatopulmonary syndrome. J Clin Gastroenterol 40:77-2006
Krowka M., Mandell S., Ransay M.: Hepatopulmonary syndrome and portopulmonary hypertension: a report of the multicenter liver transplant database. Liver Transpl 10:174-2004
Krowka M.: The dilemma of portopulmonary hypertension. En Arroyo V., Navasa M., Forns X. (Edit.): Update in treatment of liver disease. Ars Medica, Barcelona 2005
Kumar S., DeLeve L., Kamath P.: Hepatic veno-occlusive disease after hematopoietic stem cell transplantation. Mayo Clin Proc 78:589-2003
LeGall J., Klar J., Lemeshow S.: The Logistic Organ Dysfunction System. JAMA 276:802-1996LeGall J., Klar J., Lemeshow S.: How to assess organ dysfunction in the intensive care unit?. The
Logistic Organ Dysfunction System. Sepsis 1:45-1997

Lima B., Franca A., Päzin-Filho A.: Frequency, clinical characteristics, and respiratory parameters of hepatopulmonary syndrome. Mayo Clin Proc 79:42-2004
Marik P., Gayowsky T., Starzl T.: The hepatoadrenal syndrome: a common yet unrecognized clinical condition. Crit Care Med 33:1254-2005
Marshall J., Cook D., Christou N.: Multiple Organ Dysfunction Score (MODS): a reliable descriptor of a complex clinical outcome. Crit Care Med 23:1638-1995
O´Callaghan D., Gaine S.: Hepatopulmonary syndromes: treatment of liver transplantation candidates. Curr Opin Organ Transplant 7:107-2002
Paston C., Suter P.: Hepatic hemodynamics and cell functions in human and experimental sepsis. Anesth Analg 89:344-1999
Richardson P., Guinan E.: The pathology, diagnosis, and treatment of hepatic veno-occlusive disease: current status and novel approaches. Brit J Haemat 107:485-1999
Rivers E., Nguyen B., Havstad S.: Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 345:1368-2001
Rodriguez Roisin R, Krowka M., Herve P.: Pulmonary-hepatic vascular disorder (PHD). Task Force Report. Eur Respir J 24:861-2004
Swanson K., Wiesner R., Krowka M.: Long-term survival in hepatopulmonary syndrome. Chest 122:(Suppl.4):S210-2002
Taille C., Cadranel J., Bellocq A.: Liver transplantation for hepatopulmonary syndrome : a ten-year experience in Paris, France. Transplantation 79:1482-2003
Vettukattil J.: Pathogenesis of pulmonary arteriovenous malformations: role of hepatopulmonary interactions. Heart 88:561-2002
Vincent J., Moreno R., Takala J.: The SOFA (Sepsis related organ failure assessment) score to describe organ dysfunction/failure. Intensive Care Med 22:707-1996
Wadleigh M., Ho V., Momtaz P.: Hepatic veno-occlusive disease: pathogenesis, diagnosis and treatment. Curr Opin Hematol 10:451-2003





























